

Abstract
Microbial systems have been synthetically engineered to deploy therapeutic payloads in vivo1,2. With emerging evidence that bacteria naturally home in on tumours3,4 and modulate antitumour immunity5,6, one promising application is the development of bacterial vectors as precision cancer vaccines2,7. Here we engineered probiotic Escherichia coli Nissle 1917 as an antitumour vaccination platform optimized for enhanced production and cytosolic delivery of neoepitope-containing peptide arrays, with increased susceptibility to blood clearance and phagocytosis. These features enhance both safety and immunogenicity, achieving a system that drives potent and specific T cell-mediated anticancer immunity that effectively controls or eliminates tumour growth and extends survival in advanced murine primary and metastatic solid tumours. We demonstrate that the elicited antitumour immune response involves recruitment and activation of dendritic cells, extensive priming and activation of neoantigen-specific CD4+ and CD8+ T cells, broader activation of both T and natural killer cells, and a reduction of tumour-infiltrating immunosuppressive myeloid and regulatory T and B cell populations. Taken together, this work leverages the advantages of living medicines to deliver arrays of tumour-specific neoantigen-derived epitopes within the optimal context to induce specific, effective and durable systemic antitumour immunity.
Subject terms: Cancer immunotherapy, Synthetic biology
Probiotic Escherichia coli Nissle 1917 is engineered as an antitumour vaccination platform optimized for enhanced production and cytosolic delivery of neoepitope-containing peptide arrays to safely induce specific, effective and durable systemic antitumour immunity.
Main
Bacteria support activation of both innate and adaptive immunity through their inherent foreignness and immunostimulatory properties8. These features, coupled with the ease to synthetically engineer bacteria for safe delivery of immunomodulatory compounds, make them ideal vectors to augment and direct antitumour immune responses1,2. Tumour neoantigens are attractive immunotherapeutic payloads for delivery; these antigenic species are not present in other tissues, pose minimal risk for inducing autoimmunity, and are theoretically excluded from central immunologic tolerance mechanisms9. So far, a variety of tumour neoantigen vaccines have demonstrated promising immunologic responses and survival benefit in clinical trials, although benefit remains limited to only a subset of patients10–12. In this regard, programming bacteria with genetic directives to release high levels of identified tumour neoantigens thereby provides a system to precisely instruct neoantigen targeting in situ. Here we describe new microbial immunotherapy vectors that stimulate effective and durable tumour antigen-specific immunity and inhibit immunosuppressive mechanisms that may otherwise limit traditional neoantigen vaccines13.
Engineering microbial cancer vaccines
To enable effective cancer vaccination, we developed an engineered bacterial system in probiotic Escherichia coli Nissle 1917 (EcN) to enhance expression, delivery and immune-targeting of arrays of tumour exonic mutation-derived epitopes highly expressed by tumour cells and predicted to bind major histocompatibility complex (MHC) class I and II (Fig. 1a). This system incorporates several key design elements that enhance therapeutic use: (1) optimization of synthetic neoantigen construct form with (2) removal of cryptic plasmids and deletion of Lon and OmpT proteases to increase neoantigen accumulation, (3) increased susceptibility to phagocytosis for enhanced uptake by antigen-presenting cells (APCs) and presentation of MHC class II-restricted antigens, (4) expression of listeriolysin O (LLO) to induce cytosolic entry for presentation of recombinant encoded neoantigens by MHC class I molecules and T helper 1 cell (TH1)-type immunity and (5) improved safety for systemic administration due to reduced survival in the blood and biofilm formation.
Fig. 1. Engineering live microbial tumour neoantigen vaccines.

a, Design of microbial tumour neoantigen vaccines, with a Circos plot of CT26 mutanome. b, Top, an optimized synthetic neoantigen construct schematic. Middle, relative immunoblot chemiluminescent intensity of neoantigen construct MHCIIa expressed from EcN versus derivative strains (n = 3 biological replicates per group). Bottom, a representative immunoblot of neoantigen construct MHCIIa expression in a designated strain. c, Percentage of GFP+ BMDMs after incubation with EcN or EcNcΔlon/ΔompT expressing constitutive GFP (n = 3 biological replicates per group, ****P < 0.0001, two-sided unpaired Student’s t-test); Lat. A, latrunculin A. d, Left, a representative image of EcN or EcNcΔlon/ΔompT spotted on an LB agar plate after incubation in human blood. Right, a microbial burden in colony-forming units (CFU) per ml (CFU ml−1) (n = 3 biological replicates per group, **P = 0.0039, two-sided unpaired Student’s t-test with Welch’s correction). Limit of detection (LOD) was 2 × 102 CFU ml−1. e, IL-12p70 quantification in culture supernatants of pulsed BMDCs (n = 3 biological replicates per group, **P = 0.0018, ****P < 0.0001, one-way ANOVA with Tukey’s multiple comparisons test). f, Naive OT-I T cells were incubated with pulsed BMDCs. Left, IFNγ quantification of supernatants of OT-I cultures (n = 3 biological replicates per group, ****P < 0.0001, one-way ANOVA with Tukey’s multiple comparisons test). Middle, IL-2 quantification of supernatants of OT-I cultures (n = 3 biological replicates per group, ****P < 0.0001, one-way ANOVA with Tukey’s multiple comparisons test). Right, a representative histogram of carboxyfluorescein succinimidyl ester (CFSE) dilution-stimulated OT-I T cells. b–f, Data are mean ± s.e.m. Gel source data are in Supplementary Figs. 1 and 2.
To assemble a repertoire of neoantigens, we conducted exome and transcriptome sequencing of subcutaneous CT26 tumours. Neoantigens were predicted from highly expressed tumour-specific mutations using established methods14,15, with selection criteria inclusive of putative neoantigens across a spectrum of MHC affinity16,17. Given the importance of both MHC class I and MHC class II binding epitopes in antitumour immunity15,18,19, we integrated a measure of wild-type-to-mutant MHC affinity ratio—termed agretopicity17,20—for both epitope types derived from a given mutation, to help estimate the ability of adaptive immunity to recognize a neoantigen. Predicted neoantigens were selected from the set of tumour-specific mutations satisfying all criteria, notably encompassing numerous recovered, previously validated CT26 neoantigens15 (Extended Data Fig. 1a).
Extended Data Fig. 1. Neoantigen prediction and synthetic construct design.

a, Percentage of predicted CT26 neoantigens containing mutant-epitope(s) with ≤500 nM MHC-I affinity (MHC-I), MHC-II affinity (MHC-II), both MHC-I and MHC-II affinity (Shared), or no epitope meeting affinity criteria (Neither). Previously validated neoantigens within the set are labeled. b, Upper: prototype neoantigen construct design, Lower: immunoblot of EcN expressing prototype neoantigen constructs. c, Upper: immunoblot of EcN expressing prototype neoantigen constructs with or without GS-linkers. Lower: ELISA quantification of neoantigen construct in soluble fraction with or without GS-linkers in DH5α (n = 3 biological replicates per group). NeoAgp = prototype neoantigen construct, G4S1 = 5-mer GS-linker, pTacLO− = pTac without Lac operator; pTacLO+ = with Lac operator. d, Upper: neoantigen construct design with GS-linkers, Lower-left: Relative immunoblot chemiluminescent intensity for prototype construct with or without interspersing glycine-serine linkers (n = 6 biological replicates per group). Lower-right: relative expression of prototype neoantigen construct with GS-linkers under selected promoters (n = 12 biologically independent samples). e, Upper: immunoblot of EcN expressing alternate prototype neoantigen constructs. Lower: ELISA quantification of alternative neoantigen construct in soluble fraction in DH5α (n = 3 biological replicates per group). NeomE1 = minimal epitope, Neo1 = 1 neoantigen LP in construct, Neo2 = 2 neoantigen LP in construct, G8S2 = 10-mer GS-linker, CsL = immunoprotease sensitive linker. f, Upper: Immunoblot of neoantigen constructs (NeoAgp, MHCIa, MHCIIa, MHCI/IIv), expressed in BL21 or EcN. Lower-left: relative immunoblot chemiluminescent intensity for neoantigen construct expression in EcN vs. BL21 (n = 4 biological replicates per group), Lower-right: relative immunoblot chemiluminescent intensity of predicted neoantigen constructs vs. prototype in BL21 (n = 3 biologically independent samples). c–f, Data are mean ± s.e.m. Gel source data in Supplementary Fig. 1.
We then sought to create a microbial system that could accommodate the production and delivery of diverse sets of neoantigens to lymphoid tissue and the tumour microenvironment (TME). For the purpose of assessing neoantigen production capacity, a prototype gene encoding a synthetic neoantigen construct (NeoAgp) was created by concatenating long peptides encompassing linked CD4+ and CD8+ T cell mutant epitopes—previously shown as an optimal form for stimulating cellular immunity21—derived from CT26 neoantigens (Extended Data Fig. 1b and Extended Data Table 1). The construct was cloned into a stabilized plasmid22 under constitutive expression and transformed into EcN; however, both immunoblot and enzyme-linked immunosorbent assay (ELISA) assessment showed low production of the prototype construct by EcN across several tested promoters (Extended Data Fig. 1c).
Extended Data Table 1.
CT26 peptides in neoantigen constructs

For each therapeutic construct 6–7 predicted neoantigens were included. All 3 groups (MHCIa, MHCIIa, MHCI/IIv) in combination represent nAg19. 5 antigens were included in the production optimization prototype construct (NeoAgp).
Given the dependency on antigen dosage for establishing an effective and immunodominant antigen-specific immune response23–25, we developed a system for improved recombinant neoantigen construct production. The incorporation of 5-mer glycine-serine linkers between neoantigen long peptides in the prototype increased expression roughly sixfold (Extended Data Fig. 1c,d). Conversely, expressing only minimal neoepitopes, decreasing the number of neoantigen long peptides in a construct or incorporating 10-mer glycine-serine or immunoprotease-sensitive linkers did not improve production (Extended Data Fig. 1e). To evaluate the capacity of constructs with 5-mer glycine-serine linkers to accommodate production of various neoantigens, and for eventual in vivo testing, we created three more constructs with unique neoantigens from the predicted set, selected on a spectrum of predicted affinity for MHC class I and MHC class II (MHCIa, MHCIIa, MHCI/IIv) (Extended Data Table 1). Neoantigens were grouped on the basis of high predicted affinity for MHC-I (MHCIa) or MHC-II (MHCIIa) or low–moderate affinity for MHC-I or MHC-II (MHCI/IIv). Prototype and new construct expression were evaluated in EcN versus BL21, a strain that harbours chromosomal deletions of the Lon (Δlon) and OmpT (ΔompT) proteases to facilitate recombinant protein production26. Unlike BL21, EcN also bears cryptic plasmids that can suppress the copy number of transformed recombinant plasmids27. Indeed, on average, BL21 produced tenfold higher levels of neoantigen construct relative to EcN (Extended Data Fig. 1f). Thus, to further enhance construct expression in EcN, we performed sequential synthetic modifications of the microbe. Removal of the EcN cryptic plasmids led to maintenance of roughly 30-fold higher levels of therapeutic plasmid DNA (EcNc) (Extended Data Fig. 2a), with successive deletion of the Lon protease (EcNcΔlon), OmpT protease (EcNcΔompT) or both proteases (EcNcΔlon/ΔompT) allowing up to 80-fold increased production of synthetic neoantigen constructs relative to the parental EcN strain (Fig. 1b and Extended Data Fig. 2b,c).
Extended Data Fig. 2. Evaluation of microbial tumor neoantigen vaccine functioning and immunologic activity in vitro.

a, Plasmid copy number in wildtype EcN or cryptic plasmid cured EcNc (n = 3 per group). b, Upper: relative immunoblot chemiluminescent intensity of synthetic neoantigen construct MHCI/IIv expression in wildtype EcN vs. derivative strains (n = 3 biological replicates), Lower: representative immunoblot of construct MHCI/IIv expressed in wildtype EcN and derivative strains. c, Upper: relative immunoblot chemiluminescent intensity of synthetic neoantigen construct MHCIa expression in wildtype EcN vs. derivative strains (n = 3 biological replicates), Lower: representative immunoblot of construct MHCIa expressed in wildtype EcN and derivative strains. d, Biofilm formation quantified for wildtype EcN, and derivative strains by crystal violet stain assay. (****P < 0.0001, ns = P > 0.05, One-way ANOVA with Tukey’s multiple comparison test, n = 9 biological replicates for EcN, 10 for EcNc WT, 12 for other groups). e,f, MFI of e, Left: H2kb-SIINFEKL complex and Right: MHC-II, or f, Left: CD80 and Right: PD-L1 for BMDM incubated with the indicated live microbial strain or media for 6 h (*P = 0.0231, *P = 0.0414, ***P = 0.0002, ***P = 0.0005, ****P < 0.0001, ns = P > 0.05, one-way ANOVA with Tukey’s multiple comparisons test, n = 4 biological replicates per group). g, Left: sheep red blood cells (RBCs) were incubated with lysate from EcNcΔlon/ΔompT with (LLO+) or without (LLO−) cytosolic LLO expression. Absorbance at 541 nm (n = 3 biological replicates per group). Right: percentage of live BMDM after incubation with indicated live microbial strain or control for 6 h (n = 4 biological replicates per group). h, Immunoblot depicting expression of neoantigen constructs MHCIa, MHCIIa, and MHCI/IIv in EcNcΔlon/ΔompT with (LLO+) or without (LLO−) co-expression of cytosolic LLO. i, Immunofluorescence microscopy analysis of BMDM co-incubated with Left: EcNcΔlon/ΔompT OVA or, Right: EcNcΔlon/ΔompT/LLO+ OVA. j, Naïve OT-II T cells were incubated with BMDC’s pulsed with the indicated condition. Left: IFN-γ quantification in supernatant of OT-II cultures (****P < 0.0001, one-way ANOVA with Tukey’s multiple comparisons test, n = 3 biological replicates per group), Middle: IL-2 quantification in supernatant of OT-II culture (****P < 0.0001, one-way ANOVA with Tukey’s multiple comparisons test, n = 3 biological replicates per group). Right: representative histogram depicting CFSE dilution of stimulated OT-II T cells. a–g,j, Data are mean ± s.e.m. Gel source data in Supplementary Figs. 1 and 2.
As the Lon protease has been connected to capsule and biofilm regulation28,29, and OmpT with the degradation of complement30, we tested the susceptibility of the engineered vector EcNcΔlon/ΔompT to phagocytosis and blood clearance, as well as for its proficiency in biofilm formation. Notably, EcNcΔlon/ΔompT was fourfold more susceptible to phagocytosis by bone marrow-derived macrophages (BMDMs) relative to EcN (Fig. 1c). Incubation in human blood further revealed a 1000-fold greater sensitivity to blood clearance for EcNcΔlon/ΔompT versus EcN (Fig. 1d). Moreover, EcNcΔlon/ΔompT was significantly attenuated in biofilm formation, a major mechanism of microbial resistance to immunity and antimicrobial agents in humans31 (Extended Data Fig. 2d).
As an antitumour vaccine, the microbial platform must also facilitate presentation of recombinant antigens and activation of APCs. To evaluate the system in this capacity, the model antigen ovalbumin (OVA) was expressed in the cytosol of EcNcΔlon/ΔompT using a strategy analogous to that used for synthetic neoantigen constructs. BMDMs pulsed with EcNcΔlon/ΔompT OVA, but not EcN-OVA, presented the H2Kb-SIINFEKL complex, indicating efficient processing and cross-presentation of recombinant antigens from EcNcΔlon/ΔompT (Extended Data Fig. 2e). Furthermore, pulsed BMDMs upregulated MHC class II and CD80, and downregulated PD-L1, demonstrating effective APC activation by EcNcΔlon/ΔompT expressing a recombinant antigen (Extended Data Fig. 2e,f).
To refine the immune program orchestrated by APCs, we reasoned that constitutive coexpression of LLO—a pH-dependent pore-forming protein derived from Listeria that permeabilizes the phagolysosomal membrane—would facilitate cytosolic delivery of encoded neoantigens for presentation to CD8+ T cells32, enhance IL-12 production33,34 and promote induction of TH1 immunity35. Of note, engineered microbes produced functional LLO, and LLO expression did not affect viability of APCs incubated with LLO-expressing strains (EcNcΔlon/ΔompT/LLO+) or the coexpression of neoantigen constructs (Extended Data Fig. 2g,h). Immunofluorescence microscopy of BMDMs co-incubated with either live EcNcΔlon/ΔompT OVA or EcNcΔlon/ΔompT/LLO+ OVA confirmed that LLO coexpression enabled recombinant antigen escape into the cytosol (Extended Data Fig. 2i). Bone marrow-derived dendritic cells (BMDCs) pulsed with live EcNcΔlon/ΔompT/LLO+ OVA secreted threefold higher levels of IL-12p70 compared to those pulsed with EcNcΔlon/ΔompT OVA (Fig. 1e), indicating greater TH1 instruction by APCs. Moreover, BMDCs pulsed with live EcNcΔlon/ΔompT/LLO+ OVA mediated superior activation of naive OT-I and OT-II T cells, with 2–2.5-fold increased secretion of interferon-γ (IFNγ) and interleukin-2 (IL-2) from both T cell types relative to EcNcΔlon/ΔompT OVA, and marked proliferation of both OT-I and OT-II T cells (Fig. 1f and Extended Data Fig. 2j). Conversely, BMDCs pulsed with wild-type EcN-OVA induced no measurable proliferation of either T cell type, 13–15-fold lower secretion of IL-2 and IFNγ from OT-I T cells, and no detectable cytokine secretion from OT-II T cells (Fig. 1f and Extended Data Fig. 2j). Taken together, these data suggest recombinant antigens expressed in EcNcΔlon/ΔompT/LLO+ lead to potent antigen-specific activation of both naive cytotoxic and helper T cells, with incorporation of LLO facilitating both enhanced presentation to CD8+ T cells and TH1-type immunity.
Overall, synthetic neoantigen construct optimization and genetic engineering achieved a microbial platform (EcNcΔlon/ΔompT/LLO+) capable of robust production across diverse sets of tumour neoantigens, which was attenuated in immune-resistance mechanisms, effectively taken up by and proficient in activating APCs, and able to drive potent activation of T cells specific for encoded recombinant antigens to support enhanced cellular immunity.
Treatment of colorectal cancer
To assess the in vivo efficacy of the engineered system, BALB/c mice bearing advanced CT26 tumours on a single hind flank received an intratumoural injection of EcN wild-type, EcNcΔlon/ΔompT or EcNcΔlon/ΔompT/LLO+ strains. These strains were tested either without any neoantigen plasmid (NC), expressing a single neoantigen construct (MHCIa, MHCIIa, MHCI/IIv), or as a combination of the three neoantigen construct-expressing strains in equal parts: a microbial antitumour vaccine delivering 19 total unique neoantigens (nAg19). Notably, no difference in tumour colonization efficiency was observed for EcNcΔlon/ΔompT strains compared to wild-type EcN (Extended Data Fig. 3a,b). Whereas intratumoural treatment with wild-type EcN expressing any neoantigen construct did not demonstrate therapeutic benefit (Extended Data Fig. 3c,d), a single intratumoural injection of EcNcΔlon/ΔompT/LLO+ nAg19 provided strong antitumour efficacy, with a complete response observed for three out of seven tumours and the combination nAg19 more effective than each construct alone (Fig. 2a and Extended Data Fig. 3e–g). Moreover, treatment with EcNcΔlon/ΔompT and EcNcΔlon/ΔompT/LLO+ strains was well-tolerated, with significantly attenuated body weight change compared to wild-type EcN, and no significant body weight differences compared to PBS treatment over the course of observation (Extended Data Fig. 4a). Direct comparisons of intratumoural treatment with EcNcΔlon/ΔompT/LLO+ nAg19 versus EcNcΔlon/ΔompT nAg19 showed that the inclusion of LLO significantly enhanced tumour control and extended survival (Fig. 2a and Extended Data Fig. 4b–d). Assessment of intratumoural IL-12p70 levels demonstrated that inclusion of LLO also significantly enhanced IL-12p70 levels in tumours, suggestive of enhanced TH1 instruction in vivo (Fig. 2b). Thus, the combination of all synthetic modifications (EcNcΔlon/ΔompT/LLO+ nAg19) synergized to produce a microbial antitumour vaccine with favourable toxicity profile and strong therapeutic effect in vivo.
Extended Data Fig. 3. Characterization of intratumoral treatment with microbial tumor neoantigen vaccines.

a–g, BALB/c mice with established hind-flank CT26 tumors were treated when average tumor volumes were 150–200mm3. a,b, Mice received a single intratumoral injection of EcN WT, EcNcΔlon/ΔompT, or EcNcΔlon/ΔompT/LLO+. a, Representative image of tumors colonized by microbes with a genome-integrated luminescence cassette. b, Average radiance of microbe colonized tumors in designated (n = 4 mice for EcN WT 96 h, 5 for all other groups). c, Mice received intratumoral injections of PBS or EcN WT. Tumor growth curves (n = 5 mice per group, ns = P > 0.05, two-way ANOVA with Tukey’s multiple comparisons test). d, Mice received intratumoral injections of EcN WT without therapeutic (NC), expressing construct MHCIa, MHCIIa, or MHCI/IIv, or EcN nAg19. Tumor growth curves (n = 5 mice per group, ns = P > 0.05, two-way ANOVA with Tukey’s multiple comparisons test). e,f, Mice received a single intratumoral injection of PBS, EcNcΔlon/ΔompT NC, EcNcΔlon/ΔompT nAg19, or EcNcΔlon/ΔompT/LLO+ expressing construct MHCIa, MHCIIa, or MHCI/IIv, or EcNcΔlon/ΔompT/LLO+ nAg19. e, Tumor growth curves (n = 5 mice for PBS, 7 for other groups). f, Tumor growth rate (n = 5 mice for PBS and EcNcΔlon/ΔompT NC, 7 for other groups) for designated groups. g, Individual tumor trajectories after intratumoral treatment with PBS or indicated microbial therapeutic. b–f, Data are mean ± s.e.m.
Fig. 2. Efficacy of microbial tumour neoantigen vaccines in primary and metastatic colorectal carcinoma.
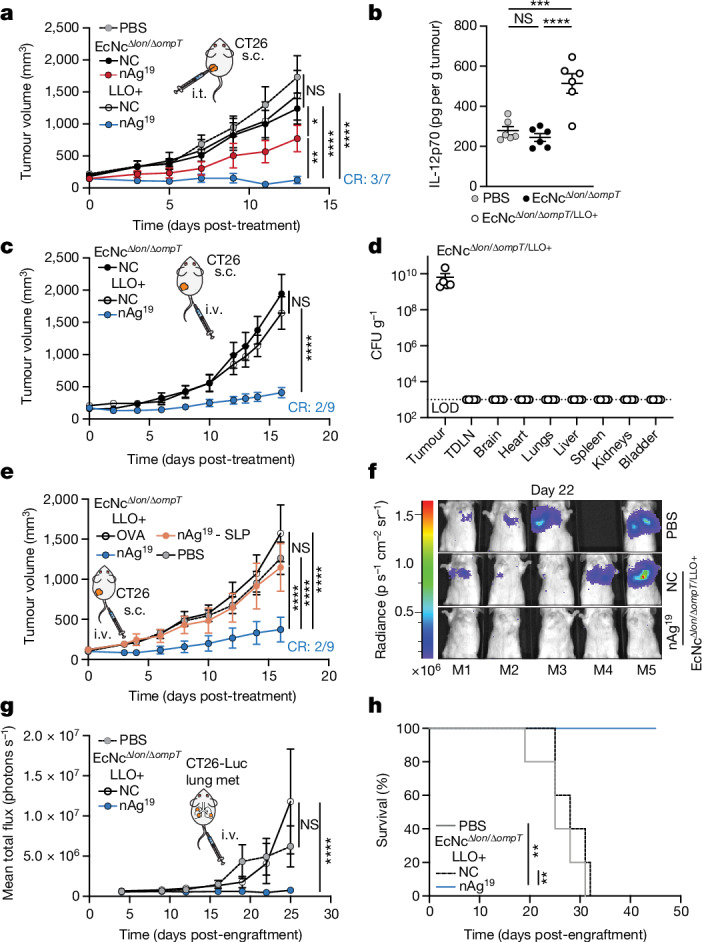
a–e, BALB/c mice with hind-flank CT26 tumours treated when average tumour volumes were 120–200 mm3. a, Intratumoural (i.t.) treatment on day 0. Tumour growth curves (n = 5 mice for PBS and EcNcΔlon/ΔompT NC, n = 7 for other groups, *P = 0.0469, **P = 0.0096, ****P < 0.0001; NS, P > 0.05, two-way ANOVA with Tukey’s multiple comparisons test). b, Intratumoural treatment. Intratumoural IL-12p70 levels (n = 6 mice per group, ***P = 0.0004, ****P < 0.0001; NS, P > 0.05, one-way ANOVA with Tukey’s multiple comparisons test). c, Intravenous (i.v.) treatment on day 0 and day 8. Tumour growth curves (n = 9 mice for nAg19, n = 8 for other groups, ****P < 0.0001; NS, P > 0.05, two-way ANOVA with Tukey’s multiple comparisons test). d, Mice (n = 5) received intravenous injection of EcNcΔlon/ΔompT/LLO+. Microbial tissue burden in CFU per gram (CFU g−1), LOD = 1 × 103 CFU g−1. e, Intravenous treatment of microbial therapies or PBS, or subcutaneous injections of SLP vaccine every 3–6 days. Tumour growth curves (n = 8 mice per group, ****P < 0.0001; NS, P > 0.05, two-way ANOVA with Tukey’s multiple comparisons test), f–h, BALB/c mice with 4 day established CT26-Luc lung metastases. Intravenous treatment every 3–5 days. f, Representative lung metastases luminescence. g, Mean total flux from lung metastases (n = 5 mice per group, ****P > 0.0001; NS, P > 0.05, two-way ANOVA with Dunnett’s multiple comparisons test). h, Kaplan–Meier survival curve for mice with CT26-Luc lung metastases (n = 5 mice per group, **P = 0.0017, **P = 0.0018, log-rank Mantel–Cox test). a–e,g, Data are mean ± s.e.m. s.c., subcutaneous. CR, complete response.
Extended Data Fig. 4. Comparative profile of intratumoral treatment with engineered microbial neoantigen vaccines.

a–d, BALB/c mice with established hind-flank CT26 tumors were treated when average tumor volumes were 150–200mm3. a,b, Mice received intratumoral injection of wildtype EcN, EcNcΔlon/ΔompT or EcNcΔlon/ΔompT/LLO+ either without neoantigen expression (NC) or the strain mixture nAg19 on day 0. a, Relative body weight of CT26 tumor-bearing mice (n = 5 mice for PBS, EcN NC, and EcN nAg19, 7 for other groups, **P = 0.0034, ****P < 0.0001, ns = P > 0.05, two-way ANOVA with Tukey’s multiple comparisons test). b–d, Mice received intratumoral injection on day 0 and 8 (n = 7 mice for EcNcΔlon/ΔompT NC and EcNcΔlon/ΔompT/LLO+ nAg19, 8 for other groups). b, Tumor growth curves (**P = 0.0020, ****P < 0.0001, two-way ANOVA with Tukey’s multiple comparisons test). c, Kaplan-Meier survival curves for CT26 tumor-bearing mice (**P = 0.0061, **P = 0.0076, Log-rank Mantel-Cox test). d, Individual tumor trajectories after intratumoral treatment with indicated microbial strain. e,f, BALB/c mice were implanted with CT26 tumors on both hind flanks. When average tumor volumes were 100–150mm3 mice received an intratumoral injection of PBS, EcNcΔlon/ΔompT/LLO+ (NC), or EcNcΔlon/ΔompT/LLO+ nAg19 into a single tumor. e, Tumor growth curves (n = 6 mice for EcNcΔlon/ΔompT/LLO+ nAg19, 5 for other groups, **P = 0.0014, ****P < 0.0001, two-way ANOVA with Tukey’s multiple comparisons test). f, CFU g−1 of tumor (n = 4 mice for EcNcΔlon/ΔompT/LLO+ nAg19, 5 for all other groups), LOD 1 ×103 CFU g−1. a,b,e,f, Data are mean ± s.e.m.
To evaluate the induction of systemic antitumour immunity after treatment with the microbial neoantigen vaccines, mice with established CT26 tumours on both hind flanks were treated with an injection of microbes into a single tumour. Whereas treatment with EcNcΔlon/ΔompT/LLO+ without neoantigen expression (NC) did not suppress tumour growth, a single injection of EcNcΔlon/ΔompT/LLO+ nAg19 induced tumour control and complete regression of two out of six treated and untreated tumours (Extended Data Fig. 4e). Microbial quantification from tumours 14 days after injection showed that microbes colonized treated tumours at high densities, with no bacteria able to be cultured from untreated tumours (Extended Data Fig. 4f). This demonstrates that the engineered neoantigen vaccines stimulate systemic antitumour immunity capable of eliminating distant tumours.
We next evaluated the efficacy of our microbial antitumour vaccination platform following intravenous administration, the preferred route of administration as to circumvent dependence on tumour accessibility. Similar to intratumoural treatment, intravenous administration of EcNcΔlon/ΔompT/LLO+ nAg19 to mice with advanced CT26 tumours provided potent antitumour efficacy and survival benefit with minimal body weight alteration (Fig. 2b and Extended Data Fig. 5a–c). After intravenous injection, the engineered microbes persisted at high density within tumours and were cleared rapidly from all other surveyed organs (Fig. 2c and Extended Data Fig. 5d).
Extended Data Fig. 5. Intravenous treatment with engineered microbial therapeutics in primary and metastatic solid tumors.

a–c, BALB/c mice with established hind-flank CT26 tumors were intravenously injected with indicated microbial therapeutics when average tumor volumes were 150–200 mm3 (n = 9 mice for EcNcΔlon/ΔompT/LLO+ nAg19, 8 for other groups). a, Kaplan-Meier survival curves for CT26-tumor-bearing mice treated with indicated therapeutic (****P < 0.0001, **P = 0.0021, Log-rank Mantel-Cox test). b, Relative body weight of mice after intravenous treatment with indicated microbial therapeutic. c, Individual tumor trajectories after intravenous treatment with the indicated microbial therapeutic. d, BALB/c mice with established hind-flank CT26 tumors were intravenously injected with EcNcΔlon/ΔompT or EcNcΔlon/ΔompT/LLO+. Upper: representative luminescent signature of tumors colonized with EcNcΔlon/ΔompT (LLO−) or EcNcΔlon/ΔompT/LLO+ (LLO + ), 48 h post-injection. Lower: average radiance of colonized tumors (n = 7 mice per group). e–f, BALB/c mice with established hind-flank CT26 tumors were intravenously injected with the indicated microbial therapeutics or PBS, or subcutaneously injected with nAg19-SLP, when average tumor volumes were 100–120 mm3. e, Kaplan-Meier survival curves for CT26 tumor-bearing mice treated with indicated therapeutic (n = 8 mice per group, **P = 0.0055, **P = 0.0018, *P = 0.0210, Log-rank Mantel-Cox test). f, Individual tumor trajectories (n = 8 mice per group) after intravenous treatment with the indicated therapeutic. g, Established hind-flank CT26 tumors were intravenously injected with the indicated microbial therapeutics or PBS, or subcutaneously injected with nAg19-SLP, when average tumor volume was 160 mm3. Tumor growth curves (n = 5 mice for PBS, 8 mice for other groups; ****P < 0.0001, two-way ANOVA with Tukey’s multiple comparisons test). h–k, BALB/c mice were injected intravenously with CT26-Luc cells. h, Upper-left: In vivo, or Upper-right: ex vivo bioluminescent images of mice (n = 3) lungs 96 h post-intravenous injection of CT26-Luc cells. Lower: Histology of metastatic lung foci 96-hours post-intravenous injection of CT26-Luc cells. i–k, Every 3–5 days mice (n = 5 per group) received intravenous injection of PBS, EcNcΔlon/ΔompT/LLO+ without therapeutic (NC), or EcNcΔlon/ΔompT/LLO+ nAg19 starting 4 days after CT26-Luc engraftment. i, Microbial tissue burden quantified as CFU g−1, LOD 4 ×102 CFU g−1 (n = 3 mice). j, Relative body weight of mice (n = 5 per group) after intravenous treatment with indicated therapeutic. k, Individual lung metastases luminescence trajectories (n = 5 mice per group). b,d,g,i,j Data are mean ± s.e.m.
Given the potent efficacy observed with intravenous treatment, we compared intravenous treatment with the microbial vaccination system to a standard antitumour vaccination strategy using synthetic long peptide (SLP) vaccination12. Mice bearing established hind-flank CT26 tumours received subcutaneous injections of SLP vaccination containing the 19 neoantigens (nAg19-SLP), or intravenous injections of PBS, EcNcΔlon/ΔompT/LLO+ expressing the strong irrelevant xenoantigen OVA or EcNcΔlon/ΔompT/LLO+ nAg19. Compared to SLP vaccination, treatment with EcNcΔlon/ΔompT/LLO+ nAg19 significantly reduced tumour growth and extended survival, with complete regression of two of eight tumours in the EcNcΔlon/ΔompT/LLO+ nAg19-treated group (Fig. 2e and Extended Data Fig. 5e,f). Increasing the dose of neoantigen SLPs and adjuvant did not enhance the efficacy of SLP vaccination in further comparison trials (Extended Data Fig. 5g).
Having observed robust efficacy in subcutaneous tumours, we then assessed therapeutic efficacy against established metastatic disease. CT26 carcinoma cells with genomically integrated firefly luciferase (CT26-Luc) were injected intravenously, which form rapidly progressive lung metastases traceable by bioluminescence imaging (Extended Data Fig. 5h). Intravenous treatment with PBS, EcNcΔlon/ΔompT/LLO+ without neoantigen expression (NC) or EcNcΔlon/ΔompT/LLO+ nAg19 was initiated 4 days after engraftment. We found that engineered microbes colonized metastases-bearing lungs and were not detectable in other tissues (Extended Data Fig. 5i). Microbially treated groups again demonstrated minimal body weight fluctuation, similarly to mice treated with PBS (Extended Data Fig. 5j). Treatment with EcNcΔlon/ΔompT/LLO+ nAg19 strongly restrained metastatic growth, with 100% of neoantigen therapeutic-treated mice surviving to 45 days after engraftment, whereas both control groups had completely succumbed to disease (Fig. 2f–h and Extended Data Fig. 5k). This demonstrates both safety and efficacy of intravenously administered EcNcΔlon/ΔompT/LLO+ nAg19 in the setting of aggressive, established metastatic disease.
Mechanisms of antitumour immunity
As the engineered microbial neoantigen vaccines are strong immunostimulants and persist within the TME, we reasoned that sustained intratumoural neoantigen production and reduced immunosuppression would facilitate enhanced activation of adaptive immunity to mediate the observed tumour control. To confirm in situ delivery of encoded neoantigens, we intravenously administered EcNcΔlon/ΔompT/LLO+ nAg19-His (wherein all three neoantigen constructs contain a C-terminal 6×His-tag) and performed immunoblots of tumour and tumour-draining lymph node (TDLN) lysates 2 days following treatment. We observed three His-tagged protein species corresponding to the three encoded neoantigen constructs (Fig. 3a). Immunophenotyping of TDLNs 2 days following intravenous treatment showed significantly higher frequencies of cDC2s in TDLNs of microbial vector-treated mice (Extended Data Fig. 6a). Enhanced frequencies of both CD80+ and CD86+ cDC1s and cDC2s (Fig. 3b,c) were also observed, demonstrating that intravenously delivered microbial vectors recruit and activate dendritic cells within the TDLN. Consistent with delivered neoantigens enhancing T cell activation, ex vivo restimulation of lymphocytes isolated from TDLNs at 8 days post-treatment with phorbol myristate acetate (PMA) and ionomycin showed increased production of IFNγ and TNF by conventional CD4+ (Foxp3−CD4+) and CD8+ T cells in mice treated with EcNcΔlon/ΔompT/LLO+ nAg19 versus those treated with PBS or control bacteria (EcNcΔlon/ΔompT) (Extended Data Fig. 6b,c).
Fig. 3. Microbial tumour neoantigen vaccines restructure the tumour immune microenvironment.
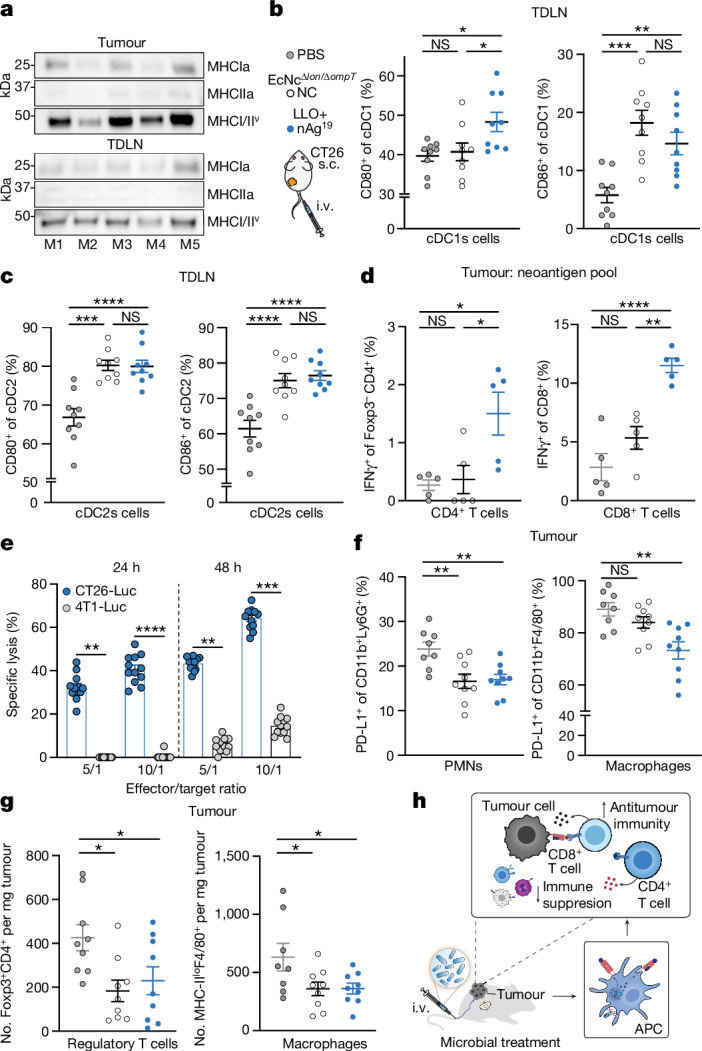
a, Immunoblot of tumours (n = 5) and TDLNs (n = 5). b, Left, the percentage CD80+ of cDC1s (*P = 0.0189, *P = 0.0415; NS, P > 0.05). Right, the percentage CD86+ of cDC1s (**P = 0.0061, ***P = 0.0002; NS, P > 0.05). c, Left, the percentage CD80+ of cDC2s (****P < 0.0001; NS, P > 0.05). Right, the percentage CD86+ of cDC2s (***P = 0.0001, ****P < 0.0001; NS, P > 0.05). d, Left, the frequency of IFNγ+Foxp3−CD4+ (n = 5 mice per group, *P = 0.0238, *P = 0.0147; NS, P > 0.05). Right, the frequency of IFNγ+CD8+ T cells (n = 5 mice per group, **P = 0.0015, ****P < 0.0001; NS, P > 0.05). e, CT26 TILs, specific lysis (n = 12 mice, **P = 0.0023, **P = 0.0027, ***P = 0.001, ****P < 0.0001; NS, P > 0.05, Kruskal–Wallis test with Dunn’s multiple comparisons test). f, Left, the percentage PD-L1+ of Ly6G+CD11b+ (n = 8 mice for PBS, n = 9 for other groups, **P = 0.0037, **P = 0.0059). Right, the percentage PD-L1+ of CD11b+F4/80+ (n = 8 mice for PBS, n = 9 for all other groups, **P = 0.0010; NS, P > 0.05). PMNs, polymorphonuclear cells. g, Left, the number of Foxp3+CD4+ T cells mg−1 (n = 9 mice per group *P = 0.0131, *P = 0.0241). Right, the number of MHC-IIlo+F4/80+ CD11b+ macrophages per mg (n = 8 mice for PBS, n = 9 for other groups, *P = 0.0385, *P = 0.0407). h, Immunologic mechanism. b–d,f,g, One-way ANOVA with a–d, Tukey’s, f, Dunnett’s or g, left, Holm–Šídák’s or right, Dunnett’s multiple comparisons test. b,c, n = 9 mice per group. b–g, Data are mean ± s.e.m. Gel source data are in Supplementary Fig. 2.
Extended Data Fig. 6. Modulation of anti-tumor immunity by engineered microbial tumor neoantigen vaccines.

a–f,h–i, BALB/c mice with established hind-flank CT26 tumors received intravenous injections of indicated therapeutic or control. a, 2 days or b–g, 8 days after treatment, tumors and TDLNs were extracted. a, Frequency of cDC2 in TDLNs (n = 9 mice per group, **P = 0.0042, **P = 0.0099, ns = P > 0.05, One-way ANOVA with Tukey’s multiple comparisons test). b–c, Lymphocytes from TDLNs were stimulated ex vivo with PMA and ionomycin in the presence of brefeldin A. b, Left: Frequency of IFN-γ+ Foxp3−CD4+ post-stimulation (n = 3 mice per group, *P = 0.0313, *P = 0.0246, ns = P > 0.05, One-way ANOVA with Tukey’s multiple comparisons test). Right: Frequency of TNF-α+ Foxp3−CD4+ T cells post-stimulation (n = 3 mice per group, *P = 0.0445, ns = P > 0.05, One-way ANOVA with Tukey’s multiple comparisons test). c, Left: Frequency of IFN-γ+ CD8+ post-stimulation (n = 3 mice per group, *P = 0.0257, ns = P > 0.05, One-way ANOVA with Tukey’s multiple comparisons test). Right: Frequency of TNF-α+ CD8+ T cells post-stimulation (n = 3 mice per group, **P = 0.0017, ***P = 0.0008, ns = P > 0.05, One-way ANOVA with Tukey’s multiple comparisons test). d, TILs were stimulated with individual 29-mer neoepitope-containing long peptides. Number IFN-γ spots (n = 8 mice per group, *P = 0.0439, *P = 0.0364, *P = 0.0281, *P = 0.0200, Kruskal-Wallis test with Dunn’s multiple comparisons test). Data are mean ± s.e.m. of background (medium control) subtracted responses. e,f, TILs were stimulated ex vivo with PMA and ionomycin in the presence of brefeldin A. e, Left: Frequency of IFN-γ+ Foxp3−CD4+ post-stimulation (n = 9 mice per group, *P = 0.0461, **P = 0.0014, ns = P > 0.05, one-way ANOVA with Tukey’s multiple comparisons test). Right: frequency of IFN-γ+ CD8+ T cells post-stimulation (n = 9 mice per group, *P = 0.0486, **P = 0.0040, ****P < 0.0001, one-way ANOVA with Tukey’s multiple comparisons test). f, Frequency of IFN-γ+ B220+ B cells post-stimulation (n = 9 mice per group, *P = 0.0351, **P = 0.0010, ns = P > 0.05, One-way ANOVA with Tukey’s multiple comparisons test). g, Left: Percentage Ki-67+ of Foxp3−CD4+ T cells in tumors (n = 9 mice per group, *P = 0.0183, ***P = 0.0008, one-way ANOVA with Dunnett’s multiple comparisons test). Right: Percentage Ki-67+ of CD8+ T cells in tumors (n = 9 mice per group, *P = 0.0453, ns = P > 0.05, One-way ANOVA with Dunnett’s multiple comparisons test). h, Naïve, tumor-free BALB/c mice were vaccinated intravenously with the designated treatment. CT26 was engrafted on a single hind-flank after the final vaccination. Tumor growth curves (n = 8 mice per group, ****P < 0.0001, two-way ANOVA with Šídák’s multiple comparisons test). i, BALB/c mice with established CT26 tumors were treated intravenously with EcNcΔlon/ΔompT/LLO+ nAg19 (as in Fig. 2e). Mice that had cleared tumors, and age-matched naïve mice, were subcutaneously rechallenged with CT26 tumor cells. Tumor growth curves (n = 5 for naïve mice, 6 for nAg19). j, Left: Frequency of FoxP3+CD4+ regulatory T cells in tumors (n = 9 mice per group, *P = 0.0491, **P = 0.0072, one-way ANOVA with Holm-Šídák’s multiple comparisons test), Right: Frequency of MHCIIloF4/80+CD11b+ macrophages in tumors (n = 8 mice for PBS, 9 for other groups, *P = 0.0173, **P = 0.0057, one-way ANOVA with Dunnett’s multiple comparisons test). k, Left: Percentage PD-L1+ of cDC1 in TDLN (n = 5 mice per group, **P = 0.0074, ns = P > 0.05, one-way ANOVA with Dunnett’s multiple comparisons test), Right: Percentage PD-L1+ of cDC2 in TDLN (n = 5 mice per group, *P = 0.0103, *P = 0.0244, one-way ANOVA with Dunnett’s multiple comparisons test). a–k, Data are mean ± s.e.m.
Next, to assess the ability for engineered neoantigen therapeutics to drive neoantigen-specific immunity, tumour-infiltrating lymphocytes (TILs) were isolated at 8 days post-treatment and restimulated ex vivo with a pool of synthetic peptides representing the 19 bacterially encoded tumour neoantigens. Flow-cytometric analysis showed increased frequencies of IFNγ secreting conventional Foxp3−CD4+ and CD8+ TILs, demonstrating that treatment with EcNcΔlon/ΔompT/LLO+ nAg19 enhanced encoded neoantigen-specific immunity (Fig. 3d). Analysis of TIL reactivity ex vivo demonstrated that several predicted MHC-I and MHC-II binding neoantigens from each neoantigen construct were targeted in EcNcΔlon/ΔompT/LLO+ nAg19-treated mice (Extended Data Fig. 6d). Furthermore, purified TILs from EcNcΔlon/ΔompT/LLO+ nAg19-treated mice were co-incubated with either CT26-Luc or irrelevant tumour cell targets (4T1-Luc) of the same MHC haplotype, demonstrating CT26 tumour cell-specific recognition and killing by generated TILs (Fig. 3e). Compared to peptide stimulation, restimulation with PMA and ionomycin showed even greater levels of IFNγ secreting Foxp3−CD4+ and CD8+ TILs in EcNcΔlon/ΔompT/LLO+ nAg19-treated tumours and IFNγ producing B220+ B cells36,37, suggestive of epitope spreading and expanded immune activation38 (Extended Data Fig. 6e,f). Furthermore, we observed increased frequencies of proliferating CD4+ and CD8+ tumour-infiltrating T cells in mice treated with EcNcΔlon/ΔompT/LLO+ nAg19 in comparison to treatment with EcNcΔlon/ΔompT or PBS (Extended Data Fig. 6g). To establish whether microbial neoantigen vaccine treatment generates tumour neoantigen-specific immune memory, we prophylactically vaccinated naive mice with EcNcΔlon/ΔompT/LLO+ OVA or nAg19 and grafted CT26 tumours post-vaccination. Tumour growth in mice prophylactically treated with EcNcΔlon/ΔompT/LLO+ nAg19 was significantly reduced compared to those treated with EcNcΔlon/ΔompT/LLO+ OVA (Extended Data Fig. 6h). As validation of immune memory formation in tumour-bearing hosts, no tumour growth was observed on rechallenge of mice that had cleared CT26 tumours after EcNcΔlon/ΔompT/LLO+ nAg19 treatment (Extended Data Fig. 6i). Together, these experiments indicate generation of tumour neoantigen-specific T cells that are proficient in tumour cell killing and establish durable immune memory.
Beyond induction of tumour antigen-specific T cell responses, treatment with EcNcΔlon/ΔompT/LLO+ nAg19 resulted in reduced frequencies of tumour-resident immunosuppressive PD-L1+Ly6G+ polymorphonuclear cells and PD-L1+F4/80+ macrophages39,40 (Fig. 3f). Bacteria-treated groups further showed reduced numbers and frequencies of Foxp3+CD4+ regulatory T cells and MHC-IIloF4/80+ tumour-associated macrophages (Fig. 3g and Extended Data Fig. 6j), two cell populations known for their roles in inhibiting antitumour immunity41,42. Moreover, within TDLNs of the neoantigen therapeutic-treated group, myeloid immunophenotyping showed a reduction of PD-L1 on cDC1s and cDC2s populations (Extended Data Fig. 6k), which has been shown to facilitate antitumour immunity43. In summary, intravenously delivered microbial neoantigen therapeutics sustain neoantigen production and availability in lymphoid tissue in vivo, recruit and activate dendritic cells, stimulate both neoantigen-specific and broad adaptive immunity, and reduce immunosuppression within the TME, shaping a more effective environment for productive antitumour immunity (Fig. 3h).
Treatment of melanoma
Neoantigens are generally unique to the individual tumour9, thus vaccination platforms must be able to flexibly incorporate and deliver diverse sets of neoantigens on the basis of the unique mutations present in a particular tumour. To evaluate the suitability of our engineered microbial platform in this regard, we performed paired exome and transcriptome sequencing on a second, more aggressive tumour cell type (B16F10 melanoma) grown orthotopically in C57BL/6 mice and designed tumour-specific therapeutics (Fig. 4a). We applied an equivalent neoantigen prediction algorithm as performed for CT26 and identified numerous putative B16F10-specific neoantigens, including many that had previously been validated by others15 (Extended Data Fig. 7a). A set of seven constructs were devised from neoantigens of varying imputed MHC-I and MHC-II affinities, with each construct containing six unique predicted neoantigens (Extended Data Table 2) and confirmed to be robustly expressed by EcNcΔlon/ΔompT/LLO+ (Fig. 4a and Extended Data Fig. 7b).
Fig. 4. Microbial antitumour vaccine efficacy in orthotopic melanoma.

a, Melanoma therapeutic design. Left, a Circos plot, mutanome B16F10. Right, an immunoblot with neoantigen constructs. b–e,g–i, C57BL/6 mice with orthotopic B16F10 tumours treated starting 9 days post-engraftment. b, Intratumoural treatment every 3–5 days. Tumour growth curves (n = 7 mice per group, ****P < 0.0001; NS, P > 0.05). c, Intravenous treatment every 3–5 days. Tumour growth curves (****P < 0.0001; NS, P > 0.05). d, Kaplan–Meier survival curves, mice from c (***P = 0.0001, log-rank Mantel–Cox test). e, Intravenous treatment, EcNcΔlon/ΔompT/LLO+ nAg42 (n = 6 mice). Microbial CFU g−1, LOD = 1 × 103 CFU g−1. f, Purified splenic T cells, specific lysis B16F10-Luc (n = 5 mice per group, ***P = 0.001, ****P < 0.0001; NS, P > 0.05). g, Intravenous treatment every 3–5 days. Tumour growth curves (n = 6 mice for OVA, n = 7 for isotype, n = 8 for other groups, **P = 0.0082, ****P < 0.0001; NS, P > 0.05). h,i, Intravenous treatment on days 9 and 12. h, Left, the number of CD103+XCR1+ cDC1 per mg (*P = 0.0103, **P = 0.0030; NS, P > 0.05). Right, the number of CD301b+ cDC2 per mg (**P = 0.0038, **P = 0.0064; NS, P > 0.05). i, Left, the number of Foxp3−CD4+ T cells per mg (**P = 0.0015, ***P = 0.0008; NS, P > 0.05). Right, the number of CD8+ T cells mg−1 (**P = 0.0022, **P = 0.0047; NS, P > 0.05). b,c,g, two-way ANOVA, or f,h,i, one-way ANOVA, with b, Šídák’s or c,f–i, Tukey’s multiple comparisons test. h,i, n = 8 mice for nAg42, n = 7 for other groups. c,d, n = 5 mice for PBS, n = 7 for other groups. b,c,e–i, Data are mean ± s.e.m. Gel source data are in Supplementary Fig. 2.
Extended Data Fig. 7. Assessment of engineered microbial neoantigen therapeutics in B16F10 melanoma.

a, Percentage of predicted B16F10 neoantigens containing mutant-epitope(s) with ≤500 nM MHC-I affinity (MHC-I), MHC-II affinity (MHC-II), both MHC-I and MHC-II affinity (Shared), or no epitope meeting affinity criteria (Neither). Previously validated neoantigens within the set are labeled. b, Immunoblot of B16F10 neoantigen construct expression in EcNcΔlon/ΔompT/LLO+. c–j, C57BL/6 mice with established hind-flank B16F10 melanoma tumors were treated 9 days after tumor engraftment. c–e, Every 3–5 days, mice received an intravenous injection of PBS, EcNcΔlon/ΔompT/LLO+ OVA, or the 7-strain combination EcNcΔlon/ΔompT/LLO+ nAg42. c, Individual tumor trajectories after intravenous treatment with indicated therapeutic. d, Relative body weight of B16F10-tumor bearing mice (n = 5 mice for PBS, 7 for other groups, ns = P > 0.05, two-way ANOVA with Tukey’s multiple comparisons test). e, Individual tumor trajectories after intravenous treatment with indicated therapeutic and intraperitoneal treatment with indicated monoclonal antibody. f–j, On day 9 and 12 post-engraftment, B16F10 tumor-bearing mice received an intravenous injection of PBS, EcNcΔlon/ΔompT/LLO+ OVA, or EcNcΔlon/ΔompT/LLO+ nAg42. Flowcytometric analysis of TILs was performed 8 days after treatment initiation (n = 8 mice for nAg42, 7 for other groups). f, Left: Frequency of CD103+XCR1+ cDC1 in tumors (*P = 0.0132, ns = P > 0.05, one-way ANOVA with Tukey’s multiple comparisons test). Right: Frequency of CD301b+ cDC2 in tumors (*P = 0.0162, ns = P > 0.05, one-way ANOVA with Tukey’s multiple comparisons test). g, Frequency of Foxp3−CD4+ T cells in tumors (***P = 0.0001, ****P < 0.0001, ns = P > 0.05, one-way ANOVA with Tukey’s multiple comparisons test). h, Frequency of CD8+ cytotoxic T cells in tumors (**P = 0.0097, ***P = 0.0002, ns = P > 0.05, one-way ANOVA with Tukey’s multiple comparisons test). i, Left: Number of NK1.1+ NK cells per mg tumor (*P = 0.0243, *P = 0.0224, ns = P > 0.05, one-way ANOVA with Tukey’s multiple comparisons test). Right: Frequency of NK1.1+ NK cells in tumors (*P = 0.0189, *P = 0.0389, ns = P > 0.05, one-way ANOVA with Tukey’s multiple comparisons test). j, Left: Number of MHCII+CD64+Ly6c+ monocytes per mg tumor (**P = 0.0041, **P = 0.0073, ns = P > 0.05, one-way ANOVA with Tukey’s multiple comparisons test). Right: Frequency of MHCII+CD64+Ly6c+ monocytes in tumors (*P = 0.0230, *P = 0.0495, ns = P > 0.05, one-way ANOVA with Tukey’s multiple comparisons test). d,f–j, Data are mean ± s.e.m. Gel source data in Supplementary Fig. 2.
Extended Data Table 2.
B16F10 peptides in neoantigen constructs

In each construct, 6 predicted neoantigens were included. All 7 groups in combination represent nAg42.
We then sought to test the antitumour efficacy of our therapeutics against advanced B16F10 tumours. When established orthotopic tumours were injected with microbial therapeutics intratumourally, tumours grew progressively after treatment with EcNcΔlon/ΔompT/LLO+ OVA, whereas treatment with the equal-parts combination of all seven construct-expressing strains—encompassing 42 unique B16F10 neoantigens (nAg42)—significantly repressed growth over the same time course (Fig. 4b). Similarly, intravenous treatment with EcNcΔlon/ΔompT/LLO+ nAg42 potently restrained orthotopic tumour growth, with 72% of nAg42-treated mice alive 50 days post-tumour engraftment, whereas all control group mice succumbed to malignancy by day 24 or 30 (Fig. 4c,d and Extended Data Fig. 7c). Treatment with intravenous microbial vaccines again induced no significant body weight change compared to PBS-treated mice (Extended Data Fig. 7d).
To evaluate the tissue biodistribution of the microbial neoantigen vaccines after systemic administration in this setting, we surveyed organs after intravenous injection of EcNcΔlon/ΔompT/LLO+ nAg42. As we observed for BALB/c mice with CT26 tumours, live microbial vectors specifically colonized the B16F10 tumour at high density without detectable presence in any other organs examined (Fig. 4e). To confirm that the microbial B16F10 neoantigen vaccine generated T cells capable of direct tumour cell killing, we treated tumour-free C57BL/6 mice intravenously with microbial therapeutics and co-incubated purified splenic T cells with B16F10 tumour cells in vitro. Indeed, T cells from mice treated intravenously with EcNcΔlon/ΔompT/LLO+ nAg42 but not EcNcΔlon/ΔompT/LLO+ OVA demonstrated enhanced killing of B16F10 tumour cells (Fig. 4f). These data verify tumour-specific colonization and antigen-specific T cell induction by microbial neoantigen vaccines in B16F10 melanoma. To assess the dependency of antitumour efficacy on CD4+ and CD8+ T cells in vivo, we depleted either CD4+ or CD8+ T cells from mice treated intravenously with EcNcΔlon/ΔompT/LLO+ nAg42. We found that depletion of either CD4+ or CD8+ T cells ablated therapeutic efficacy, indicating that both conventional CD4+ and cytotoxic CD8+ T cells are required for productive antitumour immunity in vivo (Fig. 4g and Extended Data Fig. 7e).
Immunity and metastases in melanoma
To characterize the immunologic changes associated with antitumour efficacy in this tumour model, we performed immunophenotyping of orthotopic B16F10 tumours 8 days post-intravenous microbial treatment. Tumours treated intravenously with EcNcΔlon/ΔompT/LLO+ nAg42 had significantly higher numbers and frequencies of cDC1s and cDC2s, conventional CD4+ and cytotoxic CD8+ T cells, natural killer (NK) cells and inflammatory monocytes (Fig. 4h,i and Extended Data Fig. 7f–j).
Analyses of the intratumoural lymphoid compartment showed enhanced expression of CD69 on Foxp3−CD4+ and CD8+ TILs, and significantly increased frequencies of IFNγ secreting conventional Foxp3−CD4+ and cytotoxic CD8+ TILs after restimulation with PMA and ionomycin in EcNcΔlon/ΔompT/LLO+ nAg42-treated tumours, indicating enhanced T cell activation and effector cytokine production within the TME (Fig. 5a and Extended Data Fig. 8a). Tumour-infiltrating Foxp3−CD4+ and CD8+ T cells and NK cells also expressed significantly higher levels of Granzyme-B after EcNcΔlon/ΔompT/LLO+ nAg42 treatment, suggestive of amplified cytolytic function (Fig. 5b,c). Consistent with enduring activity of antitumour immunity, we also observed higher levels of proliferating tumour-infiltrating CD4+ and CD8+ T cells and NK cells as assessed by Ki-67 staining (Extended Data Fig. 8b).
Fig. 5. Microbial neoantigen vectors restructure the tumour immune microenvironment and suppress established metastatic melanoma.

a–e, Mice with orthotopic B16F10 received an intravenous treatment on day 9 and 12 post-engraftment. a, Left, experimental schematic. Middle, the frequency of IFNγ+Foxp3−CD4+ post-stimulation (*P = 0.0335, **P = 0.0040; NS, P > 0.05). Right, the frequency of IFNγ+CD8+ T cells (****P < 0.0001; NS, P > 0.05). b, Left, the frequency of Granzyme-B+Foxp3−CD4+ (**P = 0.0024, **P = 0.0041; NS, P > 0.05). Right, the frequency of Granzyme-B+ CD8+ T cells (*P = 0.0495, **P = 0.0014; NS, P > 0.05). c, Frequency of Granzyme-B+NK1.1+ NK cells (****P < 0.0001; NS, P > 0.05). d, Left, the median fluorescence intensity (MFI) of TIM-1 on CD19+ B cells (*P = 0.0457, **P = 0.0029, ****P < 0.0001). Right, the frequency of TIM-1+CD19+ B cells of CD45+ cells (*P = 0.0442; NS, P > 0.05). e, Left, the frequency of Foxp3+CD4+ T cells of CD4+ cells (**P = 0.0035, **P = 0.0038). Right, the frequency of MHCII−Ly6c+ MDSCs of CD45+ cells (*P = 0.0440; NS, P > 0.05). f–h, C57BL/6 mice (n = 5 mice per group) with 2-day established B16F10-Luc systemic metastases. Intravenous treatment every 3–5 days. f, Representative systemic metastases luminescence. g, Mean total flux from systemic metastases (***P > 0.0003; NS, P > 0.05, two-way ANOVA with Dunnett’s multiple comparisons test). h, Kaplan–Meier survival curve for mice with B16F10-Luc systemic metastases (**P = 0.0015, log-rank Mantel–Cox test). a–e, One-way ANOVA with a–c, Tukey’s, d, left, Tukey’s or right, Dunnett’s and e, Dunnett’s multiple comparisons test. a–e, n = 8 mice for nAg42, n = 7 for other groups. a–e,g, Data are mean ± s.e.m.
Extended Data Fig. 8. Mechanisms of anti-tumor immunity and metastases suppression in melanoma.

a–e, On days 9 and 12 post-engraftment, B16F10 tumor-bearing mice received intravenous injections of PBS, EcNcΔlon/ΔompT/LLO+ OVA, or EcNcΔlon/ΔompT/LLO+ nAg42. Flow cytometric analysis was performed 8 days after treatment initiation (n = 8 mice for nAg42, 7 for other groups). a, Left: Percentage CD69+ of Foxp3−CD4+ T cells in tumors (*P = 0.0228, **P = 0.0092, ns = P > 0.05, one-way ANOVA with Tukey’s multiple comparisons test). Right: Percentage CD69+ of CD8+ T cells in tumors (**P = 0.0021, ****P < 0.0001, ns = P > 0.05, one-way ANOVA with Tukey’s multiple comparisons test). b, Left: Percentage Ki-67+ of Foxp3−CD4+ T cells in tumors (*P = 0.0188, **P = 0.0020, ns = P > 0.05, one-way ANOVA with Tukey’s multiple comparisons test). Middle: Percentage Ki-67+ of CD8+ T cells in tumors (**P = 0.0048, **P = 0.0086, ns = P > 0.05, one-way ANOVA with Tukey’s multiple comparisons test). Right: Percentage Ki-67+ of NK1.1+ NK cells in tumors (*P = 0.0366, **P = 0.0070, ****P < 0.0001, ns = P > 0.05, one-way ANOVA with Tukey’s multiple comparisons test). c, Left: Representative histogram of TIM-1 expression on CD19+ B cells. Right: Percentage Ki-67+ of CD19+B cells in tumors (**P = 0.0015, ****P < 0.0001, ns = P > 0.05, one-way ANOVA with Tukey’s multiple comparisons test). d, Frequency of MHC-IIloF4/80+ macrophages in tumors (*P = 0.0130, ns = P > 0.05, one-way ANOVA with Dunnett’s multiple comparisons test). e, Left: MHC-II MFI of CD64+Ly6c+ monocytes in tumors (*P = 0.0171, **P = 0.0041, ns = P > 0.05, one-way ANOVA with Tukey’s multiple comparisons test). Right: MHC-II MFI of CD301b+ cDC2 in tumors (**P = 0.0090, ns = P > 0.05, one-way ANOVA with Tukey’s multiple comparisons test). f–i, C57BL/6 mice were injected intravenously with B16F10-Luc cells. f, Upper-left: In vivo, or Upper-right: ex vivo bioluminescent images of mice (n = 3) lungs 48-hours post-intravenous injection of B16F10-Luc cells. Lower: Histology of metastatic lung foci 48-hours post-intravenous injection of B16F10-Luc cells. g–i, Mice received intravenous injection of either PBS, EcNcΔlon/ΔompT/LLO+ OVA or nAg42 every 3–5 days starting 2 days post intravenous injection of B16F10-Luc cells. g, Images of systemic metastases luminescence in each mouse in all groups over treatment course. h, Individual systemic metastases luminescence trajectories (n = 5 mice per group). i, Relative body weight of mice (n = 5 per group) after intravenous treatment with indicated therapeutic. a–e,i, Data are mean ± s.e.m.
In addition to the enhanced activation of tumour-infiltrating T and NK cells, treatment with EcNcΔlon/ΔompT/LLO+ nAg42 significantly reduced TIM-1 expression by tumour-infiltrating CD19+ B cells and thus the frequency of regulatory TIM-1+ B cells—an important immunosuppressive cell population in the B16F10 model44—and increased B cell proliferation (Fig. 5d and Extended Data Fig. 8c). Moreover, EcNcΔlon/ΔompT/LLO+ nAg42 vaccination reduced the frequency of immunosuppressive Foxp3+ regulatory T cells, myeloid-derived suppressor cells (MDSCs) and MHC-IIlo macrophages within tumours (Fig. 5e and Extended Data Fig. 8d). Infiltrating monocytes and dendritic cells in EcNcΔlon/ΔompT/LLO+ nAg42-treated tumours showed increased expression of MHC-II (Extended Data Fig. 8e), suggestive of enhanced antigen presentation capacity. Overall, these data demonstrate that intravenous microbial tumour neoantigen vaccination mediates immunologic restructuring within the melanoma TME, recruiting APCs and activating NK cells, and CD4+ and CD8+ T cells while diminishing immunosuppressive cell populations.
Given the robust antitumour efficacy induced by intravenous vaccination in orthotopic B16F10, we investigated the efficacy of EcNcΔlon/ΔompT/LLO+ nAg42 in established, systemic B16F10-Luc metastases (Extended Data Fig. 8f). Whereas systemic metastases rapidly progressed in PBS or EcNcΔlon/ΔompT/LLO+ OVA treated mice, EcNcΔlon/ΔompT/LLO+ nAg42 strongly inhibited metastatic growth (Fig. 5f,g and Extended Data Fig. 8g,h). Treatment with EcNcΔlon/ΔompT/LLO+ nAg42 significantly extended survival, with 60% of mice surviving to 55 days with no detectable metastases, whereas all control treated mice had died by day 27 (Fig. 5h). Again, treatment was well tolerated, with no significant weight change relative to PBS (Extended Data Fig. 8i). These data demonstrate that the microbial tumour neoantigen vaccination system stimulates productive antitumour immunity in vivo after intravenous administration in established, systemic metastatic melanoma.
Discussion
Through microbial engineering, we couple the tumour-homing and immunostimulatory nature of bacteria with precise instructions for coordinated adaptive immunity towards tumour neoantigens, achieving a platform capable of mediating control and eradication of advanced solid tumours.
Bioinformatic-based identification of neoantigens that are both immunogenic and able to generate T cells capable of tumour cell killing remains a challenge. Predicted MHC-binding affinity is a frequently used criterion in neoantigen identification, as analyses of MHC-I binding epitopes from viruses showed that immunogenic epitopes usually possess less than 500 nM affinity, but most show less than 50–200 nM affinity45. Whereas many immunogenic tumour neoantigens similarly show strong MHC-binding affinity (less than 50–500 nM)12,20, only a small fraction of predicted strong binding tumour neoepitopes are immunogenic20, and studies have revealed presentation, immunogenicity and TIL reactivity for neoantigens with low binding affinity (greater than 500 nM)17,46,47. A detailed analysis of MHC-I binding neoepitopes from human tumours showed that of the 10.5% of neoepitopes with more than 500 nM affinity, 50% contained cysteine residues, suggesting that low-affinity neoantigens may possess distinct characteristics48. Further criteria have been proposed to aid neoantigen identification, including differential binding affinity of the neoepitope versus corresponding wild-type epitope17,20, variant allele fraction of the mutation14 and expression level of the mutation-containing gene15; however, no standardized prediction algorithm has been identified20. Although we recognize the neoantigen prediction methods we use may have limitations, in this study, we used a combination of prediction criteria and primarily selected high-affinity neoantigens, with the incorporation of some low-affinity neoantigens.
We found that antigen sets encompassing both predicted MHC-I and MHC-II binding neoantigens mediated antitumour efficacy. Indeed, enhanced frequencies of neoantigen-specific CD4+ and CD8+ T cells were observed, MHC-I and MHC-II binding neoantigens were targeted by TILs in immunized mice, and both CD4+ and CD8+ T cells were required for efficacy of the microbial vaccination system in vivo. This agrees with the critical role of both CD4+ and CD8+ T cells in effective antitumour immunity, and the expanding set of verified MHC-I- and MHC-II-dependent neoantigens recognized across tumours12,15,18,46,47. Together, these results support the targeting of both MHC-I and MHC-II binding tumour neoantigens in antitumour immunotherapies.
We show that across distinct tumour models and genetic backgrounds, the antitumour effect of vaccination is accompanied by broad modulation of the immune compartment within the TME. The coordinated regulation of APCs, reduction of immunosuppressive myeloid, regulatory T and B cell populations, and activation of NK cells and CD4+ and CD8+ T cells together indicate the advantage of precisely engineered microbial platforms as next-generation antitumour vaccines that align several arms of immunity13. In agreement with previous reports regarding synthetic peptide, adenoviral and messenger RNA (mRNA) neoantigen vaccines49–51, we did not observe suppression of established CT26 tumour growth on SLP vaccination. By contrast, engineered microbial vaccines significantly reduced growth and achieved a portion of complete responses. This is consistent with a model whereby microbial vectors enable direct modulation of innate and adaptive immunity and sustained neoantigen delivery within the TME to promote effective therapeutic vaccination of established tumours.
The unique ability of microbial vaccines to directly remodel the TME may promote synergy with other forms of immunotherapy. Adoptive T cell therapy (ACT) has resulted in regression of advanced malignancy in a subset of patients, although solid tumours often show resistance52. Previous murine studies demonstrated that target-antigen vaccination amplifies ACT efficacy in solid tumours when targeting a carcinoembryonic antigen (claudin-6) with chimeric antigen receptor T (CAR-T) cell therapy53. However, in a human solid tumour trial the effect of combining claudin-6 vaccination with CAR-T cell therapy was unclear compared with CAR-T cell monotherapy54. Solid tumour resistance mechanisms to T cell-mediated immunotherapies often occur within the TME: restricted antigen availability, reduced APC infiltration and function, antitumour immune cell exclusion, upregulation of immunoinhibitory ligands and enrichment of immunosuppressive populations55. As the microbial neoantigen vectors locally increase neoantigen density, recruit and activate dendritic cells and CD4+ and CD8+ T cells, and reduce immunosuppressive populations and ligands within the TME, combination with ACT may oppose these resistance mechanisms and provide synergistic benefit.
Through extra programming of the microbial vectors and rational incorporation of other immunotherapeutics, this system may achieve reliable eradication of established solid tumours and metastases through precision cancer immunotherapy using living antitumour vaccines.
Methods
Cell lines
The B16F10 melanoma (ATCC CRL-6475), CT26 colon carcinoma (ATCC CRL-2638) and 4T1 breast cancer (ATCC CRL-2539) authenticated cell lines were purchased directly from ATCC. CT26-Luc, B16F10-Luc and 4T1-Luc cells were lentivirally transduced with luciferase. Cells were confirmed mycoplasma free. Cells were cultured in incubators at 37 °C with atmosphere of humidified 5% CO2. B16F10 and B16F10-Luc cells were grown in DMEM supplemented with 10% (vol/vol) fetal bovine serum (FBS), 1× GlutaMax, 1% (vol/vol) MEM non-essential amino acids solution (Gibco-11140050) and 100 U ml−1 penicillin–streptomycin. CT26, CT26-Luc, 4T1 and 4T1-Luc cells were grown in Roswell Park Memorial Institute medium (RPMI-1640) supplemented with 10% (vol/vol) FBS, 1× GlutaMax, 1% (vol/vol) MEM non-essential amino acids solution and 100 U ml−1 penicillin–streptomycin. No commonly misidentified cell lines were used in this study.
Exome sequencing
Paired tumour and tail DNA from BALB/c mice bearing subcutaneous CT26 tumours or C57BL/6 mice bearing subcutaneous B16F10 tumours was extracted in triplicate (n = 3 mice per tumour line) using Qiagen DNeasy Blood & Tissue Minikit following the manufacturer’s instructions. Exome capture from mouse tumour and tail DNA triplicates was conducted using Agilent SureSelectXT All Exon kit for target enrichment DNA library preparation56, according to the manufacturer’s instructions (Agilent). Genomic DNA was fragmented by acoustic shearing with a Covaris S220 instrument. Fragmented DNAs were cleaned, end-repaired and adenylated at the 3′ end. Adaptors were ligated to DNA fragments, and adaptor-ligated DNA fragments enriched with limited-cycle PCR. Adaptor-ligated DNA fragments were validated using Agilent TapeStation (Agilent) and quantified using Qubit 2.0 Fluorometer (ThermoFisher Scientific) and Real-Time PCR (KAPA Biosystems). Sequencing libraries were clustered onto a lane of a flow cell. After clustering, the flow cell was loaded on an Illumina HiSeq4000 Instrument per the manufacturer’s instructions. Samples were sequenced using 2 × 150 bp paired end configuration. Image analysis and base calling was conducted by the HiSeq Control Software. Raw sequence data (.bcl files) generated from Illumina HiSeq was converted into fastq files and de-multiplexed using Illumina bcl2fastq2.17. Sequence reads were trimmed to remove adaptor sequences and nucleotides with poor quality using Trimmomatic v.0.39 (ref. 57). Trimmed reads were aligned to the GRCm38 reference genome using the Illumina Dragen Bio-IT platform. Alignments were sorted and PCR or optical duplicates marked for generation of BAM files. Somatic single-nucleotide variants and insertion or deletion (indel) variants were called using Illumina Dragen58 and GATK Mutect2 (ref. 59). All variants from paired-normal tissue and murine variants from the dbSNP database60 were removed during the process. VCF files were left aligned and normalized, with splitting of multiallelic sites into several sites using bcftools v.1.13 (ref. 61). Only tumour-specific variants called by both algorithms were used for further analysis.
RNA sequencing
Tumour RNA from BALB/c mice bearing subcutaneous CT26 tumours or C57BL/6 mice bearing subcutaneous B16F10 tumours was extracted in triplicate using Qiagen RNeasy Minikit as per the manufacturer’s instructions. Extracted RNA samples were quantified using Qubit 2.0 Fluorometer (Life Technologies) and RNA integrity checked using Agilent TapeStation 2400 (Agilent). RNA sequencing libraries were prepared using the NEBNext Ultra RNA library Prep Kit for Illumina as per the manufacturer’s instructions (New England Biolabs). mRNAs were enriched with Oligo(dT) beads. Enriched mRNAs were fragmented for 15 min at 94 °C. First- and second-strand complementary DNAs (cDNAs) were synthesized subsequently. cDNA fragments were end-repaired and adenylated at 3′ ends, and universal adaptors ligated to cDNA fragments, followed by index addition and library enrichment by limited-cycle PCR. Sequencing libraries were validated on Agilent TapeStation (Agilent), and quantified using Qubit 2.0 Fluorometer (Invitrogen) and quantitative PCR (qPCR) (KAPA Biosystems). Library loading, sequencing and read trimming were done as described above. Trimmed reads were aligned to the mm10 reference using STAR aligner v.2.5.2b (ref. 62). Unique gene hit counts were calculated using feature counts from Subread Package v.1.5.2. Unique reads that fell within exon regions were counted. The gene hit counts table was used for expression analysis using DESeq2 v.1.20.0 (ref. 63).
Neoantigen prediction and selection
Mutation-specific RNA expression and allele fraction were added to somatic VCF files using Bam-readcount64 and VAtools (http://vatools.org). Somatic VCFs were annotated with The Ensembl Variant Effect Predictor (VEP Ensembl v.104)65. Only PASS variants from VCFs were considered. Annotated VCFs were analysed using pVacSeq for neoepitope discovery14. MHC-I affinities were predicted with NetMHCpan v.4.1 (ref. 66) and NetMHC v.4.0 (ref. 67), and MHC-II affinities were predicted with NetMHCIIpan v.4.1 (ref. 68) and NNalign v.2.0 (ref. 69). Exonic mutation-derived long peptides based on single-nucleotide polymorphisms (SNP) or indels predicted to generate mutant MHC-binding peptides were first filtered on the basis of the set of minimum criteria: (1) present in all tumour sample triplicates (DNA variant allele fraction ≥ 0.05) and none of the normal tissue triplicates, (2) non-synonymous mutation resulting from either SNP or indel, (3) confirmed exonic mutation transcription (RNA variant allele fraction ≥ 0.05) and gene expression by RNA sequencing in tumour sample triplicate (transcripts per million ≥ 1), (4) at least one predicted MHC-I or MHC-II binding epitope and (5) MHC-I or MHC-II differential binding affinity17,20 (wild-type half-maximum inhibitory concentration (IC50)/mutant IC50) ≥ 1.2. Predicted neoantigens fulfilling all previous criteria were then prioritized for inclusion and selected according to the following hierarchy: (1) high predicted affinity (MHC-I or MHC-II IC50 ≤ 500 nM), (2) moderate predicted affinity (MHC-I or MHC-II IC50 500–1,000 nM) and (3) low predicted affinity (MHC-I or MHC-II IC50 1,000–5,000 nM) (Extended Data Tables 1 and 2).
Strains and plasmids
Plasmids were constructed using restriction-enzyme mediated and Gibson assembly cloning methods. Neoantigen construct iterations were designed and created as Geneblocks (IDT) encoding a constitutive promoter and 5′ untranslated region (UTR) containing selected ribosome-binding site, followed by coding region composed of mutant-residue containing long peptides connected in tandem or by various linkers as indicated. 5′ BamHI and 3′ XbaI restriction endonuclease sites were added to constructs. Coding sequences were codon optimized for E. coli. Constructs were cloned between BamHI and XbaI restriction sites on a stabilized p246-luxCDABE plasmid where luxCDABE had been cloned out22, and flanked by 3′ λ-phage transcription terminator, with high-copy pUC origin. For protein expression assessment studies, the codon sequence for a 6×-Histidine Tag (HisTag) was added immediately before the stop codon within the neoantigen construct coding sequence by PCR amplification of full construct plasmids with oligonucleotide containing 6×-HisTag sequence followed by kinase, ligase, DpnI enzyme mix protocol (NEB). Neoantigen construct plasmids were transformed into chemically competent E. coli DH5α or BL21(DE3) (New England Biolabs), or electrocompetent EcN parental strain or genetic derivatives. The parental EcN strain and all derivatives used in this study harbour an integrated luxCDABE cassette within the genome, which also contains an erythromycin resistance gene70. Plasmid encoding constitutive LLO was constructed by cloning in the hok/sok stabilization system to pCG02-p15a backbone71, PCR amplification of backbone with SLC cloned out, and Gibson assembly of Geneblock encoding LLO under constitutive promoter and 5′ UTR containing selected ribosome-binding site. Constitutive LLO plasmids were transformed into electrocompetent EcN parental and genetic derivative strains. Strains were cultured in Luria-Bertani (LB) medium with antibiotics for plasmid retention (pUC:kanamycin 50 μg ml−1, p15a:spectinomycin 50 μg ml−1) in a 37 °C orbital incubator.
Construction of cryptic plasmid-cured EcN
EcN cryptic plasmids were cured with Cas9-mediated double-strand break, as described previously27. Briefly, EcN was transformed with pFREE or pCryptDel4.8 to cure the cryptic plasmids pMUT1 or pMUT2, respectively. The transformants were grown overnight and diluted 1:1,000 the next day into fresh LB containing 0.2% rhamnose and 0.43 μM anhydrotetracycline. After 24 h of incubation, the culture was streaked onto LB plates without antibiotics and incubated overnight in a 30 °C incubator. Colonies were screened with colony PCR to verify the loss of cryptic plasmids.
Construction of genetic knockout strains
Genetic knockouts were performed using the lambda red recombination system72. In brief, EcNc was transformed with pKD46. Transformants were grown at 30 °C in LB with ampicillin and l-arabinose, then made electrocompetent. The chloramphenicol resistance cassette with corresponding overhangs for each target gene for deletion was prepared by PCR amplification of pKD3. Electroporation was performed using 100 μl of competent cells and 50–300 ng amplified DNA. After 2 h of recovery, cells were plated on LB agar containing chloramphenicol and incubated at 37 °C overnight. Target gene deletion was verified by colony PCR. For excision of the antibiotic resistance marker, pCP20 was transformed, and the transformants were plated on fresh LB plates containing ampicillin and incubated at 30 °C overnight. Selected colonies were then inoculated onto fresh LB plates without antibiotics and cultured at 43 °C overnight for induction of flippase and plasmid curing. Clones were subsequently screened for loss of antibiotic resistance.
qPCR for PCN
Copy number variant plasmids were constructed from a high-copy pUC-GFP22 plasmid. The plasmid backbone excluding the pUC origin was PCR-amplified and Gibson assembled with sc101*, p15A or ColE1 origin of replication insert. The respective inserts were prepared from PCR amplification of template plasmid pCG02_sc101*, pCG02_p15A or pTH05_ColE1. Plasmid copy number (PCN) was determined as reported previously22, in which the relative abundance of plasmid DNA compared to genomic DNA is measured by qPCR. Briefly, strains with the plasmid of interest were grown at 37 °C overnight in fresh LB with appropriate antibiotics. Cells were collected by centrifugation at 3,000g at 4 °C for 10 min, the supernatant removed and the cell pellet resuspended in distilled water for optical density measurement at 600 nm (OD600) equal to 1. Resuspended cells were fivefold serially diluted. Samples were denatured at 95 °C for 10 min and 2 μl of each sample dilution was added into 18 μl of NEB Luna Universal qPCR Master Mix in each well of a 96-well plate. Then 25-fold diluted samples were used for the measurement of crossing point values: the cycle number when amplified sample fluorescence exceeds the background. Fivefold diluted samples were used for generation of the standard curve for PCR efficiency (E). E was defined from the slope (S) of each standard curve with the equation E = 5(−1/S) and PCN was determined with the equation PCN = (EGCTG)/(EPCTP), where respective values for genomic DNA are denoted by a subscript G and plasmid DNA by subscript P.
Immunoblot and ELISA
For immunoblot and ELISA, a C-terminal 6×-HisTag was attached to each neoantigen construct. Strains expressing neoantigen construct with C-terminal 6×-HisTag were grown overnight in LB media with appropriate antibiotics. Equalization of OD600 measurement to match colony-forming units (CFU) per ml (CFU ml−1) between all cultures was done before all sample processing. CFU-matched cultures were centrifuged at 3,000g at 4 °C for 10 min. For immunoblot, samples were resuspended in B-PER lysis reagent (ThermoFisher Scientific) containing 250 U ml−1 benzonase nuclease (Millipore Sigma) and 1 U ml−1 rLysozyme (Millipore Sigma) and placed on an orbital shaker for 15 min at room temperature. Samples were centrifuged at 10,000g for 20 min at 4 °C to separate soluble and insoluble fractions or total lysate used directly. Processed samples were mixed with SDS-loading buffer with 5 mM dithiothreitol, boiled and subject to immunoblot analysis. For relative quantification of immunoblot chemiluminescent intensity, target protein bands on the same blot were normalized to the loading control band DnaK for the same sample. DnaK loading controls were always run on the same gel as target proteins. Normalized values were divided to provide relative intensity values. Mouse anti-6×His (αTHE) was purchased from GenScript, mouse anti-DnaK was purchased from Abcam (8E2/2). αTHE and 8E2/2 antibodies were used at 1:5,000 dilution.
For HisTag ELISA, samples were resuspended in ice-cold PBS containing HALT protease inhibitor cocktail (ThermoFisher Scientific). Samples were sonicated on ice for 2 min total time. Sonicated samples were centrifuged at 10,000g for 20 min at 4 °C. Soluble sample fractions were analysed using GenScript HisTag ELISA Detection Kit as per the manufacturer’s instructions.
For ex vivo immunoblot analysis, BALB/c mice bearing established hind-flank CT26 tumours were injected intravenously with the EcNcΔlon/ΔompT/LLO+ nAg19-His strain cocktail that contains all three neoantigen constructs, in which each construct (MHCIa, MHCIIa and MHCI/IIv) contained a C-terminal 6×-HisTag. Then 48 h after treatment, tumours and TDLNs were extracted from mice and placed in B-PER lysis reagent (ThermoFisher Scientific) with 250 U ml−1 benzonase nuclease (Millipore Sigma), and homogenized using a gentleMACS tissue dissociator (Miltenyi Biotec, C-tubes). Tissue homogenate was sonicated on ice for 3 min. Sonicated samples were centrifuged at 10,000g for 20 min at 4 °C to separate soluble and insoluble fractions, and fractions subsequently resuspended and diluted in lysis buffer. Sample fractions were mixed with SDS-loading buffer with 5 mM dithiothreitol, boiled and subject to immunoblot analysis.
For ex vivo IL-12p70 ELISA analysis, BALB/c mice bearing established hind-flank CT26 tumour were injected intratumourally with PBS, EcNcΔlon/ΔompT or EcNcΔlon/ΔompT/LLO+. Then 4–24 h after treatment, tumours were extracted and placed in ice-cold PBS containing HALT protease inhibitor cocktail without dimethylsulfoxide (DMSO) (ThermoFisher Scientific). Tumours were homogenized using a gentleMACS tissue dissociator (Miltenyi Biotec, C-tubes), and centrifuged at 3,000g for 10 min at 4 °C. The supernatant was then collected and centrifuged at 10,000g for 20 min at 4 °C to separate soluble and insoluble fractions. IL-12p70 in soluble sample fractions was analysed using the Mouse IL-12p70 Quantikine ELISA Kit (R&D systems) according to the manufacturer’s instructions.
Blood bactericidal assay
EcN wild-type or EcNcΔlon/ΔompT were cultured overnight in LB media without antibiotics. Cultures were centrifuged at 3,000g for 10 min, resuspended in 1 ml of ice-cold sterile PBS and normalized to OD600 = 1. Then 50 μl of OD600 = 1 microbe suspension was added to 1 ml of single donor human whole blood (Innovative Research) in triplicate and incubated in a 37 °C stationary incubator. After 2 h of incubation, a sample was taken from each blood–microbe mixture and serial dilution was prepared in PBS. Dilutions were plated on LB agar with erythromycin (25 μg ml−1). After incubation overnight at 37 °C, colonies were quantified by spot-forming assay and CFU ml−1 blood was calculated.
Biofilm assay
Biofilm formation assays were conducted as described previously73. Briefly, EcN wild-type, cryptic plasmid-cured (EcNc), Lon knockout (EcNcΔlon), OmpT knockout (EcNcΔompT) or double protease knockout (EcNcΔlon/ΔompT) were cultured for 48 h in LB media with 25 μg ml−1 erythromycin in borosilicate glass tubes in a 30 °C stationary incubator, with tube caps wrapped with parafilm to prevent evaporation. At 48 h, cultures were discarded and borosilicate tubes were washed three times with PBS. Tubes were inverted and allowed to dry for 6 h. Biofilms left on borosilicate tubes were stained with 0.1% (vol/vol) crystal violet for 15 min. Crystal violet stain was discarded and tubes washed three times with PBS, then inverted and allowed to dry overnight. Crystal violet-stained biofilms were dissolved with 95% ethanol and transferred to 96-well plates for measurement of absorbance at 590 nm.
Phagocytosis assay
Bacterial phagocytosis assays were adapted from previous work74. Culture and isolation of murine BMDMs was performed as described previously75. Bulk femoral bone marrow cells from BALB/c or C57BL/6 mice were cultured on 15 cm non-treated cell culture Petri dishes in RPMI with 20% FBS, 25 ng ml−1 M-CSF (R&D Systems) and 100 U ml−1 penicillin–streptomycin. Media was replaced with fresh media after 4 days of culture. After 7 days of culture, plates were washed with PBS and adherent macrophages were dissociated using trypsin-EDTA. Macrophages were washed in PBS, resuspended at a density of 2 × 105 cells per ml in media and 1 ml transferred to each well of 24-well plates. The 24-well plates were incubated overnight in a 37 °C incubator with humidified 5% CO2. EcN wild-type or EcNcΔlon/ΔompT, with or without a constitutive green-fluorescent protein (GFP)-expressing plasmid were cultured overnight in LB media with appropriate antibiotics. Bacterial cultures were centrifuged at 3,000g for 10 min, washed three times with sterile PBS and resuspended at a density of 4 × 108 bacteria per ml in sterile PBS. Media from wells containing adherent macrophages was aspirated, wells washed three times with PBS and 1 ml of RPMI with 5% mouse serum added to each well. Latrunculin A was added at a concentration of 1 μM to selected wells to inhibit phagocytosis. Next, 2 × 107 CFU of microbes were added to each well with each condition tested in triplicate. Microbial strains were incubated with BMDMs for 30 min in a 37 °C incubator at 20 rpm. After 30 min, media was aspirated and wells were washed six times with sterile ice-cold PBS. Adherent macrophages were dissociated using non-enzymatic cell dissociation buffer (Gibco), resuspended in fluorescence-activated cell sorting (FACS) buffer (PBS containing 2% FBS, 2 mM EDTA and 0.09% sodium azide) and analysed by flow cytometry.
In vitro BMDM activation
BMDMs were cultured as described above for phagocytosis assays. BMDMs were washed in PBS, resuspended at a density of 2 × 105 ml−1 in media and 1 ml transferred to each well of 24-well plates. The 24-well plates were incubated overnight in a 37 °C incubator with humidified 5% CO2. Wild-type EcN or EcNcΔlon/ΔompT with constitutive expression of OVA from a pUC origin plasmid were cultured overnight in LB media with appropriate antibiotics. Cultures were centrifuged at 3,000g for 10 min, washed three times with PBS and resuspended at a density of 4 × 108 bacteria ml−1 in sterile PBS. Media from wells containing macrophages was aspirated, wells were washed three times with PBS and 1 ml of RPMI with 5% mouse serum was added to each well. Next, 1 × 107 live microbes were added to each well, with each condition replicated in triplicate. Live microbial strains were incubated with BMDMs for 6 h in a 37 °C incubator. After 6 h, media was aspirated and wells were washed six times with sterile ice-cold PBS. Adherent macrophages were dissociated using non-enzymatic cell dissociation buffer (Gibco), resuspended in FACS buffer and analysed by flow cytometry. DRAQ7 cell viability reagent was used to exclude dead cells (diluted 1:1,000 in FACS buffer). Extracellular antibodies for BMDM activation panel included CD80 (catalogue no. 16-10A1, Biolegend), MHC-II (catalogue no. M5/114.15.2, Biolegend), PD-L1 (catalogue no. 10F.9G2, Biolegend) and H2Kb-SIINFEKL (catalogue no. 25-D1.16, Biolegend), each used at 1:200 dilution.
In vitro BMDC stimulation
BMDC isolation and culture from mouse bone marrow was adapted from previous methods76. BMDCs from C57BL/6 mice were cultured on 15 cm non-treated cell culture Petri dishes in RPMI with 20% FBS, 20 ng ml−1 GM-CSF (Biolegend) and 100 U ml−1 penicillin–streptomycin. Every 1–2 days for the first 4 days, plates were gently washed and non-adherent granulocytes removed by aspirating 50% of the culture media with subsequent replacement of fresh media. On day 4, media was aspirated completely and replaced with fresh culture media with 20 ng ml−1 GM-CSF. On day 6, BMDC plates were washed with PBS and loosely adherent and non-adherent cells collected. Cells were centrifuged at 300g for 5 min, resuspended in fresh culture media and replated on 15 cm non-treated cell culture Petri dishes. On days 7–8, plates were washed with PBS and loosely adherent and non-adherent cells were collected. Cells were centrifuged at 300g for 5 min, resuspended in fresh culture media at a density of 2.5 × 105 ml−1 and 200 μl was transferred to 96-well plates and incubated overnight in a 37 °C incubator. The next day, media from wells containing BMDCs was aspirated, and 1 ml of RPMI with 5% mouse serum was added to each well. BMDCs were pulsed with live bacteria at an multiplicity of infection of 10 for 2 h. Plates were centrifuged at 300g for 5 min, media aspirated and replaced with fresh RPMI with 10% FBS, 10 μg ml−1 gentamicin and 100 U ml−1 penicillin–streptomycin. Gentamicin concentration was increased to 40 μg ml−1 after 2–4 h. Plates were incubated for 5–48 h in a 37 °C incubator, at which time the supernatant was assessed for IL-12p70 using the Mouse IL-12p70 Quantikine ELISA Kit (R&D systems) according to the manufacturer’s instructions.
OT-I and OT-II T cell stimulation and proliferation
BMDCs were cultured as above, resuspended at a density of 2.5 × 105 ml−1 and 5 × 104 BMDCs transferred to 96-well plates and incubated overnight in a 37 °C incubator. The next day, media from wells containing BMDCs was aspirated, and 1 ml of RPMI with 5% mouse serum was added to each well. BMDCs were pulsed with 2 × 106 CFU of the respective bacterial strain for 2.5 h, plates were centrifuged at 300g for 5 min, media aspirated and replaced with fresh RPMI with 10% FBS and 10 μg ml−1 gentamicin and 100 U ml−1 penicillin–streptomycin. Gentamicin concentration was increased to 40 μg ml−1 after 2–4 h. Spleens from naive OT-I and OT-II mice were extracted, filtered through 100 µm cell strainers and washed in complete RPMI (RPMI-1640 supplemented with 10% (vol/vol) FBS, 1× GlutaMax, 1% (vol/vol) MEM non-essential amino acids solution (Gibco-11140050) and 100 U ml−1 penicillin–streptomycin). OT-I and OT-II T cells were isolated from single-cell suspensions of spleens from the respective transgenic mouse using the EasySep Mouse T Cell Isolation Kit (StemCell Technologies) according to the manufacturer’s instructions. Purified OT-I and OT-II T cells were resuspended in T cell media (complete RPMI supplemented with 50 μM β-mercaptoethanol) at a density of 5 × 105 ml−1 and 5 × 104 T cells incubated with 5 × 104 BMDCs pulsed with the respective microbial strains. For cytokine secretion assessment, T cells were incubated with BMDCs for 24 h, at which time supernatant was assessed for IFNγ and IL-2 using Mouse IFNgamma Quantikine ELISA Kit and Mouse IL-2 Quantikine ELISA Kit (R&D systems) according to the manufacturer’s instructions.
Carboxyfluorescein succinimidyl ester (CFSE) proliferation assays were conducted as previously described77. Here, 1 × 107 OT-I or OT-II T cells were resuspended in 1 ml of room temperature PBS, and 1 μl of 5 mM CFSE (Biolegend) was added. T cells were incubated in CFSE solution for 5 min at room temperature protected from light, after which time staining was quenched by adding ten times the staining volume of cell culture media. T cells were centrifuged at 300g for 5 min, resuspended in T cell media at a density of 5 × 105 ml−1 and incubated for an extra 10 min at room temperature. Then 5 × 104 T cells were incubated with 5 × 104 BMDCs pulsed with the respective live microbial strains. At 48 h, 50% of the media from each well was gently aspirated so as to not disturb any cells, and replaced with fresh T cell media. At 72–96 h, OT-I and OT-II T cells were collected and analysed by flow cytometry. DRAQ7 cell viability reagent was used to exclude dead cells (diluted 1:1,000 in FACS buffer). Extracellular antibody staining for CFSE assays included antimouse CD3 (catalogue no. 17A2, Biolegend), used at 1:200 dilution.
Listeriolysin haemolytic activity assay
Sheep red blood cell (RBC) lysis by bacterial lysate was performed as described previously78. Briefly, bacteria were grown overnight in fresh LB containing appropriate antibiotics. Cultures were centrifuged at 3,000g for 10 min, supernatants discarded and the cell pellet resuspended to OD600 = 8 in 0.1% (w/w) bovine serum albumin (BSA) in sterile PBS titrated to pH of 5.25 with 1 M HCl. Bacteria were sonicated for 2 min. After sonication, the soluble fraction was isolated by centrifugation at 10,000g at 4 °C for 20 min. Sheep RBCs were washed three times with PBS and resuspended at a final concentration of 6 × 108 ml−1 in 0.1% (w/w) BSA in PBS titrated to pH of 5.25. Equal parts of bacterial lysate soluble fraction and sheep RBC suspension were mixed and incubated for 15 min at 37 °C. After incubation RBC mixtures were centrifuged at 1,000g for 1 min at 4 °C and supernatant absorbance at 541 nm was then measured to quantify RBC lysis.
Listeriolysin cytosolic access assay
BMDMs were cultured as described above for phagocytosis assays. BMDMs were washed in PBS, resuspended at a density of 2.5 × 105 ml−1 in media, 100 ml transferred to wells of an eight-well Lab-Tek Chamber Slide system (ThermoFisher) and incubated overnight in a 37 °C incubator. The next day, media from wells containing BMDMs was aspirated, and 1 ml of complete RPMI without antibiotics was added to each well. BMDMs were then pulsed with 1.25 × 106 CFU of the respective bacterial strain for 60 min. After the designated time media from each well was aspirated, wells were washed four times with PBS and media was replaced with fresh RPMI with 10% FBS and 40 μg ml−1 gentamicin and incubated in a stationary 37 °C incubator. After either 30 or 60 min of more incubation, media was aspirated and wells washed four times with ice-cold PBS. Then 100 ml of 100% methanol at −80 °C was then added to each well for fixation and allowed to incubate at room temperature for 10 min. Methanol was then removed, 100 ml of ice-cold PBS added to each well and slides incubated at 4 °C. Cells were permeabilized with 0.5% Triton X in PBS for 10 min. Blocking solution in 10% heat-inactivated horse serum and 3% BSA was added to each well for 30 min. After washing three times, primary antibodies in 1% heat-inactivated horse serum and 1% BSA were incubated overnight at 4 °C in a humidified chamber. The next day, slides were washed with PBS three times for 10 min each and secondary antibodies were applied for 1 h at room temperature in the dark. DAPI (4′6-diamidino-2-phenylindole) was applied as part of the secondary antibody cocktail for nuclear staining. Slides were washed in PBS three times before mounting coverslips with DAKO gel and stored at 4 °C until immunofluorescence analysis. Anti-ovalbumin (catalogue no. EPR27117-90, Abcam) and anti-CD11b (catalogue no. M1/70, Abcam) primary antibodies were used for staining, both at 1:200 dilution.
Animal experiments
All animal experiments were approved by the Institutional Animal Care and Use Committee (Columbia University, protocol AABQ5551). The 6–7-week-old female BALB/c, C57BL/6 and B6(Cg)-Tyrc-2J/J (Jackson Laboratories) mice were kept in accordance with all rules for animal research at Columbia University. Mice were housed in a facility with a 12 h light–dark cycle, and provided unrestricted access to both food and water. The housing facility was maintained at 21–24 °C, and kept at 40–60% humidity. Sample size was determined on the basis of our previous studies and/or pilot experiments. For subcutaneous tumour models: 5 × 106 CT26 cells in 100 μl of sterile PBS were inoculated subcutaneously on the hind flank of BALB/c mice, or 5 × 105 B16F10 melanoma cells in 100 μl of sterile PBS subcutaneously on the hind flank (orthotopic) of C57BL/6 mice using a 26G needle on a 1 cc syringe. CT26 tumours were allowed to establish as indicated for each experiment, and mice were distributed between groups to equate the average starting tumour volume before treatment. B16F10 orthotopic tumours were allowed to establish for 9 days, and initial average tumour volume equated between groups before treatment. Tumour dimensions were measured unblinded with a calliper every 1–3 days for calculating tumour volumes using the equation (a2 × b)/2 (a is width, b is length, where width is the smaller dimension). Group tumour sizes were computed as mean ± s.e.m. Body weight was measured each time tumour measurements were taken. Animals were euthanized when any of the following criteria were met: tumour burden greater than 2 cm in the largest dimension for any subcutaneous tumour, greater than 20% body weight loss, as otherwise recommended by veterinary staff or when showing clinical signs of impaired health. To examine the requirement of individual T cell populations for the efficacy of the microbial neoantigen vaccines, mice were injected intraperitoneally with 200 μg (in 100 μl of InVivoPure pH 6.5 buffer, BioXcell) of antimouse CD4 (clone GK1.5, BioXcell), 200 μg (in 100 μl of InVivoPure pH 7.0 buffer, BioXcell) of antimouse CD8β (clone Ly-3.2, BioXcell) or 200 μg (in 100 μl of InVivoPure pH 7.0 buffer, BioXcell) of IgG1 isotype control (clone HRPN, BioXcell) beginning 2 days before the initiation of therapeutic treatment and every 2–3 days thereafter until study endpoint.
In prophylactic vaccination studies, BALB/c mice received an intravenous injection of either EcNcΔlon/ΔompT/LLO+ OVA or EcNcΔlon/ΔompT/LLO+ nAg19 every 3–5 days for a total of four injections. Four days after the final injection, 1 × 106 CT26 cells in 100 μl of sterile PBS were inoculated subcutaneously on the hind flank. In rechallenge studies, BALB/c mice that had cleared subcutaneous CT26 tumours on a single hind flank were engrafted with 1 × 106 CT26 cells on the opposite hind flank 100 days after tumour clearance. Age-matched naive BALB/c mice were engrafted with 1 × 106 of the same CT26 cells on a single hind flank as controls.
For therapeutic studies in systemic metastases models, 5 × 105 CT26-Luc cells or 1.5 × 105 B16F10-Luc cells were injected in 100 μl of sterile PBS through the lateral tail vein with a 27G needle on 1 cc syringe. Metastases were allowed to establish for 4 days in Balb/C mice before treatment for CT26-Luc, and for 2 days in C57BL/6 albino mice (B6(Cg)-Tyrc-2J/J) for B16F10-Luc. Mice were randomly distributed between groups after metastases engraftment and before treatment. For in vivo luminescence tracking of metastases burden, mice were injected intraperitoneally with 125 μl of aqueous solution of d-Luciferin (50 mg ml−1) 6 min before imaging, and placed under isoflurane anaesthesia for imaging using an in vivo imaging system (IVIS), with exposure time set to 6 min. Total flux from the lungs (CT26-Luc) or body (B16F10-Luc) was used to quantify tumour burden. For evaluation of lung metastases burden at the timepoint of treatment initiation, mice were injected intraperitoneally with 250 μl of aqueous solution of d-Luciferin (50 mg ml−1) 6 min before imaging and placed under isoflurane anaesthesia for imaging using an IVIS with exposure time set to 10 min. After in vivo IVIS analysis, mice were then re-injected with 100 μl of aqueous solution of d-luciferin (50 mg ml−1) and lungs were extracted for ex vivo IVIS imaging with exposure time set to 2 min.
No formal blinding was done for in vivo experiments. For all animal experiments, intratumoural treatments were injected directly into the tumour core with care to not allow leakage of any therapeutic solution. Intravenous treatments were injected through the lateral tail vein, with care not to allow leakage of any therapeutic solution.
SLP vaccination
The formulation and administration of SLP vaccines was adapted from previous studies15,79,80. Each dose contained either 20 μg of each 29-mer CT26 neoantigen peptide (19 neoantigens, 380 μg of total peptide per dose) and 50 μg of poly I:C in 200 μl of 10% DMSO/90% PBS (vol/vol) in Fig. 2e and Extended Data Fig. 5e,f, or 25 μg of each 29-mer CT26 neoantigen peptide (19 neoantigens, 475 μg total peptide per dose) and 100 μg of poly I:C in 200 μl of 10% DMSO /90% PBS (vol/vol) in Extended Data Fig. 5g. Therapeutic SLP vaccinations were administered subcutaneously to BALB/c mice with established hind-flank CT26 tumours on the contralateral hind flank using a 29G needle. SLP vaccine groups were treated on the same days as microbial therapeutic groups.
Ex vivo lung histology
Explanted lungs from BALB/C mice bearing CT26-Luc metastases or C57BL/6 albino mice (B6(Cg)-Tyrc-2J/J) bearing B16F10-Luc metastases were washed three times in PBS and placed in 10% formalin. After at least 24 h of fixation, lungs were transferred to 70% ethanol and subsequently embedded in paraffin. Then 50 μm consecutive sections were stained with haematoxylin and eosin. Lung sections were analysed for the presence of tumour foci.
Microbial administration for in vivo experiments
For therapeutic administration, bacterial strains were grown overnight in fresh LB media containing the appropriate antibiotics. Overnight cultures were centrifuged at 3,000g at 4 °C for 10 min and washed three times with ice-cold sterile PBS. Microbes were delivered intratumourally at a concentration of 5 × 108 CFU ml−1 in sterile PBS, with 20 μl of injected using a 1 cc syringe with a 29G needle. For intravenous treatment, 100 μl of microbes were delivered at a concentration of 1 × 108 CFU ml−1 in sterile PBS, through the lateral tail vein using a 1 cc syringe with a 29G needle.
Biodistribution and in vivo bacterial dynamics
For biodistribution experiments, BALB/c mice bearing established hind-flank CT26 or lung metastatic CT26-Luc tumours were injected intravenously with 100 μl of 1 × 108 CFU ml−1 EcNcΔlon/ΔompT/LLO+. Then 96–120 h after a single i.v. injection for hind-flank tumours or at endpoint for lung metastases, tumours or tumour-bearing lungs and other organs were extracted from mice, weighed and homogenized using a gentleMACS tissue dissociator (Miltenyi Biotec, C-tubes). Homogenates were serially diluted in sterile PBS and plated on LB agar plates at 37 °C overnight. Colonies were quantified per organ and computed as CFU per gram of tissue (CFU g−1). For tracking bacterial colonization of subcutaneous tumours by microbial luminescence, tumour-bearing mice treated intratumourally or intravenously with wild-type EcN parental strain or genetic derivates were imaged using IVIS at various time points. For abscopal experiments, treated and untreated tumours were harvested 14 days after a single intratumoural bacterial injection.
Ex vivo T cell killing assay
For B16F10-Luc specific killing, naive tumour-free C57BL/6 mice were injected intravenously every 4 days with PBS, EcNcΔlon/ΔompT/LLO+ OVA or nAg42 for a total of four doses. Five days after the final dose spleens from treated mice were extracted, filtered through 100 µm cell strainers and washed in complete RPMI. T cells were isolated from single-cell suspensions of spleens from the respective mouse using the EasySep Mouse T Cell Isolation Kit (StemCell Technologies) according to the manufacturer’s instructions. Purified T cells were resuspended in T cell media for use in the specific lysis assay.
The luciferase-based killing assay was adapted from previous methods81. B16F10-Luc target cells were grown for 48 h in the presence of 100 U ml−1 murine IFNγ. Target cells were gathered and plated at 1 × 104 cells per well in a 96-well plate. After 12 h, T cells were added to each well to achieve designated effector-to-target ratios (10:1, 20:1 or 40:1). After 42 h of co-incubation, 50 U ml−1 IL-2 was added to all wells.
For CT26-Luc versus 4T1-Luc luciferase-based specific killing assay: BALB/c mice with established hind-flank CT26 tumours were treated intravenously with EcNcΔlon/ΔompT/LLO+ nAg19 on days 0 and 3. On day 8, tumours were extracted and mechanically homogenized, followed by digestion with collagenase A (1 mg ml−1, Roche) in isolation buffer (RPMI-1640 with 5% FBS, 1% l-glutamine, 1% penicillin–streptomycin and 10 mM HEPES) with gentamicin (40 μg ml−1) for 1 h at 37 °C on a shaker platform at 150 rpm. Tumour homogenates were filtered through 100 µm cell strainers and washed in T cell media. Tumour-infiltrating T cells were isolated from single-cell suspensions of tumours from mice using the EasySep Mouse T Cell Isolation Kit (StemCell Technologies) according to the manufacturer’s instructions. Purified T cells were resuspended in T cell media.
CT26-Luc or 4T1-Luc target cells were grown for 12 h in the presence of 100 U ml−1 murine IFNγ. Target cells were gathered and plated at 1 × 104 cells per well in a 96-well plate. After 12 h, T cells were added to each well to achieve designated effector-to-target ratios (5:1 or 10:1), with 50 U ml−1 IL-2 added to all wells.
Luminescence from each well was quantified after addition of One-Glo Luciferase Assay System (Promega), as per the manufacturer’s instructions, after 24–96 h of coculture. Minimum lysis wells contained only the respective luciferase-expressing tumour target cells. In maximum lysis wells, 20 μl of media was replaced with 20 μl of 3% Triton X-100 60 min before luminescence readout. Specific lysis (%) was calculated using the luminescence values of the respective conditions with the following formula: 100 − (100 × ((sample − maximum lysis)/(minimum lysis − maximum lysis))).
IFNγ ELISpot
BALB/c mice with established hind-flank CT26 tumours were treated intravenously with EcNcΔlon/ΔompT/LLO+ nAg19 on day 0 and 3. On day 8, tumours were extracted and mechanically homogenized, followed by digestion with collagenase A (1 mg ml−1, Roche) in isolation buffer (RPMI-1640 with 5% FBS, 1% l-glutamine, 1% penicillin–streptomycin and 10 mM HEPES) with gentamicin (40 μg ml−1) for 1 h at 37 °C on a shaker platform at 150 rpm. Tumour homogenates were filtered through 100 µm cell strainers and washed in RPMI containing CTL-Wash Supplement (Immunospot) and 1% l-glutamine. Splenocytes from naive BALB/c mice were isolated in the same way, without digestion. T cells were isolated from single-cell suspensions of tumours from mice using the EasySep Mouse T Cell Isolation Kit (StemCell Technologies) according to the manufacturer’s instructions. Purified T cells were resuspended in CTL-Test Medium supplemented with 1% l-glutamine for use in the enzyme-linked immunosorbent spot (ELISpot) assay.
Mouse IFNγ Single-Color ELISpot plates and kits were purchased from Immunospot. ELISpot plates were prepared as per manufacturer’s instructions. Here, 5 × 105 naive splenocytes were plated with 2 × 104 TILs per well in 200 μl CTL-Test Medium supplemented with 1% l-glutamine and gentamicin (30 μg ml−1). Then 29-mer synthetic neoantigen or negative control (OVA) peptides were added to each well at a final concentration of 5 μg ml−1. Cells were stimulated overnight in a stationary 37 °C incubator with atmosphere of humidified 5% CO2. After incubation, plates were developed as per the manufacturer’s protocol and spots quantified using a CTL Immunospot S6 Universal machine and CTL ImmunoSpot software v.7.0.24.0.
Flow cytometry immunophenotyping
For CT26 flow-cytometric immunophenotyping, BALB/c mice with hind-flank CT26 tumours received intravenous treatment with the indicated microbial therapeutic or PBS on day 0. Two or 8 days after treatment, TDLNs and/or tumours were extracted. For B16F10 flow-cytometric immunophenotyping, C57BL/6 mice with hind-flank, orthotopic B16F10 tumours received intravenous treatment with the indicated microbial therapeutic or PBS on day 0 and 3. Eight days after treatment, tumours were extracted. Lymphoid and myeloid immune subsets were isolated from tumour tissue by mechanical homogenization of tumour or TDLN tissue, followed by digestion with collagenase A (1 mg ml−1, Roche) and DNase I (0.5 µg ml−1, Roche) in isolation buffer (RPMI-1640 with 5% FBS, 1% l-glutamine, 1% penicillin–streptomycin and 10 mM HEPES) for 1 h at 37 °C for tumours or 30 min at 37 °C for TDLNs, on a shaker platform at 150 rpm. For ex vivo lymphocyte stimulation with PMA and ionomycin, TDLNs were not digested beforehand. Tumour and TDLN homogenates were filtered through 100 µm cell strainers and washed in isolation buffer. To measure overall cytokine production by T cells, cells were stimulated for 3 h with PMA (50 ng ml−1, Sigma-Aldrich) and ionomycin (1 nM, Calbiochem) in the presence of brefeldin A (1 μg ml−1). To measure neoantigen-specific cytokine production by T cells, cells were stimulated for 5 h with pools of peptides (2 μg ml−1) representing the neoantigens encoded in therapeutic strains in the presence of brefeldin A (1 μg ml−1). Cells were stained in FACS buffer. Ghost Dye cell viability reagent was used to exclude dead cells (diluted 1:1,000 in PBS). Extracellular antibodies for lymphoid immunophenotyping included: CD4 (RM4-5, Biolegend), NKp46 (29A1.4, BD Biosciences), NK1.1 (PK136, Biolegend), CD45 (30-F11, BD Biosciences), B220 (RA3-6B2, BD Biosciences), CD19 (6D5, Biolegend), CD8a (53-6.7, Biolegend), TIM-1 (RMT1-4, BD Biosciences) and CD69 (H1.2F3, BD Biosciences). After extracellular staining, cells were washed with FACS buffer, and fixed using the FOXP3/transcription factor staining buffer set (Tonbo), as per the manufacturer’s instructions. Intracellular antibodies for lymphoid immunophenotyping included: Foxp3 (FJK-16s, Thermo), CD3ε (145-2C11, Biolegend), TCRβ (H57-507, BD Biosciences), Ki-67 (SolA15, Thermo), Granzyme-B (QA16A02, Biolegend), TNF (MP6-XT22, Biolegend) and IFNγ (XMG1.2, Biolegend). For myeloid immunophenotyping, extracellular antibodies included: Ly6C (HK1.4, Biolegend), I-A/I-E (M5/114.15.2, BD Biosciences), XCR1 (ZET, Biolegend), CD11b (M1/70, Biolegend), CD103 (2E7, Biolegend), CD45 (30-F11, BD Biosciences), F4/80 (BM8, Biolegend), CD11c (HL3, BD Biosciences), CD172a/SIRPα (P84, Biolegend), Ly6G (1A8, Biolegend and BD Biosciences), PD-L1 (10 F.9G2, Biolegend), CD301b (URA-1, Biolegend), CD3 (145-2C11, Biolegend), CD19 (1D3, Biolegend), NK1.1 (PK136, Biolegend), NKp46 (29A1.4, Biolegend) CD64 (X54-5/7.1, Biolegend), CD80 (16-10A1, Biolegend) and CD86 (GL-1, BD Biosciences). All antibodies for flow cytometry were used at a 1:200 dilution. After staining, cells were washed and resuspended with FACS buffer for flow cytometry analysis using a BD LSRFortessa or Cytek Aurora cell analyser. FACS Diva or SpectroFlo software was used for data acquisition. Collected flow cytometry data were analysed using FlowJo.
Synthetic peptides
Synthetic peptides representing neoantigens for lymphocyte restimulation assays and vaccination were synthesized by and purchased from Peptide v.2.0. All peptides were above or equal to 95% purity.
Statistics and reproducibility
Statistical analyses and P value calculations were performed using GraphPad Prism v.9 and v.10. For each experiment, the particular statistical analysis is detailed in the respective figure legend. A two-tailed unpaired Student’s t-test, one-way analysis of variance (ANOVA) or two-way ANOVA with appropriate post hoc test was used for data that were roughly normally distributed. For analysis of Kaplan–Meier survival experiments, the log-rank (Mantel–Cox) test was used. All analyses were two-tailed. For all statistical analyses, NS denotes not significant, which is P > 0.05.
For Fig. 4a and Extended Data Fig. 7b, immunoblot data are representative of four independent experiments. In Extended Data Figs. 1b and 2h, immunoblot data are representative of three independent experiments. Immunofluorescence data in Extended Data Fig. 2i are representative of three independent experiments. Histology data in Extended Data Figs. 5h and 8f are representative of three independent experiments. All other results in the paper were replicated at least two to three times in independent experiments.
Biological materials availability
Reasonable requests for biological materials used in this study will be promptly reviewed by Columbia Technology Ventures to verify whether the request is subject to any intellectual property or confidentiality obligations. Any materials that can be shared will be released through a material transfer agreement.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at 10.1038/s41586-024-08033-4.
Supplementary information
This file contains all Supplementary Information for the article in Figs. 1–10. Figs. 1 and 2 contain raw immunoblots. Figs. 3–10 contain the gating strategies used for flow-cytometric immunophenotyping experiments.
Source data
Acknowledgements
Research reported in this publication was performed in the Columbia University Department of Microbiology and Immunology Flow Cytometry core facility, the Columbia Center for Translational Immunology (CCTI) and the Herbert Irving Comprehensive Cancer Center (HICCC) Flow Cytometry core facility. Grant numbers NIH/NCI R01CA249160, NIH/NCI R01CA259634, NIH/NCI U01CA247573, NIH/NIGMS T32GM145766, NIH S10OD030282, Searle Scholars Program SSP-2017-2179, Cancer Research Institute CRI 3446, Pershing Square Foundation PSSCRA CU20-0730 and a Roy and Diana Vagelos Precision Medicine Pilot grant supported the study.
Extended data figures and tables
Author contributions
A.R., T.D. and N.A. conceived the study. A.R. designed the system and experiments, conducted sequencing analysis and neoantigen selection, constructed all therapeutics and performed most experiments. A.R. and J.I. designed microbial genetic knockouts. A.R. and B.R. designed and performed all in vitro and ex vivo T cell assays, and flow-cytometric immunophenotyping. A.R., J.I., S.H., M.K., Y.J., W.S. and A.V. performed cloning. J.I., S.H., M.K. and Y.J. performed knockouts. M.R., F.L. and J.I. assisted with sample processing for immunophenotyping. J.I., W.S., Z.S., C.R.G., S.H., J.H., E.R.B., R.L.V., M.K. and A.V. assisted with some in vitro, in vivo and/or ex vivo experiments. J.I., Z.S., S.H., M.K. and Y.J. performed blood bactericidal assays and qPCR. A.R. and M.R. performed immunofluorescence studies. A.R., T.D. and N.A. analysed data and wrote the paper with input from all authors.
Peer review
Peer review information
Nature thanks Paul Robbins and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Data availability
Whole-exome sequencing and RNA sequencing data generated for this study are deposited under umbrella BioProject accession number PRJNA1025007. Whole-exome data are available on the Short Read Archive with BioProject ID PRJNA1024050. RNA sequencing data are available on National Center for Biotechnology Information Gene Expression Omnibus with accession number GSE244808. All other data are available within the article or its Supplementary Information. Datasets used for analyses in this study were: Ensembl release 102 M. musculus GRCm38 gene annotations (GRCm38, https://useast.ensembl.org/Mus_musculus/Info/Index, accession GCA_000001635.8) and dbSNP build 142 (ftp.ncbi.nih.gov/snp). Source data are provided with this paper.
Competing interests
A.R., J.I., T.D. and N.A. have filed a provisional patent application with the US Patent and Trademark Office related to this work. The other authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Andrew Redenti, Jongwon Im
Contributor Information
Tal Danino, Email: td2506@columbia.edu.
Nicholas Arpaia, Email: na2697@cumc.columbia.edu.
Extended data
is available for this paper at 10.1038/s41586-024-08033-4.
Supplementary information
The online version contains supplementary material available at 10.1038/s41586-024-08033-4.
References
- 1.Canale, F. P. et al. Metabolic modulation of tumours with engineered bacteria for immunotherapy. Nature598, 662–666 (2021). [DOI] [PubMed] [Google Scholar]
- 2.Chen, Y. E. et al. Engineered skin bacteria induce antitumor T cell responses against melanoma. Science380, 203–210 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nejman, D. et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science368, 973–980 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pawelek, J. M., Low, K. B. & Bermudes, D. Tumor-targeted Salmonella as a novel anticancer vector. Cancer Res.57, 4537–4544 (1997). [PubMed] [Google Scholar]
- 5.Naghavian, R. et al. Microbial peptides activate tumour-infiltrating lymphocytes in glioblastoma. Nature10.1038/s41586-023-06081-w (2023). [DOI] [PMC free article] [PubMed]
- 6.Kalaora, S. et al. Identification of bacteria-derived HLA-bound peptides in melanoma. Nature592, 138–143 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stark, J. C. et al. On-demand biomanufacturing of protective conjugate vaccines. Sci. Adv.7, eabe9444 (2021). [DOI] [PMC free article] [PubMed]
- 8.Iwasaki, A. & Medzhitov, R. Control of adaptive immunity by the innate immune system. Nat. Immunol.16, 343–353 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schumacher, T. N. & Schreiber, R. D. Neoantigens in cancer immunotherapy. Science348, 69–74 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Sahin, U. et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature547, 222–226 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Rojas, L. A. et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature618, 144–150 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ott, P. A. et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature547, 217–221 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sellars, M. C., Wu, C. J. & Fritsch, E. F. Cancer vaccines: building a bridge over troubled waters. Cell185, 2770–2788 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hundal, J. et al. pVACtools: a computational toolkit to identify and visualize cancer neoantigens. Cancer Immunol. Res.8, 409–420 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kreiter, S. et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature520, 692–696 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee, M. Y., Jeon, J. W., Sievers, C. & Allen, C. T. Antigen processing and presentation in cancer immunotherapy. J. Immunother. Cancer8, e001111 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duan, F. et al. Genomic and bioinformatic profiling of mutational neoepitopes reveals new rules to predict anticancer immunogenicity. J. Exp. Med.211, 2231–2248 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alspach, E. et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature574, 696–701 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kruse, B. et al. CD4+ T cell-induced inflammatory cell death controls immune-evasive tumours. Nature618, 1033–1040 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wells, D. K. et al. Key parameters of tumor epitope immunogenicity revealed through a consortium approach improve neoantigen prediction. Cell183, 818–834.e13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zwaveling, S. et al. Established human papillomavirus type 16-expressing tumors are effectively eradicated following vaccination with long peptides. J. Immunol.169, 350–358 (2002). [DOI] [PubMed] [Google Scholar]
- 22.Fedorec, A. J. H. et al. Two new plasmid post-segregational killing mechanisms for the implementation of synthetic gene networks in Escherichia coli. iScience14, 323–334 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zinkernagel, R. M. et al. Antigen localisation regulates immune responses in a dose- and time-dependent fashion: a geographical view of immune reactivity. Immunol. Rev.156, 199–209 (1997). [DOI] [PubMed] [Google Scholar]
- 24.Henrickson, S. E. et al. T cell sensing of antigen dose governs interactive behavior with dendritic cells and sets a threshold for T cell activation. Nat. Immunol.9, 282–291 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yewdell, J. W. & Bennink, J. R. Immunodominance in major histocompatibility complex class I-restricted T lymphocyte responses. Annu. Rev. Immunol.17, 51–88 (1999). [DOI] [PubMed] [Google Scholar]
- 26.Daegelen, P., Studier, F. W., Lenski, R. E., Cure, S. & Kim, J. F. Tracing ancestors and relatives of Escherichia coli B, and the derivation of B strains REL606 and BL21(DE3). J. Mol. Biol.394, 634–643 (2009). [DOI] [PubMed] [Google Scholar]
- 27.Kan, A., Gelfat, I., Emani, S., Praveschotinunt, P. & Joshi, N. S. Plasmid vectors for in vivo selection-free use with the probiotic E. coli Nissle 1917. ACS Synth. Biol.10, 94–106 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Russo, T. A. & Singh, G. An extraintestinal, pathogenic isolate of Escherichia coli (O4/K54/H5) can produce a group 1 capsule which is divergently regulated from its constitutively produced group 2, K54 capsular polysaccharide. J. Bacteriol.175, 7617–7623 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Niba, E. T. E., Naka, Y., Nagase, M., Mori, H. & Kitakawa, M. A genome-wide approach to identify the genes involved in biofilm formation in E. coli. DNA Res.14, 237–246 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Riva, R., Korhonen, T. K. & Meri, S. The outer membrane protease PgtE of Salmonella enterica interferes with the alternative complement pathway by cleaving factors B and H. Front. Microbiol.6, 63 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lewis, K. Riddle of biofilm resistance. Antimicrob. Agents Chemother.45, 999–1007 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Higgins, D. E., Shastri, N. & Portnoy, D. A. Delivery of protein to the cytosol of macrophages using Escherichia coli K-12. Mol. Microbiol.31, 1631–1641 (1999). [DOI] [PubMed] [Google Scholar]
- 33.Wallecha, A. et al. Listeria monocytogenes-derived listeriolysin O has pathogen-associated molecular pattern-like properties independent of its hemolytic ability. Clin. Vaccine Immunol.20, 77–84 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kang, S.-J., Liang, H.-E., Reizis, B. & Locksley, R. M. Regulation of hierarchical clustering and activation of innate immune cells by dendritic cells. Immunity29, 819–833 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamamoto, K. et al. Listeriolysin O, a cytolysin derived from Listeria monocytogenes, inhibits generation of ovalbumin-specific Th2 immune response by skewing maturation of antigen-specific T cells into Th1 cells. Clin. Exp. Immunol.142, 268–274 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harris, D. P. et al. Reciprocal regulation of polarized cytokine production by effector B and T cells. Nat. Immunol.1, 475–482 (2000). [DOI] [PubMed] [Google Scholar]
- 37.Ding, Q., Mohib, K., Kuchroo, V. K. & Rothstein, D. M. TIM-4 identifies IFN-γ-expressing proinflammatory B effector 1 cells that promote tumor and allograft rejection. J. Immunol.199, 2585–2595 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vanderlugt, C. L. & Miller, S. D. Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat. Rev. Immunol.2, 85–95 (2002). [DOI] [PubMed] [Google Scholar]
- 39.Gabrilovich, D. I., Ostrand-Rosenberg, S. & Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol.12, 253–268 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.He, G. et al. Peritumoural neutrophils negatively regulate adaptive immunity via the PD-L1/PD-1 signalling pathway in hepatocellular carcinoma. J. Exp. Clin. Cancer Res.34, 141 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tanaka, A. & Sakaguchi, S. Regulatory T cells in cancer immunotherapy. Cell Res.27, 109–118 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang, B. et al. Transition of tumor-associated macrophages from MHC class II(hi) to MHC class II(low) mediates tumor progression in mice. BMC Immunol.12, 43 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peng, Q. et al. PD-L1 on dendritic cells attenuates T cell activation and regulates response to immune checkpoint blockade. Nat. Commun.11, 4835 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bod, L. et al. B-cell-specific checkpoint molecules that regulate anti-tumour immunity. Nature619, 348–356 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sette, A. et al. The relationship between class I binding affinity and immunogenicity of potential cytotoxic T cell epitopes. J. Immunol.153, 5586–5592 (1994). [PubMed] [Google Scholar]
- 46.Bassani-Sternberg, M. et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat. Commun.7, 13404 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tran, E. et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science350, 1387–1390 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sachs, A. et al. Impact of cysteine residues on MHC binding predictions and recognition by tumor-reactive T cells. J. Immunol.205, 539–549 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arbelaez, C. A. et al. A nanoparticle vaccine that targets neoantigen peptides to lymphoid tissues elicits robust antitumor T cell responses. NPJ Vaccines5, 106 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.D’Alise, A. M. et al. Adenoviral vaccine targeting multiple neoantigens as strategy to eradicate large tumors combined with checkpoint blockade. Nat. Commun.10, 2688 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Salomon, N. et al. A liposomal RNA vaccine inducing neoantigen-specific CD4+ T cells augments the antitumor activity of local radiotherapy in mice. OncoImmunology9, 1771925 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rosenberg, S. A. et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res.17, 4550–4557 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reinhard, K. et al. An RNA vaccine drives expansion and efficacy of claudin-CAR-T cells against solid tumors. Science367, 446–453 (2020). [DOI] [PubMed] [Google Scholar]
- 54.Mackensen, A. et al. CLDN6-specific CAR-T cells plus amplifying RNA vaccine in relapsed or refractory solid tumors: the phase 1 BNT211-01 trial. Nat. Med.29, 2844–2853 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.O’Donnell, J. S., Teng, M. W. L. & Smyth, M. J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol.16, 151–167 (2019). [DOI] [PubMed] [Google Scholar]
- 56.Gnirke, A. et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat. Biotechnol.27, 182–189 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miller, N. A. et al. A 26-hour system of highly sensitive whole genome sequencing for emergency management of genetic diseases. Genome Med.7, 100 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Van der Auwera, G. A. & O’Connor, B. D. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra (O’Reilly Media, 2020).
- 60.Sherry, S. T. et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res.29, 308–311 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics27, 2987–2993 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol.15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Khanna, A. et al. Bam-readcount—rapid generation of basepair-resolution sequence metrics. J. Open Source Softw.7, 3722 (2022).
- 65.McLaren, W. et al. The Ensembl Variant Effect Predictor. Genome Biol.17, 122 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Reynisson, B., Alvarez, B., Paul, S., Peters, B. & Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res.48, W449–W454 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Andreatta, M. & Nielsen, M. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics32, 511–517 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Reynisson, B. et al. Improved prediction of MHC II antigen presentation through integration and motif deconvolution of mass spectrometry MHC eluted ligand data. J. Proteome Res.19, 2304–2315 (2020). [DOI] [PubMed] [Google Scholar]
- 69.Nielsen, M. & Lund, O. NN-align. An artificial neural network-based alignment algorithm for MHC class II peptide binding prediction. BMC Bioinformatics10, 296 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Danino, T. et al. Programmable probiotics for detection of cancer in urine. Sci. Transl. Med.7, 289ra84–289ra84 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gurbatri, C. R. et al. Engineered probiotics for local tumor delivery of checkpoint blockade nanobodies. Sci. Transl. Med.12, eaax0876 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Datsenko, K. A. & Wanner, B. L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl Acad. Sci. USA97, 6640–6645 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Reisner, A., Krogfelt, K. A., Klein, B. M., Zechner, E. L. & Molin, S. In vitro biofilm formation of commensal and pathogenic Escherichia coli strains: impact of environmental and genetic factors. J. Bacteriol.188, 3572–3581 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Boero, E. et al. Use of flow cytometry to evaluate phagocytosis of Staphylococcus aureus by human neutrophils. Front. Immunol.12, 635825 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang, X., Goncalves, R. & Mosser, D. M. The isolation and characterization of murine macrophages. Curr. Protoc. Immunol.Chapter 14, 14.1.1–14.1.14 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Inaba, K. et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J. Exp. Med.176, 1693–1702 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Quah, B. J. C., Warren, H. S. & Parish, C. R. Monitoring lymphocyte proliferation in vitro and in vivo with the intracellular fluorescent dye carboxyfluorescein diacetate succinimidyl ester. Nat. Protoc.2, 2049–2056 (2007). [DOI] [PubMed] [Google Scholar]
- 78.Mengaud, J. et al. Expression in Escherichia coli and sequence analysis of the listeriolysin O determinant of Listeria monocytogenes. Infect. Immun.56, 766–772 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gubin, M. M. et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature515, 577–581 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hailemichael, Y. et al. Persistent antigen at vaccination sites induces tumor-specific CD8+T cell sequestration, dysfunction and deletion. Nat. Med.19, 465–472 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fu, X. et al. A simple and sensitive method for measuring tumor-specific T cell cytotoxicity. PLoS ONE5, e11867 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This file contains all Supplementary Information for the article in Figs. 1–10. Figs. 1 and 2 contain raw immunoblots. Figs. 3–10 contain the gating strategies used for flow-cytometric immunophenotyping experiments.
Data Availability Statement
Whole-exome sequencing and RNA sequencing data generated for this study are deposited under umbrella BioProject accession number PRJNA1025007. Whole-exome data are available on the Short Read Archive with BioProject ID PRJNA1024050. RNA sequencing data are available on National Center for Biotechnology Information Gene Expression Omnibus with accession number GSE244808. All other data are available within the article or its Supplementary Information. Datasets used for analyses in this study were: Ensembl release 102 M. musculus GRCm38 gene annotations (GRCm38, https://useast.ensembl.org/Mus_musculus/Info/Index, accession GCA_000001635.8) and dbSNP build 142 (ftp.ncbi.nih.gov/snp). Source data are provided with this paper.