

Abstract

The synthesis of glycosyl-α-amino acids presents a significant challenge due to the need for precise glycosidic linkages connecting carbohydrate moieties to amino acids while maintaining stereo- and regiochemical fidelity. Classical methods relying on ionic intermediates (2e–) often involve intricate synthetic procedures, particularly when dealing with 2-N-acetamido-2-deoxyglycosides linked to α-amino acids—a crucial class of glycoconjugates that play important biological roles. Considering the growing prominence of photocatalysis, this study explores various photoredox catalytic approaches to achieving glycosylation reactions. Our focus lies on the notoriously difficult case of 2-N-acetamido-2-deoxyglycosyl-α-amino acids, which could be obtained efficiently by two methodologies that involved, on the one hand, photoredox Giese reactions using a chiral dehydroalanine (Dha) as an electron density-deficient alkene in these radical 1,4-additions and, on the other hand, photoredox glycosylations using selenoglycosides as glycosyl donors and hydroxyl groups of protected amino acids as acceptors.
Introduction
Glycopeptides and glycoproteins play crucial roles in biochemical processes1 like molecular recognition and immune response, whose studies are essential for developing biomedical applications.2 Therefore, the demand for homogeneous samples of glycopeptides and their minor units of glycosyl-α-amino acids for biological research has boomed in recent decades. Especially relevant are 2-N-acetamido-2-deoxyglycosides connected to amino acids, which are widely distributed in living organisms as glycoconjugates with important biological roles.3
In this context, O-linked-β-N-acetylglucosamine (β-O-GlcNAc), a single sugar modification of Ser and Thr residues of proteins, is an important glycosylation since, while other carbohydrates modify proteins on the cell surface, O-GlcNAc modifies nucleocytoplasmic proteins, which are involved in transcription, ubiquitination, cell cycle, and stress responses.3c In addition, O-linked-α-N-acetylgalactosamine to Ser or Thr, namely, the Tn antigen (α-O-GalNAc-l-Ser/Thr), has been the focus of numerous investigations due to their use in therapeutic vaccines against cancer or as powerful tools for early diagnosis of cancer.4 However, one potential drawback in using the native Tn antigen for vaccine design is the instability of the glycosidic linkage to glycosidases and the low immunogenicity. To overcome this problem, a plethora of structural mimetics of the Tn antigen has been used.5 Therefore, the chemical synthesis of different glycosyl-α-amino acid structures is necessary, especially α-O-GalNAc-l-Ser/Thr and β-O-GlcNAc-l-Ser/Thr along with their mimetics displaying α- or β-C-, S- or Se-glycosidic bonds. In this regard, the main synthetic challenge is to construct glycosidic linkages that connect monomeric carbohydrate units to amino acids with the appropriate stereo- and regiochemical orientation. In recent decades, many procedures for chemical glycosylation have been developed, making it a very active area of research.6 In particular, several methods have been reported for the difficult case of 2-N-acetamido-2-deoxyglycosyl-α-amino acids but all of them use precursors that need to be prepared in multiple steps, including the specific protecting/deprotecting procedures, which decreases the efficiency of synthesis.7 Although more and more syntheses of complex carbohydrates are becoming standardized,8 the complete stereochemical control of glycosylation reactions remain a challenge in carbohydrate and glycoconjugate chemistry.9
Classical methods concerning the formation of glycosidic bonds frequently use reactions involving ionic intermediates and need a methodology that requires the activation of a glycosyl donor, with most of these activating agents being sensitive to air and humidity, so they must be used under strict anhydrous conditions and at low temperatures, adding another difficulty to the synthetic process.6 Because of this, much of the interest in glycosylation reactions has therefore shifted toward the discovery of simpler procedures. In the past, some methods for carrying out this process using glycosyl radicals have also been described.10 These processes involving radical reactions are not widely used, given the disadvantages of the use of toxic tin hydrides.11 In addition, the development of radical processes has been challenging due to the numerous reduction, elimination, and nucleophilic substitution side reactions that compete with the labile glycosyl donors commonly used. More recently, an interesting radical methodology was established to achieve 2-amidoglycosylation of glycals.12
However, in recent years, due to the rise of photocatalysis13 and given the advantages of developing radical reactions promoted by photoredox catalytic processes, some methodologies have been described to synthesize carbohydrate derivatives by photoinduced radical glycosylation reactions.14 In this way, glycosylation reactions take place under mild reaction conditions at room temperature and do not require strict anhydrous conditions. Moreover, they use substoichiometric amounts of activating agents.15 Despite the versatility of photoredox catalysis in organic synthesis and its fundamental role in the development of major areas of contemporary synthetic chemistry, it has not yet been addressed in depth in carbohydrate chemistry, especially in the case of important 2-deoxy-2-(acetamido)pyranose derivatives (Figure 1a).14 In addition, although several cases have been reported for photocatalytic glycosylation reactions, very few works have been published concerning the synthesis of glycosyl-α-amino acids.16−25
Figure 1.

(a–c) Furanosyl and pyranosyl radicals, 2-N-acetamido-2-deoxyglycosyl-α-amino acid derivatives, and some O-, S-, and C-glycosyl-α-amino acids synthesized using photocatalytic glycosylation reactions.
In this field, only two O-glycosyl-α-amino acids16−18 and some examples of S-glycosyl-α-amino acids19 have been reported (Figure 1b). However, it is important to note that in this last work about the synthesis of S-glycosylated derivatives, the authors described a limitation of this method since when N-(acyl)glucosamines (protected GlcNAc bromide, for instance) are used as substrates for the C–S coupling reaction, the desired products (α-S-GlcNAc-l-Cys among others) are not formed, probably due to the lability of these reactants.19 In fact, the reaction with such substrates is not described in any of the S-glycosides formed, suggesting challenging access to this type of 2-N-acetamido-2-deoxyglycosyl-α-amino acid.
This drawback was not observed in the synthesis of α-S-GalNAc-Cys derivatives using allyl glycosyl sulfones as precursors to glycosyl radicals.20a The abovementioned drawback was also not detected for the case of α-Se-GalNAc-Sec included in peptides and proteins using a photocatalytic method for the rapid and efficient dimerization of (α-Se-GalNAcSe)2 and peptide diselenides in the presence of an iridium photocatalyst and a phosphine. However, this method uses substantial quantity of the starting diselenide (α-Se-GalNAcSe)2, which is challenging to synthesize.20b
The most explored photoredox glycosylation reactions of this type of compound involve the synthesis of C-glycosyl-α-amino acids from a radical generated in the glycosyl moiety, in most of the cases in anomeric positions, which is captured with different electrophiles in chemoselective addition reactions (Figure 1c). For instance, protected dehydroalanines (Dha)—via a Giese-type reaction—,21 α-imino esters,22 or activated glycines23 have been used as good electrophiles with nucleophilic glycosyl radicals. In other cases, the radical is generated in the amino acid moiety, and it reacts with glycosyl halides to generate the corresponding C-glycosyl-α-amino acids via a photoinduced, nickel-catalyzed reaction.24,25
Considering this background, we focused on the synthesis of glycosyl-α-amino acids, especially 2-N-acetamido-2-deoxyglycosides linked to α-amino acids, by means of some published methodologies that use photoredox catalytic processes. Thus, we envisioned the use of chiral bicyclic dehydroalanine derivatives, already reported by our group, to construct these glycosyl-α-amino acid derivatives via photoredox catalysis (Scheme 1).26
Scheme 1. Photocatalytic Giese-Type 1,4-Additions of Carboxylic Acids to Dha 1 as an Entry to a Variety of Enantiomerically Pure Unnatural Amino Acids.

Results and Discussion
α-Glycosyl-α-amino Acids by Photoredox 1,4-Additions
Initially, we focused on the challenging synthesis of important glycosyl-α-amino acids derived from C2-N-acetyl pyranoses following the above commented photoredox strategy.26 In particular, and to be used for their application in cancer vaccines,5 we are interested in the synthesis of three mimetics of the Tn antigen α-O-D-GalNAc-l-Ser incorporating nonproteinogenic α-amino acids: α-O-D-GalNAc-l-Hse, α-S-D-GalNAc-l-Hcy, and α-C-D-GalNAc-l-Abu. One of them displays a homologue of Ser amino acid, homoserine (Hse), the other one incorporates homocysteine (Hcy), and the third one corresponds to the α-C-GalNAc-glycosylated 2-aminobutanoic acid (Abu).
Based on retrosynthetic analysis, we considered the disconnection of a hitherto little explored linkage (marked in bold in Scheme 2a) to address the synthesis of the aforementioned glycosyl-α-amino acids involving 1,4-additions of alkyl radicals to dehydroalanines (Dha) mediated by photoredox processes. We prepared starting materials incorporating carboxylic acid groups to generate the corresponding decarboxylative alkyl radicals. Thus, in the first case, we installed an -O-CH2-CO2H group at the anomeric position of α-GalNAc by a well-known Koenigs–Knorr glycosylation reaction27 between 3,4,6-tri-O-acetyl-2-azido-2-deoxy-α-O-d-galactopyranosyl chloride 2 and benzyl hydroxyacetate 3 followed by conversion of azide to the N-acetyl group and further debenzylation using a hydrogenolysis reaction (Scheme 2b).
Scheme 2. Retrosynthesis of α-GalNAc-α-Amino Acids by a Photocatalytic Giese Process (a) and Synthesis of Adequate GalNAc Precursors (b).

Once compound 4O was obtained, we synthesized its partner 4S that incorporates a sulfur atom instead of oxygen at the anomeric position but now using a different methodology. Thus, the nucleophilic substitution reaction between α-S-d-GalNAc-SH 5-SH and tert-butyl bromoacetate 6 in the presence of diisopropylethyl amine (DIEA) as a base and using acetonitrile as a solvent followed by acid hydrolysis of the tert-butyl ester group with trifluoracetic acid (TFA) afforded the corresponding acid 4S (Scheme 2b).
In the third case, oxidative cleavage of the alkene group of the corresponding α-C-d-GalNAc-allyl derivative 7 by using ruthenium tetraoxide in situ generated from RuCl3·H2O and NaIO4 gave the (α-C-GalNAc)acetic acid 4C. Compound 7 was obtained from GalNAc chloride 5-Cl following the published synthetic procedure for its GlcNAc partner (Scheme 2).28 With the starting material in our hands (4O, 4S, and 4C), we tested these carboxylic acids in photocatalytic Giese-type reactions with chiral Dha 1, using the optimized conditions26 previously commented in Scheme 1. The reactions work very well when using Dha 1 (1.0 equiv), carboxylic acid 4O or 4S (1.2 equiv), Cs2CO3 (1.5 equiv) as a base, and 4CzIPN (0.05 equiv) as an organophotocatalyst in N,N-dimethylformamide (DMF) as a solvent at room temperature. Once reactions were completed after 16 h under blue-light-emitting diode (LED) irradiation, we observed the clean formation of a single diastereomer in each case, corresponding to adducts 8O and 8S, respectively (Scheme 3).
Scheme 3. α-GalNAc-α-Amino Acids by Photocatalytic Giese-Type 1,4-Additions of GalNAc-Carboxylic Acids to Chiral Dha 1.

However, when 4C was used in the same conditions, we found the expected adduct 8C in very low yield and accompanied by several nonidentified products. This fact is probably due to the instability of the corresponding alkyl radical intermediate (Scheme 3). These adducts 8O, 8S, and 8C can be regarded as protected α-glycosyl-α-amino acids in which hydroxyl groups of the carbohydrate moiety are protected as acetates, carboxylic acid group as a cyclic ester (lactone), and amino group as a cyclic carbamate, since—as described in our previous work26—all of them can be easily deprotected by acid hydrolysis. In fact, Giese adducts 8O, 8S, and 8C were hydrolyzed using a 4 M aqueous solution of HCl at 40 °C for 16 h to give glycosyl-α-amino acids 8O-AA, 8S-AA, and 8C-AA, respectively (Scheme 3). The substructure of some of these new glycosyl amino acids appear included in some peptides, and the most related one is GalNAc-l-homoserine protected as the Fmoc derivative, which was incorporated in an antitumor neoglycopeptide vaccine as a novel homoserine Tn antigen.29
The stereochemical outcome of these photocatalytic reactions between Dha 1 and carboxylic acids 4O, 4S, and 4C follows a stereoinduction mechanism similar to that previously described by us.26 As in this case, the absolute configurations (S) of the new stereocenter (carbon atom marked as a green dot in Scheme 3) corresponding to the Cα of the amino acid moiety of the glycosyl-α-amino acid derivatives 8O, 8S, and 8C formed on Giese additions were confidently assigned based on 2D NOESY experiments. These results likely indicate that reactions follow a mechanism similar to that described in our previous work (Supporting Information).
As a next step, we selected the procedure described by Wang and co-workers19 to generate S-glycosyl-α-amino acids attending to the fact that the authors developed a radical trapping study with methyl acrylate to confirm the radical engaged process. In our case, we aim to apply this method to C-glycosylation reactions. Starting from the optimized conditions used in that work, we tested different organophotocatalysts (4ClCzIPN and 4CzIPN), bases (K3PO4 and Cs2CO3), solvents (DMF and CH2Cl2), and proportions. The best conditions, which matched precisely with those described in that work, were selected to assay the photoredox reactions. Thus, some glycosyl bromides (9a–9c) were used to generate visible light-mediated anomeric glycosyl radicals by the action of 4ClCzIPN as organophotocatalyst—avoiding the use of a transition metal—and (TMS)3SiOH as a halogen atom transfer (XAT) reagent in the presence of K3PO4, as a base, and a mixture of dichloroethane (DCE) and dimethyl sulfoxide (DMSO) as a solvent. This process took place at room temperature under irradiation of blue LEDs in an inert atmosphere, and the radical was trapped with chiral Dha 1 as an acceptor alkene (Scheme 4a).
Scheme 4. Organophotocatalytic Radical C-Glycosylation Reactions Using Glycosyl Bromides 10a–10c and Dha 1 (a) and Deprotection of α-C-Glc Adduct 10b by Acid Hydrolysis to Give α-C-Glucosyl-α-amino Acid 10b-AA (b).

First, we evaluated the performance using galactosyl bromide (9a) as a radical donor for coupling with chiral Dha 1. When the reaction was irradiated with 16 W Kessil blue LEDs in an inert atmosphere at room temperature for 16 h, α-C-galactosyl adduct 10a was obtained as a solely diastereoisomer in 46% yield after purification by column chromatography (β-anomer was not detected). The process serves as a general approach since α-C-glucosyl and α-C-mannosyl-α-amino acid derivatives 10b and 10c were also obtained in 40 and 46% yield, respectively.
Unfortunately, the reaction did not take place when 3,4,6-tri-O-acetyl-2-acetamido-2-deoxy-α-O-d-galactopyranosyl bromide (OAc)3GalNAc-α-Br 9d was used, probably due to the lability of the anomeric radical formed. In these conditions, we only detected, isolated (75%), and characterized the known product 10d, corresponding to the addition of one hydroxyl group to the anomeric carbon (Scheme 5 and Scheme S10 in the Supporting Information).
Scheme 5. Organophotocatalytic Reaction of Glycosyl Bromide 9d and Dha 1.

Interestingly, total axial selectivity was observed for the three adducts 10a–10c and, most importantly, the absolute configurations of the two new stereogenic centers created in the photoredox radical reaction (highlighted as green dots in Scheme 4a) were totally controlled. As in the previous cases (Scheme 3), adducts 10a, 10b, and 10c can be regarded as protected glycosyl-α-amino acids. To illustrate this characteristic, one of these adducts—specifically, adduct 10b—underwent hydrolysis in an acidic environment, resulting in the acquisition of the corresponding fully deprotected α-C-glucosyl-α-amino acid, denoted as 10b-AA (Scheme 4b).
These absolute configurations were assessed by 2D NOESY experiments for the case of the new stereocenter (C3) corresponding to Cα of the amino acid moiety of adduct 10a formed in the Giese addition and attending to the study of the coupling contants (J) values observed for the case of anomeric carbon (C 1s). Similar structural characteristics were found in adducts 10b and 10c (Supporting Information). Alternatively, these structural features were also determined by X-ray analysis of a single crystal of compound 10a (Figure 2 and Figure S88 of the Supporting Information).
Figure 2.
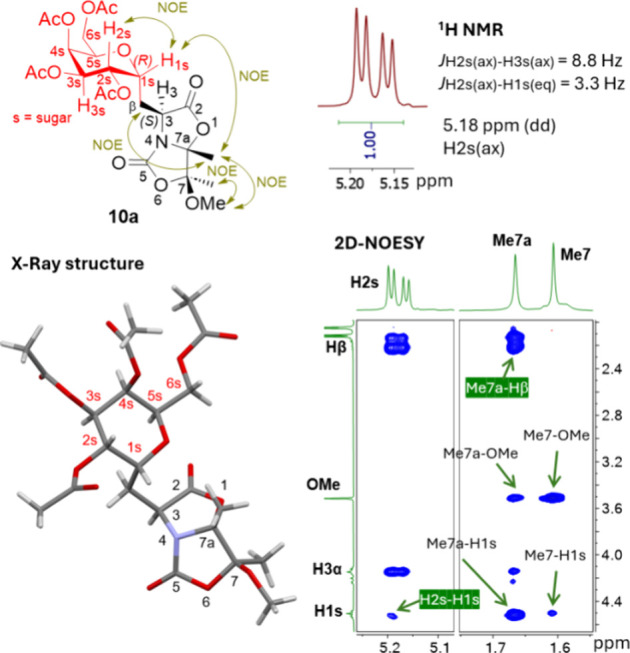
1H NMR and 2D NOESY spectra as well as X-ray structure to determine the absolute configurations of the stereogenic centers created in the photoredox reaction for compound 10a.
Therefore, the stereochemical outcome of these photocatalytic reactions between Dha 1 and glycosyl bromides 9a–9c indicates a highly conserved stereoinduction mechanism, not only at the amino acid level created in the Giese reaction26—as demonstrated in our previous work—but also at the anomeric carbon C 1s of the carbohydrate moiety, possibly due to the stabilizing anomeric effect on the anomeric radical formed in the process.30 That is, this photoredox process allows stereochemical control of the two stereogenic centers created in the photoredox radical reaction.
Based on the above-described results, we propose that these photoredox catalytic Giese reactions proceed via the mechanism shown in Scheme S10 of the Supporting Information.
β-O-Glycosyl-α-amino Acids from Selenoglycosides
Regarding the synthesis of O-GalNAc-α-amino acids using photoredox strategies, we tested the methodology published by Ragains and co-workers that involves a photocatalytic glycosylation of alcohols with selenoglycosides promoted by visible light.31 We envisioned that this mild procedure would be applied to the synthesis of challenging O-GalNAc-α-amino acids by using the visible light photocatalytic activation of easily available phenyl 3,4,6-tri-O-acetyl-2-acetamido-2-deoxy-1-seleno-α-d-galactopyranoside3211—abbreviated as α-Se-GalNAc-SePh—as a selenoglycoside donor and hydroxyl groups of protected Ser and Thr as acceptors. Starting from the optimized conditions used in that work, we tested different organophotocatalysts—(PhSe)2 and 4CzIPN—, bases (TEA and DTBMP), and proportions. The best conditions that matched those described in that work were selected. Therefore, we used diphenyldiselenide [(PhSe)2, 10 mol %] as an organocatalyst in the presence of tetrabromomethane (CBr4, 1.1 equiv) as an electron acceptor and 2,6-di-tert-butyl-4-methylpyridine (DTBMP, 1.2 equiv) as a base in dichloromethane as a solvent under blue LED irradiation. The corresponding compounds displaying hydroxyl groups were selected as glycosyl acceptors: MeOH (a) or Boc-l-Ser-OBn (b), Fmoc-l-Ser-OtBu (c), Fmoc-l-Thr-OtBu (d), and Fmoc-L-Hse-OtBu (e) were used in excess (3.0 equiv) with respect to selenoglycoside donor 11. We obtained acceptable yields of glycosylation products 12a–12e after irradiation and subsequent purification by column chromatography (Scheme 6).
Scheme 6. Organophotocatalytic O-Glycosylation Reactions Using Selenoglycosides 11 and 13 as Donors and Alcohols a–e as Acceptors to Generate Protected β-O-GalNAc- or β-O-GlcNAc-α-Amino Acids.

Reactions afford, in all cases, β-anomers with high selectivities since we do not detect the corresponding α-anomers in the crude reactions. This result is contrary to that obtained by the authors since they observed preferentially α-selectivity, but probably is due to the fact that they used β-Se-(OBn)4Gal-SePh or β-Se-(OBn)4Glc-SePh as selenoglycosides, in which the lack of 2-acetate or 2-acetamido groups as an anchimeric support does not favor β-selectivity, attending to the proposed mechanism (Supporting Information).
It is crucial to note that while β-O-GalNAc-Ser/Thr derivatives are rare in nature, this isomer of the Tn antigen (α-O-GalNAc-Ser/Thr), differing only in anomeric configuration, has been used as a mimic of the natural Tn antigen in the development of cancer vaccines,33 showing improved immunological potential with respect to the natural one. This observation underscores the idea that a straightforward alteration in the stereochemistry of the glycosidic linkage between carbohydrate and amino acid moieties could serve as a viable strategy for designing anticancer vaccines. In this sense, both a synthetic approach and a deep conformational study comparing α- and β-anomers of the Tn antigen have been carried out in the past by us.34
To validate the viability of this synthetic process and recognize the significance of this Tn antigen mimetic (β-O-GalNAc-Thr), we opted to scale up this methodology. Notably, previously reported synthetic approaches relying on classical ionic methods necessitate multiple steps or the use of harsh conditions, including mercury salts.33,34 Thus, we could obtain derivative 12d in a 23% yield on a scale of hundreds of milligrams from readily available raw material 11 (Supporting Information). These glycosylation reactions offer distinct advantages over classical ionic synthetic methodologies as they exhibit tolerance toward diverse functional groups and enable precise stereochemical control.
Considering the above results, we focus on the synthesis of important glycosylated building blocks3c consist of N-acetylglucosamine β-O-linked to Ser and Thr residues (β-O-GlcNAc-Ser/Thr). Initially, we synthesized easily available phenyl 3,4,6-tri-O-acetyl-2-acetamido-2-deoxy-1-seleno-α-d-galactopyranoside 13, abbreviated as α-Se-GlcNAc-SePh, using a modified procedure already described.35 Following similar glycosylation conditions used for their GalNAc-counterparts [(PhSe)2, CBr4, and DTBMP in CH2Cl2 under blue LEDs, the reaction of selenoglycoside donor 13 with Fmoc-l-Ser-OtBu (c) and Fmoc-l-Thr-OtBu (d) as glycosyl acceptors gave the required adequately protected β-O-GlcNAc-α-amino acids Fmoc-l-Ser[β-O-(OAc)3GlcNAc]-OtBu 14c and Fmoc-l-Thr[β-O-(OAc)3GlcNAc]-OtBu 14d in acceptable yields (48 and 28%, respectively, Scheme 7). It is important to mention that all synthesized O-Ac, CO2tBu, and NHFmoc-protected GalNAc-glycosylated α-amino acids are important building blocks adequately prepared, once the quantitatively transformed CO2tBu group is transformed to −CO2H with TFA, to be used in solid phase peptide synthesis (SPPS), allowing the obtention of interesting glycopeptides.
Scheme 7. Visible Light-Promoted O-Glycosylation Reactions, in the Presence of Ph2S2 and (CBrCl2)2, Using Selenoglycoside 11 as a Donor with Fmoc-l-Ser-OtBu (c) as Glycosyl Acceptors to Generate Protected β-O-GalNAc-l-Ser 12c and Deprotection of the tBu Group.

Building upon the proposed mechanism for this specific reaction,31 irradiation with blue LEDs initiates the homolysis of (PhSe)2,36 yielding phenylselenyl radicals (PhSe•). These radicals subsequently engage with CBr4, affording PhSeBr. Notably, this compound converts selenoglycosyl donor 11 into the active selenium specie 11-SePh+.37 The latter would further evolve into the corresponding oxonium intermediate, which undergoes conversion to a dihydrooxazolium species, although none of them species have been detected by mass spectrometry of the crude reactions. These reactive intermediates, under a basic medium, then engages the alcohol group of the Thr-derivative d via a β-attack, ultimately leading to the formation of the β-O-glycosyl-α-amino acid 12d (Scheme S11 of the Supporting Information). When the same reaction conditions were used but without blue LED irradiation or without CBr4, no reaction takes place. We emphasize that this proposed mechanism remains the subject of an ongoing investigation.
Although there is no disputing the efficacy of the CBr438 and (PhSe)239 agents in the aforementioned radical β-O-glycosylation protocol, the replacement of (PhSe)2 with the less toxic and cheaper (PhS)2 and CBr4 with (CBrCl2)2, which is less harmful and similarly priced, is warranted.40,41 These substitutions address economic, toxic, and environmental concerns in the radical β-O-glycosylation protocol. To the best of our knowledge, (CBrCl2)2 has never been used as a reagent in photoredox reactions.
We tested these new conditions in the reactions of selenoglycoside donor 11 with Fmoc-l-Ser-OtBu (c) as a glycosyl acceptor to obtain a similar (even improved) yield of glycosylation product 12c after irradiation for 16 h and subsequent purification by column chromatography (Scheme 7). In addition, to demonstrate the feasibility of this synthetic process to afford β-O-glycosyl-α-amino acids, we scaled up (1 mmol) the synthesis of the β-O-GalNAc-l-Ser 12c, including the synthetic procedure to obtain the selenoglycoside 11 (on a gram scale) from readily available raw materials (Supporting Information). Thus, using this new methodology and starting from 11 (1 mmol), β-O-GalNAc-l-Ser derivative 12c was obtained in 55% yield (392 mg). This protected glycosylamino acid 12c could be transformed easily and quantitatively to the required Fmoc-l-Ser[β-O-(OAc)3GalNAc]–OH 12c–OH, ready to be used in SPPS, by treatment with TFA in dichloromethane for 3 h at room temperature (Scheme 7). It is important to note that although several methodologies have been reported for their congeners (α-O-GalNAc-Ser, α-O-GlcNAc-Ser, or β-O-GlcNAc-Ser), very little attention has been paid to the synthesis of this protected β-O-GalNAc-Ser derivative 12c–OH.
We can conclude that this photoredox methodology can compete with the classical glycosylation methodology used in the past to obtain, in particular, glycosylamino acid 12c–OH since this compound is obtained from d-galactopyranose in seven steps with a 22% overall yield (Supporting Information and Scheme 8).
Scheme 8. Two Synthetic Methodologies of Protected β-O-GalNAc-l-Ser 12c–OH.

By contrast, Fmoc-l-Ser(β-O-D-GalNAc)–OH 12c–OH was previously obtained from 2-amino-2-deoxy-d-galactose hydrochloride in four steps that involved peracetylation, treatement with AcCl and HCl (g) to give α-GalNAc(OAc)3-Cl, glycosylation of this derivative with Fmoc-l-Ser-OBn, and further debenzylation by hydrogenolysis. 2-Amino-2-deoxy-d-galactose hydrochloride was previously synthesized from commercially available d-tagatose via the Heyns rearrangement. In summary, 12c–OH was synthesized by classical methods starting from commercially available d-tagatose using seven steps in a 10% overall yield (Supporting Information and Scheme 8).
This highly efficient strategy has advantages such as high functional group tolerance, biocompatible reaction conditions, and stereochemistry control to obtain important, adequately protected β-O-2-N-acetamido-2-deoxyglycosyl-α-amino acids. Special relevance shows GalNAc-glycosylated α-amino acids 8O-AA, 8S-AA, 8C-AA, and 12c–12e, and in the future, we will explore their behavior as mimetics of the Tn antigen by incorporation into peptides to develop cancer vaccines or diagnostic tools.
Conclusions
The ability to selectively functionalize biologically relevant structures such as amino acids with carbohydrates underlines the synthetic power of a synthetic methodology. In this sense, it is particularly noteworthy that the protocol based on the use of glycosyl bromides and the chiral dehydroalanine (Dha) electrophile 1 is amenable for the synthesis of α-C-linked carbohydrates to amino acids scaffolds, a synthetic challenge in glycosylation, as demonstrated in common hexoses to afford glucosyl-, galactosyl-, and mannosyl-α-amino acids 10a–10c, with a total stereocontrol of the two stereogenic carbon generated in the photoredox process. In addition, taking into account that the synthesis of 2-N-acetamido-2-deoxyglycosyl-α-amino acids represents a special challenge in photoredox carbohydrate chemistry, we report here a photoredox Giese reaction based on the use of the same Dha 1 with carbohydrates bearing a carboxylic acid group that allowed to synthesize α-linked O-, S-, and C-2-N-acetamido-2-deoxyglycosyl-α-amino acids 8O-AA, 8S-AA, and 8C-AA. Finally, adequately protected β-O-GalNAc- and β-O-GlcNAc-α-amino acids 12b–12e and 14c–14d, respectively, could be efficiently constructed, using α-selenoglycoside donors and (PhSe)2 or (PhS)2 as an organophotocatalyst in the presence of CBr4 or (CBrCl2)2, respectively. In summary, the results presented here demonstrate that 2-N-acetamido-2-deoxyglycosyl-α-amino acid derivatives could be obtained by photoredox methods with high stereochemistry control. We believe that the broad scope, functional group tolerance, and modularity of these photoredox synthetic approaches are likely to be of great use to chemists in both academia and industry.
Experimental Section
General and Experimental Methods
Commercial reagents were used without further purification. Analytical thin layer chromatography (TLC) was performed on Macherey-Nagel precoated aluminum sheets with a 0.20 mm thickness of silica gel 60 with the fluorescent indicator UV254. TLC plates were visualized with UV light and by staining with a potassium permanganate solution (0.75 g KMnO4, 5 g K2CO3, and 0.63 mL 10% NaOH in 100 mL of water) or a ninhydrin solution (1.5 g ninhydrin in 100 mL of n-butanol and 3.0 mL of acetic acid). Column chromatography was performed on a silica gel (230–400 mesh). 1H and 13C{1H} NMR spectra were measured with a 300 or 400 MHz spectrometer with TMS as the internal standard. Multiplicities are quoted as singlet (s), broad singlet (br s), doublet (d), doublet of doublets (dd), triplet (t), or multiplet (m). Spectra were assigned using the COSY and HSQC experiments. The results of these experiments were processed with the MestreNova software. High-resolution electrospray mass (ESI) spectra were recorded on a microTOF spectrometer; accurate mass measurements were achieved by using sodium formate as an external reference.
General Procedure for Photoredox Giese Reactions to Obtain α-O-, α-S-, and α-C-Glycosyl-α-amino Acid Derivatives 8O, 8S, and 8C
Chiral bicyclic Dha 1 (0.1 mmol, 1.0 equiv), carboxylic acids 4O, 4S, or 4C (0.12 mmol, 1.2 equiv), Cs2CO3 (0.15 mmol, 1.5 equiv), and 4CzIPN (0.005 mmol, 0.05 equiv) were added in sample vials. The tube was evacuated and backfilled with N2 (three times). Then, deoxidized DMF (1 mL, final concentration 0.1 M) was added by using a syringe. The solution was then stirred at room temperature under irradiation of blue LEDs (30 W) for 16 h. Once completed, water (1 mL) was added and extracted by ethyl acetate. The combined organic layer was dried over anhydrous Na2SO4, and the solvent was removed under vacuum. The crude mixture was purified by column chromatography (hexane/ethyl acetate) on silica gel to afford desired products 8O, 8S, and 8C.
General Procedure for the Photoredox Giese Reaction to Obtain α-C-Glycosyl-α-amino Acid Derivatives 10a–10c
Glycosyl bromide 9a–9c (0.2 mmol), chiral bicyclic Dha 1 (0.1 mmol), 4ClCzIPN (0.005 mmol), and K3PO4 (0.4 mmol) were added to sample vials. The vial was evacuated and backfilled with N2. Then, anhydrous dichloroethane (0.5 mL) and DMSO (0.5 mL) were added using a syringe, and the solution was degassed. After degassing, (TMS)3SiOH (0.15 mmol) was added through a syringe. The solution was then stirred at room temperature under irradiation with blue LEDs (16 W) for 16 h. Once completed, water (1 mL) was added, and the mixture was extracted with ethyl acetate. The combined organic layer was dried over anhydrous Na2SO4, and the solvent was removed under vacuum. The crude mixture was purified by column chromatography (hexanes/ethyl acetate) on silica gel to afford the desired products 10a–10c.
General Procedure for Photoredox β-O-Glycosylation of Selenoglycosides with Alcohols
A dried 5 mL Pyrex reactor vial was charged with the α-Se-glycosyl donor 11 or 13 (0.074 mmol, 1 equiv), (PhSe)2 (0.0074 mmol, 0.1 equiv), CBr4 (0.081 mmol, 1.1 equiv), 2,6-di-tert-butyl-4-methylpyridine (DTBMP) (0.088 mmol, 1.2 equiv), the glycosyl acceptor (0.22 mmol, 3 equiv), and 1 mL of dry CH2Cl2 as a solvent under a nitrogen atmosphere. The vial was sealed with a Teflon cap, and the reaction was stirred at room temperature under irradiation with 30 W blue LEDs. Reaction progress was monitored by TLC. After consumption of the glycosyl donor (16–72 h), the crude products were concentrated and then purified by silica gel chromatography (ethyl acetate/hexanes, 7:3) to obtain β-O-glycosyl derivatives 12a–12e and 14c and 14d.
Note: (PhSe)2 can be replaced by (PhS)2 and CBr4 by (CBrCl2)2, and the procedure has been scaled up to 1 mmol of selenoglycoside 11 to obtain 392 mg (55%) of Fmoc-l-Ser(β-O-d-GalNAc)-OtBu 12c, after purification by column chromatography.
2D NMR Experiments
Spectra were assigned using COSY and edited-HSQC experiments (blue for CH2 and red for CH and CH3 groups). NOESY experiments were recorded on a 400 MHz spectrometer at 298 K. The experiments were conducted by using phase-sensitive ge-2D NOESY spectra. The number of scans used was 16, and the mixing time was 800 ms.
X-ray Diffraction Analysis
X-ray diffraction data of compound 10a were collected on a VENTURE Bruker diffractometer using Cu Kα (λ = 1.54178 Å). A colorless arrowhead single crystal was mounted on a MiTeGen support and cooled to 100(2) K with open-flow nitrogen gas. Data were collected using ω and φ scans with a narrow frames strategy. Diffracted intensities were integrated and corrected from absorption effects with SAINT42 and SADABS43 programs, included in the APEX4 package.44 The crystal structure was solved and refined using SHELXS45 and SHELXL46 included in the Olex2 program.47 Hydrogen atoms were observed in difference Fourier maps; however, to increase the data/parameter ratio, most of them were included in the model in calculated positions and refined with a riding model. Hydrogen atoms bound to stereogenic centers were included in the model in the observed positions and freely refined. The absolute configuration was determined based on previously known internal references, and this assignment was confirmed using the Flack parameter.48
Crystal Data of 10a
C23H31NO14; Mr = 545.49; colorless arrowhead 0.025 × 0.10 × 0.11 mm3; monoclinic P21; a = 8.3147(3) Å, b = 7.0757(2) Å; c = 22.2102(6) Å, β = 92.107(2)°; V = 1305.79(7) Å3; Z = 2; Dc = 1.387 g/cm3; μ = 0.998 mm–1; min and max. absorption correction factors: 0.5584 and 0.7538; 2θmax = 149.268°; 46291 reflections measured, 5348 unique; Rint = 0.0.0539; number of data/restraint/parameters: 5348/1/374; R1 = 0.0330 [5084 reflections, I > 2σ(I)], wR(F2) = 0.0871 (all data); largest difference peak: 0.29 e Å–3; Flack parameter: −0.14(10).49
Acknowledgments
We thank the Agencia Estatal de Investigación of Spain (AEI; PID2021-127622OB-I00 and PDC2022-133725-C21 projects), the Asociación Española Contra el Cáncer (AECC, INNOVA project 2023), Universidad de La Rioja (REGI22/47 and REGI22/16 projects), and the EU (Marie-Sklodowska Curie ITN, DIRNANO, grant agreement No. 956544). C.B. and M.T. thank (AECC La Rioja) for predoctoral fellowships.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.4c07412.
Experimental procedures, characterization data, and copies of the NMR spectra (PDF)
FAIR Data is available as Supporting Information for Publication and includes the primary NMR FID files for compounds: [4C, 40, 4O-OBn, 4S, 4S-tBu, 8C, 8O, 8S, 10a, 10b, 10b-AA, 10c, 10d, 12a, 12b, 12c, 12c–OH, 12d, 12e, 14c, 14d]. See FID for Publication for additional information.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- a Varki A. Biological Roles of Oligosaccharides: All of the Theories are Correct. Glycobiology 1993, 3, 97. 10.1093/glycob/3.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Dwek R. A. Glycobiology: Toward Understanding the Function of Sugars. Chem. Rev. 1996, 96, 683. 10.1021/cr940283b. [DOI] [PubMed] [Google Scholar]; c Bertozzi C. R.; Keissling A. L. L. Chemical Glycobiology. Science 2001, 291, 2357. 10.1126/science.1059820. [DOI] [PubMed] [Google Scholar]; d Davis B. G. Synthesis of Glycoproteins. Chem. Rev. 2002, 102, 579. 10.1021/cr0004310. [DOI] [PubMed] [Google Scholar]; e OhtSubo K.; Marth J. D. Glycosylation in Cellular Mechanisms of Health and Disease. Cell 2006, 126, 855. 10.1016/j.cell.2006.08.019. [DOI] [PubMed] [Google Scholar]; f Kiessling L. L.; Splain R. A. Chemical approaches to glycobiology. Annu. Rev. Biochem. 2010, 79, 619. 10.1146/annurev.biochem.77.070606.100917. [DOI] [PubMed] [Google Scholar]
- Gupta A.; Gupta G. S. Applications of Mannose-binding Lectins and Mannan Glycoconjugates in Nanomedicine. J. Nanopart. Res. 2022, 24, 228. 10.1007/s11051-022-05594-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Helenius A.; Aebi A. M. Intracellular Functions of N-Linked Glycans. Science 2001, 291, 2364. 10.1126/science.291.5512.2364. [DOI] [PubMed] [Google Scholar]; b Schjoldager K. T.; Narimatsu Y.; Joshi H. J.; Clausen H. Global View of Human Protein Glycosylation Pathways and Functions. Nat. Rev. Mol. Cell. Biol. 2020, 21, 729. 10.1038/s41580-020-00294-x. [DOI] [PubMed] [Google Scholar]; c Ma J.; Wu C.; Hart G. W. Analytical and Biochemical Perspectives of Protein O-GlcNAcylation. Chem. Rev. 2021, 121, 1513. 10.1021/acs.chemrev.0c00884. [DOI] [PubMed] [Google Scholar]
- a Ju T.; Wang Y.; Aryal R. P.; Lehoux S. D.; Ding X.; Kudelka M. R.; Cutler C.; Zeng J.; Wang J.; Sun X.; Heimburg-Molinaro J.; Smith D. F.; Cummings R. D. Tn and Sialyl-Tn Antigens, Aberrant O-glycomics as Human Disease Markers Proteomics Clin. Appl. 2013, 7, 618. 10.1002/prca.201300024. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ju T.; Aryal R. P.; Kudelka M. R.; Wang Y.; Cummings R. D. The Cosmc Connection to the Tn Antigen in Cancer. Cancer Biomark. 2014, 14, 63. 10.3233/CBM-130375. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Kudelka M. R.; Ju T.; Heimburg-Molinaro J.; Cummings R. D. Simple Sugars to Complex Disease—mucin-type O-Glycans in Cancer. Adv. Cancer Res. 2015, 126, 53. 10.1016/bs.acr.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Stowell S. R.; Ju T.; Cummings R. D. Protein Glycosylation in Cancer. Annu. Rev. Pathol. 2015, 10, 473. 10.1146/annurev-pathol-012414-040438. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Chia J.; Goh G.; Bard F. Short O-GalNAc Glycans: Regulation and Role in Tumor Development and Clinical Perspectives. Biochim. Biophys. Acta 2016, 1860, 1623. 10.1016/j.bbagen.2016.03.008. [DOI] [PubMed] [Google Scholar]; f Matsumoto Y.; Ju T. Aberrant Glycosylation as Immune Therapeutic Targets for Solid Tumors. Cancers (Basel) 2023, 15, 3536. 10.3390/cancers15143536. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Pinto D.; Parameswaran R. Role of Truncated O-GalNAc Glycans in Cancer Progression and Metastasis in Endocrine Cancers. Cancers (Basel) 2023, 15, 3266. 10.3390/cancers15133266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Martínez-Sáez N.; Supekar N. T.; Wolfert M. A.; Bermejo I. A.; Hurtado-Guerrero R.; Asensio J. L.; Jiménez-Barbero J.; Busto J. H.; Avenoza A.; Boons G.-J.; Peregrina J. M.; Corzana F. Mucin Architecture Behind the Immune Response: Design, Evaluation and Conformational Analysis of an Antitumor Vaccine Derived from an Unnatural MUC1 Fragment. Chem. Sci. 2016, 7, 2294. 10.1039/C5SC04039F. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bermejo I. A.; Navo C. D.; Castro-López J.; Guerreiro A.; Jiménez-Moreno E.; Sánchez Fernández E. M.; García-Martín F.; Hinou H.; Nishimura S.-I.; García Fernández J. M.; Ortiz Mellet C.; Avenoza A.; Busto J. H.; Bernardes G. J. L.; Hurtado-Guerrero R.; Peregrina J. M.; Corzana F. Synthesis, Conformational Analysis and In Vivo Assays of an Anti-cancer Vaccine that Features an Unnatural Antigen based on an sp2-Iminosugar Fragment. Chem. Sci. 2020, 11, 3996. 10.1039/c9sc06334j. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Martínez-Sáez N.; Peregrina J. M.; Corzana F. Principles of Mucin Structure: Implications for the Rational Design of Cancer Vaccines Derived from MUC1-Glycopeptides. Chem. Soc. Rev. 2017, 46, 7154. 10.1039/C6CS00858E. [DOI] [PubMed] [Google Scholar]; d Bermejo I. A.; Guerreiro A.; Eguskiza A.; Martínez-Sáez N.; Lazaris F. S.; Asín A.; Somovilla V. J.; Compañón I.; Raju T. K.; Tadic S.; Garrido P.; García-Sanmartín J.; Mangini V.; Grosso A. S.; Marcelo F.; Avenoza A.; Busto J. H.; García-Martín F.; Hurtado-Guerrero R.; Peregrina J. M.; Bernardes G. J. L.; Martínez A.; Fiammengo R.; Corzana F. Structure-Guided Approach for the Development of MUC1-Glycopeptide-Based Cancer Vaccines with Predictable Responses. JACS Au 2024, 4, 150. 10.1021/jacsau.3c00587. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Somovilla V. J.; Bermejo I. A.; Albuquerque I. S.; Martínez-Sáez N.; Castro-López J.; García-Martín F.; Compañón I.; Hinou H.; Nishimura S.-I.; Jiménez-Barbero J.; Asensio J. L.; Avenoza A.; Busto J. H.; Hurtado-Guerrero R.; Peregrina J. M.; Bernardes G. J. L.; Corzana F. The Use of Fluoroproline in MUC1 Antigen Enables Efficient Detection of Antibodies in Patients with Prostate Cancer. J. Am. Chem. Soc. 2017, 139, 18255. 10.1021/jacs.7b09447. [DOI] [PubMed] [Google Scholar]; f Yin L.; Zhou Y.; Hong S.; Ding F.; Cai H. Strategies for Synthesizing and Enhancing the Immune Response of Cancer Vaccines Based on MUC1 Glycopeptide Antigens. ChemBioChem 2023, 24, e202200805 10.1002/cbic.202200805. [DOI] [PubMed] [Google Scholar]
- a Das R.; Mukhopadhyay B. Chemical O-Glycosylations: An Overview. ChemistryOpen 2016, 5, 401. 10.1002/open.201600043. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Nielsen M. M.; Pedersen C. M. Catalytic Glycosylations in Oligosaccharide Synthesis. Chem. Rev. 2018, 118, 8285. 10.1021/acs.chemrev.8b00144. [DOI] [PubMed] [Google Scholar]; and references cited therein.
- Shaik A. A.; Nishat S.; Andreana P. R. Stereoselective Synthesis of Natural and Non-natural Thomsen-nouveau Antigens and Hydrazide Derivatives. Org. Lett. 2015, 17, 2582. 10.1021/acs.orglett.5b00512. [DOI] [PubMed] [Google Scholar]
- a Mulani S. K.; Hung W. C.; Ingle A. B.; Shiau K. S.; Mong K.-K. Modulating Glycosylation with Exogenous Nucleophiles: an Overview. Org. Biomol. Chem. 2014, 12, 1184. 10.1039/c3ob42129e. [DOI] [PubMed] [Google Scholar]; b Nicolaou K. C.; Mitchell H. J. Adventures in Carbohydrate Chemistry: New Synthetic Technologies, Chemical Synthesis, Molecular Design, and Chemical Biology. Angew. Chem., Int. Ed. 2001, 40, 1576.. [DOI] [PubMed] [Google Scholar]
- Crawford C. J.; Seeberger P. H. Advances in Glycoside and Oligosaccharide Synthesis. Chem. Soc. Rev. 2023, 52, 7773. 10.1039/D3CS00321C. [DOI] [PubMed] [Google Scholar]
- Kessler H.; Wittmann V.; Köck M.; Kottenhahn M. Synthesis of C-Glycopeptides via Free Radical Addition of Glycosyl Bromides to Dehydroalanine Derivatives. Angew. Chem., Int. Ed. 1992, 31, 902. 10.1002/anie.199209021. [DOI] [Google Scholar]
- Crespi S.; Fagnoni M. Generation of Alkyl Radicals: From the Tyranny of Tin to the Photon Democracy. Chem. Rev. 2020, 120, 9790. 10.1021/acs.chemrev.0c00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang W.; Zhu C.; Peng F.; Pan Z.; Ding Y.; Xia C. Nitrogen-Centered Radical-Mediated Cascade Amidoglycosylation of Glycals. Org. Lett. 2021, 23, 1222. 10.1021/acs.orglett.0c04178. [DOI] [PubMed] [Google Scholar]
- a Shaw M. H.; Twilton J.; MacMillan D. W. C. Photoredox Catalysis in Organic Chemistry. J. Org. Chem. 2016, 81, 6898. 10.1021/acs.joc.6b01449. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Cannalire R.; Pelliccia S.; Sancineto L.; Novellino E.; Tron G. C.; Giustiniano M. Visible Light Photocatalysis in the Late-Stage Functionalization of Pharmaceutically Relevant Compounds. Chem. Soc. Rev. 2021, 50, 766. 10.1039/D0CS00493F. [DOI] [PubMed] [Google Scholar]; and references cited therein.
- a Sangwan R.; Mandal P. K. Recent Advances in Photoinduced Glycosylation: Oligosaccharides, Glycoconjugates and their Synthetic Applications. RSC Adv. 2017, 7, 26256. 10.1039/C7RA01858D. [DOI] [Google Scholar]; b Xu L.-Y.; Fan N.-L.; Hu X.-G. Recent Development in the Synthesis of C-Glycosides Involving Glycosyl Radicals. Org. Biomol. Chem. 2020, 18, 5095. 10.1039/D0OB00711K. [DOI] [PubMed] [Google Scholar]; c Ghouilem J.; De Robichon M.; Le Bideau F.; Ferry A.; Messaoudi S. Emerging Organometallic Methods for the Synthesis of C-Branched (Hetero)aryl, Alkenyl, and Alkyl Glycosides: C-H Functionalization and Dual Photoredox Approaches. Chem. - Eur. J. 2021, 27, 491. 10.1002/chem.202003267. [DOI] [PubMed] [Google Scholar]; d Yang Y.; Yu B. Recent Advances in the Chemical Synthesis of C-Glycosides. Chem. Rev. 2017, 117, 12281. 10.1021/acs.chemrev.7b00234. [DOI] [PubMed] [Google Scholar]; e Gorelik D. J.; Desai S. P.; Jdanova S.; Turner J. A.; Taylor M. S. Transformations of Carbohydrate Derivatives Enabled by Photocatalysis and Visible Light Photochemistry. Chem. Sci. 2024, 15, 1204. 10.1039/D3SC05400D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ischay M. A.; Anzovino M. E.; Du J.; Yoon T. P. Efficient Visible Light Photocatalysis of [2 + 2] Enone Cycloadditions. J. Am. Chem. Soc. 2008, 130, 12886. 10.1021/ja805387f. [DOI] [PubMed] [Google Scholar]
- Mao R.-Z.; Guo F.; Xiong D.-C.; Li Q.; Duan J.; Ye X.-S. Photoinduced C-S Bond Cleavage of Thioglycosides and Glycosylation. Org. Lett. 2015, 17, 5606. 10.1021/acs.orglett.5b02823. [DOI] [PubMed] [Google Scholar]
- Zhao G.; Wang T. Stereoselective Synthesis of 2-Deoxyglycosides from Glycals by Visible-Light-Induced Photoacid Catalysis. Angew. Chem., Int. Ed. 2018, 57, 6120. 10.1002/anie.201800909. [DOI] [PubMed] [Google Scholar]
- Liu K.-M.; Wang P.-Y.; Guo Z.-Y.; Xiong D.-C.; Qin X.-J.; Liu M.; Liu M.; Xue W.-Y.; Ye X.-S. Iterative Synthesis of 2-Deoxyoligosaccharides Enabled by Stereoselective Visible-Light-Promoted Glycosylation. Angew. Chem., Int. Ed. 2022, 61, e202114726 10.1002/anie.202114726. [DOI] [PubMed] [Google Scholar]
- Ji P.; Zhang Y.; Gao F.; Bi F.; Wang W. Direct, Stereoselective Thioglycosylation Enabled by an Organophotoredox Radical Strategy. Chem. Sci. 2020, 11, 13079. 10.1039/D0SC04136J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Wan L.-Q.; Zhang X.; Zou Y.; Shi R.; Cao J.-G.; Xu S.-Y.; Deng L.-F.; Zhou L.; Gong Y.; Shu X.; Lee G. Y.; Ren H.; Dai L.; Qi S.; Houk K. N.; Niu D. Nonenzymatic Stereoselective S-Glycosylation of Polypeptides and Proteins. J. Am. Chem. Soc. 2021, 143, 11919. 10.1021/jacs.1c05156. [DOI] [PubMed] [Google Scholar]; b Dowman L. J.; Kulkarni S. S.; Alegre-Requena J. V.; Giltrap A. M.; Norman A. R.; Sharma A.; Gallegos L. C.; Mackay A. S.; Welegedara A. P.; Watson E. E.; van Raad D.; Niederacher G.; Huhmann S.; Proschogo N.; Patel K.; Larance M.; Becker C. F. W.; Mackay J. P.; Lakhwani G.; Huber T.; Paton R. S.; Payne R. J. Site-Selective Photocatalytic Functionalization of Peptides and Proteins at Selenocysteine. Nat. Commun. 2022, 13, 6885. 10.1038/s41467-022-34530-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Constantin T.; Zanini M.; Regni A.; Sheikh N. S.; Juliá F.; Leonori D. Aminoalkyl Radicals as Halogen-Atom Transfer Agents for Activation of Alkyl and Aryl Halides. Science 2020, 367, 1021. 10.1126/science.aba2419. [DOI] [PubMed] [Google Scholar]; b Wang Q.; Lee B. C.; Tan T. J.; Jiang Y.; Ser W. H.; Koh M. J. Visible Light Activation Enables Desulfonylative Cross-Coupling of Glycosyl Sulfones. Nat. Synth. 2022, 1, 967. 10.1038/s44160-022-00162-w. [DOI] [Google Scholar]; c Sun T.; Jin R.; Yang Y.; Jia Y.; Hu S.; Jin Y.; Wang Q.; Li Z.; Zhang Y.; Wu J.; Jiang Y.; Lv X.; Liu S. Direct α-C-H Alkylation of Structurally Diverse Alcohols via Combined Tavaborole and Photoredox Catalysis. Org. Lett. 2022, 24, 7637. 10.1021/acs.orglett.2c03117. [DOI] [PubMed] [Google Scholar]; d Ji P.; Zhang Y.; Dong Y.; Huang H.; Wei Y.; Wang W. Synthesis of Enantioenriched α-Deuterated α-Amino Acids Enabled by an Organophotocatalytic Radical Approach. Org. Lett. 2020, 22, 1557. 10.1021/acs.orglett.0c00154. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Ji P.; Chen J.; Meng X.; Gao F.; Dong Y.; Xu H.; Wang W. Design of Photoredox-Catalyzed Giese-Type Reaction for the Synthesis of Chiral Quaternary α-Aryl Amino Acid Derivatives via Clayden Rearrangement. J. Org. Chem. 2022, 87, 14706. 10.1021/acs.joc.2c02029. [DOI] [PubMed] [Google Scholar]
- a Ji P.; Zhang Y.; Wei Y.; Huang H.; Hu W.; Mariano P. A.; Wang W. Visible-Light-Mediated, Chemo- and Stereoselective Radical Process for the Synthesis of C-Glycoamino Acids. Org. Lett. 2019, 21, 3086. 10.1021/acs.orglett.9b00724. [DOI] [PubMed] [Google Scholar]; b Wan I. C. S.; Witte M. D.; Minnaard A. J. From d- to l-Monosaccharide Derivatives via Photodecarboxylation-Alkylation. Org. Lett. 2019, 21, 7669. 10.1021/acs.orglett.9b03016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi R.; Wang C.; Ma Z.; Wang H.; Chen Q.; Liu L.; Pan D.; Ren X.; Wang R.; Xu Z. Visible-Light-Promoted Stereoselective C(sp3)-H Glycosylation for the Synthesis of C-Glycoamino Acids and C-Glycopeptides. Angew. Chem., Int. Ed. 2022, 61, e202200822 10.1002/anie.202200822. [DOI] [PubMed] [Google Scholar]
- Wei Y.; Wang Q.; Koh M. J. A Photoinduced, Nickel-Catalyzed Reaction for the Stereoselective Assembly of C-Linked Glycosides and Glycopeptides. Angew. Chem., Int. Ed. 2022, 62, e202214247 10.1002/anie.202214247. [DOI] [PubMed] [Google Scholar]
- Mao R.; Xi S.; Shah S.; Roy M. J.; John A.; Lingford J. P.; Gäde G.; Scott N. E.; Goddard-Borger E. D. Synthesis of C-Mannosylated Glycopeptides Enabled by Ni-Catalyzed Photoreductive Cross-Coupling Reactions. J. Am. Chem. Soc. 2021, 143, 12699. 10.1021/jacs.1c05567. [DOI] [PubMed] [Google Scholar]
- Oroz P.; Navo C. D.; Avenoza A.; Busto J. H.; Corzana F.; Jiménez-Osés G.; Peregrina J. M. Towards Enantiomerically Pure Unnatural α-Amino Acids via Photoredox Catalytic 1,4-Additions to a Chiral Dehydroalanine. J. Org. Chem. 2022, 87, 14308. 10.1021/acs.joc.2c01774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanessian S.; Qiu D.; Prabhanjan H.; Reddy G. V.; Lou B. Synthesis of Clustered D-GalNAc (Tn) and D-Galβ(1→3)GalNAc (T) Antigenic Motifs Using a Pentaerythritol Scaffold. Can. J. Chem. 1996, 74, 1738. 10.1139/v96-192. [DOI] [Google Scholar]
- Babic A.; Gobec S.; Gravier-Pelletier C.; Le Merrer Y.; Pecar S. Synthesis of 1-C-Linked Diphosphate Analogues of UDP-N-Ac-Glucosamine and UDP-N-Ac-Muramic Acid. Tetrahedron 2008, 64, 9093. 10.1016/j.tet.2008.07.009. [DOI] [Google Scholar]
- a Fuchss T.; Schmidt R. R. Synthesis of the C-analog of 2-acetylamino-2-deoxy-β-D-glucopyranosyl-L- and -D-serine. Synthesis 1998, 5, 753. 10.1055/s-1998-4490. [DOI] [Google Scholar]; Vichier-Guerre S.; Lo-Man R.; Huteau V.; Dériaud E.; Leclerc C.; Bay S. Synthesis and immunological evaluation of an antitumor neoglycopeptide vaccine bearing a novel homoserine Tn antigen. Bioorg. Med. Chem. Lett. 2004, 14, 3567. 10.1016/j.bmcl.2004.04.047. [DOI] [PubMed] [Google Scholar]; c Zhu X.; Schmidt R. R. Efficient Synthesis of S-Linked Glycopeptides in Aqueous Solution by a Convergent Strategy. Chem. - Eur. J. 2004, 10, 875. 10.1002/chem.200305163. [DOI] [PubMed] [Google Scholar]
- a Paredes M. D.; Alonso R. J. Org. Chem. 2000, 65, 2292. 10.1021/jo9912855. [DOI] [PubMed] [Google Scholar]; b Le Vaillant F.; Garreau M.; Nicolai S.; Gryn’ova G.; Corminboeuf C.; Waser J. Fine-Tuned Organic Photoredox Catalysts for Fragmentation-Alkynylation Cascades of Cyclic Oxime Ethers. Chem. Sci. 2018, 9, 5883. 10.1039/C8SC01818A. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Le C.; Chen T. Q.; Liang T.; Zhang P.; Macmillan D. W. C. A Radical Approach to the Copper Oxidative Addition Problem: Trifluoromethylation of Bromoarenes. Science 2018, 360, 1010. 10.1126/science.aat4133. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Giese B.; Dupuis J. Anomeric Effect of Radicals. Tetrahedron Lett. 1984, 25, 1349. 10.1016/S0040-4039(01)80154-4. [DOI] [Google Scholar]
- Spell M.; Wang X.; Wahba A. E.; Conner E.; Ragains J. An α-Selective, Visible Light Photocatalytic Glycosylation of Alcohols with Selenoglycosides. Carbohydr. Res. 2013, 369, 42. 10.1016/j.carres.2013.01.004. [DOI] [PubMed] [Google Scholar]
- a Grant L.; Liu Y.; Walsh K. E.; Walter D. S.; Gallagher T. Galacto, Gluco, Manno, and Disaccharide-Based C-Glycosides of 2-Amino-2-Deoxy Sugars. Org. Lett. 2002, 4, 4623. 10.1021/ol0269695. [DOI] [PubMed] [Google Scholar]; b Galashov A.; Kazakova E.; Stieger C. E.; Hackenberger C. P. R.; Seitz O. Rapid Building Block-Economic Synthesis of Long, Multi-O-GalNAcylated MUC5AC Tandem Repeat Peptides. Chem. Sci. 2024, 15, 1297. 10.1039/D3SC05006H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Wu X.; McFall-Boegeman H.; Rashidijahanabad Z.; Liu K.; Pett C.; Yu J.; Schorlemer M.; Ramadan S.; Behren S.; Westerlind U.; Huang X. Synthesis and Immunological Evaluation of the Unnatural β-Linked Mucin-1 Thomsen-Friedenreich Conjugate. Org. Biomol. Chem. 2021, 19, 2448. 10.1039/D1OB00007A. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Campo V. L.; Riul T. B.; Bortot L. O.; Martins-Teixeira M. B.; Marchiori M. F.; Iaccarino E.; Ruvo M.; Dias-Baruffi M.; Carvalho I. A Synthetic MUC1 Glycopeptide Bearing βGalNAc-Thr as a Tn Antigen Isomer Induces the Production of Antibodies Against Tumor Cells. ChemBioChem 2017, 18, 527. 10.1002/cbic.201600473. [DOI] [PubMed] [Google Scholar]
- Corzana F.; Busto J. H.; Jiménez-Osés G.; García De Luis M.; Asensio J. L.; Jiménez-Barbero J.; Peregrina J. M.; Avenoza A. Serine versus Threonine Glycosylation: the Methyl Group Causes a Drastic Alteration on the Carbohydrate Orientation and on the Surrounding Water Shell. J. Am. Chem. Soc. 2007, 129, 9458. 10.1021/ja072181b. [DOI] [PubMed] [Google Scholar]
- Czernecki S.; Ayadi E.; Randriamandimby D. Seleno glycosides. 2. Synthesis of Phenyl 2-(N-Acetylamino)- and 2-Azido-2-deoxy-1-seleno-α-D-glycopyranosides via Azido-phenylselenylation of Diversely Protected Glycals. J. Org. Chem. 1994, 59, 8256. 10.1021/jo00105a051. [DOI] [Google Scholar]
- Tsuchii K.; Doi M.; Hirao T.; Ogawa A. Highly Selective Sequential Addition and Cyclization Reactions Involving Diphenyl Diselenide, an Alkyne, and Alkenes under Visible-Light Irradiation. Angew. Chem., Int. Ed. 2003, 42, 3490. 10.1002/anie.200250790. [DOI] [PubMed] [Google Scholar]
- Tingoli M.; Tiecco M.; Testaferri L.; Temperini A. Substituted Azides from Selenium-Promoted Deselenenylation of Azido Selenides. Glycosylation Reactions of Protected 2-Azido-2-deoxy-1-selenoglycopyranoses. J. Chem. Soc., Chem. Commun. 1994, 1883. 10.1039/c39940001883. [DOI] [Google Scholar]
- Kumar S.; Shah T. A.; Punniyamurthy T. Recent Advances in the Application of Tetrabromomethane in Organic Synthesis. Org. Chem. Front. 2021, 8, 4288. 10.1039/D0QO01369B. [DOI] [Google Scholar]
- a Heredia A. A.; Bouchet L. M.; Castro-Godoy W. D.; Argüello J. E. Synthesis of Organoselenium Compounds Using Electrochemical and Photochemical Methods as Novel Approaches in Organic Chemistry. Tetrahedron 2023, 148, 133667. 10.1016/j.tet.2023.133667. [DOI] [Google Scholar]; b Shaaban S.; Ba-Ghazal H.; Al-Faiyz Y. S.; Al-Karmalawy A. A.; Amri N.; Youssef I. Recent Advances in the Synthesis of Organoselenium Heterocycle Conjugates. Tetrahedron 2024, 157, 133957 10.1016/j.tet.2024.133957. [DOI] [Google Scholar]
- Patehebieke Y. An Overview on Disulfide-Catalyzed and-Cocatalyzed Photoreactions. Beilstein J. Org. Chem. 2020, 16, 1418. 10.3762/bjoc.16.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söderman S. C.; Schwan A. L. 1, 2-Dibromotetrachloroethane: An Ozone-Friendly Reagent for the in situ Ramberg-Bäcklund Rearrangement and its Use in the Formal Synthesis of E-Resveratrol. J. Org. Chem. 2012, 77, 10978. 10.1021/jo3021852. [DOI] [PubMed] [Google Scholar]
- SAINT+, v6.01: Area-Detector Integration software; Bruker AXS: Madison, WI, 2001. [Google Scholar]
- a SADABS, v2016/2 Area Detector Absorption Program; Bruker AXS: Madison, WI, 1996. [Google Scholar]; b Krause L.; Herbst-Irmer R.; Sheldrick G. M.; Stalke D. Comparison of Silver and Molybdenum Microfocus X-Ray Sources for Single-Crystal Structure Determination. J. Appl. Crystallogr. 2015, 48, 3. 10.1107/S1600576714022985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- APEX 4 version 2021.10–0; Bruker AXS: Madison, WI, 2021. [Google Scholar]
- Sheldrick G. M. A Short History of SHELX. Acta Crystallogr. 2008, A64, 112. 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- Sheldrick G. M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. 2015, C71, 3. 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolomanov O. V.; Bourhis L. J.; Gildea R. J.; Howard J. A. K.; Puschmann H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339. 10.1107/S0021889808042726. [DOI] [Google Scholar]
- Flack H. D. On enantiomorph-polarity estimation. Acta Crystallogr. 1983, A39, 876. 10.1107/S0108767383001762. [DOI] [Google Scholar]
- Deposition number CCDC 2328241 for 10a contains the supplementary crystallographic data for this paper. These data are provided free of charge by the Cambridge Crystallographic Data Centre Access Structures service.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.