

Abstract
Protein Kinase RNA-activated (PKR) is an enzyme that plays a role in many systemic processes, including modulation of inflammation, and is implicated in neurodegenerative diseases, such as Alzheimer’s disease (AD). PKR phosphorylation results in the production of several cytokines involved in the regulation / modulation of sleep, including interleukin-1β, tumor necrosis factor-α and interferon-γ. We hypothesized targeting PKR would alter spontaneous sleep of mice, attenuate responses to sleep deprivation, and inhibit responses to immune challenge. To test these hypotheses, we determined the sleep-wake phenotype of mice lacking PKR (knockout; PKR−/−) during undisturbed baseline conditions; in responses to six hours of sleep deprivation; and after immune challenge with lipopolysaccharide (LPS). Adult male mice (C57BL/6J, n = 7; PKR−/−, n = 7) were surgically instrumented with EEG recording electrodes and an intraperitoneal microchip to record core body temperature. During undisturbed baseline conditions, PKR −/− mice spent more time in non-rapid eye movement sleep (NREMS) and rapid-eye movement sleep (REMS), and less time awake at the beginning of the dark period of the light:dark cycle. Delta power during NREMS, a measure of sleep depth, was less in PKR−/− mice during the dark period, and core body temperatures were lower during the light period. Both mouse strains responded to sleep deprivation with increased NREMS and REMS, although these changes did not differ substantively between strains. The initial increase in delta power during NREMS after sleep deprivation was greater in PKR−/− mice, suggesting a faster buildup of sleep pressure with prolonged waking. Immune challenge with LPS increased NREMS and inhibited REMS to the same extent in both mouse strains, whereas the initial LPS-induced suppression of delta power during NREMS was greater in PKR−/− mice. Because sleep regulatory and immune responsive systems in brain are redundant and overlapping, other mediators and signaling pathways in addition to PKR are involved in the responses to acute sleep deprivation and LPS immune challenge.
Keywords: PKR, Sleep, Sleep deprivation, Lipopolysaccharide, Cytokines, Inflammation, Neurodegeneration, Alzheimer’s disease
1. Introduction
Although known for its anti-viral actions during innate immune responses to viral infections (García et al., 2006), protein kinase RNA-activated (PKR) is a kinase that plays a role in multiple cellular and systemic processes, including but not limited to mRNA translation, regulation of proliferation, apoptosis and inflammatory responses (García et al., 2007; Liu et al., 2020). PKR effects in the CNS include bidirectional activation of inflammatory pathways. For example, pro inflammatory mediators such as interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α) activate PKR (Khandelwal et al., 2011). Activated PKR, in turn can simulate additional proinflammatory pathways via nuclear factor kappa beta (NFκB; Bonnet et al., 2000). Conversely, depending on the stimulus and cell type, activation of PKR can lead to the release of pro-inflammatory [TNF-α, IFN-γ, IL-1β, IL-18; (Lu et al., 2012; Zhang and Samuel, 2008)] and anti-inflammatory [IL-10; (Grolleau et al., 2000)] cytokines. As such, PKR’s role in inflammatory responses is complex and not fully understood.
Cytokines and their receptors are constitutively expressed in the central nervous system, where they contribute to the regulation / modulation of multiple physiologic and behavioral processes, including sleep-wake behavior (Irwin, 2002; Imeri and Opp, 2009; Irwin and Opp, 2017; Krueger et al., 2001). IL-1β and TNF-α are the most studied cytokines within the context of sleep, and they and their receptors are expressed in neurons in brain regions involved in the regulation of sleep-wake behavior (Breder et al., 1993, 1988; Imeri and Opp, 2009). Many studies demonstrate that these inflammatory mediators, particularly IL-1β and TNF-α, increase non-rapid eye movements sleep (NREMS) when administered centrally or peripherally (Krueger et al., 2001). IL-1β and TNF-α participate in the regulation of normal, physiological sleep in the absence of pathology; in the alterations in sleep that occur after acute or chronic sleep loss; and during inflammatory pathologies such as infections or trauma (Irwin, 2002; Irwin and Opp, 2017; Rowe and Griesbach, 2022).
Recent evidence also implicates PKR in facets of neurodegenerative diseases, including Alzheimer’s disease (AD). Disrupted sleep is prevalent in individuals with AD, with some estimates as high as 66 % of patients exhibiting some form of sleep disturbance (Holth et al., 2017; Lim et al., 2014; Musiek et al., 2015). Changes in sleep architecture and altered circadian rhythmicity result in excessive daytime sleepiness and nighttime insomnia. Nighttime sleep disturbances include reduced NREMS, reduced rapid eye movements sleep (REMS), and decreased sleep efficiency. Collectively, these changes result in fragmented nighttime sleep, with increased awakenings and more overall wakefulness (Bliwise et al., 1989; Bonanni et al., 2005). Sleep disturbances often occur early in the disease, before clinical symptoms of cognitive impairment manifest (Hita-Yañez et al., 2013; Ma et al., 2013; Westerberg et al., 2012), and worsen with disease progression (Bonanni et al., 2005; Liguori et al., 2014).
Although disrupted sleep is a hallmark manifestation of AD, and sleep disturbance are a risk factor for dementia and cognitive decline (Benedict et al., 2015; Bubu et al., 2017; Lim et al., 2013; Spira et al., 2014; Sterniczuk et al., 2013; Tsapanou et al., 2015), mechanisms underlying AD-induced alterations in sleep remain largely unknown. Because PKR is implicated in the production of cytokines involved in sleep regulation, interfering with this signaling pathway is expected to reduce these inflammatory mediators and alter subsequent sleep. A PKR knockout (KO; PKR−/−) mouse exists and represents a potential tool with which to test hypotheses of a role for sleep and pro-inflammatory mediators in the etiology of neurodegenerative disease, including AD. However, to our knowledge there are no published sleep studies in which the PKR−/− mouse has been used. As such, in this study we phenotyped the PKR−/− mouse to determine if this kinase participates in sleep regulation or modulation under conditions of sleep loss and responses to acute immune challenge. Specifically, we determined the sleep-wake phenotype of PKR−/− mice during undisturbed baseline conditions (spontaneous sleep); in responses to acute sleep deprivation; and after immune activation by lipopolysaccharide (LPS). We hypothesized that lack of PKR will reduce the magnitude of NREMS enhancement and febrile responses to LPS because of the impairment of a signaling pathway for pro-inflammatory cytokine production.
2. Methods
2.1. Animals
C57BL/6J mice were obtained from Jackson Laboratory (Bar Harbor, ME) and subsequently bred and raised in our own colony. PKR deficient mice (PKR −/−) used for this study, were generated at the Ottawa Regional Cancer Center Research Laboratories (Abraham et al., 1999). PKR deficiency was introduced by homologous recombination that interferes with exon 12, resulting in the disruption of the PKR catalytic domain. The targeting vector was originally introduced into a J1 embryonic stem cell line. Chimeric mice were obtained after microinjecting the embryonic stem cell line into BALB/c donor blastocysts that were subsequently implanted into CD1 foster mothers. PKR−/− mice have been backcrossed with C57BL/6J mice for more than 20 generations, and genotyping confirmed that experimental animals were missing Exon 12 (supplementary Fig. 1).
All mice were group housed until surgery, after which they were housed singly. Mice were maintained on a 12:12 h light:dark cycle at an ambient temperature of 25 °C (±2°C). Food and water were available ad libitum. Male PKR −/− (n = 6) and C57BL/6J (n = 7) mice were enrolled into the phenotyping protocol (see later) when their body weights were between 21–27 g. All procedures involving the use of animals were approved by the University of Colorado Institutional Animal Care and Use Committee in accordance with the U.S. Department of Agriculture Animal Welfare Act and the National Institutes of Health policy on Humane Care and Use of Laboratory Animals.
2.2. Substances
Pyrogen-free saline (PFS; Abbot Laboratories, North Chicago, IL) was used as vehicle for control injections. Lipopolysaccharide (LPS, Escherichia coli serotype 0111:B4) was purchased from Millipore Sigma (Burlington, Massachusetts), reconstituted in PFS, aliquoted, and frozen until use.
2.3. Surgical procedures
Under isoflurane anesthesia (4 % induction, 2 % maintenance), four stainless steel electroencephalogram (EEG) recording electrodes were implanted epidurally over the frontal and parietal cortices. The leads from the electrodes were soldered to pins of a plastic integrated circuit connector that was subsequently embedded in dental acrylic. During this procedure, animals were also implanted intraperitoneally (IP) with an RFID transponder that had an integrated temperature biosensor (UI Devices, Lake Villa, IL). All surgical incisions were treated with a topical analgesic (4 % lidocaine) and a triple antibiotic ointment (neomycin / polymyxin B / Bacitracin). Mice were allowed one week of surgical recovery, after which they were connected to the recording apparatus via a flexible tether for habituation.
2.4. Experimental protocol
Animals were connected to the recording system with light-weight flexible tethers for a minimum of three days adaptation. After adaptation, 48 h recordings were obtained from undisturbed animals. These recordings served to ascertain the impact of PKR deficiency on undisturbed sleep-wake behavior and as the control for sleep deprivation (Fig. 1). At light onset immediately following the baseline recording, mice were subjected to six hours of sleep deprivation by gentle handling. During sleep deprivation, mice were continuously observed and whenever behavioral signs of sleep were observed a preventive intervention was performed and noted. Interventions to prevent sleep during the deprivation period ranged from low intensity (mere presence of laboratory personnel; light tapping on the outside of the cage) to higher intensity (introducing a novel wad of paper into the cage; moving the wad of paper; gently touching the mouse with a cotton-tipped swab) as necessary, based on the behavior of the animal: the longer the deprivation the greater was the intensity and frequency of the intervention required to keep the animal awake. At dark onset on protocol day four, mice were injected intraperitoneally with vehicle (PFS) and 24-h control recordings obtained. Finally, at dark onset on protocol day five, mice were injected intraperitoneally with LPS, and recordings continued for 24 h, at which time the protocol ended (Fig. 1).
Fig. 1.

Experimental Protocol. After surgical recovery and habituation, C57BL/6J mice and PKR knockout (PKR−/−) mice were enrolled into a six-day recording protocol. Baseline recordings from undisturbed mice started at light onset and continued for 48 h. After baseline recordings, all mice were sleep deprived for six hours beginning at light onset and then left undisturbed for an 18-h recovery sleep opportunity. All mice then were injected intraperitoneally with vehicle (PFS, pyrogenfree saline) at dark onset of protocol day four and with lipopolysaccharide (LPS) 24 h later, at dark onset of protocol day five. Recordings continued for the 24-h period following LPS administration, after which the protocol ended. In this protocol each animal served as its own control. (Figure created in Biorender.com).
2.5. Determination of sleep-wake behavior
EEG and cage activity recordings were used to determine sleep-wake behavior of mice. Signals obtained from the recording electrodes were amplified (factor of 5000; model P511AC, Grass Telefactor, West Warwick, RI), filtered (line filter: notch type, 60 Hz; low band pass filter: −6 db, 0.3 Hz; high band pass filter: −6 db, 30 Hz), and recorded for offline processing using the ICELUS (written in LabView for Windows, M. Opp, University of Washington, Seattle, WA; National Instruments, Austin TX). Cage activity was detected using an observation unit with an infrared sensor (BioBserve, GmbH, Bonn, Germany). Movement detected by the infrared sensor was converted to a voltage output directly proportional to the amount of movement detected. All EEG and movement signals were converted from analog to digital (A/D board: PCI:3033E, National Instruments, TX) and stored as binary files for later analysis.
The EEG and cage activity records were visually scored in 10-s epochs to determine arousal state. Determination of sleep – wake behavior was done by a fully trained and experienced investigator who was blinded with respect to animal ID and experimental condition until all processing of EEG data was completed. Arousal state was designated as non-rapid eye movement sleep (NREMS), rapid eye movement sleep (REMS), or WAKE, as previously published (Baracchi and Opp, 2008). Briefly, NREMS was identified by large amplitude, low frequency (<5 Hz) EEG waveforms and lack of body movement. REMS was characterized by low-amplitude, 6 – 9 Hz EEG waveforms, whereas WAKE was defined based on cage activity / body movements and a mixed frequency EEG.
2.6. Lps-induced cytokine responses
Groups of male PKR−/− (n = 7) and C57BL/6J mice (n = 10) were used to determine acute cytokine responses to immune challenge. These mice were group-housed and habituated for one week in the same environmental conditions as those used for mice from which sleep – wake behavior was determined. At the end of habituation, each animal was injected intraperitoneally with 0.4 mg/kg LPS. Two hours after LPS injection, a terminal blood sample was obtained. Animals were first injected intraperitoneally with 0.05 ml of Euthasol®, and once vital signs and responses to pain were diminished blood was collected into a Vacutainer® tube containing EDTA using 1 ml syringes. All blood samples were kept on ice for less than 30 min before centrifugation (3220 × g, 10 min, 4 °C) to obtain plasma. Plasma was aliquoted and frozen at −80 °C until assay.
Concentrations of circulating interleukin (IL)-1β, IL-6, tumor necrosis factor (TNF)-α and interferon (IFN)-γ were quantified using a high sensitivity magnetic bead T cell multiplex panel (MILLIPLEX®; Millipore, Burlington, NY, Catalog ID. MHSTCMAG-70 K-02) according to the manufacturer’s instructions. A separate kit was used to determine concentrations of IL-4 and IL-10. All samples were assayed in duplicate using a Bio-Plex 200 system (BioRad Laboratories, Inc.; Hercules, CA). Cytokine data were analyzed with Bio-Plex Manager 4.1 software with seven-parameter logistic regression curve fitting. Plasma concentrations of cytokines are reported as pg/ml.
2.7. Statistical analysis
Results are presented as mean ± standard error of the mean. All statistical analyses and data figures were made in GraphPad Prism (v 9.4.0 for Windows; GraphPad Software; San Diego, CA). Comparisons between conditions were made with a two-way repeated measures ANOVA with Bonferroni’s multiple comparison test. A mixed-effects model was used for analyses of temperature data and for spectral EEG analyses (delta power during NREM sleep) because of missing data during some time points for some animals. For between strain comparisons of spontaneous (undisturbed) sleep-wake behavior, “mouse strain” was the fixed effect; outcome measures (body temperature, time spent in each sleep stage, EEG parameters) the dependent variables; and time blocks (hours) the repeated measure. For comparisons after manipulation, the manipulation (sleep deprivation; LPS administration) was the fixed effect; outcome measures (body temperature, time spent in each sleep stage, EEG parameters) the dependent variables; and time blocks (hours) the repeated measure. Data from the 12-h light period and from the 12-h dark period were analyzed independently. An α level of 0.05 was set for all tests as indicating statistically significant differences between conditions / strains / outcome measures. Test statistics for all experiments are provided in Supplemental Tables. In some cases, visual inspection of the data revealed instances when individual time blocks appeared to deviate from control values in a meaningful way. In these instances, a single time block was evaluated with a two-way ANOVA. All such cases are noted, and test statistics for these analyses are presented in the text of the results section.
3. Results
3.1. Spontaneous sleep-wake behavior
EEG and core body temperature data were recorded for 48 h, during which time mice were not disturbed. Subsequent analyses indicated that values obtained during these two 24 h periods did not differ statistically for any parameter. As such, hourly values from individual mice for each 24 h period for each parameter were averaged to create a single 24 h undisturbed baseline sleep-wake and core body temperature profile.
During the undisturbed baseline period, both mouse strains (C57BL/6J, PKR−/−) exhibited diurnal variations in sleep-wake behavior and core body temperature that are typical of laboratory rodents: more NREMS and REMS during the light period and more WAKE and higher core body temperatures during the dark period (Fig. 2). Except where otherwise noted, statistical analyses were performed on consecutive 3-h time blocks across the 24 h recording period. Subtle differences were found between the strains, specifically during the first three hours after dark onset. Relative to C57BL/6J mice, PKR−/− mice spent less time awake (C57BL/6J, 55.7 ± 2.8 min; PKR−/− 48.2 ± 4.7 min; F=14.24, P=0.0006) and more time in NREMS and REMS (NREMS: C57BL/6J, 4.0 ± 2.7 min; PKR−/−, 10.5 ± 4.0 min; F=12.69, P=0.0011; REMS: C57BL/6J, 0.31 ± 0.13 min; PKR−/−, 1.2 ± 0.6 min; F=13.24, P=0.0009) during this three hour time block (Fig. 2A, B, C).
Fig. 2.
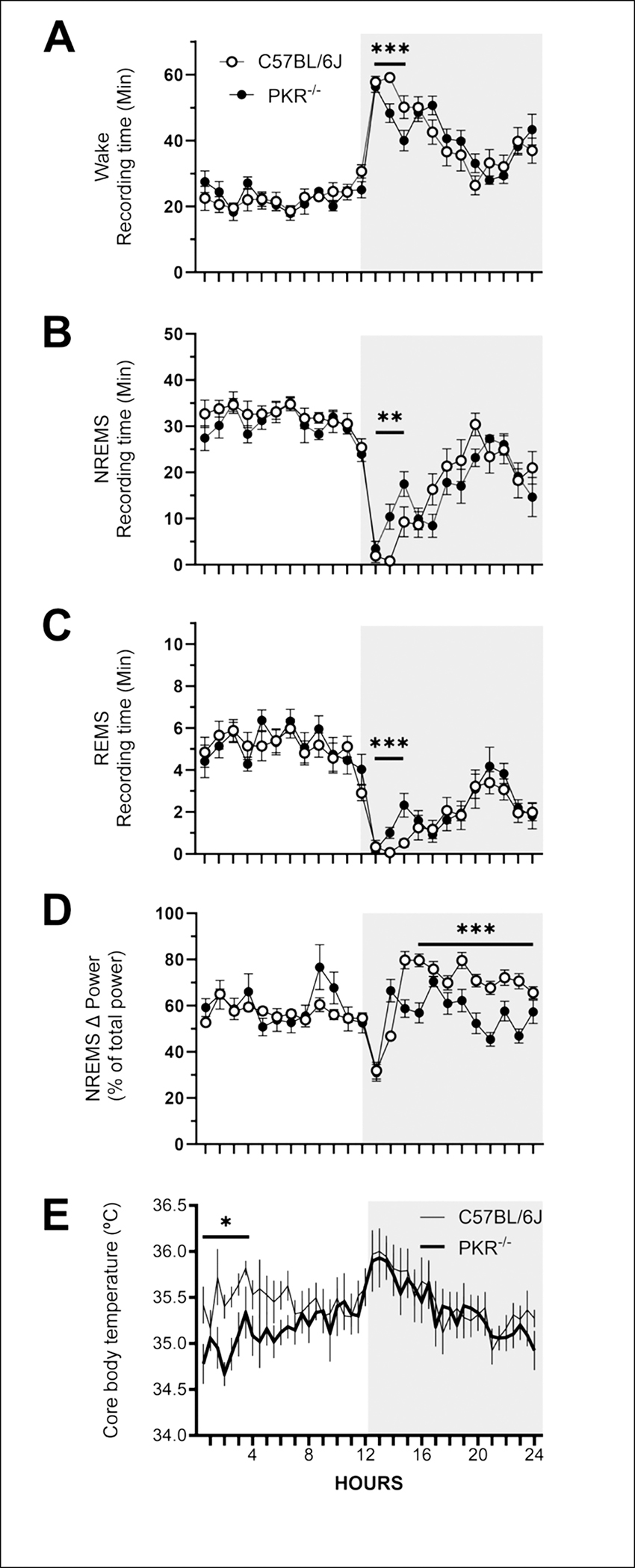
PKR knockout mice exhibit a diurnal pattern of wakefulness, non-rapid eye movement sleep (NREMS), rapid eye movement sleep (REMS) and core body temperature. Data are expressed as mean ± SEM percentage of recording time obtained from C57BL/6J mice (open symbols; n = 7) and PKR knockout mice (PKR−/−, filled symbols; n = 6) during undisturbed baseline recordings. All sleep parameters are presented as hourly values whereas core body temperature is depicted in 30 min intervals. Statistical analyses were performed on sequential 3-h time blocks. Sample sizes for core body temperature measures are n = 6 (C57BL/6J) and n = 5 (PKR−/−) due to failed temperature transponders. Statistically significant differences between C57BL/6Jmice and PKR−/− mice are depicted as: *, p < 0.05; **, p < 0.01; *** p < 0.001). The shaded portion of each panel represents the dark period of the light:dark cycle.
Because the EEG fundamentally varies among mice, values for delta power (0.5 – 5 Hz) during NREMS were normalized for each animal. Specifically, total spectral power from 0.5 – 20 Hz was calculated by summing values from all frequency bins within each 10-s epoch across the 24 h baseline period. Delta power during NREMS was then expressed as a percentage of the total spectral power for each epoch for each animal. Statistical analyses of normalized spectra did not reveal strain differences in NREMS delta power during the light period (Fig. 2D). In contrast, NREMS delta power in PKR−/− mice was lower than that of C57BL/6J mice during the last nine hours of the dark period (C57BL/6J, 72.4 ± 1.6 %; PKR−/−, 56.7 ± 2.6 %; F=85.61, P<0.0001; Fig. 2D).
All RFID transponders used to record core body temperature were calibrated prior to surgical implantation. However, because of their small size, when implanted in the peritoneal cavity in some cases the transponder can migrate through the inguinal canal to the scrotum of male mice. In these cases, the transponder does not record a ‘true’ body temperature. We determined that two transponders had migrated out of the peritoneal cavity and recorded core body temperatures that were lower than obtained from the rest of the mice. We omitted temperature data from one C57BL/6J mouse and from one PKR−/− mouse. The core body temperature data from these two mice was omitted for all conditions used in this study. In addition, although we calibrated the transponders prior to surgical implantation, there was a significant amount of variability among temperature recordings when using these RFID devices. That said, as previously reported core body temperature for both mouse strains varied in a diurnal-dependent manner; body temperature was lower during the light (inactive) period and higher during the dark (active) period. Relative to C57BL/6J mice, core body temperature values from PKR−/− mice were significantly lower during the first three hours of the light period (C57BL/6J, 35.5 ± 0.08°C; PKR−/−, 34.9 ± 0.07°C; F=7.358, P<0.02; Fig. 2E).
3.2. Responses to sleep deprivation
Our sleep deprivation method was effective as mice were kept awake for ~ 95 % of the six-hour period. Responses to sleep deprivation were analyzed in two ways. First, comparisons were made between data obtained during undisturbed baseline and corresponding values after sleep deprivation within each mouse strain. That is, data obtained from each animal after sleep deprivation were compared to baseline data from the same animal during corresponding time periods (Fig. 3). Second, difference scores were calculated for each parameter and each mouse for each hour by subtracting values obtained during undisturbed baseline recordings from those obtained after sleep deprivation at corresponding time points. These difference scores were then analyzed to determine differences between mouse strains (Fig. 4).
Fig. 3.

Sleep deprivation reduces subsequent wakefulness and increases non-rapid eye movement sleep (NREMS) and rapid eye movement sleep (REMS) in mice. Data are the mean ± SEM from C57BL/6J mice (left panels, n = 7) and PKR knockout mice (PKR−/−; right panels, n = 6) mice during undisturbed baseline recordings (open symbols) and after sleep deprivation (filled symbols). All sleep parameters are presented as hourly values whereas core body temperature is depicted in 30 min intervals. Data for sleep parameters and core body temperature are not shown during the 6-h sleep deprivation period. Statistical analyses were performed on sequential 3-h time blocks. Sample sizes for core body temperature measures are n = 6 (C57BL/6J) and n = 5 (PKR−/−) due to failed temperature transponders. Statistically significant differences between baseline and sleep deprivation are depicted as: *, p < 0.05; **, p < 0.01. The shaded portion of each panel represents the dark period of the light:dark cycle. BL=baseline undisturbed recordings; SD=sleep deprivation.
Fig. 4.

Loss of PKR function alters some aspects of sleep-wake responses to sleep deprivation. To determine the effect of genotype on responses to sleep deprivation, difference scores were calculated for each mouse for each hour by subtracting control values (undisturbed baseline) from experimental values obtained after sleep deprivation. Data are expressed as mean ± SEM from C57BL/6J mice (open symbols, n = 7) and PKR knockout mice (PKR−/−; filled symbols, n = 6) during undisturbed baseline and after 6-h of sleep deprivation. All sleep parameters are presented as hourly values whereas core body temperature is depicted in 30 min intervals. Data are not depicted for the 6-h period of sleep deprivation. Statistical analyses were performed on sequential 3-h time blocks. Sample sizes for core body temperature measures are n = 6 (C57BL/6J) and n = 5 (PKR−/−) due to failed temperature transponders. Statistically significant differences between mouse strains are: *, p < 0.05; **, p < 0.01. The shaded portion of each panel represents the dark period of the light:dark cycle.
Both mouse strains exhibited increased NREMS and REMS (e.g., sleep rebound) and reduced WAKE after sleep deprivation (Fig. 3A – H), although the timing differed between strains. Compared to corresponding baseline values, during the first six hours after sleep deprivation, C57BL/6J mice spent more time in NREMS (hours 7 – 9: baseline 32.8 ± 1.0 min; sleep deprivation 39.1 ± 1.2 min; F=19.34, P<0.0001; hours 10 – 12: baseline 28.9 ± 1.8 min; sleep deprivation 32.1 ± 3.2 min, F=4.120, P=0.0498) and REMS (hours 7 – 9: baseline 5.3 ± 0.3 min; sleep deprivation 6.5 ± 0.3 min; F=5.671, P=0.0227; hours 10 – 12: baseline mean = 4.2 ± 0.7 min; sleep deprivation mean = 5.8 ± 0.8 min; F=7.924, P=0.0079) (Fig. 3C, E). In contrast, PKR−/− mice predominantly exhibited rebounds in NREMS and REMS during the dark phase (NREMS: hours 16 – 18; baseline 12.1 ± 2.9 min; sleep deprivation 19.4 ± 4.5 min; F=7.228, P<0.0116; hours 19 – 21 baseline 22.50 ± 2.9 min; sleep deprivation 28.1 ± 2.4 min; F=4.369, P=0.0452) and REMS (REMS: hours 16 – 18; baseline 1.4 ± 0.3 min; sleep deprivation 3.4 ± 0.8 min, F=12.21, P<0.0015; hours 19 – 21 baseline 3.1 ± 0.7 min; sleep deprivation 4.9 ± 0.4 min, F=7.277, P=0.0113; Fig. 3D, F). These increases in sleep paralleled reductions in WAKE for both strains, although reductions in WAKE of C57BL/6J mice reached statistical significance during the light period (hours 7 – 9: baseline 21.5 ± 1.4 min; sleep deprivation 14.4 ± 1.4 min; F=17.40, P=0.0002; hours 10 – 12: baseline 26.5 ± 2.1 min; sleep deprivation 22.3 ± 3.9 min, F=5.302, P=0.0272) whereas PKR−/− mice expressed this reduction during the dark period (hours 16 – 18: baseline 46.6 ± 3.1 min; sleep deprivation 37.2 ± 5.2 min; F=8.527, P=0.0066; hours 19 – 21: baseline 33.6 ± 3.4 min; sleep deprivation 26.9 ± 2.7 min, F=4.699, P=0.0383) (Fig. 3A, B).
Delta power during NREMS in C57BL/6J mice increased during the first three hours after sleep deprivation (baseline, 56.9 ± 1.9 %; sleep deprivation, 64.6 ± 4.7 %; F=22.42, P<0.0001; Fig. 3G). Most of this increase in delta power during NREMS occurred during the first hour following sleep deprivation, during which there was a 30.1 % increase relative to the corresponding baseline values (Fig. 3D). Delta power during NREMS did not differ from baseline values during the remainder of the light period, post-deprivation hours 10 – 12. However, during the dark period, delta power during NREMS of C57BL/6J mice was generally reduced compared to the corresponding baseline hours (hours 16 – 18 baseline, 75.1 ± 2.8 %; sleep deprivation 58.9 ± 1.1 %, F=58.87, P<0.0001; hours 19 – 21 baseline, 72.7 ± 3.5 %; sleep deprivation 61.5 ± 3.5 %, F=28.40, P=0.0498; Fig. 3G).
In contrast to C57BL/6J mice, delta power during NREMS in PKR−/− mice increased only during the first hour after sleep deprivation (baseline, 59.0 ± 7.9 %; sleep deprivation, 89.0 ± 12.4 %, F=2.480, P=0.0005; Fig. 3H). For the remainder of the light period, delta power during NREMS was generally reduced relative to corresponding baseline values (hours 10 – 12 baseline, 60.8 ± 7.5 %; sleep deprivation 53.6 ± 2.1 %, F=8.670, P=0.0062). During the dark period, significant increases in delta power during NREMS were observed during the first six hours (hours 13 – 15 baseline, 59.9 ± 10.0 %; sleep deprivation 69.1 ± 6.0 %, F=7.514, P=0.0102; hours 16 – 18 baseline, 63.6 ± 4.1 %; sleep deprivation 83.2 ± 4.9 %, F=18.04, P=0.0002), which was followed by modest but statistically significant reductions for the remainder of the dark period (hours 19 – 21 baseline, 61.7 ± 5.0 %; sleep deprivation 57.5 ± 1.4 %, F=6.488, P=0.0162; hours 22 – 24 baseline, 59.3 ± 3.5 %; sleep deprivation 52.4 ± 1.3 %, F=10.94, P=0.0025; Fig. 3H).
There was a tendency for reduced core body temperature of C57BL/6J mice throughout the entire post-deprivation recovery period that did not reach statistical significance (Fig. 3I).
Core body temperature of PKR−/− mice was reduced after sleep deprivation and remained lower than baseline values throughout the remainder of the light period (baseline, 35.3 ± 0.23°C; sleep deprivation, 34.8 ± 0.23°C; F=5.539, P=0.05; Fig. 3J). No significant differences in core body temperatures were observed during the subsequent dark period.
To directly compare response to sleep deprivation between genotypes, we analyzed difference scores between values after sleep deprivation and corresponding values during undisturbed baseline for each hour for each mouse (Fig. 4). In general, no consistent statistically significant differences were found in the response between strains after sleep deprivation for NREMS, REMS, or WAKE (Fig. 4A, B, C). Delta power values during NREMS after sleep deprivation were greater in PKR−/− mice during the first hour after sleep deprivation (C57BL/6J: 14.4 ± 6.6 %; PKR−/− 30.3 ± 18.1 %, F=7.514, P=0.0363; Fig. 4D) and during the six hours after the beginning of the dark period (hours 13 – 15 C57BL/6J −29.5 ± 22.8 %;/PKR−/− −13.0 ± 13.1 %, F=16.86, P=0.0003; hours 16 – 18 C57BL/6J −10.4 ± 7.3 %, PKR−/− 9.6 ± 11.9 %, F=12.07, P=0.0016; Fig. 4D). Core body temperature between strains did not differ between the two strains (Fig. 4E).
3.3. Lps-induced alterations in sleep-wake behavior and core body temperature
Intraperitoneal administration of LPS at dark onset altered sleep-wake behavior and EEG parameters in both mouse strains, consistent with previous reports (Ingiosi and Opp, 2016; Morrow and Opp, 2005). Specifically, LPS administration increased NREMS, suppressed REMS, and reduced WAKE (Fig. 5A – F). The changes in WAKE and NREMS were generally limited to the 12-h dark period following LPS administration, whereas REMS suppression persisted for most of the recording period (see supplemental tables 5 and 6 for statistical values).
Fig. 5.

Lipopolysaccharide increases non-rapid eye movement sleep (NREMS), reduces wakefulness and rapid eye movement sleep (REMS) and alters core body temperature of mice. Data are expressed as mean ± SEM from C57BL/6J mice (left panels) and PKR knockout mice (PKR−/−; right panels, n = 6) after administration of vehicle (pyrogen-free saline; open symbols) and lipopolysaccharide (LPS; filled symbols). All sleep parameters are presented as hourly values whereas core body temperature is depicted in 30 min intervals. Statistical analyses were performed on sequential 3-h time blocks. Sample sizes for core body temperature measures are n = 6 (C57BL/6J) and n = 5 (PKR−/−) due to failed temperature transponders. Statistically significant differences between mouse strains are: *, p < 0.05; **, p < 0.01; ***, p < 0.001 The shaded portion of each panel represents the dark period of the light:dark cycle.
Delta power during NREMS was dramatically reduced in both mouse strains for essentially the entire 24-h recording period after LPS administration (see supplemental tables 5 and 6 for statistical values; Fig. 5G, H). In C57BL/6J mice, there was a tendency for a biphasic temperature response LPS; an initial hypothermia followed by increased core body temperature, changes of which did not achieve statistical significance (Fig. 5I). There were no apparent changes in core body temperature of PKR−/− mice after LPS administration (Fig. 5J).
To directly compare genotype responses to LPS we calculated differences scores by subtracting values obtained after vehicle administration (control; PFS) from values obtained after LPS administration (experimental). Modest differences were detected between strains for sleep-wake behavior. The initial LPS-induced reduction in wakefulness was less pronounced in PKR−/− mice during the first 3 h after injection as compared to C57BL/6J mice (C57BL/6J −18.0 ± 7.8 min; PKR−/−, 7.4 ± 7.3 min, F=5.331, P=0.0274; Fig. 6A). There were no consistent statistically significant strain differences in WAKE, NREMS or REMS for the remainder of the recording period. However, there were some strain differences in delta power during NREMS, although they were not consistent (Fig. 6D). The initial suppression of delta power during NREMS after LPS administration was of greater magnitude in PKR−/− mice than in C57BL/6J mice (hours 1 – 3 C57BL/6J –23.2 ± 9.3 %; PKR−/−, −35.9 ± 11.4 %, F=11.12, P=0.0023. Strain differences in the extent to which LPS suppressed delta power during NREMS varied during the remainder of the recording period.
Fig. 6.

Loss of PKR function reduces the impact of lipopolysaccharide on some facets of sleep-wake behavior and core body temperature. Data are expressed as mean ± SEM from C57BL/6J mice (open symbols, n = 7; core body temperature n = 7, thin line) and PKR knockout mice (PKR−/−; filled symbols, n = 6; core body temperature n = 6, thick line). All sleep parameters are presented as hourly values whereas core body temperature is depicted in 30 min intervals. Statistical analyses were performed on sequential 3-h time blocks. Sample sizes for core body temperature measures are n = 6 (C57BL/6J) and n = 5 (PKR−/−) due to failed temperature transponders. Statistically significant differences between mouse strains are: *, p < 0.05; **, p < 0.01; ***, p < 0.001. The shaded portion of each panel represents the dark period of the light:dark cycle.
Core body temperature responses to LPS administration were generally similar between strains for most of the 24 h recording period. Relative to C57BL/6J mice, there was a tendency for reduced body temperatures in PKR−/− mice during the latter half of the light period that did not reach statistical significance (.
3.4. Lps-induced cytokine expression
Intraperitoneal LPS injection into mice induces a transient systemic inflammatory response that is characterized by increased circulating cytokines. We compared plasma concentrations of IL-1β, IL-6, TNF-α, IFN-γ, IL-4 and IL-10 in samples collected two hours after LPS injection. There were no genotype differences in plasma cytokine concentrations after LPS administration (Fig. 7).
Fig. 7.

Loss of PKR function does not alter lipopolysaccharide-induced changes in pro- and anti-inflammatory cytokines. Plasma concentrations of pro-inflammatory (IL-1β, IL-6, TNFα, IFNγ) and anti-inflammatory (IL-4, IL-10) cytokines following intraperitoneal administration of lipopolysaccharide (LPS; 0.4 mg/kg) did not differ between mouse strains. (C57BL/6J; n = 10; open bars; PKR−/−; n = 7; filled bars).
4. Discussion
Results of this study demonstrate that PKR deficiency transiently alters spontaneous sleep-wake behavior after the transition from light-to-dark periods and reduces core body temperature during the light period. Although the lack of PKR increases NREMS and REMS during the early portion of the dark (active) period of mice, the sleep of these animals is of ‘poor quality’ as delta power during NREMS, often used as a measure of sleep depth, is reduced during nighttime sleep. When PKR is absent, sleep pressure during prolonged wakefulness (i.e., sleep deprivation) increases to a greater extent, as evidenced by elevated delta power during NREMS immediately after the end of sleep deprivation. Although PKR has some impact on sleep and temperature responses to LPS, its role is not as substantive as we initially hypothesized. These results demonstrate that PKR modulates some, not all, aspects of sleep-wake behavior and responses to homeostatic and immune challenge in mice.
4.1. Spontaneous sleep and responses to sleep deprivation
PKR is a ubiquitous kinase that plays a role in multiple cellular and systemic processes, including inflammation (Gal-Ben-Ari et al., 2019; García et al., 2007). Recent data suggest that PKR may be a therapeutic target to delay the initiation and progression of neurodegenerative diseases because of its central role in the production of pro- and anti-inflammatory cytokines (Couturier et al., 2010; Dumurgier et al., 2013; Gal-Ben-Ari et al., 2019; Hwang et al., 2017; Segev et al., 2015). Our interest in PKR as an inflammatory mediator stems from its ability to modulate the production of cytokines known to be involved in the etiology of neurodegenerative disease (Khandelwal et al., 2011; Lu et al., 2012) and in the regulation of sleep (Irwin and Opp, 2017). Cytokines, and their receptors are in brain regions involved in the regulation of sleep-wake behavior (Breder et al., 1993, 1988; Krueger, 2017), and there is ample evidence that interleukin-1β (IL-1β) and tumor necrosis factor alpha (TNF-α) are involved in the regulation of spontaneous physiological sleep and in the alterations in sleep that follow immune challenge (Baracchi and Opp, 2008; Ingiosi and Opp, 2016; Irwin and Opp, 2017; Krueger, 2017).
Although the duration of NREMS and REMS of PKR−/− mice transiently increases during the early portion of the dark period, this sleep does not appear to be of ‘good quality”. During the dark period, delta power during NREMS in PKR−/− mice is consistently reduced relative to that of genetically intact C57BL/6J mice. Delta power during NREMS is accepted as a measure of sleep homeostasis reflecting the depth or intensity of sleep (e.g., Davis et al., 2011). Our previous study in rats (Imeri et al., 2006) revealed that inhibiting caspase-1 activity reduces slow-wave activity during NREMS during the dark period. As such, the reduction of delta power during NREMS in spontaneously sleeping PKR−/− mice in this present study generally agrees with the impact of inhibiting (upstream) caspase-1 on this sleep parameter. Collectively, these data suggest that PKR contributes differently to behavioral aspects of sleep (increased duration of NREMS and REMS) and to EEG parameters that reflect sleep quality (reduced delta power during NREMS).
PKR deficiency impacts spontaneous sleep-wake behavior only during the early dark period of the diurnal cycle. One possible explanation for changes in sleep-wake behavior following the light-to-dark transition may be related to timing. PKR is one molecule that modulates eukaryotic initiation factor 2 and its α subunit (eIF2α). eIF2α is an essential modulator of the initiation phase of mRNA translation, which indirectly modulates per2 transcription via translational control of ATF4 (Harding et al., 2000; Pathak et al., 2019). Per2 is a component of the molecular machinery comprising the circadian clock (Albrecht et al., 1997). Since PKR is activated by molecules that have circadian expression patterns, e. g., cytokines (Gal-Ben-Ari et al., 2019), eIF2α may be phosphorylated at a different rate in PKR−/− mice, altering the transcription of per2, which in turn could affect the timing of the sleep-wake cycle. The present study does not address mechanisms by which the lack of PKR might affect circadian aspects of the timing of sleep-wake behavior and these hypotheses remain to be tested.
PKR deficiency impacts core body temperature of undisturbed mice. During the first half of the light period, core body temperature of PKR−/− mice is consistently lower than that of genetically intact C57BL/6J mice. Reduced core body temperature of PKR−/− mice during the light period is not a byproduct of activity, since sleep-wake behavior does not differ between the two strains during this diurnal phase. These data suggest that PKR plays a role in physiological thermoregulatory mechanisms that differs from those involved in sleep regulation. Several cytokines involved in thermoregulatory regulation, e.g. IL-1α, IL-1β, TNF-α, TNFβ and IL-6 (Bennett and Benson, 1953; Dinarello, 2004), exhibit diurnal rhythms (Floyd and Krueger, 1997; Nilsonne et al., 2016; Taishi et al., 1997). As briefly summarized, PKR is a component of signaling pathways that result in the induction of IL-1β, and activation of PKR is modulated by molecules expressed in a circadian manner (Chukwurah et al., 2021; Gal-Ben-Ari et al., 2019; Kang and Tang, 2012). The extent to which PKR participates in regulating core body temperature and the timing of those processes requires additional investigation.
Comparisons between undisturbed spontaneous sleep and responses to sleep deprivation reveal that both mouse strains respond similarly to this homeostatic challenge with increased NREMS and REMS, reduced wakefulness, and changes in delta power during NREMS. However, the timing of these responses differs between strains. C57BL/6J mice in the present study respond to sleep deprivation in a manner consistent with previous reports (Baracchi and Opp, 2008; Morrow and Opp, 2005b). In contrast, the increase in NREMS in PKR−/− mice after sleep deprivation occurs during the subsequent dark period, much later than observed in C57BL/6J mice. Although both mouse strains respond to sleep deprivation with increased NREMS, REMS and reduced wakefulness at various time points during the post-deprivation recovery period, the magnitude of these responses does not differ between genotypes.
Acute sleep deprivation is frequently used to determine homeostatic responses to prolonged wakefulness. The primary readout of sleep homeostasis is an EEG measure of the spectral power in the delta (0.5 – 5 Hz) frequency band. Delta power during NREMS indicates sleep depth or intensity. Because it is an intensity measure, the extent to which delta power during NREMS increases between one sleep bout and the next depends on the duration of intervening wakefulness. Therefore, this measure reflects the buildup of ‘sleep pressure’ that occurs during wakefulness and is dissipated during subsequent sleep. Both mouse strains respond to sleep deprivation with increased delta power during NREMS (rise time of sleep pressure) that declines during subsequent sleep (decay or dissipation of sleep pressure). However, during the first hour of sleep after sleep deprivation, delta power during NREMS is greater in PKR−/− mice than in C57BL/6J mice, suggesting that homeostatic sleep pressure builds up more rapidly when PKR function is inhibited. Although sleep pressure increases more quickly during prolonged wakefulness in PKR−/− mice, its dissipation during subsequent sleep is the same as that of genetically intact C57BL/J6 mice. These data suggest that although PKR deficiency may be detrimental during prolonged wakefulness (getting sleepier more quickly) recovery processes are not impaired.
4.2. Lps-induced alterations in sleep and core body temperature
Multiple immune stimuli, including bacterial and viral infections, injury, and others (Kang and Tang, 2012) activate PKR, and it is a component of signaling pathways involved in inflammation and the regulation of immunity. Immune activation alters sleep irrespective of whether the stimulus is infection with replicating pathogens or administration of immunomodulators (Imeri and Opp, 2009). Immune activation by immunomodulators alters sleep in a manner that differs from responses to a replicating pathogen. Within the context of infection, PKR is best known for its anti-viral actions, but it also plays a role in bacterial infections, including those with E. coli (e.g., Smyth and Sun, 2021). LPS is a component of Gram-negative E.coli bacteria cell walls that induces responses closely resembling those of infection without all the sequelae associated with processes driven by replicating pathogens, a so-called ‘sterile infection’. For these reasons, we used LPS to determine the impact of interfering with this signaling pathway on sleep and temperature responses to immune challenge. Previous reports demonstrate the extent to which LPS alters sleep of mice (Ingiosi and Opp, 2016; Lancel et al., 1995; Morrow and Opp, 2005a) and in this present study we hypothesized that responses to LPS would be attenuated in PKR−/− mice because of impaired PKR signaling.
We hypothesized that PKR deficiency would attenuate responses to LPS challenge because it is auto-phosphorylated in response to pathogen-associated molecular patterns (PAMPS) and damage-associated molecular patterns (DAMPS). Once phosphorylated, PKR acts on the inflammasome, which induces caspase-1 (Franchi et al., 2009). Caspase-1 then cleaves pro-IL-1β into its biologically active form. As mentioned, the role of IL-1β in regulating / modulating spontaneous physiological sleep is well documented (Baracchi and Opp, 2008; Ingiosi and Opp, 2016; Irwin and Opp 2017). We previously reported that inhibiting caspase-1 during the dark phase reduces spontaneous NREMS and REMS, and increases wakefulness of rats (Imeri et al., 2006). The impact of inhibition of caspase-1 on sleep-wake behavior presumably results from a reduction in biologically active IL-1β. Because it is upstream of caspase-1 in this signaling cascade, we hypothesized that impaired PKR function would affect sleep in a manner like caspase-1 inhibition. Contrary to our hypothesis, data herein demonstrate that PKR deficiency reduces wakefulness and increases NREMS and REMS. Mechanisms by which PKR deficiency increases, rather than decreases spontaneous sleep of mice are not clear. The literature is mixed concerning a putative role for PKR in inflammasome activation. For example, some reports demonstrate a role for PKR in activation of NFκB and caspase-1 pathways (Bonnet et al., 2000; Franchi et al., 2009; Gal-Ben-Ari et al., 2019; Lu et al., 2012; Kang and Tang, 2012), whereas others do not (He et al., 2013; Abraham et al., 1999). Our data are consistent with reports that PKR is not necessary for inflammasome / caspase-1 activation in response to LPS challenge for two reasons. First, LPS-induced alterations in sleep-wake behavior are mediated by inflammatory mediators, notably IL-1β and TNF-α, and sleep of PKR−/− mice is not dramatically altered in response to this immune challenge. Second, in our study, LPS-induced increases in plasma IL-1β, IL-6 and TNF-α are virtually identical in PKR−/− mice and in genetically intact C57BL/6J mice. That LPS-induced alterations in sleep-wake behavior and plasma pro-inflammatory cytokines are not dramatically attenuated / inhibited in PKR−/− mice suggests that activation of the inflammasome and / or the canonical NFκB pathway is not impaired in these animals.
As with other immune challenges (Krueger et al., 1994; Krueger and Opp, 2016) there are diurnal differences in responses to LPS (Mathias et al., 2000; Morrow and Opp, 2005a). In this study we administered LPS at dark onset because this is the active phase of laboratory rodents during which they sleep the least. As such, LPS administration at this time point is expected to increase NREMS. Both mouse genotypes respond similarly to LPS with a prolonged increase in NREMS and concurrent suppression of REMS and wakefulness. Sleep of C57BL/6J mice in this study was altered in a manner nearly identical to that previously reported for this mouse strain using the same dose and serotype of LPS (Ingiosi and Opp, 2016; Morrow and Opp, 2005a) with increased NREMS for 12 h and suppressed REMS for almost 20 h post injection. Because there are no significant genotype differences, these data suggest that PKR does not play a role in LPS-induced alterations in sleep-wake behavior.
As previously discussed, strain differences in sleep-wake behavior are less apparent than hypothesized based on the putative role of PKR in inflammatory signaling after LPS challenge. Although NREMS is increased to the same extent in both mouse strains, the magnitude of the initial LPS-induced suppression of wakefulness is less in PKR−/− mice. Delta power during NREMS is reduced in both mouse strains, although to a greater extent in PKR−/− mice. These observations suggest that in response to LPS, PKR plays a relatively minor role in some facets of sleep behavior (NREMS duration) and sleep quality (delta power during NREMS). We had anticipated a greater impact of PKR−/− deficiency on responses to LPS, particularly increases in NREMS. Administration of IL-1 increases NREMS when administered to laboratory rodents at dark onset. Because PKR is a component of one signaling pathway by which LPS induces IL-1 production (Lu et al., 2012), we anticipated this response would be attenuated, and as previously discussed these data suggest that other signaling pathways are more critically involved in LPS-induced alterations in sleep.
Administration of LPS impacts thermoregulatory systems in a manner that results in complex changes in brain and core body temperature. We previously reported (Morrow and Opp, 2005a; Ingiosi and Opp, 2016) that administration at dark onset into C57BL/6J mice of the dose and serotype of LPS used in this study induces a biphasic temperature response; an initial hypothermia is subsequently followed by a fever. In the present study, LPS-induced changes in core body temperature did not differ statistically from control values for either genotype. However, the ‘pattern’ of core body temperature responses of C57BL/6J mice to LPS in this study is similar to that which we have previously reported for this mouse strain (Morrow and Opp, 2005a; Ingiosi and Opp, 2016). The LPS-induced changes in sleep in this study are essentially identical to those in our previous reports, indicating the LPS was active and that mice responded appropriately. We attribute the lack of a statistically significant change in core body temperature of C57BL/6J mice in this study, at least in part to the large degree of variability among the RFID transponders. Future studies using different temperature recording devices are necessary to determine if the lack of a core body temperature response in PKR−/− mice in this study reflects biology rather than variability of the recording devices.
5. Conclusions
Collectively, the results of this study demonstrate that PKR plays a role in some facets of spontaneous sleep-wake behavior, responses to sleep loss, and responses to immune challenge. Our data suggest that PKR may play a role in the timing of sleep and in stabilizing sleep homeostasis as indicated by delta power during NREMS.
Although we did not determine the impact of PKR deficiency on brain cytokines, results of this present study suggest that PKR does not appear to play a role in circulating (peripheral) cytokine responses to LPS immune challenge. However, that PKR deficiency does not alter circulating cytokines after LPS challenge does not necessarily mean there would be a similar lack of effect on brain cytokine responses. PKR plays a role in neurodegenerative diseases, which are fundamentally inflammatory pathologies that affect the brain. For example, there is a relatively large literature implicating PKR in the etiology of Alzheimer’s disease. Immunoreactivity for phosphorylated PKR and eIF2α is detected in neurons from post-mortem brain tissue of AD patients; most of these neurons also are immunoreactive for pTAU (Chang et al., 2002). Furthermore, activated PKR is detected in cerebrospinal fluid of AD patients (Dumurgier et al., 2013) and in post-mortem brain (Taga et al., 2017). When PKR is inhibited, cytokine production by peripheral blood mononuclear cells from AD patients is reduced (Couturier et al., 2010). Pre-clinical studies demonstrate that neurons cultured from PKR−/− mice are resistant to Aβ-induced apoptosis (Gourmaud et al., 2016), and cognitive impairment in response to Aβ oligomer injection is reduced in these mice (Lourenco et al., 2013). These, and other data suggest a role for PKR in AD and that targeting PKR may be a viable therapeutic intervention for a broad spectrum of neurodegenerative diseases (Chang et al., 2002; Hwang et al., 2017). Although there is a phenotype, responses of PKR−/− mice to acute sleep loss and to LPS immune challenge are not as dramatic as hypothesized. As such, our data suggest that PKR may not play a major role in the altered sleep that occurs in AD.
Supplementary Material
Acknowledgments
The breeding pairs of PKR−/− mice from which we started our colony were kindly provided by Dr. Eric Klann, Center for Neural Science, New York University. We thank Dr. Shiying Zhou for breeding, genotyping and maintaining the PKR−/− mouse colony. The technical assistance of Amrita George and Lindsey Beauregard is gratefully acknowledged. Dr. Rachel Rowe and Dr. Tabitha Green performed the submandibular punctures for blood collection and engaged in discussions of mechanisms underlying neuroinflammation. This project was funded by award 1RF1AG064465 from the National Institute on Aging of the National Institutes of Health to MRO, CAH, and CL.
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
S. Valencia-Sanchez: Writing – original draft, Visualization, Investigation, Formal analysis, Data curation. M. Davis: Investigation. J. Martensen: Investigation. C. Hoeffer: Supervision, Resources, Project administration, Methodology, Funding acquisition, Conceptualization. C.Link: Writing – review & editing, Supervision, Project administration, Methodology, Funding acquisition, Conceptualization. M.R. Opp: Writing – review & editing, Writing – original draft, Visualization, Supervision, Project administration, Methodology, Funding acquisition, Formal analysis, Conceptualization.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bbi.2024.07.027.
Data availability
Data will be made available on request.
References
- Abraham N, Stojdl DF, Duncan PI, Méthot N, Ishii T, Dubé M, Vanderhyden BC, Atkins HL, Gray DA, McBurney MW, Koromilas AE, Brown EG, Sonenberg N, Bell JC, 1999. Characterization of transgenic mice with targeted disruption of the catalytic domain of the double-stranded RNA-dependent protein kinase, PKR. J. Biol. Chem. 274, 5953–5962. 10.1074/jbc.274.9.5953. [DOI] [PubMed] [Google Scholar]
- Albrecht U, Sun ZS, Eichele G, Lee CC, 1997. A differential response of two putative mammalian circadian regulators, mper1 and mper2, to light. Cell 91, 1055–1064. 10.1016/S0092-8674(00)80495-X. [DOI] [PubMed] [Google Scholar]
- Baracchi F, Opp MR, 2008. Sleep-wake behavior and responses to sleep deprivation of mice lacking both interleukin-1β receptor 1 and tumor necrosis factor-α receptor 1. Brain Behav. Immun. 22, 982–993. 10.1016/j.bbi.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedict C, Byberg L, Cedernaes J, Hogenkamp PS, Giedratis V, Kilander L, Lind L, Lannfelt L, Schiöth HB, 2015. Self-reported sleep disturbance is associated with Alzheimer’s disease risk in men. Alzheimer’s and Dementia 11, 1090–1097. 10.1016/j.jalz.2014.08.104. [DOI] [PubMed] [Google Scholar]
- Bennett IL, Benson PB, 1953. Studies on the pathogenesis of fever. I. The effect of injection of extracts and suspensions of uninfected rabbit tissues upon the body temperature of normal rabbits. J. Exp. Med. 98, 477–492. 10.1084/jem.98.5.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliwise DL, Yesavage JA, Tinklenberg JR, Dement WC, 1989. Sleep apnea in Alzheimer’s disease. Neurobiol. Aging 10, 343–346. 10.1016/0197-4580(89)90046-8. [DOI] [PubMed] [Google Scholar]
- Bonanni E, Maestri M, Tognoni G, Fabbrini M, Nucciarone B, Manca ML, Gori S, Iudice A, Murri L, 2005. Daytime sleepiness in mild and moderate Alzheimer’s disease and its relationship with cognitive impairment. J. Sleep Res. 14, 311–317. 10.1111/j.1365-2869.2005.00462.x. [DOI] [PubMed] [Google Scholar]
- Bonnet MC, Weil R, Dam E, Hovanessian AG, Meurs EF, 2000. PKR Stimulates NF-κB Irrespective of Its Kinase Function by Interacting with the IκB Kinase Complex. Mol. Cell Biol. 20, 4532–4542. 10.1128/mcb.20.13.4532-4542.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breder CD, Dinarello CA, Saper CB, 1988. Interleukin-1 Immunoreactive Innervation of the Human Hypothalamus Published by: American Association for the Advancement of Science Stable URL : https://www.jstor.org/stable/1701601 REFERENCES Linked references are available on JSTOR for this article : 240, 321–324. [DOI] [PubMed] [Google Scholar]
- Breder CD, Tsujimoto M, Terano Y, Scott DW, Saper CB, 1993. Distribution and characterization of tumor necrosis factor-α-like immunoreactivity in the murine central nervous system. J Comp Neurol 337, 543–567. 10.1002/cne.903370403. [DOI] [PubMed] [Google Scholar]
- Bubu OM, Brannick M, Mortimer J, Umasabor-Bubu O, Sebastiaõ YV, Wen Y, Schwartz S, Borenstein AR, Wu Y, Morgan D, Anderson WM, 2017. Sleep, Cognitive impairment, and Alzheimer ‘s disease: A Systematic Review and Meta-Analysis. Sleep 40, 1–18. [DOI] [PubMed] [Google Scholar]
- Chang RCC, Suen KC, Ma CH, Elyaman W, Ng HK, Hugon J, 2002. Involvement of double-stranded RNA-dependent protein kinase and phosphorylation of eukaryotic initiation factor-2α in neuronal degeneration. J. Neurochem. 83, 1215–1225. 10.1046/j.1471-4159.2002.01237.x. [DOI] [PubMed] [Google Scholar]
- Chukwurah E, Farabaugh KT, Guan B, Ramakrishnan P, Hatzoglou M, 2021. A tale of two proteins: PACT and PKR and their roles in inflammation. FEBS J. 288, 6365–6391. 10.1111/febs.15691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couturier J, Page G, Morel M, Gontier C, Lecron JC, Pontcharraud R, Fauconneau B, Paccalin M, 2010. Inhibition of double-stranded RNA-dependent protein kinase strongly decreases cytokine production and release in peripheral blood mononuclear cells from patients with Alzheimer’s disease. Journal of Alzheimer’s Disease 21, 1217–1231. 10.3233/JAD-2010-100258. [DOI] [PubMed] [Google Scholar]
- Davis CJ, Clinton JM, Jewett KA, Zielinski MR, Krueger JM, 2011. Delta wave power: An independent sleep phenotype or epiphenomenon? J. Clin. Sleep Med. 7, 7–9. 10.5664/JCSM.1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello CA, 2004. Infection, fever, and exogenous and endogenous pyrogens: Some concepts have changed. J. Endotoxin Res. 10, 201–222. 10.1179/096805104225006129. [DOI] [PubMed] [Google Scholar]
- Dumurgier J, Mouton-Liger F, Lapalus P, Prevot M, Laplanche JL, Hugon J, Paquet C, 2013. Cerebrospinal Fluid PKR Level Predicts Cognitive Decline in Alzheimer’s Disease. PLoS One 8, 1–5. 10.1371/journal.pone.0053587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd RA, Krueger JM, 1997. Diurnal variation of TNFα in the rat brain. Neuroreport 8, 915–918. 10.1097/00001756-199703030-00020. [DOI] [PubMed] [Google Scholar]
- Franchi L, Eigenbrod T, Muñoz-Planillo R, Nuñez G, 2009. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 10, 241–247. 10.1038/ni.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gal-Ben-Ari S, Barrera I, Ehrlich M, Rosenblum K, 2019. PKR: A kinase to remember. Front. Mol. Neurosci. 10.3389/fnmol.2018.00480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García MA, Gil J, Ventoso I, Guerra S, Domingo E, Rivas C, Esteban M, 2006. Impact of Protein Kinase PKR in Cell Biology: from Antiviral to Antiproliferative Action. Microbiol Mol Biol Rev 70, 1032–1060. 10.1128/mmbr.00027-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García MA, Meurs EF, Esteban M, 2007. The dsRNA protein kinase PKR: Virus and cell control. Biochimie 89, 799–811. 10.1016/j.biochi.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Gourmaud S, Mouton-Liger F, Abadie C, Meurs EF, Paquet C, Hugon J, 2016. Dual Kinase Inhibition Affords Extended in vitro Neuroprotection in Amyloid-β Toxicity. Journal of Alzheimer’s Disease 54, 1659–1670. 10.1016/j.neurobiolaging.2020.03.011. [DOI] [PubMed] [Google Scholar]
- Grolleau A, Kaplan MJ, Hanash SM, Beretta L, Richardson B, 2000. Impaired translational response and increased protein kinase PKR expression in T cells from lupus patients. J. Clin. Investig. 106, 1561–1568. 10.1172/JCI9352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D, 2000. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 6, 1099–1108. 10.1016/S1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- He Y, Franchi L, Núñez G, 2013. The protein kinase PKR is critical for LPS-induced iNOS production but dispensable for inflammasome activation in macrophages. Eur. J. Immunol. 43, 1147–1152. 10.1002/eji.201243187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hita-Yañez E, Atienza M, Cantero JL, 2013. Polysomnographic and subjective sleep markers of mild cognitive impairment. Sleep 36, 1327–1334. 10.5665/sleep.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holth JK, Patel TK, Holtzman DM, 2017. Sleep in Alzheimer’s Disease-Beyond Amyloid. Neurobiol Sleep Circadian Rhythms 2, 4–14. 10.1016/j.nbscr.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang KD, Bak MS, Kim SJ, Rhee S, Lee YS, 2017. Restoring synaptic plasticity and memory in mouse models of Alzheimer’s disease by PKR inhibition. Mol. Brain 10, 1–10. 10.1186/s13041-017-0338-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imeri L, Bianchi S, Opp MR, 2006. Inhibition of caspase-1 in rat brain reduces spontaneous nonrapid eye movement sleep and nonrapid eye movement sleep enhancement induced by lipopolysaccharide. Am. J. Physiol. Regul. Integr. Comp. Physiol. 291, 197–204. 10.1152/ajpregu.00828.2005. [DOI] [PubMed] [Google Scholar]
- Imeri L, Opp MR, 2009. How (and why) the immune system makes us sleep. Nat. Rev. Neurosci. 10, 199–210. 10.1038/nrn2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingiosi AM, Opp MR, 2016. Sleep and immunomodulatory responses to systemic lipopolysaccharide in mice selectively expressing interleukin-1 receptor 1 on neurons or astrocytes. Glia 64, 780–791. 10.1002/glia.22961. [DOI] [PubMed] [Google Scholar]
- Irwin M, 2002. Effects of sleep and sleep loss on immunity and cytokines. Brain Behav Immun 16, 503–512. 10.1016/S0889-1591(02)00003-X. [DOI] [PubMed] [Google Scholar]
- Irwin MR, Opp MR, 2017. Sleep Health: Reciprocal Regulation of Sleep and Innate Immunity. Neuropsychopharmacology. 10.1038/npp.2016.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang R, Tang D, 2012. PKR-dependent inflammatory signals. Sci. Signal. 5 10.1126/scisignal.2003511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandelwal PJ, Herman AM, Moussa CEH, 2011. Inflammation in the early stages of neurodegenerative pathology. J. Neuroimmunol. 238, 1–11. 10.1016/j.jneuroim.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger J, 2017. Cytokines in Immune Function Ans Sleep Re 4, 229–240. 10.1016/B978-0-444-52006-7.00015-0.Cytokines. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger JM, Obál F, Fang J, Kubota T, Taishi P, 2001. The role of cytokines in physiological sleep regulation. Ann. n. y. Acad. Sci. 933, 211–221. 10.1111/j.1749-6632.2001.tb05826.x. [DOI] [PubMed] [Google Scholar]
- Krueger JM, Opp MR, 2016. Sleep and Microbes. Physiol. Behav. 207–225. 10.1016/bs.irn.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger JM, Toth LA, Floyd R, Fang J, Kapás L, Bredow S, Obál F Jr., 1994. Sleep, Microbes and Cytokines. Neuroimmunomodulation 1, 100–109. 10.1159/000097142. [DOI] [PubMed] [Google Scholar]
- Lancel M, Cronlein J, Muller-Preuss P, Holsboer F, 1995. Lipopolysaccharide increases EEG delta activity within non-REM sleep and disrupts sleep continuity in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 268 10.1152/ajpregu.1995.268.5.r1310. [DOI] [PubMed] [Google Scholar]
- Liguori C, Romigi A, Nuccetelli M, Zannino S, Sancesario G, Martorana A, Albanese M, Mercuri NB, Izzi F, Bernardini S, Nitti A, Sancesario GM, Sica F, Marciani MG, Placidi F, 2014. Orexinergic system dysregulation, sleep impairment, and cognitive decline in Alzheimer disease. JAMA Neurol. 71, 1498–1505. 10.1001/jamaneurol.2014.2510. [DOI] [PubMed] [Google Scholar]
- Lim MM, Gerstner JR, Holtzman DM, 2014. The sleep-wake cycle and Alzheimer’s disease: what do we know? Neurodegener Dis Manag 4, 351–362. 10.2217/nmt.14.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim ASP, Kowgier M, Yu L, Buchman AS, Bennett DA, 2013. Sleep fragmentation and the risk of incident Alzheimer’s disease and cognitive decline in older persons. Sleep 36, 1027–1032. 10.5665/sleep.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wang M, Cheng A, Yang Q, Wu Y, Jia R, Liu M, Zhu D, Chen S, Zhang S, Zhao XX, Huang J, Mao S, Ou X, Gao Q, Wang Y, Xu Z, Chen Z, Zhu L, Luo Q, Liu Y, Yu Y, Zhang L, Tian B, Pan L, Rehman MU, Chen X, 2020. The role of host eIF2α in viral infection. Virol. J. 17, 1–15. 10.1186/s12985-020-01362-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourenco MV, Clarke JR, Frozza RL, Bomfim TR, Forny-Germano L, Batista AF, Sathler LB, Brito-Moreira J, Amaral OB, Silva CA, Freitas-Correa L, Espírito-Santo S, Campello-Costa P, Houzel JC, Klein WL, Holscher C, Carvalheira JB, Silva AM, Velloso LA, Munoz DP, Ferreira ST, De Felice FG, 2013. TNF-α mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s β-amyloid oligomers in mice and monkeys. Cell Metab. 18, 831–843. 10.1016/j.cmet.2013.11.002. [DOI] [PubMed] [Google Scholar]
- Lu B, Nakamura T, Inouye K, Li J, Tang Y, Lundback P, Valdes-Ferrer SI, Olofsson PS, Kalb T, Roth J, Zou Y, Erlandsson-Harris H, Yang H, Ting J-P-Y, Wang H, Andersson U, Antoine DJ, Chavan SS, Hotamisligil GS, Tracey KJ, 2012. Novel role of PKR in inflammasome activation and HMGB1 release. Nature 488, 670–674. 10.1038/nature11290.Novel. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q, Teter B, Ubeda OJ, Morihara T, Dhoot D, Nyby MD, Tuck ML, Frautschy SA, Cole GM, 2013. Sleep quality and preclinical Alzheimer Disease. JAMA Neurol. 70, 587–593. 10.1001/jamaneurol.2013.2334.Sleep. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathias S, Schiffelholz T, Linthorst ACE, Pollmächer T, Lancel M, 2000. Diurnal variations in lipopolysaccharide-induced sleep, sickness behavior and changes in corticosterone levels in the rat. Neuroendocrinology 71, 375–385. 10.1159/000054558. [DOI] [PubMed] [Google Scholar]
- Morrow JD, Opp MR, 2005a. Diurnal variation of lipopolysaccharide-induced alterations in sleep and body temperature of interleukin-6-deficient mice. Brain Behav. Immun. 19, 40–51. 10.1016/j.bbi.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Morrow JD, Opp MR, 2005b. Sleep-wake behavior and responses of interleukin-6-deficient mice to sleep deprivation. Brain Behav. Immun. 19, 28–39. 10.1016/j.bbi.2004.02.003. [DOI] [PubMed] [Google Scholar]
- Musiek ES, Xiong DD, Holtzman DM, 2015. Sleep, circadian rhythms, and the pathogenesis of Alzheimer disease. Exp. Mol. Med. 47, e148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsonne G, Lekander M, Åkerstedt T, Axelsson J, Ingre M, 2016. Diurnal variation of circulating interleukin-6 in humans: A meta-analysis. PLoS One 11, 1–17. 10.1371/journal.pone.0165799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak SS, Liu D, Li T, de Zavalia N, Zhu L, Li J, Karthikeyan R, Alain T, Liu AC, Storch KF, Kaufman RJ, Jin VX, Amir S, Sonenberg N, Cao R, 2019. The eIF2α Kinase GCN2 Modulates Period and Rhythmicity of the Circadian Clock by Translational Control of Atf4. Neuron 104, 724–735.e6. 10.1016/j.neuron.2019.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe RK, Griesbach GS, 2022. Immune-endocrine interactions in the pathophysiology of sleep-wake disturbances following traumatic brain injury: A narrative review. Brain Res. Bull. 185, 117–128. 10.1016/j.brainresbull.2022.04.017. [DOI] [PubMed] [Google Scholar]
- Segev Y, Barrera I, Ounallah-Saad H, Wibrand K, Sporild I, Livne A, Rosenberg T, David O, Mints M, Bramham CR, Rosenblum K, 2015. PKR inhibition rescues memory deficit and atf4 overexpression in apoe ε4 human replacement mice. J. Neurosci. 35, 12986–12993. 10.1523/JNEUROSCI.5241-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth R, Sun J, 2021. Protein Kinase R in Bacterial Infections: Friend or Foe. Front. Immunol. 12, 702142 10.3389/fimmu.2021.702142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spira AP, Chen-Edinboro LP, Wu MN, Yaffe K, 2014. Impact of Sleep on the Risk of Cognitive Decline and Dementia. Curr. Opin. Psychiatry 27, 478–483. 10.1097/YCO.0000000000000106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterniczuk R, Theou O, Rusak B, Rockwood K, 2013. Sleep Disturbance is Associated with Incident Dementia and Mortality. Curr. Alzheimer Res. 10, 1–9. 10.2174/15672050113109990134. [DOI] [PubMed] [Google Scholar]
- Taga M, Minett T, Classey J, Matthews FE, Brayne C, Ince PG, Nicoll JAR, Hugon J, Boche D, 2017. Metaflammasome components in the human brain: a role in dementia with Alzheimer’s pathology? Brain Pathology 27, 266–275. 10.1111/bpa.12388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taishi P, Bredow S, Guha-Thakurta N, Obál FO, Krueger JM, 1997. Diurnal variations of interleukin-1β mRNA and β-actin mRNA in rat brain. J. Neuroimmunol. 75, 69–74. 10.1016/S0165-5728(97)00002-7. [DOI] [PubMed] [Google Scholar]
- Tsapanou A, Gu Y, Manly J, Schupf N, Tang MX, Zimmerman M, Scarmeas N, Stern Y, 2015. Daytime Sleepiness and Sleep Inadequacy as Risk Factors for Dementia. Dement Geriatr Cogn Dis Extra 5, 286–295. 10.1159/000431311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerberg CE, Bryce AM, Florczak SM, Weintraub S, Mesulam M, Zee PC, Paller KA, 2012. Concurrent Impairments in Sleep and Memory in Amnestic Mild Cognitive Impairment. Journal of the International Neurophychological Society 18, 490–500. 10.1017/S135561771200001X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Samuel CE, 2008. Induction of protein kinase PKR-dependent activation of interferon regulatory factor 3 by vaccinia virus occurs through adapter IPS-1 signaling. J. Biol. Chem. 283, 34580–34587. 10.1074/jbc.M807029200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.