

SUMMARY
Although antibody variable regions mediate antigen-specific binding, they can also mediate non-specific interactions with non-cognate antigens, impacting diverse immunological processes and the efficacy, safety, and half-life of antibody therapeutics. To understand the molecular basis of antibody non-specificity, we sorted two dissimilar human naïve antibody libraries against multiple reagents to enrich for variants with different levels of polyreactivity. Sequence analysis of >300,000 paired antibody variable regions revealed that the heavy chain primarily mediates human antibody polyreactivity, and this is due to the high positive charge, high hydrophobicity, and combinations thereof in the corresponding complementarity-determining regions, which can be predicted using a machine learning model developed in this work. Notably, a subset of the most important features governing antibody non-specific interactions, namely those that contain tyrosine, also govern specific antigen recognition. Our findings are broadly relevant for understanding fundamental aspects of antibody molecular recognition and the applied aspects of antibody-drug design.
Graphical Abstract
In brief
Chen et al. sorted diverse human antibody libraries against various multiple polyreactivity reagents, revealing that heavy-chain variable regions primarily mediate non-specificity. In addition, a machine learning model was developed to predict human antibody polyreactivity, revealing key features relevant to antibody recognition and drug design.
INTRODUCTION
Antibody recognition of a target antigen typically involves a delicate balance of different molecular interactions, including ionic, hydrophobic, and hydrogen-bonding interactions, collectively mediating high affinity and specificity. However, antigen-specific antibodies also display variable and difficult-to-predict levels of non-specific binding mediated by their variable regions,1-3 referred to herein as polyreactivity.4 Antibody polyreactivity is involved in diverse immunological processes, ranging from weak antibody binding during early antibody maturation5-7 to conferring a competitive advantage during long-term maintenance of memory B cells.6,8 In contrast, antibody polyreactivity typically compromises the therapeutic potential of monoclonal antibodies, as high levels of polyreactivity are linked to fast anti-body clearance in vivo.2,9-15
Despite the broad importance of human antibody polyreactivity, it has proven challenging to study and ultimately predict for several reasons. First, there is no unique definition of high or low polyreactivity, and it depends on the reagent(s) used to evaluate these interactions. Second, previously reported datasets that link human antibody sequences to their corresponding levels of polyreactivity are typically insufficient in terms of size and, even more importantly, diversity of germlines, frameworks, and complementarity-determining regions (CDRs).16-19 Third, even in rare cases where large human antibody polyreactivity datasets have been reported, these datasets are limited in their use for model development because they lack diversity in the light chain and define polyreactive antibodies as those that bind specific antigens as well as polyreactivity reagents.20
We sought to address each of these previous limitations in order to evaluate four outstanding questions related to human antibody polyreactivity. First, what molecular features of human antibodies are most strongly linked—both positively and negatively—to antibody polyreactivity? Second, are specific regions within the variable fragment (Fv) most important in mediating human antibody polyreactivity? Third, which (if any) antibody molecular features linked to polyreactivity are also involved in mediating antigen-specific binding? Fourth, to what extent can human antibody polyreactivity be predicted? Herein, we report the generation of large (>300,000 variants) and self-consistent antibody sequence/polyreactivity datasets for two diverse naïve human single-chain (scFv) antibody libraries enriched against four different polyreactivity reagents (Figure 1A). We use these large datasets to identify molecular features and regions within Fv involved in mediating polyreactive interactions and, in some cases, affinity interactions as well. We also use these datasets to demonstrate that human antibody polyreactivity can be predicted with high accuracy using a relatively simple model that only uses the amino acid sequences of the antibody Fv regions.
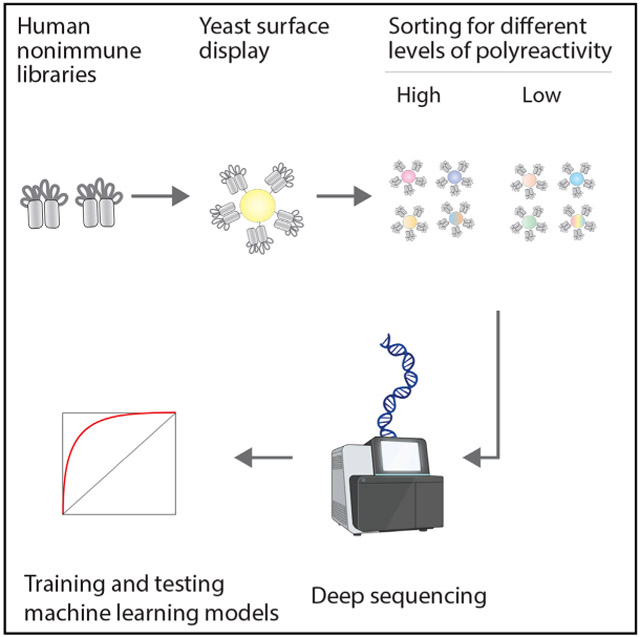
Figure 1. Overview of the library sorting, deep sequencing, and model training methods used to generate machine learning models for predicting human antibody polyreactivity.
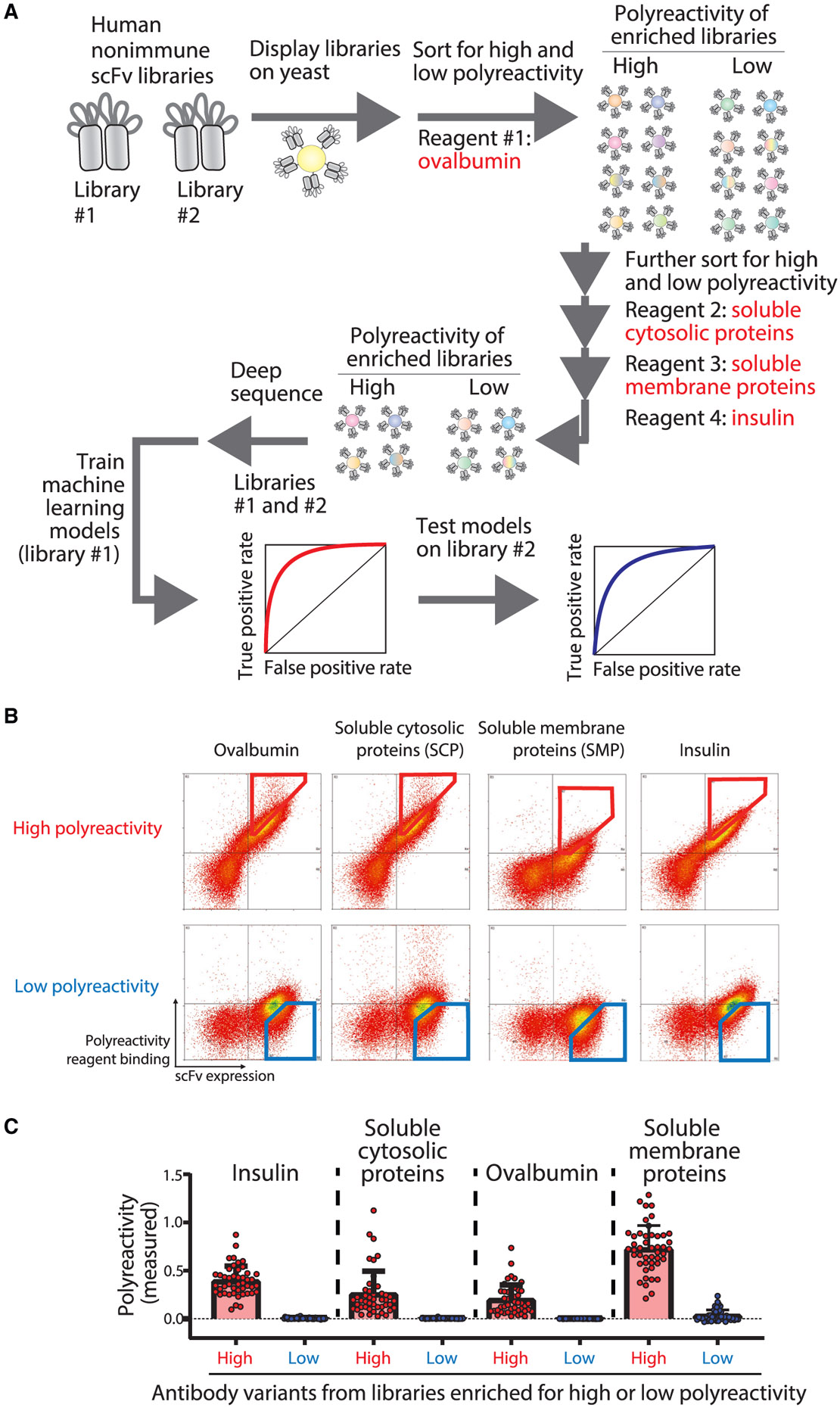
(A) Two human single-chain variable fragment (scFv) libraries displayed on the surface of yeast were sorted for positive or negative binding to multiple poly-specificity reagents. The enriched libraries were deep sequenced to generate large datasets of antibody sequences and corresponding classifications for either high or low polyspecificity. One of the large human scFv datasets was used along with a smaller dataset for clinical-stage antibodies to train machine learning models to predict antibody polyspecificity. Finally, the models were tested on the second large human scFv dataset (not used for training) and additional independent datasets for preclinical and clinical-stage antibodies.
(B) The human scFv library (library #1)21 was displayed on the surface of yeast and sorted successively against ovalbumin (0.13 mg/mL [2.9 μM]; fluorescence-activated cell sorting [FACS] sort #1), soluble cytosolic proteins (SCPs) from CHO cells (0.13 mg/mL; FACS sort #2), soluble membrane proteins (SMPs) from CHO cells (0.13 mg/mL; FACS sort #3), and insulin (0.13 mg/mL [22.4 μM]; FACS sort #4). FACS cytograms are shown for FACS sort #5, which was performed using the output libraries from FACS sort #4 to collect samples for deep sequencing analysis. The FACS cytograms report the antibody (scFv) expression on the x axis and non-specific binding on the y axis. The positive and negative non-specific binding populations that were selected for deep sequencing analysis are shown in red (high non-specific binding) and blue (low non-specific binding) gates.
(C) Individual scFv variants were isolated after FACS sort #4, and their levels of non-specific binding were evaluated using yeast surface display and flow cytometry. The four polyspecificity reagents were used as described in (B) except at a lower concentration (0.026 mg/mL). The reported levels of non-specific binding were first normalized to their scFv expression levels and then normalized between two scFv standards (FN4 for the negative control and FP3 for positive control; Dataset S3). In (C), the data are averages of two biological replicates, and the error bars are standard errors.
RESULTS
Generation of deep sequencing datasets for human antibodies with high and low polyreactivity
To better understand and ultimately predict human antibody polyreactivity, we first sought to generate large datasets of human antibodies enriched for high and low levels of polyreactivity. We used two previously reported human single-chain antibody libraries, namely libraries #121 and #2,22 displayed on the yeast surface for our analysis (Figure S1). Human antibody library #1 was cloned from human spleen and lymph node samples from 58 adults.21 Human library #2 was cloned from three samples of human spleen cells and two samples from human peripheral blood lymphocytes.22 Both libraries contain significant diversity in their variable heavy (VH) and variable light (VL) domain germ-lines, frameworks, and CDRs.
To ensure the libraries would be sufficiently enriched or depleted for polyreactive antibodies, we sorted the libraries against four different polyspecificity reagents, namely ovalbumin, soluble cytosolic proteins (SCPs; from CHO cells), soluble membrane proteins (SMPs; from CHO cells), and insulin. We first selected the top 10% and the bottom 10% of each library against the first reagent (ovalbumin) and then subsequently sorted each enriched population (ovalbumin+ and ovalbumin−) against SCPs (top 10% of the ovalbumin+ library and bottom 10% of the ovalbumin− sample) to generate libraries doubly enriched for non-specific binding (ovalbumin+/SCP+) or a lack thereof (ovalbumin−/SCP−). This process was repeated two more times against SMPs and insulin, which resulted in libraries generally enriched for high (ovalbumin+/SCP+/SMP+/insulin+) or low (ovalbumin−/SCP−/SMP−/insulin−) polyreactivity. After four rounds of sorting, the final enriched libraries were sorted one final time against each of the four reagents to collect high- and low-polyreactivity samples for deep sequencing analysis, as shown for library #1 in Figure 1B.
To evaluate the quality of the sorted library samples, we randomly isolated 48 high-polyreactivity and 48 low-polyreactivity antibodies from library #1. Encouragingly, the levels of anti-body binding to the four polyreactivity reagents were much higher for antibodies enriched for high polyreactivity than those enriched for low polyreactivity (Figure 1C). These results demonstrate that the enriched libraries display the expected differences in polyreactivity and are well suited for further analysis of the determinants of antibody polyreactivity.
Identification of Fv molecular features strongly linked to antibody polyreactivity
Next, the enriched antibody library samples were deep sequenced as paired VH/VL amplicons, which resulted in the generation of two large antibody Fv sequence datasets, namely Dataset S1 for library #1, including 131,255 antibodies with low polyreactivity and 115,038 antibodies with high polyreactivity, and Dataset S2 for library #2, including 34,137 antibodies with low polyreactivity and 93,080 anti-bodies with high polyreactivity. Each library contained diverse germline families (Figures 2A-2D). Most (10 out of 11) germline families displayed consistent enrichment in either high or low polyreactivity for both libraries. For example, VH3, VL1, VL2, and VL3 were enriched in both libraries for low polyreactivity, while VH4, VH5, VH6, VK1, VK3, VK4, VK6, and VL6 were enriched in both libraries for high polyreactivity. Moreover, one germline family, namely VH1, showed opposite behaviors across the two libraries.
Figure 2. Germline family and sequence comparisons for antibody libraries #1 and #2 after enrichment for high and low levels of poly-reactivity.
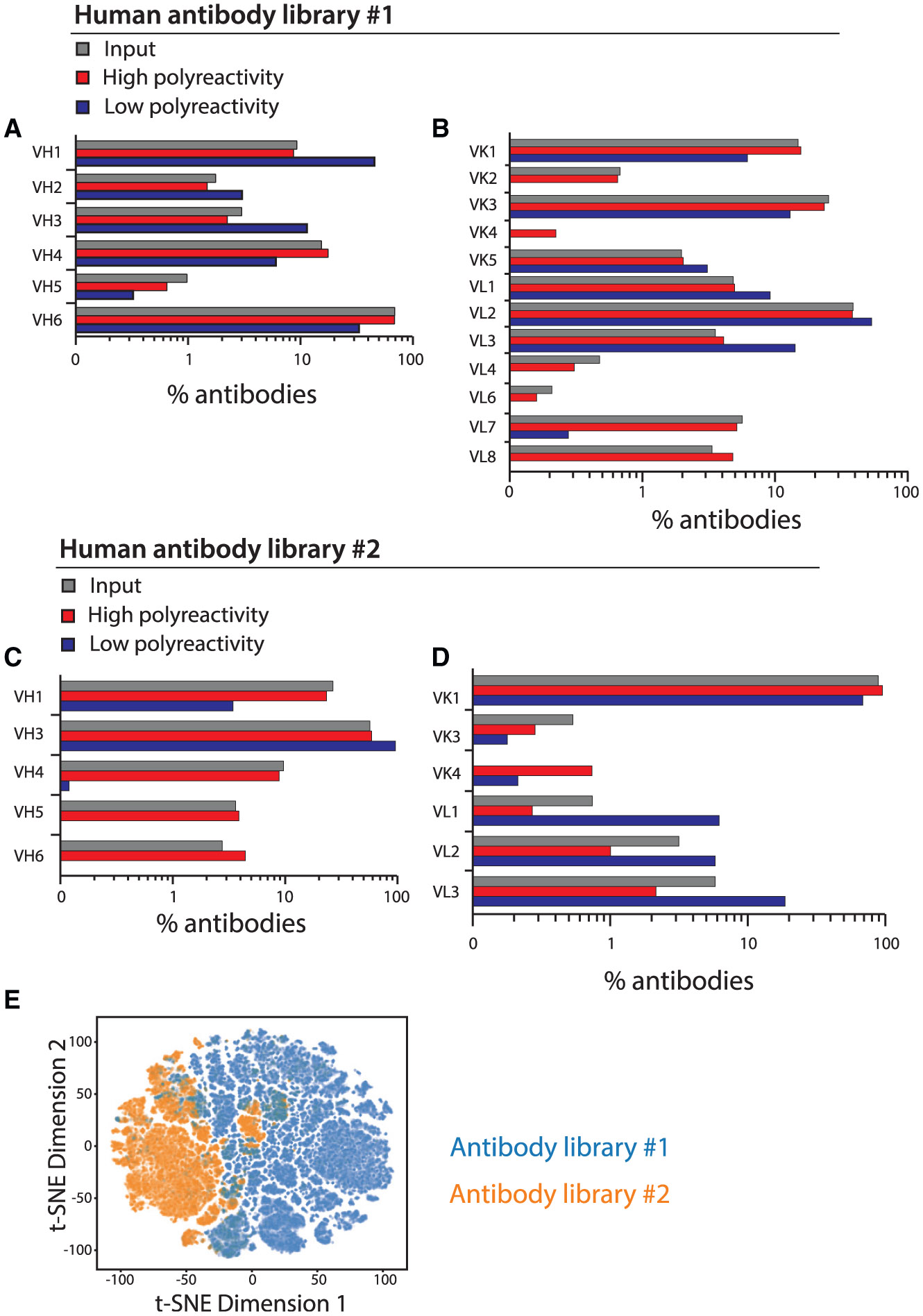
(A–D) Distribution of germline families for antibody (A and B) library #1 and (C and D) library #2.
(E) Analysis of sequence differences between the two libraries. The sequences were OneHot encoded, subjected to dimensionality reduction via a truncated singular value decomposition, and embedded into two-dimensional space for visualization with t-distributed stochastic neighbor embedding (t-SNE).
We also evaluated the polyreactivity of the most common germlines in each library (Tables S1 and S2). Some notable heavy-chain germlines displayed consistent levels of polyreactivity in both libraries, including low polyreactivity for VH3-23 and high polyreactivity for VH6-1 (Table S1). Notably, only 42% of the most common heavy-chain germlines were consistent in their levels of polyreactivity across the two libraries. Conversely, several notable light-chain germlines displayed consistent levels of polyreactivity in both libraries, including VK1-33 (Table S2). Unlike the heavy-chain germlines, most (85%) of the common light-chain germlines displayed consistent levels of polyreactivity across the two libraries. Finally, the most common VH/VL germline pairs and their corresponding levels of polyreactivity are summarized in Table S3.
Next, we performed a standard dimensionality reduction analysis of the antibody sequences for the two libraries (Figure 2E). This analysis revealed that library #2 displays larger differences between high- and low-polyreactivity anti-bodies (Figure S2). Sequence similarity analysis of the most common heavy-chain germline families in both libraries, namely VH1–VH4 and VH6 for library #1 and VH1 and VH3 for library #2, revealed that high-poly-reactivity antibodies show higher sequence similarity within each germline family than low-polyreactivity antibodies (Figure S3). Similar analysis of the most common light-chain germline families, namely VK1, VK3, VK5, and VL1–VL3 for library #1 and VK1,VL2, and VL3 for library #2, revealed that high-polyreactivity antibodies show higher sequence similarity within each germline family for library #1 and low-polyreactivity antibodies show higher sequence similarity within each germline family for library #2.
Next, we reasoned that the large differences in the sequence spaces sampled by the two libraries provided an opportunity to identify common and potentially general molecular features that mediate antibody polyreactivity. Therefore, we first evaluated diverse sequence-based (SB) Fv features that may mediate antibody polyreactivity, including the net charge and the number of specific individual amino acids and combinations thereof, and identified the most important features that were consistent in both libraries (Figures 3A and 3B; Table S4). Notably, the most important feature was the Fv net charge (pH 7.4), as higher positive charge was linked to increased polyreactivity. Interestingly, the Fv net charge distributions of both input libraries were similar to those of the corresponding libraries enriched for high polyreactivity (Figures 3A and 3B).
Figure 3. Molecular features that strongly differentiate between human antibodies with high and low polyreactivity.
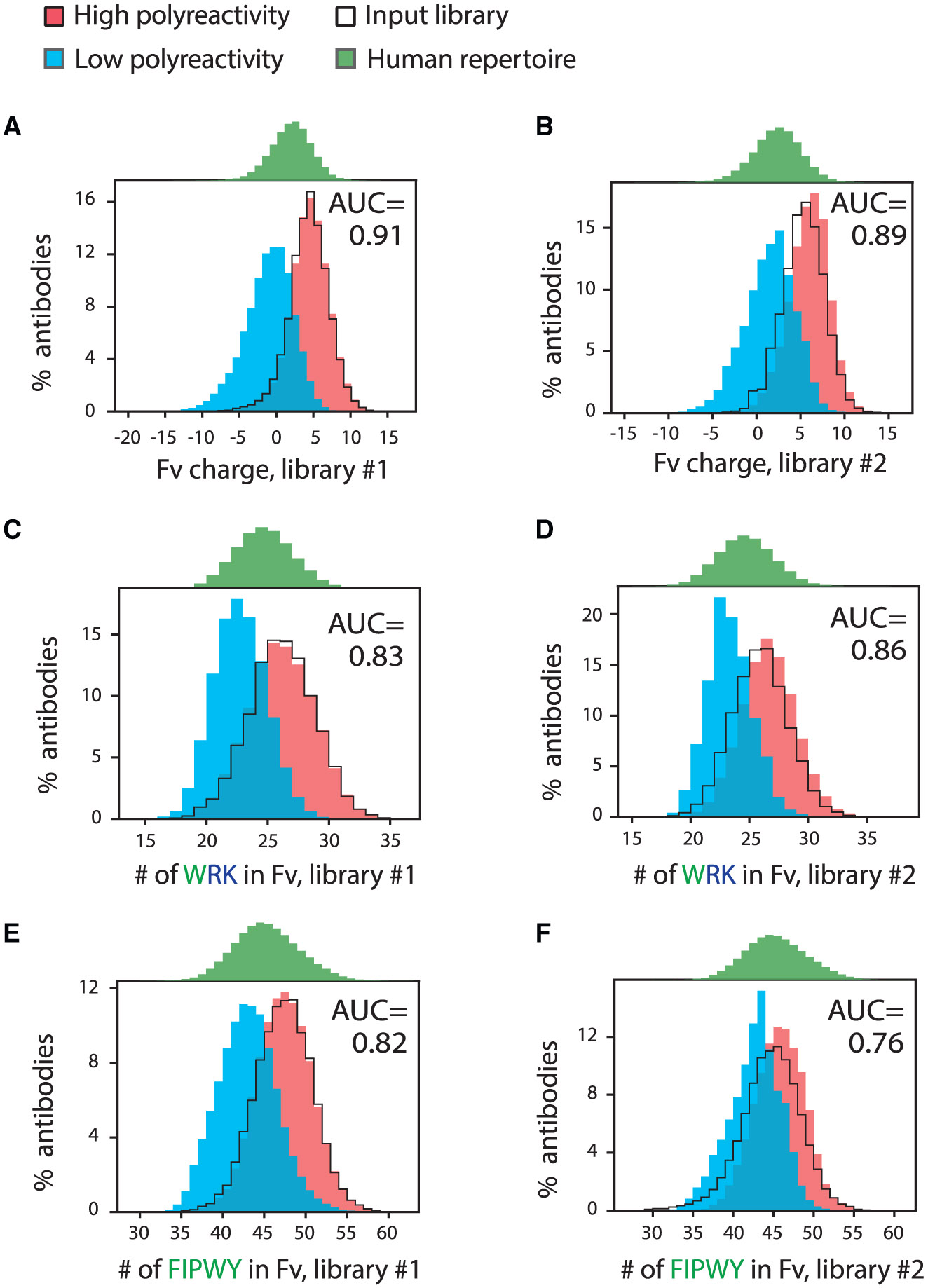
Distributions of key molecular features linked to polyreactivity for human antibodies (libraries #1 and #2) and their corresponding area under the ROC curve (AUC) values calculated using logistic regression analysis. The same features for the input antibody libraries and a human repertoire dataset were also calculated.
(A and B) Fv charge (pH 7.4) distribution for (A) library #1 and (B) library #2.
(C and D) Distributions of the number of tryptophan, arginine, and lysine residues in Fv for (C) library #1 and (D) library #2.
(E and F) Distributions of the number of phenylalanine, isoleucine, proline, tryptophan, and tyrosine residues in Fv for (E) library #1 and (F) library #2.
In (A) and (B), the net charge (pH 7.4) was calculated using charges of +1 for Arg and Lys, +0.1 for His, and −1 for Asp and Glu.
The next most significant set of molecular features, which had area under the receiver operating characteristic (ROC) curve (AUC) values > 0.8 for both libraries, were those that describe the combined numbers of positively charged and hydrophobic residues in the Fv region (Figures 3C and 3D; Table S4). Of the ten features composed of positively charged and hydrophobic residues with AUC values > 0.8, arginine, lysine, and tryptophan were the most common residues. Additional residues observed in these combined features included isoleucine, tyrosine, and proline.
Interestingly, molecular features that describe hydrophobicity alone in the Fv region were less significant (Figures 3E and 3F; Table S4). For example, only six such features were identified across the two libraries with AUC values > 0.7. Tryptophan was the only individual hydrophobic residue that alone led to AUC values > 0.7 for both libraries (0.74 for library #1 and 0.73 for library #2).
We also sought to identify any Fv molecular features negatively associated with antibody polyreactivity (Table S4). This analysis led to the identification of five such features with AUC values > 0.7. Four features contained aspartic acid alone or in combination with glutamate and at least one polar residue (asparagine or glutamine). This finding reveals that increased levels of these residues in Fv are associated with reduced polyreactivity. The other feature negatively linked to antibody polyreactivity was the number of glycine residues in Fv.
Heavy-chain CDRs of human antibodies primarily govern polyreactivity
Our identification of the most significant and general Fv molecular features in human antibodies mediating polyreactivity naturally raises the question about which variable domain (VH vs. VL), or even which subsets of the CDRs, is most important. Therefore, we further analyzed the ability of each key molecular feature, when limited only to a subset of Fv residues, to differentiate between high- and low-polyreactivity antibodies in both libraries (Table S4; Figure 4). Notably, this analysis revealed that the VH domain was more important than the VL domain in mediating polyreactivity. For example, for the most significant Fv features (AUC > 0.8), the VH AUC values are an average of 0.13 larger than the VL AUC values (Table S4). This behavior was observed for the hydrophobicity features, as the VH AUC values are 0.22 larger on average than the VL values. Interestingly, there is little difference between the VH and VL AUC values for features negatively correlated with polyreactivity.
Figure 4. Charge and hydrophobicity features based on antibody regions that include the heavy-chain CDRs strongly differentiate between antibodies with high and low levels of polyreactivity.
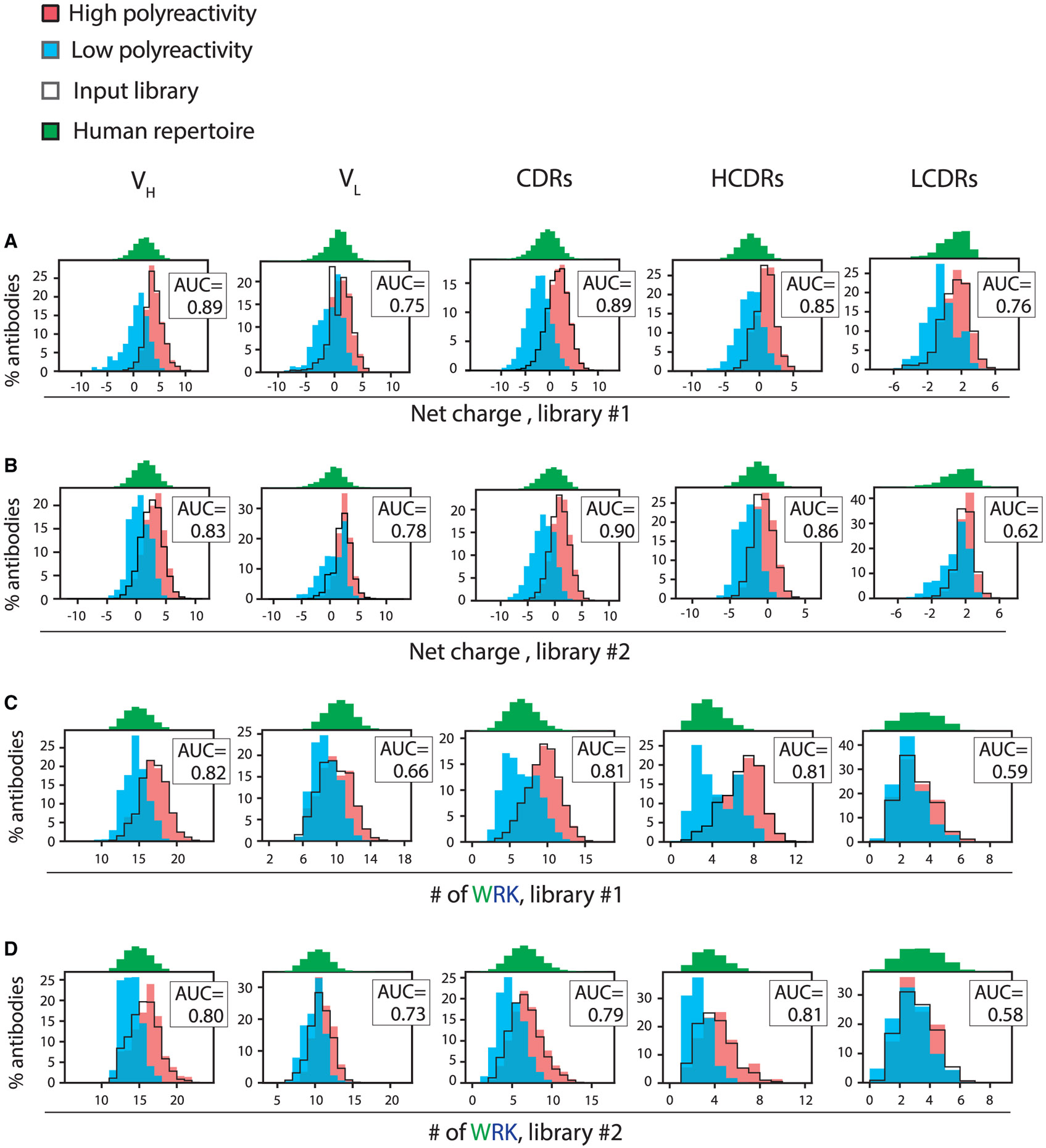
Distributions of key molecular features for different antibody regions in Fv linked to polyreactivity for human antibodies (libraries #1 and #2) and their corresponding AUC values. The same features for the input libraries and a human repertoire dataset were also calculated.
(A and B) Net charge (pH 7.4) distributions for (A) library #1 and (B) library #2.
(C and D) Distributions of the number of tryptophan, arginine, and lysine residues for (C) library #1 and (D) library #2. The antibody regions are noted as VH (variable heavy domain), VL (variable light domain), CDRs (six CDRs for the heavy and light chains), HCDRs (three heavy-chain CDRs), and LCDRs (three light-chain CDRs).
We also performed a similar analysis of the CDRs, which revealed that they contribute significantly to the observed AUC values (Table S4; Figure 4). For example, the Fv AUC values are 0.91 (library #1) and 0.89 (library #2) for the net charge feature, which are close to the corresponding AUC values for the CDRs (0.89 for library #1 and 0.90 for library #2). Moreover, the heavy-chain CDRs generally displayed higher AUC values than those for light-chain CDRs (average difference of 0.18 for features with Fv AUC values > 0.8; Table S4; Figure 4).
We also observed similar patterns of behavior for the individual CDRs, although the results are less significant (Table S5). Notably, the most important features for any single CDR, as defined by AUC values > 0.7 for both human libraries, were associated with HCDR2. There were five HCDR2 features positively correlated with polyreactivity, including the net charge (AUC of 0.71–0.78), the combined numbers of positively charged (arginine and lysine) and hydrophobic (especially tyrosine and proline) residues (AUC values of 0.70–0.81; three features), and the numbers of hydrophobic residues (tryptophan, tyrosine, and isoleucine; Table S5; Figure S4). There were also two HCDR2 features negatively correlated with polyreactivity, namely the number of glycine residues (AUC values of 0.71–0.77) and the combined number of negatively charged and polar residues (glutamate, aspartate, asparagine, and glutamine; AUC values of 0.70–0.77; Table S5).
In contrast, the only other feature for a different CDR with AUC values > 0.7 in both libraries was the net charge of HCDR3 (AUC values of 0.72–0.73), which is positively correlated with polyreactivity. Given the variable length of HCDR3, we also analyzed subsets of antibodies with fixed HCDR3 lengths from each library. We found that relatively long (15-residue) HCDR3s had higher AUC values (0.86–0.94) relative to shorter HCDR3s (5–12 residues; AUC values of 0.55–0.82) when considering the net charge of HCDR3 (Table S6). In addition, the ability of HCDR2 features to distinguish between high- and low-polyreactivity antibodies varies for the specific VH germline genes (Table S7). For example, the net charge in HCDR2 is positively linked to high polyreactivity for the VH3-7 germline. In contrast, the number of asparagine, glutamine, aspartate, and glutamate residues in HCDR2 is linked to low polyreactivity for the VH4-4 germline (AUC values > 0.8 for both human libraries).
We also analyzed the eleven most similar pairs of high- and low-polyreactivity antibodies from library #1 to determine if charge and hydrophobicity features explain the differences in polyreactivity for closely related antibodies (Table S8). Notably, we found that most of these antibodies show an increased net charge in VH (9 out of 11), Fv(11 out of 11), and HCDRs (9 out of 11), as well as increased numbers of the tryptophan, tyrosine, arginine, and lysine residues in VH (90%) and phenylalanine, tryptophan, arginine, and lysine residues in Fv (80%). These results support our findings that increased levels of positively charged and hydrophobic residues are linked to increased polyreactivity.
Given the critical role of the CDRs, especially the heavy-chain CDRs, in mediating polyreactivity, we wondered if the most important features that govern antibody polyreactivity also contribute to antigen-specific binding. To evaluate this question, we isolated 468 antibody/antigen complexes from the Protein Data Bank (PDB; Dataset S7), identified the paratope residues (<5 Å from the antigen), and evaluated whether each feature was enriched in the paratope vs. non-paratope regions in the VH and VL regions (Table S9; Figure 5). Interestingly, some of the features that mediate polyreactivity are strongly depleted or enriched in antibody paratopes, while others are not. For example, the net charge (pH 7.4) of antibody paratopes is reduced by approximately one charge unit relative to that of non-paratope residues (VH and VL; Figures 5A and 5B). The depletion of positive charge in the paratopes of antigen-specific antibodies is similar to the reduction in positive charge (or increase in negative charge) in diverse variable regions and CDRs of antibodies with low polyreactivity, revealing similar mechanisms for increasing antibody specificity.
Figure 5. A subset of molecular features that differentiate between high- and low-poly-reactivity antibodies also differentiate between paratope and non-paratope residues.
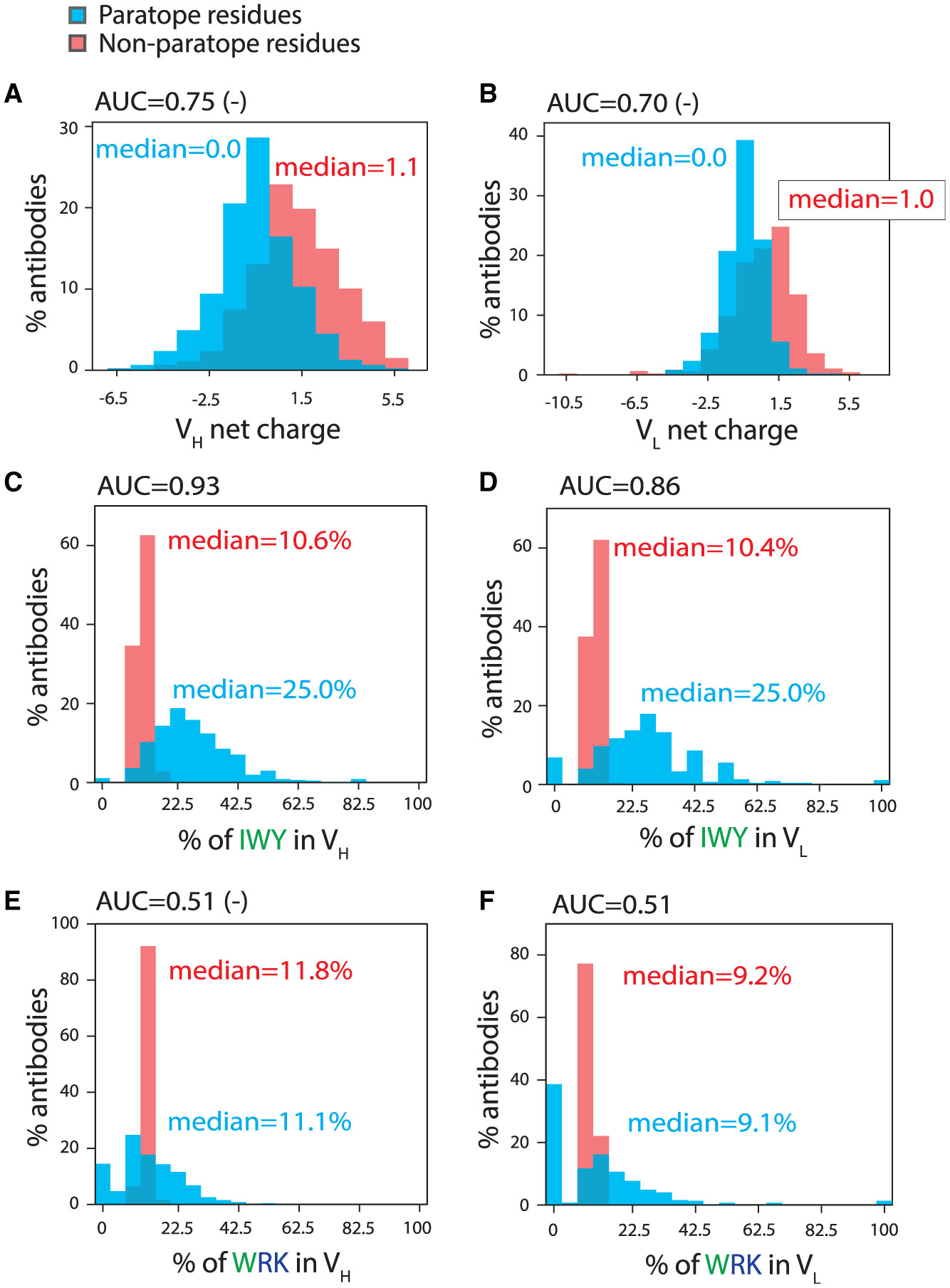
Distributions of molecular features for paratope and non-paratope residues in VH and VL for 468 anti-bodies and their corresponding AUC values.
(A and B) Net charge (pH 7.4) distributions for (A) VH and (B) VL.
(C and D) Distributions of the number of isoleucine, tryptophan, and tyrosine residues for (C) VH and (D) VL.
(E and F) Distributions of the number of tryptophan, arginine, and lysine residues for (E) VH and (F) VL. AUC values with a negative sign next to them signify that the feature values are depleted in the antibody paratope or non-paratope residues.
We also observed similar behavior, but in the opposite direction, for other features that were both linked to increased polyreactivity and strongly enriched in the paratopes of antigen-specific antibodies (Table S9; Figure 5). For example, one of the hydrophobicity features (combined isoleucine, tryptophan, and tyrosine content) was strongly enriched in antibody paratopes, especially in VH (AUC of 0.93; Figure 5C). Given that this same feature for VH and the heavy-chain CDRs is also linked to high polyreactivity (AUC values of 0.75–0.88), our findings suggest that increased hydrophobicity is involved in mediating both antigen-specific and polyreactive interactions, highlighting the delicate balance between specific and non-specific interactions.
Importantly, we also identified molecular features linked to increased antibody polyreactivity that were not strongly enriched or depleted in antibody paratopes (Table S9). An example is one of the combined positive charge and hydrophobicity features, which is the number of tryptophan, arginine, and lysine residues in the variable regions (Figures 5E and 5F). This feature displayed AUC values for the paratope analysis close to 0.5 (0.52 [negative correlation] for VH and 0.51 [positive correlation] for VL), indicating a lack of ability to distinguish between enrichment and depletion of such residues in antibody paratopes. Given that this feature bears the strongest connection to increased antibody polyreactivity (AUC values of 0.80–0.82 for VH and heavy-chain CDRs; Table S4; Figure 5), this is a striking example of a molecular feature typically involved in mediating polyreactive interactions without mediating antigen-specific interactions. Interestingly, other combined positive charge and hydrophobicity features that included tyrosine displayed strong paratope enrichment (AUC values of 0.84–0.88 for VH), while those without tyrosine showed little enrichment in the paratopes (AUC values of 0.52–0.61 for VH). The same was observed for the hydrophobicity features, as those with tyrosine showed strong paratope enrichment (0.81–0.94), while those without tyrosine showed little paratope enrichment (0.50–0.59). This observation is notable given that tyrosine content alone was not significantly enriched in polyreactive antibodies (AUC values of 0.52–0.67 [Fv], 0.59–0.71 [VH], and 0.62–0.76 [heavy-chain CDRs]; Table S4) but is enriched in antibody paratopes (AUC values of 0.97 [Fv], 0.91 [VH], and 0.80 [heavy-chain CDRs]; Table S9). This reveals that tyrosine is important for mediating both specific and non-specific binding, the latter of which appears limited to antibody variable regions with sufficiently high levels of hydrophobicity and/or positive charge.
Random forest models for classifying antibody polyreactivity
We next sought to evaluate if molecular features linked to anti-body polyreactivity could be combined into a random forest model to accurately predict this property. We used antibody library #1 for model training and antibody library #2 as a holdout set for testing because library #1 is both better balanced in terms of high and low polyreactive antibodies and much more diverse (Figure S2). For example, library #1 was generated from more unique human samples (58 for library #1 vs. 5 for library #2), it is more evenly distributed across germline families in both high- and low-polyreactivity samples (Figures 2A and 2B), and there are more unique antibodies in the deep sequencing datasets after enrichment for high (115,038 for library #1 vs. 93,080 for library #2) and low (131,255 for library #1 vs. 34,137 for library #2) polyreactivity.
Model training was conducted in two steps. In the first step, we generated an initial model using the 31 most significant SB features and 12 previously reported SB features weighted by site-specific solvent accessibilities23 (SB-SE features; Table S10). In the second step, we generated the final model using up to 10 of the most important SB features in the initial model and up to 12 SB-SE features. The training of both models used standard 10-fold cross-validation (80% training and 20% testing for antibody set #1; see STAR Methods for more detail).
The performance of our final model with 18 features (Table S11) is summarized in Table 1 and Figure S5. The training and validation accuracies were >95% (antibody set #1; 246,293 antibodies). As a control, we confirmed that the model accuracies were poor (<55%) when the experimental labels for high- or low-polyreactivity antibodies were randomized in the training set. The relative importance of the 18 features revealed that the two most important ones were the net charge of VH (rank #1) and Fv (rank #2). Moreover, the combined tryptophan, tyrosine, arginine, and lysine content (WYRK) in VH was also important (rank #3). Notably, the model also performed well when applied to the second antibody library (antibody set #2; 127,217 anti-bodies), with an accuracy of 86.5%, even though the second library was not used for training.
Table 1.
Performance of different classifier models for predicting antibody polyreactivity
| Model | Ab. set #1 (scFvs) | Ab. set #2 (scFvs) | Ab. set #3 (scFvs) | Ab. sets #4 and #6 (scFvs) |
Ab. sets #4 and #5 (mAbs) |
|---|---|---|---|---|---|
| Random forest #1 (7 SB, 11 SB-SE features) | 0.956 | 0.865 | 0.951 | 0.928 | 0.819 |
| Random forest #2 (10 SB, 12 SB-SE features) | 0.971 | 0.856 | 0.966 | 0.851 | 0.788 |
| Random forest #3 (10 ESM-2, 12 SB-SE features) | 0.964 | 0.792 | 0.971 | 0.797 | 0.797 |
| Random forest #4 (10 SB features) | 0.899 | 0.823 | 0.958 | 0.803 | 0.762 |
| Random forest #5 (10 ESM-2 features) | 0.866 | 0.858 | 0.887 | 0.746 | 0.620 |
| Random forest #6 (12 SB-SE features) | 0.941 | 0.695 | 0.935 | 0.708 | 0.758 |
| Random forest #7 (31 SB features) | 0.938 | 0.842 | 0.971 | 0.912 | 0.756 |
| Random forest #8 (320 ESM-2 features) | 0.927 | 0.757 | 0.924 | 0.912 | 0.692 |
| Fv: net charge >+2.1 | 0.838 | 0.774 | 0.929 | 0.674 | 0.671 |
| VH: net charge >+2.0 | 0.829 | 0.750 | 0.892 | 0.804 | 0.707 |
| Fv: WYRK >36 | 0.788 | 0.730 | 0.832 | 0.587 | 0.621 |
| SVC: PSSM | 0.863 | 0.699 | 0.842 | 0.575 | 0.616 (−) |
| SGD: OneHot | 0.988 | 0.748 | 0.950 | 0.826 | 0.534 |
| AIMS: SB features | 0.951 | 0.810 | 0.956 | 0.690 | 0.580 |
The performance values of the best random forest model in this work for different antibody (Ab.) sets were compared to those for random forest models based on different feature sets, as well as a support vector classifier (SVC) model that uses position-specific scoring matrix (PSSM) features, a stochastic gradient descent (SGD) model that uses one-hot encoding (OneHot) features, and an automated immune molecule separator (AIMS) model with sequence-based (SB) features. In addition, the performance levels of three single molecular features, in the form of rules, were also evaluated for predicting antibody polyreactivity. The antibody sets that were evaluated are defined in the STAR Methods section. For antibody sets #1 and #2, the performance metrics are accuracies, while the performance metrics are AUC values for the other antibody sets.
We also evaluated whether this model could predict the level of polyreactivity for additional antibody datasets that were smaller but better characterized (Figures 6 and S6). Encouragingly, we found that the model performs well for 88 single-chain human antibodies randomly isolated from library #1 (antibody set #3; Figure 6A), a set of 47 single-chain human preclinical and clinical-stage antibodies (antibody sets #4 and #6; Figure 6B), a set of 20 clinical-stage immunoglobulin IgGs that were held out during model training (antibody set #4; Figure 6C), a set of 80 clinical-stage IgGs (Dataset S8) evaluated in a separate study (Figure 6D),24 and a set of 15 bococizumab variants (Dataset S9; Figure 6E).25 Moreover, we observed strong model performance for predicting non-specific binding for a set of 125 emibetuzumab variants and a library of 4,000 emibetuzumab variants (Figure S6).26 Finally, given that previous work has reported that antibody repertoires for naïve B cells are more polyreactive than those for memory or plasma B cells,27 we next evaluated the predicted polyreactivity of human antibodies from a large sequence database, namely the Observed Antibody Space.28 Notably, we found that a greater percentage of naïve antibodies are predicted to be polyreactive (49.6% ± 0.3%) relative to those from memory B cells (28.4% ± 1.9%) or plasma cells (24.4% ± 2.4%).
Figure 6. Evaluation of machine learning model for predicting human antibody polyreactivity.
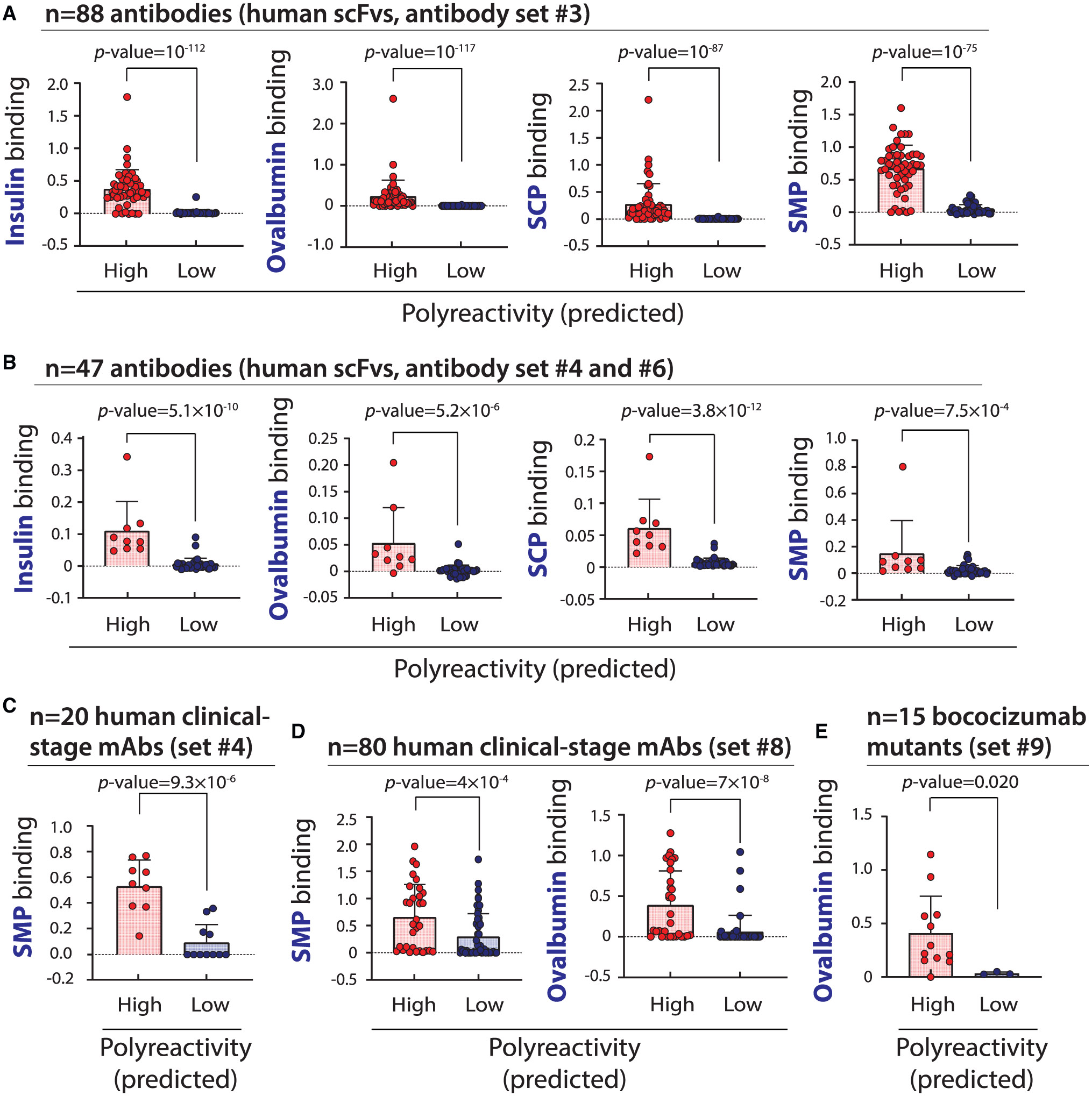
The ability of the machine learning (random forest) model developed in this work to predict the level of human antibody polyreactivity was tested using several sets of antibodies with experimentally defined levels of non-specific binding to multiple reagents. The model predictions were tested on the following antibody sets: (A) antibody set #3 (88 human scFvs), (B) antibody sets #4 and #6 (47 human scFvs), (C) antibody set #4 (20 human clinical-stage IgGs), (D) antibody set #8 (80 clinical-stage IgGs),24 and (E) 15 bococizumab variants with HCDR2 and HCDR3 mutations (antibody set #9).25 The reported p values were calculated using the Anderson-Darling test. In (A), (B), (D), and (E), the data are mean values, the errors are standard deviations, and the numbers of biological replicates are (A) two, (B) three, (D) three, and (E) two. In (C), the data are from a previous publication,16 and the number of biological replicates is unknown.
Next, we compared our model to several related models trained on the first library (antibody dataset #1) using different types of models and features (Table 1). Interestingly, we could not identify another model that was able to generalize across the different datasets with the same level of performance. For example, removing the 11 SB-SE features from our model and replacing them with additional SB features resulted in lower performance, especially for antibody sets #4–#6. Moreover, random forest models (models #3, #5, and #8) trained with protein language model (ESM-229) features alone or in combination with SB features also failed to generalize across diverse datasets as well as our optimal model (model #1). As expected, simpler models based on single features were also unable to generalize across the different datasets while maintaining high performance. We also found that other types of models, such as a support vector classifier (SVC) model trained on position-specific scoring matrix (PSSM) features or a stochastic gradient descent (SGD) model trained one-hot encoded (OneHot) features, resulted in modest prediction performance. Moreover, we evaluated a previously reported model for predicting antibody polyreactivity—an automated immune molecule separator (AIMS) model with SB features,17 and observed modest performance despite that this model used more features (>50) relative to our best model (18 features). Overall, these findings demonstrate the ability of our random forest model to predict human antibody polyreactivity more accurately than other related models.
We also tested the ability of the Therapeutic Antibody Profiler (TAP)—which provides five developability guidelines for selecting antibodies with appropriate biophysical properties30—to identify polyreactive antibodies (Table S12). However, we found that TAP flagged antibodies with high and low polyreactivity at relatively low frequencies (2%–35%) and typically flagged low polyreactive antibodies at a higher frequency. For example, TAP flags low polyreactive antibodies more than high polyreactive ones based on CDR length, CDR patches of surface hydrophobicity, CDR patches of negative charge, and structural Fv charge symmetry parameters, while it flags high polyreactive antibodies more for CDR patches of positive charge. More generally, this highlights the challenge of predicting specific antibody biophysical properties, such as polyreactivity, using general guidelines developed from clinical-stage antibodies regardless of their specific biophysical properties.
Finally, we also analyzed the distribution of molecular features linked to polyreactivity for antibody paratopes and the corresponding epitopes for a set of 468 antibody/antigen complexes from the PDB and their predicted polyreactivities (Table S13). First, we found that 42% of the PDB antibodies were predicted to be polyreactive, and the top five antibodies with the highest polyreactivity probabilities are HIV broadly neutralizing anti-bodies (Dataset S7), which is notable given that these types of antibodies are known to have a high risk for being polyreactive.31-34 Second, some molecular features, such as paratope charge at pH 7.4, were positively associated with polyreactivity (Figure S7). In contrast, the same feature for the corresponding epitopes showed the opposite correlation. The increased positive charge in paratopes is more common in high-polyreactivity antibodies, while the increased positive charge in epitopes is more common for low polyreactive antibodies. In addition, high-polyreactivity antibodies are linked to large positive differences in charge between the paratope relative to the epitope, while the opposite is true for low-polyreactivity antibodies. This results in a negative correlation between paratope charge vs. epitope charge for antibodies with high and low predicted polyreactivity. We also observed similar results for a second molecular feature, namely the combined numbers of tryptophan, tyrosine, arginine, and lysine residues. However, the correlations between the number of such residues in the paratope and epitope were weaker than those for net charge, and the correlation for high-polyreactivity antibodies was not significant.
DISCUSSION
One of the most interesting findings of our study is that the heavy-chain CDRs govern human antibody polyreactivity. This finding, based on the analysis of >300,000 antibodies across two highly dissimilar human antibody libraries, is notable because it explains mutational results from several previous studies using much smaller panels of preclinical or clinical-stage antibodies. Of the HCDRs, we find that HCDR2 and, to a lesser extent, HCDR3 are most strongly linked to polyreactivity, although the importance of HCDR3 was length dependent (Table S6).
For example, mutations that reduce non-specific binding for emibetuzumab are located in the heavy-chain CDRs, and the most effective sets of mutations include those in HCDR2.23 Likewise, mutations that reduce bococizumab non-specific binding are also located in HCDR2.25,35 Moreover, a preclinical antibody (MEDI-1912) with high non-specific binding was optimized by introducing mutations into two heavy-chain CDRs (HCDR1 and HCDR2).10 Similar findings were observed for a human antibody repertoire enriched for high polyreactivity, which led to the strong enrichment of VH6 antibodies whose polyreactivity was governed by HCDR2.19 These and related studies19,27,36 highlight the key role of the VH domain and heavy-chain CDRs in mediating human antibody polyreactivity.
Our study also identified several key molecular features of heavy-chain CDRs mediating human antibody polyreactivity. Our observation that increased positive charge in the Fv, VH, CDRs, and heavy-chain CDRs is linked to increased polyreactivity is consistent with many previous studies, as summarized in multiple reviews.1,2,27 However, our study also demonstrates that the charge of heavy-chain CDRs is generally more important in mediating human antibody polyreactivity than that of the light-chain CDRs, and this behavior is primarily due to HCDR2 and, to a lesser degree, HCDR3. These findings are supported by observations that positively charged mutations in these heavy-chain CDRs commonly increase polyreactivity, while negatively charged mutations in the same CDRs commonly reduce polyreactivity.19,23,25,35,37-43
Our finding that increased levels of the combined numbers of positively charged residues—especially arginine and lysine— and hydrophobic residues—especially tryptophan, tyrosine, isoleucine, and proline—in the heavy-chain CDRs are linked to increased antibody polyreactivity deserves further consideration. These specific combined features are particularly interesting because they are more significant than any other features in our study except for the charge features. While positively charged and aromatic residues have been individually linked to polyreactivity,23,25,35,37,41,42 we show that the combination is much more significantly linked to polyreactivity. For individual amino acids, our observation that increased tryptophan content in the heavy-chain CDRs is most strongly linked to increased polyreactivity is consistent with several previous studies.18,19,39 Conversely, our finding that increased aspartic acid content in VH and the heavy-chain CDRs is linked to reduced polyreactivity is also consistent with several previous studies.23,25,35,37,38,40,44 Moreover, the fact that reduced glycine content in VH and the heavy-chain CDRs is linked to increased polyreactivity is notable because previous studies using smaller and/or less diverse datasets have found both the same and opposite results.19,26,39 The specific context of glycine residues in VH and the heavy-chain CDRs likely determines their impact on polyreactivity, but our study highlights that increased glycine content, like increased aspartic acid content, is generally beneficial for reducing polyreactivity for human antibodies.
We also demonstrate that a subset of molecular features linked to increased human antibody polyreactivity are also enriched in antibody paratopes, especially in the VH domain. While this may seem contradictory, it is notable that all of the amino acid features linked to both increased polyreactivity and enrichment in antibody paratopes contain tyrosine and positively charged and/or hydrophobic residues. Importantly, we found that tyrosine content alone was not strongly linked to polyreactivity, as reported previously,41 and it was the most strongly enriched feature in antibody paratopes. Therefore, it appears that tyrosine, in the context of hydrophobic and/or positively charged residues, can enhance polyreactivity. This finding has interesting implications not only for understanding the natural mechanisms of antibody evolution but also for improving the design of antibody mutations and libraries for enhancing antibody affinity and specificity.45-49
Our reported model for predicting human antibody polyreactivity also deserves further consideration. First, our best model contains 18 features, but the top 6 features that contribute most (>75%) to the model performance are sequenced-based features based on the VH and Fv regions and not specifically on the CDRs (Table S11). While the performance of our model did improve by also employing features based only on CDRs, as 7 of 18 of our model features are only based on CDR composition, we found that models based only on CDR features were unable to generalize across diverse datasets with high accuracy. Second, our model validation for predicting human anti-body polyreactivity is notable because it was tested against the largest and most diverse antibody holdout sets reported to date, including a diverse human antibody library (131,255 antibodies in library #2). This is particularly important, as previous attempts to develop models that predict human polyreactivity have used much smaller datasets,17 datasets that are strongly biased toward single human antibody families (e.g., VH6),18 or datasets that lack diversity in light chains and use non-standard definitions of polyreactivity.20 Third, we intentionally limited our models to a relatively small number of features (<20) despite the large size of our training and testing sets to increase their potential generality and simplify their use and interpretability. The fact that our model only uses 18 features is notable because this is less than that used in recently reported models (≥46 features) trained and tested using a single dataset (~19,000 antibodies)18 and a second reported model that appears to use >50 features and is trained on much smaller datasets (~1,000 antibodies).17 Fourth, although we found that random forest models trained using features from a protein language model (ESM-2) were unable to outperform those trained on SB features due to a lack of ability to generalize across diverse datasets, it will be important in the future to explore the potential of antibody-specific protein language models50,51 to further improve model performance and generalization.
Our study also opens the door to several exciting future research directions. First, we expect that our model for predicting antibody polyreactivity—which has been trained on diverse human germlines—will enable the analysis of human antibody repertoires to evaluate how polyreactivity changes during anti-body maturation at much larger scales than possible previously. While experimental analysis of germline and matured anti-bodies has revealed a general trend toward reduced polyreactivity for matured antibodies,5,6,19,27 these conclusions are based on relatively small datasets and a limited set of germlines. Second, we also expect that our model will be useful for investigating the potential role of polyreactivity in mediating the ability of rare antibodies to broadly neutralize different viral variants for HIV, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), and other viruses. These broadly neutralizing antibodies, at least in some cases, accumulate large numbers of mutations, and it remains incompletely understood if polyreactivity contributes to or is associated with their unique ability to recognize different viral variants.31,52-57 Third, we expect that our model—and the identified molecular features linked to polyreactivity—can be used to guide the design of antibody libraries with low polyreactivity. This is particularly important for isolating lead antigen-specific antibodies from non-immune libraries using in vitro display technologies, such as phage and yeast surface display, because it is relatively common to isolate antibodies with higher levels of polyreactivity using such display methods than for antibodies generated using immunization.1,16 Fourth, we expect that our model will be useful during antibody humanization to guide the selection of frameworks resulting in humanized variants with low polyreactivity. This possibility is particularly exciting given emerging approaches for generating humanized antibodies with frameworks that have diverse physicochemical properties.58 Finally, we expect that our model will be useful for re-engineering preclinical and clinical-stage anti-bodies to reduce polyreactivity, which is especially important in cases where high polyreactivity is linked to fast antibody clearance.2,9-15
Limitations of the study
There are limitations to this work that will need to be addressed in the future. First, our random forest model for predicting antibody polyreactivity requires antibody sequences to be aligned and numbered using methods such as ANARCI59 via a standard anti-body numbering scheme, such as the Kabat numbering used in this work. This process is needed because some of our features (SB-SE) require predictions of amino acid solvent accessibilities.23 This process is relatively inefficient, and future work should address this limitation. Second, our models are limited to human antibodies, and future work is needed to develop similar types of models for single-domain antibodies, such as nanobodies (VHHs) with diverse frameworks in addition to diverse CDRs. Third, our models permit the classification of high and low polyreactivity, and future work should extend our findings to regression models that would be even more useful for multi-objective antibody optimization.26
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Peter M. Tessier (ptessier@umich.edu).
Materials availability
Antibody sequences used for training and testing in this study are published in Datasets S1, S2, S3, S4, S5, S6, S7, S8, and S9. All sequences of antibodies were verified via Sanger sequencing.
Data and code availability
Antibody sequences are provided in the Tessier lab GitHub repository: https://zenodo.org/doi/10.5281/zenodo.13387056.
Codes to generate the random forest model are available in the Tessier lab GitHub repository: https://zenodo.org/doi/10.5281/zenodo.13387056.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
STAR★METHODS
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Cell lines
The yeast strain EBY10061 was used for display of antibody libraries by fusing them to the Aga2 protein. EBY100 was transformed using a standard media solution [40% (w/v) PEG 3350, 100 mM Lithium acetate, 10 mM Tris (pH 7.5), 0.4 mM EDTA]. Yeast harboring the plasmids were then recovered in SDCAA medium with shaking at 30°C. Yeast surface display of the scFvs was induced by transferring mid-exponential phase cells (OD600 at 2 to 4) into SD-GCAA medium with shaking at 30°C for 20 h before flow analysis. The subsequent analysis for the library sorting and characterization of individual antibody variants is described in the methods details.
METHOD DETAILS
Human scFv library sorting and clone evaluation
To generate large datasets of scFvs with low and high levels of non-specific binding, two human scFv libraries (human library #121 and #222 were displayed on yeast (Aga2-VH-linker-VL) and sorted first for full-length scFv display by MACS and then progressively against four different polyspecificity reagents by FACS. The initial sort using MACS (sort 1) was conducted with cMyc antibody (Cell Signaling Technology, 2276S; library #1) or V5 antibody (Abcam, ab27671; library #2) to select full-length scFvs. To perform the MACS selections, 2 × 109 cells were incubated with 80 nM cMyc or V5 antibody in PBSB (1 g/L BSA, PBS) with 1% milk at room temperature (3 h). Afterward, the cells were washed once with PBSB and incubated with Protein G beads and gently rocked for 30 min at 4°C. The cells suspension was then passed through a MACS column for bound cells. After washing, the cell/bead complexes were transferred into SDCAA media (20 g dextrose, 5 g casamino acids, 6.7 g yeast nitrogen base without amino acids, 16.75 g sodium citrate, and 4 g anhydrous citric acid per liter) and grown at 30°C for 2 d.
Next, the scFv libraries were progressively sorted for positive and negative binding to four biotinylated polyspecificity reagents (0.13 mg/mL), namely ovalbumin (Sigma Aldrich, A5503; sort 2), soluble cytosolic proteins from Chinese hamster ovary (CHO) cells (SCP60; sort 3), soluble membrane proteins from CHO cells (SMP60; sort 4) and insulin (Sigma Aldrich, I9278; sort 5). The SCP and SMP reagents were prepared using CHO cell lysates.60 CHO cells were cultured, pelleted and washed sequentially with PBSB and Buffer B (50 mM HEPES, 0.15 M NaCl, 2 mM CaCl2, 5 mM KCl, 5 mM MgCl2, 10% glycerol, pH 7.2). Buffer B was then supplemented with protease inhibitor (Sigma Aldrich, 4693159001) for all subsequent experiments, as denoted as Buffer B+. The cells were pelleted again and resuspended in 5 mL of Buffer B+. The resuspended cells were homogenized for four cycles (30 s per cycle) and then sonicated for three cycles (30 s per cycle). The suspension was then centrifuged at 40,000× g for 1 h. The supernatant, containing the SCP fraction, and the pellet, containing the SMP fraction, were collected, biotinylated, and used for evaluating antibody polyreactivity.
The libraries were sorted via a FACS MoFlo Astrios (Beckman) using standard protocols.63 Yeast cells were incubated with the polyspecificity reagents for 3 h at room temperature (ovalbumin, SCP and insulin) or 20 min on ice (SMP). The polyspecificity reagents were biotinylated and detected via streptavidin Alexa Fluor 647 secondary (1:1000; Invitrogen, S21374). The top 10% and bottom 10% of the scFv binding population of yeast cells that expressed human scFvs were collected separately and cultured for the next round of positive or negative selection. The enriched library samples for high non-specific binding to ovalbumin in sort 2 were next selected for high non-specific binding to SCP in sort 3, while the enriched libraries for low non-specific binding to ovalbumin in sort 2 were next selected for low non-specific binding to SCP in sort 3. This process was repeated in successive sorts to generate enriched scFv library samples that were either broadly polyreactive or non-polyreactive.
Yeast plasmids were extracted from the positive and negative populations from sort 5 via yeast minipreps (Zymo, D2004), and transformed into DH5-α component cells. Single colonies were picked, and scFv coding fragments was confirmed via Sanger sequencing. The sequenced scFv plasmids were transformed into yeast cells (EBY100), as described above. Non-specific binding of the polyspecificity reagents to the scFvs was evaluated using 26 μg/mL biotinylated polyspecificity reagents and streptavidin Alexa Fluor 647 conjugate (1:1000; Invitrogen). Human scFv expression was detected using mouse cMyc (1:1000; Cell Signaling Technology) and goat anti-mouse Alexa Fluor 488 secondary (1:200; Invitrogen). To compare the non-specific binding levels of different scFvs, the median binding signals were normalized for each scFv between two control scFvs. The median value for each scFv was normalized between a positive control (FP3) with a score of 1 and a negative scFv (FN4) with a score of 0.
Deep sequencing of human libraries
Plasmids (pCTcon262) from the sorted libraries were isolated using yeast mini preps (Zymo, D2004). The variable light and heavy domains were amplified from the plasmids and purified with a 1% agarose gel. The products were then gel purified using a QIAquick Gel Extraction Kit (Qiagen, 28704). The concentrations of the purified amplicons were then determined using a Qubit 4 Fluorometer. Each sample (50 μL at 10 ng/μL) was sent to the University of Wisconsin Sequencing Core for PacBio sample preparation and sequencing. The output bam files from PacBio sequencing were first converted to fastq files via BedTools and then converted into a fasta format for ease of processing.64 The fasta files were evaluated and full-length sequences without stop codons were collected into a dictionary containing the number of occurrences of each sequence.
Human library #1 scFv dataset
The deep sequencing data from antibody set #1 was analyzed to identified scFvs in human library samples sorted either for binding or a lack of binding to ovalbumin, insulin, CHO soluble cytosolic proteins (SCP) and CHO soluble membrane proteins (SMP). The resulting library samples, which are referred to as either positive or negative samples against each reagent, were collected in three independent experiments. The antibody sequences were pooled for the twelve antibody library samples (four reagents with three biological replicates) sorted positively for non-specific binding, while the corresponding twelve antibody library samples sorted for a lack of non-specific binding were also pooled. Next, the two sets of pooled library sequences were processed to eliminate duplicates with each pooled sequence sets and any duplicates between the two different sequence sets. Afterward, the remaining variable heavy (VH) and variable light (VL) domains in each pooled sequence set were aligned in ANARCI using a database of Hidden Markov Models,59 and scFvs were eliminated from further consideration if VH and VL germlines could not be assigned. Finally, scFvs were eliminated if the number of amino acids in specific antibody regions exceeded maximum limits: 134 residues for VH; 12 residues for HCDR1; 19 residues for HCDR2; 21 residues for HCDR3; 122 residues for VL; 17 residues for LCDR1; 11 residues for LCDR2; and 12 residues for LCDR3. The antibody CDRs were defined using a combination of Chothia and Kabat numbering. This resulted in a final set of 131,255 scFv sequences selected for low non-specific binding and 115,038 scFv sequences selected for high non-specific binding, which together (246,293 scFvs) is referred to as antibody set #1 (Dataset S1).
Human library #2 scFv dataset
The deep sequencing data for antibody set #2 was processed in the same manner as the corresponding data for set #1. The library samples were collected by sorting against the same four polyspecificity reagents as used for set #1, and two biological replicates were performed. After processing the sequencing data, the final sets of antibodies included 34,137 scFv sequences selected for low non-specific binding and 93,080 scFv sequences selected for high non-specific binding, which together (127,217 scFvs) is referred to as antibody set #2 (Dataset S2).
Additional antibody datasets with experimental measurements
The models developed in this work were trained or evaluated using four additional sets of antibodies. Antibody set #3 contained 96 selected clones from human library #1 and were identified using Sanger sequencing from (Dataset S3). Of the 96 selected clones, 88 of these met the sequence selection requirements and were used for testing the models. Antibody set #4 contained 20 clinical-stage antibodies that were selected based on displaying either generally high or generally low levels of non-specific binding (Dataset S4).16 These antibodies were selected based on the previously reported non-specific binding values from three assays, namely the polyspecificity reagent (PSR) assay using soluble membrane and cytosolic proteins65 and two ELISAs using either baculovirus particles15,32 or a mixture of proteins, lipid and nucleic acid reagents.16 Each of the selected antibodies displayed either higher or lower values for all three assays than the published threshold values for high levels of non-specific binding. Two additional mAbs were initially identified in this analysis (crenezumab and natalizumab), but their VH and VL germlines were not observed in antibody set #1 and were excluded from further analysis. A second set of clinical-stage antibodies was also used during the training process (antibody set #5). This set included the variable regions of 103 mAbs and their corresponding values of non-specific binding to the PSR reagent (Dataset S5). Finally, a set of 27 scFvs provided by Boehringer Ingelheim (antibody set #6) was used for model evaluation (Dataset S6).
Identification of molecular features linked to antibody polyreactivity
A diverse panel of molecular features were evaluated for their ability to discriminate between antibodies with high and low levels of non-specific binding in antibody set #1 and set #2. The features were evaluated for the antibody VH, VL and Fv, as well as individual CDRs, sets of three HCDRs or LCDRs, and the entire set of six CDRs, and were based on two categories of features, namely: (i) sequence-based (SB) features; and (ii) sequence-based features weighted by site-specific solvent accessibilities (SB-SE).23 For evaluating SB features, two sub-categories were evaluated: (i) number of amino acids (e.g., number of Asp and Glu residues in VH); and (ii) theoretical net charge at pH 7.4 (+1 for Lys and Arg, +0.1 for His, and −1 for Asp and Glu).
For the features in category (i) of the SB features, the number of amino acids were evaluated in terms of seven groups based on their properties: (a) hydrophobic amino acid (Phe, Ile, Leu, Pro, Val, Trp and Tyr); (b) positively charged amino acid (Arg, Lys and His); (c) hydrophobic and positively charged amino acid (Phe, Ile, Leu, Pro, Val, Trp, Tyr, Arg, Lys and His); (d) hydrophilic amino acid (Gln, Ser, Thr and Asn); (e) negatively charged amino acid (Asp and Glu); (f) hydrophilic and negatively charge amino acid (Gln, Ser, Thr, Asn, Asp and Glu); (g) the twenty amino acids. In each group, only certain number of residues were selected, generating various subgroups of SB features: 2–5 residues for feature type (a); 2–3 residues for feature type (b); 2–7 residues for feature type (c); 2–4 residues for feature type (d); 2 residues for feature type (e); 2–5 residues for feature type (f); and one residue for feature type (g).
The SB-SE features required evaluating the site-specific solvent accessibilities of residues in the VH and VL regions. This was performed using a published Random Forest machine learning model trained on over 900 antibodies in the Protein Data Bank66 and analyzed via RStudio package.
The most significant features that discriminated between antibodies with low and high levels of non-specific binding (antibody sets #1 and #2) were identified by calculating the area under of the ROC curve (AUC) for logistic regression analysis. A subset of the most significant features is reported in Table S4 based on different AUC cutoffs due to different extents of significance for different feature groups: (i) AUC>0.8 for SB feature types (a) and (b); (ii) AUC>0.75 for SB feature types (c), (d) and the number of tryptophan; (iii) AUC>0.70 for SB feature type (f), the number of aspartic acid and the number of glycine residues. The number of tyrosine residues is also given for reference. The AUC values for these features in six different antibody regions (Fv, VH, VL, CDR, HCDR, and LCDR) were analyzed for comparison (Table S4). The individual CDRs were also evaluated and reported in Table S5.
Additionally, significant sequence-based features in individual HCDRs for discriminating between high and low polyreactivity were also analyzed for a subset of antibodies with different fixed HCDR3 lengths. Antibodies with 5, 8, 10, 12, and 15 residues in HCDR3 were evaluated using AUC analysis for both library #1 and #2.
Molecular features linked to antibody affinity interactions
The same molecular (SB) features listed in Table 1 were evaluated for their contributions to affinity interactions. For the dataset of 468 antibody-antigen complexes from the Protein Data Bank (Dataset S7), the number of residues for each SB feature in category (i) was identified in VH and VL domains as well as paratope (<5 Å from the antigen) and non-paratope regions in VH and VL domains for each antibody. Next, the percentage of paratope residues for each feature in each variable region (e.g., % of WRK residues in VH paratope) was calculated by dividing the number of each feature in the paratope (e.g., # of WRK residues in the VH paratope) by the length of corresponding paratope (e.g., # of VH paratope residues) for each antibody. As a comparison, the percentage of non-paratope residues for the same feature in each variable region (e.g., % of WRK residues in the non-paratope region of VH) was calculated by dividing the feature value in the non-paratope region (e.g., # of WRK residues in VH non-paratope region) by the number of non-paratope residues in the corresponding variable region. As for the features in category (ii) of the SB features, the theoretical net charge (pH 7.4) in the paratope was compared with the corresponding non-paratope region in each variable region. The ability of these features to distinguish between enrichment or depletion from the paratope was evaluated using AUC analysis (Table S9). The median values for paratope and non-paratope regions in the VH and VL domains are also reported.
Germline polyreactivity analysis
The most common VH germlines, VL germlines and VH/VL germline pairs for library #1 and library #2 were evaluated for polyreactivity if there are at least a total of 50 sequences in high and low polyreactivity antibodies. The number of high and low polyreactivity anti-bodies, the total number of antibodies and the ratio of high to low polyreactivity for each listed VH germline gene, VL germline gene, and VH/VL germline gene pair in both libraries are reported in Tables S1, S2, and S3, respectively. In addition, the ability of significant sequence-based features in HCDR2 to distinguish between high and low polyreactivity antibodies was evaluated for the VH germlines listed in Table S1. AUC of the features were calculated for both library #1 and #2 and reported in Table S7.
Related to Figure S3, up to 1000 antibody sequences were randomly selected from each of the most common germline families for library #1 and #2 (2000 antibody sequences total). The sequence similarity per germline family was calculated by averaging the sequence similarities of all antibody pairs within the randomly selected 1000 sequences for each germline family. The sequence similarity of each antibody pair was computed using the sum of blocks substitution matrix score (BLOSUM62) for each amino acid pair of the aligned sequences.
Analysis of similar antibodies with high and low polyreactivity
For library #1, 1000 antibody sequences were randomly selected from the libraries enriched for high and low polyreactivity (2000 antibodies total). Each antibody in the high polyreactivity set was paired with each one in the low polyreactivity set. The similarity of each antibody pair was computed using the BLOSUM62 scoring matrix. Of all possible pairs within the selected set, 11 of the most similar antibody pairs were sampled. The net charge and key molecular features were calculated and compared.
Random forest model generation and analysis
The molecular (SB) features were preprocessed by normalizing feature values from 0 to 1 using MinMaxScaler function in the scikit-learn preprocessing package in Python. The antibodies with high and low polyreactivity in antibody set #1 (Dataset S1) were split into training (80%) and test (20%) sets using stratified sampling. The training set was further divided into ten random partitions (folds) for 10-fold cross-validation, nine of which were used for training and the other for validation (and this was repeated ten times). The first-generation model was trained using the random forest classifier package in the scikit-learn ensemble package in Python. The model was optimized by tuning the hyperparameters and selecting the best model based on two criteria: (i) minimum coefficient of variation for the ten validation accuracies calculated for antibody set #1 (80% training set); and (ii) area under the ROC curve >0.75 (logistic regression) for predicting the relative polyreactivity (as judged by PSR experimental measurements16) for 123 clinical-stage anti-bodies (antibody sets #4 and #5). The parameters of the best first-generation random forest model were n_estimator of 5, max_depth of 20, min_sample_split of 20 and min_samples_leaf of 1.
The first-generation model was used to identify the most important molecular features for predicting antibody polyreactivity. Next, up to 18 SB features were combined with up to 11 SB-SE features,23 for training the second-generation random forest model using a total of 11–18 features per model. The models were trained as described for the first-generation model except the best model was chosen based on the following three criteria: (i) > 90% accuracy for the average of the ten validation accuracies calculated for anti-body set #1 (80% training set); (ii) minimum coefficient of variation for the corresponding calculations in (i); and (iii) area under the ROC curve >0.8 (logistic regression) for predicting the relative polyreactivity for antibody set #5. The parameters of the best second-generation random forest model (referred to as random forest model #1 in Table 1) were n_estimator of 15, max_depth of 15, min_sample_split of 100 and min_samples_leaf of 1.
Additional model generation and analysis
For the SVC model, two PSSMs were generated for antibodies with low and high polyreactivity in antibody set #1 (PSSM1 for low polyreactivity antibodies and PSSM2 for high polyreactivity antibodies). Each antibody sequence was given two scores, one from PSSM1 and the other from PSSM2. Next, for the SVC model, an optimized radial basis function, was trained using antibody set #1 to differentiate between antibodies with low and high polyreactivity using a C parameter of 10. For the SGD model, a multiple sequence alignment of the antibodies in antibody set #1 was created using ANARCI.59 The antibodies in set #1 were one-hot encoded using all amino acids, including gaps, and the positions observed in antibody set #1 alignment. Positions observed in other antibody sets, but not antibody set #1, were removed. The one-hot encoding resulted in a matrix with 5,355 columns. The one-hot encoded matrix based on antibody set #1 was concatenated and an 80/20% training/test split was made to train the SGD model using α = 0.00001 and a modified huber loss function.
For the AIMS model,17 the IMGT CDRs of each antibody in set #1 were retrieved and the model was trained using standard cross validation (80/20% training/test splits). The Jupyter notebook for the original AIMS model were used for model development. Minor modifications of the original code were performed to enable use of a model trained on one dataset to make predictions on other data-sets. The AIMS model calculates several sequence-based properties for each position in the CDRs for use in a linear discriminant analysis algorithm, which resulted in over 5,600 features for antibody set #1. The model was optimized with respect to the total number of features (N). This was done by evaluating the test accuracies for antibody sets #1 and #2 for a range of the numbers of features (500–900 features). A value of N of 700 was chosen to balance the maximum accuracy for antibody dataset #2. The accuracy for antibody dataset #1 could be further improved by using even more features, but this also resulted in reduced model generality, as judged by the accuracy for antibody set #2.
Analysis of paratopes and epitopes of predicted polyreactive antibodies
First, the paratope residues for each antibody and the corresponding epitope residues in each antigen were identified. In the paratopes and epitopes, the number of residues for each feature was calculated and normalized by the length of the paratopes and epitopes, respectively. The difference for the two quantities (paratope-epitope) was also calculated. The distributions of the three quantities were visualized for both predicted high and low polyreactivity, with the AUC value representing the separation for the two groups. The correlation between paratope and epitope properties was evaluated using a bivariate plot with the Spearman coefficient. Similar analysis was also performed for net charge in paratopes and epitopes.
QUANTIFICATION AND STATISTICAL ANALYSIS
The areas under the ROC curve in Figures 3 and 4 were calculated using Python. The p-value analyses in Figure 6 were performed in MATLAB using the Anderson-Darling test.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-myc-tag (9B11) antibody | Cell Signaling Technology | Cat#2276S; RRID:AB_331783 |
| Mouse monoclonal anti-V5 tag antibody | Abcam | Cat#Ab27671; RRID:AB_471093 |
| Goat anti-mouse IgG Alexa Fluor 488 | Invitrogen | Cat#A11001; RRID:AB_2534069 |
| Bacterial and virus strains | ||
| DH5α competent cells | Thermo Fisher Scientific | Cat#18265017 |
| Chemicals, peptides, and recombinant proteins | ||
| 10x Phosphate Buffered Saline (PBS) | Fisher Scientific | Cat# BP39920 |
| Bovine Serum Albumin | Fisher Scientific | Cat#BP9706100 |
| Poly(ethylene glycol) 3350 | Sigma Aldrich | Cat#P4338 |
| Lithium acetate | Fisher Scientific | Cat#AC268640010 |
| Trizma base | Sigma Aldrich | Cat#T1503-1KG |
| Ethylenediaminetetraacetic Acid | Fisher Scientific | Cat#O2793-500 |
| Agarose | Fisher Scientific | Cat#BP160-500 |
| TAE Buffer | Fisher Scientific | Cat#BP13324 |
| Ovalbumin | Sigma Aldrich | Cat#A5503 |
| Biotinylated Soluble cytosolic proteins from CHO cells | Makowski et al.60 | N/A |
| Biotinylated Soluble membrane proteins from CHO cells | Makowski et al.60 | N/A |
| Insulin | Sigma Aldrich | Cat#I9278 |
| Sulfo-NHS-LC-biotin | Thermo Fisher Scientific | Cat#21335 |
| Streptavidin Alexa Fluor 647 | Invitrogen | Cat# S32357 |
| NeutrAvidin PE | Invitrogen | Cat#A2660 |
| Dextrose (D-Glucose) | Fisher Scientific | Cat#D16-10 |
| Yeast Nitrogen Base without Amino Acids | Fisher Scientific | Cat#DF0919-15-3 |
| Casamino Acids | Fisher Scientific | Cat#BP1424-500 |
| Sodium Citrate Dihydrate | Fisher Scientific | Cat#S279-500 |
| Citric Acid | Fisher Scientific | Cat#A940-500 |
| D(+)-Galactose | Fisher Scientific | Cat#AC150610051 |
| Sodium Phosphate Monobasic Monohydrate | Fisher Scientific | Cat#S369-500 |
| Sodium Phosphate Dibasic Dihydrate | Fisher Scientific | Cat#S472-500 |
| Lithium acetate | Fisher Scientific | Cat# AC268640010 |
| Critical commercial assays | ||
| Zymoprep Yeast Plasmid Miniprep II | Zymo Research | Cat#D2004 |
| QIAquick Gel Extraction Kit | QIAGEN | Cat# 28704 |
| Non-fat dry milk | Kroger | Cat#0001111008733 |
| Deposited data | ||
| Human Antibodies | This paper | Datasets S1, S2, S3, S4, S5, S6, S7, S8, and S9 |
| Original Code | This paper | https://doi.org/10.5281/zenodo.13387057 |
| Experimental models: cell lines | ||
| Yeast: EBY100 | Julian et al.61 | N/A |
| Recombinant DNA | ||
| pCTcon2 | Chao et al.62 | Addgene Cat#41843 |
| Software and algorithms | ||
| Python version 3.9 | Python Software Foundation | https://www.python.org |
| RStudio Desktop version 2024.4.1.748 | Posit team | https://posit.co/download/rstudio-desktop |
| MATLAB R2019a | MathWorks | https://www.mathworks.com/products/matlab.html |
| Others | ||
| Beckman Coulter MoFlo Astrios | Beckman Coulter | Cat#B52102 |
| Protein G Dynabeads | Invitrogen | Cat# S311-500 |
Highlights.
Generation of large polyreactivity datasets for diverse human antibody repertoires
Antibody polyreactivity is primarily linked to the heavy-chain CDRs
Some molecular features are linked to both specific and non-specific antibody interactions
Development of a machine learning model for predicting human antibody polyreactivity
ACKNOWLEDGMENTS
We thank the Tessier lab for providing valuable feedback on multiple versions of this manuscript. This work was supported by Boehringer Ingelheim (including through postdoctoral fellowships to N.R. and G.L.), the National Institutes of Health (R35GM136300, RF1AG059723, R01AG080016, and R21AI171844 to P.M.T.), the National Science Foundation (Graduate Research Fellowship to M.D.S. [DGE 1256260]), and the Albert M. Mattocks Chair (to P.M.T.).
Footnotes
DECLARATION OF INTERESTS
N.R., G.L., M.S.M., and S.K. are current or former employees of the company (Boehringer Ingelheim) that funded this research. P.M.T. is a member of the scientific advisory boards for Nabla Bio, Aureka Biotechnologies, and Dualitas Therapeutics.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.114801.
REFERENCES
- 1.Starr CG, and Tessier PM (2019). Selecting and engineering monoclonal antibodies with drug-like specificity. Curr. Opin. Biotechnol 60, 119–127. 10.1016/j.copbio.2019.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ausserwöger H, Schneider MM, Herling TW, Arosio P, Invernizzi G, Knowles TPJ, and Lorenzen N (2022). Non-specificity as the sticky problem in therapeutic antibody development. Nat. Rev. Chem 6, 844–861. 10.1038/s41570-022-00438-x. [DOI] [PubMed] [Google Scholar]
- 3.Dostalek M, Prueksaritanont T, and Kelley RF (2017). Pharmacokinetic de-risking tools for selection of monoclonal antibody lead candidates. mAbs 9, 756–766. 10.1080/19420862.2017.1323160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cunningham O, Scott M, Zhou ZS, and Finlay WJJ (2021). Polyreactivity and polyspecificity in therapeutic antibody development: risk factors for failure in preclinical and clinical development campaigns. mAbs 13, 1999195. 10.1080/19420862.2021.1999195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, and Nussenzweig MC (2003). Predominant Autoantibody Production by Early Human B Cell Precursors. Science 301, 1374–1377. 10.1126/science.1086907. [DOI] [PubMed] [Google Scholar]
- 6.Koelsch K, Zheng N-Y, Zhang Q, Duty A, Helms C, Mathias MD, Jared M, Smith K, Capra JD, and Wilson PC (2007). Mature B cells class switched to IgD are autoreactive in healthy individuals. J. Clin. Invest 117, 1558–1565. 10.1172/JCI27628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dimitrov JD, Planchais C, Roumenina LT, Vassilev TL, Kaveri SV, and Lacroix-Desmazes S (2013). Antibody Polyreactivity in Health and Disease: Statu Variabilis. J. Immunol 191, 993–999. 10.4049/jimmunol.1300880. [DOI] [PubMed] [Google Scholar]
- 8.Tiller T, Tsuiji M, Yurasov S, Velinzon K, Nussenzweig MC, and Wardemann H (2007). Autoreactivity in Human IgG+ Memory B Cells. Immunity 26, 205–213. 10.1016/j.immuni.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Datta-Mannan A, Thangaraju A, Leung D, Tang Y, Witcher DR, Lu J, and Wroblewski VJ (2015). Balancing charge in the complementarity-determining regions of humanized mAbs without affecting pI reduces non-specific binding and improves the pharmacokinetics. mAbs 7, 483–493. 10.1080/19420862.2015.1016696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dobson CL, Devine PWA, Phillips JJ, Higazi DR, Lloyd C, Popovic B, Arnold J, Buchanan A, Lewis A, Goodman J, et al. (2016). Engineering the surface properties of a human monoclonal antibody prevents self-association and rapid clearance in vivo. Sci. Rep 6, 38644. 10.1038/srep38644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grinshpun B, Thorsteinson N, Pereira JN, Rippmann F, Nannemann D, Sood VD, and Fomekong Nanfack Y (2021). Identifying biophysical assays and in silico properties that enrich for slow clearance in clinical-stage therapeutic antibodies. mAbs 13, 1932230. 10.1080/19420862.2021.1932230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Datta-Mannan A, Lu J, Witcher DR, Leung D, Tang Y, and Wroblewski VJ (2015). The interplay of non-specific binding, target-mediated clearance and FcRn interactions on the pharmacokinetics of humanized antibodies. mAbs 7, 1084–1093. 10.1080/19420862.2015.1075109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kelly RL, Yu Y, Sun T, Caffry I, Lynaugh H, Brown M, Jain T, Xu Y, and Wittrup KD (2016). Target-independent variable region mediated effects on antibody clearance can be FcRn independent. mAbs 8, 1269–1275. 10.1080/19420862.2016.1208330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kelly RL, Sun T, Jain T, Caffry I, Yu Y, Cao Y, Lynaugh H, Brown M, Vásquez M, Wittrup KD, and Xu Y (2015). High throughput cross-interaction measures for human IgG1 antibodies correlate with clearance rates in mice. mAbs 7, 770–777. 10.1080/19420862.2015.1043503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hötzel I, Theil F-P, Bernstein LJ, Prabhu S, Deng R, Quintana L, Lutman J, Sibia R, Chan P, Bumbaca D, et al. (2012). A strategy for risk mitigation of antibodies with fast clearance. mAbs 4, 753–760. 10.4161/mabs.22189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jain T, Sun T, Durand S, Hall A, Houston NR, Nett JH, Sharkey B, Bobrowicz B, Caffry I, Yu Y, et al. (2017). Biophysical properties of the clinical-stage antibody landscape. Proc. Natl. Acad. Sci. USA 114, 944–949. 10.1073/pnas.1616408114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boughter CT, Borowska MT, Guthmiller JJ, Bendelac A, Wilson PC, Roux B, and Adams EJ (2020). Biochemical patterns of antibody polyreactivity revealed through a bioinformatics-based analysis of CDR loops. Elife 9, e61393. 10.7554/eLife.61393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lim H, and No KT (2022). Prediction of polyreactive and nonspecific single-chain fragment variables through structural biochemical features and protein language-based descriptors. BMC Bioinf. 23, 520. 10.1186/s12859-022-05010-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kelly RL, Zhao J, Le D, and Wittrup KD (2017). Nonspecificity in a nonimmune human scFv repertoire. mAbs 9, 1029–1035. 10.1080/19420862.2017.1356528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Éliás S, Wrzodek C, Deane CM, Tissot AC, Klostermann S, and Ros F (2024). Prediction of polyspecificity from antibody sequence data by machine learning. Front. Bioinform 3, 1286883. 10.3389/fbinf.2023.1286883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feldhaus MJ, Siegel RW, Opresko LK, Coleman JR, Feldhaus JMW, Yeung YA, Cochran JR, Heinzelman P, Colby D, Swers J, et al. (2003). Flow-cytometric isolation of human antibodies from a nonimmune Saccharomyces cerevisiae surface display library. Nat. Biotechnol 21, 163–170. 10.1038/nbt785. [DOI] [PubMed] [Google Scholar]
- 22.Sheets MD, Amersdorfer P, Finnern R, Sargent P, Lindquist E, Schier R, Hemingsen G, Wong C, Gerhart JC, and Marks JD (1998). Efficient construction of a large nonimmune phage antibody library: The production of high-affinity human single-chain antibodies to protein antigens. Proc. Natl. Acad. Sci. USA 95, 6157–6162. 10.1073/pnas.95.11.6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y, Wu L, Gupta P, Desai AA, Smith MD, Rabia LA, Ludwig SD, and Tessier PM (2020). Physicochemical Rules for Identifying Monoclonal Antibodies with Drug-like Specificity. Mol. Pharm 17, 2555–2569. 10.1021/acs.molpharmaceut.0c00257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Makowski E, Wang T, Zupancic J, Huang J, Wu L, Schardt J, De Groot A, Elkins S, Martin W, and Tessier P (2024). Optimization of therapeutic antibodies for reduced self-association and non-specific binding via interpretable machine learning. Nat. Biomed. Eng 8, 45–56. 10.1038/s41551-023-01074-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Makowski EK, Chen H, Lambert M, Bennett EM, Eschmann NS, Zhang Y, Zupancic JM, Desai AA, Smith MD, Lou W, et al. (2022). Reduction of therapeutic antibody self-association using yeast-display selections and machine learning. mAbs 14, 2146629. 10.1080/19420862.2022.2146629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Makowski EK, Kinnunen PC, Huang J, Wu L, Smith MD, Wang T, Desai AA, Streu CN, Zhang Y, Zupancic JM, et al. (2022). Co-optimization of therapeutic antibody affinity and specificity using machine learning models that generalize to novel mutational space. Nat. Commun 13, 3788. 10.1038/s41467-022-31457-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shehata L, Maurer DP, Wec AZ, Lilov A, Champney E, Sun T, Archambault K, Burnina I, Lynaugh H, Zhi X, et al. (2019). Affinity Maturation Enhances Antibody Specificity but Compromises Conformational Stability. Cell Rep. 28, 3300–3308.e4. 10.1016/j.celrep.2019.08.056. [DOI] [PubMed] [Google Scholar]
- 28.Olsen TH, Boyles F, and Deane CM (2022). Observed Antibody Space: A diverse database of cleaned, annotated, and translated unpaired and paired antibody sequences. Protein Sci. 31, 141–146. 10.1002/pro.4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin Z, Akin H, Rao R, Hie B, Zhu Z, Lu W, Smetanin N, Verkuil R, Kabeli O, Shmueli Y, et al. (2023). Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130. 10.1126/science.ade2574. [DOI] [PubMed] [Google Scholar]
- 30.Raybould MIJ, Marks C, Krawczyk K, Taddese B, Nowak J, Lewis AP, Bujotzek A, Shi J, and Deane CM (2019). Five computational developability guidelines for therapeutic antibody profiling. Proc. Natl. Acad. Sci. USA 116, 4025–4030. 10.1073/pnas.1810576116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prigent J, Jarossay A, Planchais C, Eden C, Dufloo J, Kök A, Lorin V, Vratskikh O, Couderc T, Bruel T, et al. (2018). Conformational Plasticity in Broadly Neutralizing HIV-1 Antibodies Triggers Polyreactivity. Cell Rep. 23, 2568–2581. 10.1016/j.celrep.2018.04.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mouquet H, Scheid JF, Zoller MJ, Krogsgaard M, Ott RG, Shukair S, Artyomov MN, Pietzsch J, Connors M, Pereyra F, et al. (2010). Polyreactivity increases the apparent affinity of anti-HIV antibodies by heteroligation. Nature 467, 591–595. 10.1038/nature09385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mengfei L, Guang Y, Kevin W, I NN, A VN, Wes R, Mattia B, Munir AS, Jingyun G, F HB, et al. (2014). Polyreactivity and Autoreactivity among HIV-1 Antibodies. J. Virol 89, 784–798. 10.1128/jvi.02378-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liao H, and Zhang Z (2018). Polyreactive Antibodies in Anti-HIV-1 Responses. Curr. Mol. Med 18, 126–133. 10.2174/1566524018666180720165406. [DOI] [PubMed] [Google Scholar]
- 35.Dyson MR, Masters E, Pazeraitis D, Perera RL, Syrjanen JL, Surade S, Thorsteinson N, Parthiban K, Jones PC, Sattar M, et al. (2020). Beyond affinity: selection of antibody variants with optimal biophysical properties and reduced immunogenicity from mammalian display libraries. mAbs 12, 1829335. 10.1080/19420862.2020.1829335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gupta P, Makowski EK, Kumar S, Zhang Y, Scheer JM, and Tessier PM (2022). Antibodies with Weakly Basic Isoelectric Points Minimize Trade-offs between Formulation and Physiological Colloidal Properties. Mol. Pharm 19, 775–787. 10.1021/acs.molpharmaceut.1c00373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rabia LA, Zhang Y, Ludwig SD, Julian MC, and Tessier PM (2018). Net charge of antibody complementarity-determining regions is a key predictor of specificity. Protein Eng. Des. Sel 31, 409–418. 10.1093/protein/gzz002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sakhnini LI, Greisen PJ, Wiberg C, Bozoky Z, Lund S, Wolf Perez A-M, Karkov HS, Huus K, Hansen J-J, Bülow L, et al. (2019). Improving the Developability of an Antigen Binding Fragment by Aspartate Substitutions. Biochemistry 58, 2750–2759. 10.1021/acs.biochem.9b00251. [DOI] [PubMed] [Google Scholar]
- 39.Lecerf M, Kanyavuz A, Lacroix-Desmazes S, and Dimitrov JD (2019). Sequence features of variable region determining physicochemical properties and polyreactivity of therapeutic antibodies. Mol. Immunol 112, 338–346. 10.1016/j.molimm.2019.06.012. [DOI] [PubMed] [Google Scholar]
- 40.Schaefer ZP, Bailey LJ, and Kossiakoff AA (2016). A polar ring endows improved specificity to an antibody fragment. Protein Sci. 25, 1290–1298. 10.1002/pro.2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Birtalan S, Zhang Y, Fellouse FA, Shao L, Schaefer G, and Sidhu SS (2008). The Intrinsic Contributions of Tyrosine, Serine, Glycine and Arginine to the Affinity and Specificity of Antibodies. J. Mol. Biol 377, 1518–1528. 10.1016/j.jmb.2008.01.093. [DOI] [PubMed] [Google Scholar]
- 42.Tiller KE, Li L, Kumar S, Julian MC, Garde S, and Tessier PM (2017). Arginine mutations in antibody complementarity-determining regions display context-dependent affinity/specificity trade-offs. J. Biol. Chem 292, 16638–16652. 10.1074/jbc.M117.783837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Birtalan S, Fisher RD, and Sidhu SS (2010). The functional capacity of the natural amino acids for molecular recognition. Mol. Biosyst 6, 1186–1194. 10.1039/B927393J. [DOI] [PubMed] [Google Scholar]
- 44.Bellesia G, Jewett AI, and Shea J-E (2010). Sequence periodicity and secondary structure propensity in model proteins. Protein Sci. 19, 141–154. 10.1002/pro.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang B, Gallolu Kankanamalage S, Dong J, and Liu Y (2021). Optimization of therapeutic antibodies. Antib. Ther 4, 45–54. 10.1093/abt/tbab003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fischman S, and Ofran Y (2018). Computational design of antibodies. Curr. Opin. Struct. Biol 51, 156–162. 10.1016/j.sbi.2018.04.007. [DOI] [PubMed] [Google Scholar]
- 47.Tiller KE, and Tessier PM (2015). Advances in Antibody Design. Annu. Rev. Biomed. Eng 17, 191–216. 10.1146/annurev-bioeng-071114-040733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Makowski EK, Wu L, Gupta P, and Tessier PM (2021). Discovery-stage identification of drug-like antibodies using emerging experimental and computational methods. mAbs 13, 1895540. 10.1080/19420862.2021.1895540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rojas G. (2022). Understanding and Modulating Antibody Fine Specificity: Lessons from Combinatorial Biology. Antibodies 11, 48. 10.3390/antib11030048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Olsen TH, Moal IH, and Deane CM (2022). AbLang: an antibody language model for completing antibody sequences. Bioinform. Adv 2, vbac046. 10.1093/bioadv/vbac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leem J, Mitchell LS, Farmery JHR, Barton J, and Galson JD (2022). Deciphering the language of antibodies using self-supervised learning. Patterns 3, 100513. 10.1016/j.patter.2022.100513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jardine JG, Sok D, Julien J-P, Briney B, Sarkar A, Liang C-H, Scherer EA, Henry Dunand CJ, Adachi Y, Diwanji D, et al. (2016). Minimally Mutated HIV-1 Broadly Neutralizing Antibodies to Guide Reductionist Vaccine Design. PLoS Pathog 12, e1005815. 10.1371/journal.ppat.1005815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Corti D, Cameroni E, Guarino B, Kallewaard NL, Zhu Q, and Lanzavecchia A (2017). Tackling influenza with broadly neutralizing antibodies. Curr. Opin. Virol 24, 60–69. 10.1016/j.coviro.2017.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kelsoe G, and Haynes BF (2017). Host controls of HIV broadly neutralizing antibody development. Immunol. Rev 275, 79–88. 10.1111/imr.12508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guthmiller JJ, Lan LY-L, Fernández-Quintero ML, Han J, Utset HA, Bitar DJ, Hamel NJ, Stovicek O, Li L, Tepora M, et al. (2020). Polyreactive Broadly Neutralizing B cells Are Selected to Provide Defense against Pandemic Threat Influenza Viruses. Immunity 53, 1230–1244.e5. 10.1016/j.immuni.2020.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Burton DR, Poignard P, Stanfield RL, and Wilson IA (2012). Broadly Neutralizing Antibodies Present New Prospects to Counter Highly Anti-genically Diverse Viruses. Science 337, 183–186. 10.1126/science.1225416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reyes-Ruiz A, and Dimitrov JD (2021). How can polyreactive anti-bodies conquer rapidly evolving viruses? Trends Immunol. 42, 654–657. 10.1016/j.it.2021.06.008. [DOI] [PubMed] [Google Scholar]
- 58.Tennenhouse A, Khmelnitsky L, Khalaila R, Yeshaya N, Noronha A, Lindzen M, Makowski EK, Zaretsky I, Sirkis YF, Galon-Wolfenson Y, et al. (2024). Computational optimization of antibody humanness and stability by systematic energy-based ranking. Nat. Biomed. Eng 8, 30–44. 10.1038/s41551-023-01079-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dunbar J, and Deane CM (2016). ANARCI: antigen receptor numbering and receptor classification. Bioinformatics 32, 298–300. 10.1093/bioinformatics/btv552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Makowski EK, Wu L, Desai AA, and Tessier PM (2021). Highly sensitive detection of antibody nonspecific interactions using flow cytometry. mAbs 13, 1951426. 10.1080/19420862.2021.1951426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Julian MC, Rabia LA, Desai AA, Arsiwala A, Gerson JE, Paulson HL, Kane RS, and Tessier PM (2019). Nature-inspired design and evolution of anti-amyloid antibodies. J. Biol. Chem 294, 8438–8451. 10.1074/jbc.RA118.004731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chao G, Lau WL, Hackel BJ, Sazinsky SL, Lippow SM, and Wittrup KD (2006). Isolating and engineering human antibodies using yeast surface display. Nat. Protoc 1, 755–768. 10.1038/nprot.2006.94. [DOI] [PubMed] [Google Scholar]
- 63.Van Deventer JA, and Wittrup KD (2014). Yeast Surface Display for Antibody Isolation: Library Construction, Library Screening, and Affinity Maturation. In Monoclonal Antibodies: Methods and Protocols, Ossipow V and Fischer N, eds. (Humana Press; ), pp. 151–181. 10.1007/978-1-62703-992-5_10. [DOI] [PubMed] [Google Scholar]
- 64.Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xu Y, Roach W, Sun T, Jain T, Prinz B, Yu T-Y, Torrey J, Thomas J, Bobrowicz P, Vásquez M, et al. (2013). Addressing polyspecificity of antibodies selected from an in vitro yeast presentation system: a FACS-based, high-throughput selection and analytical tool. Protein Eng. Des. Sel 26, 663–670. 10.1093/protein/gzt047. [DOI] [PubMed] [Google Scholar]
- 66.Jain T, Boland T, Lilov A, Burnina I, Brown M, Xu Y, and Vásquez M (2017). Prediction of delayed retention of antibodies in hydrophobic interaction chromatography from sequence using machine learning. Bioinformatics 33, 3758–3766. 10.1093/bioinformatics/btx519. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Antibody sequences are provided in the Tessier lab GitHub repository: https://zenodo.org/doi/10.5281/zenodo.13387056.
Codes to generate the random forest model are available in the Tessier lab GitHub repository: https://zenodo.org/doi/10.5281/zenodo.13387056.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.