

Summary
Understanding the molecular mechanisms of epicardial epithelial-to-mesenchymal transition (EMT), particularly in directing cell fate toward epicardial derivatives, is crucial for regenerative medicine using human induced pluripotent stem cell (iPSC)-derived epicardium. Although transforming growth factor β (TGF-β) plays a pivotal role in epicardial biology, orchestrating EMT during embryonic development via downstream signaling through SMAD proteins, the function of SMAD proteins in the epicardium in maintaining vascular homeostasis or mediating the differentiation of various epicardial-derived cells (EPDCs) is not yet well understood. Our study reveals that TGF-β-independent SMAD3 expression autonomously predicts epicardial cell specification and lineage maintenance, acting as a key mediator in promoting the angiogenic-oriented specification of the epicardium into cardiac pericyte progenitors. This finding uncovers a novel role for SMAD3 in the human epicardium, particularly in generating cardiac pericyte progenitors that enhance cardiac microvasculature angiogenesis. This insight opens new avenues for leveraging epicardial biology in developing more effective cardiac regeneration strategies.
Keywords: SMAD3, EMT, hiPSC, NG2, CD105, cardiac pericyte, epicardium
Graphical abstract
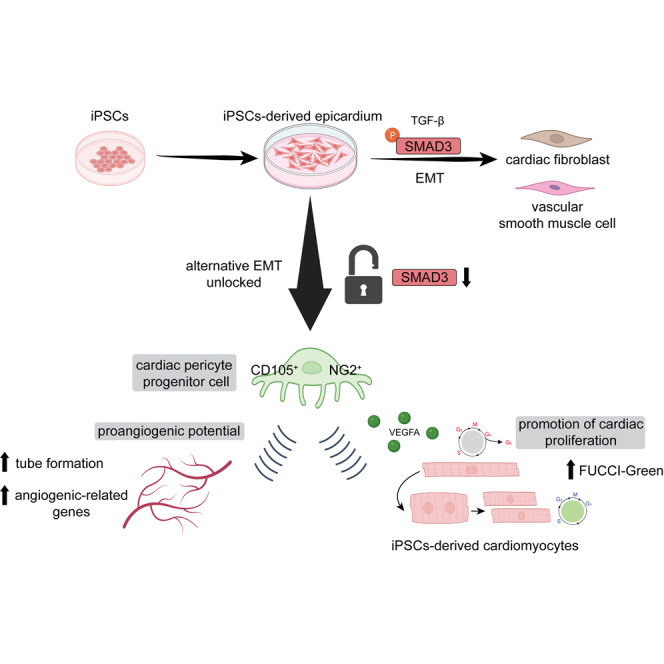
Highlights
-
•
SMAD3 is an independent indicator of successful epicardial lineage establishment
-
•
Loss of SMAD3 in epicardial cells promoted EMT into cardiac pericyte lineage
-
•
SMAD3-downregulating epicardial cells revealed pro-angiogenic potential
-
•
SMAD3-downregulating epicardial cells promoted the proliferation of cardiomyocytes
Miyoshi et al. show that silencing SMAD3 in iPSC-derived epicardial cells promotes epithelial-to-mesenchymal transition (EMT) toward the cardiac pericyte lineage with pro-angiogenic potential. SMAD3-downregulated epicardial cells cooperate with endothelial cells to enhance angiogenesis. Furthermore, these SMAD3-downregulated epicardial cells secrete more VEGFA and promote the proliferation of iPSC-derived cardiomyocytes in a paracrine manner.
Introduction
During cardiac development, the epicardium plays an essential role in the establishment of blood vessels within the heart, a process referred to as coronary angiogenesis (Olivey and Svensson, 2010). The epicardium gives rise to specialized cells known as epicardial-derived cells (EPDCs) (Smits et al., 2018), which contribute significantly to the development of the coronary vasculature (Lim, 2021). These EPDCs possess the remarkable ability to differentiate into various cell types through distinct cell fate signaling pathways that determine cell lineage decisions (Gittenberger-de Groot et al., 2010). These cell types include vascular smooth muscle cells (Iyer et al., 2015), endothelial cells (Quijada et al., 2021), and fibroblasts (Fang et al., 2016), some of which will form the cardiac vasculature. Moreover, some EPDCs can secrete growth factors and signaling molecules that act as potent stimulants for angiogenesis in cardiac repair, fostering the establishment of a functional vasculature (Volz et al., 2015).
Upon cardiac injury, the epicardium swiftly responds by initiating a process known as epicardial activation (van Wijk et al., 2012). In this scenario, activated epicardial cells proliferate and migrate toward the injured site. These cells secrete growth factors, most notably vascular endothelial growth factor (VEGF), which significantly promotes angiogenesis—an essential part of the cardiac healing process (Chung et al., 2015).
Crucial to this process is epicardial epithelial-to-mesenchymal transition (EMT), which serves as a fundamental mechanism for generating EPDCs (Streef and Smits, 2021). This transition is broadly mediated by transforming growth factor β (TGF-β) signaling via structured responses in coordination with phosphorylated SMAD protein cascades (Sridurongrit et al., 2008). The mechanisms of EMT that operate independently of the canonical TGF-β pathway, however, remain largely unexplored. While the SMAD-mediated epicardial EMT is well understood in a global context, with a primary focus on well-represented EPDCs such as cardiac fibroblasts and smooth muscle cells, it is essential to gain a comprehensive understanding of the intricate mechanisms that underlie epicardial EMT-mediated angiogenesis. This knowledge is critical for devising therapeutic strategies aimed at enhancing cardiac repair and recovery following injury.
Human induced pluripotent stem cells (hiPSCs) (Takahashi and Yamanaka, 2006; Takahashi et al., 2007) have revolutionized the field of regeneration and development by providing an accurate model to recreate the biogenesis of challenging-to-study tissues, such as the human epicardium (Witty et al., 2014). Recently, researchers have successfully differentiated hiPSCs into high-quality epicardial cells, thereby opening avenues for exploring epicardial functionality (Bao et al., 2016; Junghof et al., 2022).
In this context, we have utilized hiPSC-derived epicardium to recreate epicardiogenesis in vitro and elucidate the role of SMAD proteins in conditioning epicardial EMT during cell fate decisions. Notably, our findings have unveiled numerous TGF-β-independent functions of SMAD3 in epicardial biology and the priming of cell fate toward angiogenic responses. SMAD3 expression emerged as an independent predictor of successful epicardial differentiation, from the pro-epicardial stage to the full establishment of the fetal program. More importantly, loss of SMAD3 in the fetal epicardium led to a disruption in the epicardial program, initiating an EMT decoupled from TGF-β and SMAD3 phosphorylation. Consequently, this resulted in a pro-angiogenic cell pool enriched in neural/glial antigen 2 (NG2), CD105, CD13, and CD248-expressing cells—functioning as cardiac pericyte progenitor cells (Alex et al., 2022; 2023). These cells exhibited enhanced functionality in supporting primary endothelial cells and promoting increased proliferation of hiPSC-derived ventricular cardiomyocytes.
These findings shed light on novel SMAD3 functions within the human epicardium, endorsing its regenerative and TGF-β-independent capabilities. Furthermore, our research paves the way for the development of engineered epicardial tissues (EETs) primed for enhanced angiogenesis and vascular regeneration—an exciting prospect in the field of cardiac regeneration and tissue engineering.
Results
TGF-β-independent expression of SMAD3 autonomously predicts the development of epicardial cells from induced pluripotent stem cells
To understand the expression dynamics of SMAD genes during TGF-β induction, we first retrospectively analyzed publicly available RNA sequencing (RNA-seq) data (GSE122200) with relevant transcriptomic profiling on epicardial EMT in a mouse epicardial cell line (MEC1). Whereas Smad2 and Smad4 showed none to mild transcriptomic upregulation, Smad3 levels showed a consistent downregulation tendency regardless of the genetic background on the regulatory epicardial EMT axis involving p53-PRMT1, suggesting that despite phospho-Smad3 levels are canonically increased during the onset of the EMT, total gene expression downregulated, indicating an uncoupled TGF-β response with biological implications to be elucidated (Figure 1A). Elevation of some of the TGF-β/SMAD target genes regulating gene expression in the canonical response to EMT initiation was confirmed by the upregulated expression of Skil, Runx3, and Ski (Figure 1B).
Figure 1.

Characterization of SMAD gene expression dynamics in epicardial biology using mouse, human, and hiPSC-derived cellular platforms
(A and B) Retrospective analysis using dataset GSE122200 to examine changes in Smad3, Smad2, and Smad4 (A) and Skil, Runx3, and Ski (B) in response to TGF-β in MEC1 genetic backgrounds with silenced p53 or both Prmt1 and p53.
(C and D) Retrospective analysis of dataset GSE84085 comparing gene expression of SMAD3, SMAD2, and SMAD4 (C) and SKIL, RUNX3, and SKI (D) between human pluripotent stem cell (hPSC)-derived fetal epicardial cells and adult epicardium.
(E) Schematic workflow for the differentiation of epicardial cells from human induced pluripotent stem cells (hiPSCs).
(F) Phase contrast images displaying the cobblestone-like morphology of hiPSC-derived epicardial cells (EPI cells) at day 12 and day 24.
(G) Immunocytochemistry results for WT1, TBX18, and ZO-1 in EPI cells at day 24.
(H) Flow cytometry-derived histograms for WT1 and TBX18 expression with positive ratio in EPI cells at day 24 (red), with gray indicating the unstained control (n = 3; mean ± standard error of the mean [SEM]).
(I) Quantitative reverse-transcription PCR (qRT-PCR) analysis showing upregulation of WT1 and TBX18 through epicardial induction from day 12 to day 24 (n = 3; ∗∗∗p < 0.001).
(J) Protein analysis for WT1 and TBX18 expression at day 12 and day 24.
(K) qRT-PCR analysis indicating the transition of SMAD3, SMAD2, and and SMAD4 at day 12 and day 24 (n = 3; ∗∗∗p = 0.0003; ns, not significant).
(L) Western blot analysis of total and phosphorylated SMAD3 at day 12, day 24, and a positive control (day 24+TGF-β for 1 h). All error bars represent SEM; the graph plots are derived from experimental replicates, obtained from independent batches; statistical analysis was performed using a two-sided unpaired Student’s t test; scale bars equal 100 μm, unless otherwise indicated; see also Figure S1.
Next, to contextualize the expression dynamics of the SMAD genes during epicardial development and maturation, we systemically analyzed the GSE84085 dataset in a differential expression context from pluripotent stem cell (PSC)-derived epicardium compared to donor samples from freshly isolated human adult epicardium. Remarkably, only SMAD3 mRNA expression showed a significant upregulation in the adult samples compared to fetal PSC-derived samples (Figure 1C), indicating that SMAD3 expression is correlative to the degree of maturation of the tissue in a context of inactive EMT and TGF-β blockage, as indicated by the unchanged expression of SMAD targets (Figure 1D).
Next, in order to understand how SMAD3 expression influences the development and maturation of the human embryonic epicardium, we conducted a systematic study on SMAD gene expression dynamics using hiPSCs to create epicardial-like cells.
We initiated the differentiation process by generating epicardial-like cells from hiPSCs (referred to as EPI cells). This was achieved by exposing the cells to Activin A and BMP4, leading to the formation of cardiac mesoderm over the course of days 1–3.5. Subsequently, we induced the specification of epicardial fate by activating the WNT signaling pathway from day 3.5 to day 8. To maintain the characteristic features of epicardial cells and enable the long-term culture of EPI cells without spontaneous differentiation, we inhibited TGF-β signaling using SB431542 starting from days 3.5 (Figure 1E). These cells exhibited the typical cobblestone-like morphology expected by day 24 of differentiation (Figure 1F). Notably, by day 24, EPI cells expressed nuclear WT1 and TBX18, which was confirmed through immunofluorescence co-staining with ZO1 (Figure 1G) and flow cytometry (Figure 1H). We further revealed the upregulation of key markers of epicardial cells, such as WT1 and TBX18, during the differentiation process at both the bulk mRNA level (Figure 1I) and total protein level (Figures 1J and S1A). Finally, we confirmed that only SMAD3 showed a steady progressive expression from the pro-epicardial stage (day 12) to the fetal stage (day 24) (Figures 1K and S1B) in the context of inactive EMT, peaking on this day, and maintaining stable long-term expression with no activation of phosphorylated SMAD3 over time (Figures 1L, S1A, S1C, and S1D). hiPSC-derived epicardial monolayers expressed higher SMAD3 levels than their undifferentiated, pluripotent counterparts (hiPSCs) (Figure S1E). The removal of SB431542 led to a time-dependent loss of epicardial identity, which was irreversible; however, no short-term decrease in SMAD3 was observed. This indicates that the time-dependent increase in SMAD3 within the epicardial lineage was specific to lineage specification and not a compensatory regulation driven by TGF-β inhibitory conditions (Figures S1F and S1G).
Altogether, our data indicate that SMAD3 has TGF-β-independent contributions to epicardial biology and could represent an independent predictor of epicardial maturity.
Next, we wanted to evaluate the specific responses of SMAD3 expression in the context of epicardial EMT onset initiated by TGF-β. To that end, we treated hiPSC-derived epicardium at day 24 with 5 ng/mL of TGF-β for 72 h (Figure 2A) and then, we evaluated gene and protein expression at the endpoint, where epicardial monolayers had visibly undergone a morphological transition to EPDCs (Figure 2B). We confirmed the transcriptomic departure from the epicardial fate by the loss of WT1 and TBX18 (Figure 2C). Given the developmental-driven changes in transcriptomics depending on cell types, we evaluated three different housekeeping controls and confirmed GAPDH as the preferred one for downstream analyses (Figure S2A). This loss of expression in epicardial markers is consistent with the widely characterized cadherin switch during EMT initiation (Kang and Massagué, 2004; Loh et al., 2019) (Figure 2D). We then confirmed the significant expression of some key EPDC markers ACTA2, CNN1, PDGFRA, and TAGLN, both at mRNA and protein levels (Figures 2E, 2F, and S2B). Finally, while we observed unchanged expression in SMAD genes (Figure 2G), a mild decrease in total SMAD3 was noted, as expected via TGF-β-induced ubiquitination mechanisms (Figures 2H and S2B). Importantly, we confirmed an increase in phospho-SMAD3, consistent with a TGF-β-dependent response (Figures 2H and S2B).
Figure 2.

SMAD3 expression dynamics during EMT in hiPSC-derived epicardial monolayers
(A) Description of the experimental workflow for epithelial-to-mesenchymal transition (EMT) induction using TGF-β and the control agent SB431542.
(B) Observations of morphological differences in hiPSC-derived EPI cells treated with TGF-β and SB431542 for 3 days, post day 24.
(C and D) Comparative qRT-PCR analysis of epicardial markers (WT1 and TBX18) (C) and genes CDH1 and CDH2 (D) in cells treated with TGF-β versus SB431542 treatment (n = 3; ∗p < 0.05, ∗∗∗p < 0.001).
(E) qRT-PCR analysis comparing the expression of epicardial-derived cell (EPDC) markers (ACTA2, CNN1, PDGFRA, and TAGLN) between TGF-β and SB431542 treatments (n = 4; ∗∗p = 0.0028, ∗∗∗p < 0.001).
(F) Western blot (Wes) analysis of EPDC markers (α-SMA, CNN1, and TAGLN) comparing TGF-β and SB431542 treatments.
(G) Comparison of qRT-PCR results for SMAD3, SMAD2, and SMAD4 between cells treated with TGF-β and SB431542 (n = 3; ns, not significant).
(H) Western blot analysis of total and phosphorylated SMAD3 in cells treated with TGF-β versus SB431542 (TGF-β treatment for 1 h is used as a positive control). All error bars represent SEM; the graph plots are derived from experimental replicates, obtained from independent batches; statistical analysis was performed using a two-sided unpaired Student’s t test; scale bars equal 100 μm, unless otherwise indicated; see also Figure S2.
SMAD3 overexpression maintains a functional state in fetal epicardial cells without independently promoting either EMT or epicardial maturation
To investigate the role of SMAD3 in epicardial differentiation and maturation, we conducted a 7-day forced expression experiment in day 24 hiPSC-derived epicardial monolayers using a lentivirus expression system (Figure 3A). Following successful transcriptional overexpression confirmation of total SMAD3 and its phosphorylation capacity upon TGF-β treatment (Figures 3B, 3D, and S3A–S3C), we monitored the epicardial morphology throughout the experiment. Interestingly, we did not observe any significant morphological changes either during the initial days of overexpression or at the endpoint (Figure 3C).
Figure 3.

Characterization of hiPSC-derived epicardium during ectopic SMAD3 overexpression
(A) Description of the experimental process for inducing SMAD3 overexpression using a lentivirus.
(B) Validation of SMAD3 overexpression through qRT-PCR analysis (n = 4; ∗∗p = 0.0023).
(C) Time-lapse phase contrast imaging of hiPSC-derived EPI cells at 3 days and 7 days following SMAD3 overexpression.
(D) Western blot analysis of total and phosphorylated SMAD3 in SMAD3-overexpressing EPI cells.
(E) qRT-PCR analysis of epicardial markers, including WT1, TBX18, ALDH1A2, and BNC1 (n = 4; ns, not significant).
(F) Flow cytometry assessment of epicardial markers WT1 and TBX18, showing no significant difference between SMAD3 overexpression (red) and control (gray).
(G) qRT-PCR analysis for genes CDH1 and CDH2 (n = 4; ns, not significant, ∗p = 0.020).
(H) qRT-PCR analysis for mature epicardial markers: UPK3B, MSLN, ITLN1, EFEMP1, and C3 (n = 4; ns, not significant). All error bars represent SEM; the graph plots are derived from experimental replicates, obtained from independent batches; statistical analysis was performed using a two-sided unpaired Student’s t test; scale bars equal 100 μm, unless otherwise indicated; see also Figure S3.
We found unchanged expression levels of epicardial markers such as WT1, TBX18, ALDH1A2, and BNC1, indicating no perturbation in epicardial identity (Figure 3E). Although we confirmed consistent protein expression patterns for WT1 and TBX18 (Figure 3F), several EMT markers (CDH1, SNAI1, SNAI2) remained unchanged, while CDH2 and ZEB1 showed mild upregulation with no observed morphological changes (Figures 3G and S3D). These changes may reflect the effects of SMAD3 overexpression, but they are incomplete as an EMT phenotype. This suggests that SMAD3-driven CDH2 and ZEB1 upregulation could contribute to greater tissue homeostasis in overexpression.
Subsequently, we investigated the markers associated with epicardial maturation—defined by a quiescent tissue with lost EMT functions—and found no significant alterations in UPK3B, MSLN, ITLN1, EFEMP1, and C3 (Du et al., 2023; Knight-Schrijver et al., 2022) (Figure 3H).
Since SMAD3 protein levels are relatively constant from day 24 onward, we also performed SMAD3 overexpression on day 12 monolayers to avoid the peak expression time point. However, we did not find any upregulation in epicardial maturation markers (Figures S3E–S3G) indicative of epicardial quiescence or loss of EMT functionality, as cells remained proliferative and responsive to TGF-β. This suggests that the autonomous expression of SMAD3 alone did not induce epicardial maturation. Therefore, our findings indicate that while sustained SMAD3 expression may signify progress in epicardial specification and maturation, it is insufficient to promote maturation on its own. This implies that epicardial maturation requires a more complex interplay of factors during its developmental process.
Loss of SMAD3 in epicardial cells triggers a poised EMT that leads to the derivation of cardiac pericyte progenitors
To investigate the functional role of SMAD3 in maintaining the epicardial program, we employed a strategy to downregulate SMAD3 expression in day 24 hiPSC-derived epicardial monolayers while maintaining TGF-β blockage (SB431542) to prevent the spontaneous activation of epicardial EMT (Figure 4A). The downregulation of SMAD3 resulted in viable cells (Figure S4A) with noticeable morphological changes with a subset of cells transitioning to an elongated shape and losing the characteristic cobblestone-like morphology, indicative of a shift toward mesenchymal cells (Figure 4B). We confirmed a significant reduction in SMAD3 levels within this cell subset, both at the mRNA and protein levels, 7 days after siRNA expression (Figures 4C, 4D, and S4B). Interestingly, the phosphorylated levels of SMAD3 remained unchanged during these morphological dynamics, suggesting that the cell transition was driven by a direct on-target mechanism of SMAD3 rather than initiating a canonical TGF-β-SMAD pathway-dependent epicardial EMT (Figure 4D).
Figure 4.

Loss of SMAD3 in hiPSC-derived epicardium causes EMT toward cardiac pericyte progenitor cells
(A) Overview of the siRNA transfection experiment and subsequent analysis steps.
(B) Phase contrast microscopy images captured on day 27 (day 24 + 3) and day 31 (day 24 + 7) highlighting spindle-like morphology in hiPSC-derived EPI cells with silenced SMAD3, as indicated by blue arrowheads.
(C) Evidence of SMAD3 downregulation post-siRNA transfection, as shown by qRT-PCR (n = 3; ∗∗∗∗p < 0.0001).
(D) Western blot (Wes) analysis revealing a decrease in total SMAD3 levels with no change in phosphorylated SMAD3.
(E) Immunocytochemistry results for WT1 and ZO-1 7 days following siRNA transfection.
(F) Reduction in WT1 expression due to SMAD3 silencing, as determined by qRT-PCR (n = 3; ∗∗∗p = 0.0006).
(G) Western blot results showing decreased protein expression of WT1 and TBX18 by SMAD3 silencing.
(H) qRT-PCR analysis for ENG (CD105) (n = 5; ∗p < 0.05) and NG2 (n = 5; ∗∗p < 0.01).
(I) Western blot analysis indicating increased levels of CD105, NG2, CD248, and CD13 in SMAD3-silenced EPI cells.
(J and K) Immunocytochemistry images for ENG (CD105) (J) and NG2 (K) comparing SMAD3-silenced and control (scramble) EPI cells 7 days after siRNA transfection.
(L and M) Flow cytometry contour plots analyzing CD105 (L) and NG2 (M) expression.
(N and O) Flow cytometry-derived histograms for CD105 expression (N) and NG2 expression (O) (gray represents the unstained control).
(P) Heatmap displaying a curated gene set associated with the cardiac pericyte lineage in cells with downregulated SMAD3 (log fold change). All error bars represent SEM; the graph plots are derived from experimental replicates, obtained from independent batches; statistical analysis was performed using a two-sided unpaired Student’s t test; scale bars equal 100 μm, unless otherwise indicated; scrambled siRNA was used as a control; see also Figures S4 and S5.
Further confirmation of the departure from epicardial cells was evident through the loss of epicardial markers WT1, ZO-1, and TBX18 (Figures 4E–4G and S4B). Notably, we observed a steady time-dependent transition in cadherin expression and the upregulation of endpoint EMT activation markers such as SNAI1, SNAI2, and ZEB1 at the transcriptional level throughout SMAD3 downregulation, indicating the initiation of EMT (Figure S4C).
To delve deeper into the newly acquired identity of EPDCs, we systematically examined the mRNA expression levels of major epicardial EPDC subtypes, including cardiac fibroblasts, smooth muscle cells, and cardiac pericytes. While markers such as ACTA2, CNN1, TAGLN, and POSTN, as well as the EMT axis p53-PRMT1, showed either unchanged or downregulated expression patterns (Figure S4D), we observed significant overexpression of endoglin (CD105) and CSPG4 (NG2) at both the gene expression and protein levels (Figures 4H, 4I, and S4B). Additionally, enhanced protein expression of CD248 and CD13 (Zhu et al., 2022) was confirmed in SMAD3-downregulating cells (Figures 4I and S4B). These are all known markers associated with the cardiac pericyte fate. Protein expression of CD105 (Figure 4J) and NG2 (Figure 4K) in SMAD3-downregulating bulk monolayers was further verified. Quantitative analysis using fluorescence-activated cell sorting revealed that 8.2% of cells expressed CD105, while 12.9% expressed NG2 (Figures 4L, 4M, and S4E), and these data were plotted in their respective histogram plots (Figures 4N and 4O). The modest representation of cardiac pericyte markers at the protein level may indicate a premature developmental stage of cardiac pericyte progenitor cells, with the bulk population transitioning toward the full establishment of this transcriptional lineage landscape.
To expand our understanding, we conducted a comprehensive transcriptional analysis of SMAD3-downregulating cells, focusing on a curated selection of cardiac pericyte markers to confirm that our condition was significantly enriched in DLK1, CD13, CD146, and CD248, further supporting the presence of cardiac pericyte hallmarks (Figure 4P). Importantly, to dissociate this phenotype from the TGF-β-driven EPDC differentiation into hiPSC-derived cardiac fibroblasts (iCFs) and smooth muscle cells (iSMCs), we verified that the cell pools resulting from TGF-β and basic fibroblast growth factor (bFGF) treatments do not express either CD248 or NG2, while EPDC markers are highly expressed in differentiation condition toward iCF and iSMC cell fates. This confirms that the enrichment of a cardiac pericyte-like fate is indeed an on-target mechanism driven by SMAD3 downregulation (Figures S4F and S4G). Mechanistically, TGF-β can activate both the ALK1 (activin receptor-like kinase 1) and ALK5 (activin receptor-like kinase 5) pathways. TGF-β signals through a family of serine/threonine kinase receptors, which include type I receptors such as ALK1 and ALK5. While the ALK5 pathway usually activates phospho-SMAD2 and 3, the ALK1 pathway is mainly associated with the activation of phospho-SMAD1, 5, and 9. We confirmed that phospho-SMAD2 does not compensate for the activation of the ALK5 pathway in the absence of phospho-SMAD3 (Figures S5A and S5B). Additionally, we confirmed that the ALK1 pathway is not compensating, neither at the mRNA nor protein levels (Figures S5A, S5C, and S5D). Our data support that the nature of this EMT and the increased expression of CD105 are not caused by the relative activation of either the ALK1 or ALK5 pathways, which are key players in TGF-β-mediated signaling.
We also investigated whether the concurrent downregulation of SMAD3 and TGF-β induction could modify cell fate decisions. While TGF-β led the fate trajectory to produce a pool of cells expressing markers of iSMC and iCF as expected, adding siSMAD3 to the experimental condition included CD105, NG2, CD13, and CD248-expressing cells (progenitors of the cardiac pericyte lineage) in the pool (Figures S5E and S5F). In summary, our findings suggest that the loss of SMAD3 disrupts the embryonic epicardial program, initiating a TGF-β-independent EMT.
SMAD3-downregulating cells signal reparative factors and exhibit pro-angiogenic properties improving functions of primary endothelial cells
To comprehensively understand the cellular functionality of SMAD3-downregulating cells, we conducted whole mRNA sequencing (RNA-seq) to dissect the transcriptomic landscape of these cells in comparison to other known EPDC trajectories, including iSMCs and iCFs. Principal component analysis demonstrated that SMAD3-downregulating cells exhibited distinct transcriptomic profiles compared to other EPDCs (Figure 5A), further confirming their emergence as cardiac pericyte progenitor cells with a unique cellular identity.
Figure 5.

SMAD3-downregulating epicardial cells show pro-angiogenic properties in transcriptomic analysis and functional assay with endothelial cells
(A) Principal component analysis (PCA) plot comparing control hiPSC-derived EPI cells at day 1 and day 7, SMAD3-silenced EPI cells from day 1 to day 7, iPSC-derived smooth muscle cells (iSMCs), and iPSC-derived cardiac fibroblasts (iCFs) ( control, n = 2;
control, n = 2;  siSMAD3, n = 7;
siSMAD3, n = 7;  iSMC, n = 1;
iSMC, n = 1;  iCF, n = 1).
iCF, n = 1).
(B) Gene Ontology (GO) analysis focusing on the biological processes of genes upregulated in SMAD3-silenced EPI cells.
(C) Heatmaps illustrating genes that are upregulated and downregulated in SMAD3-silenced EPI cells, specifically those associated with the positive regulation of angiogenesis and regulation of response to wounding, compared with control EPI cells.
(D) Heatmap depicting the upregulation of angiogenic-related genes in SMAD3-silenced EPI cells.
(E) Flow cytometry analysis revealing an increase in CD44 expression in SMAD3-silenced EPI cells (light blue) (gray represents the unstained control).
(F) Time-course upregulation of SDF-1, VEGFA, ANGPT1, and IL-6 in response to SMAD3 silencing, as observed in RNA sequencing data.
(G) Description of the experimental workflow, including siRNA transfection, EGFP overexpression, and co-culture in an endothelial tube formation assay.
(H) Phase contrast and immunofluorescent imaging of the endothelial tube formation assay at 6 h, comparing co-cultured human aortic endothelial cells (HAECs) with control and SMAD3-silenced EPI cells.
(I) Quantitative analysis showing an increased number of junctions in HAEC when co-cultured with SMAD3-silenced EPI cells (n = 3; ∗∗∗∗p < 0.0001).
(J and K) Observation of GFP-positive tubules, indicated by blue arrowheads, exclusively in HAEC co-cultured with SMAD3-silenced EPI cells. All error bars represent SEM; the graph plots are derived from experimental replicates, obtained from independent batches; statistical analysis was performed using a two-sided unpaired Student’s t test; scale bars equal 100 μm, unless otherwise indicated; scrambled siRNA was used as a control; see also Figure S6.
Gene Ontology analysis focusing on biological processes unveiled terms related to angiogenesis, wound repair, and the regulation of blood vessels (Figure 5B). As an illustrative example, we generated a heatmap displaying genes associated with the first two terms: (1) positive regulation of angiogenesis and (2) regulation of response to wounding. Notable genes contributing to both processes, such as CXCL8 and SERPINE1 for angiogenesis regulation (Hooglugt et al., 2020) and IL33 and PLAU for response to wounding regulation (Yin et al., 2013), were prominently expressed (Figure 5C).
Subsequently, we highlighted a curated selection of genes potentially implicated in cardiac angiogenesis, with a particular emphasis on CD44, VEGFA, and PDGFRA (Figure 5D). CD44, a marker indicative of mesenchymal stemness and heightened pro-angiogenic potential (Zhang et al., 2022), exhibited enhanced expression in SMAD3-downregulating cells (Figures 5E and S6A).
We further examined the temporal expression patterns of candidate genes encoding secreted proteins that could contribute to the regenerative milieu. Notably, we observed increased expression of SDF-1, VEGFA, angiopoietin-1 (ANGPT1), and IL-6, suggesting their potential involvement in enhanced functionality within a reparative context (Figure 5F).
To assess the reparative potential of siSMAD3 cells as cardiac pericyte progenitors, we transduced lentiviruses carrying EGFP for monitoring and co-cultured them with a primary cell line of endothelial cells (human aortic endothelial cell [HAEC]) (Figure 5G). In vitro endothelial tube formation assays were performed to evaluate endothelial cell functionality. Following optimization of cell ratios (data not shown), we observed a substantial enhancement in the tubule formation capacity of HAEC when co-cultured with siSMAD3 cells after 6 h (Figure 5H), resulting in a significant increase in the number of formed junctions (Figure 5I). Importantly, we observed physical interactions suggestive of cardiac pericyte-like behavior exhibited by siSMAD3 cells as they are closely associated with HAECs (Figures 5J and 5K). Collectively, our data endorse the pro-angiogenic functionality of siSMAD3 cells, potentially attributed to their physical association with endothelial cells, thereby improving their overall functionality within a reparative context.
SMAD3-downregulating cells release soluble factors that enhance paracrine-driven proliferative reactivation of hiPSC-derived cardiomyocytes
VEGFA and ANGPT1 are well-known factors that promote angiogenesis and activate proliferative pathways in the functional myocardium following myocardial infarction. Cardiomyocyte proliferation relies, in part, on the CDK6/CCND1 axis. Enhanced expression of CDK6 can induce proliferation in cardiomyocytes, which are typically quiescent and exhibit proliferative arrest, presenting a significant hurdle in cardiac repair (Rhee and Wu, 2018).
To investigate cardiac differentiation, we employed a reporter induced pluripotent stem cell line (FUCCI [fluorescent ubiquitination-based cell cycle indicator]) (Kasamoto et al., 2023) to differentiate ventricular cardiomyocytes (hiPSC-CMs) and monitor the proliferative state of hiPSC-CMs in response to siSMAD3 supernatant (Figure 6A).
Figure 6.

Characterization of siSMAD3-derived secretome in hiPSC-CM proliferation
(A) Schematic illustration of the co-culture experiment, involving the treatment of hiPSC-derived cardiomyocytes (hiPSC-CMs) with the secretome derived from siSMAD3 treatment, to monitor cardiomyocyte proliferation using FUCCI (fluorescent ubiquitination-based cell cycle indicator) cells.
(B) Flow cytometry analysis to identify cTnT-positive cells in hiPSC-CMs at differentiation day 16 (n = 3; mean ± SEM).
(C) Fluorescence microscopy to observe FUCCI-red (non-proliferative) and FUCCI-green (proliferative) hiPSC-CMs.
(D) Representative flow cytometry contour plots analyzing FUCCI-green percentage.
(E) FUCCI-green percentage in hiPSC-CMs following treatment with the siSMAD3-derived supernatant compared to control (n = 3; ∗p = 0.044).
(F) qRT-PCR analysis for TNNI1, CCND1, CDK4, and CDK6 gene expression in hiPSC-CMs following treatment with the siSMAD3-derived supernatant (n = 3; ∗p = 0.044, ∗∗∗p = 0.0010, ns = not significant).
(G) Protein analysis in treated hiPSC-CMs for the detection of TNNI1, CDK4, and CDK6 proteins.
(H) ELISA showing that VEGFA concentration in the supernatant from SMAD3-silenced EPI cells was 49.5 pg/mL, 1.59 times compared to control (n = 3; ∗∗p = 0.010). All error bars represent SEM; the graph plots are derived from experimental replicates, obtained from independent batches; statistical analysis was performed using a two-sided unpaired Student’s t test; scale bars equal 100 μm, unless otherwise indicated; scrambled siRNA was used as a control; see also Figure S6.
Following the epicardial differentiation and ectopic silencing of SMAD3 to divert EMT toward cardiac pericyte progenitor cells, we co-cultured ventricular cardiomyocytes in monolayers (cTnT+) (Figure 6B) with siSMAD3 cell supernatant to observe changes in the proliferative state using the FUCCI system. After 36 h of culture, a noticeable increase in FUCCI-green cells was observed under similar cardiomyocyte cell density (Figures 6C–6E), indicating reactivation of proliferation due to secreted factors present in the supernatant collected from siSMAD3 cells. Notably, gene expression analysis of treated hiPSC-CMs showed increased mRNA expression of fetal cardiac troponin I (TNNI1), possibly indicating the emergence of de novo fetal hiPSC-CMs (Figure 6F). There was a significant increase in CCND1 levels and an upward trend in CDK4 and CDK6 mRNA levels, suggesting proliferative activity in hiPSC-CMs (Figure 6F). In addition, protein analysis revealed an increasing tendency of TNNI1, CDK4, and CDK6 (Figures 6G and S6B). Finally, we performed immunodetection analysis (ELISA) in siSMAD3-derived supernatant to identify mechanistically the causative effector of increased hiPSC-CM proliferation. While we failed to detect the difference in secreted SDF-1 or ANGPT1 proteins, we detected a significant upregulation in secreted VEGFA in the supernatant of siSMAD3 EPI cells compared to control hiPSC-derived epicardial monolayers (Figures 6H and S6C). In summary, our findings demonstrate that the siSMAD3 secretome can enhance cardiac proliferation in an extrinsic manner, partially through the paracrine secretion of VEGFA that targets the CDK6/CCND1 axis, partially activating the cell cycle of hiPSC-CMs.
Discussion
Leveraging epicardial EMT mechanisms to guide cell fate decisions in regeneration holds promise as a therapeutic strategy for constructing EETs to repair injured hearts and transplant regenerative cells for myocardial healing (Jackman et al., 2018; Tan et al., 2021). The reconstruction of the human epicardium using induced pluripotent stem cells (iPSCs) has emerged as a compelling approach, reactivating the adult primary tissue from a quiescent state to stimulate in situ regeneration involving unknown mechanisms (Suffee et al., 2020).
Our hiPSC-derived epicardium serves as a robust source of cardiac progenitor cells with high plasticity for regenerative phenotypes, as previously demonstrated (Junghof et al., 2022). While the differentiation of human epicardium into EPDCs has been well documented for certain populations like cardiac fibroblasts and vascular smooth muscle cells, it typically involves trigger molecules such as bFGF or TGF-β, a potent inducer of EMT (Whitehead et al., 2022).
TGF-β is a multifunctional cytokine that plays a crucial role in various cellular processes, including cell growth, differentiation, and tissue homeostasis (Tzavlaki and Moustakas, 2020). TGF-β exerts its biological effects by binding to specific cell surface receptors (Heldin and Moustakas, 2016). Two important receptors involved in TGF-β signaling are ALK1 and ALK5 (Goumans et al., 2003). Although our study indicated a potential pathway connection involving endoglin (CD105) expression, which is a significant mediator in the ALK1 pathway (Roman and Hinck, 2017), the loss of SMAD3 had no impact on this pathway. This finding supports the idea that the observed CD105 expression was more likely associated with a new cellular identity rather than being a TGF-β-response element (Rossi et al., 2019). It is increasingly recognized that signaling pathways like TGF-β have pleiotropic effects, and the downstream SMAD protein signaling is highly context dependent (Derynck and Zhang, 2003). It can vary based on the cellular environment, the presence of other signaling molecules, and developmental timing. Our study has unveiled a novel role for SMAD3 in epicardial biology, indicating that SMAD3 expression can independently signify epicardial differentiation and stability over time.
However, SMAD3 overexpression alone appeared insufficient to autonomously induce a maturation phenotype from fetal epicardium. The effects of SMAD3 overexpression in hiPSC-derived epicardial monolayers may exhibit non-linear or biphasic behavior, making it challenging to replicate the physiological expression pattern of SMAD3 during development. Our study suggests that SMAD3 in the human epicardium fulfills a broader spectrum of functions (Dennler et al., 1998).
Cardiac pericytes are crucial for vessel stabilization, angiogenesis, and tissue homeostasis (Quijada et al., 2023). They are known for their transcriptional heterogeneity and can express various markers depending on factors like their location, developmental origin, and specific organ (Avolio et al., 2024).
Common pericyte markers include PDGFRβ (platelet-derived growth factor receptor-beta) and NG2 (also known as CSPG4) (Smyth et al., 2022). However, not all pericytes express both markers simultaneously (Yamazaki and Mukouyama, 2018). Our SMAD3-downregulating cells exhibited no protein expression of PDGFRβ, even though they displayed characteristics of cardiac pericytes in our experimental setup. This variability can be influenced by factors such as the developmental stage or the presence of specific signaling molecules.
Our study suggests that SMAD3-downregulating cells may represent an immature pericyte state with the potential to differentiate into mature pericyte-like cells (Bouacida et al., 2012). During this differentiation process, the expression of pericyte markers may change. As these cells mature and integrate into the cardiac microvasculature, their marker expression may become more pronounced or diversified (Lerman et al., 2018).
A recent study that differentiated cardiac pericytes from hiPSCs (Shen et al., 2023) indicated that exogenous supplementation of PDGF-BB (platelet-derived growth factor BB), primarily secreted by endothelial cells, is necessary to yield PDGFRβ-expressing cells. Since our study primarily focuses on characterizing the autonomous expression of SMAD3 in epicardial biology and the specification of cardiac pericyte progenitor cells, without investigating factors secreted by other cardiac cell types, it is possible that PDGF-BB could trigger a pericyte maturation program in SMAD3-downregulating cells.
Angiogenesis is a pivotal process in tissue reconstitution during cardiac repair (Kobayashi et al., 2017; Li et al., 2022), and the human epicardium can assess the extent of damage and the specific cellular response required based on external cues.
Loss of SMAD3 has been linked to various functions related to vascular remodeling (Cobb et al., 2022; Zabini et al., 2018), angiogenesis, and the maintenance of microvascular homeostasis (Feinberg et al., 2004). SMAD transcription factors occupy a central position in a highly adaptable cytokine signaling pathway that remains incompletely understood (Massagué et al., 2005). Angiotensin II can activate the SMAD pathway in vascular smooth muscle cells (Carvajal et al., 2008), similar to the way TGF-β does. It is conceivable that the human epicardium has evolved to preserve a broader spectrum of differentiation capabilities beyond our current knowledge. SMAD3 specifically regulates a differentiation process leading to a pro-angiogenic phenotype (Nakagawa et al., 2004), highlighting its significance in cardiac vascular processes.
SMAD3-downregulating cells exhibited an inherent ability to interact with primary endothelial cells, facilitating enhanced microvasculature formation in vitro, mimicking the process of vascular development in vivo (Bergers and Song, 2005; Geevarghese and Herman, 2014; Yuan et al., 2016). Our study demonstrated that SMAD3 regulates the secretion of regenerative factors involved in at least two critical processes during cardiac repair: (1) vascular repair and (2) stimulation of functional myocardial regeneration. While our study did not identify the specific secreted factors promoting hiPSC-CM proliferation, promising candidates include SDF-1, VEGFA, and ANGPT1, which are known to stimulate cardiomyocyte proliferation (Chen et al., 2015; Eschenhagen et al., 2017; Lin and Pu, 2014; Renko et al., 2018; Tao et al., 2011). Although a few conceptual studies have shown that cardiomyocytes can re-enter the cell cycle through small molecules or genetic approaches (Kasamoto et al., 2023), our research suggests that EPDCs play a crucial role in cardiac repair by promoting functional myocardial regeneration.
Our experiments using SMAD3-downregulating cell secretome showed a mild increase in FUCCI-green, indicating that cardiomyocyte cell cycle reactivation involves a broader network of genes beyond CDK6/CCND1 (Li et al., 2021; Murganti et al., 2022). This reactivation possibility offers the potential to generate highly engraftable hiPSC-CMs. Proliferative cardiomyocytes can integrate more effectively into host hearts, as demonstrated in a recent study that induced cardiac proliferation using Am80, a retinoic acid receptor agonist (Kasamoto et al., 2023).
In summary, our study sheds light on the manipulation of epicardial EMT, with SMAD3 playing a vital role in the specification and maintenance of the epicardial program independently. The mechanism by which the loss of SMAD3 overrides the effects of SB431542 (Bao et al., 2016) in blocking TGF-β to initiate an alternative epicardial EMT remains unclear, and further research on SMAD3 DNA interactions during epicardiogenesis is warranted. In conclusion, our work defines a biological function of SMAD3 in the epicardial context, opening avenues for the manipulation and therapeutic modulation of the active epicardium for cardiac repair and regeneration. This includes the generation of cardiac pericyte progenitor cells capable of interacting with and promoting in situ regeneration of primary tissue, potentially aiding in the recovery of injured myocardium and cardiac microvasculature.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Yoshinori Yoshida (yoshinor@cira.kyoto-u.ac.jp).
Materials availability
All unique and stable reagents generated in this study are available from the lead contact upon completion of a Materials Transfer Agreement.
Data and code availability
All data presented in this study are available within the main text, in the supplemental items file, and can be obtained from the corresponding authors upon request. The RNA-seq data have been deposited in the GEO database under the accession code GSE165450.
Experimental procedures
Cell lines and culture conditions
We used two hiPSC lines (409B2 and 201B7-FUCCI), both of which were reprogrammed using Yamanaka factors via retroviral methods. Both cell lines were cultured in ReproCell Primate ES cell media supplemented with 4 ng/mL recombinant human bFGF on irradiated MEF feeder cells. In the timing of passage or induction, the feeder layers were eliminated by CTK. Experiments using human iPSCs were approved by the Ethics Committee of Kyoto University.
The human aortic endothelial cell line (ScienCell, # SCR-6100-1) was cultured in endothelial cell medium (ScienCell, # 1001) following the manufacturer’s manual.
Regular mycoplasma testing was conducted to avoid contamination for all cell lines.
Cell transfection
We diluted the following Silencer Select siRNAs (Thermo Fisher Scientific) to a 10 μmol/L stock solution: siSMAD3 (Cat #: 4392420, ID: s535081) and negative control (scrambled siRNA) (Cat #: 4390844). For silencing experiments, the cells were seeded to be 60%–70% confluent one day before the transfection. To silence the cells, 10 μL of the siRNA stock and 10 μL of Lipofectamine RNAiMAX reagent were separately diluted in 500 μL of Opti-MEM. These two solutions were mixed and after 15 min incubation at room temperature, the final solution was added dropwise, 500 μL silencing solution into one well of 6-well plate with 2 mL maintenance media. At 24 h after transfection, the media was exchanged and cells were continued to be cultured for 7 days with changing the media containing SB431542 (epicardial maintenance media) every 2–3 days.
Overexpression
For SMAD3 overexpression, human SMAD3 ORF (NM_001407011.1) was amplified by PCR, subcloned into pENTR-D-TOPO (Invitrogen, #K2400-20), and transferred to pLenti6.3/V5-DEST (Invitrogen, #V533-06), as the protocol instructed (the vector map is shown in Figure S6D). For EGFP overexpression, we used control vector in the kit.
The cells were seeded to be 60%–70% confluent one day before the transduction. After optimization of the concentration of lentiviruses, 1.0 × 107 IFU/mL lentiviruses and 4 μg/mL polybrene were transduced to the cells. Thereafter, centrifugation (32°C, 1200 × g, 90 min) was performed to increase transduction efficiency. At 24 h after transduction, the media was exchanged and cells were continued to be cultured for 7 days as aforementioned.
Bioinformatics and RNA-seq
A retrospective analysis of SMAD3 gene expression was conducted using the GSE122200 dataset from GEO for studying EMT dynamics in the mouse epicardium, and GSE84085 for analyzing PSC-derived epicardium and adult primary human epicardium from donors.
For the in-house RNA-seq data generated for this manuscript, normalization was performed using the NOISeq package. The processed data and raw fastq files from this study were submitted to the Gene Expression Omnibus (GEO) under the accession code GSE165450. Further analysis of the raw data was conducted in RStudio.
Gene expression data were read, explored, and pre-processed using the Bioconductor package NOISeq was used for differential expression analysis of RNA-seq. A hierarchical cluster dendrogram was generated using the hclust function from the stats package and the agnes function from the cluster package, with distances assessed by the Manhattan city-block distance algorithm. K-means clustering was performed using the kmeans function. Distance and correlation matrices were computed and plotted using the get_dist and fviz_dist functions from the factoextra package, while the fviz_cluster function was used for cluster scatterplots. Heatmaps were generated to visualize differentially expressed genes using R script.
Statistical analysis and visualization of gene functional profiles and clusters for Gene Ontology terms were carried out using the DOSE and clusterProfiler packages. Ingenuity Pathway Analysis was employed for upstream pathway analysis and identifying pathway activity patterns.
Acknowledgments
We want to thank all members of the Yoshida laboratory for their constructive feedback on this study. This work was supported by a grant from the Leducq Foundation (18CVD05) and JSPS KAKENHI grants (22K16137 [A.L.-C.], 24K11267 [A.L.-C.], and 21H02912 [Y.Y.]). Research Center Network also provides funding for Realization of Regenerative Medicine, Japan Agency for Medical Research and Development (AMED) (JP19bm0104001 and JP20bm0804022) (Y.Y.), Acceleration Program of R&D and implementation for Regenerative Medicine and Cell and Gene Therapy, AMED (JP23bm1423011 and JP23bm132300) (Y.Y.), the Research on Regulatory Science of Pharmaceuticals and Medical Devices, AMED (JP22mk0101189 and JP22mk0101241) (Y.Y.), the Translational Research grant, AMED (JP22ym0126091) (Y.Y.), and the iPS Cell Research Fund (A.L.-C and Y.Y.). We want to express our gratitude to Rumi Fujihara, Tomomi Gibson, and Hiroko Sata for their administrative support.
Author contributions
A.L.-C. and Y.Y. conceived the study and designed the project. Y. Miyoshi, Y.T., Y. Matsumura, K.T., M. Nishikawa, and A.L.-C. performed the experiments. M. Narita assisted in RNA sequencing. Y. Miyoshi and A.L.-C. analyzed and interpreted the data. Y.Y. and A.L.-C. provided funding and supervision. A.L.-C., Y. Miyoshi, and Y.Y. wrote the manuscript. All authors discussed the results.
Declaration of interests
Y.Y. is a scientific advisor of Orizuru Therapeutics and receives grants from Takeda Pharmaceutical Company and Altos Labs, Inc. outside the submitted work.
Published: September 26, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.stemcr.2024.08.008.
Contributor Information
Antonio Lucena-Cacace, Email: a.lucena.prime@osaka-u.ac.jp.
Yoshinori Yoshida, Email: yoshinor@cira.kyoto-u.ac.jp.
Supplemental information
References
- Alex L., Tuleta I., Harikrishnan V., Frangogiannis N.G. Validation of Specific and Reliable Genetic Tools to Identify, Label, and Target Cardiac Pericytes in Mice. J. Am. Heart Assoc. 2022;11 doi: 10.1161/JAHA.121.023171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alex L., Tuleta I., Hernandez S.C., Hanna A., Venugopal H., Astorkia M., Humeres C., Kubota A., Su K., Zheng D., Frangogiannis N.G. Cardiac Pericytes Acquire a Fibrogenic Phenotype and Contribute to Vascular Maturation After Myocardial Infarction. Circulation. 2023;148:882–898. doi: 10.1161/CIRCULATIONAHA.123.064155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avolio E., Campagnolo P., Katare R., Madeddu P. The role of cardiac pericytes in health and disease: therapeutic targets for myocardial infarction. Nat. Rev. Cardiol. 2024;21:106–118. doi: 10.1038/s41569-023-00913-y. [DOI] [PubMed] [Google Scholar]
- Bao X., Lian X., Hacker T.A., Schmuck E.G., Qian T., Bhute V.J., Han T., Shi M., Drowley L., Plowright A., et al. Long-term self-renewing human epicardial cells generated from pluripotent stem cells under defined xeno-free conditions. Nat. Biomed. Eng. 2016;1 doi: 10.1038/s41551-016-0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergers G., Song S. The role of pericytes in blood-vessel formation and maintenance. Neuro Oncol. 2005;7:452–464. doi: 10.1215/S1152851705000232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouacida A., Rosset P., Trichet V., Guilloton F., Espagnolle N., Cordonier T., Heymann D., Layrolle P., Sensébé L., Deschaseaux F. Pericyte-like progenitors show high immaturity and engraftment potential as compared with mesenchymal stem cells. PLoS One. 2012;7 doi: 10.1371/journal.pone.0048648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvajal G., Rodríguez-Vita J., Rodrigues-Díez R., Sánchez-López E., Rupérez M., Cartier C., Esteban V., Ortiz A., Egido J., Mezzano S.A., Ruiz-Ortega M. Angiotensin II activates the Smad pathway during epithelial mesenchymal transdifferentiation. Kidney Int. 2008;74:585–595. doi: 10.1038/ki.2008.213. [DOI] [PubMed] [Google Scholar]
- Chen D., Xia Y., Zuo K., Wang Y., Zhang S., Kuang D., Duan Y., Zhao X., Wang G. Crosstalk between SDF-1/CXCR4 and SDF-1/CXCR7 in cardiac stem cell migration. Sci. Rep. 2015;5 doi: 10.1038/srep16813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung H.-J., Kim J.-T., Kim H.-J., Kyung H.-W., Katila P., Lee J.-H., Yang T.-H., Yang Y.-I., Lee S.-J. Epicardial delivery of VEGF and cardiac stem cells guided by 3-dimensional PLLA mat enhancing cardiac regeneration and angiogenesis in acute myocardial infarction. J. Control. Release. 2015;205:218–230. doi: 10.1016/j.jconrel.2015.02.013. [DOI] [PubMed] [Google Scholar]
- Cobb M.S., Tao S., Shortt K., Girgis M., Hauptman J., Schriewer J., Chin Z., Dorfman E., Campbell K., Heruth D.P., et al. Smad3 promotes adverse cardiovascular remodeling and dysfunction in doxorubicin-treated hearts. Am. J. Physiol. Heart Circ. Physiol. 2022;323:H1091–H1107. doi: 10.1152/ajpheart.00312.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennler S., Itoh S., Vivien D., ten Dijke P., Huet S., Gauthier J.-M. Direct binding of Smad3 and Smad4 to critical TGFβ-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 1998;17:3091–3100. doi: 10.1093/emboj/17.11.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck R., Zhang Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- Du J., Yuan X., Deng H., Huang R., Liu B., Xiong T., Long X., Zhang L., Li Y., She Q. Single-cell and spatial heterogeneity landscapes of mature epicardial cells. J. Pharm. Anal. 2023;13:894–907. doi: 10.1016/j.jpha.2023.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eschenhagen T., Bolli R., Braun T., Field L.J., Fleischmann B.K., Frisén J., Giacca M., Hare J.M., Houser S., Lee R.T., et al. Cardiomyocyte Regeneration: A Consensus Statement. Circulation. 2017;136:680–686. doi: 10.1161/CIRCULATIONAHA.117.029343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang M., Xiang F.-L., Braitsch C.M., Yutzey K.E. Epicardium-derived fibroblasts in heart development and disease. J. Mol. Cell. Cardiol. 2016;91:23–27. doi: 10.1016/j.yjmcc.2015.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg M.W., Shimizu K., Lebedeva M., Haspel R., Takayama K., Chen Z., Frederick J.P., Wang X.-F., Simon D.I., Libby P., et al. Essential Role for Smad3 in Regulating MCP-1 Expression and Vascular Inflammation. Circ. Res. 2004;94:601–608. doi: 10.1161/01.RES.0000119170.70818.4F. [DOI] [PubMed] [Google Scholar]
- Geevarghese A., Herman I.M. Pericyte-Endothelial Cross-Talk: Implications and Opportunities for Advanced Cellular Therapies. Transl. Res. 2014;163:296–306. doi: 10.1016/j.trsl.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittenberger-de Groot A.C., Winter E.M., Poelmann R.E. Epicardium-derived cells (EPDCs) in development, cardiac disease and repair of ischemia. J. Cell Mol. Med. 2010;14:1056–1060. doi: 10.1111/j.1582-4934.2010.01077.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goumans M.J., Valdimarsdottir G., Itoh S., Lebrin F., Larsson J., Mummery C., Karlsson S., ten Dijke P. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling. Mol. Cell. 2003;12:817–828. doi: 10.1016/s1097-2765(03)00386-1. [DOI] [PubMed] [Google Scholar]
- Heldin C.-H., Moustakas A. Signaling Receptors for TGF-β Family Members. Cold Spring Harb. Perspect. Biol. 2016;8 doi: 10.1101/cshperspect.a022053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooglugt A., van der Stoel M.M., Boon R.A., Huveneers S. Endothelial YAP/TAZ Signaling in Angiogenesis and Tumor Vasculature. Front. Oncol. 2020;10 doi: 10.3389/fonc.2020.612802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer D., Gambardella L., Bernard W.G., Serrano F., Mascetti V.L., Pedersen R.A., Talasila A., Sinha S. Robust derivation of epicardium and its differentiated smooth muscle cell progeny from human pluripotent stem cells. Development. 2015;142:1528–1541. doi: 10.1242/dev.119271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman C.P., Ganapathi A.M., Asfour H., Qian Y., Allen B.W., Li Y., Bursac N. Engineered cardiac tissue patch maintains structural and electrical properties after epicardial implantation. Biomaterials. 2018;159:48–58. doi: 10.1016/j.biomaterials.2018.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junghof J., Kogure Y., Yu T., Verdugo-Sivianes E.M., Narita M., Lucena-Cacace A., Yoshida Y. CDH18 is a fetal epicardial biomarker regulating differentiation towards vascular smooth muscle cells. NPJ Regen. Med. 2022;7:14. doi: 10.1038/s41536-022-00207-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y., Massagué J. Epithelial-Mesenchymal Transitions: Twist in Development and Metastasis. Cell. 2004;118:277–279. doi: 10.1016/j.cell.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Kasamoto M., Funakoshi S., Hatani T., Okubo C., Nishi Y., Tsujisaka Y., Nishikawa M., Narita M., Ohta A., Kimura T., Yoshida Y. Am80, a retinoic acid receptor agonist, activates the cardiomyocyte cell cycle and enhances engraftment in the heart. Stem Cell Rep. 2023;18:1672–1685. doi: 10.1016/j.stemcr.2023.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight-Schrijver V.R., Davaapil H., Bayraktar S., Ross A.D.B., Kanemaru K., Cranley J., Dabrowska M., Patel M., Polanski K., He X., et al. A single-cell comparison of adult and fetal human epicardium defines the age-associated changes in epicardial activity. Nat. Cardiovasc. Res. 2022;1:1215–1229. doi: 10.1038/s44161-022-00183-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K., Maeda K., Takefuji M., Kikuchi R., Morishita Y., Hirashima M., Murohara T. Dynamics of angiogenesis in ischemic areas of the infarcted heart. Sci. Rep. 2017;7:7156. doi: 10.1038/s41598-017-07524-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerman D.A., Diaz M., Peault B. Changes in coexpression of pericytes and endogenous cardiac progenitor cells from heart development to disease state. Eur. Heart J. 2018;39 doi: 10.1093/eurheartj/ehy565.P1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Wang Z., Yang F., Huang J., Hu X., Deng S., Tian M., Si X. miR-449a-5p suppresses CDK6 expression to inhibit cardiomyocyte proliferation. Mol. Med. Rep. 2021;23:14. doi: 10.3892/mmr.2020.11652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Zhao Y., Zhu W. Targeting angiogenesis in myocardial infarction: Novel therapeutics (Review) Exp. Ther. Med. 2022;23:64. doi: 10.3892/etm.2021.10986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim G.B. Heterogeneity of the post-infarct epicardium. Nat. Rev. Cardiol. 2021;18:612. doi: 10.1038/s41569-021-00596-3. [DOI] [PubMed] [Google Scholar]
- Lin Z., Pu W.T. Strategies for cardiac regeneration and repair. Sci. Transl. Med. 2014;6 doi: 10.1126/scitranslmed.3006681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh C.-Y., Chai J.Y., Tang T.F., Wong W.F., Sethi G., Shanmugam M.K., Chong P.P., Looi C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells. 2019;8:1118. doi: 10.3390/cells8101118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué J., Seoane J., Wotton D. Smad transcription factors. Genes Dev. 2005;19:2783–2810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- Murganti F., Derks W., Baniol M., Simonova I., Trus P., Neumann K., Khattak S., Guan K., Bergmann O. FUCCI-Based Live Imaging Platform Reveals Cell Cycle Dynamics and Identifies Pro-proliferative Compounds in Human iPSC-Derived Cardiomyocytes. Front. Cardiovasc. Med. 2022;9 doi: 10.3389/fcvm.2022.840147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T., Li J.H., Garcia G., Mu W., Piek E., Böttinger E.P., Chen Y., Zhu H.J., Kang D.-H., Schreiner G.F., et al. TGF-β induces proangiogenic and antiangiogenic factorsvia parallel but distinct Smad pathways1. Kidney Int. 2004;66:605–613. doi: 10.1111/j.1523-1755.2004.00780.x. [DOI] [PubMed] [Google Scholar]
- Olivey H.E., Svensson E.C. Epicardial-Myocardial Signaling Directing Coronary Vasculogenesis. Circ. Res. 2010;106:818–832. doi: 10.1161/CIRCRESAHA.109.209197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quijada P., Trembley M.A., Misra A., Myers J.A., Baker C.D., Pérez-Hernández M., Myers J.R., Dirkx R.A., Cohen E.D., Delmar M., et al. Coordination of endothelial cell positioning and fate specification by the epicardium. Nat. Commun. 2021;12:4155. doi: 10.1038/s41467-021-24414-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quijada P., Park S., Zhao P., Kolluri K.S., Wong D., Shih K.D., Fang K., Pezhouman A., Wang L., Daraei A., et al. Cardiac pericytes mediate the remodeling response to myocardial infarction. J. Clin. Invest. 2023;133 doi: 10.1172/JCI162188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renko O., Tolonen A.-M., Rysä J., Magga J., Mustonen E., Ruskoaho H., Serpi R. SDF1 gradient associates with the distribution of c-Kit+ cardiac cells in the heart. Sci. Rep. 2018;8:1160. doi: 10.1038/s41598-018-19417-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee J.-W., Wu J.C. Cardiac Cell Cycle Activation as a Strategy to Improve iPSC-Derived Cardiomyocyte Therapy. Circ. Res. 2018;122:14–16. doi: 10.1161/CIRCRESAHA.117.312287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman B.L., Hinck A.P. ALK1 signaling in development and disease: new paradigms. Cell. Mol. Life Sci. 2017;74:4539–4560. doi: 10.1007/s00018-017-2636-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi E., Bernabeu C., Smadja D.M. Endoglin as an Adhesion Molecule in Mature and Progenitor Endothelial Cells: A Function Beyond TGF-β. Front. Med. 2019;6 doi: 10.3389/fmed.2019.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen M., Liu C., Zhao S.R., Manhas A., Sundaram L., Ameen M., Wu J.C. Stepwise Generation of Human Induced Pluripotent Stem Cell-Derived Cardiac Pericytes to Model Coronary Microvascular Dysfunction. Circulation. 2023;147:515–518. doi: 10.1161/CIRCULATIONAHA.122.061770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits A.M., Dronkers E., Goumans M.-J. The epicardium as a source of multipotent adult cardiac progenitor cells: Their origin, role and fate. Pharmacol. Res. 2018;127:129–140. doi: 10.1016/j.phrs.2017.07.020. [DOI] [PubMed] [Google Scholar]
- Smyth L.C.D., Highet B., Jansson D., Wu J., Rustenhoven J., Aalderink M., Tan A., Li S., Johnson R., Coppieters N., et al. Characterisation of PDGF-BB:PDGFRβ signalling pathways in human brain pericytes: evidence of disruption in Alzheimer’s disease. Commun. Biol. 2022;5:235. doi: 10.1038/s42003-022-03180-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridurongrit S., Larsson J., Schwartz R., Ruiz-Lozano P., Kaartinen V. Signaling via the Tgf-β type I receptor Alk5 in heart development. Dev. Biol. 2008;322:208–218. doi: 10.1016/j.ydbio.2008.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streef T.J., Smits A.M. Epicardial Contribution to the Developing and Injured Heart: Exploring the Cellular Composition of the Epicardium. Front. Cardiovasc. Med. 2021;8 doi: 10.3389/fcvm.2021.750243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suffee N., Moore-Morris T., Jagla B., Mougenot N., Dilanian G., Berthet M., Proukhnitzky J., Le Prince P., Tregouet D.A., Pucéat M., Hatem S.N. Reactivation of the Epicardium at the Origin of Myocardial Fibro-Fatty Infiltration During the Atrial Cardiomyopathy. Circ. Res. 2020;126:1330–1342. doi: 10.1161/CIRCRESAHA.119.316251. [DOI] [PubMed] [Google Scholar]
- Takahashi K., Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Tan J.J., Guyette J.P., Miki K., Xiao L., Kaur G., Wu T., Zhu L., Hansen K.J., Ling K.-H., Milan D.J., Ott H.C. Human iPS-derived pre-epicardial cells direct cardiomyocyte aggregation expansion and organization in vitro. Nat. Commun. 2021;12:4997. doi: 10.1038/s41467-021-24921-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Z., Chen B., Tan X., Zhao Y., Wang L., Zhu T., Cao K., Yang Z., Kan Y.W., Su H. Coexpression of VEGF and angiopoietin-1 promotes angiogenesis and cardiomyocyte proliferation reduces apoptosis in porcine myocardial infarction (MI) heart. Proc. Natl. Acad. Sci. USA. 2011;108:2064–2069. doi: 10.1073/pnas.1018925108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzavlaki K., Moustakas A. TGF-β Signaling. Biomolecules. 2020;10:487. doi: 10.3390/biom10030487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volz K.S., Jacobs A.H., Chen H.I., Poduri A., McKay A.S., Riordan D.P., Kofler N., Kitajewski J., Weissman I., Red-Horse K. Pericytes are progenitors for coronary artery smooth muscle. Elife. 2015;4 doi: 10.7554/eLife.10036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead A.J., Hocker J.D., Ren B., Engler A.J. Improved epicardial cardiac fibroblast generation from iPSCs. J. Mol. Cell. Cardiol. 2022;164:58–68. doi: 10.1016/j.yjmcc.2021.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wijk B., Gunst Q.D., Moorman A.F.M., van den Hoff M.J.B. Cardiac regeneration from activated epicardium. PLoS One. 2012;7 doi: 10.1371/journal.pone.0044692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witty A.D., Mihic A., Tam R.Y., Fisher S.A., Mikryukov A., Shoichet M.S., Li R.-K., Kattman S.J., Keller G. Generation of the epicardial lineage from human pluripotent stem cells. Nat. Biotechnol. 2014;32:1026–1035. doi: 10.1038/nbt.3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki T., Mukouyama Y.S. Tissue Specific Origin, Development, and Pathological Perspectives of Pericytes. Front. Cardiovasc. Med. 2018;5 doi: 10.3389/fcvm.2018.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H., Li X., Hu S., Liu T., Yuan B., Gu H., Ni Q., Zhang X., Zheng F. IL-33 accelerates cutaneous wound healing involved in upregulation of alternatively activated macrophages. Mol. Immunol. 2013;56:347–353. doi: 10.1016/j.molimm.2013.05.225. [DOI] [PubMed] [Google Scholar]
- Yuan K., Orcholski M.E., Huang N.F., de Jesus Perez V.A. In Vivo Study of Human Endothelial-Pericyte Interaction Using the Matrix Gel Plug Assay in Mouse. J. Vis. Exp. 2016;54617 doi: 10.3791/54617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabini D., Granton E., Hu Y., Miranda M.Z., Weichelt U., Breuils Bonnet S., Bonnet S., Morrell N.W., Connelly K.A., Provencher S., et al. Loss of SMAD3 Promotes Vascular Remodeling in Pulmonary Arterial Hypertension via MRTF Disinhibition. Am. J. Respir. Crit. Care Med. 2018;197:244–260. doi: 10.1164/rccm.201702-0386OC. [DOI] [PubMed] [Google Scholar]
- Zhang Q., Chen L., Huang L., Cheng H., Wang L., Xu L., Hu D., He C., Fu C., Wei Q. CD44 promotes angiogenesis in myocardial infarction through regulating plasma exosome uptake and further enhancing FGFR2 signaling transduction. Mol. Med. 2022;28:145. doi: 10.1186/s10020-022-00575-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S., Chen M., Ying Y., Wu Q., Huang Z., Ni W., Wang X., Xu H., Bennett S., Xiao J., Xu J. Versatile subtypes of pericytes and their roles in spinal cord injury repair, bone development and repair. Bone Res. 2022;10:30. doi: 10.1038/s41413-022-00203-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data presented in this study are available within the main text, in the supplemental items file, and can be obtained from the corresponding authors upon request. The RNA-seq data have been deposited in the GEO database under the accession code GSE165450.