

Abstract
Background
A common genetic variant at the glutamate-ammonia ligase (GLUL) locus has been previously associated with an increased risk of coronary artery disease (CAD) as well as alterations of glutamic acid metabolism and the γ-glutamyl cycle in individuals with type 2 diabetes (T2D). Here we investigated whether less frequent variants in GLUL and 15 additional genes in these pathways are associated with differences in CAD risk in T2D.
Methods
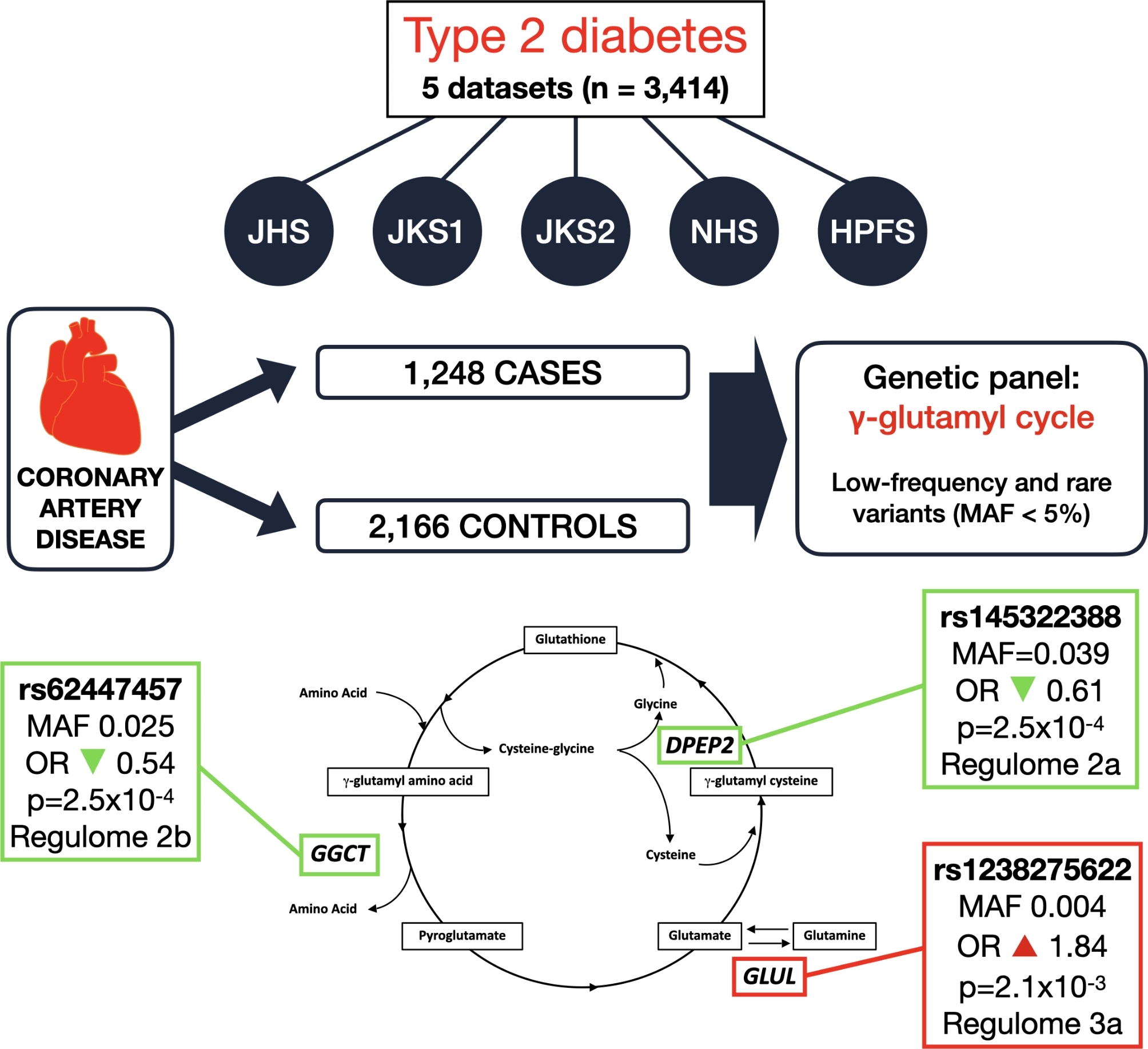
Coding sequences and regulatory elements of these genes were sequenced in 2,394 individuals with T2D from three CAD case/control sets.
Results
Ninety-six variants with minor allele frequency [MAF]< 0.05 were identified as being nominally associated with CAD status. One of these variants (rs62447457, MAF 0.025), placed in a non-coding region flanking the γ-glutamylcyclotransferase (GGCT) gene, showed nominal evidence of replication in two other cases-control sets (n = 1,132), with summary OR of 0.54 (p = 2.5 × 10–4). Another variant (rs145322388, MAF = 0.039), flanking the dipeptidase 2 (DPEP2) gene, showed association with CAD status across discovery and replications sets (summary OR 0.61, p = 2.5 × 10–4). A third variant (rs1238275622, MAF 0.004), flanking the GLUL gene, was associated with increased risk of CAD (summary OR 1.84, p-value 2.1 × 10–3). Based on their Regulome scores (2b, 2a, and 3a, respectively), all three variants are very likely to have regulatory functions.
Conclusions
In summary, we have identified low-frequency variants associated with CAD in T2D at two loci involved in glutamic acid metabolism and the γ-glutamyl cycle. These findings provide further evidence for a role of these pathways in the link between T2D and CAD.
Graphical Abstract
Supplementary Information
The online version contains supplementary material available at 10.1186/s12933-024-02442-5.
Keywords: Diabetes, Coronary artery disease, Genetics, Gamma-glutamyl cycle
Background
Individuals with type 2 diabetes (T2D) are at increased risk of coronary artery disease (CAD), which can be present in about 20% of this population [1]. In recent years, a global declining trend has been observed in the prevalence [2, 3] of CAD and associated mortality [4]. This decline has been more pronounced in individuals with T2D than in the population without diabetes, but the former are still at a higher risk of CAD. While better control of glucose levels and associated risk factors such as dyslipidemia and hypertension [4] have contributed to this declining trend, additional interventions are needed to fully neutralize the adverse cardiovascular effects of T2D whenever its onset cannot be prevented. In order to develop such new treatments, a better understanding of the causal pathways through which diabetes increases cardiovascular risk is required.
Several mechanisms have been proposed as underlying the atherogenic effects of diabetes: lipid alterations specific to the hyperglycemic state, inflammation, oxidative stress, and the effect of hyperglycemia on advanced glycation end product production [5, 6]. Structural heart alterations are also possible explanations of the excess mortality seen in T2D [7]. Since the described mechanisms do not fully explain the increased CAD risk seen in T2D, one approach that has been pursued to expand knowledge in this field is through genetic studies. While over 350 loci have been found to be associated with coronary artery disease (CAD) in the general population [8–16], a distinctive goal has been to look for genetic loci that are associated with CAD specifically among diabetic subjects, since these are the genes that can tell us the most about the unique mechanisms linking diabetes to cardiovascular disease [17].
Following this approach, through a genome-wide association study (GWAS), we identified a common genetic variant (rs10911021) that is associated with an increased risk of CAD exclusively among individuals with diabetes [18]—a finding that has been independently replicated [19]. The variant is adjacent to the GLUL gene, which codes for the enzyme glutamate-ammonia ligase catalyzing the conversion of glutamic acid and ammonia into glutamine. Two lines of evidence point to a possible mechanism for the association between rs10911021 and CAD. First, this common variant was found to be associated with lower GLUL expression and lower pyroglutamic-to-glutamic acid ratio in plasma of individuals with T2D in the GWAS [18]. Second, human umbilical vein endothelial cells (HUVEC) that are homozygous for the risk allele of this variant were found to have decreased intracellular glutathione-to-glutamate ratio and increased levels of the atherogenic compound methylglyoxal (MG), which is normally detoxified by glutathione through the glyoxylase system [20]. Since pyroglutamic acid is an intermediate of the γ-glutamyl cycle and this pathway is responsible for generating the natural antioxidant glutathione (Fig. 1) [21], these two lines of evidence, taken together, suggest that an impairment of the γ-glutamyl cycle may underlie the association between variant rs10911021 and increased CAD risk. If this hypothesis is true, genetic variants at other loci involved in the γ-glutamyl cycle might also influence CAD risk.
Fig. 1.

Schematic representation of the γ-glutamyl cycle and genes coding key enzymes in this metabolic pathway
In the present study, we aimed to determine the extent to which variability in the genes involved in glutamate metabolism and the γ-glutamyl cycle modulate CAD risk in diabetes, reasoning that identification of other variants at these loci would provide further proof of the importance of these pathways in the pathogenesis of CAD in diabetes. We specifically focused our study on low-frequency and rare variants since these may contribute to the etiology of complex traits [22, 23], but are not adequately captured by the GWAS arrays that were used in previous studies.
Methods
Study subjects
The coding regions of GLUL and 15 other genes shown in Fig. 1, along with their flanking regulatory regions, were sequenced in subjects with available DNA from three discovery sets of CAD cases and controls, all with type 2 diabetes (T2D). One set was from the Joslin Heart Study (JHS), the other two from the Joslin Kidney Study (JKS1 and JKS2). A detailed description of these sets can be found in published reports [18, 24, 25]. Briefly, the JHS is a series of 1,486 non-Hispanic White CAD cases and controls, all with T2D, recruited at the Joslin Diabetes Center and Beth Israel Deaconess Medical Center. Cases are T2D individuals with angiographic evidence of significant stenosis of the coronary artery; controls are T2D individuals with a negative cardiovascular history and a normal exercise treadmill test; a total of 731 cases and 755 controls were included in the present study. The Joslin Kidney Study (JKS) is a longitudinal observational study of the natural history of declining renal function in T1D and T2D. Set JKS1 consisted of 137 individuals with T2D only who died of cardiovascular causes and 342 subjects who did not; set JKS2 included another 117 subjects with T2D only who experienced fatal or non-fatal CAD events during follow-up along with a random sample of 312 JKS participants who did not experience these events.
Replication of the strongest associations was sought by two methods. First, we utilized data from two ongoing prospective cohort studies in the US (the Nurses’ Health Study [NHS] and Health Professionals Follow-up Study [HPFS]). The NHS enrolled 121,700 registered nurses aged 30–55 years in 1976, and the HPFS enrolled 51,529 male health professionals aged 40–75 in 1986. Biennial follow-up questionnaires were sent to participants to identify newly diagnosed medical conditions, including T2D and CAD. Newly reported cases were confirmed according to protocols described in detail previously [26, 27]. We genotyped two case–control datasets including individuals with confirmed T2D from the NHS (n = 724; 169 cases and 555 controls) and from the HPFS (n = 296; 94 cases and 202 controls) [18]; both are nested case–control studies including non-Hispanic White participants who had T2D and experienced a fatal or non-fatal CAD event (CAD cases) or were free of CAD events (CAD controls) during follow-up. Only individuals with available DNA were included. Second, we browsed association data in the Type 2 Diabetes Knowledge Portal (t2d.hugeamp.org) concerning the “Cardiovascular disease in type 2 diabetes” phenotype derived from up to 10,717 individuals with T2D.
Target enrichment
To fully interrogate functional genetic variation in the regions of the 16 genes described in Fig. 1, we leveraged data made available by the ENCODE Project [28] and performed targeted next-generation sequencing (NGS) of all candidate functional elements at these loci along with each gene’s corresponding coding sequences. The candidate functional elements included all ENCODE-defined chromatin accessibility, histone modification, and transcription factor binding sites identified within 250 Kb up- and downstream of the 16 genes in a variety of cell types relevant to CAD, including umbilical cord endothelial cells, B-lymphocytes, CD4 + monocytes, aortic adventitial fibroblast cells, and aortic smooth muscle cells. These regulatory elements spanned a total of 1.57 Mb. Using these data, we designed a custom Agilent SureSelectXT Target Enrichment System Capture (Agilent, Santa Clara, CA) based on 5X tiling of 120-mer RNA baits across all the regulatory regions, as defined above, and the entire coding sequences of the 16 genes. A complete list of genomic coordinates for all 2,787 regions sequenced is provided in Additional file 1.
Library preparation and sequencing
Genomic DNA (750 ng per sample) from the three discovery sets was sheared on a Covaris S2 focused-ultrasonicator (Covaris, Woburn, MA), aiming at a 150-bp median fragment length. After end-repairing and A-tailing, DNA fragments were ligated to unique 6-bp barcodes, allowing library pooling from 96 individuals, and to Illumina paired-end sequencing adapters according to previously published protocols [29] using the Quick Ligation™ kit (New England Biolabs). Libraries were then amplified with Herculase II Fusion DNA Polymerase (Agilent) and pooled in sets of 96 comprising similar proportions of CAD-positive cases and CAD negative controls. After undergoing target enrichment using the custom Agilent SureSelectXT system described above, libraries were sequenced at the Bauer Core Facility of Harvard University, on an Illumina HiSeq 2500 (Illumina Inc., San Diego, CA) instrument, employing 75-bp long paired-end reads in a High Output (Standard) v4 flow-cell.
Bioinformatics analyses
Fastq files obtained from the sequencing facility were demultiplexed with fastq-multx. Sequenced reads were then aligned to the human genome (hg19) with the Burrows-Wheeler Aligner (bwa-mem) version 0.7.17 [30]. Aligned reads were then sorted, deduplicated, and recalibrated using Genome Analysis Toolkit (GATK) version 4.2.0 [31]. Depth of coverage was assessed by mosdepth version 0.2.3 [32]. GATK’s HaplotypeCaller was used for joint variant calling. Variants were annotated by ANNOVAR [33] and RegulomeDB, version 2.2 for genome assembly hg19 [34]. In brief, ANNOVAR provides variant annotation regarding location, predicted function, and MAF, whereas Regulome assigns a score to each variant denoting the likelihood of it bearing regulatory function. The score is based on the variant seating in one or more of the following: expression quantitative trait locus (eQTL), transcription factor (TF) binding, TF motif, DNAse footprint, and DNAse peak. Additionally, variants were searched for significant cis-eQTLs on one of the 16 corresponding target genes in our panel in the eQTLGen Consortium database, which aggregates data on the expression of genes in whole-blood samples (eqtlgen.org).
Variant genotyping
Variants prioritized for replication were genotyped in the HPFS and NHS cohorts using the TaqMan Open Array Genotyping System along with custom-made oligonucleotide positive controls to allow the accurate genotyping of infrequent alleles as previously done for other projects [35].
Statistical analysis
Since the present analysis was focused on low-frequency and rare variants, variants with minor allele frequency (MAF) ≥ 5% were filtered out. To account for the small number of minor allele carriers, single variant analysis was carried out by means of Fisher’s tests as implemented in PLINK [36] separately for each discovery dataset. Results were then meta-analyzed by Fisher’s method as implemented in Metal [37] and in the meta R package [38]. Summary odds-ratios (ORs) were calculated by the Cochran-Mantel–Haenszel method. Variants with p-values < 0.01 for association with CAD and those with p-values < 0.05 that had a high Regulome score (1 or 2) were further investigated in nested case–control studies from the NHS and HPFS cohorts [35]. Association of each variant with CAD was analyzed by Fisher’s test as described above. Results were then meta-analyzed along with the results from the discovery stage to obtain summary statistics. A p-value smaller than 0.05 corrected for the 22 comparisons that were carried out at this stage (p < 2.27 × 10–3) across discovery and replication datasets was considered as significant evidence of association.
All variants prioritized for replication were also searched for association with the “Cardiovascular disease in type 2 diabetes” phenotype in the datasets available in the Type 2 Diabetes Knowledge Portal (T2DKP; t2d.hugeamp.org). Results were meta-analyzed along with our original data to provide further evidence of replication. Association of the same SNPs with hepatic phenotypes aspartate aminotransferase (AST), alanine aminotransferase (ALT), γ-glutamyl transferase (GGT), and alkaline phosphatase (ALP) was also searched for to provide further evidence of the involvement of the SNPs in the γ-glutamyl cycle.
Power
The number of cases and controls in the discovery sets provided at least 80% power to detect nominally significant (α = 0.05) associations with CAD with OR ranging from 0.4 (MAF = 0.01) to 0.65 (MAF = 0.05) for protective variants and from 1.45 (MAF = 0.05) to 2.2 (MAF = 0.01) for predisposing variants. The combined discovery sets and T2DKP datasets provided > 80% power to detects association with CAD of those magnitudes at an alpha error accounting for the number of variants for which replication was sought (α = 0.05/22 = 2.27 × 10–3).
Data and resource availability
The data and resources generated or analyzed in this study are available from the corresponding author upon reasonable request.
Human subjects research approval
The study protocols of the JHS, JKS1, and JKS2 were approved by the Joslin Committee on Human Studies (protocol numbers 99-20, 01-38 and 08-31). The study protocols of the NHS and HPFS were approved by the Institutional Review Boards of the Brigham and Women's Hospital and the Harvard T.H. Chan School of Public Health (protocol number 2000P002221).
Results
Study subjects
Clinical characteristics of individuals included in the three discovery stage datasets are shown in Table 1. In the JHS dataset, CAD cases were older, had a higher frequency of hypertension, were more often taking lipid-lowering medications, and were more frequently smokers than controls. They also had lower eGFR and total, LDL, and HDL-cholesterol. In the JKS1 dataset, CAD cases were older and had longer diabetes duration than controls. They were also more frequently hypertensive, had lower eGFR, and were less likely to be taking oral glucose-lowering medications and more likely to be on insulin therapy. In the JKS2 dataset, cases had higher BMI and were less likely to be taking lipid-lowering medications than controls.
Table 1.
Baseline characteristics of discovery cohorts
| JHS | JKS1 | JKS2 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Cases | Controls | p-value* | Cases | Controls | p-value* | Cases | Controls | p-value* | |
| n | 731 | 755 | 137 | 342 | 117 | 312 | |||
| Age (years) | 64.8 (7.1) | 63.9 (5.9) | 0.0071 | 60.6 (7.7) | 56.1 (10.1) | < 0.0001 | 58.3 (5.5) | 58.3 (5.7) | 0.9497 |
| Duration (years) | 12.2 (8.7) | 12.5 (6.3) | 0.5313 | 17.0 (7.8) | 12.7 (7.8) | < 0.0001 | 15.2 (7.9) | 14.6 (8.0) | 0.5170 |
| BMI (kg/m2) | 32.4 (5.9) | 32.4 (5.6) | 0.8530 | 30.2 (6.8) | 29.7 (6.2) | 0.4356 | 33.5 (7.2) | 31.5 (6.9) | 0.0115 |
| eGFR (ml/min) | 70.3 (24.4) | 76.3 (21.7) | < 0.0001 | 77.5 (33.4) | 92.4 (30.0) | < 0.0001 | 72.7 (21.4) | 74.2 (20.3) | 0.5070 |
| Female (%) | 26.2 | 39.2 | < 0.0001 | 39.4 | 46.9 | 0.1546 | 29.1 | 35.9 | 0.2086 |
| Hypertension (%) | 88.5 | 74.8 | < 0.0001 | 72.3 | 56 | 0.0009 | 85.1 | 76.6 | 0.1046 |
| Lipid medication (%) | 84.8 | 72.1 | < 0.0001 | NA | NA | NA | 68.4 | 81.7 | 0.0039 |
| A1c (%) | 7.4 (1.4) | 7.4 (1.3) | 0.6912 | 8.7 (1.6) | 8.4 (1.7) | 0.0593 | 8.2 (1.7) | 8.1 (1.4) | 0.4843 |
| Oral antidiabetics (%) | 66.4 | 68.5 | 0.4053 | 20.4 | 37.2 | 0.0003 | 59 | 52.6 | 0.2765 |
| Insulin (%) | 49.3 | 50.7 | 0.3725 | 77.4 | 51.9 | < 0.0001 | 65 | 66.4 | 0.8194 |
| Smoking (%) | 65.8 | 44.3 | < 0.0001 | NA | NA | NA | 62.7 | 55.7 | 0.2159 |
| Total cholesterol (mg/dL) | 155 (38) | 169 (36) | < 0.0001 | 236 (54) | 227 (46) | 0.1161 | 177 (42) | 174 (39) | 0.5210 |
| HDL (mg/dL) | 40 (12) | 47 (18) | < 0.0001 | NA | NA | NA | 42 (12) | 48 (17) | 0.0010 |
| LDL (mg/dL) | 85 (31) | 94 (33) | < 0.0001 | NA | NA | NA | 97 (37) | 93 (30) | 0.2245 |
| Triglycerides (mg/dL) | 181 (130) | 175 (114) | 0.3387 | NA | NA | NA | 188 (117) | 165 (143) | 0.1194 |
JHS Joslin Heart Study, JKS Joslin Kidney Study, BMI: body mass index, eGFR estimated glomerular filtration rate, NA not available
*p value for comparison between cases and controls, within each dataset
Variants associated with CAD in the discovery stage
The median depth of coverage of the genomic regions sequenced in the discovery stage was 27X (interquartile range 17–34), with 72.5% of regions being covered at ≥ 20X (Supplementary Figure S1). After filtering out variants with less than 10X coverage, 7,102 single nucleotide variants (SNV) with MAF below 5% were identified. Of those, 1,036 (14.6%) were low-frequency (1% ≤ MAF < 5%) and 6,066 (85.4%) were rare (MAF < 1%) (Fig. 2A). Most SNVs were in non-coding regions (51.4% intronic, 23.7% intergenic, 10% intronic in non-coding RNA [ncRNA]), with only 5.4% placed in exons (Fig. 2B). Available Regulome scores for all but one [7,101] of the SNVs are shown in Fig. 2C, with 1,012 (14.3%) showing scores ≤ 2, which indicate a high likelihood of being functional.
Fig. 2.
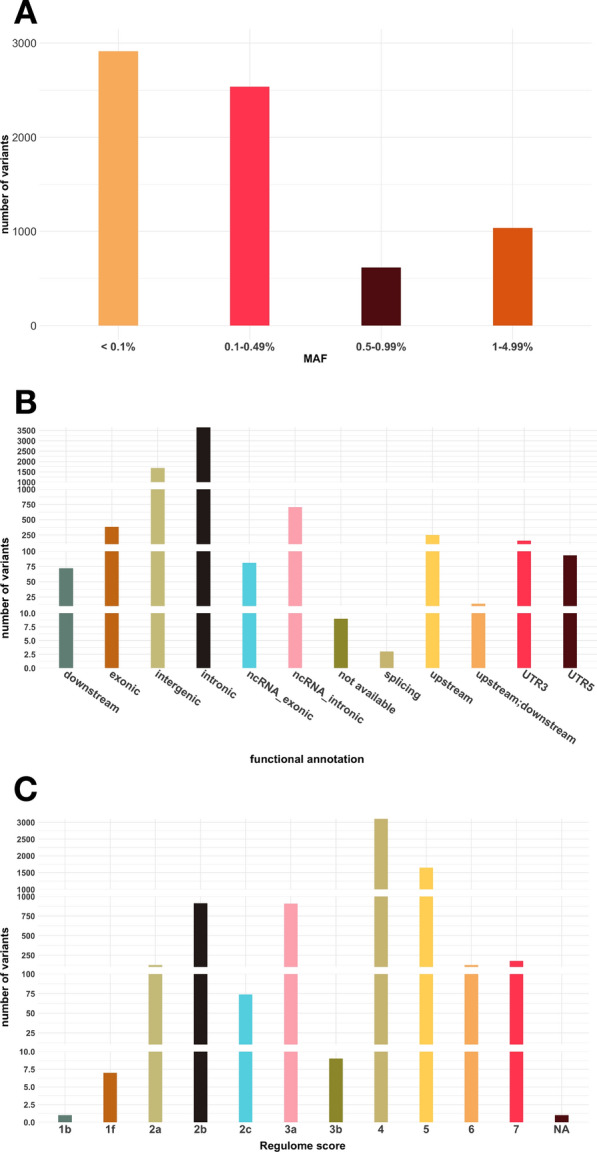
Descriptive statistics of the 7,102 variants found in the discovery stage. A. Distribution according to minor allele frequency (MAF); B: Distribution according to functional annotation (using ANNOVAR); C. Distribution according to Regulome Score. NA: not available
Plots of each genomic region, showing all variants detected in the discovery stage with genomic coordinates, p-value for association with CAD, and Regulome score, are shown in Fig. 3. Ninety-six of the 7,102 SNVs showed a nominally significant association with CAD in a meta-analysis of the discovery datasets (Supplementary Table S1).
Fig. 3.

Plots of each locus included in the study showing genomic positions and log p-values for association with CAD of individuals variants found in the discovery stage. Circle sizes denote MAF and colors represent Regulome scores. A: GCLM (chr1); B: GLUL (chr1); C: GAD1 (chr2); D: GLS (chr2); E: GCLC (chr6); F: GGCT (chr 7); G: GSR (chr 8); H: OPLAH (chr 8); I: GAD2 (chr 10); J: ALDH18A1 (chr 10); K: GOT1 (chr 10); L: GLS2 (chr 12); M: GOT2 (chr 16); N: DPEP2 (chr 16); O: GSS (chr 20); P: GGT1 (chr 22)
Variants associated with CAD in the replication stage
Of the variants identified in the discovery stage as being nominally associated with CAD, 22 were selected for replication according to the criteria described in the Methods: 10 were selected for having p-values < 0.01 for association with CAD and 12 for having p-values in the 0.01–0.05 range along with Regulome scores ≤ 2 (Supplementary Table S1). Three additional variants that met these criteria were excluded from the replication analysis: one because it was in complete linkage disequilibrium with another variant selected for replication and two because of failure of the genotyping assay.
Table 2 shows the association of the 22 variants with CAD in the discovery and replication stages, as well as in the two stages combined. The strongest evidence of association was observed for variants rs62447457 and rs145322388. Variant rs62447457 is placed in the γ-glutamylcyclotransferase [GGCT] locus on chromosome 7. The A allele (MAF in Europeans = 2.5%) was associated with protection from CAD in the discovery stage (OR 0.56, p-value 0.016), in the replication stage (OR 0.44, p-value 0.023), and in the two stages combined (OR 0.54, p-value of 2.5 × 10–4, p-value for heterogeneity 0.79). This variant had a Regulome score of 2b based on the following evidence: 68 ChIP-Seq peaks (the most notable being a peak for the transcription factor CTCF in cardiac muscle cells); 127 chromatin state marks (including 3 for Weak transcription in heart); 20 DNA-accessibility marks (also in HUVEC and aortic smooth muscle cells); 2 matched motifs. Variant rs145322388 (placed in the dipeptidase 2 (DPEP2) locus on chromosome 16) showed a nominally significant association with CAD in the replication stage that went in the same direction as in the discovery stage, with the A allele (MAF in Europeans = 3.9%) being protective for CAD (discovery OR 0.69, p-value 0.016; replication OR 0.43, p value 5.0 × 10–3; overall OR 0.61 p-value 2.5 × 10–4, p-value for heterogeneity 0.52). This variant, placed about 140 kilobase pairs (Kb) 5’ of the transcription start of the DPEP2 gene, was a significant cis-eQTL for DPEP2 in whole-blood (p-value 2.0 × 10–5) and had a Regulome score of 2a based on the following evidence: presence of 318 ChIP-Seq peaks (transcription factor binding sites); 127 chromatin state marks, among which 1 weak transcription mark and 3 active TSS (transcription start site) marks in heart; 35 DNA accessibility marks (most notably in HUVEC and aortic smooth muscle cells); 9 matched motifs (computationally predicted DNA binding motifs with one or more matched transcription factors).
Table 2.
Association between low-frequency/rare variants and CAD in candidate genes related to glutamate metabolism
| SNP | Chr | Position | Candidate gene | Distance from candidate gene (kb)* | Regulome score | MAF Euro | Joslin Cohorts (n = 2394) | HSPH Cohorts (n = 1020) | Joslin + HSPH (n = 3414) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| JHS | JKS1 | JKS2 | ALL | NHS | HPFS | ALL | ALL | |||||||||||
| OR | OR | OR | OR | P | OR | OR | OR | P | OR | P | Het P | |||||||
| rs62447457 | 7 | 30,588,417 | GGCT | 44 | 2b | 0.025 | 0.57 | 0.56 | 0.44 | 0.56 | 1.63E-02 | 0.44 | 0.88 | 0.53 | 2.30E-02 | 0.54 | 2.55E-04 | 7.89E-01 |
| rs145322388 | 16 | 67,880,412 | DPEP2 | 140.9 | 2a | 0.039 | 0.74 | 0.60 | 0.56 | 0.69 | 1.69E-02 | 0.52 | 0.29 | 0.43 | 5.01E-03 | 0.61 | 2.53E-04 | 5.17E-01 |
| rs1238275622 | 1 | 182,269,861 | GLUL | 81 | 3a | 0.004 | 1.82 | 1.63 | 1.97 | 1.80 | 4.17E-03 | 0 | ∞ | 2.93 | 3.44E-01 | 1.84 | 2.07E-03 | 7.62E-01 |
| rs150255305 | 10 | 101,124,009 | GOT1 | 32.6 | 4 | 0.007 | 2.09 | 2.34 | 11.70 | 2.52 | 7.43E-03 | 1.45 | 2.18 | 1.68 | 3.05E-01 | 2.25 | 3.67E-03 | 6.06E-01 |
| rs534659781 | 1 | 182,169,679 | GLUL | 181.2 | 4 | 0.002 | 6.26 | ∞ | ∞ | 10.15 | 7.05E-03 | 0 | 4.30 | 1.73 | 4.98E-01 | 4.60 | 3.92E-03 | 6.99E-01 |
| rs2676140 | 2 | 171,878,953 | GAD1 | 161.3 | 3a | 0.002 | 1.80 | 1.99 | 1.47 | 1.80 | 7.05E-03 | 1.29 | 1.09 | 1.19 | 7.29E-01 | 1.72 | 5.45E-03 | 9.63E-01 |
| rs187364908 | 22 | 25,031,034 | GGT1 | 6.1 | 2b | NA | 1.98 | 1.40 | 3.89 | 2.04 | 3.07E-02 | 0 | ∞ | 5.84 | 3.46E-01 | 2.21 | 9.92E-03 | 6.92E-01 |
| rs141886579 | 10 | 97,320,916 | ALDH18A1 | 44.8 | 2a | 0.012 | 0.55 | 0.24 | 0.50 | 0.50 | 4.23E-02 | 0.78 | 0.46 | 0.64 | 5.60E-01 | 0.54 | 1.08E-02 | 9.01E-01 |
| rs62508274 | 8 | 30,672,919 | GSR | 87.5 | 2b | 0.046 | 0.69 | 1.04 | 0.51 | 0.70 | 4.65E-02 | 0.80 | 0.58 | 0.70 | 2.58E-01 | 0.70 | 1.27E-02 | 8.03E-01 |
| rs78980639 | 7 | 30,634,361 | GGCT | 89.9 | 2b | 0.006 | 3.65 | 2.36 | ∞ | 3.59 | 4.43E-02 | 2.40 | 0.71 | 1.77 | 2.38E-01 | 2.50 | 1.48E-02 | 7.36E-01 |
| rs150362059 | 10 | 26,761,006 | GAD2 | 167.5 | 4 | 0.016 | 1.52 | 1.83 | 6.05 | 1.84 | 2.23E-03 | 0.35 | 1.54 | 0.75 | 4.76E-01 | 1.55 | 1.98E-02 | 6.40E-02 |
| rs150416223 | 20 | 33,687,170 | GSS | 143.6 | 2b | 0.008 | 3.15 | 2.39 | 1.95 | 2.87 | 6.03E-03 | 0.67 | 0.95 | 0.80 | 7.87E-01 | 1.89 | 2.20E-02 | 3.77E-01 |
| rs62179904 | 2 | 191,859,054 | GLS | 28.8 | 3a | 0.004 | 0.18 | 1.16 | 0 | 0.27 | 9.06E-03 | 6.59 | 0.72 | 1.89 | 2.24E-01 | 0.46 | 2.26E-02 | 8.67E-02 |
| rs1175583158 | 1 | 182,269,865 | GLUL | 81 | 3a | 0.001 | 6.30 | 1.78 | 2.96 | 3.70 | 2.07E-03 | 0.50 | ∞ | 0.73 | 9.39E-01 | 2.14 | 2.47E-02 | 1.79E-01 |
| rs11465742 | 12 | 56,731,664 | GLS2 | 133.1 | 2a | 0.002 | ∞ | 3.56 | ∞ | 11.54 | 7.42E-04 | 0.26 | 3.35 | 0.45 | 9.66E-03 | 0.67 | 5.00E-02 | 5.65E-06 |
| rs117658070 | 10 | 97,234,713 | ALDH18A1 | 131 | 2c | 0.016 | 2.90 | 1.55 | 0.41 | 2.13 | 3.66E-02 | 1.44 | 0.71 | 1.12 | 6.50E-01 | 1.58 | 6.29E-02 | 2.75E-01 |
| rs191582687 | 16 | 67,880,106 | DPEP2 | 141.2 | 2b | 0.002 | ∞ | 2.36 | 0 | 5.34 | 2.55E-02 | 0 | 2.17 | 1.24 | 7.21E-01 | 2.95 | 7.05E-02 | 6.53E-01 |
| rs144013746 | 8 | 30,411,844 | GSR | 123.7 | 2b | 0.004 | 2.79 | 0.94 | 2.93 | 2.23 | 4.34E-02 | 0.36 | 4.32 | 0.88 | 9.31E-01 | 1.75 | 8.82E-02 | 3.59E-01 |
| rs17111645 | 10 | 101,156,407 | GOT1 | 0.2 | 4 | 0.045 | 0.77 | 0.38 | 0.49 | 0.66 | 9.14E-03 | 1.68 | 1.83 | 1.74 | 4.69E-02 | 0.84 | 2.08E-01 | 1.90E-02 |
| rs2490283 | 10 | 101,196,080 | GOT1 | 5.7 | 2b | 0.029 | 0.81 | 0.31 | 0.60 | 0.70 | 3.95E-02 | 1.68 | 1.83 | 1.74 | 4.69E-02 | 0.91 | 5.17E-01 | 3.78E-02 |
| rs111358122 | 10 | 101,286,886 | GOT1 | 96.5 | 2b | 0.006 | 3.49 | 2.31 | ∞ | 3.65 | 2.14E-02 | 0.78 | 1.47 | 0.82 | 5.71E-01 | 0.98 | 8.83E-01 | 1.09E-01 |
| rs77316565 | 8 | 30,710,566 | GSR | 125.1 | 2b | 0.026 | 1.64 | 1.12 | 1.48 | 1.50 | 4.22E-02 | 0.39 | 1.77 | 0.61 | 4.62E-02 | 1.00 | 9.89E-01 | 2.50E-03 |
*Based on Ensembl canonical transcript, MAF Euro minor allele frequency in Europeans, JHS Joslin Heart Study, JKS1 Joslin Kidney Study (cohort 1), JKS2 Joslin Kidney Study (cohort 2), HSPH Harvard School of Public Health, NHS Nurses Health Study, HPFS Health Professionals Follow-up Study, Het P P-value for heterogeneity
A third variant, rs1238275622 (MAF in Europeans = 0.4%), flanking the GLUL gene (on chromosome 1), while not associated with CAD in the replication datasets, showed a significant p-value (p < 2.3 × 10–3) in the discovery/replication aggregate results. The T allele was associated with increased risk of CAD (OR 1.80, p-value 4.2 × 10–3 in the discovery stage; OR 2.93, p-value 0.34 in the replication stage; OR 1.84, p-value 2.1 × 10–3 in the two stages combined, p-value for heterogeneity 0.76). This variant had a Regulome score 3a, based on the following findings: 28 ChIP-Seq peaks; 127 chromatin state marks (including Quiescent/Low in heart/aorta and Low Transcription in heart); 13 DNA-accessibility marks; 2 unmatched motifs (computationally predicted DNA binding motifs without a matched transcription factors).
None of the other 19 SNVs showed evidence of replication, including the top variant in the discovery stage (rs11465742 at the GLS2 locus), which showed an association with CAD in opposite direction in the replication stage (Table 2).
Data from the T2D knowledge portal
Aggregate statistics about genetic variants associated with CAD in other populations with T2D were retrieved from the T2DKP. Data concerning the phenotype “Cardiovascular disease in type 2 diabetes” in individuals of European ancestry were available from the AMP-T2D UCSD Diabetic Complications Study-European dataset for 17 of the 22 SNV selected for replication, as described in Table 3. According to these data, the replicated variant rs62447457 showed an association with CAD in the AMP-T2D that went in the same direction (OR = 0.83) as in the discovery and replication stages, although without reaching statistical significance (p = 0.08). A meta-analysis of these data along with our findings from the discovery and replication stages yielded a p-value of 8.0 × 10–4, with an I2 of 0 for heterogeneity (p = 0.34) (Table 3). Association of rs145322388 with the phenotype was not replicated in T2DKP. No phenotype data were available for rs1238275622. Another variant, rs191582687, which had not been replicated in our study, showed a p-value of 0.004 for association with CAD in the T2DKP datasets that went in the same direction as in the discovery and replication stages. A meta-analysis of our data with those from the portal yielded a p-value of 2.9 × 10–3, also with low heterogeneity (I2 = 0, p = 0.95) (Table 3). This variant has Regulome score 2b, based on the finding of 39 ChIP-Seq peaks; 127 chromatin state marks (including 3 active TSS and 1 flanking active TSS in heart, as well as 1 enhancer mark in aorta); 12 DNA-accessibility marks (most notably in heart and HUVEC); 2 matched motifs.
Table 3.
Association of variants with the phenotype “Cardiovascular disease in type 2 diabetes” in the Type 2 Diabetes Knowledge Portal
| SNP | Joslin + HSPH | T2DKP | Joslin + HSPH + T2DKP | ||||||
|---|---|---|---|---|---|---|---|---|---|
| OR | P-value | OR | P-value | same direction | n | P-value | HetISq | HetPVal | |
| rs62447457 | 0.54 | 2.55E-04 | 0.83 | 8.40E-02 | yes | 3,032 | 8.01E-04 | 0.0% | 3.35E-01 |
| rs191582687 | 2.95 | 7.05E-02 | 1.65 | 4.00E-03 | yes | 9,010 | 2.93E-03 | 0.0% | 9.52E-01 |
| rs150362059 | 1.55 | 1.98E-02 | 1.12 | 9.10E-02 | yes | 9,814 | 1.67E-02 | 59.5% | 3.03E-02 |
| rs2490283 | 0.91 | 5.17E-01 | 0.91 | 3.90E-02 | yes | 10,544 | 3.90E-02 | 51.1% | 6.92E-02 |
| rs17111645 | 0.84 | 2.08E-01 | 0.93 | 7.70E-02 | yes | 10,717 | 5.14E-02 | 58.0% | 3.61E-02 |
| rs150255305 | 2.25 | 3.67E-03 | 1.06 | 6.00E-01 | yes | 9,010 | 1.49E-01 | 36.6% | 1.63E-01 |
| rs117658070 | 1.58 | 6.29E-02 | 1.07 | 3.10E-01 | yes | 9,814 | 1.49E-01 | 31.6% | 1.99E-01 |
| rs144013746 | 1.75 | 8.82E-02 | 1.12 | 4.00E-01 | yes | 9,010 | 1.62E-01 | 20.0% | 2.83E-01 |
| rs111358122 | 0.98 | 8.83E-01 | 1.20 | 1.00E-01 | no | 9,506 | 2.18E-01 | 43.0% | 1.35E-01 |
| rs150416223 | 1.89 | 2.20E-02 | 1.01 | 9.00E-01 | yes | 9,506 | 4.93E-01 | 40.2% | 1.37E-01 |
| rs78980639 | 2.50 | 1.48E-02 | 0.98 | 8.60E-01 | no | 9,010 | 6.97E-01 | 27.3% | 2.40E-01 |
| rs77316565 | 1.00 | 9.89E-01 | 0.97 | 5.50E-01 | yes | 10,515 | 6.97E-01 | 70.4% | 4.71E-03 |
| rs62179904 | 0.46 | 2.26E-02 | 1.02 | 8.90E-01 | no | 9,010 | 7.66E-01 | 59.1% | 4.43E-02 |
| rs62508274 | 0.70 | 1.27E-02 | 1.02 | 6.50E-01 | no | 10,544 | 7.86E-01 | 34.4% | 1.79E-01 |
| rs187364908 | 2.21 | 9.92E-03 | 0.98 | 7.90E-01 | no | 10,276 | 8.06E-01 | 50.4% | 1.09E-01 |
| rs145322388 | 0.61 | 2.53E-04 | 1.00 | 9.10E-01 | no | 10,622 | 9.10E-01 | 66.6% | 1.05E-02 |
| rs141886579 | 0.54 | 1.08E-02 | 1.00 | 9.90E-01 | no | 9,814 | 9.90E-01 | 26.6% | 2.35E-01 |
HSPH harvard school of public health, T2DKP: Type 2 Diabetes Knowledge Portal, HetISq: I-squared for heterogeneity, HetPVal: p-value for heterogeneity
Data about the association with hepatic markers aspartate transaminase (AST), alanine transaminase (ALT), γ-glutamyltransferase (GGT), and alkaline phosphatase (ALP) were available in the T2DKP for 18 of the 22 SNPs. Variant rs145322388 was associated with all four markers: AST (p-value 6.76 × 10–5), ALT (p-value 1.07 × 10–2), GGT (p-value 7.85 × 10–3), and ALP (p-value 1.77 × 10–6). No association of those phenotypes with rs62447457 was seen. Six other variants (rs11465742, rs141886579, rs150255305, rs187364908, rs191582687, and rs2490283) showed significant associations (Supplementary table S2).
Discussion
The last two decades have seen unprecedented progress in identifying common genetic variants associated with common diseases. The GWAS approach, studying millions of common variants throughout the genome, has been widely successful in identifying significant associations with CAD across diverse populations [15, 16]. However, some limitations of this approach can specifically impact the understanding of CAD in T2D. First, GWASs are usually performed for one phenotype at the time. Studying cross-phenotype associations can be challenging, especially when phenotypes are causally connected such as T2D and CAD [39]. Second, findings from GWASs have fallen short of entirely explaining genetic predisposition to disease, thereby challenging the common disease-common variant hypothesis and pointing to a potential role of rare genetic variation in common multifactorial disorders, such as CAD in T2D [40].
More than a decade ago, we were able to address the first of the limitations described above by designing a GWAS of CAD exclusively among individuals with T2D, which led to the identification of a common variant at the GLUL locus that is specifically associated with an increased risk of CAD in this population—an effect that appeared to be mediated by alterations of the γ-glutamyl cycle [18]. In the present study, we tried to address the second limitation, by using deep resequencing to investigate whether less frequent genetic variants at the GLUL locus and 15 other loci involved in the regulation of the γ-glutamyl cycle also affect the risk of CAD among individuals with T2D. Through this effort, we have found robust evidence of association with CAD for a low-frequency variant at the GGCT locus (rs62447457) as well as suggestive evidence for two low-frequency variants at the DPEP2 (rs145322388) and GLUL (rs1238275622) loci.
Variant rs62447457 showed the strongest overall evidence of association with CAD, with a p-value of 8 × 10–4 in a combined analysis of our datasets with data from the T2DKP. The minor allele A was protective for CAD, with an OR of 0.54 in the combined discovery and replication stages and 0.84 in T2DKP. Association of this variant with other phenotypes has not been reported in literature. GGCT—the gene adjacent to this variant—codes for the enzyme γ-glutamylcyclotransferase, catalyzing the formation of pyroglutamate from γ-glutamyl dipeptides. This metabolite is a precursor of glutamate, which is the substrate of the GLUL enzyme (Fig. 1). In Regulome, this variant is associated with regulatory elements in relevant cell types such as left ventricle and aortic smooth muscle. Since this allele is protective for CAD, we can hypothesize that, by increasing the availability of pyroglutamate in the γ-glutamyl cycle, this variant increases glutathione (GSH) synthesis, thereby decreasing CAD risk. No eQTL data are currently available to corroborate this hypothesis.
Variant rs145322388, located at the DPEP2 locus, showed the strongest evidence of association with CAD in the discovery and replication stages of our study (summary OR = 0.61), but this finding was not supported by the T2DKP data (OR = 1.0). It is a significant eQTL in whole-blood, with the A allele being associated with increased expression of DPEP2 (z-score 4.27, p-value = 2.0 × 10–5). This gene codes the enzyme dipeptidase 2, which cleaves cysteine-glycine dipeptides into single amino acids. Cysteine is then combined with glutamate to form γ-glutamyl cysteine, which in turn is combined with glycine to form GSH. Since the A allele is protective for CAD, our working hypothesis is that overexpression of DPEP2 improves functioning of the γ-glutamyl cycle by increasing the breakdown of cysteine-glycine and consequently glycine availability, thereby enhancing GSH synthesis. While no association with CAD was found for this variant in the T2DKP, the finding of an association with serum levels of AST, ALT, GGT, and ALP in that same dataset provides support for the involvement of this variant in the γ-glutamyl cycle. Of note, another rare variant at the DPEP2 locus (rs191582687) that was nominally associated with CAD in the discovery sets, although not in the replication sets, showed evidence of association with CAD in T2DKP. In this case, the minor allele increased the risk of CAD (OR 2.79 in discovery + replication stages and 1.65 in T2DKP).
Allele T of the rs1238275622 variant, flanking the GLUL gene, was associated with increased risk of CAD (summary OR = 1.84). The C allele of the common variant rs10911021, previously associated with increased risk of CAD in individuals with diabetes, was associated with 32% lower expression of GLUL in homozygotes [18]. While no eQTL data are available for rs1238275622, we hypothesize that rs1238275622 may be also associated with lower expression of GLUL, thereby increasing the risk of CAD by similar mechanisms. The finding of association with quiescent chromatin states in relevant tissues supports this hypothesis.
Overall, these results provide further support to the hypothesis that dysregulation of the γ-glutamyl cycle and GSH synthesis contribute to CAD in T2D. This hypothesis was initially suggested by metabolomic data [18] and then corroborated by our studies in cultured HUVEC showing that the allele C of the common GLUL variant rs10911021, which is associated with an increased risk of CAD, is also associated with lower expression of GLUL, lower glutathione-to-glutamate ratio, and higher levels of MG—an atherogenic precursor of advanced glycation end-products (AGEs) that is normally detoxified by glutathione [20]. Similar studies will be needed to investigate the impact of the rare variants identified in this report on these intracellular processes, although these are hampered by the low frequencies of these variants, making the identification of endothelial cells naturally carrying these sequence differences, as it was done for rs10911021, quite challenging. An alternative may be to introduce these variants by genome editing through CRISPR, but since this technique cannot be easily used in primary cell cultures such as HUVEC, the pros and cons of introducing rare mutations in cell types less relevant to the pathophysiological processes studied here should be weighed against each other.
While the variants that we have identified show evidence of replication in independent datasets, their statistical significance in the meta-analysis of discovery and replication sets did not reach genome-wide criteria. This is consistent with the results of other studies investigating the association of rare/infrequent variants with common disorders, many of which were only able to demonstrate nominal significance. For instance, Ahituv et al. did not find individual rare variants in a panel of obesity-associated genes to be associated with extreme obesity in comparison with lean individuals. When grouping variants together, they found non-synonymous variants to be more common in obese individuals, reaching nominal significance [41]. Another example is the study by Bonnefond et al., which analyzed the association of rare variants in the MTNR1B gene with T2D. No significant association of single variants with the phenotype were found, but when pooling together 36 variants with MAF < 0.1%, a significant association was seen, with a p-value in the 10–4 order of magnitude as in our study [42]. Similar findings were obtained in the lipid field regarding HDL-cholesterol and hypertriglyceridemia [43]. These examples illustrate the challenges of studying the association of rare variants with clinical phenotypes. In our study, we addressed this problem by focusing on genomic regions that were adjacent to the candidate loci and showed evidence of functional relevance. Although regulatory regions can be located many thousands of base pairs distant from target genes [44], this approach reduced the possibility of false positives by minimizing the number of comparisons that were made and maximizing the prior probability of a functional effect for the variants included in the analysis.
In addition to the emphasis on functional regions, another strength of our study was the focus on individuals selected for having been diagnosed with T2D before the ascertainment of CAD phenotypes. As shown in our previously published GWAS, this design is effective in capturing genetic predisposition to CAD that is unique to diabetes [17]. Nonetheless, some limitations of our study should be acknowledged. First, the sample size of our discovery sets provided us enough power to detect only relatively large genetic effects. While these were consistent with the magnitude that one would expect for infrequent variants, we may have missed some weaker, but still relevant genetic effects. Second, for those variants with discovery stage p values in the 0.01 to 0.05 range, we attempted replication only for those with strong evidence of a potential functional role (Regulome score ≤ 2), in order to contain the number of variants tested at the replication stage. By doing so, we may have missed true associations concerning variants with weaker functional evidence. Third, our study included only White subjects, making the generalizability of results to subjects of different ancestries unknown. This reflects the challenges of finding suitable studies including individuals of non-White ancestries characterized for two phenotypes, one of which (T2D) must have temporally preceded the other one (CAD). Fourth, the association between genetic variants and CAD may have been influenced by measured or unmeasured population characteristics or environmental factors. Imbalances in these characteristics between cases and controls may have led to false positives and/or false negatives, and differences in these factors among cohorts may have been responsible for some of the observed discrepancies in the association between identified variants and CAD. The requirement for replication to consider a variant as being genuinely associated with CAD attenuated the concern for false positives but did not address the possibility of false negatives. Fifth, only individuals with available DNA were included, which may impact the generalizability of our findings. Finally, no eQTL data were available for some of the replicated variants, which limited our ability to make inferences about their functional relevance. However, those variants for which eQTL data were not available showed several other regulatory marks captured by their high Regulome scores, indicating a high likelihood of being functional.
Conclusions
In summary, we have identified low-frequency genetic variants associated with coronary artery disease in individuals with T2D that are placed in non-coding regions with a high probability of regulating the expression of genes involved in glutamate metabolism and the γ-glutamyl cycle. While further studies are necessary to confirm the functional role of these variants, these findings provide further support to the hypothesis of a dysregulation of the γ-glutamyl cycle as playing an important role in the etiology of CAD in T2D.
Supplementary Information
Acknowledgements
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations
- AGE
Advanced glycation end-product
- ALP
Alkaline phosphatase
- ALT
Alanine aminotransferase
- AST
Aspartate aminotransferase
- CAD
Coronary artery disease
- CRISPR
Clustered regularly interspaced short palindromic repeats
- ENCODE
Encyclopedia of DNA elements
- eGFR
estimated glomerular filtration rate
- eQTL
Expression quantitative trait locus
- GATK
Genome analysis toolkit
- GGT
Gamma-glutamyl transferase
- GLUL
Glutamate-ammonia ligase
- GWAS
Genome-wide association study
- HPFS
Health Professionals follow-up study
- HUVEC
Human umbilical vein cells
- JHS
Joslin heart study
- JKS
Joslin kidney study
- MAF
Minor allele frequency
- NGS
Next-generation sequencing
- NHS
Nurses’ health study
- OR
Odds ratio
- SNV
Single-nucleotide variant
- T2D
Type 2 diabetes
- T2DKP
Type 2 diabetes knowledge portal
Author contributions
AD designed the study; FMAG and AD drafted the manuscript; CM, MGP, SKR, HSS, QS, and YT critically revised the final version of the manuscript for important intellectual input; CM and FMAG prepared genomic libraries for sequencing; FMAG, SGF, and MGP designed the bioinformatics pipeline and performed the analyses; SKR and QS performed data collection and analysis for the replication stage; FMAG, HSS, YT, and AD performed statistical analysis. AD and FMAG are the guarantors of this work and, as such, had full access to all data of the study and take full responsibility for the integrity of the data and the accuracy of data analysis. AD and FMAG are joint corresponding authors.
Funding
Funded by NIH grants 5R01HL132254 (AD), R01HL034594, R01HL35464, R01CA49449, U01CA167552, and UM1CA186107. SKR is supported by a Postdoctoral Fellowship from the Canadian Institutes of Health Research.
Availability of data and materials
Individual data generated and/or analyzed during the current study cannot be share due to ethical restrictions. Aggregate data are available from the corresponding authors on reasonable request.
Declarations
Ethics approval and consent to participate
The study protocols of the JHS, JKS1, and JKS2 were approved by the Joslin Committee on Human Studies. The study protocols of the NHS and HPFS were approved by the Institutional Review Boards of the Brigham and Women's Hospital and the Harvard T.H. Chan School of Public Health.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Co-senior authors: Fernando M. A. Giuffrida and Alessandro Doria.
Contributor Information
Fernando M. A. Giuffrida, Email: fernando.giuffrida@me.com
Alessandro Doria, Email: alessandro.doria@joslin.harvard.edu.
References
- 1.Einarson TR, Acs A, Ludwig C, Panton UH. Prevalence of cardiovascular disease in type 2 diabetes: a systematic literature review of scientific evidence from across the world in 2007–2017. Cardiovasc Diabetol. 2018;17(1):83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gregg EW, Li Y, Wang J, Burrows NR, Ali MK, Rolka D, et al. Changes in diabetes-related complications in the United States, 1990–2010. N Engl J Med. 2014;370(16):1514–23. [DOI] [PubMed] [Google Scholar]
- 3.Jung CH, Chung JO, Han K, Ko SH, Ko KS, Park JY, et al. Improved trends in cardiovascular complications among subjects with type 2 diabetes in Korea: a nationwide study (2006–2013). Cardiovasc Diabetol. 2017;16(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aidin R, Araz R, Stefan F, Björn E, Ann-Marie S, Mervete M, et al. Mortality and cardiovascular disease in Type 1 and Type 2 diabetes. N Engl J Med. 2017;376(15):1407–18. [DOI] [PubMed] [Google Scholar]
- 5.Bahiru E, Hsiao R, Phillipson D, Watson KE. Mechanisms and treatment of dyslipidemia in diabetes. Curr Cardiol Rep. 2021;23(4):26. [DOI] [PubMed] [Google Scholar]
- 6.Poznyak A, Grechko AV, Poggio P, Myasoedova VA, Alfieri V, Orekhov AN. The diabetes mellitus-atherosclerosis connection: the role of lipid and glucose metabolism and chronic inflammation. Int J Mol Sci. 2020;21(5):1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ritchie RH, Abel ED. Basic mechanisms of diabetic heart disease. Circ Res. 2020;126(11):1501–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McPherson R, Pertsemlidis A, Kavaslar N, Stewart A, Roberts R, Cox DR, et al. A common allele on chromosome 9 associated with coronary heart disease. Science. 2007;316(5830):1488–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Helgadottir A, Thorleifsson G, Manolescu A, Gretarsdottir S, Blondal T, Jonasdottir A, et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science. 2007;316(5830):1491–3. [DOI] [PubMed] [Google Scholar]
- 10.Consortium WTCC, Consortium C, Trégouët DA, König IR, Erdmann J, Munteanu A, et al. Genome-wide haplotype association study identifies the SLC22A3-LPAL2-LPA gene cluster as a risk locus for coronary artery disease. Nat Genet. 2009;41(3):283–5 [DOI] [PubMed]
- 11.Group IA Thrombosis, and Vascular Biology Working, Consortium MIG, Consortium WTCC, Consortium C, Erdmann J, Großhennig A, et al. New susceptibility locus for coronary artery disease on chromosome 3q22.3. Nat Genet 2009; 41(3):280–2 [DOI] [PMC free article] [PubMed]
- 12.Schunkert H, König IR, Kathiresan S, Reilly MP, Assimes TL, Holm H, et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. 2011;43(4):333–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Consortium TCardi, Consortium D, Consortium C, Consortium M, Consortium WTCC, Deloukas P, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. (2013); 45(1):25–33 [DOI] [PMC free article] [PubMed]
- 14.Cole JB, Florez JC. Genetics of diabetes mellitus and diabetes complications. Nat Rev Nephrol. 2020;16(7):377–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aragam KG, Jiang T, Goel A, Kanoni S, Wolford BN, Atri DS, et al. Discovery and systematic characterization of risk variants and genes for coronary artery disease in over a million participants. Nat Genet. 2022;54(12):1803–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tcheandjieu C, Zhu X, Hilliard AT, Clarke SL, Napolioni V, Ma S, et al. Large-scale genome-wide association study of coronary artery disease in genetically diverse populations. Nat Med. 2022;28(8):1679–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doria A. Leveraging Genetics to Improve Cardiovascular Health in Diabetes: the 2018 Edwin Bierman Award Lecture. Diabetes. 2019;68(3):479–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qi L, Qi Q, Prudente S, Mendonca C, Andreozzi F, di Pietro N, et al. Association between a genetic variant related to glutamic acid metabolism and coronary heart disease in individuals with type 2 diabetes. JAMA. 2013;310(8):821–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Look AHEAD Research Group. Prospective association of GLUL rs10911021 with cardiovascular morbidity and mortality among individuals with type 2 diabetes: The Look AHEAD Study. Diabetes. 2015; 65(1):db150890 [DOI] [PMC free article] [PubMed]
- 20.Pipino C, Shah H, Prudente S, Pietro ND, Zeng L, Park K, et al. Association of the 1q25 diabetes-specific coronary heart disease locus with alterations of the γ-Glutamyl cycle and increased methylglyoxal levels in endothelial cells. Diabetes. 2020;69(10):2206–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krebs HA. Metabolism of amino-acids. Biochem J. 1935;29(8):1951–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lange LA, Hu Y, Zhang H, Xue C, Schmidt EM, Tang ZZ, et al. Whole-exome sequencing identifies rare and low-frequency coding variants associated with LDL cholesterol. Am J Hum Genetics. 2014;94(2):233–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grarup N, Sandholt CH, Hansen T, Pedersen O. Genetic susceptibility to type 2 diabetes and obesity: from genome-wide association studies to rare variants and beyond. Diabetologia. 2014;57(8):1528–41. [DOI] [PubMed] [Google Scholar]
- 24.Niewczas MA, Sirich TL, Mathew AV, Skupien J, Mohney RP, Warram JH, et al. Uremic solutes and risk of end stage renal disease in type 2 diabetes. Kidney Int. 2014;85(5):1214–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shah HS, Moreno LO, Morieri ML, Tang Y, Mendonca C, Jobe JM, et al. Serum orotidine: a novel biomarker of increased CVD risk in Type 2 diabetes discovered through metabolomics studies. Diabetes Care. 2022;45(8):1882–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu Y, Li Y, Sampson L, Wang M, Manson JE, Rimm E, et al. Lignan intake and risk of coronary heart disease. J Am Coll Cardiol. 2021;78(7):666–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee DH, Li J, Li Y, Liu G, Wu K, Bhupathiraju S, et al. Dietary inflammatory and insulinemic potential and risk of type 2 diabetes: results from three prospective us cohort studies. Diabetes Care. 2020;43(11):2675–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lazaro-Guevara J, Fierro-Morales J, Wright AH, Gunville R, Simeone C, Frodsham SG, et al. Targeted next-generation sequencing identifies pathogenic variants in diabetic kidney disease. Am J Nephrol. 2021;52(3):239–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv.org [Internet]. 2013;q-bio.GN. Available from: arXiv.org
- 31.der Auwera GAV, Carneiro MO, Hartl C, Poplin R, del Angel G, Levy-Moonshine A, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Current protocols bioinformatics. 2013. 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pedersen BS, Quinlan AR. Mosdepth: quick coverage calculation for genomes and exomes. Bioinformatics. 2017;34(5):867–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164–e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dong S, Boyle AP. Predicting functional variants in enhancer and promoter elements using RegulomeDB. Hum Mutat. 2019;40(9):1292–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yaghootkar H, Lamina C, Scott RA, Dastani Z, Hivert MF, Warren LL, et al. Mendelian randomization studies do not support a causal role for reduced circulating adiponectin levels in insulin resistance and type 2 diabetes. Diabetes. 2013;62(10):3589–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics (Oxford, England). 2010;26(17):2190–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Balduzzi S, Rücker G, Schwarzer G. How to perform a meta-analysis with R: a practical tutorial. Évid Based Ment Heal. 2019;22(4):153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li X, Zhu X. Statistical human genetics, methods and protocols. Methods Mol Biol. 2017;1666:455–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sazonovs A, Barrett JC. Rare-variant studies to complement genome-wide association studies. Annu Rev Genom Hum G. 2018;19(1):1–16. [DOI] [PubMed] [Google Scholar]
- 41.Ahituv N, Kavaslar N, Schackwitz W, Ustaszewska A, Martin J, Hébert S, et al. Medical sequencing at the extremes of human body mass. Am J Hum Genetics. 2007;80(4):779–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.(MAGIC) TMA of G and IRTC, Bonnefond A, Clément N, Fawcett K, Yengo L, Vaillant E, et al. Rare MTNR1B variants impairing melatonin receptor 1B function contribute to type 2 diabetes. Nat Genet. 2012;44(3):297–301 [DOI] [PMC free article] [PubMed]
- 43.Cohen JC, Kiss RS, Pertsemlidis A, Marcel YL, McPherson R, Hobbs HH. Multiple rare alleles contribute to low plasma levels of hdl cholesterol. Science. 2004;305(5685):869–72. [DOI] [PubMed] [Google Scholar]
- 44.Zuk O, Schaffner SF, Samocha K, Do R, Hechter E, Kathiresan S, et al. Searching for missing heritability: designing rare variant association studies. Proc National Acad Sci. 2014;111(4):E455–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Individual data generated and/or analyzed during the current study cannot be share due to ethical restrictions. Aggregate data are available from the corresponding authors on reasonable request.