

Abstract

A cascade transformation of C2-quaternary indoxyls leading to an efficient assembly of complex (dihydro)indolo[1,2-a]quinolin-5-one ring systems is reported. The method involves the gram-scale preparation of 2-(2-aryl-3-oxoindolin-2-yl)-2-phenylacetonitriles which are then converted with methyl ketones to the corresponding 2-(2-oxo-2-aryl(alkyl)ethyl)-2-phenylindolin-3-ones. The latter can either be isolated with good yields (75–96%) or, in the case of o-nitroacetophenone, used in situ for further base-assisted intramolecular SNAr cyclization resulting in indoxyl-fused quinolone-4 hybrids (up to 95%).
1. Introduction
While primarily known for its antibacterial properties,1−3 the quinolone-4 core has long been recognized as an omnipotent pharmacological motif demonstrating a wide range of biological activities including anticancer, antimalarial, antifungal, anti-inflammatory, and antiviral effects.4,5 In this view, the exploration6,7 of new structurally diverse quinolone derivatives remains highly valuable for the development of novel drugs across various therapeutic areas. Apart from structure–activity relationship (SAR)-driven8 selective functionalization9 of the quinolone skeleton at specific positions, another approach to achieving diversity lies in combining the quinolone moiety with other pharmacophores, including ring-fused ones. In recent years, these hybrid molecules10,11 have attracted particular interest due to their potential ability to act on different drug targets (dual mode of action) and to overcome some of the drawbacks (toxicity, drug resistance, side effects, etc.) associated with each of the substructural domains.
Herein, we would like to present an effective, one-pot synthesis of previously unknown tetracyclic quinolones 1 and 2 (Figure 1) in which the potent indoxyl entity (shaded blue) found in many natural alkaloids12 is embedded within the quinolinone framework (highlighted in black).
Figure 1.

Indolo[1,2-a]quinolinones 1 and 2 prepared by a single-step, cascade annulation reaction as described here.
It should be noted that there are numerous examples (Figure 2) of ring-fused 4-quinolones,7,13 both naturally occurring14−16 and synthetic.17−19 For instance, the tricyclic alkaloid 3, a member of the Penicillium-derived quinolactacin family,15 has been shown to inhibit acetylcholinesterase (AChE), whereas floxacrine 4 displays antimalarial activity.20 Third-generation fluoroquinolone 5 is one of the most commonly prescribed antibiotics2 and the compound 6(21) exhibits potential antitumor effects.
Figure 2.
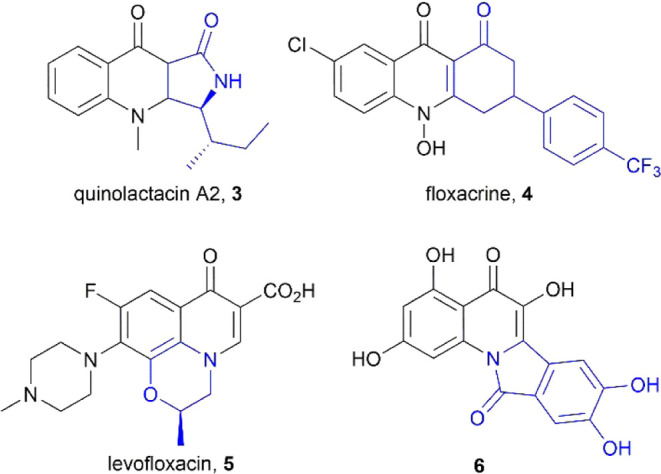
Selected examples of ring-fused biologically active 4-quinolones.
In contrast, only a small number21−25 of the polycyclic quinolone derivatives annulated at face a (as in isoindolo[2,1-a]quinolinone 6) have been described so far, making such templates generally underrepresented in drug discovery. Therefore, unlike the much more common indolo[1,2-a]quinolines,26,27 to our knowledge, no examples of indolo[1,2-a]quinolinone-2-like structures have been reported in the literature, and to date, only one representative of quinolone 1 is known.28
2. Results and Discussion
Some years ago, we developed several convenient protocols29,30 for the gram-scale synthesis of 2-(3-oxoindolin-2-yl)-2-arylacetonitriles 7 (Scheme 1) starting from 2-substituted indoles 8 and nitroalkenes 9. One of these methods29 involves a direct, single-step, acid-catalyzed preparation of acetonitriles 7 (Scheme 1b) while the other two proceed through chromatographically isolated intermediates—spirocyclic indolines 10(31,32) or nitroethylindoles 11(30) (Scheme 1a,c, respectively).
Scheme 1. Synthetic Pathways for Preparation of Acetonitriles 7 from Indoles 8 and Nitroalkenes 9.

It is worth noting that target compounds 7 belong to a subclass of 2,2-disubstituted indolin-3-ones (also referred to as pseudoindoxyls) which is both interesting from the synthetic33,34 (as a versatile building block to related heterocycles) and the medicinal chemistry viewpoint as they are structural parts of many biologically active natural products.12 For us, subsequent studies of such C2-quaternary indolin-3-ones 7 indeed turned out to be a fruitful area of research, leading to the discovery of several unexpected rearrangements35−37 (Scheme 2).
Scheme 2. Cascade Rearrangements Involving Acetonitriles 7.

We speculated back then that the KOH-assisted transformation35 of N-unsubstituted 2-(3-oxoindolin-2-yl)acetonitriles 7 to 3-hydroxyindolin-2-ones 13 (Scheme 2b) likely occurs through intermediates 17 (Scheme 3), which are essentially activated C-acylimines known38 for their diverse reactivity and ambiphilic properties.
Scheme 3. Plausible Mechanism of the Acetonitriles 7 to Indolin-2-Ones 13 Conversion.

Hence, for example, chemoselective nucleophilic addition at the imine carbon of such preformed indolin-3-ones 17(39,40) is one of the two most common synthetic approaches to 2,2-disubstituted indoxyls 20 (Scheme 4a) followed by direct oxidative dearomatization on 2-substituted indoles 8 with various nucleophiles41,42 (Scheme 4b).
Scheme 4. General Approaches to 2,2-Disubstituted Indoxyls 20.

Based on the preceding considerations, it was hypothesized that under specific base/reagent conditions, iminium intermediates 17 generated in situ from acetonitriles 7 (Scheme 3) could be trapped with a suitable nucleophilic reagent leading to the target C2-quaternary indolin-3-ones 20 (Scheme 4). We selected simple ketones 21 as model coupling substrates because, upon a reaction with acetonitriles 22, they are expected to form the corresponding indolin-3-ones 23 featuring a β-carbonyl functional group at the C2 position (Scheme 5l). This kind of functionalized pseudoindoxyls has been shown28,43 to be useful in the synthesis of other N-heterocyclic derivatives. However, only a few number of efficient, practical approaches28,43−52 to such derivatives is known (Scheme 5a–k).
Scheme 5. Synthetic Pathways to 2,2-Disubstituted Indoxyls 23: Previous and Present Work.

The results of the screening experiments are presented in Table 1. They were very encouraging, confirming the validity of our initial concept while the identified reaction conditions—1.5 equiv of cesium carbonate in 1,4-dioxane at 100 °C (entry 8)—consistently delivered an excellent yield (up to 95%) of the target indolinone 23aa.
Table 1. Results of Screening Experiments to Afford 2,2-Disubstituted Indolin-3-one 23aaa.

| entry | solvent | base (equiv) | T (°C) | yield (%)b |
|---|---|---|---|---|
| 1 | xylene | MeONa (1) | reflux | 30 |
| 2 | DMF | NaH (1) | 100 | 49 |
| 3 | DMSO | Cs2CO3 (1) | 100 | 57 |
| 4 | DMSO | DBU (1) | 100 | 15 |
| 5 | DMSO | KOH (1) | 100 | 0 |
| 6 | THF | Cs2CO3 (1) | reflux | 71 |
| 7 | dioxane | Cs2CO3 (1) | reflux | 89 |
| 8 | dioxane | Cs2CO3 (1.5) | reflux | 95 |
| 9 | dioxane | Cs2CO3 (3) | reflux | 95 |
| 10 | dioxane | K2CO3 (1.5) | reflux | 82 |
Reaction conditions: 22a (1.0 mmol), 21a (1.0 mmol), base (× equiv) in solvent (2.0 mL) at the given temperature for 1 h.
Isolated yield.
Having these at hand, synthesizing a set of 16 indolinone 23 samples (Scheme 6) to assess the substrate scope of the method (five indolinones 22 vs 11 ketones 21) was straightforward.
Scheme 6. Substrate Scope in Cs2CO3-Assisted Reaction of Acetonitriles 22 with Various Ketones 21.

The presented data highlights excellent yields (75–96%) across a wide range of starting reagents. That, coupled with the fact that nitriles 22 are readily available in one step29 on a gram scale from the corresponding 2-substituted indoles 8 and nitroalkenes 9 (Scheme 1), makes the given procedure a valuable addition to already existing methods for the preparation of such C2-quaternary indoxyls 23 (Scheme 5a–k). As expected, the latter in our case were formed as racemates except for the compound 23al which gave a diastereomeric mixture of about 1:1. The structure and purity of all new compounds were unambiguously established using NMR and high-resolution mass spectrometry, and the identity of the known indolinones 23 was confirmed by comparison of their physical and spectral data with those previously described.
The plausible mechanism for this transformation appears relatively straightforward (Scheme 7). It likely proceeds, as in case35 of hydroxyl ion in the role of a nucleophile (Scheme 3), through the ejection of a stabilized benzyl cyanide ion 25 as a good leaving group.
Scheme 7. Proposed Mechanism to Product 23aa.

As previously noted, C2-disubstituted indolin-3-ones 23 containing β-carbonyl functions can undergo further useful synthetic transformations28,43 (Scheme 8).
Scheme 8. Known Synthetic Applications Involving Indolinones 23.

We were particularly intrigued by the sequence (h–i) in Scheme 5 because it offers access not only to virtually unknown indoxyl-fused 4-quinolinones 31 but also, upon further carbonyl group reduction, to tetracyclic tetrahydroindolo[1,2-a]quinolines 32. The structural core of these compounds is found in a large family53 of plant-derived monoterpene indole alkaloids (Figure 3).
Figure 3.

Some polycyclic indole-based alkaloids isolated from Kopsia plants.
The literature review revealed that dihydroindolo[1,2-a]quinoline-5,7-dione 31 is apparently the only reported representative of its class, so the development of practical approaches to the latter is imperative for the search for novel therapeutic candidates among such N-condensed 4-quinolones. It should be noted that around the same time, we were also working on a separate project54 focused on the intramolecular cyclization of 2′-nitrochalcone derivatives via ipso-substitution of the −NO2 group. This experience led us to hypothesize that the ortho-nitro group might be more effective compared to the bromine one in the desired annulation reaction of o-substituted indolinone 30 (Scheme 8h). Furthermore, o-nitroacetophenones 21 needed to afford the corresponding indolinones 23 are generally more readily available than their Br-substituted counterparts. And, since we already knew that o-nitroindoxyl 23af (Scheme 6) could be obtained with near-quantitative yield, it was decided from the very beginning to pursue the development of a one-pot, tandem transformation leading to the target tetracyclic quinolones 1 (Scheme 9).
Scheme 9. Proposed Single-Step, Cascade Pathway for Preparation of Fused 4-quinolones 1 from Acetonitriles 22.

As previously discussed, the optimal conditions for SNAr intramolecular cyclization involving ipso-substitution of the −NO2 group which proved successful in our prior project54 was a combination of DBU (2 equiv) at 80 °C in dimethyl sulfoxide (DMSO) as the solvent. So, we first tried to carry out the intended annulation reaction in the presence of DBU and Cs2CO3 but in a dioxane medium for convenience. This did not work, yielding only the expected indolinone 23af and not the target quinolone 1af. However, replacing dioxane with DMSO and running the reaction sequentially, first with 1.5 equiv of Cs2CO3 at 100 °C for 1 h and then adding 2 equiv of DBU and heating at 100 °C for another hour, the desired product 1af was obtained with an excellent yield of 95%. At this point, we decided to focus on creating a small library of these N-bridged heterocycles 1 (Scheme 10), as we did not see much potential in searching for alternative options. In a process, we accidentally made another interesting discovery when a laboratory hot plate malfunctioned, causing the oil bath temperature to exceed 150 °C. This resulted in the formation of a previously unknown compound 2af via, supposedly, 1,2-aryl shift. We took advantage of this unexpected finding and prepared another set of such indolo[1,2-a]quinolinones 2 (Scheme 10). Interestingly, the 1,2-shift in the tert-butyl derivative 2lf occurred at a temperature as low as 100 °C which can be attributed to the high mobility of the t-Bu group.
Scheme 10. Preparation of (Dihydro)indolo[1,2-a]quinolinones 1 and 2.

As can be seen, the yields of both tetracyclic heterocycles 1 and 2 are quite good (51–95%), which makes the described approach to these compounds practical. A complete set of NMR and high-resolution mass spectrometry (HRMS) data confirms their identity and, additionally, the lattice parameters and crystal structure of the compounds 1gf and 2df were obtained by X-ray diffraction analysis (Figures 4 and 5, respectively).
Figure 4.

ORTEP plot of 1gf (CCDC #2352893) showing atom numbering schemes and 50% probability ellipsoids.
Figure 5.

ORTEP plot of 2df (CCDC #2352894) showing atom numbering schemes and 50% probability ellipsoids.
The mechanism of this reaction is believed to be a SNAr intramolecular cyclization, as outlined below (Scheme 11).
Scheme 11. Mechanistic Rationale for the Formation of Products 1af and 2af.

The reaction begins with NH-deprotonation to produce anion 33, followed by nucleophilic attack at the ipso position of the −NO2 group. Our limited study indicates that the solvent-base combination may influence the stability of the sigma complex 34 since the DMSO–DBU pair provided the best results. Elimination of NO2 anion then triggers rearomatization leading to the formation of product 1af. An increase in temperature accompanied by a 4-fold excess of bases, leads to initially an anion 35 which is then rearranged through a 1,2-aryl shift into the more stable intermediate 36 and then into the product 2af. The above-mentioned 1,2-shift in indolin-3-one systems has already been observed35 by us (Scheme 3) where it resulted in the formation of a more stable cyclic amide 19. The main driving force behind this rearrangement is the aromatization of the quinolone moiety.
3. Conclusions
This paper presents a new, efficient Cs2CO3-assisted reaction for preparing 2-(2-oxo-(aryl/alkyl)ethyl)-2-(aryl/alkyl)indolin-3-ones. Although many approaches to the latter have been proposed, the given procedure starts with readily available on a gram scale in a single step 2-(2-aryl-3-oxoindolin-2-yl)-2-phenylacetonitriles and can therefore be considered a practical and valuable alternative to the existing pathways. With this discovery, we developed a robust, one-pot synthetic protocol that led to previously largely unknown classes of tetracyclic N-fused indoloquinolines that combine two of the most potent pharmacophores within a single molecule.
4. Experimental Part
4.1. General Information
NMR spectra, 1H, 13C, and 19F were measured in solutions of CDCl3 or DMSO-d6 on Bruker AVANCE-III HD instrument (at 400, 101, and 376 MHz, respectively). Residual solvent signals were used as internal standards, in DMSO-d6 (2.50 ppm for 1H, and 40.45 ppm for 13C nuclei) or CDCl3 (7.26 ppm for 1H, and 77.16 ppm for 13C nuclei). HRMS spectra were measured on Bruker maXis impact (electrospray ionization, in MeCN solutions, employing HCO2Na–HCO2H for calibration). IR spectra were measured on FT-IR spectrometer Shimadzu IRAffinity-1S equipped with an ATR sampling module. Reaction progress, purity of isolated compounds, and Rf values were monitored with thin-layer chromatography (TLC) on Silufol UV-254 plates. Column chromatography was performed on silica gel (32–63 μm, 60 Å pore size). Melting points were measured with the Stuart SMP30 apparatus. All acetonitriles 22 except novel 22l,e,i,n were synthesized according to the previously reported procedure29 and were identical to those described. All reagents and solvents were purchased from commercial vendors.
4.2. Preparation of Novel Acetonitriles 22l,e,i,n (General Procedure)29
Wheaton microreactor equipped with magnetic spin-vane and Mininert valve was charged with a mixture of (2-nitrovinyl)benzene (2.0 mmol), corresponding 2-substituted indole (2.0 mmol), phosphorus acid (2.0 g), and formic acid (2.0 g). The mixture was vigorously stirred for 2 h at room temperature, while it turned dark red and homogenized. Then, the mixture was poured into water (50 mL) and the formed crude spirane precipitate was collected and washed with water (4 × 20 mL), dried, and dissolved in ethanol (4 mL). Triethylamine (204 mg, 2.0 mmol) was added, and the resulting solution was stirred at room temperature for 3 h. The crystalline precipitate of crude product was formed, which was collected and purified by preparative column chromatography on silica gel, eluting with ethyl acetate/hexane mixture (v/v).
4.2.1. 2-(2-(tert-Butyl)-3-oxoindolin-2-yl)-2-phenylacetonitrile (22l)
This compound was prepared by general procedure employing 2-(tert-butyl)-1H-indole (346 mg, 2.0 mmol) (346 mg, 2.0 mmol) and (2-nitro vinyl)benzene (298 mg, 2.0 mmol). Purification by column chromatography (EtOAc/hexane, 1:8, v/v). The titled compound was obtained as a yellowish solid, mp 206–207 °C, Rf 0.22 (EtOAc/hexane, 1:6, v/v). Yield 444 mg (1.46 mmol, 73%). 1H NMR (400 MHz, DMSO-d6) δ 7.62 (s, 1H), 7.53–7.46 (m, 2H), 7.41–7.35 (m, 2H), 7.33–7.27 (m, 3H), 6.86 (d, J = 8.1 Hz, 1H), 6.64 (t, J = 7.3 Hz, 1H), 4.66 (s, 1H), 0.85 (s, 9H). 13C {1H} NMR (101 MHz, DMSO-d6) δ 201.5, 161.8, 137.4, 133.5, 129.6 (2C), 128.4, 128.3 (2C), 123.3, 120.4, 119.6, 117.5, 111.5, 73.5, 40.1, 38.0, 25.3 (3C); FTIR, vmax: 3256, 2978, 2249, 1658, 1615, 1471, 1329, 1258, 1165, 1056 cm–1; HRMS (ESI TOF) m/z calcd. for C20H20N2NaO [M + Na]+: 327.1468, found: 327.1469 (−0.3 ppm).
4.2.2. 2-(5,6-Dimethoxy-3-oxo-2-phenylindolin-2-yl)-2-phenylacetonitrile (22e)
This compound was prepared by general procedure employing 5,6-dimethoxy-2-phenyl-1H-indole (506 mg, 2.0 mmol) and (E)-(2-nitrovinyl)benzene (298 mg, 2.0 mmol). Purification by column chromatography (EtOAc/hexane, 1:2, v/v). The titled compound was obtained as a brown solid, mp 271–274 °C, Rf 0.11 (EtOAc/hexane, 1:2, v/v). Yield 614 mg (1.6 mmol, 80%). 1H NMR (400 MHz, DMSO-d6) δ 7.88 (s, 1H), 7.54 (d, J = 7.3 Hz, 2H), 7.32–7.19 (m, 8H), 6.87 (s, 1H), 6.68 (s, 1H), 5.21 (s, 1H), 3.90 (s, 3H), 3.69 (s, 3H). 13C {1H} NMR (101 MHz, DMSO-d6) δ 196.1, 159.6, 158.8, 143.8, 135.4, 132.0, 129.4 (2C), 128.3 (2C), 128.2 (3C), 128.0, 126.1 (2C), 119.1, 108.6, 104.3, 94.6, 73.5, 55.9, 55.7, 43.5. FTIR, vmax: 3226, 2311, 2286, 1703, 1665, 1558, 1487, 1464, 1339, 1279, 1159 cm–1; HRMS (ESI TOF) m/z calcd. for C24H20N2NaO3 [M + Na]+: 407.1336, found: 407.1338 (−0.5 ppm).
4.2.3. 2-(6-Methoxy-3-oxo-2-phenylindolin-2-yl)-2-phenylacetonitrile (22i)
This compound was prepared by general procedure employing 6-methoxy-2-phenyl-1H-indole (446 mg, 2.0 mmol) and (E)-(2-nitrovinyl)benzene (298 mg, 2.0 mmol). Purification by column chromatography (EtOAc/hexane, 1:2, v/v). The titled compound was obtained as a yellow solid, mp 256–258 °C, Rf 0.22 (EtOAc/hexane, 1:2, v/v). Yield 552 mg (1.56 mmol, 78%). 1H NMR (400 MHz, DMSO-d6) δ 8.17 (s, 1H), 7.55 (d, J = 7.1 Hz, 2H), 7.37 (d, J = 8.6 Hz, 1H), 7.28–7.23 (m, 8H), 6.58 (d, J = 1.9 Hz, 1H), 6.37 (dd, J = 8.6, 2.0 Hz, 1H), 5.23 (s, 1H), 3.86 (s, 3H). 13C {1H} NMR (101 MHz, DMSO-d6) δ 196.4, 168.2, 164.4, 135.7, 132.2, 129.8 (2C), 128.76 (2C), 128.74 (3C), 128.5, 126.6 (3C), 119.4, 111.5, 109.3, 94.8, 73.9, 56.1, 43.9. FTIR, vmax: 3226, 2331, 1711, 1649, 1575, 1481, 1450, 1339, 1253, 1147 cm–1; HRMS (ESI TOF) m/z calcd. for C23H18N2NaO2 [M + Na]+: 377.1260, found: 377.1261 (−0.3 ppm).
4.2.4. 2-(5-Fluoro-2-(naphthalen-2-yl)-3-oxoindolin-2-yl)-2-phenylacetonitrile (22n)
This compound was prepared by general procedure employing 5-fluoro-2-(naphthalen-2-yl)-1H-indole (522 mg, 2.0 mmol), (E)-(2-nitrovinyl)benzene (298 mg, 2.0 mmol). Purification by column chromatography (EtOAc/hexane, 1:3, v/v). The titled compound was obtained as a yellow solid, mp 238–239 °C, Rf 0.21 (EtOAc/hexane, 1:4, v/v). Yield 564 mg (1.44 mmol, 72%). 1H NMR (400 MHz, DMSO-d6) δ 8.28 (s, 1H), 7.97 (s, 1H), 7.93–7.82 (m, 3H), 7.77 (d, J = 8.4 Hz, 1H), 7.53–7.45 (m, 3H), 7.32–7.25 (m, 3H), 7.25–7.17 (m, 4H), 5.48 (s, 1H). 13C {1H} NMR (101 MHz, DMSO-d6) δ 198.5, 158.7, 155.6 (d, J = 236.9 Hz), 132.4, 132.3, 132.0, 131.5, 129.4 (2C), 128.4 (3C), 128.2, 127.9, 127.5, 126.7, 126.6, 126.5 (d, J = 27.5 Hz), 125.2, 124.1, 118.8, 117.7 (d, J = 7.6 Hz), 114.1 (d, J = 7.7 Hz), 109.3 (d, J = 22.5 Hz), 74.4, 43.3. 19F NMR (376 MHz, DMSO-d6) δ −125.27. FTIR, vmax: 3349, 2273, 1711, 1647, 1511, 1476, 1337, 1251, 1209 cm–1; HRMS (ESI TOF) m/z calcd. for C26H17FN2NaO [M + Na]+: 415.1217, found: 415.1214 (0.7 ppm).
4.3. Preparation of Indolinones 23 (Typical Procedure A)
Starting acetonitrile 22 (1.0 mmol), corresponding methylketone (21) (1.0 mmol), and 2 mL of 1,4-dioxane were charged in a 10 mL round-bottom flask. Then, Cs2CO3 (489 mg, 1.5 mmol) was added and the resulting mixture was stirred at 100 °C for 1 h. The reaction progress was monitored by TLC. After completion reaction mixture was poured into water and extracted with EtOAc (4 × 25 mL). The combined extracts were dried over Na2SO4, concentrated under reduced pressure, and the residue was purified by preparative column chromatography on silica gel, eluting with ethyl acetate/hexane mixture (v/v).
4.3.1. 2-(2-Oxo-2-phenylethyl)-2-phenylindolin-3-one (23aa)28,55
This compound was prepared by typical procedure A employing 2-(3-oxo-2-phenylindolin-2-yl)-2-phenylacetonitrile 22a (324 mg, 1.0 mmol), acetophenone 21a (120 mg, 1.0 mmol). Purification by column chromatography (EtOAc/hexane, 1:5, v/v). The titled compound was obtained as a pale-yellow solid, mp 186–187 °C, (lit.55 mp 147–149 °C), Rf 0.38 (EtOAc/hexane, 1:4, v/v). Yield 311 mg (0.95 mmol, 95%). 1H NMR (400 MHz, DMSO-d6) δ 8.00–7.91 (m, 3H), 7.63 (t, J = 7.3 Hz, 1H), 7.56 (d, J = 7.5 Hz, 2H), 7.53–7.44 (m, 3H), 7.41 (d, J = 7.6 Hz, 1H), 7.32 (t, J = 7.5 Hz, 2H), 7.28–7.21 (m, 1H), 7.03 (d, J = 8.2 Hz, 1H), 6.72 (t, J = 7.3 Hz, 1H), 4.22 (d, J = 18.1 Hz, 1H), 3.75 (d, J = 18.1 Hz, 1H). 13C {1H} NMR (101 MHz, DMSO-d6) δ 200.7, 196.3, 161.2, 139.3, 137.0, 136.4, 133.5, 128.8 (2C), 128.4 (2C), 128.1 (2C), 127.3, 125.5 (2C), 124.4, 118.3, 117.4, 111.9, 68.8, 45.7. FTIR, vmax: 3246, 1682, 1617, 1483, 1221, 1059, 756, 691 cm–1; HRMS (ESI TOF) m/z calcd. for C22H17NNaO2 [M + Na]+: 350.1151, found: 350.1154 (−0.9 ppm).
4.3.2. 2-(2-(4-Methoxyphenyl)-2-oxoethyl)-2-phenylindolin-3-one (23ab)28,55
This compound was prepared by typical procedure A employing 2-(3-oxo-2-phenylindolin-2-yl)-2-phenylacetonitrile 22a (324 mg, 1.0 mmol), 1-(4-methoxyphenyl)ethanone 21b (150 mg, 1.0 mmol). Purification by column chromatography (EtOAc/hexane, 1:5, v/v, EtOAc). The titled compound was obtained as a pale-yellow solid, mp 208–210 °C (lit.55 mp 205–206 °C), Rf 0.27 (EtOAc/hexane, 1:4, v/v). Yield 343 mg (0.96 mmol, 96%). 1H NMR (400 MHz, DMSO-d6) δ 7.94 (d, J = 8.6 Hz, 3H), 7.54 (d, J = 7.6 Hz, 2H), 7.46 (t, J = 7.6 Hz, 1H), 7.40 (d, J = 7.6 Hz, 1H), 7.31 (t, J = 7.5 Hz, 2H), 7.24 (t, J = 7.2 Hz, 1H), 7.02 (dd, J = 8.5, 3.2 Hz, 3H), 6.71 (t, J = 7.3 Hz, 1H), 4.16 (d, J = 17.9 Hz, 1H), 3.82 (s, 3H), 3.66 (d, J = 17.9 Hz, 1H). 13C {1H} NMR (101 MHz, DMSO-d6) δ 200.7, 194.6, 163.3, 161.1, 139.4, 137.0, 130.5 (2C), 129.4, 128.4 (2C), 127.2, 125.5 (2C), 124.3, 118.2, 117.3, 113.9 (2C), 111.9, 68.9, 55.6, 45.3. FTIR, vmax: 3327, 1684, 1557, 1487, 1249, 1171, 821, 754, 702 cm–1; HRMS (ESI TOF) m/z calcd. for C23H19NNaO3 [M + Na]+: 380.1257, found: 380.1252 (1.3 ppm).
4.3.3. 2-(2-(3-Methoxyphenyl)-2-oxoethyl)-2-phenylindolin-3-one (23ac)28
This compound was prepared by typical procedure A employing 2-(3-oxo-2-phenylindolin-2-yl)-2-phenylacetonitrile 22a (324 mg, 1.0 mmol), 1-(3-methoxyphenyl)ethanone 21c (150 mg, 1.0 mmol). Purification by column chromatography (EtOAc/hexane, 1:5, v/v). The titled compound was obtained as a pale-yellow solid, mp 138–140 °C (lit.28 mp 97–99 °C), Rf 0.32 (EtOAc/hexane, 1:4, v/v). Yield 321 mg (0.90 mmol, 90%). 1H NMR (400 MHz, DMSO-d6) δ 7.92 (s, 1H), 7.59–7.51 (m, 3H), 7.49–7.38 (m, 4H), 7.32 (t, J = 7.4 Hz, 2H), 7.25 (t, J = 7.2 Hz, 1H), 7.20 (dd, J = 8.1, 2.0 Hz, 1H), 7.01 (d, J = 8.3 Hz, 1H), 6.71 (t, J = 7.3 Hz, 1H), 4.18 (d, J = 18.2 Hz, 1H), 3.79 (s, 3H), 3.76 (d, J = 18.2 Hz, 1H). 13C {1H} NMR (101 MHz, DMSO-d6) δ 200.6, 196.1, 161.2, 159.4, 139.3, 137.7, 137.0, 129.9, 128.4 (2C), 127.2, 125.5 (2C), 124.3, 120.7, 119.7, 118.3, 117.4, 112.3, 111.9, 68.8, 55.3, 45.8. FTIR, vmax: 3373, 1701, 1682, 1613, 1494, 1322, 1008, 750 cm–1; HRMS (ESI TOF) m/z calcd. for C23H19NNaO3 [M + Na]+: 380.1257, found: 380.1259 (−0.5 ppm).
4.3.4. 2-(2-(2-Methoxyphenyl)-2-oxoethyl)-2-phenylindolin-3-one (23ad)
This compound was prepared by typical procedure A employing 2-(3-oxo-2-phenylindolin-2-yl)-2-phenylacetonitrile 22a (324 mg, 1.0 mmol), 1-(2-methoxyphenyl)ethanone 21d (150 mg, 1.0 mmol). Purification by column chromatography (EtOAc/hexane, 1:5, v/v). The titled compound was obtained as a pale-yellow solid, mp 131–133 °C, Rf 0.27 (EtOAc/hexane, 1:4, v/v). Yield 321 mg (0.93 mmol, 93%). 1H NMR (400 MHz, DMSO-d6) δ 7.95 (s, 1H), 7.53 (t, J = 7.9 Hz, 1H), 7.47 (dd, J = 13.1, 7.8 Hz, 4H), 7.37 (d, J = 7.6 Hz, 1H), 7.31 (t, J = 7.5 Hz, 2H), 7.23 (t, J = 7.2 Hz, 1H), 7.16 (d, J = 8.4 Hz, 1H), 7.02 (d, J = 8.3 Hz, 1H), 6.98 (t, J = 7.5 Hz, 1H), 6.70 (t, J = 7.3 Hz, 1H), 4.09 (d, J = 18.1 Hz, 1H), 3.88 (s, 3H), 3.57 (d, J = 18.1 Hz, 1H).13C {1H} NMR (101 MHz, DMSO-d6) δ 200.4, 197.4, 161.1, 158.4, 139.3 (2C), 137.0, 134.1, 129.5, 128.4 (2C), 127.1, 125.3 (2C), 124.3, 120.5, 118.0, 117.3, 112.5, 111.8, 69.0, 55.8, 50.6. FTIR, vmax: 3231, 1682, 1556, 1489, 1322, 1242, 1177, 1012, 763 cm–1; HRMS (ESI TOF) m/z calcd. for C23H19NNaO3 [M + Na]+: 380.1257, found: 380.1259 (−0.5 ppm).
4.3.5. 2-(2-Oxo-2-(p-tolyl)ethyl)-2-phenylindolin-3-one (23ae)28,55
This compound was prepared by typical procedure A employing 2-(3-oxo-2-phenylindolin-2-yl)-2-phenylacetonitrile 22a (324 mg, 1.0 mmol), 1-(p-tolyl)ethanone 21e (134 mg, 1.0 mmol). Purification by column chromatography (EtOAc/hexane, 1:5, v/v). The titled compound was obtained as a pale-yellow solid, mp 190–191 °C (lit.55 mp 177–179 °C), Rf 0.43 (EtOAc/hexane, 1:4, v/v). Yield 310 mg (0.91 mmol, 91%). 1H NMR (400 MHz, DMSO-d6) δ 7.94 (s, 1H), 7.85 (d, J = 8.1 Hz, 2H), 7.53 (d, J = 7.6 Hz, 2H), 7.46 (t, J = 7.6 Hz, 1H), 7.39 (d, J = 7.7 Hz, 1H), 7.31 (t, J = 7.6 Hz, 4H), 7.24 (t, J = 7.2 Hz, 1H), 7.02 (d, J = 8.2 Hz, 1H), 6.71 (t, J = 7.4 Hz, 1H), 4.17 (d, J = 18.1 Hz, 1H), 3.69 (d, J = 18.0 Hz, 1H), 2.36 (s, 3H). 13C {1H} NMR (101 MHz, DMSO-d6) δ 200.7, 195.8, 161.2, 143.9, 139.3, 137.0, 133.9, 129.3 (2C), 128.4 (2C), 128.2 (2C), 127.2, 125.5 (2C), 124.4, 118.2, 117.4, 111.9, 68.8, 45.6, 21.2. FTIR, vmax: 3399, 1686, 1621, 1561, 1490, 1326, 1180, 1048, 895, 756 cm–1; HRMS (ESI TOF) m/z calcd. for C23H19NNaO2 [M + Na]+: 364.1308, found: 364.1311 (−0.8 ppm).
4.3.6. 2-(2-(2-Nitrophenyl)-2-oxoethyl)-2-phenylindolin-3-one (23af)
This compound was prepared by typical procedure A employing 2-(3-oxo-2-phenylindolin-2-yl)-2-phenylacetonitrile 22a (324 mg, 1.0 mmol), 1-(2-nitrophenyl)ethanone 21f (165 mg, 1.0 mmol). Purification by column chromatography (EtOAc/hexane, 1:5, v/v). The titled compound was obtained as an orange solid, mp 160–161 °C, Rf 0.19 (EtOAc/hexane, 1:4, v/v). Yield 357 mg (0.96 mmol, 96%). 1H NMR (400 MHz, DMSO-d6) δ 8.04 (d, J = 7.6 Hz, 2H), 7.84–7.77 (m, 1H), 7.77–7.69 (m, 2H), 7.56–7.45 (m, 3H), 7.39–7.29 (m, 3H), 7.26 (t, J = 7.2 Hz, 1H), 7.06 (d, J = 8.1 Hz, 1H), 6.72 (t, J = 7.3 Hz, 1H), 3.96 (d, J = 17.8 Hz, 1H), 3.75 (d, J = 17.8 Hz, 1H).13C {1H} NMR (101 MHz, DMSO-d6) δ 199.9, 197.9, 161.3, 146.3, 138.6, 137.3, 134.7, 133.9, 132.0, 128.7, 128.5 (2C), 127.4, 125.4 (2C), 124.5, 124.2, 117.8, 117.6, 112.0, 68.7, 48.4. FTIR, vmax: 3396, 1686, 1523, 1490, 1345, 1221, 1045, 903, 742 cm–1; HRMS (ESI TOF) m/z calcd. for C22H16N2NaO4 [M + Na]+: 395.1002, found: 395.1001 (0.3 ppm).
4.3.7. 2-(2-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-2-oxoethyl)-2-phenylindolin-3-one (23ag)
This compound was prepared by typical procedure A employing 2-(3-oxo-2-phenylindolin-2-yl)-2-phenylacetonitrile 22a (324 mg, 1.0 mmol), 1-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)ethanone 21g (178 mg, 1.0 mmol). Purification by column chromatography (EtOAc/hexane, 1:5, v/v). The titled compound was obtained as a pale-yellow solid, mp 160–162 °C, Rf 0.16 (EtOAc/hexane, 1:4, v/v). Yield 366 mg (0.95 mmol, 95%). 1H NMR (400 MHz, DMSO-d6) δ 7.92 (s, 1H), 7.53 (d, J = 7.7 Hz, 2H), 7.51–7.41 (m, 3H), 7.39 (d, J = 7.6 Hz, 1H), 7.31 (t, J = 7.5 Hz, 2H), 7.24 (t, J = 7.2 Hz, 1H), 7.02 (d, J = 8.2 Hz, 1H), 6.94 (d, J = 8.5 Hz, 1H), 6.71 (t, J = 7.4 Hz, 1H), 4.35–4.23 (m, 4H), 4.12 (d, J = 18.0 Hz, 1H), 3.64 (d, J = 18.0 Hz, 1H). 13C {1H} NMR (101 MHz, DMSO-d6) δ 200.6, 194.6, 161.2, 148.1, 143.2, 139.3, 137.0, 130.1, 128.4 (2C), 127.2, 125.5 (2C), 124.4, 122.2, 118.2, 117.4, 117.12, 117.07, 111.9, 68.9, 64.6, 63.9, 45.3. FTIR, vmax: 3269, 1684, 1619, 1494, 1329, 1295, 1069, 750 cm–1; HRMS (ESI TOF) m/z calcd. for C24H19NNaO4 [M + Na]+: 408.1206, found: 408.1201 (1.2 ppm).
4.3.8. 2-(2-Oxo-2-(pyridin-2-yl)ethyl)-2-phenylindolin-3-one (23ah)
This compound was prepared by typical procedure A employing 2-(3-oxo-2-phenylindolin-2-yl)-2-phenylacetonitrile 22a (324 mg, 1.0 mmol), 1-(pyridin-2-yl)ethanone 21h (121 mg, 1.0 mmol). Purification by column chromatography (EtOAc/hexane, 1:5, v/v). The titled compound was obtained as a pale-yellow solid, mp 150–151 °C, Rf 0.16 (EtOAc/hexane, 1:4, v/v). Yield 272 mg (0.83 mmol, 83%). 1H NMR (400 MHz, DMSO-d6) δ 8.74 (d, J = 4.1 Hz, 1H), 7.97 (d, J = 7.5 Hz, 2H), 7.86 (d, J = 7.8 Hz, 1H), 7.71–7.64 (m, 1H), 7.50 (dd, J = 13.5, 7.9 Hz, 3H), 7.41 (d, J = 7.7 Hz, 1H), 7.32 (t, J = 7.5 Hz, 2H), 7.25 (t, J = 7.2 Hz, 1H), 7.00 (d, J = 8.2 Hz, 1H), 6.72 (t, J = 7.3 Hz, 1H), 4.60 (d, J = 18.1 Hz, 1H), 3.64 (d, J = 18.1 Hz, 1H). 13C {1H} NMR (101 MHz, DMSO-d6) δ 200.7, 197.6, 161.2, 152.5, 149.3, 139.2, 137.7, 137.1, 128.5 (2C), 128.1, 127.3, 125.4 (2C), 124.4, 121.3, 118.2, 117.5, 111.9, 68.7, 44.6. FTIR, vmax: 3373, 1697, 1682, 1617, 1557, 1490, 1328, 1001, 765 cm–1; HRMS (ESI TOF) m/z calcd. for C21H16N2NaO2 [M + Na]+: 351.1104, found: 351.1106 (−0.6 ppm).
4.3.9. 2-(2-Oxo-2-(thiophen-2-yl)ethyl)-2-phenylindolin-3-one (23aj)28
This compound was prepared by typical procedure A employing 2-(3-oxo-2-phenylindolin-2-yl)-2-phenylacetonitrile 22a (324 mg, 1.0 mmol), 1-(thiophen-3-yl)ethanone 21j (121 mg, 1.0 mmol). Purification by column chromatography (EtOAc/hexane, 1:5, v/v). The titled compound was obtained as a pale-yellow solid, mp 206–207 °C (lit.28 mp 115–117 °C), Rf 0.35 (EtOAc/hexane, 1:4, v/v). Yield 317 mg (0.95 mmol, 95%). 1H NMR (400 MHz, DMSO-d6) δ 8.05 (d, J = 3.7 Hz, 1H), 7.99 (d, J = 4.9 Hz, 1H), 7.96 (s, 1H), 7.54 (d, J = 7.7 Hz, 2H), 7.46 (t, J = 7.6 Hz, 1H), 7.39 (d, J = 7.6 Hz, 1H), 7.32 (t, J = 7.5 Hz, 2H), 7.28–7.21 (m, 2H), 7.01 (d, J = 8.2 Hz, 1H), 6.71 (t, J = 7.4 Hz, 1H), 4.13 (d, J = 17.5 Hz, 1H), 3.69 (d, J = 17.5 Hz, 1H). 13C {1H} NMR (101 MHz, DMSO-d6) δ 200.3, 189.3, 161.2, 143.5, 139.1, 137.1, 135.3, 134.1, 128.9, 128.5 (2C), 127.3, 125.5 (2C), 124.4, 118.2, 117.5, 111.9, 68.8, 45.9. FTIR, vmax: 3317, 1680, 1490, 1251, 1048, 953, 863, 752 cm–1; HRMS (ESI TOF) m/z calcd. for C20H15NNaO2S [M + Na]+: 356.0716, found: 356.0717 (−0.3 ppm).
4.3.10. 2-(2-(2-Aminophenyl)-2-oxoethyl)-2-phenylindolin-3-one (23ai)
This compound was prepared by typical procedure A employing 2-(3-oxo-2-phenylindolin-2-yl)-2-phenylacetonitrile 22a (324 mg, 1.0 mmol), 1-(2-aminophenyl)ethanone 21i (135 mg, 1.0 mmol). Purification by column chromatography (EtOAc/hexane, 1:5, v/v). The titled compound was obtained as a yellow solid, mp 155–157 °C, Rf 0.45 (EtOAc/hexane, 1:4, v/v). Yield 280 mg (0.82 mmol, 82%). 1H NMR (400 MHz, DMSO-d6) δ 7.89 (s, 1H), 7.81 (d, J = 7.9 Hz, 1H), 7.52 (d, J = 7.4 Hz, 2H), 7.45 (t, J = 8.1 Hz, 1H), 7.38 (d, J = 7.5 Hz, 1H), 7.31 (t, J = 7.5 Hz, 2H), 7.23 (t, J = 7.9 Hz, 2H), 7.04 (s, 2H), 7.02 (d, J = 8.3 Hz, 1H), 6.70 (t, J = 8.0 Hz, 2H), 6.52 (t, J = 7.3 Hz, 1H), 4.12 (d, J = 17.8 Hz, 1H), 3.62 (d, J = 17.7 Hz, 1H). 13C {1H} NMR (101 MHz, DMSO-d6) δ 200.8, 197.8, 161.1, 151.1, 139.5, 136.9, 134.3, 131.5, 128.4 (2C), 127.1, 125.5 (2C), 124.4, 118.3, 117.3, 116.9, 116.3, 114.5, 111.9, 69.0, 46.0. FTIR, vmax: 3327, 1671, 1568 1424, 1249, 1127, 1019, 924, 851 cm–1; HRMS (ESI TOF) m/z calcd. for C22H18N2NaO2 [M + Na]+: 365.1260, found: 365.1257 (0.8 ppm).
4.3.11. 2-(3,3-Dimethyl-2-oxobutyl)-2-phenylindolin-3-one (23ak)
This compound was prepared by typical procedure A employing 2-(3-oxo-2-phenylindolin-2-yl)-2-phenylacetonitrile 22a (324 mg, 1.0 mmol), 3,3-dimethylbutan-2-one 21k (100 mg, 1.0 mmol). Purification by column chromatography (EtOAc/hexane, 1:7, v/v). The titled compound was obtained as a pale-yellow solid, mp 136–137 °C, Rf 0.54 (EtOAc/hexane, 1:4, v/v). Yield 230 mg (0.75 mmol, 75%). 1H NMR (400 MHz, DMSO-d6) δ 7.85 (s, 1H), 7.48–7.42 (m, 3H), 7.35 (d, J = 7.7 Hz, 1H), 7.30 (t, J = 7.5 Hz, 2H), 7.23 (t, J = 7.2 Hz, 1H), 7.01 (d, J = 8.2 Hz, 1H), 6.68 (t, J = 7.4 Hz, 1H), 3.69 (d, J = 18.1 Hz, 1H), 3.17 (d, J = 18.1 Hz, 1H), 1.05 (s, 9H). 13C {1H} NMR (101 MHz, DMSO-d6) δ 211.5, 200.5, 161.1, 139.2, 136.9, 128.3 (2C), 127.2, 125.4 (2C), 124.3, 118.0, 117.3, 111.8, 68.7, 44.2, 43.5, 25.9 (3C). FTIR, vmax: 3396, 1701, 1684, 1557, 1492, 1328, 1251, 1068, 750 cm–1; HRMS (ESI TOF) m/z calcd. for C20H21NNaO2 [M + Na]+: 330.1465, found: 330.1466 (−0.3 ppm).
4.3.12. 2-(2-Oxocyclohexyl)-2-phenylindolin-3-one (23al)44
This compound was prepared by typical procedure A employing 2-(3-oxo-2-phenylindolin-2-yl)-2-phenylacetonitrile 22a (324 mg, 1.0 mmol), cyclohexanone 21l (98 mg, 1.0 mmol). Purification by column chromatography (EtOAc/hexane, 1:6, v/v). The titled compound was obtained as a pale-yellow solid, mp 204–205 °C, Rf 0.37 (EtOAc/hexane, 1:4, v/v). Yield 268 mg (0.88 mmol, 88%). 1H NMR (400 MHz, DMSO-d6) δ 7.80 (s, 1H), 7.53 (d, J = 7.2 Hz, 2H), 7.41 (t, J = 7.3 Hz, 1H), 7.31 (d, J = 7.0 Hz, 3H), 7.26 (d, J = 6.7 Hz, 1H), 6.95 (d, J = 8.0 Hz, 1H), 6.67 (t, J = 7.1 Hz, 1H), 3.60 (d, J = 7.7 Hz, 1H), 2.39 (dt, J = 13.1, 6.6 Hz, 1H), 2.14 (d, J = 14.1 Hz, 1H), 1.95 (s, 1H), 1.73 (s, 1H), 1.54 (dd, J = 35.2, 8.5 Hz, 4H). 13C {1H} NMR (101 MHz, DMSO-d6) δ 207.4, 200.4, 160.4, 138.3, 135.9, 128.5 (2C), 127.4, 125.7 (2C), 123.9, 119.9, 117.1, 111.5, 71.7, 57.8, 41.4, 27.9, 26.1, 24.1. FTIR, vmax: 3361, 1699, 1682, 1617, 1489, 1322, 954, 750 cm–1; HRMS (ESI TOF) m/z calcd. for C20H19NNaO2 [M + Na]+: 328.1308, found: 328.1311 (−0.9 ppm).
4.3.13. 2-(Naphthalen-2-yl)-2-(2-oxo-2-phenylethyl)indolin-3-one (23da)
This compound was prepared by typical procedure A employing 2-(3-oxo-2-(2-(naphthalen-2-yl)-3-oxoindolin-2-yl)-2-phenylacetonitrile 22d (374 mg, 1.0 mmol), acetophenone 21a (120 mg, 1.0 mmol). Purification by column chromatography (EtOAc/hexane, 1:6, v/v). The titled compound was obtained as a pale-yellow solid, mp 214–216 °C, Rf 0.34 (EtOAc/hexane, 1:4, v/v). Yield 343 mg (0.91 mmol, 91%). 1H NMR (400 MHz, DMSO-d6) δ 8.09 (s, 2H), 7.98 (d, J = 7.4 Hz, 2H), 7.87 (d, J = 8.6 Hz, 3H), 7.73–7.66 (m, 1H), 7.63 (t, J = 7.3 Hz, 1H), 7.56–7.44 (m, 5H), 7.42 (d, J = 7.7 Hz, 1H), 7.08 (d, J = 8.2 Hz, 1H), 6.74 (t, J = 7.3 Hz, 1H), 4.34 (d, J = 18.2 Hz, 1H), 3.87 (d, J = 18.2 Hz, 1H). 13C {1H} NMR (101 MHz, DMSO-d6) δ 200.6, 196.4, 161.3, 137.1, 136.9, 136.4, 133.5, 132.8, 132.2, 128.8 (2C), 128.1 (2C), 128.0, 127.9, 127.4, 126.3, 126.0, 124.4, 124.1, 123.9, 118.3, 117.5, 112.1, 68.9, 45.7. FTIR, vmax: 3386, 1701, 1680, 1619, 1489, 1324, 1102, 1056, 756 cm–1; HRMS (ESI TOF) m/z calcd. for C26H19NNaO2 [M + Na]+: 400.1308, found: 400.1306 (0.5 ppm).
4.3.14. 2-(2-Oxo-2-phenylethyl)-2-(5,6,7,8-tetrahydronaphthalen-2-yl)indolin-3-one (23ba)
This compound was prepared by typical procedure A employing 2-(3-oxo-2-(5,6,7,8-tetrahydronaphthalen-2-yl)indolin-2-yl)-2-phenylacetonitrile 22b (378 mg, 1.0 mmol), acetophenone 21a (120 mg, 1.0 mmol). Purification by column chromatography (EtOAc/hexane, 1:6, v/v). The titled compound was obtained as a pale-yellow solid, mp 152–153 °C, Rf 0.49 (EtOAc/hexane, 1:4, v/v). Yield 312 mg (0.82 mmol, 82%). 1H NMR (400 MHz, DMSO-d6) δ 7.95 (d, J = 7.6 Hz, 2H), 7.86 (s, 1H), 7.62 (t, J = 7.1 Hz, 1H), 7.53–7.42 (m, 3H), 7.39 (d, J = 7.6 Hz, 1H), 7.26–7.20 (m, 2H), 7.03–6.94 (m, 2H), 6.70 (t, J = 7.3 Hz, 1H), 4.15 (d, J = 18.1 Hz, 1H), 3.69 (d, J = 18.0 Hz, 1H), 2.64 (s, 4H), 1.67 (s, 4H).13C {1H} NMR (101 MHz, DMSO-d6) δ 200.8, 196.3, 161.1, 136.9, 136.5, 136.4, 136.2, 135.3, 133.4, 129.0, 128.7 (2C), 128.1 (2C), 125.8, 124.3, 122.7, 118.4, 117.3, 111.9, 68.6, 45.8, 29.1, 28.4, 22.8, 22.7. FTIR, vmax: 3331, 3070, 1772, 1699, 1684, 1559, 1510, 1054, 828 cm–1; HRMS (ESI TOF) m/z calcd. for C26H23NNaO2 [M + Na]+: 404.1621, found: 404.1624 (−0.7 ppm).
4.3.15. 5-Fluoro-2-(2-oxo-2-phenylethyl)-2-phenylindolin-3-one (23ca)55
This compound was prepared by typical procedure A employing 2-(5-fluoro-3-oxo-2-phenylindolin-2-yl)-2-phenylacetonitrile 22c (342 mg, 1.0 mmol), acetophenone 21a (120 mg, 1.0 mmol). Purification by column chromatography (EtOAc/hexane, 1:6, v/v). The titled compound was obtained as a pale-yellow solid, mp 154–155 °C (lit.55 mp 151–153 °C), Rf 0.37 (EtOAc/hexane, 1:4, v/v). Yield 293 mg (0.85 mmol, 85%). 1H NMR (400 MHz, DMSO-d6) δ 7.95 (d, J = 7.4 Hz, 2H), 7.87 (s, 1H), 7.64 (t, J = 7.3 Hz, 1H), 7.52 (dt, J = 15.4, 7.6 Hz, 4H), 7.38 (td, J = 9.1, 2.7 Hz, 1H), 7.33 (t, J = 7.4 Hz, 2H), 7.26 (t, J = 7.1 Hz, 1H), 7.18 (dd, J = 7.6, 2.4 Hz, 1H), 7.04 (dd, J = 8.9, 3.9 Hz, 1H), 4.21 (d, J = 18.1 Hz, 1H), 3.81 (d, J = 18.1 Hz, 1H). 13C {1H} NMR (101 MHz, DMSO-d6) δ 200.5, 196.2, 158.1, 155.1 (d, J = 235.1 Hz), 138.9, 136.2, 133.4, 124.9 (d, J = 25.4 Hz), 128.7 (2C), 128.4 (2C), 128.1 (2C), 127.3, 125.5 (2C), 118.4 (d, J = 7.4 Hz), 113.2 (d, J = 7.6 Hz), 108.9 (d, J = 22.2 Hz), 69.8, 45.9. 19F NMR (376 MHz, DMSO-d6) δ −127.07. FTIR, vmax: 3399, 1684, 1563, 1489, 1247, 1196, 1048, 878, 823 cm–1; HRMS (ESI TOF) m/z calcd. for C22H16FNNaO2 [M + Na]+: 368.1057, found: 368.1058 (−0.3 ppm).
4.3.16. 5,6-Dimethoxy-2-(2-oxo-2-phenylethyl)-2-phenylindolin-3-one (23ea)
This compound was prepared by typical procedure A employing 2-(5,6-dimethoxy-3-oxo-2-phenylindolin-2-yl)-2-phenylacetonitrile 22e (384 mg, 1.0 mmol), acetophenone 21a (120 mg, 1.0 mmol). Purification by column chromatography (EtOAc/hexane, 1:6, v/v). The titled compound was obtained as a pale-yellow solid, mp 218–219 °C, Rf 0.12 (EtOAc/hexane, 1:3, v/v). Yield 348 mg (0.90 mmol, 90%). 1H NMR (400 MHz, DMSO-d6) δ 7.95 (d, J = 7.6 Hz, 2H), 7.69 (s, 1H), 7.62 (t, J = 7.3 Hz, 1H), 7.50 (t, J = 7.8 Hz, 4H), 7.29 (t, J = 7.4 Hz, 2H), 7.22 (t, J = 7.1 Hz, 1H), 6.82 (s, 1H), 6.62 (s, 1H), 4.21 (d, J = 18.0 Hz, 1H), 3.85 (s, 3H), 3.68 (s, 3H), 3.57 (d, J = 18.0 Hz, 1H). 13C {1H} NMR (101 MHz, DMSO-d6) δ 198.2, 196.6, 159.0, 158.2, 143.1, 139.9, 136.6, 133.4, 128.8 (2C), 128.3 (2C), 128.0 (2C), 127.0, 125.4 (2C), 108.7, 104.7, 94.4, 69.4, 55.8, 55.8, 45.1. FTIR, vmax: 3346, 1667, 1623, 1559, 1492, 1246, 1217, 1048, 851 cm–1; HRMS (ESI TOF) m/z calcd. for C24H21NNaO4 [M + Na]+: 410.1363, found: 410.1360 (0.7 ppm).
4.4. Preparation of Quinolones 1 (Typical Procedure B)
Starting acetonitrile 22 (1.0 mmol), corresponding acetophenone 21f (1.0 mmol), and 2 mL of DMSO were charged in a 10 mL round-bottom flask. Then, Cs2CO3 (489 mg, 1.5 mmol) was added and the resulting mixture was stirred at 100 °C for 1 h. After this, DBU (2.0 mmol, 304 mg) was added and the reaction mixture was stirred for another hour. The reaction progress was monitored by TLC. After completion reaction mixture was poured into water and extracted with EtOAc (4 × 25 mL). The combined extracts were dried over Na2SO4, concentrated under reduced pressure, and the residue was purified by preparative column chromatography on silica gel, eluting with ethyl acetate/hexane mixture.
4.4.1. 6a-Phenyl-6,6a-dihydroindolo[1,2-a]quinoline-5,7-dione (1af)28
This compound was prepared by typical procedure B employing 2-(3-oxo-2-phenylindolin-2-yl)-2-phenylacetonitrile 22a (324 mg, 1.0 mmol), 1-(2-nitrophenyl)ethanone 21f (165 mg, 1.0 mmol). Purification by column chromatography (EtOAc/hexane, 1:6, v/v). The titled compound was obtained as a yellow solid, mp 246–247 °C (lit.28 mp 204–206 °C), Rf 0.32 (EtOAc/hexane, 1:4, v/v). Yield 309 mg (0.95 mmol, 95%). 1H NMR (400 MHz, DMSO-d6) δ 8.00 (d, J = 8.3 Hz, 1H), 7.94 (d, J = 8.5 Hz, 1H), 7.85–7.78 (m, 1H), 7.78–7.70 (m, 3H), 7.35 (d, J = 7.0 Hz, 2H), 7.33–7.25 (m, 3H), 7.13 (q, J = 7.6 Hz, 2H), 3.55 (d, J = 16.5 Hz, 1H), 3.43 (d, J = 16.6 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 197.4, 191.1, 154.3, 141.7, 138.5, 136.1, 134.8, 129.1 (2C), 128.4, 127.3, 126.3 (2C), 125.8, 122.8, 122.7, 121.5, 120.3, 120.2, 111.0, 72.5, 43.2. FTIR, vmax: 3300, 2952, 1657, 1598, 1513, 1402, 1261, 1133, 1025 cm–1; HRMS (ESI TOF) m/z calcd. for C22H15NNaO2 [M + Na]+: 348.0995, found: 348.0997 (−0.6 ppm).
4.4.2. 6a-(Naphthalen-2-yl)-6,6a-dihydroindolo[1,2-a]quinoline-5,7-dione (1df)
This compound was prepared by typical procedure B employing 2-(2-(naphthalen-2-yl)-3-oxoindolin-2-yl)-2-phenylacetonitrile 22d (374 mg, 1.0 mmol), 1-(2-nitrophenyl)ethenone 21f (165 mg, 1.0 mmol). Purification by column chromatography (EtOAc/hexane, 1:6, v/v). The titled compound was obtained as a yellow solid, mp 258–261 °C, Rf 0.29 (EtOAc/hexane, 1:4, v/v). Yield 337 mg (0.90 mmol, 90%). 1H NMR (400 MHz, DMSO-d6) δ 8.04 (d, J = 8.1 Hz, 1H), 8.01–7.94 (m, 2H), 7.91–7.78 (m, 4H), 7.74 (t, J = 9.3 Hz, 3H), 7.52–7.44 (m, 2H), 7.42–7.35 (m, 1H), 7.16 (t, J = 7.4 Hz, 1H), 7.09 (t, J = 7.5 Hz, 1H), 3.73 (d, J = 16.6 Hz, 1H), 3.51 (d, J = 16.7 Hz, 1H). 13C {1H} NMR (101 MHz, DMSO-d6) δ 197.5, 191.2, 154.5, 141.6, 138.6, 136.0, 132.8, 132.5, 132.4, 129.0, 128.0, 127.4, 127.3, 126.8, 126.7, 126.2, 125.8, 123.3, 122.9, 122.8, 121.5, 120.5, 120.3, 111.1, 72.8, 43.0. FTIR, vmax: 3229, 1699, 1686, 1617, 1490, 1318, 1288, 1244, 811, 748 cm–1; HRMS (ESI TOF) m/z calcd. for C26H17NNaO2 [M + Na]+: 398.1151, found: 398.1147 (1.0 ppm).
4.4.3. 6a-(4-Methoxyphenyl)-6,6a-dihydroindolo[1,2-a]quinoline-5,7-dione (1ff)
This compound was prepared by typical procedure B employing 2-(2-(4-methoxyphenyl)-3-oxoindolin-2-yl)-2-phenylacetonitrile 22f (324 mg, 1.0 mmol), 1-(2-nitrophenyl)ethanone 21f (165 mg, 1.0 mmol). Purification by column chromatography (EtOAc/hexane, 1:6, v/v). The titled compound was obtained as a yellow solid, mp 231–234 °C, Rf 0.27 (EtOAc/hexane, 1:4, v/v). Yield 266 mg (0.75 mmol, 75%). 1H NMR (400 MHz, DMSO-d6) δ 7.98 (d, J = 8.1 Hz, 1H), 7.92 (d, J = 8.5 Hz, 1H), 7.80 (t, J = 7.7 Hz, 1H), 7.74 (t, J = 9.2 Hz, 3H), 7.23 (d, J = 8.7 Hz, 2H), 7.13 (q, J = 7.0 Hz, 2H), 6.84 (d, J = 8.7 Hz, 2H), 3.65 (s, 3H), 3.48 (d, J = 16.5 Hz, 1H), 3.39 (d, J = 16.5 Hz, 1H). 13C {1H} NMR (101 MHz, DMSO-d6) δ 197.8, 191.2, 159.3, 154.2, 141.6, 138.5, 136.0, 127.6 (2C), 127.3, 126.3, 125.8, 122.9, 122.6, 121.4, 120.3, 120.3, 114.5 (2C), 111.0, 72.2, 55.1, 43.1. FTIR, vmax: 3316, 2909, 1696, 1558, 1479, 1431, 1349, 1298, 971, 799 cm–1; HRMS (ESI TOF) m/z calcd. for C23H17NNaO3 [M + Na]+: 378.1101, found: 378.1104 (0.8 ppm).
4.4.4. 6a-(p-Tolyl)-6,6a-dihydroindolo[1,2-a]quinoline-5,7-dione (1gf)
This compound was prepared by typical procedure B employing 2-(3-Oxo-2-(p-tolyl)indolin-2-yl)-2-phenylacetonitrile 22g (338 mg, 1.0 mmol), 1-(2-nitrophenyl)ethanone 21f (165 mg, 1.0 mmol). Purification by column chromatography (EtOAc/hexane, 1:6, v/v). The titled compound was obtained as a yellow solid, mp 224–226 °C, Rf 0.38 (EtOAc/hexane, 1:4, v/v). Yield 308 mg (0.91 mmol, 91%). 1H NMR (400 MHz, DMSO-d6) δ 7.98 (d, J = 8.5 Hz, 1H), 7.93 (d, J = 8.5 Hz, 1H), 7.81 (t, J = 7.7 Hz, 1H), 7.77–7.68 (m, 3H), 7.21 (d, J = 8.2 Hz, 2H), 7.16–7.06 (m, 4H), 3.50 (d, J = 16.5 Hz, 1H), 3.39 (d, J = 16.6 Hz, 1H), 2.18 (s, 3H). 13C {1H} NMR (101 MHz, DMSO-d6) δ 197.6, 191.2, 154.2, 141.7, 138.5, 137.9, 136.0, 131.8, 129.7 (2C), 127.3, 126.2 (2C), 125.8, 122.9, 122.6, 121.4, 120.3, 120.3, 111.0, 72.4, 43.1, 20.5. FTIR, vmax: 3396, 1699, 1563, 1475, 1460, 1362, 1246, 928, 744 cm–1; HRMS (ESI TOF) m/z calcd. for C23H17NNaO2 [M + Na]+: 362.1151, found: 362.1149 (0.6 ppm).
4.4.5. 6a-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-6,6a-dihydroindolo[1,2-a]quinoline-5,7-dione (1hf)
This compound was prepared by typical procedure B employing 2-(2-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-3-oxoindolin-2-yl)-2-phenylacetonitrile 22h (382 mg, 1.0 mmol), 1-(2-nitrophenyl)ethenone 21f (165 mg, 1.0 mmol). Purification by column chromatography (EtOAc/hexane, 1:4, v/v). The titled compound was obtained as a yellow solid, mp 266–269 °C, Rf 0.19 (EtOAc/hexane, 1:4, v/v). Yield 314 mg (0.82 mmol, 82%). 1H NMR (400 MHz, DMSO-d6) δ 7.98 (d, J = 8.2 Hz, 1H), 7.91 (d, J = 8.5 Hz, 1H), 7.83–7.69 (m, 4H), 7.13 (t, J = 7.4 Hz, 2H), 6.77 (s, 3H), 4.15 (s, 4H), 3.45 (d, J = 16.6 Hz, 1H), 3.35 (d, J = 16.4 Hz, 1H).13C {1H} NMR (101 MHz, DMSO-d6) δ 197.5, 191.2, 154.1, 143.7, 141.7, 138.5, 136.1, 127.4, 127.4, 125.8, 122.9 (2C), 122.7, 121.4, 120.3, 120.3, 119.1, 117.7, 115.0, 111.0, 72.0, 64.0, 64.0, 43.0. FTIR, vmax: 3354, 17724, 1701, 1686, 1561, 1506, 1469, 1364, 1244, 1069, 759 cm–1; HRMS (ESI TOF) m/z calcd. for C24H17NNaO4 [M + Na]+: 406.1050, found: 406.1047 (0.7 ppm).
4.4.6. 10-Methoxy-6a-phenyl-6,6a-dihydroindolo[1,2-a]quinoline-5,7-dione (1if)
This compound was prepared by typical procedure B employing 2-(6-methoxy-3-oxo-2-phenylindolin-2-yl)-2-phenylacetonitrile 22i (354 mg, 1.0 mmol), 1-(2-nitrophenyl)ethenone 21f (165 mg, 1.0 mmol). Purification by column chromatography (EtOAc/hexane, 1:5, v/v). The titled compound was obtained as a yellow solid, mp 118–120 °C, Rf 0.21 (EtOAc/hexane, 1:4, v/v). Yield 316 mg (0.89 mmol, 89%). 1H NMR (400 MHz, DMSO-d6) δ 8.04 (d, J = 8.4 Hz, 1H), 7.75 (t, J = 7.3 Hz, 2H), 7.64 (d, J = 8.6 Hz, 1H), 7.33–7.24 (m, 6H), 7.13 (t, J = 7.5 Hz, 1H), 6.76–6.70 (m, 1H), 3.98 (s, 3H), 3.52 (d, J = 16.6 Hz, 1H), 3.40 (d, J = 16.6 Hz, 1H).13C {1H} NMR (101 MHz, DMSO-d6) δ 195.0, 191.3, 168.0, 156.3, 141.5, 136.2, 135.2, 129.0 (2C), 128.3, 127.4, 127.3, 126.2 (2C), 123.0, 122.9, 120.5, 113.4, 110.3, 94.4, 73.2, 56.1, 43.4. FTIR, vmax: 3365, 1774, 1701, 1686, 1561, 1508, 1358, 1249, 1115, 730 cm–1; HRMS (ESI TOF) m/z calcd. for C23H17NNaO3 [M + Na]+: 378.1101, found: 378.1102 (−0.3 ppm).
4.5. Preparation of Quinolones 2 (Typical Procedure C)
Starting acetonitrile 22 (1.0 mmol), corresponding acetophenone 21f (1.0 mmol), and 2 mL of DMSO were charged in a 10 mL round-bottom flask. Then, Cs2CO3 (489 mg, 1.5 mmol) was added and the resulting mixture was stirred at 100 °C for 1 h. After this, DBU (2.0 mmol, 304 mg) was added and the reaction mixture was stirred for another hour. The reaction temperature was increased to 150 °C and the reaction mixture was stirred for 30 min more. The reaction progress was monitored by TLC. After completion reaction mixture was poured into water and extracted with EtOAc (4 × 25 mL). The combined extracts were dried over Na2SO4, concentrated under reduced pressure, and the residue was purified by preparative column chromatography on silica gel, eluting with ethyl acetate/hexane mixture.
4.5.1. 7-Hydroxy-7-(naphthalen-2-yl)indolo[1,2-a]quinolin-5(7H)-one (2df)
This compound was prepared by typical procedure C employing 2-(2-(naphthalen-2-yl)-3-oxoindolin-2-yl)-2-phenylacetonitrile 22d (374 mg, 1.0 mmol), 1-(2-nitrophenyl)ethenone 21f (165 mg, 1.0 mmol). Purification by column chromatography (EtOAc/hexane, 1:3, v/v). The titled compound was obtained as a gray solid, mp 276–278 °C, Rf 0.51 (EtOAc/hexane, 1:1, v/v). Yield 229 mg (0.61 mmol, 61%). 1H NMR (400 MHz, DMSO-d6) δ 8.66 (d, J = 8.6 Hz, 1H), 8.35 (d, J = 8.3 Hz, 1H), 8.28 (d, J = 6.9 Hz, 1H), 8.10 (s, 1H), 7.98–7.89 (m, 2H), 7.88–7.84 (m, 1H), 7.82 (d, J = 8.7 Hz, 1H), 7.60–7.49 (m, 4H), 7.42 (d, J = 7.2 Hz, 1H), 7.32–7.25 (m, 3H), 6.20 (s, 1H). 13C {1H} NMR (101 MHz, DMSO-d6) δ 176.8, 162.0, 141.4, 140.5, 137.9, 137.7, 133.1, 132.6, 132.4, 130.2, 128.2 (2C), 127.5, 126.47, 126.46, 126.4, 126.1, 125.7, 125.5, 124.5, 123.6, 123.6, 117.1, 114.3, 107.3, 79.6. FTIR, vmax: 3105, 1867, 1735, 1686, 1557, 1506, 1244, 1186, 863 cm–1; HRMS (ESI TOF) m/z calcd. for C26H17NNaO2 [M + Na]+: 398.1151, found: 398.1153 (−0.5 ppm).
4.5.2. 7-Hydroxy-7-(4-methoxyphenyl)indolo[1,2-a]quinolin-5(7H)-one (2ff)
This compound was prepared by typical procedure C employing 2-(2-(4-methoxyphenyl)-3-oxoindolin-2-yl)-2-phenylacetonitrile 22f (354 mg, 1.0 mmol), 1-(2-nitrophenyl)ethenone 21f (165 mg, 1.0 mmol). Purification by column chromatography (EtOAc/hexane, 1:3, v/v). The titled compound was obtained as a gray solid, mp 295–297 °C, Rf 0.46 (EtOAc/hexane, 1:1, v/v). Yield 231 mg (0.65 mmol, 65%). 1H NMR (400 MHz, DMSO-d6) δ 8.61 (d, J = 8.6 Hz, 1H), 8.27 (t, J = 8.0 Hz, 2H), 7.88 (t, J = 7.6 Hz, 1H), 7.58–7.49 (m, 2H), 7.40 (d, J = 7.2 Hz, 1H), 7.33–7.24 (m, 3H), 7.06 (s, 1H), 6.88 (d, J = 8.6 Hz, 2H), 6.17 (s, 1H), 3.71 (s, 3H). 13C {1H} NMR (101 MHz, DMSO-d6) δ 176.7, 162.4, 158.8, 141.2, 138.2, 137.6, 135.2, 133.1, 130.0, 126.5 (3C), 126.1, 125.6, 125.4, 124.4, 117.0, 114.1, 113.8 (2C), 107.0, 79.2, 55.1. FTIR, vmax: 3269, 1739, 1685, 1534, 1498, 1329, 1224, 1039, 861 cm–1; HRMS (ESI TOF) m/z calcd. for C23H17NNaO3 [M + Na]+: 378.1101, found: 378.1104 (−0.8 ppm).
4.5.3. 7-Hydroxy-7-(p-tolyl)indolo[1,2-a]quinolin-5(7H)-one (2gf)
This compound was prepared by typical procedure C employing 2-(3-oxo-2-(p-tolyl)indolin-2-yl)-2-phenylacetonitrile 22g (338 mg, 1.0 mmol), 1-(2-nitrophenyl)ethenone 21f (165 mg, 1.0 mmol). Purification by column chromatography (EtOAc/hexane, 1:3, v/v). The titled compound was obtained as a gray solid, mp 268–272 °C, Rf 0.25 (EtOAc/hexane, 1:1, v/v). Yield 237 mg (0.70 mmol, 70%). 1H NMR (400 MHz, DMSO-d6) δ 8.61 (d, J = 8.6 Hz, 1H), 8.28 (dd, J = 11.4, 8.4 Hz, 2H), 7.89 (t, J = 7.3 Hz, 1H), 7.53 (q, J = 8.0 Hz, 2H), 7.38 (d, J = 7.1 Hz, 1H), 7.29 (t, J = 7.4 Hz, 1H), 7.23 (d, J = 8.0 Hz, 2H), 7.13 (d, J = 7.9 Hz, 2H), 7.07 (s, 1H), 6.14 (s, 1H), 2.25 (s, 3H). 13C {1H} NMR (101 MHz, DMSO) δ 176.7, 162.3, 141.2, 140.4, 138.2, 137.6, 137.0, 133.1, 130.0, 129.0 (2C), 126.4, 126.1, 125.6, 125.4, 125.1 (2C), 124.4, 117.0, 114.1, 107.1, 79.4, 20.6. FTIR, vmax: 3392, 1680, 1636, 1556, 1490, 1468, 1301, 1102, 1069, 748 cm–1; HRMS (ESI TOF) m/z calcd. for C23H17NNaO2 [M + Na]+: 362.1151, found: 362.1148 (0.8 ppm).
4.5.4. 7-Hydroxy-7-phenylindolo[1,2-a]quinolin-5(7H)-one (2af)
This compound was prepared by typical procedure C employing 2-(3-oxo-2-phenylindolin-2-yl)-2-phenylacetonitrile 22a (324 mg, 1.0 mmol), 1-(2-nitrophenyl)ethenone 21f (165 mg, 1.0 mmol). Purification by column chromatography (EtOAc/hexane, 1:3, v/v). The titled compound was obtained as a gray solid, mp 276–277 °C, Rf 0.51 (EtOAc/hexane, 1:1, v/v). Yield 244 mg (0.75 mmol, 75%). 1H NMR (400 MHz, DMSO-d6) δ 8.62 (d, J = 8.7 Hz, 1H), 8.30 (d, J = 8.3 Hz, 1H), 8.26 (d, J = 8.0 Hz, 1H), 7.90 (t, J = 7.2 Hz, 1H), 7.59–7.49 (m, 2H), 7.40 (d, J = 7.2 Hz, 1H), 7.37–7.26 (m, 6H), 7.14 (s, 1H), 6.15 (s, 1H).13C {1H} NMR (101 MHz, DMSO-d6) δ 176.7, 162.2, 143.2, 141.2, 138.0, 137.6, 133.1, 130.1, 128.5 (2C), 127.8, 126.4, 126.0, 125.6, 125.4, 125.1 (2C), 124.4, 117.1, 114.1, 107.1, 79.5. FTIR, vmax: 3396, 1701, 1636, 1573, 1559, 1466, 1293, 1102, 1068, 752 cm–1; HRMS (ESI TOF) m/z calcd. for C22H15NNaO2 [M + Na]+: 348.0995, found: 348.0991 (1.2 ppm).
4.5.5. 7-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-7-hydroxyindolo[1,2-a]quinolin-5(7H)-one (2hf)
This compound was prepared by typical procedure C employing 2-(2-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-3-oxoindolin-2-yl)-2-phenylacetonitrile 22h (382 mg, 1.0 mmol), 1-(2-nitrophenyl)ethanone 21f (165 mg, 1.0 mmol). Purification by column chromatography (EtOAc/hexane, 1:3, v/v). The titled compound was obtained as a gray solid, mp 270–271 °C, Rf 0.17 (EtOAc/hexane, 1:1, v/v). Yield 264 mg (0.69 mmol, 69%). 1H NMR (400 MHz, DMSO-d6) δ 8.60 (d, J = 8.6 Hz, 1H), 8.26 (t, J = 7.4 Hz, 2H), 7.88 (t, J = 7.3 Hz, 1H), 7.58–7.49 (m, 2H), 7.41 (d, J = 7.2 Hz, 1H), 7.30 (t, J = 7.4 Hz, 1H), 7.05 (s, 1H), 6.86 (d, J = 1.9 Hz, 1H), 6.78 (d, J = 8.5 Hz, 1H), 6.70 (dd, J = 8.5, 2.0 Hz, 1H), 6.16 (s, 1H), 4.20 (s, 4H).13C {1H} NMR (101 MHz, DMSO-d6) δ 176.7, 162.2, 143.1, 143.0, 141.2, 138.0, 137.6, 136.3, 133.1, 130.1, 126.4, 126.1, 125.5, 125.4, 124.4, 118.1, 117.1, 117.0, 114.1, 113.9, 107.0, 79.1, 64.08, 64.05. FTIR, vmax: 3388, 1682, 1638, 1559, 1506, 1284, 1108, 1062, 893 cm–1; HRMS (ESI TOF) m/z calcd. for C24H17NNaO4 [M + Na]+: 406.1050, found: 406.1046 (1.0 ppm).
4.5.6. 9-Fluoro-7-hydroxy-7-phenylindolo[1,2-a]quinolin-5(7H)-one (2cf)
This compound was prepared by typical procedure C employing 2-(5-fluoro-3-oxo-2-phenylindolin-2-yl)-2-phenylacetonitrile 22c (342 mg, 1.0 mmol), 1-(2-nitrophenyl)ethenone 21f (165 mg, 1.0 mmol). Purification by column chromatography (EtOAc/hexane, 1:3, v/v). The titled compound was obtained as a gray solid, mp 302–303 °C, Rf 0.52 (EtOAc/hexane, 1:1, v/v). Yield 209 mg (0.61 mmol, 61%). 1H NMR (400 MHz, DMSO-d6) δ 8.57 (d, J = 8.6 Hz, 1H), 8.33 (dd, J = 8.7, 3.2 Hz, 1H), 8.26 (d, J = 7.8 Hz, 1H), 7.89 (t, J = 7.7 Hz, 1H), 7.53 (t, J = 7.4 Hz, 1H), 7.40–7.25 (m, 8H), 6.17 (s, 1H). 13C {1H} NMR (101 MHz, DMSO-d6) δ 176.6, 162.2, 159.6 (d, J = 243.6 Hz), 142.7, 140.5 (d, J = 7.8 Hz), 137.5, 137.3, 133.1, 128.6 (2C), 128.0, 126.4, 126.0, 125.1 (2C), 124.5, 116.8, 116.4 (d, J = 23.5 Hz), 115.6 (d, J = 8.2 Hz), 112.78 (d, J = 24.4 Hz), 107.3, 79.3. 19F NMR (376 MHz, DMSO-d6) δ −117.03. FTIR, vmax: 3346, 1873, 1732, 1699, 1684, 1556, 1471, 1184, 876 cm–1; HRMS (ESI TOF) m/z calcd. for C22H14FNNaO2 [M + Na]+: 366.0901, found: 366.0902 (−0.3 ppm).
4.5.7. 7-Hydroxy-9-isopropyl-7-phenylindolo[1,2-a]quinolin-5(7H)-one (2kf)
This compound was prepared by typical procedure C employing 2-(5-isopropyl-3-oxo-2-phenylindolin-2-yl)-2-phenylacetonitrile 22k (366 mg, 1.0 mmol), 1-(2-nitrophenyl)ethenone 21f (165 mg, 1.0 mmol). Purification by column chromatography (EtOAc/hexane, 1:3, v/v). The titled compound was obtained as a gray solid, mp 148–151 °C, Rf 0.37 (EtOAc/hexane, 1:1, v/v). Yield 198 mg (0.54 mmol, 54%). 1H NMR (400 MHz, DMSO-d6) δ 8.59 (d, J = 8.6 Hz, 1H), 8.28–8.24 (m, 1H), 8.21 (d, J = 8.5 Hz, 1H), 7.91–7.86 (m, 1H), 7.51 (t, J = 7.5 Hz, 1H), 7.43–7.40 (m, 1H), 7.38–7.30 (m, 5H), 7.27 (s, 1H), 7.14 (s, 1H), 6.15 (s, 1H), 2.98–2.89 (m, 1H), 1.17 (dd, J = 9.0, 7.0 Hz, 6H).13C {1H} NMR (101 MHz, DMSO-d6) δ 176.7, 162.4, 145.9, 143.4, 139.4, 138.2, 137.5, 133.0, 128.5 (2C), 127.8, 127.7, 126.4, 126.0, 125.1 (2C), 124.3, 123.3, 117.0, 114.0, 107.0, 79.6, 32.9, 24.0, 23.7. FTIR, vmax: 3350, 1734, 1697, 1607, 1559, 1477, 1374, 1142, 1121, 934 cm–1; HRMS (ESI TOF) m/z calcd. for C25H21NNaO2 [M + Na]+: 390.1465, found: 390.1463 (0.5 ppm).
4.5.8. 9-Chloro-7-hydroxy-7-phenylindolo[1,2-a]quinolin-5(7H)-one (2mf)
This compound was prepared by typical procedure C employing 2-(5-chloro-3-oxo-2-phenylindolin-2-yl)-2-phenylacetonitrile 22m (359 mg, 1.0 mmol), 1-(2-nitrophenyl)ethenone 21f (165 mg, 1.0 mmol). Purification by column chromatography (EtOAc/hexane, 1:3, v/v). The titled compound was obtained as a gray solid, mp 302–304 °C, Rf 0.28 (EtOAc/hexane, 1:1, v/v). Yield 183 mg (0.51 mmol, 51%). 1H NMR (400 MHz, DMSO-d6) δ 8.55 (d, J = 7.5 Hz, 1H), 8.29 (dd, J = 22.0, 7.1 Hz, 2H), 7.89 (s, 1H), 7.61–7.52 (m, 2H), 7.41–7.26 (m, 7H), 6.17 (s, 1H).13C {1H} NMR (101 MHz, DMSO-d6) δ 176.7, 161.9, 142.6, 140.3, 140.0, 137.3, 133.2, 129.8, 129.3, 128.6 (2C), 128.0, 126.5, 126.0, 125.3, 125.1 (2C), 124.6, 117.0, 115.7, 107.3, 79.3. FTIR, vmax: 3403, 3120, 1699, 1684, 1636, 1512, 1469, 1248, 1182, 746 cm–1; HRMS (ESI TOF) m/z calcd. for C22H14ClNNaO2 [M + Na]+: 382.0605, found: 382.0609 (−1.0 ppm).
4.5.9. 9-Fluoro-7-hydroxy-7-(naphthalen-2-yl)indolo[1,2-a]quinolin-5(7H)-one (2nf)
This compound was prepared by typical procedure C employing 2-(5-fluoro-2-(naphthalen-2-yl)-3-oxoindolin-2-yl)-2-phenylacetonitrile 22n (392 mg, 1.0 mmol), 1-(2-nitrophenyl)ethenone 21f (165 mg, 1.0 mmol). Purification by column chromatography (EtOAc/hexane, 1:3, v/v). The titled compound was obtained as a gray solid, mp 305–308 °C, Rf 0.51 (EtOAc/hexane, 1:1, v/v). Yield 216 mg (0.55 mmol, 55%). 1H NMR (400 MHz, DMSO-d6) δ 8.61 (d, J = 8.6 Hz, 1H), 8.37 (dd, J = 8.9, 3.6 Hz, 1H), 8.27 (d, J = 7.3 Hz, 1H), 8.10 (s, 1H), 7.98–7.81 (m, 4H), 7.57–7.48 (m, 3H), 7.46–7.36 (m, 2H), 7.34–7.26 (m, 2H), 6.21 (s, 1H). 13C {1H} NMR (101 MHz, DMSO-d6) δ 176.6, 162.1, 159.6 (d, J = 243.7 Hz), 140.5 (d, J = 7.7 Hz), 139.9, 137.6 (d, J = 1.8 Hz) 137.4, 133.2, 132.6, 132.5, 128.4, 128.3, 127.5, 126.5, 126.5 (2C), 126.0, 124.5, 123.7, 123.5, 116.9, 116.5 (d, J = 23.2 Hz), 115.7 (d, J = 8.3 Hz), 112.9 (d, J = 24.5 Hz), 107.5, 79.4. 19F NMR (376 MHz, DMSO-d6) δ −117.00. IR, vmax: 3281, 11696, 1665, 1515, 1268, 1139, 1025, 838 cm–1; HRMS (ESI TOF) m/z calcd. for C26H16FNNaO2 [M + Na]+: 416.1057, found: 416.1055 (0.5 ppm).
4.5.10. 7-(tert-Butyl)-7-hydroxyindolo[1,2-a]quinolin-5(7H)-one (2lf)
This compound was prepared by typical procedure C employing 2-(2-(tert-butyl)-3-oxoindolin-2-yl)-2-phenylacetonitrile 22l (304 mg, 1.0 mmol), 1-(2-nitrophenyl)ethenone 21f (165 mg, 1.0 mmol). Purification by column chromatography (EtOAc/hexane, 1:3, v/v). The titled compound was obtained as a white solid, mp 244–245 °C, Rf 0.28 (EtOAc/hexane, 1:1, v/v). Yield 213 mg (0.70 mmol, 70%). 1H NMR (400 MHz, DMSO-d6) δ 8.43 (d, J = 8.6 Hz, 1H), 8.25 (d, J = 7.9 Hz, 1H), 8.08 (d, J = 8.2 Hz, 1H), 7.84 (t, J = 7.8 Hz, 1H), 7.60 (d, J = 7.4 Hz, 1H), 7.53–7.45 (m, 2H), 7.28 (t, J = 7.5 Hz, 1H), 6.37 (s, 1H), 6.35 (s, 1H), 0.92 (s, 9H). 13C {1H} NMR (101 MHz, DMSO-d6) δ 176.0, 162.5, 141.8, 137.1, 135.8, 132.8, 129.5, 126.4, 126.3, 126.0, 124.3, 123.9, 117.5, 112.3, 108.5, 83.5, 38.7, 24.0 (3C). FTIR, vmax: 3388, 1871, 1735, 1699, 1636, 1515, 1456, 1295, 1240, 851 cm–1; HRMS (ESI TOF) m/z calcd. for C20H19NNaO2 [M + Na]+: 328.1308, found: 328.1312 (−1.2 ppm).
Acknowledgments
This work was supported by the Russian Science Foundation (grant #21-73-20051; https://rscf.ru/en/project/21-73-20051/).
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.4c07691.
Copies of the NMR spectra of all products and starting compounds 22; X-ray crystallographic structure determination for compounds 1fg and 2df (PDF)
Author Contributions
N. A. Arutiunov—investigation, validation; A.M.Z.—investigation; A. A. Aksenova—investigation; N.A.A.—conceptualization, supervision, validation; D.A.A.—investigation; A.V.L.—writing (original draft, review, and editing); A.V.A.—funding acquisition, supervision, validation. All authors have read and agreed to the published version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Kumar A.; Saxena N.; Mehrotra A.; Srivastava N. Review: Studies on the Synthesis of Quinolone Derivatives with Their Antibacterial Activity (Part 1). Curr. Org. Chem. 2020, 24, 817–854. 10.2174/1385272824999200427082108. [DOI] [Google Scholar]
- Pham T. D. M.; Ziora Z. M.; Blaskovich M. A. T. Quinolone Antibiotics. MedChemComm 2019, 10, 1719–1739. 10.1039/C9MD00120D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millanao A. R.; Mora A. Y.; Villagra N. A.; Bucarey S. A.; Hidalgo A. A. Biological Effects of Quinolones: A Family of Broad-Spectrum Antimicrobial Agents. Molecules 2021, 26, 7153. 10.3390/molecules26237153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dube P. S.; Legoabe L. J.; Beteck R. M. Quinolone: A Versatile Therapeutic Compound Class. Mol. Divers. 2023, 27, 1501–1526. 10.1007/s11030-022-10581-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena N.; Shankhdhar S.; Kumar A.; Srivastava N. Part 2, Studies on the Synthesis of Quinolone Derivatives with Their Biological Activity. Curr. Org. Chem. 2024, 28, 185–212. 10.2174/0113852728271272231124042138. [DOI] [Google Scholar]
- Dine I.; Mulugeta E.; Melaku Y.; Belete M. Recent Advances in the Synthesis of Pharmaceutically Active 4-Quinolone and Its Analogues: A Review. RSC Adv. 2023, 13, 8657–8682. 10.1039/D3RA00749A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh G.; Devi V.; Monga V. Recent Developments in the Synthetic Strategies of 4-Quinolones and Its Derivatives. ChemistrySelect 2020, 5, 14100–14129. 10.1002/slct.202003570. [DOI] [Google Scholar]
- Sharma V.; Das R.; Kumar Mehta D.; Gupta S.; Venugopala K. N.; Mailavaram R.; Nair A. B.; Shakya A. K.; Kishore Deb P. Recent Insight into the Biological Activities and SAR of Quinolone Derivatives as Multifunctional Scaffold. Bioorg. Med. Chem. 2022, 59, 116674 10.1016/j.bmc.2022.116674. [DOI] [PubMed] [Google Scholar]
- Ghosh P.; Das S. Synthesis and Functionalization of 4-Quinolones - A Progressing Story. Eur. J. Org. Chem. 2019, 2019, 4466–4516. 10.1002/ejoc.201900452. [DOI] [Google Scholar]
- Gao F.; Zhang X.; Wang T.; Xiao J. Quinolone Hybrids and Their Anti-Cancer Activities: An Overview. Eur. J. Med. Chem. 2019, 165, 59–79. 10.1016/j.ejmech.2019.01.017. [DOI] [PubMed] [Google Scholar]
- Hu Y.-Q.; Zhang S.; Xu Z.; Lv Z.-S.; Liu M.-L.; Feng L.-S. 4-Quinolone Hybrids and Their Antibacterial Activities. Eur. J. Med. Chem. 2017, 141, 335–345. 10.1016/j.ejmech.2017.09.050. [DOI] [PubMed] [Google Scholar]
- Dhote P. S.; Patel P.; Vanka K.; Ramana C. V. Total Synthesis of the Pseudoindoxyl Class of Natural Products. Org. Biomol. Chem. 2021, 19, 7970–7994. 10.1039/D1OB01285A. [DOI] [PubMed] [Google Scholar]
- Mekheimer R. A.; Al-Sheikh M. A.; Medrasi H. Y.; Sadek K. U. Advancements in the Synthesis of Fused Tetracyclic Quinoline Derivatives. RSC Adv. 2020, 10, 19867–19935. 10.1039/D0RA02786C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoi R.; Saito Y.; Miura Y.; Nakagawa-Goto K. Total Synthesis of Waltherione A, a Quinolone Alkaloid Fused with Oxabicyclo[3.2.1]Octane. Org. Lett. 2023, 25, 4755–4758. 10.1021/acs.orglett.3c01837. [DOI] [PubMed] [Google Scholar]
- da Silva G.; Moreira R.; Silva A. M. S.. Bioactive Quinolactacins and Structurally Related Pyrroloquinolones. In Studies in Natural Products Chemistry; Elsevier B.V, 2019; Vol. 62, pp 433–453. [Google Scholar]
- Gao H.; Zhang L.; Zhu T.; Gu Q.; Li D. Unusual Pyrrolyl 4-Quinolinone Alkaloids from the Marine-Derived Fungus Penicillium Sp. Ghq208. Chem. Pharm. Bull. 2012, 60, 1458–1460. 10.1248/cpb.c12-00487. [DOI] [PubMed] [Google Scholar]
- Khalifa M. M.; Philkhana S. C.; Golden J. E. Synthesis of Ring-Fused, N-Substituted 4-Quinolinones Using pKa-Guided, Base-Promoted Annulations with Isatoic Anhydrides: Total Synthesis of Penicinotam. J. Org. Chem. 2020, 85, 464–481. 10.1021/acs.joc.9b02541. [DOI] [PubMed] [Google Scholar]
- Tsoung J.; Bogdan A. R.; Kantor S.; Wang Y.; Charaschanya M.; Djuric S. W. Synthesis of Fused Pyrimidinone and Quinolone Derivatives in an Automated High-Temperature and High-Pressure Flow Reactor. J. Org. Chem. 2017, 82, 1073–1084. 10.1021/acs.joc.6b02520. [DOI] [PubMed] [Google Scholar]
- Wang M.; Ma J.; Wang J.; Lu W.; Pan B. Base Promoted Synthesis of 4-Quinolones Fused with Medium-Sized Rings. Eur. J. Org. Chem. 2023, 26, e202300519 10.1002/ejoc.202300519. [DOI] [Google Scholar]
- Beteck R. M.; Smit F. J.; Haynes R. K.; N’Da D. D. Recent Progress in the Development of Anti-Malarial Quinolones. Malar. J. 2014, 13, 339 10.1186/1475-2875-13-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui Z.; Altom J.; Nguyen V. N.; Fernandez J.; Bernstein J. I.; Hiliard J. J.; Barrett J. F.; Podlogar B. L.; Ohemeng K. A. Synthesis and Inhibitory Activity of Novel Tri- and Tetracyclic Quinolines against Topoisomerases. Bioorg. Med. Chem. 1998, 6, 735–742. 10.1016/S0968-0896(98)00030-3. [DOI] [PubMed] [Google Scholar]
- Li X.; Bian Y.; Chen X.; Zhang H.; Wang W.; Ren S.; Yang X.; Lu C.; Chen C.; Peng J. Tunable Synthesis of Quinolinone-Fused Isoquinolines through Sequential One-Pot Nucleophilic Addition and Palladium-Catalyzed Intramolecular C–H Alkenylation. Org. Biomol. Chem. 2019, 17, 321–332. 10.1039/C8OB02437E. [DOI] [PubMed] [Google Scholar]
- Halim R.; Aurelio L.; Scammells P. J.; Flynn B. L. Scaffold-Divergent Synthesis of Ring-Fused Indoles, Quinolines, and Quinolones via Iodonium-Induced Reaction Cascades. J. Org. Chem. 2013, 78, 4708–4718. 10.1021/jo400125p. [DOI] [PubMed] [Google Scholar]
- Cincinelli R.; Musso L.; Beretta G.; Dallavalle S. 4-Quinolone Fused Heterocyclic Ring Systems by Intramolecular Reactions of 4-Quinolone-2-Carboxamides. Tetrahedron 2014, 70, 9797–9804. 10.1016/j.tet.2014.11.018. [DOI] [Google Scholar]
- Brambilla E.; Gugiatti M.; Rizzato S.; Abbiati G.; Pirovano V. One-Pot Gold/Acid-Catalyzed Synthesis of Indolo[1,2-a]Quinolin-5(6H)-ones from 1-(2-Ethynylphenyl)-1H-indoles. Eur. J. Org. Chem. 2024, 27, e202400083 10.1002/ejoc.202400083. [DOI] [Google Scholar]
- Alnamer Y. A.; Ibrahim M. A.; Ahmed A.; Ali N. M.; Badran A.-S. Synthetic Approaches for Annulated Quinolines at Face a. Polycyclic Aromat. Compd. 2023, 2863–2886. 10.1080/10406638.2023.2225674. [DOI] [Google Scholar]
- Darbandizadeh S. A.; Balalaie S. Recent Advances in the Synthesis of Fused-Cyclic Quinolines. Asian J. Org. Chem. 2024, 13, e202400041 10.1002/ajoc.202400041. [DOI] [Google Scholar]
- Li J.-S.; Liu Y.-J.; Li S.; Ma J.-A. Chiral Phosphoric Acid-Catalyzed Direct Asymmetric Mannich Reaction of Cyclic C-Acylimines with Simple Ketones: Facile Access to C2-Quaternary Indolin-3-Ones. Chem. Commun. 2018, 54, 9151–9154. 10.1039/C8CC05125A. [DOI] [PubMed] [Google Scholar]
- Aksenov A. V.; Aksenov D. A.; Aksenov N. A.; Aleksandrova E. V.; Rubin M. Preparation of Stereodefined 2-(3-Oxoindolin-2-yl)-2-Arylacetonitriles via One-Pot Reaction of Indoles with Nitroalkenes. J. Org. Chem. 2019, 84, 12420–12429. 10.1021/acs.joc.9b01874. [DOI] [PubMed] [Google Scholar]
- Aksenov A. V.; Aksenov N. A.; Aleksandrova E. V.; Aksenov D. A.; Grishin I. Y.; Sorokina E. A.; Wenger A.; Rubin M. Direct Conversion of 3-(2-Nitroethyl)-1H-Indoles into 2-(1H-Indol-2-yl)Acetonitriles. Molecules 2021, 26, 6132. 10.3390/molecules26206132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aksenov A. V.; Aksenov N. A.; Aksenov D. A.; Khamraev V. F.; Rubin M. Nitrostyrenes as 1,4-CCNO-Dipoles: Diastereoselective Formal [4 + 1] Cycloaddition of Indoles. Chem. Commun. 2018, 54, 13260–13263. 10.1039/C8CC07451H. [DOI] [PubMed] [Google Scholar]
- Aksenov A. V.; Aksenov D. A.; Arutiunov N. A.; Aksenov N. A.; Aleksandrova E. V.; Zhao Z.; Du L.; Kornienko A.; Rubin M. Synthesis of Spiro[Indole-3,5′-Isoxazoles] with Anticancer Activity via a Formal [4 + 1]-Spirocyclization of Nitroalkenes to Indoles. J. Org. Chem. 2019, 84, 7123–7137. 10.1021/acs.joc.9b00808. [DOI] [PubMed] [Google Scholar]
- Xu F.; Smith M. W. A General Approach to 2,2-Disubstituted Indoxyls: Total Synthesis of Brevianamide A and Trigonoliimine C. Chem. Sci. 2021, 12, 13756–13763. 10.1039/D1SC03533A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F.; Smith M. W. 2,2-Disubstituted Indoxyls via Oxidative Dearomatization: Generalization to 2-Alkylindoles and Application to Alkaloid Synthesis. Synlett 2023, 34, 1539–1548. 10.1055/a-2020-8717. [DOI] [Google Scholar]
- Aksenov A. V.; Aleksandrova E. V.; Aksenov D. A.; Aksenova A. A.; Aksenov N. A.; Nobi M. A.; Rubin M. Synthetic Studies toward 1,2,3,3a,4,8b-Hexahydropyrrolo[3,2-b]Indole Core. Unusual Fragmentation with 1,2-Aryl Shift. J. Org. Chem. 2022, 87, 1434–1444. 10.1021/acs.joc.1c02753. [DOI] [PubMed] [Google Scholar]
- Aksenov A. V.; Arutiunov N. A.; Zatsepilina A. M.; Aksenova A. A.; Aleksandrova E. V.; Aksenov N. A.; Leontiev A. V.; Aksenov D. A. Novel Two-Step Synthesis of N-Alkylated 2,3-Diaryl-4-Quinolones. Synthesis 2024, 56, 435–444. 10.1055/s-0042-1751530. [DOI] [Google Scholar]
- Aksenov A. V.; Kirilov N. K.; Arutiunov N. A.; Aksenov D. A.; Kuzminov I. K.; Aksenov N. A.; Turner D. N.; Rogelj S.; Kornienko A.; Rubin M. Reductive Cleavage of 4′H-Spiro[Indole-3,5′-Isoxazoles] En Route to 2-(1H-Indol-3-yl)Acetamides with Anticancer Activities. J. Org. Chem. 2022, 87, 13955–13964. 10.1021/acs.joc.2c01627. [DOI] [PubMed] [Google Scholar]
- Pareek A.; Singh G.; Yaragorla S. Synthetic Applications of Ambiphilic C-acylimines in Organic Synthesis. Asian J. Org. Chem. 2022, 11, e202200395 10.1002/ajoc.202200395. [DOI] [Google Scholar]
- Wu X.; Ma T.; Qiao X.; Zou C.; Li G.; He Y.; Zhao X. Enantioselective Alkynylation of 2-Aryl-3H-indol-3-ones via Cooperative Catalysis of Copper/Chiral Phosphoric Acid. Chem. – Asian J. 2023, 18, e202300526 10.1002/asia.202300526. [DOI] [PubMed] [Google Scholar]
- Zhang Q.; Li Y.; Wang J.; Yang C.; Liu C.; Li X.; Cheng J. B(C6F5)3/Chiral Phosphoric Acid Catalyzed Ketimine–Ene Reaction of 2-Aryl-3H-indol-3-ones and α-Methylstyrenes. Angew. Chem., Int. Ed. 2020, 59, 4550–4556. 10.1002/anie.201915226. [DOI] [PubMed] [Google Scholar]
- Jiang X.; Zhu B.; Lin K.; Wang G.; Su W.-K.; Yu C. Metal-Free Synthesis of 2,2-Disubstituted Indolin-3-Ones. Org. Biomol. Chem. 2019, 17, 2199–2203. 10.1039/C8OB03057J. [DOI] [PubMed] [Google Scholar]
- Shimizu M.; Imazato H.; Mizota I.; Zhu Y. A Facile Approach to 2-Alkoxyindolin-3-One and Its Application to the Synthesis of N-Benzyl Matemone. RSC Adv. 2019, 9, 17341–17346. 10.1039/C9RA02204J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Y.; Zeng Y.-M.; Ji J.-Y.; Sun X.-Q.; Yang H.-T.; Miao C.-B. The Cs2CO3–Catalyzed Reaction of 2-Oxindoles with Enones for the Preparation of Indolin-3-Ones and Their Further Transformation. J. Org. Chem. 2016, 81, 12443–12450. 10.1021/acs.joc.6b01993. [DOI] [PubMed] [Google Scholar]
- Suneel Kumar C. V.; Ramana C. V. Ru-Catalyzed Redox-Neutral Cleavage of the N–O Bond in Isoxazolidines: Isatogens to Pseudoindoxyls via a One-Pot [3 + 2]-Cycloaddition/N–O Cleavage. Org. Lett. 2015, 17, 2870–2873. 10.1021/acs.orglett.5b00837. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Li P.; Lyu C.; Yong W.; Li J.; Pan X.; Zhu X.; Rao W. Palladium-Catalyzed One-Pot Synthesis of C2-Quaternary Indolin-3-ones via 1H-indole-3-sulfonates Generated in Situ from 2-Alkynyl Arylazides and Sulfonic Acids. Adv. Synth. Catal. 2017, 359, 4147–4152. 10.1002/adsc.201700838. [DOI] [Google Scholar]
- Fan H.; Xu Y.; Yang F.; Xu S.; Zhao X.; Zhang X. PPh3-Mediated Wittig-Like/Mannich Tandem Reactions of 2-Alkynylnitrobenzenes with Ketones for the Synthesis of 2,2-Disubstituted Indolin-3-Ones. Adv. Synth. Catal. 2022, 364, 2358–2363. 10.1002/adsc.202200323. [DOI] [Google Scholar]
- Dong C.-L.; Ding X.; Huang L.-Q.; He Y.-H.; Guan Z. Merging Visible Light Photocatalysis and L/D-Proline Catalysis: Direct Asymmetric Oxidative Dearomatization of 2-Arylindoles To Access C2-Quaternary Indolin-3-Ones. Org. Lett. 2020, 22, 1076–1080. 10.1021/acs.orglett.9b04613. [DOI] [PubMed] [Google Scholar]
- Ding X.; Dong C.; Guan Z.; He Y. Concurrent Asymmetric Reactions Combining Photocatalysis and Enzyme Catalysis: Direct Enantioselective Synthesis of 2,2-Disubstituted Indol-3-ones from 2-Arylindoles. Angew. Chem., Int. Ed. 2019, 58, 118–124. 10.1002/anie.201811085. [DOI] [PubMed] [Google Scholar]
- Lu F.-Y.; Chen Y.-J.; Chen Y.; Ding X.; Guan Z.; He Y.-H. Highly Enantioselective Electrosynthesis of C2-Quaternary Indolin-3-Ones. Chem. Commun. 2020, 56, 623–626. 10.1039/C9CC09178E. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; An J.; Yang F.; Guan X.; Fu X.; Li Z.; Wang D.; Zhou M.; Yang Y.; He B. One-Pot Asymmetric Oxidative Dearomatization of 2-Substituted Indoles by Merging Transition Metal Catalysis with Organocatalysis to Access C2-Tetrasubstituted Indolin-3-Ones. Adv. Synth. Catal. 2022, 364, 1277–1285. 10.1002/adsc.202101498. [DOI] [Google Scholar]
- Liu X.; Yan X.; Yu J.-H.; Tang Y.-D.; Wang K.; Zhang H. Organocatalytic Asymmetric Dearomative Oxyalkylation of Indoles Enables Access to C2-Quaternary Indolin-3-Ones. Org. Lett. 2019, 21, 5626–5629. 10.1021/acs.orglett.9b01965. [DOI] [PubMed] [Google Scholar]
- Li J.-S.; Liu Y.-J.; Zhang G.-W.; Ma J.-A. Catalytic Asymmetric Mukaiyama–Mannich Reaction of Cyclic C-Acylimines with Difluoroenoxysilanes: Access to Difluoroalkylated Indolin-3-Ones. Org. Lett. 2017, 19, 6364–6367. 10.1021/acs.orglett.7b03213. [DOI] [PubMed] [Google Scholar]
- Hop N. Q.; Son N. T. A Comprehensive Review on Phytochemistry and Pharmacology of Genus Kopsia: Monoterpene Alkaloids – Major Secondary Metabolites. RSC Adv. 2022, 12, 19171–19208. 10.1039/D2RA01791A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aksenov N. A.; Aksenov D. A.; Ganusenko D. D.; Kurenkov I. A.; Aksenov A. V. A Diastereoselective Assembly of Tetralone Derivatives via a Tandem Michael Reaction and Ipso-Substitution of the Nitro Group. J. Org. Chem. 2023, 88, 5639–5651. 10.1021/acs.joc.3c00134. [DOI] [PubMed] [Google Scholar]
- Liu J.; Huang J.; Jia K.; Du T.; Zhao C.; Zhu R.; Liu X. Direct Oxidative Dearomatization of Indoles with Aromatic Ketones: Rapid Access to 2,2-Disubstituted Indolin-3-Ones. Synthesis 2020, 52, 763–768. 10.1055/s-0039-1691528. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.