

Graphical abstract

Inhaled MK-5475 is proposed to directly stimulate soluble guanylate cyclase (sGC), independently of and synergistically with nitric oxide (NO), to increase levels of intracellular cyclic guanosine monophosphate (cGMP). Treatment with inhaled MK-5475 at 100 µg and 380 μg significantly reduced pulmonary vascular resistance (PVR) in participants with pulmonary arterial hypertension (PAH). Change in 6-min walk distance (6MWD) was not significant. Treatment with MK-5475 at all three doses was well tolerated. Error bars in graphs represent 95% confidence intervals. AE: adverse event; AEOI: adverse event of interest; GTP: guanosine triphosphate; PDE5: phosphodiesterase 5; SAE: serious adverse event.
Abstract
Background
MK-5475 is an investigational inhaled soluble guanylate cyclase stimulator hypothesised to avoid most side-effects of systemic vasodilation.
Methods
The phase 2 INSIGNIA-PAH (NCT04732221) trial randomised adults with pulmonary arterial hypertension (PAH) on stable background therapy 1:1:1:1 to once-daily dosing with placebo, MK-5475 32 µg, 100 µg or 380 µg via dry powder inhalation for 12 weeks.
Objectives
The objectives were to evaluate pulmonary vascular resistance (PVR; primary), 6-min walk distance (6MWD; secondary), additional selected haemodynamic parameters, and safety and tolerability in participants with PAH.
Results
168 participants were randomised to placebo (n=41), MK-5475 32 µg (n=42), 100 µg (n=44), and 380 µg (n=41). Median age was 51 years. Most participants were female (73.8%), diagnosed with idiopathic PAH (63.7%), receiving concomitant phosphodiesterase type 5 inhibitors (PDE5i; 93.5%), and treated with double or triple combination therapy (85.1%). At week 12, the placebo-corrected changes in PVR by least-squares means were −9.2% (95% CI −21.3%, 2.9%; p=0.068) with 32 µg, −22.0% (95% CI −33.7%, −10.3%; p<0.001) with 100 µg, and −19.9% (95% CI −33.4%, −6.4%; p=0.002) with 380 µg MK-5475. No treatment differences versus placebo were observed in 6MWD. Treatment-related adverse events and serious adverse events were similar across treatment groups. Three participants died: two on placebo and one on MK-5475 100 µg. One participant had symptomatic hypotension and one had haemoptysis (both on MK-5475 100 µg).
Conclusions
In participants with PAH on stable background therapy, including PDE5i, inhaled MK-5475 reduced PVR and was well tolerated, without evidence of systemic side-effects such as hypotension, suggesting a pulmonary selective pharmacodynamic effect.
Shareable abstract
The inhaled soluble guanylate cyclase stimulator MK-5475 reduced PVR and was well tolerated in patients with PAH, without evidence of systemic side-effects such as hypotension, suggesting a pulmonary selective pharmacodynamic effect https://bit.ly/4dJ2nAs
Introduction
Pulmonary arterial hypertension (PAH) is a debilitating, progressive disease marked by the remodelling and narrowing of the pulmonary vasculature [1–4]. Approved treatments for PAH primarily target the endothelin, prostacyclin or nitric oxide pathways, either to reduce vasoconstriction (e.g. endothelin receptor antagonists) or to promote vasodilation (e.g. prostacyclin analogues, prostacyclin receptor agonists, phosphodiesterase type 5 inhibitors (PDE5i) and soluble guanylate cyclase (sGC) stimulators) [5]. Most PAH therapies are administered systemically by oral, subcutaneous or intravenous routes, which may result in systemic side-effects that limit their use or dose increases. Approved inhaled treatment options exist but require frequent administration and can be associated with systemic side-effects.
Drugs that inhibit PDE5 or stimulate sGC both act on the nitric oxide–sGC–cyclic guanosine monophosphate (NO–sGC–cGMP) pathway to increase the production of cGMP, which has an important physiological effect on tissue relaxation in vascular smooth muscle cells [6, 7]. Riociguat is an oral sGC stimulator that has been clinically validated for the treatment of PAH and chronic thromboembolic pulmonary hypertension [8, 9]. The combined use of riociguat and oral PDE5i is contraindicated due to the reported increased risk of hypotensive adverse events (AEs) [10]. The inability to co-administer some PAH therapies limits the therapeutic options for a patient population that often requires combination regimens for maximum effectiveness [5]. Thus, a pulmonary selective sGC stimulator may represent a beneficial therapy option for patients with PAH.
MK-5475 is a small molecule stimulator of sGC formulated for delivery by dry powder inhalation. The inhaled administration of MK-5475 optimises the deposition of the drug deep in lung tissue at the site of action, potentially minimising the occurrence of side-effects associated with systemic vasodilation. In a phase 1 study of MK-5475 (NCT03744637) in participants with PAH, there were no reports of hypotension or serious AEs among participants who received a single inhaled dose of MK-5475 (n=19) versus placebo (n=6) [11]. Furthermore, reductions in pulmonary vascular resistance (PVR) were observed across a range of MK-5475 doses from 120 µg to 360 µg. Based on the route of administration and pulmonary selective attributes of MK-5475, it is hypothesised that the drug may have a favourable benefit–risk profile.
Here we report the phase 2 results of a randomised, controlled study of inhaled MK-5475 for the treatment of PAH in participants on stable background PAH therapy.
Methods
Study design
INSIGNIA-PAH (NCT04732221) was a two-part phase 2/3, multicentre, randomised, double-blind, placebo-controlled study that evaluated the efficacy and safety of MK-5475, an inhaled sGC stimulator, for the treatment of PAH (figure 1 and supplementary table S1). This report describes the phase 2 part of the INSIGNIA-PAH study. The date of data cut-off was 4 January 2024, after the last participant enrolled completed the 12-week base period. The phase 2 part comprised a 4-week screening period, 12-week base period, an extension period up to 40 months, and a 2-week follow-up safety period. In the base period, participants were randomised 1:1:1:1 to once-daily dosing with placebo or one of three doses of MK-5475 (32 µg, 100 µg or 380 µg) administered via dry powder inhalation. Participants were trained by site staff during the screening period on the proper use of the dry powder inhaler. Participants self-administered the first dose of study intervention under the guidance of site staff, and then self-administered at home for the rest of the study. Intervention randomisation was stratified by World Health Organization (WHO) functional class at the time of randomisation (class II or class III/IV). The primary objective was to evaluate the effect of MK-5475 versus placebo on pulmonary vascular resistance (PVR) at week 12. PVR assessments were conducted using right heart catheterisation (RHC) during screening and centrally reviewed.
FIGURE 1.

INSIGNIA-PAH PN007 phase 2 study schema. PAH: pulmonary arterial hypertension; R: initial randomisation; R2: re-randomisation at the end of the base period; WHO FC: World Health Organization functional class.
Secondary objectives included evaluation of treatment effect on 6-min walk distance (6MWD) and haemodynamic parameters of mean right atrial pressure (mRAP), cardiac index and stroke volume index (SVI) at week 12. Assessment of safety and tolerability was also a secondary objective and was evaluated by clinical review of relevant parameters, including AEs and discontinuation due to AEs. AEs associated with symptomatic hypotension, pulmonary haemorrhage, haemoptysis and drug-induced liver injury were prespecified events of clinical interest.
Exploratory objectives included, among others (supplementary table S2), the evaluation of change from baseline in distribution of WHO functional class and change from baseline in N-terminal prohormone of brain natriuretic peptide (NT-proBNP) levels. Health-related quality of life was measured with a disease-specific patient-reported outcome instrument, the Living with Pulmonary Hypertension (LPH) questionnaire [12], with change from baseline at 12 weeks for the Physical Dimension subscale and the total LPH scores assessed. Anchor-based responder definitions have been calculated to be an absolute value score change of 1.48–3.69 in the LPH Physical Dimension score and 4.41–11.02 in the LPH total score [12]. Post hoc analyses were carried out to identify differences between intervention groups on additional haemodynamic parameters.
In the extension period, participants who received placebo during the base period were re-randomised 1:1:1 to one of three doses of MK-5475 (32 µg, 100 µg or 380 µg). Participants who received MK-5475 during the base period continued at the same dose and were assigned new randomisation numbers to maintain study blinding. The objective of the blinded extension period was to evaluate longer-term safety and tolerability of MK-5475. After the last study visit in the extension period, a telephone follow-up visit was made for AE monitoring (follow-up safety period).
The study was conducted in compliance with global standards, local and/or national regulations (including all applicable data protection laws and regulations), the International Council for Harmonisation (ICH) Good Clinical Practice, the ICH General Considerations for Clinical Studies, and in accordance with the ethical principles per the Declaration of Helsinki. All participants provided written informed consent prior to any study-related procedure.
Study participants
Eligible participants were 18–75 years of age (inclusive) with a body mass index of 18.5–40 kg·m−2 and a diagnosis of Group 1 PAH (idiopathic, heritable, drug- or toxin-associated, or associated with connective tissue disease, infection with human immunodeficiency virus, or corrected congenital heart disease) of WHO functional class II–IV. All participants were on stable background PAH-specific therapy with no change in therapy within 90 days or in dosage within 30 days prior to or during the screening period. At screening, participants were required to have a 6MWD between 150 m and 500 m and pre-capillary pulmonary hypertension with mean pulmonary artery pressure (mPAP) ≥25 mmHg, PVR of ≥3 Wood units, and pulmonary capillary wedge pressure or left ventricular end diastolic pressure (LVEDP) ≤15 mmHg. A full list of inclusion and exclusion criteria is provided in supplementary table S3.
Right heart catheterisation
RHC was performed at screening after at least 90 days of stable background PAH therapy and within 30 days prior to screening for eligibility. RHC was also performed at week 12. The RHC procedure included a complete haemodynamic profile including cardiac output (CO), arterial oxygen saturation and mixed venous oxygen saturation (SvO2). If pulmonary artery wedge pressure (PAWP) was deemed unreliable, then left heart catheterisation was performed to measure LVEDP. CO was assessed preferably by the thermodilution method (mean values of at least three consecutive measurements). RHC pressure tracings were anonymised to ensure blinding and subsequently analysed by a blinded independent central reviewer. Prior to transmission of haemodynamic tracing data for central review, sites ensured that all tracings were accompanied by a simultaneous ECG tracing, containing at least three entire respiratory cycles recorded during spontaneous breathing and with the scale of the tracing adjusted to the size of measurement for the respective pressure. PVR was calculated according to the formula PVR=(mPAP−PAWP)/CO using the site-measured averaged CO.
Statistical methods
A sample of 41 participants per group was calculated to be sufficient to provide more than 85% power to demonstrate superiority for each of the MK-5475 treatment comparisons using a one-sided alpha=0.025 and the following assumptions: true percent change from baseline in PVR at week 12 is 30% for each MK-5475 dose versus 0% for placebo, standard deviation of the difference of percent change from baseline in PVR at week 12 between each MK-5475 and placebo is 0.4, and 10% of participants dropped out before week 12.
Efficacy and safety analyses included all randomly assigned participants who received at least one dose of study treatment. Safety analyses included all data through the 12-week base period and the time between the week-12 visit and the start of treatment in the extension period, if enrolled in the extension period, or all data through the 12-week base period and the 2-week post-treatment follow-up period, if not enrolled in the extension period.
The primary analyses of efficacy occurred when all participants completed (or discontinued prior to the end of) the base period. Robust regression modelling [13], adjusted for WHO functional class (II versus III/IV), was used to estimate the difference in percent change from baseline and 95% confidence intervals in PVR (primary endpoint), mean change from baseline and 95% confidence intervals in 6MWD (secondary endpoint) and additional haemodynamic parameters (secondary endpoints and post hoc analyses). Missing observations at week 12 from participants in all treatment groups were imputed based on the observed observations at week 12 from the placebo group. A p-value for the comparison of MK-5475 versus placebo <0.025 (one-sided) was considered statistically significant. The p-value for the primary hypothesis was contingent upon the multiplicity strategy of sequential comparison of the MK-5475 dose groups (in descending order) versus placebo and stopping when statistical significance was not reached. Nominal p-values for secondary endpoints and post hoc analyses were provided as an assessment of strength of evidence. Summary statistics were provided for other haemodynamic parameters. Subgroup analyses were performed to adjust the between-group treatment effect (nominal 95% confidence interval) for the primary endpoint of PVR, by age, sex, PAH subtype, number and type of background PAH therapies, baseline 6MWD, and baseline PVR.
For the safety analysis, between-group differences in percentage of participants with AEs (with 95% confidence intervals) were analysed using the Miettinen and Nurminen method [14]. AEs of interest (symptomatic hypotension, pulmonary haemorrhage, haemoptysis and drug-induced liver injury) were summarised by treatment group, with point estimates and 95% confidence intervals for between-group differences. Additional information on the statistical analyses is provided in the supplementary material.
Results
Participant disposition and baseline characteristics
A total of 168 participants were randomised in the phase 2 base period to placebo (n=41), MK-5475 32 µg (n=42), 100 µg (n=44) and 380 µg (n=41). All randomised participants received at least one dose of study treatment and were included in the analysis population for efficacy and safety. Baseline demographics and clinical characteristics are summarised in table 1. The treatment groups were well-balanced in terms of baseline disease characteristics, except for the relatively low proportion of participants with PAH associated with connective tissue disease in the placebo group. Also, participants in the MK-5475 100 µg group had higher mean levels of NT-proBNP at baseline, and participants in the placebo group had higher mean 6MWD, lower mean PVR and lower mean NT-proBNP at baseline, than the other groups. Mean±sd age at baseline was 49.9±15.0 years. Most participants (64.3%) were in WHO functional class II. At baseline, all participants were on stable background therapy that included a PDE5i (93.5%), an endothelin receptor antagonist (83.9%), a prostacyclin receptor agonist (23.2%) or a parenteral prostacyclin (24.4%). Most participants (85.1%) were on combination PAH treatment, of whom 39.9% were on triple therapy, and 45.2% on double therapy.
TABLE 1.
Demographics and baseline clinical characteristics
| Placebo (n=41) | MK-5475 32 µg (n=42) | MK-5475 100 µg (n=44) | MK-5475 380 µg (n=41) | Total (N=168) | |
|---|---|---|---|---|---|
| Age, years | |||||
| Median (range) | 51.0 (19–73) | 50.5 (18–75) | 51.0 (19–74) | 48.0 (18–75) | 51.0 (18–75) |
| Mean±sd | 52.1±12.7 | 48.1±16.9 | 51.7±14.2 | 47.6±15.9 | 49.9±15.0 |
| Sex | |||||
| Female | 28 (68.3) | 34 (81.0) | 32 (72.7) | 30 (73.2) | 124 (73.8) |
| Male | 13 (31.7) | 8 (19.0) | 12 (27.3) | 11 (26.8) | 44 (26.2) |
| Race | |||||
| White | 32 (78.0) | 34 (81.0) | 41 (93.2) | 36 (87.8) | 143 (85.1) |
| Black or African American | 2 (4.9) | 3 (7.1) | 1 (2.3) | 1 (2.4) | 7 (4.2) |
| Asian | 3 (7.3) | 1 (2.4) | 0 | 0 | 4 (2.4) |
| Native American or Alaska native | 3 (7.3) | 0 | 0 | 0 | 3 (1.8) |
| Multiple | 1 (2.4) | 4 (9.5) | 2 (4.5) | 4 (9.8) | 11 (6.5) |
| Body mass index, kg·m−2 | 26.5# (18.7–40.6) | 29.3 (17.7–39.6) | 25.4 (18.5–38.0) | 25.9 (19.3–39.2) | 26.2 (17.7–40.6) |
| Time since PAH diagnosis, years | 5.0 (0.0–34.0) | 5.5 (0.0–27.0) | 4.6 (0.3–26.0) | 4.0 (0.3–36.0) | 4.8 (0.0–36.0) |
| Classification of PAH | |||||
| Idiopathic | 32 (78.0) | 25 (59.5) | 27 (61.4) | 23 (56.1) | 107 (63.7) |
| Heritable | 4 (9.8) | 5 (11.9) | 5 (11.4) | 4 (9.8) | 18 (10.7) |
| Drug- or toxin-induced | 0 | 3 (7.1) | 0 | 1 (2.4) | 4 (2.4) |
| CTD-PAH | 3 (7.3) | 7 (16.7) | 11 (25.0) | 10 (24.4) | 31 (18.5) |
| CHD-PAH | 2 (4.9) | 2 (4.8) | 1 (2.3) | 3 (7.3) | 8 (4.8) |
| WHO functional class | |||||
| II | 27 (65.9) | 27 (64.3) | 28 (63.6) | 26 (63.4) | 108 (64.3) |
| III/IV¶ | 14 (34.1) | 15 (35.7) | 16 (36.4) | 15 (36.6) | 60 (35.7) |
| Background PAH therapy | |||||
| Monotherapy | 8 (19.5) | 8 (19.0) | 6 (13.6) | 3 (7.3) | 25 (14.9) |
| Double therapy | 14 (34.1) | 15 (35.7) | 23 (52.3) | 24 (58.5) | 76 (45.2) |
| Triple therapy | 19 (46.3) | 19 (45.2) | 15 (34.1) | 14 (34.1) | 67 (39.9) |
| PDE5i | 38 (92.7) | 39 (92.9) | 42 (95.5) | 38 (92.7) | 157 (93.5) |
| ERA | 32 (78.0) | 33 (78.6) | 38 (86.4) | 38 (92.7) | 141 (83.9) |
| Oral prostacyclin | 11 (26.8) | 12 (28.6) | 9 (20.5) | 7 (17.1) | 39 (23.2) |
| i.v./s.c. prostacyclin | 12 (29.3) | 11 (26.2) | 8 (18.2) | 10 (24.4) | 41 (24.4) |
| 6MWD, m | 406.9±71.0 | 378.7±77.1 | 403.1±65.1 | 389.5±71.2 | 394.6±71.4 |
| NT-proBNP, pg·mL−1 | 604.9±902.7 | 621.0±1419.9 | 859.3±1796.9 | 632.0±1134.6 | 682.1±1354.1 |
| PVR, WU | 7.8±3.6 | 8.6±3.7 | 9.0±4.7 | 9.1±5.8 | 8.7±4.5 |
| Mean RAP, mmHg | 8.0±3.3 | 7.7±4.0 | 8.1±4.1 | 8.9±5.8 | 8.1±4.4 |
| Mean PAP, mmHg | 46.3±11.6 | 48.9±11.9 | 49.9±14.6 | 49.8±17.6 | 48.8±14.1 |
| PAWP, mmHg | 10.6±3.3 | 9.9±3.9 | 10.8±3.3 | 10.6±3.0 | 10.5±3.4 |
| Cardiac output, L·min−1 | 5.0±1.3 | 4.8±1.4 | 4.7±1.2 | 5.0±1.7 | 4.9±1.4 |
| Cardiac index, L·min−1·m−2 | 2.8±0.8 | 2.6±0.9 | 2.6±0.7 | 2.8±0.9 | 2.7±0.8 |
| Stroke volume index, mL·m−2 | 37.1±10.6 | 36.0±11.8 | 36.6±8.8 | 38.8±12.3 | 37.1±10.9 |
| PAC, mL·mmHg−1 | 1.5±0.5 | 1.5±0.6 | 1.4±0.4 | 1.8±1.1 | 1.6±0.7 |
| SVR, WU | 16.8±4.8 | 17.7±5.0 | 17.7±5.6 | 16.8±6.7 | 17.3±5.6 |
| SvO2, % | 69.3±7.8 | 70.2±6.3+ | 68.8±7.2+ | 68.4±7.3 | 69.2±7.1 |
| SaO2, % | 94.4±4.4§ | 94.4±3.1§ | 94.3±3.5§ | 93.8±5.4 | 94.2±4.2 |
| SBP, mmHg | 112.5±13.5 | 113.1±15.6 | 114.6±11.0 | 108.2±12.7 | 112.1±13.4 |
Data are presented as n (%), median (range) or mean±sd, unless otherwise indicated. PAH: pulmonary arterial hypertension; CHD-PAH: congenital heart disease-associated PAH; CTD-PAH: connective tissue disease-associated PAH; WHO: World Health Organization; PDE5i: phosphodiesterase type 5 inhibitors; ERA: endothelin receptor antagonists; 6MWD: 6-min walk distance; NT-proBNP: N-terminal prohormone of brain natriuretic peptide; PVR: pulmonary vascular resistance; WU: Wood units; RAP: right atrial pressure; PAP: pulmonary arterial pressure; PAWP: pulmonary artery wedge pressure; PAC: pulmonary arterial compliance; SVR: systemic vascular resistance; SvO2: mixed venous oxygen saturation; SaO2: arterial oxygen saturation; SBP: systolic blood pressure. #: one participant in the placebo group had missing data for body mass index; ¶: none of the participants had WHO functional class IV at the time of randomisation; +: one participant in the MK-5475 32 µg and two participants in the MK-5475 100 µg groups had missing data for SvO2; §: three participants in the placebo, two participants in the MK-5475 32 µg, and two participants in the MK-5475 100 µg groups had missing data for SaO2.
As of the date of data cut-off, 4 January 2024, 164 (97.6%) participants completed the 12-week base period (four (2.4%) of the 168 enrolled participants discontinued from the study during the base period), and 135 (80.4%) participants enrolled in the extension period (supplementary table S4). This report describes findings of the 12-week base period. At this time, the extension period is ongoing, and those findings will be described in a future report.
Primary endpoint of pulmonary vascular resistance
We hypothesised that at least one MK-5475 dose would be superior to placebo in reducing PVR from baseline at week 12. The differences in percent change in PVR between MK-5475 and placebo from baseline at week 12 were significant with MK-5475 dosing at 100 µg (−22.0%, 95% CI −33.7%, −10.3%; p<0.001) and 380 µg (−19.9%, 95% CI −33.4%, −6.4%; p=0.002) but not 32 µg (−9.2%, 95% CI −21.3%, 2.9%; p=0.068) (figure 2 and table 2). The between-group differences in percent change in PVR from baseline at week 12 in subgroups defined by baseline characteristics, including age, sex, WHO functional class, background therapy and other clinical characteristics, generally showed a treatment effect with MK-5475 across subgroups, particularly at the 100 µg and 380 µg doses (supplementary figure S1).
FIGURE 2.
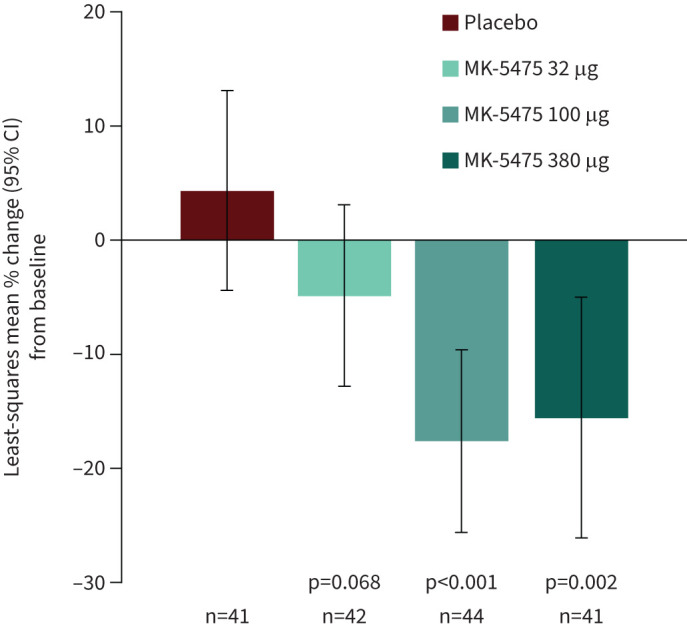
Least-squares mean percent change (with 95% confidence intervals) from baseline at week 12 for the primary endpoint of pulmonary vascular resistance.
TABLE 2.
Primary and secondary efficacy endpoints and other haemodynamic parameters obtained from right heart catheterisation
| Placebo (n=41) | MK-5475 32 µg (n=42) | MK-5475 100 µg (n=44) | MK-5475 380 µg (n=41) | |
|---|---|---|---|---|
| Primary efficacy endpoint | ||||
| PVR, WU | ||||
| LS mean (se) percent change# from baseline, % | 4.3 (4.45) | −4.9 (4.07) | −17.6 (4.07) | −15.6 (5.39) |
| Between-group difference versus placebo (95% CI) | −9.2 (−21.3, 2.9) | −22.0 (−33.7, −10.3) | −19.9 (−33.4, −6.4) | |
| p-value | 0.068 | <0.001 | 0.002 | |
| Secondary efficacy endpoints | ||||
| 6MWD, m | ||||
| LS mean (se) change# from baseline | 14.4 (8.97) | 26.3 (7.02) | 8.8 (7.38) | 9.3 (5.52) |
| Between-group difference versus placebo (95% CI) | 11.9 (−9.9, 33.6) | −5.6 (−25.8, 14.5) | −5.1 (−25.3, 15.1) | |
| p-value | 0.143 | 0.708 | 0.689 | |
| Mean RAP, mmHg | ||||
| LS mean (se) change# from baseline | −0.4 (0.65) | 0.8 (0.62) | −0.6 (0.68) | 0.0 (0.56) |
| Between-group difference versus placebo (95% CI) | 1.2 (−0.5, 2.9) | −0.3 (−2.1, 1.5) | 0.4 (−1.3, 2.0) | |
| p-value | 0.915 | 0.385 | 0.675 | |
| Cardiac index, L·min −1 ·m−2 | ||||
| LS mean (se) change# from baseline | −0.0 (0.09) | 0.1 (0.09) | 0.2 (0.09) | 0.1 (0.12) |
| Between-group difference versus placebo (95% CI) | 0.1 (−0.1, 0.3) | 0.2 (0.0, 0.5) | 0.2 (−0.1, 0.5) | |
| p-value | 0.212 | 0.024 | 0.121 | |
| Stroke volume index, mL·m−2 | ||||
| LS mean (se) change# from baseline | 1.3 (1.14) | 1.8 (1.51) | 3.3 (1.63) | 1.0 (1.74) |
| Between-group difference versus placebo (95% CI) | 0.4 (−3.1, 3.9) | 2.0 (−1.7, 5.7) | −0.3 (−4.3, 3.7) | |
| p-value | 0.403 | 0.148 | 0.563 | |
| Other haemodynamic parameters | ||||
| Mean PAP, mmHg | ||||
| LS mean (se) change# from baseline | 2.5 (1.27) | −1.3 (1.39) | −4.8 (1.13) | −3.5 (1.71) |
| Between-group difference versus placebo (95% CI) | −3.8 (−7.6, −0.0) | −7.4 (−10.6, −4.1) | −6.0 (−10.3, −1.7) | |
| p-value | 0.024 | <0.001 | 0.003 | |
| PAWP, mmHg | ||||
| LS mean (se) change# from baseline | 0.2 (0.65) | 0.9 (0.74) | −0.4 (0.62) | 0.6 (0.96) |
| Between-group difference versus placebo (95% CI) | 0.7 (−1.3, 2.8) | −0.6 (−2.3, 1.1) | 0.4 (−1.9, 2.6) | |
| p-value | 0.233 | 0.755 | 0.378 | |
| Cardiac output, L·min−1 | ||||
| LS mean (se) change# from baseline | −0.1 (0.15) | 0.1 (0.15) | 0.4 (0.17) | 0.2 (0.20) |
| Between-group difference versus placebo (95% CI) | 0.1 (−0.3, 0.5) | 0.4 (−0.0, 0.9) | 0.3 (−0.2, 0.8) | |
| p-value | 0.299 | 0.027 | 0.129 | |
| PAC, mL·mmHg −1 | ||||
| LS mean (se) change# from baseline | 0.1 (0.11) | 0.1 (0.10) | 0.2 (0.09) | −0.1 (0.09) |
| Between-group difference versus placebo (95% CI) | 0.0 (−0.2, 0.3) | 0.1 (−0.2, 0.4) | −0.1 (−0.4, 0.1) | |
| p-value | 0.375 | 0.241 | 0.806 | |
| SVR, WU | ||||
| LS mean (se) change# from baseline | −0.6 (0.61) | −0.8 (0.78) | −0.7 (0.86) | −0.1 (0.76) |
| Between-group difference versus placebo (95% CI) | −0.2 (−2.0, 1.6) | −0.2 (−2.2, 1.9) | 0.5 (−1.4, 2.4) | |
| p-value | 0.416 | 0.438 | 0.693 | |
| SvO2, % | ||||
| LS mean (se) change# from baseline | −3.3 (1.13) | 0.5 (0.82) | 0.2 (0.82) | −0.4 (1.07) |
| Between-group difference versus placebo (95% CI) | 3.7 (1.1, 6.4) | 3.5 (0.7, 6.3) | 2.8 (−0.0, 5.7) | |
| p-value | 0.003 | 0.006 | 0.026 | |
| SaO2, % | ||||
| LS mean (se) change# from baseline | −1.3 (0.89) | 0.2 (0.55) | 0.1 (0.46) | −1.2 (0.57) |
| Between-group difference versus placebo (95% CI) | 1.5 (−0.6, 3.6) | 1.4 (−0.6 3.3) | 0.1 (−1.9, 2.1) | |
| p-value | 0.921 | 0.918 | 0.528 | |
| SBP, mmHg | ||||
| LS mean (se) change# from baseline | −3.1 (2.12) | −1.6 (1.86) | −1.6 (1.64) | −0.0 (1.38) |
| Between-group difference versus placebo (95% CI) | 1.6 (−3.7, 6.9) | 1.5 (−3.5, 6.6) | 3.1 (−1.8, 8.0) | |
| p-value | 0.717 | 0.722 | 0.893 | |
PVR: pulmonary vascular resistance; WU: Wood units; 6MWD: 6-min walk distance; RAP: right atrial pressure; PAP: pulmonary artery pressure; PAWP: pulmonary artery wedge pressure; PAC: pulmonary arterial compliance; SVR: systemic vascular resistance; SvO2: mixed venous oxygen saturation; SaO2: arterial oxygen saturation; SBP: systolic blood pressure. #: by least-squares means obtained from fitting a robust regression estimate based on a Huber-type M estimator including terms for treatment and World Health Organization functional class at baseline. Missing data at week 12 due to death were imputed using the worst observed value and other missingness were imputed using jump-to-reference method. Nominal p-values <0.025 were considered significant.
Secondary and tertiary efficacy endpoints and other haemodynamic parameters
The decline in PVR observed with MK-5475 treatment was mainly driven by significant reductions in mPAP. There were modest non-significant increases in CO and no changes in PAWP. In addition, there were no clinically meaningful effects of MK-5475 on mRAP, cardiac index, SVI, SvO2 or pulmonary arterial compliance. There was also no discernible impact on systemic blood pressure and systemic vascular resistance (table 2).
The mean changes from baseline at week 12 in 6MWD were similar across all treatment arms (figure 3). Relative to placebo, the between-group differences with any of the MK-5475 treatment groups were not significant (table 2).
FIGURE 3.
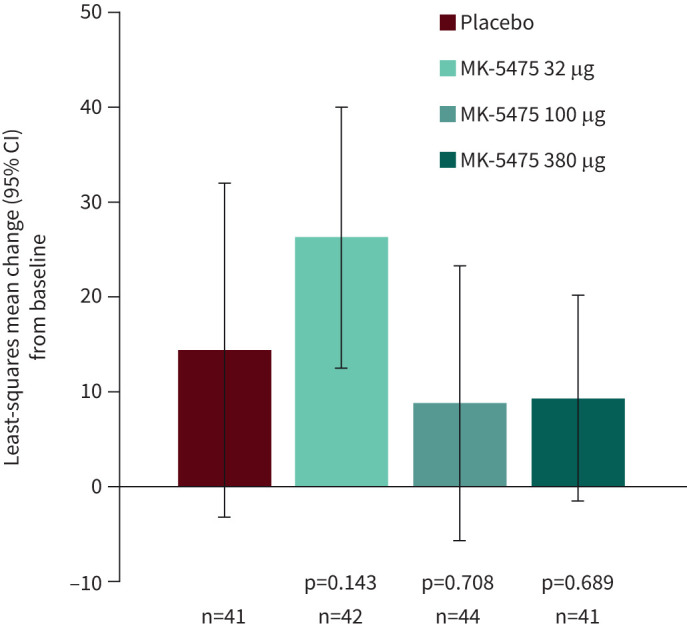
Least-squares mean change (with 95% confidence intervals) from baseline at week 12 for the secondary endpoint of 6-min walk distance.
Most participants had no change in WHO functional class from baseline to week 12, irrespective of treatment group (supplementary table S5).
Compared with placebo, there were non-significant reductions from baseline at week 12 in NT-proBNP levels at all three dose levels (supplementary table S5 and supplementary figure S2).
Safety and tolerability
A summary of treatment-emergent AEs occurring during the 12-week base period is shown in table 3. The incidence of AEs during the base period was similar across treatment arms, with approximately two-thirds of participants in each treatment group reporting one or more AEs. In 19.5% of participants on placebo and 20.5% of participants on MK-5475 (all doses combined), the AEs were determined by the investigator to be related to treatment. One participant on MK-5475 32 µg had an AE of myalgia that led to treatment discontinuation. The most frequent AEs in the study included headache (placebo versus MK-5475 all doses combined: 9.8% versus 13.4%), dyspnoea (9.8% versus 3.1%), diarrhoea (7.3% versus 5.5%), dizziness (7.3% versus 3.1%), vomiting (2.4% versus 4.7%) and nausea (0% versus 5.5%) (table 4).
TABLE 3.
Summary of treatment-emergent adverse events (AEs) during the base period
| Placebo (n=41) | MK-5475 32 µg (n=42) | MK-5475 100 µg (n=44) | MK-5475 380 µg (n=41) | |
|---|---|---|---|---|
| One or more AE# | 27 (65.9) | 28 (66.7) | 28 (63.6) | 27 (65.9) |
| Related¶ to treatment | 8 (19.5) | 12 (28.6) | 8 (18.2) | 6 (14.6) |
| Leading to treatment discontinuation | 0 | 1 (2.4) | 0 | 0 |
| Serious AE+ | 5 (12.2) | 4 (9.5) | 3 (6.8) | 4 (9.8) |
| Related¶ to treatment | 0 | 0 | 0 | 0 |
| Leading to treatment discontinuation | 0 | 0 | 0 | 0 |
| Leading to death | 2 (4.9) | 0 | 1 (2.3) | 0 |
| AE of interest | 0 | 0 | 2 (4.5) | 0 |
| Symptomatic hypotension§ | 0 | 0 | 1 (2.3) | 0 |
| Pulmonary haemorrhage | 0 | 0 | 0 | 0 |
| Haemoptysis | 0 | 0 | 1 (2.3) | 0 |
| Drug-induced livery injury | 0 | 0 | 0 | 0 |
| Serious AE of interest | 0 | 0 | 0 | 0 |
Data are presented as n (%). #: AEs included events occurring during the base period and the 14-day post-treatment follow-up if participants did not continue into the extension period; ¶: determined by the investigator to be related to treatment; +: serious AEs included events occurring during the base period and all post-treatment follow-up if participants did not continue into the extension period; §: symptomatic hypotension is defined as a physical finding of systolic blood pressure <90 mmHg or systolic blood pressure decrease of >40 mmHg below the participant's baseline and accompanied by at least one symptom, such as presyncope, dizziness, mental status changes, etc., as reported by the participant or otherwise witnessed.
TABLE 4.
Adverse events# with ≥5% incidence in any treatment arm
| Placebo (n=41) | MK-5475 32 µg (n=42) | MK-5475 100 µg (n=44) | MK-5475 380 µg (n=41) | Total (N=168) | |
|---|---|---|---|---|---|
| Headache | 4 (9.8) | 3 (7.1) | 8 (18.2) | 6 (14.6) | 21 (12.5) |
| Dyspnoea | 4 (9.8) | 3 (7.1) | 0 | 1 (2.4) | 8 (4.8) |
| Diarrhoea | 3 (7.3) | 2 (4.8) | 2 (4.5) | 3 (7.3) | 10 (6.0) |
| Dizziness | 3 (7.3) | 2 (4.8) | 2 (4.5) | 0 | 7 (4.2) |
| Fatigue | 2 (4.9) | 1 (2.4) | 3 (6.8) | 0 | 6 (3.6) |
| Accidental overdose | 1 (2.4) | 3 (7.1) | 2 (4.5) | 1 (2.4) | 7 (4.2) |
| Vomiting | 1 (2.4) | 3 (7.1) | 1 (2.3) | 2 (4.9) | 7 (4.2) |
| Chest pain | 1 (2.4) | 3 (7.1) | 1 (2.3) | 0 | 5 (3.0) |
| Anaemia | 1 (2.4) | 0 | 3 (6.8) | 0 | 4 (2.4) |
| Myalgia | 0 | 3 (7.1) | 0 | 1 (2.4) | 4 (2.4) |
| COVID-19 | 0 | 1 (2.4) | 3 (6.8) | 3 (7.3) | 7 (4.2) |
| Nausea | 0 | 1 (2.4) | 3 (6.8) | 3 (7.3) | 7 (4.2) |
#: adverse event terms are from the Medical Dictionary for Regulatory Activities (MedDRA) Version 26.1. Participants are counted a single time for each applicable row and column.
Prespecified AEs of interest occurred in two participants in the MK-5475 100 µg treatment arm: one participant had symptomatic hypotension and one had haemoptysis (table 3). Neither of these events was serious, severe, or considered to be related to study treatment. Both events resolved after treatment interruption and did not recur when study treatment was re-introduced. No participant had AEs of clinical interest of pulmonary haemorrhage or drug-induced liver injury.
16 participants experienced a total of 19 serious AEs, including six events occurring in five (12.2%) participants in the placebo arm, four events occurring in four (9.5%) participants, four events occurring in three (6.8%) participants, and five events occurring in four (9.8%) participants in the MK-5475 32 µg, 100 µg and 380 µg arms, respectively (supplementary table S6). None of the serious AEs were related to treatment in the opinion of the investigator and none led to treatment discontinuation. Three of the 16 participants with serious AEs died, two in the placebo arm and one in the MK-5475 100 µg arm. The deaths were attributed to sudden death (placebo), acute respiratory failure (placebo) and malignant lung neoplasm (MK-5475 100 µg arm). None of the deaths were considered related to treatment in the opinion of the investigator.
Health-related quality of life
The physical limitations and other impacts of PAH were evaluated at baseline and week 12 using the disease-specific LPH questionnaire (supplementary table S7). LPH Physical Dimension scores decreased in all three MK-5475 treatment arms, indicating improvements in participants’ physical functioning at 12 weeks, while the placebo group scores increased slightly, indicating a small decline in physical functioning. Improvements in total LPH scores were observed for all study arms, with larger mean change from baseline at 12 weeks for the treatment groups than for the placebo group.
Discussion
The phase 2 part of the INSIGNIA-PAH study evaluated the efficacy, safety and tolerability of placebo versus MK-5475 at 32 µg, 100 µg and 380 µg administered once daily by dry powder inhalation. This study met its primary success criterion: in adults with PAH on stable background therapy for PAH, treatment with inhaled MK-5475 at doses of 100 µg and 380 µg once daily for 12 weeks resulted in significant reductions in PVR from baseline, compared with placebo treatment. A prior study of MK-5475 treatment in participants with PAH showed reductions in PVR after a single administration of the drug at doses ranging from 120 µg to 360 µg [15]. The INSIGNIA-PAH findings expand on prior observations to show the durability of improvements in PVR with multi-day MK-5475 dosing, without negatively affecting the safety profile. The reductions in PVR following MK-5475 treatment in this study were due to significant reductions in mPAP, suggesting improved blood flow in the pulmonary circulation. This study did not find meaningful differences between the placebo and MK-5475 treatment arms in secondary endpoints of 6MWD, mRAP, cardiac index and SVI, or other haemodynamic parameters, such as systemic vascular resistance and systolic blood pressure, suggesting that the effect of MK-5475 treatment is localised to the pulmonary circulation.
These results provide further evidence that MK-5475 is a pulmonary selective vasodilator, having demonstrated a pulmonary pharmacodynamic effect without systemic vasodilatory effects, despite dosing of MK-5475 on top of PDE5i in the vast majority of study participants. Although treatment with MK-5475 in this study did not improve exercise capacity for PAH participants already well-treated with vasodilators, the pulmonary selective pharmacodynamic effect of MK-5475 after inhalation supports the potential utility of the drug in secondary forms of pulmonary hypertension, especially those with limited treatment options and concerns about perfusion/ventilation mismatch, such as pulmonary hypertension associated with COPD [15, 16].
Several factors should be considered when interpreting the lack of discernible differences between the placebo and MK-5475 treatment arms in the secondary efficacy endpoint of 6MWD in this study. In the phase 3 PATENT-1 clinical trial of participants with PAH comparing riociguat versus placebo, improvements in 6MWD (primary endpoint) were reported with active treatment [8]. The PATENT-1 trial, however, differed from the INSIGNIA-PAH study in its inclusion of treatment-naïve participants, accounting for roughly half the study population, and its exclusion of participants on background PDE5i therapy. PATENT-1 was a larger study with sufficient power to evaluate change in 6MWD; the phase 2 INSIGNIA-PAH, a dose-ranging study, was not. Consistent with having systemic vasodilatory effects, treatment with riociguat was associated with the occurrence of hypotension (10% versus 2% with placebo).
Numerous factors, including age, sex, height, weight and comorbidities, are known to influence the results of 6-min walk testing [17]. Researchers have observed also that patients with low baseline 6MWD (<165 m) experience larger gains in 6MWD than those with higher baseline 6MWD (165–440 m and >440 m) [18]. In the INSIGNIA-PAH study, the mean±sd 6MWD for the study population was 394.6±71.4 m at baseline. It is possible that this level of exercise capacity at baseline in a population already on stable background therapy for PAH affected the potential for substantial further gains in 6MWD. Moreover, the placebo group also had a lower proportion of participants with connective tissue disease-associated PAH, which may have impacted the 6MWD results in that group. There remains room for improvement in 6MWD for these individuals, as recently demonstrated in the STELLAR phase 3 study of add-on sotatercept therapy in patients with PAH receiving one, two or three drugs approved for PAH [19].
Regarding exploratory endpoints, most participants had no change in WHO functional class and no significant reductions in NT-proBNP at all three dose levels, compared with placebo. With regards to patient-reported outcomes, there were no clinically meaningful placebo versus treatment arm differences in LPH score change from baseline. Mean changes in LPH score from baseline at week 12 for all the MK-5475 treatment groups fell within the established responder definition range for the physical dimension, indicating improvement [12]. Likewise, the mean change from baseline in LPH total score showed clinically meaningful improvement in the MK-5475 32 µg and 380 µg groups, but not the 100 µg group (although the standard deviation for the total score change was within the anchor-based range for meaningful change). The LPH total score includes an emotional impacts scale, and although this scale has adequate psychometric properties, it has been found to be less associated with clinical measures and less able to discriminate between PAH severity levels [12]. This, along with the small sample sizes of the subgroups, may have contributed to observed lack of dose response in this patient-reported outcome measure and correspondence with haemodynamic results.
Treatment with MK-5475 in the INSIGNIA-PAH study was well-tolerated. The incidences of serious AEs were similar across treatment groups with no discernible pattern. No serious AEs were considered related to treatment in the opinion of the investigator, and no participants discontinued the study or study treatment due to a serious AE. There were three deaths, two in the placebo arm and one in the MK-5475 100 µg arm. None of the deaths was considered related to treatment. Regarding AEs of interest, there was only one occurrence each of symptomatic hypotension and haemoptysis, and none of pulmonary haemorrhage and drug-induced liver injury. The notable infrequency of symptomatic hypotension events with MK-5475 suggests a lack of unwanted systemic vasodilatory effects, which is consistent with the pulmonary selective attributes of inhaled MK-5475. Airway symptoms such as cough that may occur from the direct irritating effects of the treatment on the airways were infrequent (<5%) and observed at the same frequency with MK-5475 and placebo. Beyond its inhaled route of administration, MK-5475 has been shown to have a short half-life, rapid clearance, and minimal accumulation in the systemic circulation with multi-day dosing, suggesting limited systemic exposure [15]. Inhaled MK-5475, therefore, may allow use of an sGC stimulator with an oral PDE5i, a combination that is currently contraindicated for riociguat, an oral sGC stimulator.
The results of the INSIGNIA-PAH study should be interpreted in the context of the following limitations. Participants were responsible for self-reporting their treatment compliance. Treatment compliance was reportedly high in the study (>90% of participants self-reported >95% compliance), and this was reconciled by site staff based on doses remaining in the dry powder inhaler. Treatment compliance was similar across treatment groups. Baseline characteristics were overall well-balanced across treatment arms, although the placebo group had lower mean PVR and mean NT-proBNP levels, and higher mean 6MWD at baseline than other groups, suggesting that the placebo group may have had less severe disease than the MK-5475 groups.
In conclusion, the INSIGNIA-PAH phase 2 study met its primary success criterion. MK-5475 at doses of 100 µg and 380 µg significantly improved PVR, relative to placebo. MK-5475 treatment at all doses studied was well-tolerated, without meaningful systemic side-effects, such as symptomatic hypotension. These findings suggest a pulmonary selective pharmacodynamic effect of MK-5475, an inhaled sGC stimulator, on top of background PAH therapies including PDE5 inhibitors.
Supplementary material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material ERJ-01110-2024.Supplement (455.9KB, pdf)
Shareable PDF
Acknowledgements
The authors thank Anna Lau and Alan Meehan for providing manuscript development support, and Michele McColgan and Jennifer Rotonda for providing administrative assistance. A. Lau, A. Meehan, M. McColgan and J. Rotonda are employees of Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA.
Footnotes
This article has an editorial commentary: https://doi.org/10.1183/13993003.01658-2024
This clinical trial was prospectively registered at ClinicalTrials.gov with identifier NCT04732221.
Ethics approval: The study was conducted in compliance with global standards, local and/or national regulations (including all applicable data protection laws and regulations), the International Council for Harmonisation (ICH) Good Clinical Practice, the ICH General Considerations for Clinical Studies, and in accordance with the ethical principles per the Declaration of Helsinki. All participants provided written informed consent prior to any study-related procedure.
Conflict of interest: M. Humbert reports grants/contracts from Gossamer Bio and Merck & Co., Inc., Rahway, NJ, USA, consultancy for 35 Pharma, Aerovate Therapeutics, Inc., AOP Orphan Pharmaceuticals, Bayer, Chiesi Farmaceutici SpA, Ferrer Internacional SA, Gossamer Bio, Janssen Pharmaceuticals, Keros Therapeutics, Liquidia Corp., Merck & Co., Inc., Rahway, NJ, USA, Novartis, Respira Therapeutics, Roivant Sciences Ltd and United Therapeutics Corp., honoraria from Janssen Pharmaceuticals and Merck & Co., Inc., Rahway, NJ, USA, and participation on a data safety monitoring or advisory board for 35 Pharma, Aerovate Therapeutics, Inc., Janssen Pharmaceuticals, Keros Therapeutics, Merck & Co., Inc., Rahway, NJ, USA, Novartis and United Therapeutics Corp. P.M. Hassoun reports participation on a scientific steering committee for MSD and participation on a scientific advisory board for ARIA-CV. K.M. Chin reports fees for work on steering, advisory or adjudication committees from Gossamer Bio, Janssen, Merck & Co., Inc., Rahway, NJ, USA, and United Therapeutics, and research support to institution for clinical studies overseen by her from Altavant, Gossamer Bio, Janssen, Merck & Co., Inc., Rahway, NJ, USA, and United Therapeutics. G. Bortman reports consultancy for Biosidus Argentina, Tuteur SA, Aerovate Therapeutics Inc., Baliarda Argentina, Merck & Co, Inc., Glaxo Inc. and Tecnopharma/Raffo, Inc. M.J. Patel, C. La Rosa, W. Fu and M.J. Loureiro are current employees of Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA, and may hold stock and/or stock options in Merck & Co., Inc., Rahway, NJ, USA. M.M. Hoeper reports consultancy for Acceleron Pharma, Inc., Actelion Pharmaceuticals, Aerovate, AOP Orphan Pharmaceuticals, Bayer HealthCare, Ferrer Internacional SA, Gossamer Bio, Janssen Global Services, LLC, Keros, Merck & Co., Inc., Rahway, NJ, USA, and Novartis.
Support statement: The study and analyses conducted in this report were funded by Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA. Funding information for this article has been deposited with the Crossref Funder Registry.
Data availability
The data sharing policy, including restrictions, of Merck & Co., Inc., Rahway, NJ, USA is available at https://engagezone.msd.com/ds_documentation.php. Requests for access to the clinical study data can be submitted through the EngageZone site or via email to dataaccess@merck.com
References
- 1.Bousseau S, Sobrano Fais R, Gu S, et al. Pathophysiology and new advances in pulmonary hypertension. BMJ Med 2023; 2: e000137. doi: 10.1136/bmjmed-2022-000137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Humbert M, Guignabert C, Bonnet S, et al. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur Respir J 2019; 53: 1801887. doi: 10.1183/13993003.01887-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leopold JA, Maron BA. Molecular mechanisms of pulmonary vascular remodeling in pulmonary arterial hypertension. Int J Mol Sci 2016; 17: 761. doi: 10.3390/ijms17050761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schermuly RT, Ghofrani HA, Wilkins MR, et al. Mechanisms of disease: pulmonary arterial hypertension. Nat Rev Cardiol 2011; 8: 443–455. doi: 10.1038/nrcardio.2011.87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Humbert M, Kovacs G, Hoeper MM, et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J 2023; 61: 2200879. doi: 10.1183/13993003.00879-2022 [DOI] [PubMed] [Google Scholar]
- 6.Derbyshire ER, Marletta MA. Structure and regulation of soluble guanylate cyclase. Annu Rev Biochem 2012; 81: 533–559. doi: 10.1146/annurev-biochem-050410-100030 [DOI] [PubMed] [Google Scholar]
- 7.Benza RL, Grunig E, Sandner P, et al. The nitric oxide-soluble guanylate cyclase-cGMP pathway in pulmonary hypertension: from PDE5 to soluble guanylate cyclase. Eur Respir Rev 2024; 33: 230183. doi: 10.1183/16000617.0183-2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ghofrani HA, Galie N, Grimminger F, et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med 2013; 369: 330–340. doi: 10.1056/NEJMoa1209655 [DOI] [PubMed] [Google Scholar]
- 9.Ghofrani HA, D'Armini AM, Grimminger F, et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med 2013; 369: 319–329. doi: 10.1056/NEJMoa1209657 [DOI] [PubMed] [Google Scholar]
- 10.Galie N, Muller K, Scalise AV, et al. PATENT PLUS: a blinded, randomised and extension study of riociguat plus sildenafil in pulmonary arterial hypertension. Eur Respir J 2015; 45: 1314–1322. doi: 10.1183/09031936.00105914 [DOI] [PubMed] [Google Scholar]
- 11.Bajwa EK, Cislak D, Palcza J, et al. Effects of an inhaled soluble guanylate cyclase (sGC) stimulator MK-5475 in pulmonary arterial hypertension (PAH). Respir Med 2023; 206: 107065. doi: 10.1016/j.rmed.2022.107065 [DOI] [PubMed] [Google Scholar]
- 12.Bonner N, Abetz L, Meunier J, et al. Development and validation of the Living with Pulmonary Hypertension questionnaire in pulmonary arterial hypertension patients. Health Qual Life Outcomes 2013; 11: 161. doi: 10.1186/1477-7525-11-161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huber PJ. Robust regression: asymptotics, conjectures and Monte Carlo. Ann Statist 1973; 1: 799–821. [Google Scholar]
- 14.Miettinen O, Nurminen M. Comparative analysis of two rates. Stat Med 1985; 4: 213–226. doi: 10.1002/sim.4780040211 [DOI] [PubMed] [Google Scholar]
- 15.Bajwa EK, Cislak D, Kumar A, et al. Phase 1 study of MK-5475, an inhaled soluble guanylate cyclase stimulator, in participants with pulmonary hypertension associated with chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis 2024; 19: 1105–1121. doi: 10.2147/COPD.S454905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.ClinicalTrials.gov. MK-5475-013 INSIGNIA-PH-COPD: a study of the efficacy and safety of MK-5475 (an Inhaled sGC Stimulator) in adults with PH-COPD. Date last accessed: 16 July 2024. Date last updated: 9 August 2024. https://clinicaltrials.gov/study/NCT05612035
- 17.ATS Committee on Proficiency Standards for Clinical Pulmonary Function Laboratories . ATS statement: guidelines for the six-minute walk test. Am J Respir Crit Care Med 2002; 166: 111–117. doi: 10.1164/ajrccm.166.1.at1102 [DOI] [PubMed] [Google Scholar]
- 18.Moutchia J, McClelland RL, Al-Naamani N, et al. Minimal clinically important difference in the 6-minute-walk distance for patients with pulmonary arterial hypertension. Am J Respir Crit Care Med 2023; 207: 1070–1079. doi: 10.1164/rccm.202208-1547OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoeper MM, Badesch DB, Ghofrani HA, et al. Phase 3 trial of sotatercept for treatment of pulmonary arterial hypertension. N Engl J Med 2023; 388: 1478–1490. doi: 10.1056/NEJMoa2213558 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material ERJ-01110-2024.Supplement (455.9KB, pdf)
This PDF extract can be shared freely online.
Shareable PDF ERJ-01110-2024.Shareable (1MB, pdf)
Data Availability Statement
The data sharing policy, including restrictions, of Merck & Co., Inc., Rahway, NJ, USA is available at https://engagezone.msd.com/ds_documentation.php. Requests for access to the clinical study data can be submitted through the EngageZone site or via email to dataaccess@merck.com