

Summary
Translating genetic findings for neurodevelopmental and psychiatric disorders (NPDs) into actionable disease biology would benefit from large-scale and unbiased functional studies of NPD genes. Leveraging the cytosine base editing (CBE) system, we developed a pipeline for clonal loss-of-function (LoF) allele mutagenesis in human induced pluripotent stem cells (hiPSCs) by introducing premature stop codons (iSTOP) that lead to mRNA nonsense-mediated decay (NMD) or protein truncation. We tested the pipeline for 23 NPD genes on 3 hiPSC lines and achieved highly reproducible, efficient iSTOP editing in 22 genes. Using RNA sequencing (RNA-seq), we confirmed their pluripotency, absence of chromosomal abnormalities, and NMD. Despite high editing efficiency, three schizophrenia risk genes (SETD1A, TRIO, and CUL1) only had heterozygous LoF alleles, suggesting their essential roles for cell growth. We found that CUL1-LoF reduced neurite branches and synaptic puncta density. This iSTOP pipeline enables a scaled and efficient LoF mutagenesis of NPD genes, yielding an invaluable shareable resource.
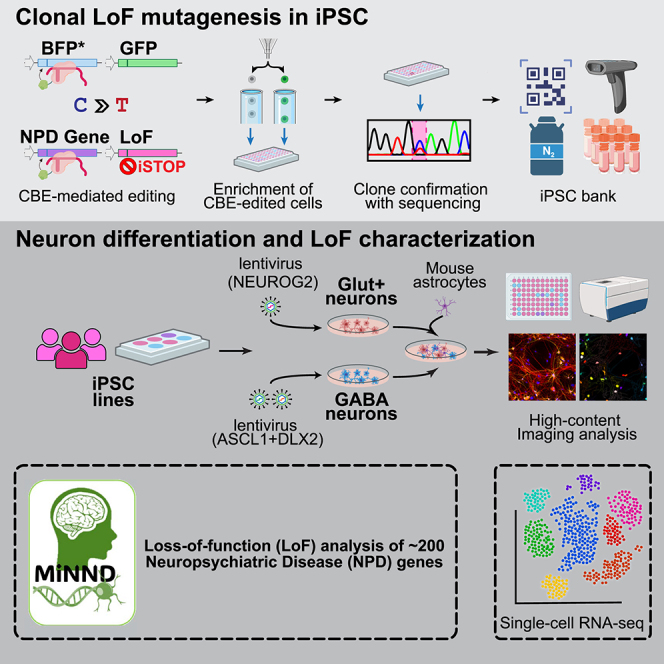
Graphical abstract
Highlights
-
•
An efficient pipeline to generate LoF alleles in hiPSC using DNA base editing
-
•
22 LoF alleles of neuropsychiatric risk genes were generated on 3 hiPSC lines
-
•
The engineered LoF alleles undergo nonsense-mediated decay or protein truncation
-
•
The SSPsyGene MiNND project provides a rich resource of LoF cell lines and data
As part of the NIMH SSPsyGene Consortium, Duan and colleagues leverage the cytosine base editing system to develop a pipeline for clonal loss-of-function (LoF) allele mutagenesis in hiPSC by introducing premature stop codons that lead to mRNA nonsense-mediated decay or protein truncation. They demonstrate its efficiency by successfully generating 22 LoF alleles on multiple genetic backgrounds, as a first installment of creating ∼200 LoF alleles for neurodevelopmental and psychiatric disorders.
Introduction
In the past decade, genome-wide association studies (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014; Grove et al., 2019; Howard et al., 2019; Meng et al., 2024; Mullins et al., 2021; Purcell et al., 2009; Ripke et al., 2011, 2013; Shi et al., 2009; Stahl et al., 2019; Stefansson et al., 2009; Trubetskoy et al., 2022; Wray et al., 2018) and whole-exome sequencing studies (Satterstrom et al., 2020; Singh et al., 2022) on neurodevelopmental and psychiatric disorders (NPDs), such as schizophrenia (SZ), autism spectrum disorder, bipolar disorder, and major depression, have identified a growing number of risk genes. However, translating these exciting genetic discoveries into translational actionable biology has been impeded by our limited knowledge of gene function and related disease mechanisms. A bottleneck in the field is that genes are often studied individually, slowing the progress and posing potential bias in functional interpretation. To overcome such limitations, the NIMH (National Institute of Mental Health)-initiated SSPsyGene (Scalable and Systematic Neurobiology of Psychiatric and Neurodevelopmental Disorder Risk Genes) Consortium (sspsygene.ucsc.edu) aims to functionally characterize the contribution of 150–250 NPD genes. The selected NPD genes mostly have disease-associated rare protein-truncating variants (PTVs) that likely cause gene loss of function (LoF) (Palmer et al., 2022; Satterstrom et al., 2020; Singh et al., 2022) and have strong effect sizes (sspsygene.ucsc.edu/resources), which will help interpret their individual biological relevance and determine any convergent or divergent biology across disorders. A large-scale, unbiased, and parallel study of these NPD genes in disease-relevant model systems will substantially deepen our understanding of the pathophysiology of NPDs.
Human induced pluripotent stem cells (hiPSCs) and their derived neural cells empowered by CRISPR-mediated gene editing provide promising cellular models for studying NPD genes (De Los Angeles et al., 2021; Duan, 2023; Michael Deans and Brennand, 2021; Muhtaseb and Duan, 2022; Wang et al., 2020; Wen et al., 2016) and for scaling up the assay. A “cell village” approach (Wells et al., 2023) enables the co-culture of tens to hundreds of hiPSC lines in a dish together, followed by assaying a specific cellular phenotype and being able to genetically infer individual cell identity. Such cell village approach may be combined with pooled screening using CRISPRi (Holtzman and Gersbach, 2018) or CRISPRoff (Nunez et al., 2021) to scale up the number of targeted genes for LoF assay. While an invaluable approach, the pooled CRISPR screening in hiPSC-derived neural models is limited by cell line-specific or LoF allele-specific unequal cellular growth, possible non-autonomous effects, and restrictive phenotypes amenable for screening.
CRISPR-Cas9 editing can be used to systematically create small DNA insertions or deletions (indels) or exon deletions in protein-coding regions through non-homologous end joining repair of double-strand breaks (DSBs) (Ran et al., 2013), resulting in protein-truncating mutations. Alternatively, LoF mutation can be generated by using CRISPR-based cytosine base editors (CBEs) to introduce premature protein stop codons (i.e., nonsense mutations; an iSTOP approach) that lead to mRNA nonsense-mediated decay (NMD) and/or protein truncates (Billon et al., 2017; Cuella-Martin et al., 2021; Hanna et al., 2021; Xu et al., 2021). Compared to the traditional CRISPR-Cas9 gene editing system, the CBE makes “C” to “T” changes in DNAs without creating cell-toxic DSBs as the Cas9 nuclease does (Ran et al., 2013) and with minimized potential off-target DNA editing (Billon et al., 2017; Cuella-Martin et al., 2021; Hanna et al., 2021; Xu et al., 2021). Furthermore, compared to the CRISPR-Cas9 editing-induced small indels that may or may not disrupt a protein sequence reading frame, a CBE can precisely introduce a premature stop codon, which makes the clonal LoF allelic confirmation more straightforward and cost-effective in a scaled LoF mutagenesis workflow. Finally, the CBE-engineered premature stop codon mutations are more reminiscent of the rare patient-specific PTVs or LoF mutations associated with NPD (Satterstrom et al., 2020; Singh et al., 2022). Recently, a DNA base editing reporter gene system has been developed to enrich the edited cells, thereby increasing the base editing efficiency of a target gene (Standage-Beier et al., 2019), including in hiPSCs (Tekel et al., 2021). However, the use of a CBE in editing hiPSC lines has been scarce (Sürün et al., 2020), and its usefulness in developing a scaled and efficient clonal LoF mutagenesis in hiPSCs has not been tested.
As part of the SSPsyGene Consortium, our Assay and Data Generation Center for the Model of iPSC-derived Neurons for NPD (MiNND) aims to employ the CBE-based iSTOP approach to generate isogenic hiPSC lines carrying LoF alleles for about 150–200 NPD genes on multiple donor genetic backgrounds. Here, leveraging an improved reporter gene editing enrichment system that can substantially increase the CBE iSTOP editing efficiency in hiPSCs, we established a semi-automated pipeline for parallel and efficient clonal LoF mutagenesis of a large number of genes. We tested the workflow on 23 NPD genes with 3 donor hiPSC lines (KOLF2.2J, CW20107, and MGS_CD14). We obtained high and reproducible iSTOP editing efficiency across all three hiPSC lines. We systematically characterized the engineered isogenic iSTOP hiPSC lines for pluripotency, karyotyping, neuron differentiation capacity, and the expected NMD and LoF.
Results
The CBEmax DNA base-editing enriching system substantially increases “C” to “T” editing in hiPSCs
A key for generating LoF alleles by using a CBE to introduce premature stop codons (C to T changes; i.e., iSTOP approach) (Billon et al., 2017; Popp and Maquat, 2016) on a large scale is to have sufficiently high gene editing efficiency. Although DNA base editors have high SNP editing efficiency (>50%) in some commonly used cell lines such as HEK293 (Rees and Liu, 2018), hiPSCs are less tested. We opted to employ a base editing reporter gene system to enrich the gene-edited cells (Standage-Beier et al., 2019), thereby increasing iSTOP editing efficiency of a target gene in selected cells. In this CBE editing enriching system (CBEmax_Enrich), a blue fluorescent protein (BFP) reporter on the reporter plasmid pEF-BFP will turn into a functional EGFP reporter when it is edited from CAC (H66) to TAC (Y66) in cells co-transfected with pEF-AncBE4max and single-guide RNAs (sgRNAs) (Figure 1A). We first individually tested the two iSTOP sgRNAs (Table S1) that target the apolipoprotein E (APOE) gene in HEK293 cells. We transiently co-transfected HEK293 cells with CBEmax_Enrich and sgRNA construct, followed by fluorescence-activated cell sorting (FACS) to enrich GFP+ cells (i.e., with the reporter gene edited) for testing editing efficiency by Sanger sequencing (Figure 1B). For each sgRNA, we found a substantial increase in the target gene editing efficiency (C to T) in FACS-sorted GFP+ cells compared to the transfected BFP+ cells (from 42% to 86% and from 29% to 81%, respectively) (Figure 1B).
Figure 1.

Improved iSTOP base editing efficiency by enriching cells with the reporter gene edited
(A) BFP cassette of the CBEmax_Enrich reporter vector. C to T change turns BFP to EGFP in cells undergoing base editing. APOBEC, apolipoprotein B mRNA editing catalytic polypeptide-like, the base editing enzyme.
(B) High C to T editing efficiency of two iSTOP sgRNAs (iSTOP#1 on the left and iSTOP#2 on the right) in HEK293 cells upon enrichment. Left: the cell gating patterns of the dissociated single cells (transfected; upper) and the editing-enriched GFP+ cells (lower).
(C) Improved C to T editing efficiency in hiPSCs for the same iSTOP sgRNAs as in (B). Red arrow: intended C to T editing; black arrow: unintended by-standing C to T editing. Editing efficiency in (B) and (C) was calculated by using EditR (Kluesner et al., 2018) that displayed DNA base composition under each sequencing trace peak.
(D) Target gene (APOE) expression knockdown in hiPSC lines homozygous or heterozygous for the T allele after editing using the iSTOP1 and iSTOP2 sgRNAs. Two donor lines are shown, and the mRNA expression was quantified by qPCR and normalized to GAPDH expression. N = 3 independent cell cultures. Data are presented as Mean±S.E.M.
Next, we similarly tested for the iSTOP editing efficiency in two hiPSC lines (Figure 1C) and whether the introduced iSTOP codons led to the expected NMD (i.e., LoF) (Figure 1D). For both iSTOP sgRNAs, we observed a robust increase, although to a less extent than in HEK293, of the target gene editing efficiency in FACS-sorted GFP+ cells compared to the transfected BFP+ cells (from 24% to 66% and from 5% to 31%, respectively) (Figure 1C). More importantly, as expected from the iSTOP-mediated NMD of mRNAs, we found 86% and 98% of APOE expression reduction in hiPSC clones homozygous for iSTOP1 and iSTOP2, respectively, and ∼50% expression reduction in hiPSC clones heterozygous for iSTOP mutations (Figure 1D).
Taken together, these results show that the CBEmax_Enrich system can significantly increase the iSTOP editing efficiency, which enables us to generate LoF alleles on a large scale by introducing premature stop codons.
A scalable workflow for efficiently deriving clonal LoF alleles in hiPSCs using CBEmax_Enrich
Our goal is to develop an efficient pipeline that involves single hiPSC cell sorting for deriving clonal LoF alleles of hundreds of NPD genes in multiple hiPSC lines. To achieve this goal, a key is to obtain a relatively high single hiPSC clonal survival rate after FACS of the enriched GFP+ cells (Figures 1B and 1C). It has been recently shown that the CEPT small molecular cocktail can increase single hiPSC cloning efficiency compared to ROCK inhibitor (Y-27632; ROCK-I) (Tristan et al., 2023). We thus tested the performance of CEPT by treating the hiPSCs with CEPT both during CBEmax_Enrich transfection (for iSTOP sgRNAs of 4 genes) and the FACS sorting of single cells into 96-well plates 48 h post-transfection. However, we observed a very low single hiPSC clonal survivability (∼5%) despite a high editing efficiency (∼70%) (Figures S1A and S1B). Combining routine ROCK-I treatment of hiPSCs at transfection with CEPT treatment during 48 h post-transfection cell sorting gave us a much higher single hiPSC clonal survivability (∼27%), and even higher survivability (∼35%) when we sorted cells 72 h post-transfection while maintaining high gene editing efficiency (Figures S1A and S1B).
After these optimizations to achieve high gene editing efficiency and single hiPSC clonal survivability, we designed a semi-automated pipeline for deriving clonal LoF alleles in hiPSCs for 23 NPD genes for each batch (Figure 2A). Briefly, the CBEmax_Enrich vector, the reporter BFP plasmid, and the sgRNA vector carrying the reporter sgRNA and a targeting sgRNA were transiently transfected into hiPSCs in a 24-well plate, each well with one of the 23 targeted LoF mutations or a non-transfection (sgRNA)-control (NTC) for 1 donor hiPSC line. We then sorted out GFP+ cells that were enriched for base editing and distributed 96 single cells per gene/LoF in a 96-well plate. A handful of single hiPSC colonies from each 96-well plate were further subjected to Sanger sequencing to verify the C to T changes (LoF allele). Then 2–3 hiPSC clones, preferably homozygous for a LoF allele, were banked. The selected hiPSC clones were also subjected to RNA sequencing (RNA-seq) to confirm the absence of chromosomal abnormality by eSNP-Karyotyping and pluripotency test. With this pipeline, we have generated LoF alleles, mostly homozygous, for 22 of the 23 selected SSPsyGene Consortium-prioritized NPD genes (no editing found for HERC1, Table S3), including 9 (ARID1B, CACNA1G, CHD8, DLL1, GABRA1, KMT2C, SCN2A, SHANK3, and SMARCC2) out of the 10 “capstone genes” (genes prioritized by the SSPsyGene Consortium to be tested for technical consistency across all consortium sites) for which we could design sgRNAs, on 3 donor lines of European ancestry (KOLF2.2J, CW20107, and MGS_CD14) (Figure 2B; Table S2). Our MiNND project within the SSPsyGene Consortium aims to produce LoF alleles for about 150–200 NPD genes on 6 different hiPSC lines. The derivation of many iSTOP LoF alleles enables us to systematically evaluate the performance of iSTOP base editing on hiPSCs and its efficiency in leading to LoF.
Figure 2.

The efficient iSTOP base editing pipeline introduces LoF alleles on a large scale
(A) The workflow that enables the iSTOP editing in batches of 24 (23 target genes +1 control). The hiPSC transfection was performed on a 24-well plate, followed by single hiPSC sorting, single clone expansion on a 96-well plate, clonal sequencing confirmation, and hiPSC banking.
(B) Genes and hiPSC lines were used in the current study. Nine prioritized genes (capstone genes) by the SSPsyGene Consortium are listed in the gray box. Other genes are those NPD risk genes selected by the SSPsyGene Consortium to have the strongest disease associations and highest priorities for creating LoF alleles.
iSTOP CBE base editing in hiPSCs is efficient and reproducible in different hiPSC lines
The performance of the CBE in hiPSCs, especially in the context of the iSTOP design and with reporter gene editing enrichment, has not been systematically evaluated. With data from the iSTOP base editing of 23 genes across 3 donor hiPSC lines (Figure 2; Table S3), we found, on average, the post-editing single-cell clonal viability to be 35%–47% (Figure 3A) and the reporter gene editing efficiency to be 31%–50% (Figure S1C). After reporter gene editing enrichment, the average target gene editing efficiency was ∼60%, with a strong correlation among different cell lines (Pearson’s R = 0.91–0.95) (Figures 3B, S1D, and S1E), demonstrating the highly efficient and reproducible iSTOP CBE editing across all three hiPSC lines. About half of the genes showed editing efficiency higher than 90%, and only 5 genes with editing efficiency less than 10% (including the one without editing) (Figure 3B). Despite the robust increase of target gene editing efficiency after reporter gene editing enrichment (Figure 1), there was a weak correlation between reporter gene editing efficiency and target gene editing efficiency (Figure S1F), suggesting target gene editing efficiency is predominately determined by gene-specific sgRNA performance.
Figure 3.

High and reproducible C to T base editing efficiencies across genes and hiPSC lines
(A) High rate of single hiPSC clonal survivability after post-transfection cell sorting.
(B) High iSTOP LoF allele editing efficiency and reproducibility. The genotypes were confirmed by Sanger sequencing for the selected individual hiPSC clones from each gene editing.
(C) Examples of Sanger sequencing traces to confirm LoF alleles of two genes, SCN2A (left) and TRIO (right), in all three hiPSC lines. The shown sequencing traces are near the iSTOP-sgRNA region, with the PAM sequence highlighted in transparent yellow boxes and the genotype of the LoF mutation site marked in red box.
Overall, we obtained clonal hiPSC lines carrying putative LoF alleles for 22 targeted genes (no editing found for HERC1), of which 15 are homozygous (Figure 3B). We found that although the genes with heterozygous LoF alleles tended to have low editing efficiency (<10%) (ANKRD11, KMT2C, GABRA1, and AKAP11), some had high editing efficiency (SETD1A with 29%–38%, TRIO with 46%–71%, and CUL1 with 33%–50%) (Figure 3B), suggesting that for some NPD genes homozygous LoF alleles may have deleterious effects on hiPSC survival or growth. It is noteworthy that all three genes (SETD1A, TRIO, and CUL1) with only heterozygous LoF clones, despite their relatively high editing efficiency, are top-ranking SZ risk genes found by the SZ Exome Sequencing Meta-Analysis (SCHEMA) Consortium to have rare and highly penetrant SZ-associated PTVs (Singh et al., 2022). Of these genes, TRIO was found to initially have homozygous hiPSC clones grown in the 96-well plate post sorting; however, only heterozygous clones (Figure 3C) were found to show sustained normal hiPSC growth, which is consistent with the known necessary role of TRIO for cell migration and growth (Deinhardt et al., 2011; Seipel et al., 1999).
The CBE-edited iSTOP hiPSC clones are pluripotent and have minimal chromosomal abnormalities
Next, we characterized the selected iSTOP hiPSC clones for stem cell pluripotency, chromosomal abnormalities, and neuron differentiation capability. Immunofluorescence staining of stem cell pluripotency markers (OCT4, SOX2, and TRA-1-60) of the engineered hiPSC lines for 6 selected LoF alleles confirmed their pluripotency (Figures 4A and S2A). To further evaluate the pluripotency of all the selected hiPSC LoF clones at the genomic and molecular level, we carried out RNA-seq for each hiPSC clone and used CellNet to quantify how closely the engineered hiPSC populations transcriptionally resembled human embryonic stem cells (ESCs) compared to other non-ESC somatic cells (Cahan et al., 2014). All hiPSC clones exhibited high stemness scores (0.93–0.97) and no traces of other somatic cell types (Figures 4B, S2B, and S2C). With the same RNA-seq data, we also confirmed the absence of large chromosomal abnormalities due to base editing using eSNP-Karyotyping (Weissbein et al., 2016; Zhang et al., 2023) (Figures 4C and S3) by analyzing the moving average of the allelic ratio of each expressed SNP along the genome (left panel) and stretches of common SNP heterozygosity on each chromosome for each batch of hiPSC lines. In total, for the assayed 69 hiPSC lines of all three batches, eSNP-Karyotyping identified only one hiPSC line from batch 3 (NTC line with barcode V4167) showing chromosomal abnormality on Chr12 (57759165–76048101 bp; hg38) (Figure S3 and https://zenodo.org/records/11591445; Table S4). Finally, we confirmed that all the selected iSTOP hiPSC lines (n = 6) could be successfully induced into neurons (MAP2+/Syn+) after Ngn2 transduction (Figure 4D).
Figure 4.

Characterization of isogenic base-edited hiPSC lines carrying iSTOP LoF alleles
(A) The iSTOP mutant lines were stained positive for pluripotent stem cell markers (OCT4, SOX2, and TRA-1-60). Scale bar: 50 μm.
(B) CellNet analysis of RNA-seq data of hiPSC lines confirmed their pluripotency. Pluripotency scores showed transcriptional similarity of the edited iSTOP LoF hiPSC lines to ESC but not other non-ESC cell types. Only one batch of hiPSC lines is shown, and the data for the other two batches are in Figure S2.
(C) eSNP-Karyotyping showed no large chromosomal abnormalities. Left: moving average of the allelic ratio of each expressed SNP along the genome; right: stretches of homozygosity on each chromosome. An example of one hiPSC line is shown. Data for all the lines are available at https://zenodo.org/records/11591445.
(D) Some selected iSTOP LoF hiPSC lines were successfully differentiated into excitatory neurons (Syn+/MAP2+). Images were taken with two different magnifications (20× and 63×). Scale bar: 20 μm. NTC, non-transfected control line. In (A) and (C), the gene name for the LoF allele and the cell line number (starting with “V”) were listed.
Most iSTOP hiPSC lines show the expected mRNA or protein reduction with the confirmation of SHANK3 LoF phenotype in Ngn2-induced neurons
Because we have employed the iSTOP approach to introduce premature stop codons that are predicted to cause NMD (Table S1), we first tested whether we could observe the expected expression reduction for each NPD gene in the engineered hiPSC lines using RNA-seq data. Compared to the unedited cell line, the iSTOP lines for about 12 genes showed partial or near-complete expression knockdown (KD) (compared to the unedited line) as expected for NMD (Figure 5A). Comparing the Z-scored expression values (counts per million reads) of each gene in each iSTOP LoF hiPSC line for all three batches (Figure S4) showed similar patterns of reduced gene expression of those 12 genes as in Figure 5A. The strongest expression KD was observed for hiPSC lines homozygous for SCN2A, CHD8, and CACNA1G iSTOP LoF alleles, exhibiting a 70%–90% expression reduction. The lack of the expected NMD for some genes may be due to possible cell type-specific NMD regulation (Huang et al., 2011), incomplete mRNA degradation, or inaccurate NMD prediction in sgRNA design. Our qPCR further confirmed the incomplete mRNA degradation or even increased mRNA production (e.g., HCN4, SP4) for genes that did not show the expected NMD in RNA-seq (Figure 5B).
Figure 5.

Characterization of NMD and LoF for iSTOP LoF hiPSC lines
(A) Heatmap of relative expression (Z-scored) of each mutant line (vs. NTC) using normalized RNA-seq expression value (CPM, count per million). The values in the diagonal boxes show the fold change of a specific line with LoF mutation. The fold change values in red fonts indicate those showing NMD.
(B) qPCR confirmation of the expression fold change in RNA-seq (vs. NTC).
(C) Strong correlation of expression fold changes (vs. NTC) between RNA-seq and qPCR data.
(D) Western blots showed the expected reduction of protein abundance for the LoF alleles of 5 selected genes that did not exhibit NMD in (A) and (B). NTC, non-transfected control. Mouse brain protein extracts were used as a positive control for each blot. Note that non-specific signals below 250 kDa, expected sizes for ARID1B, were apparent in the blots; only the putative signal of ARID1B was quantified. Each lane represents a cell lysate from 4 independent cell cultures of 2 different passages (2 for each passage). The parent line is CW20107. Data are presented as: Mean±S.E.M.
Regardless of any detectable NMD from RNA-seq or qPCR, we expected those premature stop codons at the first half of a target gene would result in protein truncations (i.e., the loss of full-length proteins). To confirm this hypothesis, we performed western blotting for 5 selected genes that did not show the expected mRNA NMD (CUL1, ANKRD11, SP4, RB1CC1, and ARID1B) (Figures 5A and 5B) using cell lysates of their respective iSTOP hiPSC lines (CUL1+/−, ANKRD11+/−, SP4−/−, RB1CC1−/−, and ARID1B−/−) (Figure 5D). Here, we have included both homozygous and heterozygous lines to examine whether we can observe the expected dosage-dependent LoF effect on protein expression. We found that compared to the unedited hiPSC line (NTC), all 5 LoF lines showed the expected protein reduction based on their genotype, with heterozygous LoF lines showing ∼50% decrease of the intact proteins (CUL1 and ANKRD11). In contrast, homozygous LoF lines exhibited near-complete KD (SP4, ARID1B, and RB1CC1) (Figure 5D). To confirm whether the antibody used for western blot targets the major transcript isoforms that did not show the expected NMD of mRNA, we re-analyzed the RNA-seq data using a pseudo-alignment-based model (Kallisto) (Bray et al., 2016) to obtain isoform expression in each hiPSC line (Table S5). We found that each antibody targets all major isoforms that did not show the expected NMD for each gene except for SP4, in which the antibody targets only one major isoform (Figure S5; Table S5); however, the SP4 transcript isoform targeted by the antibody accounts for ∼61% of the SP4 expression and also showed an increase of RNA expression in the SP4 −/− line (Figures 5A and 5B). Thus, despite the absence of the expected NMD for CUL1, ANKRD11, SP4, RB1CC1, and ARID1B, the detected protein KD of these genes was unlikely due to alternative isoforms not detected by an antibody. All together, these results strongly suggest that most LoF alleles engineered by our iSTOP base editing approach effectively led to the expected gene expression KD or complete abolishment of the gene expression, i.e., a LoF effect, at the protein level.
To further corroborate the LoF effect by the introduced iSTOP mutation, we assessed whether we could replicate the previously reported morphological phenotypes in SHANK3-deficient human neurons (Yi et al., 2016). We derived excitatory (Ex) and inhibitory (Inh)-induced neurons by ectopic expression of the Ngn2 or Ascl1/Dlx2 transcription factors (Halikere et al., 2020; McGowan et al., 2018; Yang et al., 2017; Zhang et al., 2013) (see Methods) from the hiPSC lines that carry homozygous iSTOP LoF allele of SHANK3. We also included an edited hiPSC line that carried the iSTOP LoF allele of CUL1, a strong SZ risk gene (Singh et al., 2022) that only had heterozygous clones (Figure 3B). With the co-cultures of Ex- and Inh neurons, we assayed the neurite outgrowth, branches, and synaptic puncta density of the tdTomato-labeled Ex neurons using high content imaging (HCI). The built-in neurite outgrowth module on the ImageXpress system was used for cell segmentation for assaying neurite outgrowth and branches (Figure S6A), and a customized synaptic puncta module with binary masks was used for assaying puncta density (Figure S6B). Compared to the Ex neurons from the isogenic control hiPSC line, the SHANK3 iSTOP LoF line showed ∼2/3 reduction of neurite outgrowth and branches but no significant change of synaptic puncta (Synapsin1+) density (Figures 6 and S6), which are partially consistent with the reported cellular phenotypes of SHANK3-haploinsufficiency in human neurons: SHANK3−/− neurons showed a reduction of both neurite length/branches and synaptic puncta, while SHANK3+/− neurons only showed a reduction of neurite length/branches (Yi et al., 2016). For the SZ risk gene CUL1, we observed a significant reduction of Ex neuronal neurite outgrowth and branches by ∼60% as well as a reduced synaptic puncta (Synapsin1+) density by ∼40% (Figures 6 and S6). It is noteworthy that neurons with Cul1 deficiency exhibit severe dendrite pruning defects in Drosophila (Wong et al., 2013), while small interfering RNA KD of Cul1 in rat hippocampal neurons increases synaptic F-actin but decreases Synapsin1 (Falkovich et al., 2023).
Figure 6.

High-content imaging of excitatory and inhibitory neurons co-cultured with mouse glia for iSTOP LoF hiPSC lines
(A) Representative images of neural cultures with hiPSC-differentiated excitatory neurons labeled by tdTomato (red). Neurons were stained for MAP2, SYN1, tdTomato, and DAPI. TdTomato and DAPI staining were used to quantify neurite growth/branches; tdTomato, MAP2, and SYN1 staining were used to analyze synaptic puncta. Scale bar: 20 μm.
(B) Summarized imaging result for neurite outgrowth and branches and synaptic puncta (left to right) for SHANK3 (−/−) and CUL1 (+/−) LoF alleles on donor hiPSC line CW20107. Each data point represents the measurement of an independent well of neuron culture on a 96-well plate; N = 8 wells. The upper limit of the horizontal line is 5%, and the lower limit is 95%; the box has three limites: 25%, 50% and 75%. All %s are fractions as defined by the Box and Whisker plot function in Prism 7.
To validate the observed neurite deficits (Figure 6) in a sparse neuronal culture, we performed an independent experiment with a lower cell density (Figures S6C and S6D). We found that the patterns of the neurite outgrowth/branch differences for CUL1 and SHANK3 LoF lines (compared to NTC line) were overall similar between “higher” and “lower” density neuron groups (Figures S6C and S6D). However, the phenotypes were less pronounced in lower density cultures, highlighting the importance of specific experimental conditions for detecting the dramatic deficits observed in higher density cultures in HCI. Taken together, our observed NMD of mRNA, protein expression reduction, and neural phenotypic changes in the assayed iSTOP hiPSC lines collectively suggest that most iSTOP hiPSC lines for NPD genes are expected to show LoF effect on protein expression.
Discussion
Although CRISPR editing of individual NPD risk genes/variants in hiPSCs has been widely used in the past decade (De Los Angeles et al., 2021; Duan, 2023; Michael Deans and Brennand, 2021; Muhtaseb and Duan, 2022; Wang et al., 2020; Wen et al., 2016; Zhang et al., 2020, 2023), a scaled and efficient pipeline for clonal LoF mutagenesis in hiPSCs has not been established. Our reported CBE iSTOP editing workflow benefited from the improved gene editing efficiency of the CBEmax_Enrich system, the precision of iSTOP mutagenesis, the streamlined RNA-seq-based assays for pluripotency, eSNP-Karyotyping, and iSTOP-mediated NMD and/or LoF. These factors simplified the workflow and made it more amenable for automation to increase throughput while keeping the pipeline cost-effective. Moreover, because our pipeline only involved transient transfection of hiPSCs, the engineered iSTOP hiPSC lines were genome-integration-free, as opposed to CRISPR pooled screening that often entails hiPSC genome integration with exogenous virus fragments that may confound downstream phenotypic assay readouts. The derived iSTOP hiPSC lines carrying LoF alleles for the current list of 22 (out of 23) edited genes on 3 donor hiPSC lines, and many more engineered LoF hiPSC lines to be generated, will be an invaluable sharable resource for the NPD genetics research community.
We observed high editing efficiency for most genes that were highly reproducible across all three hiPSC lines, suggesting CBE iSTOP performance was not hiPSC line-specific but rather mainly determined by sgRNAs. As expected, genes with high editing efficiency tended to have more clones homozygous for iSTOP LoF alleles. Interestingly, for three strong SZ risk genes (SETD1A, CUL1, and TRIO) identified by SCHEMA, we only obtained heterozygous LoF hiPSC clones despite high editing efficiency, suggesting the likely deleterious effect of LoF on stem cell survival. Indeed, possible lethal effects of LoF of the three genes are supported by the existing body of literature: Setd1a, encoding a histone methyltransferase, was found to be required for embryonic and neural stem cell survival (Bledau et al., 2014), and only heterozygous SETD1A-haploinsufficiency hiPSC lines (Chong et al., 2022; Wang et al., 2022; West et al., 2019) or mouse models (Mukai et al., 2019; Nagahama et al., 2020) have been reported for functional characterization; CUL1 has E3 ubiquitin-protein ligase activity and homozygous deletion of Cul1 in mice causes arrest in early embryogenesis and embryonic lethality at E6.5 (Wang et al., 1999); and TRIO functions as a guanosine diphosphate to guanosine triphosphate exchange factor and is necessary for cell migration and growth (Deinhardt et al., 2011; Seipel et al., 1999). It is noteworthy that highly penetrant patient-specific PTVs in these NPD genes are all heterozygous; thus, the obtained heterozygous LoF hiPSC clones can be valuable for ascertaining more disease-relevant cellular phenotypes.
Our clonal LoF mutagenesis pipeline leverages the precise control of the CBE iSTOP editing approach to introduce a premature stop codon that is predicted to cause NMD of mRNAs or lead to a protein truncation. However, only 12 out of the 22 edited NPD genes showed partial or nearly complete NMD in hiPSC. Other than possible inaccuracy of NMD prediction, the lack of the expected NMD for some genes is likely due to: (1) RNA-seq may have still detected partially degraded mRNAs; (2) an intricate feedback network maintains both RNA surveillance and the homeostasis of normal gene expression in mammalian cells (Huang et al., 2011); and (3) some cells do escape NMD either by translational readthrough at the premature stop codon or by a failure of mRNA degradation after successful translation termination (Sato and Singer, 2021). However, our western blotting analysis showed that most iSTOP mutations, even without detectable NMD, likely led to LoF by yielding a truncated protein too short to be detected. Although a truncated protein may arguably have normal function or even gain of function, the sgRNA in our iSTOP design often targets the protein N-terminal, thus more likely to show LoF. It is reassuring that even for genes that do not show NMD (e.g., CUL1) or even increased mRNA level (likely due to negative feedback regulation, e.g., SP4) in hiPSCs, our western blotting showed the expected protein expression KD, suggesting most iSTOP mutant alleles are likely to show LoF in neurons.
Among other limitations, our iSTOP design relies on NMD or protein truncation to achieve LoF, where the extent of LoF may not be as complete as obtained from complete knockout (KO) using CRISPR-Cas9. However, a complete gene KO may need multiple sgRNAs targeting multiple genomic regions, which poses a challenge for clonal LoF mutagenesis on a large scale and, more importantly, may cause large chromosomal arrangements. Moreover, although our scaled pipeline is suitable for studying gene LoF, it may not apply to some patient-specific mutations (or natural alleles) or copy-number variants that do not involve C to T changes but are also important for NPD. Furthermore, we acknowledge the limitation of our eSNP-Karyotyping in its inability to detect indels of non-coding regions or balanced translocations as well as its low sensitivity for genes with low expression in hiPSCs, which may leave some genetic lesions undetected in our present study. Future G-band karyotyping or genome-wide genotyping will provide a more comprehensive characterization of different types of genomics abnormalities in these hiPSC lines. Lastly, our clonal iSTOP LoF mutagenesis pipeline may be complemented by pooled CRISPR screening in combination with the rapidly evolving spatial transcriptomics and phenotyping to ascertain LoF allelic effects at single-neuron resolution simultaneously. Albeit limitations, our scaled and efficient clonal LoF mutagenesis pipeline showed robust performance in generating easily sharable individual hiPSC lines carrying LoF alleles for a large number of NPD genes. In addition to engineering LoF alleles, the pipeline can be easily adopted for precise SNP editing (C to T changes by CBE, and A to G changes by adenine base editors [ABE]) in hiPSC for studying coding or noncoding disease risk variants. The scaled LoF mutagenesis pipeline thus empowers hiPSC as a promising cellular model for understanding the disease biology of NPD and other complex genetic disorders.
Experimental procedures
Methods
hiPSC lines and cell culture
CW20107 was from the California Institute for Regenerative Medicine. KOLF2.2J was an updated version of KOLF2.1J (Pantazis et al., 2022) and is available at The Jackson Laboratory via special request. The other hiPSC line (CD14) is specific to the MiNND project and is from the Duan lab (Shi et al., 2009; Zhang et al., 2020, 2023). CD14 was originally derived from the lymphocytes of a healthy donor of the Molecular Genetics of Schizophrenia cohort (Shi et al., 2009). Detailed information is in Table S2. The institutional review board of NorthShore University HealthSystem approved the study.
Gene selection and iSTOP base editing design
The reported 23 genes were part of the ∼250 NPD genes selected by SSPsyGene Consortium (sspsygene.ucsc.edu). For designing iSTOP sgRNAs, the iSTOP web tool was used (Billon et al., 2017).
iSTOP base editing pipeline
The LoF mutagenesis was performed in batches, each containing 23 genes and an NTC on a 24-well plate format. The sorted single cells were cultured on a 96-well plate for Sanger sequencing genotyping.
hiPSC characterization
LoF mutant lines were characterized for stem cell pluripotency by both immunofluorescence staining and by using CellNet analysis of RNA-seq data (Cahan et al., 2014). For chromosomal abnormality, we used eSNP-Karyotyping (Weissbein et al., 2016) as described (Zhang et al., 2020, 2023).
Neuron differentiation from hiPSCs
We used the two commonly used methods: NGN2 + rtTA for excitatory neuron differentiation (Zhang et al., 2013) and ASCL1 + DLX2 + rtTA for inhibitory neuron differentiation (Yang et al., 2017).
Neuron morphological characterization
The neurons were imaged using Molecular Devices’ (San Jose, CA) ImageXpress Micro Confocal High-Content Imaging System. The neurite phenotypes were analyzed using the built-in module. We used a customized synaptic assay module to assay synapse density.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jubao Duan (jduan@uchicago.edu).
Materials availability
The hiPSC lines will be made available as part of the SSPsyGene Consortium to fulfill the NIMH (National Institute of Mental Health) material/data-sharing commitment.
Data and code availability
All the reported data and code used during analysis, including the full hiPSC eSNP-Karyotyping results are deposited at https://doi.org/10.5281/zenodo.13273149. The RNA-seq data’s GEO accession number is GSE262442.
Acknowledgments
Data were generated as part of the SSPsyGene Consortium, supported by RM1MH133065 awarded to Z.P.P., J.D., and J.G.M. We thank SSPsyGene Consortium members for selecting NPD genes and providing hiPSC lines CW20107 (from CIRM) and KOLF2.2J (from the Jackson Lab). We thank Molecular Genetics of SZ (MGS) investigators for collecting samples that were used to derive MGS hiPSC lines. Funding was provided by NIH grants R01AA023797 and R01MH125528 (to Z.P.P.), R01MH106575, R01MH116281, and R01AG063175 (to J.D.), and RM1MH133065 (to Z.P.P., J.D., and J.G.M.). A full BioRender license to the University of Chicago was used for visual depiction and graph making.
Author contributions
H.Z., L.P., and A.M. performed the main experiments; S.D.d.L.G. performed the neuron differentiation and characterization; S.Z. analyzed the RNA-seq data and created the figures, tables, and the graphic abstract; P.G. and M.E. performed neuron differentiation and characterization; D.S. and G.T. performed the neuron imaging analyses; A.D. assisted with RNA-seq data analysis; W.G.W. and B.J. assisted with hiPSC derivation and culture; R.P. assisted with the project coordination; R.P.H., C.N.P., J.G.M., and A.R.S. assisted with project design and result interpretation; Z.P.P. and J.D. conceived and supervised the project and wrote the manuscript. All authors contributed to the manuscript writing and editing.
Declaration of interests
The authors declare no competing interests.
Published: September 12, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.stemcr.2024.08.003.
Contributor Information
Zhiping P. Pang, Email: pangzh@rwjms.rutgers.edu.
Jubao Duan, Email: jduan@uchicago.edu.
Supplemental information
References
- Billon P., Bryant E.E., Joseph S.A., Nambiar T.S., Hayward S.B., Rothstein R., Ciccia A. CRISPR-Mediated Base Editing Enables Efficient Disruption of Eukaryotic Genes through Induction of STOP Codons. Mol. Cell. 2017;67:1068–1079.e4. doi: 10.1016/j.molcel.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bledau A.S., Schmidt K., Neumann K., Hill U., Ciotta G., Gupta A., Torres D.C., Fu J., Kranz A., Stewart A.F., Anastassiadis K. The H3K4 methyltransferase Setd1a is first required at the epiblast stage, whereas Setd1b becomes essential after gastrulation. Development. 2014;141:1022–1035. doi: 10.1242/dev.098152. [DOI] [PubMed] [Google Scholar]
- Bray N.L., Pimentel H., Melsted P., Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016;34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- Cahan P., Li H., Morris S.A., Lummertz da Rocha E., Daley G.Q., Collins J.J. CellNet: network biology applied to stem cell engineering. Cell. 2014;158:903–915. doi: 10.1016/j.cell.2014.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong Z.-S., Khong Z.J., Tay S.H., Ng S.-Y. Metabolic contributions to neuronal deficits caused by genomic disruption of schizophrenia risk gene SETD1A. Schizophrenia. 2022;8:115. doi: 10.1038/s41537-022-00326-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuella-Martin R., Hayward S.B., Fan X., Chen X., Huang J.W., Taglialatela A., Leuzzi G., Zhao J., Rabadan R., Lu C., et al. Functional interrogation of DNA damage response variants with base editing screens. Cell. 2021;184:1081–1097.e19. doi: 10.1016/j.cell.2021.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Los Angeles A., Fernando M.B., Hall N.A.L., Brennand K.J., Harrison P.J., Maher B.J., Weinberger D.R., Tunbridge E.M. Induced Pluripotent Stem Cells in Psychiatry: An Overview and Critical Perspective. Biol. Psychiatr. 2021;90:362–372. doi: 10.1016/j.biopsych.2021.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deinhardt K., Kim T., Spellman D.S., Mains R.E., Eipper B.A., Neubert T.A., Chao M.V., Hempstead B.L. Neuronal growth cone retraction relies on proneurotrophin receptor signaling through Rac. Sci. Signal. 2011;4 doi: 10.1126/scisignal.2002060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan J. Human stem cell modeling of neuropsychiatric disorders: from polygenicity to convergence. Med. Rev. 2023;3:347–350. doi: 10.1515/mr-2023-0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkovich R., Danielson E.W., Perez de Arce K., Wamhoff E.C., Strother J., Lapteva A.P., Sheng M., Cottrell J.R., Bathe M. A synaptic molecular dependency network in knockdown of autism- and schizophrenia-associated genes revealed by multiplexed imaging. Cell Rep. 2023;42 doi: 10.1016/j.celrep.2023.112430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grove J., Ripke S., Als T.D., Mattheisen M., Walters R.K., Won H., Pallesen J., Agerbo E., Andreassen O.A., Anney R., et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 2019;51:431–444. doi: 10.1038/s41588-019-0344-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halikere A., Popova D., Scarnati M.S., Hamod A., Swerdel M.R., Moore J.C., Tischfield J.A., Hart R.P., Pang Z.P. Addiction associated N40D mu-opioid receptor variant modulates synaptic function in human neurons. Mol. Psychiatr. 2020;25:1406–1419. doi: 10.1038/s41380-019-0507-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna R.E., Hegde M., Fagre C.R., DeWeirdt P.C., Sangree A.K., Szegletes Z., Griffith A., Feeley M.N., Sanson K.R., Baidi Y., et al. Massively parallel assessment of human variants with base editor screens. Cell. 2021;184:1064–1080.e20. doi: 10.1016/j.cell.2021.01.012. [DOI] [PubMed] [Google Scholar]
- Holtzman L., Gersbach C.A. Editing the Epigenome: Reshaping the Genomic Landscape. Annu. Rev. Genom. Hum. Genet. 2018;19:43–71. doi: 10.1146/annurev-genom-083117-021632. [DOI] [PubMed] [Google Scholar]
- Howard D.M., Adams M.J., Clarke T.K., Hafferty J.D., Gibson J., Shirali M., Coleman J.R.I., Hagenaars S.P., Ward J., Wigmore E.M., et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci. 2019;22:343–352. doi: 10.1038/s41593-018-0326-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L., Lou C.H., Chan W., Shum E.Y., Shao A., Stone E., Karam R., Song H.W., Wilkinson M.F. RNA homeostasis governed by cell type-specific and branched feedback loops acting on NMD. Mol. Cell. 2011;43:950–961. doi: 10.1016/j.molcel.2011.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluesner M.G., Nedveck D.A., Lahr W.S., Garbe J.R., Abrahante J.E., Webber B.R., Moriarity B.S. EditR: A Method to Quantify Base Editing from Sanger Sequencing. CRISPR J. 2018;1:239–250. doi: 10.1089/crispr.2018.0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan H., Mirabella V.R., Hamod A., Karakhanyan A., Mlynaryk N., Moore J.C., Tischfield J.A., Hart R.P., Pang Z.P. hsa-let-7c miRNA Regulates Synaptic and Neuronal Function in Human Neurons. Front. Synaptic Neurosci. 2018;10:19. doi: 10.3389/fnsyn.2018.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X., Navoly G., Giannakopoulou O., Levey D.F., Koller D., Pathak G.A., Koen N., Lin K., Adams M.J., Rentería M.E., et al. Multi-ancestry genome-wide association study of major depression aids locus discovery, fine mapping, gene prioritization and causal inference. Nat. Genet. 2024;56:222–233. doi: 10.1038/s41588-023-01596-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael Deans P.J., Brennand K.J. Applying stem cells and CRISPR engineering to uncover the etiology of schizophrenia. Curr. Opin. Neurobiol. 2021;69:193–201. doi: 10.1016/j.conb.2021.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhtaseb A.W., Duan J. Modeling common and rare genetic risk factors of neuropsychiatric disorders in human induced pluripotent stem cells. Schizophr. Res. 2022 doi: 10.1016/j.schres.2022.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai J., Cannavò E., Crabtree G.W., Sun Z., Diamantopoulou A., Thakur P., Chang C.-Y., Cai Y., Lomvardas S., Takata A., et al. Recapitulation and Reversal of Schizophrenia-Related Phenotypes in Setd1a-Deficient Mice. Neuron. 2019;104:471–487.e12. doi: 10.1016/j.neuron.2019.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins N., Forstner A.J., O'Connell K.S., Coombes B., Coleman J.R.I., Qiao Z., Als T.D., Bigdeli T.B., Børte S., Bryois J., et al. Genome-wide association study of more than 40,000 bipolar disorder cases provides new insights into the underlying biology. Nat. Genet. 2021;53:817–829. doi: 10.1038/s41588-021-00857-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahama K., Sakoori K., Watanabe T., Kishi Y., Kawaji K., Koebis M., Nakao K., Gotoh Y., Aiba A., Uesaka N., Kano M. Setd1a Insufficiency in Mice Attenuates Excitatory Synaptic Function and Recapitulates Schizophrenia-Related Behavioral Abnormalities. Cell Rep. 2020;32 doi: 10.1016/j.celrep.2020.108126. [DOI] [PubMed] [Google Scholar]
- Nunez J.K., Chen J., Pommier G.C., Cogan J.Z., Replogle J.M., Adriaens C., Ramadoss G.N., Shi Q., Hung K.L., Samelson A.J., et al. Genome-wide programmable transcriptional memory by CRISPR-based epigenome editing. Cell. 2021;184:2503–2519.e17. doi: 10.1016/j.cell.2021.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer D.S., Howrigan D.P., Chapman S.B., Adolfsson R., Bass N., Blackwood D., Boks M.P.M., Chen C.Y., Churchhouse C., Corvin A.P., et al. Exome sequencing in bipolar disorder identifies AKAP11 as a risk gene shared with schizophrenia. Nat. Genet. 2022;54:541–547. doi: 10.1038/s41588-022-01034-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantazis C.B., Yang A., Lara E., McDonough J.A., Blauwendraat C., Peng L., Oguro H., Kanaujiya J., Zou J., Sebesta D., et al. A reference human induced pluripotent stem cell line for large-scale collaborative studies. Cell Stem Cell. 2022;29:1685–1702.e22. doi: 10.1016/j.stem.2022.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp M.W., Maquat L.E. Leveraging Rules of Nonsense-Mediated mRNA Decay for Genome Engineering and Personalized Medicine. Cell. 2016;165:1319–1322. doi: 10.1016/j.cell.2016.05.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Schizophrenia Consortium. Purcell S.M., Wray N.R., Stone J.L., Visscher P.M., O'Donovan M.C., Sullivan P.F., Sklar P. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees H.A., Liu D.R. Base editing: precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018;19:770–788. doi: 10.1038/s41576-018-0059-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripke S., O'Dushlaine C., Chambert K., Moran J.L., Kähler A.K., Akterin S., Bergen S.E., Collins A.L., Crowley J.J., Fromer M., et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat. Genet. 2013;45:1150–1159. doi: 10.1038/ng.2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripke S., Sanders A.R., Kendler K.S., Levinson D.F., Sklar P., Holmans P.A., Lin D.Y., Duan J., Ophoff R.A., Andreassen O.A., et al. Genome-wide association study identifies five new schizophrenia loci. Nat. Genet. 2011;43:969–976. doi: 10.1038/ng.940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato H., Singer R.H. Cellular variability of nonsense-mediated mRNA decay. Nat. Commun. 2021;12:7203. doi: 10.1038/s41467-021-27423-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satterstrom F.K., Kosmicki J.A., Wang J., Breen M.S., De Rubeis S., An J.Y., Peng M., Collins R., Grove J., Klei L., et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell. 2020;180:568–584.e23. doi: 10.1016/j.cell.2019.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seipel K., Medley Q.G., Kedersha N.L., Zhang X.A., O'Brien S.P., Serra-Pages C., Hemler M.E., Streuli M. Trio amino-terminal guanine nucleotide exchange factor domain expression promotes actin cytoskeleton reorganization, cell migration and anchorage-independent cell growth. J. Cell Sci. 1999;112:1825–1834. doi: 10.1242/jcs.112.12.1825. [DOI] [PubMed] [Google Scholar]
- Shi J., Levinson D.F., Duan J., Sanders A.R., Zheng Y., Pe'er I., Dudbridge F., Holmans P.A., Whittemore A.S., Mowry B.J., et al. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature. 2009;460:753–757. doi: 10.1038/nature08192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh T., Poterba T., Curtis D., Akil H., Al Eissa M., Barchas J.D., Bass N., Bigdeli T.B., Breen G., Bromet E.J., et al. Rare coding variants in ten genes confer substantial risk for schizophrenia. Nature. 2022;604:509–516. doi: 10.1038/s41586-022-04556-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl E.A., Breen G., Forstner A.J., McQuillin A., Ripke S., Trubetskoy V., Mattheisen M., Wang Y., Coleman J.R.I., Gaspar H.A., et al. Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat. Genet. 2019;51:793–803. doi: 10.1038/s41588-019-0397-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standage-Beier K., Tekel S.J., Brookhouser N., Schwarz G., Nguyen T., Wang X., Brafman D.A. A transient reporter for editing enrichment (TREE) in human cells. Nucleic Acids Res. 2019;47 doi: 10.1093/nar/gkz713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson H., Ophoff R.A., Steinberg S., Andreassen O.A., Cichon S., Rujescu D., Werge T., Pietiläinen O.P.H., Mors O., Mortensen P.B., et al. Common variants conferring risk of schizophrenia. Nature. 2009;460:744–747. doi: 10.1038/nature08186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sürün D., Schneider A., Mircetic J., Neumann K., Lansing F., Paszkowski-Rogacz M., Hänchen V., Lee-Kirsch M.A., Buchholz F. Efficient Generation and Correction of Mutations in Human iPS Cells Utilizing mRNAs of CRISPR Base Editors and Prime Editors. Genes. 2020;11 doi: 10.3390/genes11050511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tekel S.J., Brookhouser N., Standage-Beier K., Wang X., Brafman D.A. Cytosine and adenosine base editing in human pluripotent stem cells using transient reporters for editing enrichment. Nat. Protoc. 2021;16:3596–3624. doi: 10.1038/s41596-021-00552-y. [DOI] [PubMed] [Google Scholar]
- Tristan C.A., Hong H., Jethmalani Y., Chen Y., Weber C., Chu P.H., Ryu S., Jovanovic V.M., Hur I., Voss T.C., et al. Efficient and safe single-cell cloning of human pluripotent stem cells using the CEPT cocktail. Nat. Protoc. 2023;18:58–80. doi: 10.1038/s41596-022-00753-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trubetskoy V., Pardiñas A.F., Qi T., Panagiotaropoulou G., Awasthi S., Bigdeli T.B., Bryois J., Chen C.-Y., Dennison C.A., Hall L.S., et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature. 2022;604:502–508. doi: 10.1038/s41586-022-04434-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M., Zhang L., Gage F.H. Modeling neuropsychiatric disorders using human induced pluripotent stem cells. Protein Cell. 2020;11:45–59. doi: 10.1007/s13238-019-0638-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Rhijn J.-R.v., Akkouh I., Kogo N., Maas N., Bleeck A., Ortiz I.S., Lewerissa E., Wu K.M., Schoenmaker C., et al. Loss-of-function variants in the schizophrenia risk gene SETD1A alter neuronal network activity in human neurons through the cAMP/PKA pathway. Cell Rep. 2022;39 doi: 10.1016/j.celrep.2022.110790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Penfold S., Tang X., Hattori N., Riley P., Harper J.W., Cross J.C., Tyers M. Deletion of the Cul1 gene in mice causes arrest in early embryogenesis and accumulation of cyclin E. Curr. Biol. 1999;9:1191–1194. doi: 10.1016/S0960-9822(00)80024-X. [DOI] [PubMed] [Google Scholar]
- Weissbein U., Schachter M., Egli D., Benvenisty N. Analysis of chromosomal aberrations and recombination by allelic bias in RNA-Seq. Nat. Commun. 2016;7 doi: 10.1038/ncomms12144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells M.F., Nemesh J., Ghosh S., Mitchell J.M., Salick M.R., Mello C.J., Meyer D., Pietilainen O., Piccioni F., Guss E.J., et al. Natural variation in gene expression and viral susceptibility revealed by neural progenitor cell villages. Cell Stem Cell. 2023;30:312–332.e13. doi: 10.1016/j.stem.2023.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Z., Christian K.M., Song H., Ming G.L. Modeling psychiatric disorders with patient-derived iPSCs. Curr. Opin. Neurobiol. 2016;36:118–127. doi: 10.1016/j.conb.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West S., Zhang H., Zhang S., Kozlova A., Sanders A., Pang Z., Gejman P., Duan J. 46 modelling the schizophrenia-associated loss-of-function mutation of SETD1A in human stem cell-derived BRAIN organoids. Eur. Neuropsychopharmacol. 2019;29:S84–S85. doi: 10.1016/j.euroneuro.2019.07.187. [DOI] [Google Scholar]
- Wong J.J.L., Li S., Lim E.K.H., Wang Y., Wang C., Zhang H., Kirilly D., Wu C., Liou Y.C., Wang H., Yu F. A Cullin1-based SCF E3 ubiquitin ligase targets the InR/PI3K/TOR pathway to regulate neuronal pruning. PLoS Biol. 2013;11 doi: 10.1371/journal.pbio.1001657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray N.R., Ripke S., Mattheisen M., Trzaskowski M., Byrne E.M., Abdellaoui A., Adams M.J., Agerbo E., Air T.M., Andlauer T.M.F., et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 2018;50:668–681. doi: 10.1038/s41588-018-0090-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P., Liu Z., Liu Y., Ma H., Xu Y., Bao Y., Zhu S., Cao Z., Wu Z., Zhou Z., Wei W. Genome-wide interrogation of gene functions through base editor screens empowered by barcoded sgRNAs. Nat. Biotechnol. 2021;39:1403–1413. doi: 10.1038/s41587-021-00944-1. [DOI] [PubMed] [Google Scholar]
- Yang N., Chanda S., Marro S., Ng Y.H., Janas J.A., Haag D., Ang C.E., Tang Y., Flores Q., Mall M., et al. Generation of pure GABAergic neurons by transcription factor programming. Nat. Methods. 2017;14:621–628. doi: 10.1038/nmeth.4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi F., Danko T., Botelho S.C., Patzke C., Pak C., Wernig M., Südhof T.C. Autism-associated SHANK3 haploinsufficiency causes Ih channelopathy in human neurons. Science. 2016;352 doi: 10.1126/science.aaf2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S., Zhang H., Forrest M.P., Zhou Y., Sun X., Bagchi V.A., Kozlova A., Santos M.D., Piguel N.H., Dionisio L.E., et al. Multiple genes in a single GWAS risk locus synergistically mediate aberrant synaptic development and function in human neurons. Cell Genom. 2023;3 doi: 10.1016/j.xgen.2023.100399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S., Zhang H., Zhou Y., Qiao M., Zhao S., Kozlova A., Shi J., Sanders A.R., Wang G., Luo K., et al. Allele-specific open chromatin in human iPSC neurons elucidates functional disease variants. Science. 2020;369:561–565. doi: 10.1126/science.aay3983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Pak C., Han Y., Ahlenius H., Zhang Z., Chanda S., Marro S., Patzke C., Acuna C., Covy J., et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron. 2013;78:785–798. doi: 10.1016/j.neuron.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the reported data and code used during analysis, including the full hiPSC eSNP-Karyotyping results are deposited at https://doi.org/10.5281/zenodo.13273149. The RNA-seq data’s GEO accession number is GSE262442.