

Abstract
Purpose
CLN3 Batten disease (also known as juvenile neuronal ceroid lipofuscinosis) is a lysosomal storage disorder that typically initiates with retinal degeneration but is followed by seizure onset, motor decline and premature death. Patient-derived CLN3 disease induced pluripotent stem cell–RPE cells show defective phagocytosis of photoreceptor outer segment (POS). Because modifier genes are implicated in CLN3 disease, our goal here was to investigate a direct link between CLN3 mutation and POS phagocytosis defect.
Methods
Isogenic control and CLN3 mutant stem cell lines were generated by CRISPR-Cas9-mediated biallelic deletion of exons 7 and 8. A transgenic CLN3Δ7–8/Δ7–8 (CLN3) Yucatan miniswine was also used to study the impact of CLN3Δ7–8/Δ7–8 mutation on POS phagocytosis. POS phagocytosis by cultured RPE cells was analyzed by Western blotting and immunohistochemistry. Electroretinogram, optical coherence tomography and histological analysis of CLN3Δ7–8/Δ7–8 and wild-type miniswine eyes were carried out at 6, 36, or 48 months of age.
Results
CLN3Δ7 – 8/Δ7 – 8 RPE (CLN3 RPE) displayed decreased POS binding and consequently decreased uptake of POS compared with isogenic control RPE cells. Furthermore, wild-type miniswine RPE cells phagocytosed CLN3Δ7–8/Δ7–8 POS less efficiently than wild-type POS. Consistent with decreased POS phagocytosis, lipofuscin/autofluorescence was decreased in CLN3 miniswine RPE at 36 months of age and was followed by almost complete loss of photoreceptors at 48 months of age.
Conclusions
CLN3Δ7 – 8/ Δ7 – 8 mutation (which affects ≤85% of patients) affects both RPE and POS and leads to photoreceptor cell loss in CLN3 disease. Furthermore, both primary RPE dysfunction and mutant POS independently contribute to impaired POS phagocytosis in CLN3 disease.
Keywords: CLN3 disease, phagocytosis, lipofuscin, neuronal ceroid lipofuscinoses, stem cells
CLN3 disease is the most common form of Batten disease (neuronal ceroid lipofuscinoses) that develops in early childhood. Symptoms include progressive loss of vision and neurodegeneration of the brain, ultimately leading to premature death.1,2 Most affected CLN3 disease patients (approximately 75%) have a 966-bp deletion that entirely removes exons 7 and 8 from the CLN3 transcript.3 Despite this, there is significant variability in the disease phenotype of CLN3 disease patients, including those harboring the same common CLN3 mutation.4–6 The lack of genotype–phenotype relationship in CLN3 disease has suggested a contribution of modifier genes to the disease development and progression.4
Murine models have been widely used as an in vivo model of CLN3 disease.7–11 However, despite some evidence of molecular and pathological alterations, there is poor recapitulation of retinal degeneration phenotype in these models.12–14 To overcome the lack of visual degeneration of the existing CLN3 models, a transgenic miniswine model was developed that carries the orthologous common 966-bp deletion found in most human patients. Importantly, CLN3Δ7–8/Δ7–8 Yucatan miniswine (CLN3 miniswine) show progressive neuronal loss, motor dysfunction, and vision impairment phenotypes similar to those seen in individuals with CLN3 disease.15
Although retinal degeneration is a prominent pathology in CLN3 disease, there is limited knowledge of the disease mechanism(s) underlying retinal degeneration in CLN3 disease. Consistent with the accumulation of autofluorescent lysosomal storage material ceroid and degeneration of neuronal cells in CLN3 disease,16,17 clinical and histopathological studies on CLN3 disease retina have shown accumulation of autofluorescent ceroid lipopigment in retinal neurons and degeneration of multiple retinal cell layers, including the RPE.18–20 However, it is important to highlight that in contrast to lysosomal accumulation of autofluorescent ceroid lipopigment in neuronal cells in CLN3 disease; RPE cells in CLN3 disease display decreased accumulation of autofluorescent lipofuscin.21–23 Note that, although both RPE lipofuscin and ceroid observed in CLN3 disease cells are autofluorescent and have similar spectral properties,24 the autofluorescent lipofuscin within RPE cells is a consequence of partial digestion of phagocytosed photoreceptor outer segment (POS) as opposed to the autofluorescent ceroid that mainly constitutes of mitochondrial ATPase subunit C, saponins, amyloid-beta, and neutral lipids.24
Recently, we have replicated decreased RPE autofluorescence in CLN3 disease donor eyes in patient-derived human induced pluripotent stem cell (hiPSC)–RPE cells (harboring the common 966-bp deletion) after chronic daily exposure to POS.22 However, because this study compared CLN3 disease patient-derived hiPSC-RPE cells with nonisogenic control hiPSC-RPE cells, it is unclear if the observed POS phagocytosis defect in CLN3 disease is a direct consequence of CLN3 mutation. Note that, as previously mentioned, modifier genes have been implicated strongly in molecular alterations observed in CLN3 disease.4
CRISPR-based gene editing has been routinely used for the in vitro correction of genetic mutations in human pluripotent stem cells, including hiPSCs and human embryonic stem cells (hESCs).25–27 In fact, CRISPR-based gene editing of hiPSCs and hESCs have been used routinely to decipher the genotype–phenotype relationship in several retinal diseases.28–31 For example, we have previously used gene-corrected hiPSC lines from patients with Doyne honeycomb retinal dystrophy to show that R345W mutation in EFEMP1 is causal for drusen accumulation in Doyne honeycomb retinal dystrophy.32 Similarly, CRISPR-Cas9–mediated repair of CLCN2 mutation rescued dysfunctional POS phagocytosis by iPSC–RPE cells from patients with CLCN2-associated retinal degeneration.33 Also, neural retina-specific leucine zipper protein–deficient hESC–retinal organoids engineered via CRISPR-Cas9 gene editing showed a direct role of neural retina-specific leucine zipper protein in rod photoreceptor specification.34 Overall, gene editing strategies including both correction of genetic defect in patient hiPSCs and introduction of disease-associated mutation in control hESC/hiPSCs provides a suitable approach to definitively link the independent contribution of genetic variants to molecular and pathological defects in a human-based cellular model.
Therefore, to link the disease-causing CLN3 mutation to the observed impaired phagocytosis in CLN3 disease, here, we used CRISPR-Cas9 gene editing to generate isogenic H9 hESC lines carrying homozygous deletion of exons 7 and 8 in CLN3 (referred to herein as CLN3Δ7–8/Δ7–8). Additionally, we used a large animal model of CLN3 disease to study the impact of common 966 bp CLN3 deletion mutation on POS phagocytosis and lipofuscin-like autofluorescent material accumulation.
Materials and Methods
Availability of Data and Materials
Requests for original data files and reagents and materials used in this study will be fulfilled by contacting the corresponding author, Dr Ruchira Singh.
Procurement and Use of hESC Lines
H9 hESCs were obtained from WiCell and used under approval from the Tasmania Health and Medical Human Research Ethics Committee (#13502) and Institutional Regulatory Board (RSRB00056538) at the University of Rochester and conformed with the ethical norms and the declaration of Helsinki.
Animals
Wild-type (WT) and transgenic (CLN3Δ7–8/Δ7–8) miniswine8 used in these studies were obtained from Exemplar Genetics and all animal studies were conducted following the approval of the Institutional Animal Care and Use Committees at Exemplar Genetics (Protocol # MRP2018-004).
CRISPR-Cas9–Mediated Editing of CLN3
Editing of CLN3 to delete exons 7 and 8 was done as previously described.35,36 Briefly, 8 × 105 H9 hESCs in single-cell suspension were resuspended in 100 µL electroporation buffer from the Human Stem Cell Nucleofector Solution 2 kit (Lonza, Basel, Switzerland; VPH-5022). Alt-R Cas9 Electroporation enhancer (1.1 µM) (IDT, #1075915) and assembled CRISPR-Cas ribonucleoprotein consisting of 1.4 µM dual crRNA (Table 1) (IDT) and 1.2 µM Alt-R S.p. Hifi Cas9 Nuclease V3 (IDT, #1081060) were mixed with cells and transferred to a nucleofection cuvette. Electroporation was conducted using Amaxa Nucleofector 2b Device (Lonza) with Nucleofector program B-016. Electroporated cells were seeded at low density (2000 cells/well) in Matrigel (Corning, Corning, NY, USA; FAL354277)-coated six-well plates with CloneR (STEMCELL Technologies, Vancouver, British Columbia, Canada; #05889)/mTeSRTM1 media (STEMCELL Technologies, #85850). When single-cell colonies were approximately 200 µm in diameter, each colony was picked and seeded into 2 wells of duplicate Matrigel-coated 96-well plates with 1 plate for cryopreservation and the other for genotyping.
Table 1.
List of CRISPR Guides and Primers Used to Confirm Deletion of Exons 7 and 8 in the CLN3 Gene
| Oligo Name | Sequence | Expected PCR Band Size |
|---|---|---|
| sg1 | ATGAAGGGGCAGAGACATCA | NA |
| sg2 | CCTCCCTTCACAGCAAGGTA | NA |
| CLN3 F1 | GGATGAATTAGATGGAGATTGAGG | Deleted: 600–800 bp |
| CLN3 R1 | CTCATCCTACTTCTAATCACCTTG | Undeleted: 1529 bp |
| CLN3 F2 | TCTGTCTCTACGGCTGCTGTGC | Deleted: 578 bp |
| CLN3 R2 | GAACACCAGGTTGAGGCACTGC | Undeleted: 795 bp |
PCR Screening and Genotyping
When cells from the genotyping 96-well plate were 50% to 60% confluent, DNA extraction was done using QuickExtract DNA Extraction Solution (Epicentre, #E09050) according to manufacturer's instructions. Subsequently, 50 ng of DNA extract was amplified through PCR reaction using KAPA HotStart PCR Kit, with dNTPs (Millenium Science, Mulgrave, Victoria, Australia; ROC-07958897001), and CLN3 F1 and CLN3 R1 primers that span the targeted region (Table 1).
Characterization of Pluripotency
ESC colonies at 80% confluency were dissociated with ReLeSR (STEMCELL Technologies, #05872). Cell aggregates were transferred into ultra-low attachment 6-well plates in Complete KSR EB medium (KnockOut SR (Life Technologies, Carlsbad, CA, USA; #10828-028) 20%, DMEM/F-12 78%, GlutaMAX supplement (Life Technologies, #10565018) 1%, MEM Non-Essential Amino Acids solution 1% (Life Technologies, #11140050), 2-mercaptoethanol 0.1 mM (Sigma, St Louis, MO, USA; M6250) for embryoid body (EB) formation. Medium was refreshed every 2 days. On day 4, EBs were plated onto Matrigel-coated wells for further differentiation in Complete KSR EB medium for 16 days. Pluripotency markers were analyzed by quantitative real-time PCR and immunocytochemistry.
For quantitative real-time PCR analyses, total RNA was isolated from positive clones with the RNeasy Plus Mini Kit (Qiagen, Hilden, Germany; #74134) according to the manufacturer's protocol. Reverse transcription of 1 µg RNA was performed using Omniscript RT kit (Qiagen, #205111). CLN3 transcript was amplified, and PCR products were sequenced with primers, CLN3 F2 and CLN3 R2 that span exons 6 through 9 (Table 1).
For immunocytochemical analyses, hESC colonies were passaged and cultured on Matrigel-coated coverslips in mTeSRTM1 medium until 60% confluency. Cells were then fixed with 4% paraformaldehyde in PBS for 20 minutes at room temperature. Fixed cells were incubated for one hour in blocking solution (5% fetal bovine serum (FBS) in PBS and 5% FBS with 0.1% Triton X-100 in PBS for extracellular and intracellular markers, respectively) followed by incubation with the primary antibodies for NANOG, OCT4, TRA-1-60, and SSEA4 overnight at 4°C. This was followed by incubation in host-specific secondary antibody for 1 hour at room temperature and thereafter mounting and visualization by fluorescent microscopy. Further details of primary antibody concentration and source can be found in Table 2.
Table 2.
List of Primary Antibodies Used in This Study
| Antibody | Manufacturer | Cat. # | Dilution Immunocytochemistry | Dilution (WB) |
|---|---|---|---|---|
| ACTN | Cell Signaling | 4970 | 1:500 | |
| ACTN | Santa Cruz | SC-47778 | 1:500 | |
| BEST1 | Millipore | MAB5466 | 1:500 | |
| CRALBP | Abcam | AB15051 | 1:500 | |
| EZR | Cell Signaling | 3145S | 1:100 | 1:1000 |
| MERTK | Abcam | AB52968 | 1:500 | |
| RHO | Millipore | MABN15 | 1:100 | 1:500 |
| OCT4 | Cell Signaling Technology | StemLight Pluripotency Antibody Kit #9656 | 1:200 | |
| NANOG | Cell Signaling Technology | StemLight Pluripotency Antibody Kit #9656 | 1:200 | |
| RPE65 | Millipore | MAB5428 | 1:500 | |
| TRA-1-60 | Cell Signaling Technology | StemLight Pluripotency Antibody Kit #9656 | 1:200 | |
| SSEA4 | Cell Signaling Technology | StemLight Pluripotency Antibody Kit #9656 | 1:200 | |
| ZO1 | Life Technologies | 61-7300 | 1:100 | |
| ZO1 | Invitrogen | 33-9100 | 1:100 |
Virtual Karyotyping
Genomic DNA (200 ng) from isogenic H9 CLN3Δ7–8/Δ7–8 cell lines were analyzed for copy number variation using Illumina HumanCytoSNP-12 beadchip array. B allele frequency and log R ratio of each single nucleotide polymorphism marker were collected from GenomeStudio (Illumina, San Diego, CA, USA). These were used for copy number variation analyses, which was performed using PennCNV37 and QuantiSNP38 with default parameter settings. Genomic regions having at least 20 contiguous single nucleotide polymorphisms or genomic regions with single nucleotide polymorphisms spanning at least 1 MB were designated as chromosomal copy number variation.39
RPE Differentiation and Culture From hESCs
Differentiation of hESCs to RPE was performed as previously described.22,40 Briefly, EBs were generated from colonies of hESCs cultured in either mTeSR or mTeSR Plus (STEMCELL Technologies). On day 6 of EB culture, EBs were plated onto laminin-coated tissue culture plates and fed with neural induction medium whose composition is described in.22,40 The cell culture medium was switched to retinal differentiation medium on day 14. Retinal differentiation medium composition is described in.22,40 hiPSC–RPE cells were dissected in patches from adherent cultures around day 60-90 when characteristic RPE morphology could be observed. The RPE patches at passage P0 were seeded and passaged onto either a nonpermeable plastic 24-well plate or onto transwell (Corning) inserts (0.4 µm pore size). RPE cells at < passage 4 were used in all experiments.
Primary Miniswine RPE Culture
Primary miniswine RPE cultures were performed as previously described.41,42 Briefly, after enucleation, eyes were placed in 0.2% povidone-iodine solution for 10 minutes on ice followed by five rinses in 1000 U/mL penicillin-streptomycin solution. The anterior portion of the eye and the retina was next removed (note the retina was used for POS isolation) and eyecups were transferred to six-well plates filled with 1 mL of prewarmed 0.5% trypsin with 5.3 mM EDTA in Hank's Balanced Salt Solution without calcium and magnesium. Subsequently, the six-well plates were placed in a 37°C incubator for 30 minutes. At the end of the incubation, RPE cells were collected in a 15-mL tube containing prewarmed media (1× DMEM with 4.5 g/L glucose, L-glutamine, and sodium pyruvate and 10% FBS) and gently centrifuged (300 rcf) to collect the RPE cell pellet. The RPE cell pellet was resuspended and plated in a T25 flask in culture media (1× DMEM with 4.5 g/L glucose, L-glutamine, and sodium pyruvate, 1% NEAA, 1% penicillin-streptomycin) containing 10% FBS. Once the RPE cells reached confluence, the FBS concentration in the culture media was reduced to 1% and subsequently RPE cells were passaged and replated onto laminin-coated 24-well plates or transwells.
POS Phagocytosis Assay
POS phagocytosis assay in vitro was performed as previously described.22 Bovine POS were obtained commercially from InVision BioResources (Cat. #98740, Seattle, WA, USA) and porcine POS were prepared from WT and CLN3 miniswine as previously described.41–43 Note that POS were isolated from eyes from three WT and four CLN3 miniswine of approximately 30 to 36 months of age.
For evaluating POS uptake, as described elsewhere,22,44 mature monolayer of RPE cells in culture were fed unlabeled POS or FITC-labeled POS (approximately 10–40 POS/RPE cell)2 for 2 hours at 37°C with 10% FBS media supplementation. Thereafter, to remove POS (unbound) on the RPE cell surface, RPE cells were thoroughly washed with 1× PBS. Subsequently, RPE cells were either harvested for Western blotting or processed for immunocytochemistry experiments. Quantitative analysis of the number of FITC-POS phagocytosed included a consideration of FITC particle size. Specifically, a threshold of less than 5 µm allowed us to evaluate the amount of bound, ingested POS, but eliminate aggregated POS from the analyses.
For evaluating POS binding and POS internalization, we used a previously published protocol44 that was also used to evaluate POS binding vs. internalization in CLN3 disease hiPSC-RPE.22 Briefly, the position of FITC particles relative to ZO1 was used to evaluate the amount of bound vs. internalized FITC-POS particles.44 Specifically, to analyze the relative position of the FITC-POS within the cell, FITC-POS fed RPE cells after immunocytochemical labeling with ZO1 antibody were imaged as confocal z-stacks spanning the entire height of the RPE cells. The images were subsequently analyzed with ImageJ software to determine the number of apical vs. basal FITC-POS particles relative to ZO1.
POS phagocytosis using rod phagosome quantification in situ was performed as described previously.45–47 Briefly, posterior eyecup wholemounts were prepared from WT miniswine at approximately the same time of sacrifice (e.g., approximately 1.0–1.5 hours after light onset). The eyecup wholemounts were immunostained with rhodopsin antibody (MABN15, Burlington, MA, USA) and a ZO1 antibody (Catalog # 61-7300, Invitrogen, Waltham, MA, USA) and subsequently Alexa-Fluor–conjugated secondary antibodies. Confocal z-stack images were captured using an Eclipse Ti2 Nikon microscope and rod phagosomes of size of less than 5 µm were analyzed using maximal projections of z-stacks in ImageJ.
RPE Autofluorescence Measurements
As previously described,22 for in vitro experiments evaluating autofluorescence accumulation, RPE cells were fed POS daily (approximately 20–40/RPE cells/day) for 14 days. At day 14, to remove any unbound POS, RPE monolayer was thoroughly washed with 1× PBS. RPE cells were then immediately fixed and processed by immunocytochemistry where the red channel (ex, 546 nm; em, 560–615 nm) was used exclusively for the evaluation of RPE autofluorescence without any antibody staining. Quantitative analyses of autofluorescent particles were performed per DAPI-stained cells for count and per viewing area for the area covered by the autofluorescent particles using ImageJ/FIJI and Microsoft Excel software.
For analysis of autofluorescence in miniswine eye samples, miniswine RPE whole mounts were used. Miniswine eyes were injected with 4% PFA after enucleation for 1 hour before dissection to remove the anterior portion and vitreous. The remaining retina/RPE eyecup was submerged in 4% PFA for a fixation of at least 4 hours at 4°C. After two 1× PBS washes, samples were prepared for retina sectioning and wholemount staining by carefully removing the RPE/choriocapillaris from the sclera. The RPE wholemount was then permeabilized/blocked in blocking buffer (1% BSA; ImmunoReagents, Inc. [Chapel Hill, NC, USA], 0.2% Triton-X-100, 0.2% Tween-20) for 1 hour at room temperature. Subsequently, RPE samples were washed and incubated with DAPI dye (Hoechst 33342; Life Technologies) for 15–30 minutes at room temperature. After one last wash with 1× PBS, RPE samples were mounted onto slides with Prolong Gold (Life Technologies P36930) and cover slipped and imaged by confocal microscopy in the red channel (ex, 546 nm; em, 560–615 nm).
Western Blot
Western blotting was performed as previously described.22 Briefly, RPE cells were lysed in radioimmunoprecipitation assay buffer containing protease inhibitor cocktail (Sigma-Aldrich). Subsequently, protein quantification was performed with Pierce BCA assay and equal amount of protein was loaded for each sample and resolved on 4% to 20% Tris-HCl gradient gels, followed by transfer onto polyvinylidene fluoride membranes. The polyvinylidene fluoride membrane was subsequently blocked with 5% dry milk in 1× PBS or commercially bought blocking buffer (LI-COR) for 1h at room temperature followed by primary antibody incubation overnight at 4°C. After primary antibody incubation, polyvinylidene fluoride membrane was washed with 0.1% Tween-PBS and incubated in host-specific horseradish peroxidase or IRDye-conjugated secondary antibody (1:10,000) in 5% dry milk 0.1% Tween-PBS for 1 hour at room temperature. Azure C500 (Azure Biosystem, Dublin, CA, USA) and Image Studio were used for analysis and quantification of the Western blot images. All primary antibodies used for Western blotting analyses are listed in Table 2.
Immunocytochemistry
Immunocytochemical analysis was performed as previously described.22,48 Briefly, RPE on transwells were permeabilized and blocked in blocking buffer (10% normal donkey serum, 0.1% Triton-X-100) for 1 hour at room temperature. This was followed by overnight incubation in primary antibody in 0.5× blocking buffer at 4°C. The next day, RPE in transwells were washed and subsequently incubated with host-specific Alexa-conjugated secondary antibody (1:500) in 0.5× blocking buffer for 1 hour at room temperature. The RPE in transwells were next incubated with Hoechst 33342 (Life Technologies) for 15 minutes to stain the nuclei, then washed in 1× PBS for 5 minutes. Subsequently, transwell membranes were cut out and mounted onto slides with Prolong Gold (Life Technologies) and imaged with a confocal microscope (LSM 510 META, Zeiss [Jena, Germany], or Eclipse Ti2, Nikon [Tokyo, Japan]). In a subset of experiments, miniswine RPE whole mounts at 6 months of age were used. Following immunostaining, samples were briefly incubated with TrueBlack (Biotium, Inc. Fremont, CA, USA) following manufacturer's instructions to quench autofluorescence signal prior to mounting onto slides with Prolong Gold (Life Technologies) and cover slipped. Confocal microscopy images were analyzed with ImageJ/FIJI software. All primary antibodies used can be found in Table 2.
Histological Analyses of Miniswine Retina
Whole eyes were fixed for approximately 3 weeks in 10% neutral buffered formalin. Subsequently, the retina was dissected and the midperiphery region of the retina was used for processing and analyses. Specifically, the dissected retina tissue was further fixed at room temperature in 4% PFA for 1 hour. After 1× PBS washes, the retina was processed for plastic embedding (Technovit 7100 kit; Electron Microscopy Sciences, Hatfield, PA, USA), as previously described.15 Retina sections were cut at 3 µm thick and dried on a slide warmer. Multiple Stain Solution (Polysciences, Inc., Warrington, PA, USA) was used to stain the retina sections on slides, which were then mounted with permount (Thermo Fisher Scientific, Waltham, MA, USA) and imaged using Keyence BZ-X800 Epifluorescence Microscope.
Transmission Electron Microscopy Analyses
Transmission electron microscopy was performed as previously described.22 Briefly, RPE grown on transwells were fixed overnight in a solution containing 0.1 M sodium cacodylate, 2.5% glutaraldehyde, and 4% paraformaldehyde. Next, RPE samples on transwell membrane were embedded in epoxy resin and processed to obtain 60-nm-thick RPE sections, then imaged at the Electron Microscopy Core at the University of Rochester using a Hitachi H-7650 Transmission Electron Microscope.
Transepithelial Resistance (TER) Measurements
EVOM2 volt-ohm meter (World Precision Instruments, Sarasota, FL, USA) was used to measure TER of RPE grown in transwell inserts as described.49 A TER measurements were reported as resistance per area or Ω.cm−2 after blank subtraction.
ERG and OCT Measurements
Miniswine were tested for retinal function using a flash ERG at 3 and 6 months of age as previously described.15 Dark-adapted ERG experiments were performed after 20 minutes of dark adaptation, with external light eliminated, shades over all light sources, and a piece of blackout fabric over the eyes of the pig while being anesthetized. Only a dim red light was used as needed to set up the test. Because light exposure was controlled carefully, ambient light in the room was not measured. Light-adapted testing was performed after minimum of 10 minutes exposure to artificial light, that is, standard operating room illumination. For the procedure, animals were anesthetized with 14 mg/kg ketamine (intramuscular) and anesthesia was maintained with 1% to 2% isoflurane. One drop of 1% tropicamide ophthalmic solution (according to the quality standards of the United States Pharmacopeia) was placed in each eye to cause dilation. Reference electrodes were connected to each ear and a ground electrode to the midline forehead. One drop of 0.5% proparacaine hydrochloride ophthalmic solution United States Pharmacopeia was placed in each eye as a local anesthetic and eye speculums were placed inside each eye to fix the eyelids open. The smaller sticky pads of a DTL Plus Electrode (LKC Technologies, Gaithersburg, MD, USA) were attached to the rostral side and larger sticky pads to caudal side of each eye. Each respective DTL electrode (right and left) was connected to the respective extension lead of the right or left reference electrode, and the ground and both reference electrodes were connected to a RETeval device (LKC Technologies).
The rabbit/minipig, photopic two-step light-adapted protocol was used for each eye and produced an 8.0 cd s/m2 flash at 2.0 Hz flash followed by an 8.0 cd s/m2 flicker at 28.3 Hz. After photopic testing of both eyes, all lights in the room were extinguished, the RETeval device was calibrated for dark adaptation and the animals were allowed to adapt to the dark for 20 minutes. After dark adaptation, the rabbit/minipig, scotopic four-step protocol was used for each eye. The first step produced a 0.06 cd s/m2 flash at 0.5 Hz (dark-adapted rod only response), followed by an 8.0 cd s/m2 flash at 0.1 Hz (dark-adapted mixed rod and cone response), followed by a 25 cd s/m2 flash at 0.05 Hz (dark-adapted mixed -rod and cone response to higher intensity flash). Raw (unsmoothed) data values were used to calculate amplitudes. The a-wave amplitude was recorded as the prestimulus baseline to an a-wave trough, and the b-wave amplitude was measured from an a-wave trough to the highest waveform peak. Amplitude data from left and right eyes, as well as latency data from left and right eyes, were pooled together for each sex/genotype/time point. The a- and b-wave amplitudes/peak times for 8.0 cd s/m2 photopic flash responses and 8.0 cd s/m2 scotopic mixed rod/cone responses were analyzed using GraphPad Prism 8.0 and a mixed effect analysis with the Bonferroni post hoc test.
Retinal layer thickness of miniswine via OCT was assessed using the Leica instrument SD-OCT system (Bioptigen Envisu R2200; Leica, Danaher, Washington, DC, USA). Before imaging, eyes were dilated to increase the field of view using topical tropicamide 1% and ophthalmic 2.5% phenylephrine eye drops. For each eye, high density scans consisted of 1000 × 100 × 1 (A scans × B scans × repeated B scans) for averaging, with a 12 mm × 12 mm volume scan centered on the retina superior to the optic nerve. Multiple images were obtained from each eye for analysis. OCT analysis to access retinal layer thickness was performed on three scans per eye using Bioptigen InVivoVue and ImageJ software.
Experimental SetUp, Data Analyses, and Statistical Testing
For all experiments comparing isogenic control and CLN3 RPE (with the exception of immunocytochemistry for RPE signature proteins), parallel age-matched cultures of RPE monolayer in transwell inserts that displayed a TER of greater than 150 Ω.cm−2 were used. Data are presented as mean ± SEM. A two-tailed unpaired Student's t test was used to compute significance and Microsoft Excel (Microsoft Corp, Redmond, WA, USA) was used to plot the graphs. For quantitative analysis of confocal microscopy data, at least five distinct images (different viewing areas) per sample were used in each experiment. In each individual figure n refers to the biological replicate sample size for data from either isogenic control vs. CLN3Δ7–8/Δ7–8 hESC-RPE cells derived from distinct EB differentiations, or ex vivo or in vivo analyses of age- and sex-matched WT and CLN3 miniswine retina and RPE.
Results
Generation and Characterization of Isogenic Control and CLN3Δ7–8/Δ7–8 (CLN3) hRPE Cells
To generate CLN3Δ7–8/Δ7–8 isogenic cell lines, we used a well-characterized hESC line, H9.50 The location of dual guides included CLN3Δ7–8/Δ7–8 intron 6 and intron 8, which are spaced 968 bp apart in the CLN3 gene (Fig. 1a). Our first attempt of CRISPR editing yielded a H9 CLN3+/Δ7-8 cell line with monoallelic deletion of exons 7 and 8 in CLN3 gene (Fig. 1b). To obtain a biallelic deletion of exons 7 and 8 in the CLN3 gene, we repeated CRISPR editing on the H9 CLN3+/Δ7–8 cell line. Subsequent PCR revealed two truncated DNA bands (Fig. 1b) that, despite having different deletion sizes, had complete deletion of exons 7 and 8 from both CLN3 alleles. The successful biallelic deletion of exons 7 and 8 was confirmed through sequencing of the CLN3 transcript using cDNA and primers targeting regions outside exons 7 and 8 in the homozygous H9 CLN3Δ7–8/Δ7–8 cell line (Fig. 1c).
Figure 1.

Generation and characterization of isogenic control and CLN3 hESC RPE. (a) Schematic depicting CRISPR-Cas9 deletion of exons 7 and 8 of CLN3 in H9 hESC. Note the CLN3 locus and the location of dual guides to excise exons 7 and 8 from the CLN3 gene. (b) Identification of exons 7 and 8 deletion in CLN3 gene in positive clones. Genomic PCR of clones with homozygous deletion (lanes 1 and 2) and heterozygous deletion (lane 3). RT-PCR analysis of homozygous 1kb deletion clone (lane 4) and control (lane 5). (c) Sequencing alignment of cDNA for control (top) and CLN3Δ7-8/Δ7-8 (bottom) cell lines shows successful biallelic deletion of exons 7 and 8 reflected as splicing of exon 6 to exon 9 (indicated by an arrow). (d) Representative light microscopy images (top) and electron microscopy images (bottom) showing the expected RPE morphology and apical microvilli in isogenic control and CLN3Δ7–8/Δ7–8 hESC RPE (CLN3 hRPE) (scale bar, 100 µm). (e) Representative confocal microscopy images showing similar and expected localization of tight junction protein, ZO1 (red, top), RPE microvilli protein, EZR (red, middle), and basolaterally expressed RPE protein, BEST1 (green, bottom) in control and CLN3 hRPE cells. Nuclei were stained with DAPI (blue) (scale bar, 10 µm). (f) Representative Western blot images showing expected presence of RPE signature proteins and ACTN in both control and CLN3 hRPE cells. (g) Representative light microscopy images showing evidence of transepithelial fluid flux with presence of fluid domes in polarized monolayers of control and CLN3 hRPE cells. Note the presence of RPE both within the fluid dome (focal plane 1, top) and outside the fluid dome (focal plane 2, bottom) (scale bar, 50 µm). (h) TER measurements showing evidence of a tight epithelial monolayer in both control and CLN3 hRPE cultures. Note the dotted line represents the known TER threshold for RPE cells in vivo53. n ≥ 3 for all experiments in Figure 1.
Genome-wide copy number variation analysis demonstrated that the H9 CLN3Δ7–8/Δ7–8 isogenic cells have normal diploid karyotypes (46,XX). No chromosomal alterations were detected in the edited cells when compared with the unedited control cells based on analysis of the B allele frequency and log R ratio (Supplementary Fig. S1).
The pluripotency of isogenic H9 and H9 CLN3Δ7–8/Δ7–8 cell lines was confirmed by positive reactivity for pluripotency markers OCT4, NANOG, TRA-1-60, and SSEA4 (Supplementary Fig. S2). Quantitative real-time PCR analysis of EBs also demonstrated expression of genes specific for all three germ layer markers (endoderm, ectoderm, and mesoderm) (Supplementary Fig. S2).
Isogenic control and CLN3Δ7–8/Δ7–8 H9 ESC lines successfully differentiated to RPE cells using our previously described protocols.22,32,40,51 Light microscopy and transmission electron microscopy of control RPE and CLN3Δ7–8/Δ7–8 RPE (referred to henceforth as CLN3 hRPE) showed the expected cell morphology including presence of elongated RPE microvilli in both control and CLN3 hRPE cultures (Fig. 1d). Confocal microscopy imaging postimmunocytochemical analyses also showed similar and expected localization of tight junction protein, ZO1, in both control and CLN3 hRPE cultures (Fig. 1e). Similarly, consistent with polarized expression of specific proteins in RPE cells, an orthogonal view of confocal microscopy images of control and CLN3 hRPE showed apical localization of microvilli protein, EZR, and basolateral localization of BEST1 (Fig. 1e). Qualitative Western blotting analyses showed robust presence of RPE signature proteins, BEST1, CRALBP, EZR, MERTK, and RPE65, as well as cytoskeletal protein ACTN in control and CLN3 hRPE cells (Fig. 1f). In addition, in agreement with transepithelial fluid movement and formation of a polarized epithelial monolayer,22,52 light microscopy images showed presence of fluid domes in both control and CLN3 hRPE cultures (Fig. 1g). Last, TER recordings (Fig. 1h) showed formation of a tight epithelial barrier with both control and CLN3 hRPE developing physiological TER of approximately 150 Ω*cm−2 expected for RPE monolayer in vivo.53
Overall, using CRISPR-Cas9 editing, we were able to generate pluripotent isogenic control and CLN3Δ7–8/Δ7–8 H9 hESC lines. Notably, isogenic control vs. CLN3 hRPE cells showed similar morphology with presence of several RPE signature proteins and the expected RPE cell polarity and tight junction characteristics akin to native human RPE in vivo.
Disease-Causing CLN3 Mutation Leads to Reduced POS Binding and Consequently Decreased POS Uptake by RPE Cells
To link CLN3 function to POS phagocytosis by RPE cells, we evaluated uptake of nondiseased POS by isogenic control vs. CLN3 hRPE cells. Confocal microscopy imaging postimmunocytochemical analyses of control and CLN3 mutant RPE cells that were fed FITC-labeled POS (approximately 20–40 POS/RPE cell) for 2 hours showed fewer number of phagocytosed POS in CLN3 hRPE cells compared with parallel cultures of control RPE cells (Figs. 2a–2c). Autofluorescence accumulation (lipofuscin) in RPE cells is a consequence of POS phagocytosis and we have previously been able to show disease-associated reduction in lipofuscin-like autofluorescence in CLN3 disease patient-derived hiPSC-RPE cells after chronic (14 day) daily supplementation of POS by measuring autofluorescence levels in the red channel (ex, 546 nm; em, 560–615 nm). Consistently, confocal microscopy analyses of autofluorescence levels in the red channel in POS-fed RPE cells (approximately 40 POS/RPE cell/day for 14 days) showed decreased accumulation of lipofuscin-like autofluorescent material (count, area) in the CLN3 hRPE cells compared with control RPE cells (Figs. 2d–2f).
Figure 2.

Evaluation of POS phagocytosis and RPE autofluorescence by control vs. CLN3 hRPE cells. (a) Schematic of experimental assay used to measure POS uptake by control and CLN3 hRPE cells after feeding a physiological dose of POS (approximately 20–40 POS/RPE cell) for 2 hours. (b, c) Representative confocal microscopy images (b; scale bar, 10 µm) and quantitative analyses (c) showing decreased phagocytosis of FITC-POS particles (green, b), but similar localization of tight junction protein (ZO1) in CLN3 hRPE cells compared with control hRPE cells. Nuclei were stained with DAPI (blue) (scale bar, 10 µm). ***P ≤ 0.001. (d) Schematic of experimental assay used to measure RPE autofluorescence after daily chronic POS feeding (approximately 20–40 POS/RPE cells/day) for a duration of 14 days by control and CLN3 hRPE cells after feeding a physiologic dose of POS (approximately 20–40 POS/RPE cell) for 2 hours. (e, f) Representative confocal microscopy images (e; scale bar, 10 µm) and quantitative analyses (f, f′) showing decreased RPE autofluorescence (count, area) in the red channel (ex, 546 nm; em, 560–615 nm), but similar localization of tight junction protein (ZO1) in CLN3 hRPE cells fed daily with POS compared with control hRPE cells fed daily with POS. Nuclei were stained with DAPI (blue) (scale bar, 10 μm). ***P ≤ 0.001. n ≥ 3 for all experiments in Figure 2.
To confirm that reduced POS phagocytosis by CLN3 mutant RPE cells is a direct consequence of defective POS binding by CLN3 mutant RPE cells, we next used a previously published protocol22,44 to analyze bound vs. internalized FITC-labeled POS (approximately 20–40 POS/RPE cell) at 2 hours after POS feeding in isogenic control vs. CLN3 hRPE cells (Fig. 3). Compared with parallel cultures of control RPE cells, CLN3 mutant hRPE that displayed decrease in total POS uptake (Figs. 2b, 2c and 3a) also showed decrease in both the amount of apically localized (bound) and basally localized (internalized) FITC-POS particles relative to ZO1 (Figs. 3b–3e). However, no difference in the internalized FITC-POS relative to bound FITC-POS was seen in control vs. CLN3 hRPE (Fig. 3f).
Figure 3.
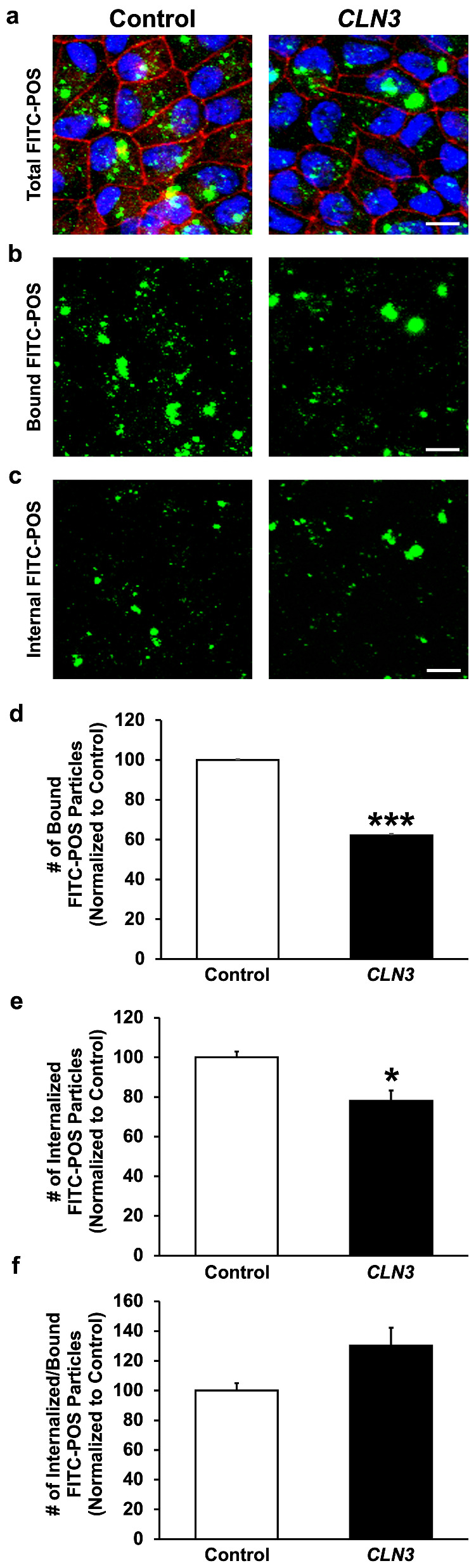
Evaluation of POS binding vs. internalization by control vs. CLN3 hRPE cells. (a–c) Representative confocal images of FITC-POS fed (2 hours) RPE monolayers showing decreased number of total FITC-POS particles (a), apical FITC-POS compared with ZO1 (consistent with reduced FITC-POS binding) (b), and basal FITC-POS compared with ZO1 (consistent with reduced FITC-POS internalization) (c) in CLN3 hRPE cells compared with control hRPE cells. Note that a published protocol (PMID: 29456184)44 that used the position of ZO-1 relative to FITC-POS to estimate bound vs. internalized POS was used in these experiments (scale bar, 10 µm). (d–f) Quantitative analyses showing decreased number of both bound FITC-POS (d) and internalized FITC-POS (e) in CLN3 hRPE cells compared with control hRPE cells. Consistent with a POS binding defect, the number of internalized FITC-POS relative to number of bound FITC-POS was unchanged between control and CLN3 hRPE cells (f). *P ≤ 0.05, ***P ≤ 0.001. n ≥ 3 for all experiments in Figure 3.
Overall, these data confirm that CLN3 mutation is independently sufficient to promote reduced POS binding and consequently defective POS phagocytosis by CLN3 disease RPE cells.
CLN3 Mutant POS are Phagocytosed Less Efficiently by WT RPE Cells
Our published paper22 and the results presented in this study (Figs. 2, 3) show that disease-causing CLN3 mutation leads to impaired POS phagocytosis by RPE cells. However, these data lack the consideration of diseased POS to the phagocytosis process. Note that POS disorganization is an early pathological hallmark of CLN3 disease retina in living human eye.2,18,21,54 Furthermore, several studies have implicated inflammation and oxidative stress in CLN3 disease55–60 and it has been shown previously that oxidized POS and POS exposed to free radicals are phagocytosed less efficiently by RPE cells.61,62 Therefore, we next investigated the phagocytosis of control vs. CLN3 mutant POS.
We chose to use CLN3 mutant POS isolated from CLN3Δ7–8/Δ7–8 Yucatan miniswine in these experiments because it is well-established that POS in stem cell-derived retina organoids lack reproducibility and consistency potentially owing to the lack of RPE.63,64 Parallel cultures of WT miniswine primary RPE were fed either unlabeled WT miniswine POS or CLN3 miniswine POS (approximately 10 POS/RPE cell) for 2 hours (Figs. 4a, 4b). Subsequently, phagocytosis of POS was measured by quantitative Western blotting analyses measuring the levels of RHO, a POS-specific protein, within the WT miniswine RPE cells (Figs. 4c, 4d).
Figure 4.

Evaluation of POS uptake of WT vs. CLN3 POS by RPE cells. (a, b) Schematic of experimental assay used to measure POS phagocytosis by WT primary miniswine RPE after feeding of WT miniswine POS vs. CLN3 pig POS for 2 hours. (c, d) Representative Western blot image (c) and quantification (d) showing decreased phagocytosis of CLN3 miniswine POS by WT miniswine RPE compared with WT miniswine POS by WT miniswine RPE. ***P ≤ 0.001. n ≥ 3 for Figure 4 experiment.
CLN3 miniswine POS were phagocytosed less efficiently by WT miniswine RPE cultures compared with WT miniswine POS (Figs. 4c, 4d). In fact, WT miniswine POS were phagocytosed greater than five-fold more efficiently than the CLN3 miniswine POS (Figs. 4c, 4d). These data suggest that CLN3-mutant POS contribute to defective POS phagocytosis by RPE cells in CLN3 disease.
Decreased POS Phagocytosis and Subsequent Decrease in Autofluorescence/Lipofuscin Levels Precedes Loss of the Outer Nuclear Layer (ONL) in the CLN3 Miniswine Model
To validate POS phagocytosis defect in vivo, we used a previously published protocol,45,46 to compare RHO levels in RPE flatmounts from WT miniswine and CLN3 miniswine retina at 6 months of age. Consistent with fewer number of phagocytosed POS in CLN3 hRPE cells (Figs. 2a–2c), CLN3 miniswine RPE showed decreased number of RHO-positive phagosomes compared with WT miniswine RPE (Figs. 5a, 5b).
Figure 5.

Longitudinal comparison of RPE autofluorescence levels and retina histology in CLN3 miniswine eye. (a, b) Representative confocal microscopy images in the planar view (a, top) and orthogonal (yz) view (a, bottom) and corresponding quantification (b) showing decreased number of RHO-positive rod phagosomes in CLN3 miniswine RPE flatmounts compared with WT miniswine RPE flatmounts at 6 months of age. Scale bar, 10 µm. **P ≤ 0.01. (c–e). Representative confocal images (c, d) and quantitative analyses (e, e′) showing similar levels of autofluorescence accumulation in RPE of WT and CLN3 miniswine at 6 months of age. In contrast, decreased autofluorescence accumulation was observed in CLN3 miniswine RPE compared with WT miniswine RPE at 36 months of age. Scale bar, 10 µm. **P ≤ 0.01. (f) Representative light microscopy images of WT retina sections (36 months) and CLN3 miniswine retina sections at 36 and 48 months of age showing the presence of POS in CLN3 miniswine retina at 36 months of age, but absence of POS with significant loss of the photoreceptor cell nuclei/ONL at 48 months of age. The retina laminae are labeled in WT retina image. GCL, ganglion cell layer; INL, inner nuclear layer; IPL, inner plexiform layer; NFL, nerve fiber layer; OPL, outer plexiform layer; PR, photoreceptors. Scale bar, 100 µm. n ≥ 3 for all experiments in Figure 5.
Lipofuscin, the autofluorescence accumulation that accumulates in RPE cells because of incomplete digestion of POS by RPE cells, is reduced in CLN3 disease donor RPE cells.18,23 CLN3 disease and CLN3 hRPE cells phagocytose (uptake) less POS and consequently have reduced lipofuscin-like RPE autofluorescence compared with control RPE cells22 (Figs. 2e, 2f). Thus, we next used autofluorescence levels in the spectral wavelength consistent with lipofuscin (ex, 546 nm; em, 560–615 nm) to evaluate the impact of impaired POS phagocytosis by RPE cells in CLN3 miniswine retina on lipofuscin/autofluorescence accumulation.
Longitudinal analyses of autofluorescence levels (ex, 546 nm; em, 560–615 nm) in the RPE cells of age- and sex-matched WT vs. CLN3 miniswine retina showed similar RPE autofluorescence in WT and CLN3 miniswine retina at 6 months of age (Figs. 5c–5e). In contrast, there was reduced lipofuscin in the CLN3 miniswine RPE compared with WT miniswine RPE at 36 months of age (Figs. 5c–5e). These results suggest that decreased RPE autofluorescence in CLN3 disease retina are likely a chronic consequence of decreased POS phagocytosis.
Note that, in our previous study,15 CLN3 miniswine had shown reduced photopic and scotopic a-wave amplitude at approximately 30 to 36 months of age. ERG analyses of CLN3 miniswine retina at 3 and 6 months of age, when there is no decrease in RPE lipofuscin/autofluorescence showed similar photopic and scotopic a-wave amplitudes in WT and CLN3 miniswine retina (Supplementary Fig. S3). Similarly, photopic and scotopic b-wave recordings were similar in WT and CLN3 miniswine retina at 3 and 6 months of age (Supplementary Fig. S3).
Longitudinal histological analyses of CLN3 miniswine retina at the 36- and 48-month timepoints showed the presence of POS. In contrast, POS were almost entirely gone with a near-complete loss of ONL in CLN3 miniswine retina at 48 months of age (Fig. 5f). Note that OCT analyses of WT vs. CLN3 miniswine retina showed no significant changes in any of the retina cell layers including the ONL at 6 months of age, although the nerve fiber layer was slightly reduced when measurements were taken 1 mm from the optic nerve (Supplementary Fig. S4).
Overall, these data show that a decrease in RPE lipofuscin/autofluorescence coincides with photopic and scotopic visual deficits and precedes overt lack of POS and ONL loss in CLN3 miniswine retina.
Discussion
CLN3 disease is the most common form of neuronal ceroid lipofuscinoses. Apart from neurological symptoms, vision loss owing to retinal degeneration is an early clinical hallmark of CLN3 disease. In a previous study, we used a CLN3 disease patient-derived hiPSC-RPE to show impaired POS phagocytosis.22 Because gene modifiers have been implicated strongly in CLN3 disease,4 we generated isogenic control and CLN3 H9 hESC lines (hESCs; H9), as well as used a CLN3 miniswine model, to show that impaired POS phagocytosis is a direct consequence of disease-causing CLN3 mutation (CLN3Δ7–8/Δ7–8), which contributes to reduced lipofuscin-like autofluorescence in RPE cells in CLN3 disease.21,22
Histopathological and clinical characterizations of CLN3 disease have suggested that retinal degeneration starts at the POS.2,65 Furthermore, CLN3 disease donor eyes show decreased RPE lipofuscin/autofluorescence.21 Despite this finding, there has been limited investigation of POS phagocytosis in CLN3 disease.12,13,22 This is presumably due to the postulation that photoreceptor cell death and the consequent lack of POS lead to reduced lipofuscin accumulation in RPE cells.21 Here, we provide evidence that POS phagocytosis dysfunction and subsequently reduced accumulation of lipofuscin-like autofluorescent material in RPE in CLN3 disease is a consequence of both primary RPE dysfunction and POS alterations. Specifically, by comparing the uptake of nondiseased POS by parallel cultures of isogenic control and CLN3 hRPE cells (Figs. 2, 3), we show that cell autonomous RPE dysfunction owing to CLN3 mutation (CLN3Δ7–8/Δ7–8) is sufficient for reduced POS phagocytosis in CLN3 disease. Similarly, by comparing the uptake of WT vs. CLN3 POS by WT RPE cells (Fig. 4), we show that POS alterations in CLN3 disease also independently contribute to decreased POS phagocytosis by RPE cells in CLN3 disease.
A limitation of our study is that we did not validate the role of POS alterations in impaired POS phagocytosis by RPE cells in the human-relevant hESC model. Similarly, we did not characterize extensively the POS phagocytosis defect in vivo in the CLN3 miniswine model. This was due to consideration of the most optimal experimental approach and reduction in the use of tissue from a large animal model of the disease. The hESC-RPE, which have been shown consistently to possess properties of human RPE cells in vivo,66,67 provided a suitable model system to investigate the ability of control vs. CLN3 RPE cells to phagocytose shed POS. Similarly, the availability of in vitro protocols to assess POS binding vs. internalization22,44 allowed us to interrogate the precise role of POS binding vs. POS internalization in impaired POS phagocytosis by CLN3 hRPE cells (Fig. 3). Furthermore, our results showed that post-daily feeding of POS (approximately 20–40 POS/RPE cell), CLN3 hRPE cells accumulate less autofluorescent lipofuscin-like material compared with parallel cultures of isogenic control hRPE cells. However, these data need to be interpreted with caution owing to the limitation that although bisretinoids are known constituents of RPE lipofuscin in humans and animal models, the composition of the lipofuscin-like material in cultured RPE after daily feeding with POS is not known.
In contrast with RPE cells, the reproducibility of POS differentiation and maturation in the stem cell-derived retinal organoid is variable.63,64 Therefore, to avoid confounding variability owing to experimental model system, we relied on primary POS isolated from WT vs. CLN3 miniswine in experiments evaluating the role of diseased POS on the efficiency of POS phagocytosis by RPE cells in CLN3 disease. Note that recent advances in protocol for POS isolation from native retina allowed us to obtain sufficient POS from three to four miniswines. Furthermore, we were able to use RPE from the animals sacrificed for POS isolation to compare autofluorescence accumulation between WT vs. CLN3 miniswine RPE cells.
Reduced lipofuscin/autofluorescence accumulation preceded the loss of POS and photoreceptors in the CLN3 miniswine model and coincided with the earliest timepoint of scotopic and photopic visual deficit in CLN3 miniswine (Fig. 5, Supplementary Fig. S3).15 This finding is consistent with pathological studies of human eyes that have shown neuronal depletion in CLN3 disease retina starting at the outer segments of the photoreceptors and proceeding inwards to the ganglion cell layer.2,65 It is important to emphasize that POS phagocytosis defect is not the sole cause of vision loss in CLN3 disease as multiple cell layers are impacted in the disease. Based on our results, we propose that POS phagocytosis defect independently contributes to photoreceptor degeneration in CLN3 disease. However, it remains to be determined whether unphagocytosed autofluorescent debris that accumulates at the RPE-photoreceptor cell interface contributes to disease in the CLN3 miniswine model.
Altogether, apart from highlighting a central role of photoreceptor-RPE interaction in CLN3 disease (that needs further investigation), these data further highlight the value of a CLN3 miniswine model for studying the retina pathobiology of CLN3 disease.
Conclusions
Using both in vivo (CLN3 miniswine) and in vitro (isogenic hESC control and CLN3 lines) models, we show that the most common disease-causing mutation in CLN3 disease (CLN3Δ7–8/Δ7–8) leads to POS alterations and primary RPE dysfunction that contribute to defective POS phagocytosis and reduced RPE autofluorescence/lipofuscin in CLN3 disease. Furthermore, decreased RPE autofluorescence/lipofuscin coincides with the earliest timepoint of scotopic and photopic visual deficit and precedes the loss of POS and photoreceptors in a large animal model of CLN3 disease.
Supplementary Material
Acknowledgments
The authors thank Exemplar Genetics staff in conducting studies (ERG, OCT, and tissue recovery) that were performed at the Exemplar Genetics facilities.
Supported by R01EY028167 (R.S.), R01EY030183 (R.S.), R01EY033192 (R.S.), funding from ForeBatten Foundation to both Singh and Weimer laboratories, and a Research to Prevent Blindness Unrestricted Challenge Grant to Department of Ophthalmology at University of Rochester and University Research Award given to R.S. by the University of Rochester. We also acknowledge the Batten Disease Support and Research Association Australia (RGP204), assistance and Royal Hobart Hospital Research Foundation (17-205) to A.L.C. and A.W.H.
Author Contributions:
Conceptualization and Methodology: A.L.C., A.W.H., J.M.W., R.S.
Experiment and Analyses: A.L.C., A.W.H., C.B., C.R.T., J.H., J.M.W., J.T., S.C., S.D., R.S.
Writing: Initial draft: R.S.
Writing – Review & Editing: A.L.C., A.W.H., C.B., C.R.T., J.H., J.M.W., J.T., S.C., S.D., R.S.
Resources: A.L.C., A.W.H., J.M.W., R.S.
Disclosure: J. Han, None; S. Chear, None; J. Talbot, None; V. Swier, None; C. Booth, None; C. Reuben-Thomas, None; S. Dalvi, None; J.M. Weimer, None; A.W. Hewitt, None; A.L. Cook, None; R. Singh, None
References
- 1. Haltia M, Goebel HH.. The neuronal ceroid-lipofuscinoses: a historical introduction. Biochim Biophys Acta. 2013; 1832(11): 1795–1800. [DOI] [PubMed] [Google Scholar]
- 2. Anderson GW, Goebel HH, Simonati A.. Human pathology in NCL. Biochim Biophys Acta. 2013; 1832(11): 1807–1826. [DOI] [PubMed] [Google Scholar]
- 3. Mole SE, Williams RE, Goebel HH.. Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics. 2005; 6(3): 107–126. [DOI] [PubMed] [Google Scholar]
- 4. Lebrun AH, Moll-Khosrawi P, Pohl S, et al.. Analysis of potential biomarkers and modifier genes affecting the clinical course of CLN3 disease. Mol Med. 2011; 17(11–12): 1253–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sakti DH, Cornish EE, Fraser CL, et al.. Early recognition of CLN3 disease facilitated by visual electrophysiology and multimodal imaging. Doc Ophthalmol. 2023; 146(3): 241–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schulz A, Kohlschütter A, Mink J, Simonati A, Williams R.. NCL diseases - clinical perspectives. Biochim Biophys Acta. 2013; 1832(11): 1801–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mitchison HM, Bernard DJ, Greene NDE, et al.. Targeted disruption of the Cln3 gene provides a mouse model for Batten disease. Neurobiol Dis. 1999; 6(5): 321–334. [DOI] [PubMed] [Google Scholar]
- 8. Finn R, Kovács AD, Pearce DA.. Altered sensitivity of cerebellar granule cells to glutamate receptor overactivation in the Cln3(Δex7/8)-knock-in mouse model of juvenile neuronal ceroid lipofuscinosis. Neurochem Int. 2011; 58(6): 648–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cotman SL, Vrbanac V, Lebel LA, et al.. Cln3(Deltaex7/8) knock-in mice with the common JNCL mutation exhibit progressive neurologic disease that begins before birth. Hum Mol Genet. 2002; 11(22): 2709–2721. [DOI] [PubMed] [Google Scholar]
- 10. Weimer JM, Benedict JW, Getty AL, et al.. Cerebellar defects in a mouse model of juvenile neuronal ceroid lipofuscinosis. Brain Res. 2009; 1266: 93–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pontikis CC, Cella CV, Parihar N, et al.. Late onset neurodegeneration in the Cln3-/- mouse model of juvenile neuronal ceroid lipofuscinosis is preceded by low level glial activation. Brain Res. 2004; 1023(2): 231–242. [DOI] [PubMed] [Google Scholar]
- 12. Zhong Y, Mohan K, Liu J, et al.. Loss of CLN3, the gene mutated in juvenile neuronal ceroid lipofuscinosis, leads to metabolic impairment and autophagy induction in retinal pigment epithelium. Biochim Biophys Acta Mol Basis Dis. 2020; 1866(10): 165883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wavre-Shapton ST, Calvi AA, Turmaine M, et al.. Photoreceptor phagosome processing defects and disturbed autophagy in retinal pigment epithelium of Cln3Δex1-6 mice modelling juvenile neuronal ceroid lipofuscinosis (Batten disease). Hum Mol Genet. 2015; 24(24): 7060–7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Seigel GM, Lotery A, Kummer A, et al.. Retinal pathology and function in a Cln3 knockout mouse model of juvenile neuronal ceroid lipofuscinosis (Batten disease). Mol Cell Neurosci. 2002; 19(4): 515–527. [DOI] [PubMed] [Google Scholar]
- 15. Swier VJ, White KA, Johnson TB, et al.. A novel porcine model of CLN3 Batten disease recapitulates clinical phenotypes. Dis Model Mech. 2023; 16(8): dmm050038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Seehafer SS, Pearce DA.. You say lipofuscin, we say ceroid: defining autofluorescent storage material. Neurobiol Aging. 2006; 27(4): 576–588. [DOI] [PubMed] [Google Scholar]
- 17. Kohlschütter A, Schulz A.. Towards understanding the neuronal ceroid lipofuscinoses. Brain Dev. 2009; 31(7): 499–502. [DOI] [PubMed] [Google Scholar]
- 18. Bensaoula T, Shibuya H, Katz ML, et al.. Histopathologic and immunocytochemical analysis of the retina and ocular tissues in Batten disease. Ophthalmology. 2000; 107(9): 1746–1753. [DOI] [PubMed] [Google Scholar]
- 19. Mole SE, Williams RE.. Neuronal Ceroid-Lipofuscinoses – RETIRED CHAPTER, FOR HISTORICAL REFERENCE ONLY. 2001 Oct 10 [Updated 2013 Aug 1]. In: Adam MP, Feldman J, Mirzaa GM, et al., eds. GeneReviews [Internet]. Seattle, WA: University of Washington, Seattle; 1993–2024. [Google Scholar]
- 20. Katz ML, Johnson GS, Tullis GE, Lei B.. Phenotypic characterization of a mouse model of juvenile neuronal ceroid lipofuscinosis. Neurobiol Dis. 2008; 29(2): 242–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ouseph MM, Kleinman ME, Wang QJ.. Vision loss in juvenile neuronal ceroid lipofuscinosis (CLN3 disease). Ann N Y Acad Sci. 2016; 1371(1): 55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tang C, Han J, Dalvi S, et al.. A human model of Batten disease shows role of CLN3 in phagocytosis at the photoreceptor-RPE interface. Commun Biol. 2021; 4(1): 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bozorg S, Ramirez-Montealegre D, Chung M, Pearce DA.. Juvenile neuronal ceroid lipofuscinosis (JNCL) and the eye. Surv Ophthalmol. 2009; 54(4): 463–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Seehafer SS, Pearce DA.. You say lipofuscin, we say ceroid: defining autofluorescent storage material. Neurobiology of Aging. 2006; 27(4): 576–588. [DOI] [PubMed] [Google Scholar]
- 25. Bassett AR. Editing the genome of hiPSC with CRISPR/Cas9: disease models. Mamm Genome. 2017; 28(7–8): 348–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Findlay GM. Linking genome variants to disease: scalable approaches to test the functional impact of human mutations. Hum Mol Genet. 2021; 30(R2): R187–R197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim HS, Kweon J, Kim Y.. Recent advances in CRISPR-based functional genomics for the study of disease-associated genetic variants. Exp Mol Med. 2024; 56(4): 861–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kurzawa-Akanbi M, Tzoumas N, Corral-Serrano JC, et al.. Pluripotent stem cell-derived models of retinal disease: elucidating pathogenesis, evaluating novel treatments, and estimating toxicity. Prog Retin Eye Res. 2024; 100: 101248. [DOI] [PubMed] [Google Scholar]
- 29. Afanasyeva TAV, Athanasiou D, Perdigao PRL, et al.. CRISPR-Cas9 correction of a nonsense mutation in. Mol Ther Methods Clin Dev. 2023; 29: 522–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nolan ND, Cui X, Robbings BM, et al.. CRISPR editing of anti-anemia drug target rescues independent preclinical models of retinitis pigmentosa. Cell Rep Med. 2024; 5(4): 101459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lim RR, Shirali S, Rowlan J, et al.. CFH Haploinsufficiency and complement alterations in early-onset macular degeneration. Invest Ophthalmol Vis Sci. 2024; 65(4): 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Galloway CA, Dalvi S, Hung SSC, et al.. Drusen in patient-derived hiPSC-RPE models of macular dystrophies. Proc Natl Acad Sci USA. 2017; 114(39): E8214–E8223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xu P, Chen Z, Ma J, et al.. Biallelic CLCN2 mutations cause retinal degeneration by impairing retinal pigment epithelium phagocytosis and chloride channel function. Hum Genet. 2023; 142(4): 577–593. [DOI] [PubMed] [Google Scholar]
- 34. Cuevas E, Holder DL, Alshehri AH, Tréguier J, Lakowski J, Sowden JC. NRL. Stem Cells. 2021; 39(4): 414–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang Q, Chear S, Wing K, et al.. Use of CRISPR/Cas ribonucleoproteins for high throughput gene editing of induced pluripotent stem cells. Methods. 2021; 194: 18–29. [DOI] [PubMed] [Google Scholar]
- 36. Chear S, Perry S, Wilson R, et al.. Lysosomal alterations and decreased electrophysiological activity in CLN3 disease patient-derived cortical neurons. Dis Model Mech. 2022; 15(12): dmm049651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang K, Li M, Hadley D, et al.. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007; 17(11): 1665–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Colella S, Yau C, Taylor JM, et al.. QuantiSNP: an objective Bayes hidden-Markov model to detect and accurately map copy number variation using SNP genotyping data. Nucleic Acids Res. 2007; 35(6): 2013–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Neavin D, Nguyen Q, Daniszewski MS, et al.. Single cell eQTL analysis identifies cell type-specific genetic control of gene expression in fibroblasts and reprogrammed induced pluripotent stem cells. Genome Biol. 2021; 22(1): 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Manian KV, Galloway CA, Dalvi S, et al.. 3D iPSC modeling of the retinal pigment epithelium-choriocapillaris complex identifies factors involved in the pathology of macular degeneration. Cell Stem Cell. 2021; 28(5): 846–862.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Toops KA, Tan LX, Lakkaraju A.. A detailed three-step protocol for live imaging of intracellular traffic in polarized primary porcine RPE monolayers. Exp Eye Res. 2014; 124: 74–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Parinot C, Rieu Q, Chatagnon J, Finnemann SC, Nandrot EF.. Large-scale purification of porcine or bovine photoreceptor outer segments for phagocytosis assays on retinal pigment epithelial cells. J Vis Exp. 2014;(94): e52100, doi: 10.3791/52100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Singh R, Shen W, Kuai D, et al.. iPS cell modeling of Best disease: insights into the pathophysiology of an inherited macular degeneration. Hum Mol Genet. 2013; 22(3): 593–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Müller C, Charniga C, Temple S, Finnemann SC.. Quantified F-actin morphology is predictive of phagocytic capacity of stem cell-derived retinal pigment epithelium. Stem Cell Rep. 2018; 10(3): 1075–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mazzoni F, Dun Y, Vargas JA, Nandrot EF, Finnemann SC.. Lack of the antioxidant enzyme methionine sulfoxide reductase A in mice impairs RPE phagocytosis and causes photoreceptor cone dysfunction. Redox Biol. 2021; 42: 101918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vargas JA, Finnemann SC.. Probing photoreceptor outer segment phagocytosis by the RPE in vivo: models and methodologies. Int J Mol Sci. 2022; 23(7): 3661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sethna S, Finnemann SC.. Analysis of photoreceptor rod outer segment phagocytosis by RPE cells in situ. Methods Mol Biol. 2013; 935: 245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dalvi S, Galloway CA, Winschel L, et al.. Environmental stress impairs photoreceptor outer segment (POS) phagocytosis and degradation and induces autofluorescent material accumulation in hiPSC-RPE cells. Cell Death Discov. 2019; 5: 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kennedy BP, Payette P, Mudgett J, et al.. A natural disruption of the secretory group II phospholipase A2 gene in inbred mouse strains. J Biol Chem. 1995; 270(38): 22378–22385. [DOI] [PubMed] [Google Scholar]
- 50. Amit M, Carpenter MK, Inokuma MS, et al.. Clonally derived human embryonic stem cell lines maintain pluripotency and proliferative potential for prolonged periods of culture. Dev Biol. 2000; 227(2): 271–278. [DOI] [PubMed] [Google Scholar]
- 51. Galloway CA, Dalvi S, Shadforth AMA, et al.. Characterization of human iPSC-RPE on a prosthetic Bruch's membrane manufactured from silk fibroin. Invest Ophthalmol Vis Sci. 2018; 59(7): 2792–2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mircheff AK, Miller SS, Farber DB, Bradley ME, O'Day WT, Bok D.. Isolation and provisional identification of plasma membrane populations from cultured human retinal pigment epithelium. Invest Ophthalmol Vis Sci. 1990; 31(5): 863–878. [PubMed] [Google Scholar]
- 53. Sonoda S, Spee C, Barron E, Ryan SJ, Kannan R, Hinton DR.. A protocol for the culture and differentiation of highly polarized human retinal pigment epithelial cells. Nat Protoc. 2009; 4(5): 662–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Radke J, Stenzel W, Goebel HH. Human NCL neuropathology. Biochim Biophys Acta. 2015; 1852(10, Part B): 2262―2266. [DOI] [PubMed] [Google Scholar]
- 55. Vidal-Donet JM, Cárcel-Trullols J, Casanova B, Aguado C, Knecht E.. Alterations in ROS activity and lysosomal pH account for distinct patterns of macroautophagy in LINCL and JNCL fibroblasts. PLoS One. 2013; 8(2): e55526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Benedict JW, Sommers CA, Pearce DA.. Progressive oxidative damage in the central nervous system of a murine model for juvenile Batten disease. J Neurosci Res. 2007; 85(13): 2882–2891. [DOI] [PubMed] [Google Scholar]
- 57. Tuxworth RI, Chen H, Vivancos V, Carvajal N, Huang X, Tear G.. The Batten disease gene CLN3 is required for the response to oxidative stress. Hum Mol Genet. 2011; 20(10): 2037–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dannhausen K, Möhle C, Langmann T.. Immunomodulation with minocycline rescues retinal degeneration in juvenile neuronal ceroid lipofuscinosis mice highly susceptible to light damage. Dis Model Mech. 2018; 11(9): dmm033597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Groh J, Berve K, Martini R.. Fingolimod and teriflunomide attenuate neurodegeneration in mouse models of neuronal ceroid lipofuscinosis. Mol Ther. 2017; 25(8): 1889–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Brooks AI, Chattopadhyay S, Mitchison HM, Nussbaum RL, Pearce DA.. Functional categorization of gene expression changes in the cerebellum of a Cln3-knockout mouse model for Batten disease. Mol Genet Metab. 2003; 78(1): 17–30. [DOI] [PubMed] [Google Scholar]
- 61. Gupta P, Tirgan N, Kalariya NM, Godley BF, Motamedi M.. Kinetics of oxidized rod outer segment phagocytosis under chronic oxidative stress in an in-vitro model of age-related macular degeneration. Invest Ophthalmol Vis Sci. 2010; 51(13): 494–494. [Google Scholar]
- 62. Becquet F, Goureau O, Soubrane G, Coscas G, Courtois Y, Hicks D.. Superoxide inhibits proliferation and phagocytic internalization of photoreceptor outer segments by bovine retinal pigment epithelium in vitro. Exp Cell Res. 1994; 212(2): 374–382. [DOI] [PubMed] [Google Scholar]
- 63. Singh RK, Nasonkin IO.. Limitations and promise of retinal tissue from human pluripotent stem cells for developing therapies of blindness. Front Cell Neurosci. 2020; 14: 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Capowski EE, Samimi K, Mayerl SJ, et al.. Reproducibility and staging of 3D human retinal organoids across multiple pluripotent stem cell lines. Development. 2018; 146: dev171686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Radke J, Stenzel W, Goebel HH. Human NCL neuropathology. Biochim Biophys Acta. 2015; 1852(10 Pt B): 2262–2266. [DOI] [PubMed] [Google Scholar]
- 66. Rowland TJ, Blaschke AJ, Buchholz DE, Hikita ST, Johnson LV, Clegg DO.. Differentiation of human pluripotent stem cells to retinal pigmented epithelium in defined conditions using purified extracellular matrix proteins. J Tissue Eng Regen Med. 2013; 7(8): 642–653. [DOI] [PubMed] [Google Scholar]
- 67. Dalvi S, Galloway CA, Singh R.. Pluripotent stem cells to model degenerative retinal diseases: the RPE perspective. Adv Exp Med Biol. 2019; 1186: 1–31. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Requests for original data files and reagents and materials used in this study will be fulfilled by contacting the corresponding author, Dr Ruchira Singh.