

Abstract
The human armpit microbiome is metabolically entangled with skin cell physiology. This “meta-organism” symbiotic mutualism results in sweat either with or without odor (osmidrosis), depending on host ABCC11 gene haplotypes. Apocrine metabolism produces odorless S-glutathione conjugate that is transferred by ABCC11 transporters into secretory vesicles, deglutamylated to S-Cys-Gly-3M3SH thiol, and exuded to skin surface. An anthropogenic clade of skin bacteria then takes up the thiol and bioconverts it to malodorous 3-methyl-3-sulfanylhexan-1-ol (3M3SH). We hypothesized a familial meta-organism association of human ABCC11 gene non-synonymous SNP rs17822931 interplaying with skin microbiome 3M3SH biosynthesis. Subjects were genotyped for ABCC11 SNPs, and their haplotypes were correlated with axilla microbiome DNA sequencing profiles and predicted metagenome functions. A multigeneration family pedigree revealed a Mendelian autosomal recessive pattern: the C allele of ABCC11 correlated with bacterial Cys-S-conjugate β-lyase (PatB) gene known for Staphylococcus hominis biosynthesis of 3M3SH from human precursor; PatB was rescinded in hosts with homozygous TT alleles encoding ABCC11 loss-of-function mutation. We posit that a C allele encoding functional ABCC11 is key to delivering host conjugate precursors that shape heritable skin niche conditions favorable to harboring Staphylococcus having genomics of odor thiol production. This provides existential insights into human evolution and global regional population ancestries.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-78711-w.
Subject terms: Microbiome, Skin manifestations, Coevolution
Introduction
It is increasingly appreciated in the literature that humans and other multicellular organisms are comprised of their own eukaryote cells plus niche communities of resident microbial cells1–4. Environmental pressures over millennia have given rise to the co-evolution of such cross-kingdom mutualistic “meta-organism” entities, reflected in familial microbiome phenotypes in Homo sapiens ancestral groups from distinct global regions1–4. This interactive relationship steers physiological, medical, and neuropsychological phenomena involving a variety of host organ systems, notably the skin and the gut-brain axis1–6. The present study centers on the relationship of human host ABCC11 gene variants interplaying with axillary skin anthropogenic microbiome functional genomics of malodor thiols.
Armpit odor (osmidrosis) phenotype is attributable to apocrine sweat molecules being biotransformed by axilla skin niche microbiota into volatile organic compounds. The early literature proposed roles for steroids and volatile fatty acids in malodor, while recent studies support the prevailing notion that a particular thioalcohol, methyl-3-sulfanylhexan-1-ol (3M3SH), is a primary factor dominating the malodor phenotype due to 3M3SH concentration prominence on skin and its significantly lower olfactory threshold compared to other axillary molecules7–19.
The metabolic pathway yielding 3M3SH involves a cross-kingdom biochemical route. It begins as an odorless S-glutathione thiol conjugate molecule generated by human host apocrine gland metabolism. Apocrine ABCC11 transporters transfer this conjugate into secretory vesicles, whereupon it is deglutamylated into S-Cys-Gly-3M3SH which is then exuded to the skin surface; subsequently, particular commensal skin bacteria take up this thiol and finalize the bioconversion of odorless S-Cys-Gly-3M3SH into 3M3SH7–15,20.
Malodor vs. non-odor phenotype is determined by niche microbiome community composition, with Staphylococcus spp. and Corynebacterium spp as the most prevalent taxa found on axillary skin of humans and non-human primates7,8,10–13,15–18,21–25. Of critical importance in determining axillary thiol odor, the Cys-S-conjugate β-lyase (PatB) gene characteristic of Staphylococcus hominis, in particular, is both necessary and sufficient to enzymatically convert human S-Cys-Gly-3M3SH precursor into microbial product 3M3SH10–12.
Host ABCC11 transporter activity is an obligatory step early in the above described chain of events leading to 3M3SH7–9,11,15. ABCC11 (“ATP-Binding Cassette, sub-family C, member 11”; EC 7.6.2.2; NCBI gene ID 85320) is a member of the ABC transporter superfamily of membrane bound ATP-dependent primary active transport proteins that catalyze efflux of cGMP, cAMP, metabolic products, and cytotoxic/xenobiotic compounds from cells or intracellular compartments20. In humans there are 49 known ABC transporters grouped into seven subfamilies. Nine members of the ABCC[1–13] subfamily, notably ABCC11, confer xenobiotic and multidrug resistance transporter activity26,27. ABCC11 is a monomer polypeptide encoded by a 82.7 kb gene consisting of 39 exons located at human chromosome 16q12.120. ABCC11 exhibits pleiotropic physiological functions in a variety of cell and tissue types throughout the body. Pioneering studies identified ABCC11 prominently expressed in apocrine glands of skin axillary and groin tissues, ceruminous ear canal structures, and mammary tissues15,28–30. Single cell RNA sequencing31 further indicates expression in sebaceous glands, liver, testes, prostate, and a prominent role in glandular epithelial cells of breast relating to physiology of lactation/colostrum and pathophysiology of breast cancer26,27. Lower expression in single cell types also includes hepatocytes, spermatids, visceral and cutaneous adipocytes, and astrocytes32. ABCC11 allele locus c.538 C encodes the wild type functional polypeptide with residue p.Gly180 required for transporter activity, with post-translational N-linked glycosylation sites at p.Asn838 and p.Asn844 required for trafficking, collectively leading to ABCC11 expression in membranes and participation in a spectrum of cell physiological processes and phenotypes8,10,11,29,33,34. Notable high-affinity substrates for human ABCC11 include S-glutathione conjugated thiols7–14,18,20, as germane to the present study.
In contrast with the roles of wild type ABCC11 described above, a non-transporting mutant variant of dysfunctional ABCC11 transporter proteins blocks the axillary apocrine gland’s obligatory step of S-glutathione conjugate transport in the chain of events that ultimately lead to 3M3SH7–9,11,15. The single nucleotide polymorphism (SNP) rs17822931 at locus c.538 C>T in the human ABCC11 gene encodes the variant non-synonymous loss-of-function translated polypeptide mutation at p.Gly180Arg20,35. The initial discovery of this SNP was associated with dry earwax (T alleles expressing non-functional ABCC11 with p.Arg180) vs. wet earwax (C alleles expressing functional p.Gly180) from ear canal ceruminous glands as differentiated between ethnic groups36,37. This fortuitously led to identifying the distinguishable thiol osmidrosis phenotypes of axillary odor as a risk factor of ABCC11 haplotypes C vs. T alleles, as associated with taxonomy differences in skin microbiome metabolism of molecules originating from axillary gland secretions15. In an interesting historical context, ABCC11 SNP rs17822931 represents the first documented non-synonymous SNP variant reported to translate as a grossly distinctive phenotype binary shift of a heritable trait35. Note that in the present study, alleles are reported to represent the forward orientation, per standard convention of NCBI dbSNP35.
The present study was undertaken in order to shed light on better understanding the heritable confluence of human ABCC11 transporter gene missense variant rs17822931 with axillary skin microbial functional genomics required for microbial biosynthesis of 3M3SH. We hypothesized that at least one copy of the host ABCC11 wild type “C” allele—in contrast with “T” allele of SNP rs17822931 that encodes mutant ABCC11 transporter loss-of-function—is sufficient and necessary for the manifestation of axillary skin Staphylococcus PatB gene, an essential step in the 3M3SH bioconversion pathway, as coupled within a multigeneration family pedigree. Understanding such a meta-organism mutualism of Homo sapiens with anthropogenic commensal microbes can provide clues for insights with existential implications in better understanding human evolution and global regional population ancestries7,17,38.
Results
Microbiome beta diversity reflects host cohorts’ SNP haplotypes
Microbiome beta diversity was examined by principal coordinate analysis ordination (PCoA), as shown in the data of Fig. 1. PCoA revealed: (1) cohort clustering of CC and of CT; (2) overlapping clustering of CC with CT; (3) clustering of TT; and (4) each CC and CT segregated from the clustering of TT. Results in Fig. 1 are based on N = 50 individual left and right axillae samples paired from each subject (pairings shown by dashed lines), which were similar to results based on pooled left + right samples (N = 25) from each subject (data not shown). To further test if the clusters observed in the PCoA could be explained by the haplotypes, the denoised rarefied ASV dataset was analyzed by Permutational Multivariate ANOVA (PERMANOVA, Table 1). Overall, the ABCC11 variant haplotypes were responsible for 5% of the variance in the axillary microbiota (R2 = 0.05, FDR-adjusted p = 0.001). The pairwise comparisons, as shown in the data of Table 1, revealed significant differences in subjects with homozygous TT vs. homozygous CC (p = 0.001), in TT vs. heterozygous CT (p = 0.006), and in TT vs. subjects with a C on any allele (i.e., a cohort with pooled metadata [CC + CT], p = 0.0001), while CC vs. CT was not significantly different (p = 0.125). Altogether, the results demonstrated the microbiome community of TT was segregated from that of CC and CT.
Fig. 1.
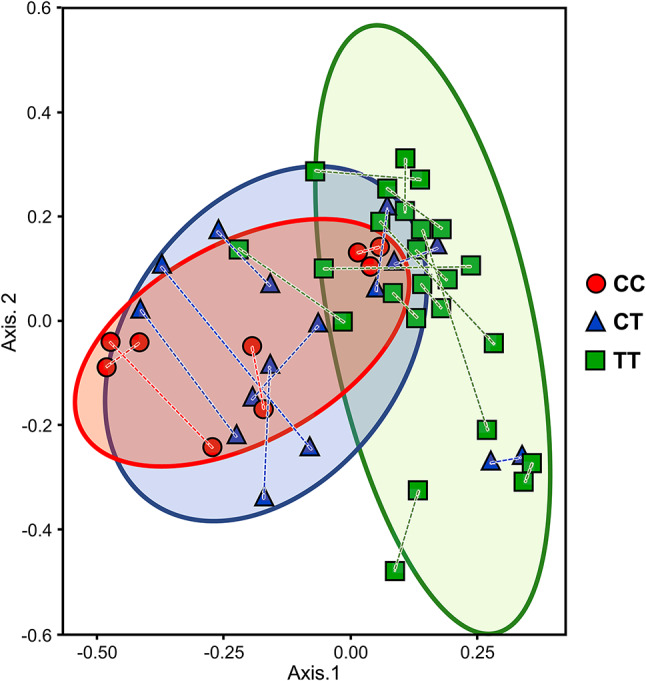
Principal coordinate analysis ordination of axillary skin microbiome ASVs for ABCC11 metadata. Beta diversity PCoA Bray–Curtis dissimilarities ordination of 9324 microbiome DADA2 denoised rarefied ASVs was obtained for human host metadata haplotypes CC (red circles), CT (blue triangles), and TT (green squares) allele combination of ABCC11 wild type and SNP rs17822931 cohorts. N = 50 samples individually representing both axillae from N = 25 subjects. Dashed lines connect paired samples of left and right armpits from each given subject. The CC and CT cohort samples were clustered within and amongst themselves, while collectively distinguishable from the clustering of homozygous TT cohort samples; detailed PERMANOVA comparisons are described in Table 1.
Table 1.
PERMANOVA of microbiome ASVs comparing human host ABCC11 haplotypes.
| Haplotype cohorts | Df | SS | R 2 | F | p (FDR adj.) | |
|---|---|---|---|---|---|---|
| TT vs. CC | 1 | 1.1214 | 0.07798 | 2.6217 | 0.001 | *** |
| Residual | 31 | 13.2602 | 0.92202 | |||
| Total | 32 | 14.3817 | 1.00000 | |||
| TT vs. CT | 1 | 0.7497 | 0.04333 | 1.7211 | 0.006 | *** |
| Residual | 38 | 16.5525 | 0.95667 | |||
| Total | 39 | 17.3022 | 1.00000 | |||
| CC vs. CT | 1 | 0.5402 | 0.05360 | 1.3027 | 0.125 | N.S. |
| Residual | 23 | 9.5371 | 0.94640 | |||
| Total | 24 | 10.0773 | 1.00000 | |||
| TT vs. (CC + CT) | 1 | 1.1027 | 0.05173 | 2.5637 | 0.0001 | *** |
| Residual | 47 | 20.2151 | 0.94827 | |||
| Total | 48 | 21.3178 | 1.00000 |
Permutational multivariate ANOVA (PERMANOVA) pairwise comparisons of skin microbiome ASV abundances differences assigned to human cohort allele combinations; p was adjusted for FDR; all permutations = 999. Human subject cohort wild type C alleles and SNP rs17822931 T allele combinations were: homozygous TT; homozygous CC; or heterozygous CT. Comparison was also assessed for TT vs. subjects with a C on any allele (i.e., a cohort of subjects with pooled allele combinations (CC + CT)). Microbiome dataset incorporated all 9324 DADA2 denoised rarefied ASVs assembled from total pool of all subjects. Data represent both armpit samples obtained from N = 25 subjects.
Host transporter SNP alleles differentiate microbiome taxonomic skin distributions
Microbiome taxonomic distributions were assigned to the ABCC11 transporter metadata haplotypes. Figure 2A shows the relative abundances ranked distributions of taxa derived from RDB database of the Illumina DADA2 denoised rarefied ASV data. Here, for TT cohort the fractional composition of the total taxa was 29% Staphylococcus and 57% Corynebacterium; for CT it was 57% Staphylococcus and 29% Corynebacterium; and for CC it was 65% Staphylococcus and 1% Corynebacterium. Figure 2B revealed taxa prevalences at the level of species within these samples, based on Nanopore 4.5 kb reads of 16S-ITS-23S rRNA genes; note the high prevalences of Staphylococcus hominis, Staphylococcus epidermidis, and Corynebacterium tuberculostearicum. The data in Fig. 2 represent a hybrid sequencing approach combining Illumina plus Nanopore analyses. Sequencing results for 16S variable regions V1V2 and V3V4 (Fig. 2A) provided comprehensive coverage of diverse microbial communities, enhancing the broad and accurate identification of bacterial taxa, reducing the reliance on specific primer pairs, minimizing single primer-induced biases, and providing the input for the PICRUSt2 necessary to generate predicted functional genomics. However, the read length constraints of Illumina limit the accuracy of taxonomic assignment at the species level. To overcome this limitation, it is essential to sequence larger regions of the rRNA gene operons. To address this, we further examined 4.5 kb contiguous long reads of the rRNA operon using Nanopore sequencing. By combining the strengths of Illumina for the quantification of microbial abundances plus Nanopore technology for the prevalence of species, we achieved a more robust and precise characterization of bacterial communities.
Fig. 2.

Taxonomic distributions. (A) Relative abundances of the ranked top 10 most abundant bacteria taxa at the genus level are shown for human host cohort metadata haplotypes CC, CT, and TT of ABCC11 wild type or SNP rs17822931 alleles. Data were based on Illumina DADA2 rarefied ASVs with RDB database. (B) Prevalences at the species level represented within the dataset. Data were derived from Nanopore 4.5 kb long-read DNA sequencing of the 16S-ITS-23S rRNA genes. The detection threshold 0.01 was used to remove species under 1% abundance. Data represent both armpit samples from N = 25 subjects.
In Fig. 3, FDR-adjusted mixed-effects model ANOVA analysis of the log-transformed abundance data indicated that Staphylococcus in CC and in CT cohorts were each significantly greater than in the TT cohort (p < 0.05), with no significant differences comparing CC vs. CT (p > 0.05). For Corynebacterium, a mixed-effects model ANOVA analysis (Fig. 3) revealed that the CC cohort had significantly lower values compared to both the TT and CT cohorts (p < 0.05), while there was no significant difference between the TT and CT cohorts (p > 0.05). In concert with SparCC network analysis indicating no significant correlations between these two taxa (p-values ranged from 0.12 to 0.92 for all allele combinations), the data collectively imply that these microbial distribution differences are due to skin niche preferences rather than competition for resources.
Fig. 3.

Distribution of Staphylococcus and Corynebacterium comparing cohorts of human host subjects expressing haplotypes CC, CT, or TT of ABCC11 wild type or SNP rs17822931 alleles. Box plot of total abundances of Staphylococcus and Corynebacterium at the taxonomic level of the genus are shown log-transformed. ANOVA means with the same letter are not significantly different (FDR adjusted p > 0.05), while different letters are significantly different (FDR p < 0.05). Data represent both armpit samples from N = 25 subjects.
Predicted functional genomics of microbiome thiol metabolism enzymes
The PICRUSt2 pipeline output results were comprised of 2332 EC numbers, 7734 KEGG KO orthologs, and MetaCyc metabolic features parsed into 3670 biochemical reactions mapped into 432 pathways. These data were then collectively filtered to isolate enzymatic steps germane to biosynthesis of axillary odor molecules of microbial origin reported in the literature7–13,16–18,20. The resulting consensus of the pooled PICRUSt2 data yielded the key genes of Staphylococcus hominis specifically required in the pathway leading to 3-methyl-3-sulfanylhexan-1-ol (3M3SH) biosynthesis from starting point precursor S-Cys-Gly-3M3SH conjugate7,10–12; namely, cysteine-S-conjugate β-lyase (PatB, ShPatB, EC4.4.1.13, K01760), Aminopeptidase A (PepA, EC3.4.11.1, K01255), and proton H+-dependent oligopeptide transporter (POT, K03305).
The data of Fig. 4 indicated that homozygous CC or heterozygous CT subjects exhibited microbiome PatB at levels significantly greater than homozygous TT subjects (p < 0.05), while not significantly different between CC vs. CT (p > 0.05). Further in Fig. 4, there were no significant differences (p > 0.05) among all haplotypes for microbial expression of both PepA and POT, as consistent with the universal expression of those enzymes amongst other microbial members of the anthropogenic clade of skin commensal bacteria10. Thus, microbiome PatB was the principal factor differentiating the allele cohorts in Fig. 4.
Fig. 4.

Key thiol-conjugate pathway enzymes of axilla skin microbiome. The PICRUSt2 pipeline yielded key microbiome enzymes PatB and PepA that are required for bioconversion of S-Cys-Gly-3M3SH into volatile 3M3SH odor molecules. Host apocrine metabolism is the source of S-Cys-Gly-3M3SH on the skin surface, which is transported into microbiota by POT10,11. Cohort comparisons include CC, CT, and TT haplotypes of ABCC11 wild type or SNP rs17822931 alleles. Box plots of enzyme abundances data are shown log-transformed. In each panel, ANOVA means with the same letter are not significantly different (FDR adjusted p > 0.05), while different letters are significantly different (FDR p <0.05). Data represent both armpit samples from N = 25 subjects.
Intertwined metabolic pathway of microbial 3M3SH malodor biosynthesis steered by host precursor inherited in ABCC11 haplotypes
A family pedigree of subjects spanning 3 generations was parsed from a subset of the study metadata pool. All family members self-identified as being of European/American modern era geosocial ancestry. The data of Fig. 5 revealed a Mendelian autosomal recessive distribution pattern of ABCC11 haplotypes within this pedigree (Fig. 5A), as further correlated with Mendelian-like inheritance of their axillary skin’s microbiome enzymes for 3M3SH bioconversion (Fig. 5B). Data in the heatmap of Fig. 5B indicated that tagged data of individual family members reflected their personal SNP-dependent microbiome enzyme abundance distributions, as being consistent within the aggregated metadata pool that encompassed non-family members.
Fig. 5.

Multigeneration human host ABCC11 haplotype pedigree and heatmap of skin microbiome genes critical for 3M3SH bioconversion. (A) Family pedigree of subjects (N = 9) spanning 3 generations, obtained from a subset of the study metadata pool. ABCC11 wild type and SNP rs17822931 allele assignments CC, CT, TT are shown. All family members self-identified as being of American/European modern era geosocial ancestry. (B) Heatmap of PICRUSt2 pipeline results for critical 3M3SH bioconversion pathway enzymes (see Fig. 4). Data are clustered by metadata ABCC11 wild type and SNP rs17822931 allele cohort assignments CC, CT, or TT. Data represent both armpit samples, also indicating the metadata subset of family pedigree N = 9 subjects from Panel A. Relative abundances are centered and scaled from 100 to -100.
Discussion
The main finding of this study is that human ABCC11 gene variant haplotypes are engaged in an interplay with 3M3SH odor bioconversion functional genomics of armpit skin niche microbiome communities. This implicates the involvement of a meta-organism symbiotic mutualism in steering thiol osmidrosis phenotype. Collectively the Results support our hypothesis that at least one copy of the ABCC11 c.538 C allele is both sufficient and necessary for familial manifestation of axillary skin microbial genes essential for the thioalcohol odor bioconversion pathway, in particular the cysteine-S-conjugate β‑lyase (PatB gene) of Staphylococcus hominis. Further, this pattern of mutualism was rescinded in hosts homozygous for SNP rs17822931 TT alleles of ABCC11, which significantly diminished prevalence and relative abundance of Staphylococcus, and instead favored the abundance of Corynebacterium. The findings were reinforced by a multigeneration family pedigree metadata subset that revealed a Mendelian autosomal recessive familial relationship.
Assimilation of host/microbe meta-organism interaction events
The Results (Figs. 1, 2, 3, 4 and 5; Table 1, Supplementary information S1) are assimilated into the Fig. 6 consensus cartoon diagram of host/microbe interactions. This is described in the following narrative, as supported by the literature7–15,17,20. In this model, malodorous volatile 3M3SH has its precursor roots beginning with the first step originating as the odorless S-glutathione conjugated molecule generated by human axillary apocrine gland metabolism. Gland intracellular membrane ABCC11 transporters transfer S-glutathione conjugate into secretory vesicles where it is deglutamylated into S-Cys-Gly-3M3SH. Sloughed vesicles then exude odorless S-Cys-Gly-3M3SH onto the skin surface, becoming available for bacterial uptake via proton-dependent oligopeptide transporter POT10,11. Host axillae niche suitability to favorably harbor particular species of Staphylococcus on its skin depends on the presence of this starting thiol delivered by ABCC11 transporters; in particular, a unique anthropogenic clade of Staphylococcus exploits the host-supplied S-Cys-Gly-3M3SH substrate to generate pyruvate as an energy source10. Volatile 3M3SH is the dominating thiol microbial byproduct of such metabolism, thereby contributing to malodor phenotype particularly attributable to Staphylococcus hominis expression of thiol biotransformation enzymes characteristic of this clade7–14,16,18. The next intermediate enzymatic step in Fig. 6, namely S-Cys-Gly-3M3SH → Cys-3MSH + Glycine, has been previously assigned to PepA10–12 in Staphylococcus—e.g., EC3.4.11.1 or K01255 in our PICRUSt2 Results (Figs. 4 and 5). Other labs8,15,17 found this conversion step could also be alternatively catalyzed potentially by a Corynebacterium enzyme TpdA (EC3.4.13.23 / K23980); however note that TpdA was not detected in the present study’s skin samples dataset. Furthermore, an alternative thiol odor pathway described by Natch’s group8,15 involving N-acyl glutamine aminoacylase (EC 3.5.1.133, K23726, NAGA, agaA) was also not detected in any of our samples. Therefore, pursuit of a Corynebacterium spp thiol odor pathway was abandoned in the present study.
Fig. 6.

Meta-organism entangled metabolic pathway of microbial 3M3SH malodor biotransformation steered by human precursor thiol conjugate substrate in ABCC11 haplotypes. Results assimilated into a functional cartoon model of skin microbiome/human mutualism that entangles human ABCC11 transporter activity and microbial pyruvate + 3M3SH biosynthesis in osmidrosis. Steps are as follows. Host axillary apocrine gland metabolism initially generates S-glutathione-3M3SH conjugate molecules that are transported into intracellular vesicles by ABCC11 membrane transporters. Within the vesicles, S-glutathione-3M3SH is converted to S-Cys-Gly-3M3SH by a gamma-glutamyl transferase step9. Shedding of fragmented vesicles onto the skin surface exposes the S-Cys-Gly-3M3SH, becoming available for commensal bacteria uptake transport via POT10,11. Relative magnitude concentrations of the human thiols on the skin surface are proportional to ABCC11 functional transporter activity, as positively correlated with expression by copy numbers of the wild type C allele vs. mutant T allele SNP rs17822931 (CC ~ CT > TT). A unique anthropogenic clade of Staphylococcusexploits host-supplied thiol substrates to generate pyruvate as an energy source along with 3M3SH byproduct10,12. Niche suitability of host axillae to adapt Staphylococcus residence harbored on its skin depends on the presence of the starting S-Cys-Gly-3M3SH conjugated thiol precursor supplied by host’s shed apocrine gland vesicles. Thus, relative abundance of Staphylococcus harbored on axillae skin is positively correlated with copies of the C allele (CC ~ CT > TT). Volatile 3M3SH is the dominating thiol byproduct of the pyruvate biosynthesis, contributing to axillary malodor phenotype, in particular attributable to Staphylococcus hominis expression of these enzymes7–14,16,18. The meta-organism mutualism genomics of odor binary phenotype manifests as a host Mendelian autosomal recessive familial trait, as in Fig. 5. The microbial transporter and enzymes are described in Fig. 4. Key: “S-glutathione-3M3SH” is glutathione-S-3-methyl-3-sulfanylhexan-1-ol conjugate; “S-Cys-Gly-3M3SH” is (S-(6-hydroxy-4-methylhexan-4-yl)-L-cysteinylglycine); “Cys-3M3SH” is (S-(6-hydroxy-4-methylhexan-4-yl)-L-cysteine); “3M3SH” is (3-methyl-3-sulfanylhexan-1-ol).
As depicted in Fig. 6 (reflecting the Results), PatB lyase is the critical enzymatic step finalizing biosynthesis of 3M3SH + pyruvate from S-Cys-3M3SH, in particular attributed to Staphylococcus hominis10–12 while lacking in Staphylococcus epidermidis10–12. In the final step of Fig. 6, 3M3SH is exported out of the bacteria onto the skin surface via simple passive diffusion10. The 3M3SH pathway steps of Fig. 6 are shown occurring in subjects with the CC or CT haplotypes that express at least one copy of functional ABCC11 transporter protein. This is in contrast with non-odor phenotype hosts who possess the SNP rs17822931 homozygous TT haplotype that encodes defective non-transporting ABCC11 protein mutants (Fig. 6). Mutant ABCC11 is the rate-limiting step that fails to export the conjugated thiol substrates out of axillae gland cells onto the skin, and thus TT haplotype thwarts the entire thiol odor generating process of Fig. 6.
The Results data collectively signify that hosts with at least one C allele provide axillary skin niche conditions of metabolic substrates advantageous to harboring Staphylococcus that possess the requisite enzymes of the biosynthesis pathway that generates pyruvate + 3M3SH. This relationship is positively correlated with copy number (CC ~ CT > TT). The present Results are consistent with the prevailing consensus reports of osmidrosis phenotype and abundances of microbiome taxonomic designations in axilla of humans7,8,10–13,15–18,22,24,25 and non-human primates21,23 including chimpanzees, gorillas, and baboons. Our Methods in the present study uniquely deployed DADA2 denoised rarefied ASVs obtained with primers spanning the entire V1-V4 hypervariable region of 16S rRNA gene and the RDP reference database. Nevertheless, some reports39–45 conflict with the majority consensus, ostensibly attributable to various alternative techniques that have included differences in skin sampling methods, culture-based bacterial analyses, DNA isolation, library preparations, 16S rRNA gene primers restricted to V3V4 or only V4, relying on OTUs instead of denoised ASVs, and choosing GreenGenes reference databases with reported limitations.
The above interpretation of Results data has been inferred from analyses of microbiome 16S rRNA genes (see Methods), whether from Nanopore 4.5 kb long reads of the rrn operon, or derived from PICRUSt2 pipeline using denoised rarefied DADA2 ASVs based on V1-V4 primers. Validation and extension of our model could be further explored in future projects using approaches that deploy other techniques including metagenomics, qPCR, and/or RNAseq.
Osmidrosis as a meta-organism phenomenon
Axillary osmidrosis (sweat odor) represents the first documented autosomal recessive trait whereby an evolutionarily conserved eukaryote single non-synonymous SNP of high penetrance genetics reflects the familial merger of a prokaryotic anthropogenic commensal niche microecosystem community composition coupled with a concomitant distinctive binary shift in eukaryotic phenotype8,10,11,15,20,35,36. People are multicellular organisms composed of their own human eukaryote cells plus prokaryote cells inextricably intertwined with the functional genomics of a personal anthropogenic prokaryotic microbial ecosystem inhabiting various anatomical niches throughout the host. This “meta-organism” arrangement is the existential consequence of co-evolutionary advantage adaptations shaping such a symbiotic mutualism. In this arrangement the microbiome provides benefits by steering human host physiology, behaviors and phenotypes of medical and social consequences, while the host reciprocates by providing housing, metabolic necessities, and social structure as a person-to-person communicable vehicle for propagating their prokaryote buddies5,6. Thus, some human familial traits are not wholly eukaryotic genetics, but rather they are microbiome traits manifested by sociological interactions and environmental factors that facilitate propagation of particular bacteria taxa by their hosts1–6. This is revealed in the data of Fig. 5, in which the host ABCC11 SNP rs17822931 haplotype family pedigree parallels transmission of particular skin niche microbiome genomics.
Apocrine odor phenotype and geosocial meta-organism relationships
The Results collectively raise the question: how did the meta-organism pedigree pattern of Fig. 5 possibly arise in this study’s family whose members all self-identified as being of modern era American/European geosocial ancestry? Clues to address this question come from evolutionary microbial phylogeny data coupled with human anthropology migration patterns.
Rudden and colleagues10 have argued that the emergence and diversification of an ancient unique anthropogenic clade of malodorous thioalcohol-producing skin Staphylococcus spp appeared within the timeframe of ancestral humanoid divergence from primates. In particular, Staphylococcus hominis in this clade arose with a novel genome expressing a unique cysteine-S-conjugate β-lyase, PatB, within its battery of enzymes; this exploited the biotransformation of its eukaryotic hosts’ apocrine exported “waste” thiol substrates of ABCC11 into pyruvate as a microbial energy source, with 3M3SH as the byproduct10. The modern-day metabolic pathway of this niche adaptation is encapsulated in Fig. 6. This co-evolution entangling of the functional genomics of ABCC11 in socially-interactive Homo sapiens with its uniquely human Staphylococcus spp. microbial community (Figs. 2 and 3), is consistent with meta-organism mutualism1–6 that tracks human behaviors and global migration patterns over time.
Such migrations originated with ABCC11 wild type homozygous CC alleles in the prehistoric out-of-Africa diaspora, branching initially northward into Asia, then migrating westward to regions of Europe and southward to Oceania. ABCC11 has been designated as a “gold standard” experimentally-validated marker of human migration admixture gene flow in the Fine-Mapping of Adaptive Variation (FineMAV)46 project. The ABCC11 c.538 C > T (p.Gly180Arg) mutation initially arose ~ 40,000 years ago, roughly 2000 generations (1023–3901, 95% CI)23. HapMap studies of global ancestry groups suggest that this mutation arose in an ancestral individual in a glacier-isolated cold climate region of Mongolia, whose peoples then initially migrated throughout the world heavily dominated by easterly movements into regions currently populated by modern peoples of Han Chinese and Korean ancestry23,28,29,34,36,47. Modern individuals in East Asia currently display ABCC11 homozygous TT at a frequency of virtually 100% and nearly 0% CT or CC23,29,36,47. This East Asian regionalization clustering is in contrast to modern individuals of generationally ancient sub-Saharan African ancestry who are virtually 100% homozygous CC and nearly 0% TT or CT23,36,47. Gunther’s group48 identified the ABCC11 SNP rs17822931 T allele variant in genomic sequences of four human remains of Scandinavian Mesolithic hunter-gatherers dating in the early postglacial migration period between 9500 and 6000 years ago, implicating an additional east to west migration from East Asia into Mesolithic Europe. Among modern humans self-identifying as being of European ancestry, the T allele exists at a rate of roughly 1–10%23,29,36,47. Thus, the autosomal recessive genetics pattern in the American/European family pedigree metadata of the present study Results (Fig. 5) is consistent with this collection of migration patterns.
Studies of ratios of nonsynonymous : synonymous nucleotide substitutions intimate that the emergence, global latitude and altitude migration admixtures, and penetrance of the ABCC11 rs17822931 loss of function C > T mutation became concentrated among geosocial clusterings of modern humans whom are the most generationally distant from original African regions, thereby discarding their prehistoric evolutionary selective allelic constraints23,28,29,34,36,47. Subsequent migrations back towards the equator then putatively re-engaged the adaptive benefits of the C allele. Roughly 10,000–25,000 years ago, migration from Eurasia across the Bering land bridge into the Americas has resulted in modern indigenous Native American descendants who exhibit rs17822931 T allele frequency linearly positively correlated with absolute latitude of their tribes23. Polynesian genome analyses29,49 indicate that a wave of migrations of East Asian ancestors (homozygous TT with 0% C alleles) southward into Oceania about 3000 years ago rapidly re-established ABCC11 C alleles into the population during island colonization, as introduced through Papuan admixtures. Contemporaries of Asians ancestors that migrated to the Melanesian island of Gidra now display a 92% C allele frequency, notably approaching the 100% frequency of C alleles in Africans at the same latitude. It has been posited that ABCC11 C alleles impart a positive selective advantage in tropical climates burdened by opportunistic pathogens, through benefits of augmented apocrine colostrum and skin bacteriostatic peptides from niche microbial communities50 provided by mothers to their newborns49 .
The full spectrum of teleological advantages to anthropomorphic bacteria and their host phenotypes resulting from the global migration patterns and modern distribution of ABCC11 SNP variants, remains to be explored17. Possible precipitating factors include global latitude or elevation adaptations to climate and temperature, circadian sun exposure of skin to UV, trade route influences on endogamous and exogamous genomics, enhanced immunity factors of colostrum, ear wax viscosity, olfactory-driven body odor/pheromone influences on localized group culture acceptance/loyalty/mate preference38, etc. There is credence in climate-olfactory theories, given that Staphylococcus hominis replete with PatB is concentrated in several moist apocrine body sites of humans25, including inguinal crease, plantar heel and toe web of feet, and antecubital crease, in addition to axillae. Alternative theories to explain the global patterns of ABCC11 distributions in humans and various animal species, such as genetic convergence, are discussed in the attached Supplementary text S1.
Who is the evolutionary driver: people or microbes?
Within a given meta-organism entity, the cross-talk assimilation of eukaryote genomics entangled with prokaryote genomics synergistically enhances the odds of a mutualistic evolutionary survival advantage3,4. In the light of this meta-organism concept, one conceivable alternative explanation for the observed human ancestral groups’ distribution pattern of ABCC11 SNPs could be that the selective pressure might be actually coming from the axillary skin bacteria taxonomic community niche ecosystem itself to propagate their prokaryotic populations. Thus, in this scenario, commensal microbes engineer communicable opportunities by steering social interactions and close contacts among their hosts who are relegated as mere Trojan horse delivery vessels and incubators for their microbial companions. As discussed above, the selective advantages of ABCC11 transporter activity in various tissues, as correlated with geography and climate, addresses phenotypes relating to apocrine physiology of sweat osmidrosis, ear canal cerumen viscosity, and breast functions. However, beyond apocrine tissues, this leaves open unexplored pleiotropic roles for evolutionary and migration selective advantages of ABCC11 variants as manifested in the physiology of various other cell types throughout the body8,10,11,15,16,20,26–29,31–34.
Odor phenotype governed by one amino acid residue of a single non-synonymous SNP
The host’s rate-limiting step in the thiol malodor chain (Fig. 6) is predicated on apocrine transporter functionality of wild type ABCC11 protein possessing the requisite p.Gly180 residue expressed by the c.538 C allele35. Our AlphaFold atomic structure modeling of ABCC11 (Supplementary Fig. S1) shows that such wild type expression forms a transmembrane functional conduit pore through the polypeptide, arranged from 12 transmembrane alpha helices, translocating glutathione conjugates into apocrine cell vesicles. In contrast, in the non-malodor phenotype29,51 the non-synonymous missense variant c.538 C>T allele of SNP rs17822931 encodes loss-of-function mutant ABCC11 transporter protein with p.180Gly > Arg substitution8,10,11,20,34,35 (Supplementary Fig. S1) which is trafficked for proteasome degradation.
Statement on plasticity of human ABCC11-microbiome relationships
It must be emphasized that the authors of the present paper are sensitive to deploying the most appropriate and responsible use of population descriptors. Building on UNESCO’s ethics declarations and meta-analysis discussions in the literature52, we ascribe the 3M3SH/osmidrosis phenotype of ABCC11 haplotypes as a meta-organism symbiotic mutualism phenomenon that is plastically influenced by environmental factors across time throughout geosocial locations within a common denominator of the Homo sapiens genome. We articulate this in the light of population descriptors in evolutionary genomics52 as being potentially misinterpreted by rogue social media sensationalizing and in irresponsible training of artificial intelligence (AI) and machine-learning models, with nuances potentially perverted to fit prejudicial agendas. To be clear, our position is that C and T alleles of ABCC11 haplotypes steer skin physiology niche conditions in a mutualistic coexistence with particular microbes as shaped by the environment, within the human genome backdrop of Homo sapiens as a global single species.
Concluding remarks
It is concluded that human ABCC11 gene C alleles, which encode apocrine cell wild type functional transporters of S-glutathione conjugates, are correlated with co-occurrence of axillary skin select commensal Staphylococcus hominis thiol conjugate bioconversion genomics crucial for osmidrosis; and furthermore, this pattern of meta-organism mutualism is rescinded by host T allele non-synonymous SNP rs17822931 that encodes mutant non-functional ABCC11 transporters. This relationship implies that human apocrine thiol conjugate metabolites transported by wild type ABCC11 shape axillary skin niche conditions as favorable to harboring familial microbiome communities of thiol osmidrosis. The present study provides a gateway for future projects exploring existential/evolutionary understandings of Homo sapiens, as shaped by (i) environmental selective advantages that influence migration-associated endogamous and exogamous genomics of population ancestry; (ii) climate-related health fitness; and (iii) olfactory-driven pheromone/bacterial odor influences on behaviors such as community cultural acceptance/fidelity and/or mate selection7,17,38.
Methods
Axillary skin microbiome sampling
The axillary skin surface microbiomes of each human host subject (≥ 18 y.o., N = 25, 13 males and 12 females) were examined by collecting and isolating independent sterile skin swabs of the right and left axillae53. Before sampling, subjects refrained from using deodorant or antiperspirants for 3 days, bathing for 24 h, and antibiotic use for 30 days. Samples swabbed from each subject’s left and right axillae were simultaneously and independently processed in parallel; DNA extraction, purification, and sequencing incorporated N = 50 axilla samples (25 subjects x 2 axillae per subject). Subsequent downstream data analyses treated each of the 50 axillae as a separate sample. Still, as microbiome composition from the same subject is likely to be more similar compared to samples from different individuals, we used paired analysis methods or mixed-effects models, when appropriate, where right or left axillae are treated as a random effect, allowing us to account for the fact that the two samples from each subject are not independent. Microbiota DNA was isolated using the DNAGenotek OMNIgene system (DNAGenotek, Ontario, Canada) according to the manufacturer’s instructions, with total DNA extraction and purification using the QIAmp PowerFecal Pro DNA kit (Qiagen LLC, Germantown, MD USA) deploying SpeedVac concentration and bead beating.
Microbiome amplicons
For each armpit sample, 2 × 300 cycles QIAseq 16S rRNA gene primer kit panels (Qiagen) were deployed to separately target hypervariable region V1V2 and region V3V4 of 16S rRNA gene separately. For each of the 50 axillary samples the pooled libraries from PCR products of both primer panels were then sequenced in one run using the Illumina platform with MiSeq41,54 software (v4.1.0; https://www.illumina.com), thereby generating data spanning V1-V4. Sequenced amplicon denoising was executed using the R package Divisive Amplicon Denoising Algorithm 2 (DADA2)54 pipeline (version 1.16; https://benjjneb.github.io/dada2/index.html) as described by us6. Briefly, paired-end raw reads were inspected and quality filtered, allowing a maximum expected error of 1 base per sequence. Forward and reverse reads were truncated at 250 bases, and the primer sequence was removed. High-quality forward and reverse readings were merged, chimeras were removed using the consensus method, and a table of amplicon sequence variants (ASV) was created. The taxonomy assignment was performed using the RDP database55,56 with RDP taxonomic training dataset 18 (https://zenodo.org/records/4310151). The resulting tables were converted into a phyloseq object in R for downstream analyses (phyloseq57 release 3.19; 10.18129/B9.bioc.phyloseq). ASVs assigned to eukaryotes, mitochondria, and chloroplasts were removed from the dataset, and data were rarefied57,58 to the minimum library size using R package microbiome (https://microbiome.github.io/tutorials/Installation.html) (Supplementary Information Table S1). After rarefaction at 85,486 sequences per sample, all samples exhibited coverage > 99% and thus were considered representative of the microbial community. The final working dataset of DADA2 sequences comprised abundances for 9324 ASVs that were used for subsequent downstream analyses.
Bioinformatic and statistical multivariate analyses
Prokaryotic metagenome predictions were generated from the pool of the 9324 ASVs using the Phylogenetic Investigation of Communities by Reconstruction of Unobserved States-2 (PICRUSt2) pipeline59 (https://github.com/picrust/picrust2.git). The PICRUSt2 output dataset yielded abundances for 2332 Enzyme Classification (EC) numbers, 7734 KEGG orthologs (KO), and MetaCyc metabolic features60 (https://metacyc.org) parsed into 3670 biochemical reactions mapped into 432 pathways. Based on these PICRUSt2 results, enzymes and metabolic pathways with attending intermediary metabolism organic compounds were deconstructed as being germane and salient to microbial odor production7–13,16–18,20.
Beta diversity differences among samples were measured by Permutational Multivariate ANOVA (PERMANOVA) analysis using the adonis function from the vegan R package61 (https://github.com/vegandevs/vegan). The dataset with 50 samples was stratified in two blocks, each comprising 25 samples from the left and right armpits. The stratified dataset was analyzed as a blocking factor to account for within-subject variation while analyzing the between-subject differences. PCoA ordination was deployed with the Bray-Curtis dissimilarity distance analysis within the R package phyloseq57. A network analysis was carried out using the SparCC (Sparse Correlations for Compositional data) method62 embedded in the microeco R package (v1.9.0; https://cran.r-project.org/web/packages/microeco/index.html) to test for co-occurrence patterns between microbes. For contrasting the abundance of individual microbes within the dataset, the total read counts were first log-transformed, and differences in means were tested by mixed-effects model analysis of variance followed by the estimated marginal means using the lme4 package in R (ver. 1.1–35.5; https://github.com/lme4/lme4). Individual microbes were specified as the dependent variable. CC, CT, and TT alleles were defined as the fixed effect, and the left and right armpit were specified as the random effect. We computed the estimated marginal means using the function emmeans within the lme4 R package and adjusted the p-values using the Bonferroni correction to interpret the model results.
Statistical multivariate analyses, data visualizations, and pedigree heatmaps of taxa and enzyme distributions were conducted using phyloseq57, Morpheus63 (https://software.broadinstitute.org/morpheus), and MicrobiomeAnalyst 2.0 modules64 (https://www.microbiomeanalyst.ca/MicrobiomeAnalyst/home.xhtml). Organic molecules’ nomenclature and structures were obtained from PubChem65 (10.1093/nar/gkac956). The above listed R packages were run using R software66 for macOS and Windows (ver 4.2.2–4.4.2; https://www.r-project.org ).
Nanopore operon sequencing prevalences
While Illumina rarefied ASV data were used to quantify and analyze relative abundances at the level of genus, the positive identification of taxon prevalences at the level of species was confirmed by sequencing the near-entire rrn operon of bacteria (4.5 kb reads encompassing the 16S-ITS-23S genes) using nanopore sequencing (Oxford Nanopore Technologies, New York). Library preparation, nanopore long-read DNA sequencing, and RESCUE pipeline bioinformatics (https://github.com/josephpetrone/RESCUE) were executed as previously described by us67.
Human ABCC11 SNP genotyping of cohorts
Human genomic DNA was extracted and analyzed from saliva samples collected with the Oragene-DISCOVER Kit (OGR-500; DNA Genotek, Ontario Canada). Genotyping68 examined alleles at 16q12.1 relating to missense variant ABCC11 single nucleotide polymorphism SNP rs17822931 (NM_001370497.1(ABCC11):c.538 C > T (p.Gly180Arg)). The PSQ HS 96 Pyromark pyrosequencing system (Qiagen) was deployed for the allele analyses. In the present study, alleles are reported to represent the forward orientation, per standard convention of NCBI dbSNP35. The ensuing metadata represented subject cohorts representing allele combinations of (i) homozygous CC; (ii) heterozygous CT; (iii) homozygous TT; and (iv) and a merged cohort denoted “[CC + CT]” which possessed at least one C on any allele.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
Acknowledgment. We thank Kent Stevens for inspiring insights.
Author contributions
B.R.S. conceived and designed the study, performed microbiome and human genomics data analyses, wrote the main manuscript and prepared all figures. L.F.W.R. contributed to microbiome data compilation and analyses. Both authors read and approved the manuscript.
Data availability
Data availability. The datasets supporting the conclusions of this article are available in the NCBI Sequence Read Archive (SRA) repository under BioProject Accession RJNA1077601 (https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1077601).
Declarations
Competing interests
The authors declare no competing interests.
Ethics declaration of IRB approval for human subjects
The project protocol was approved as a non‑intervention observational study of samples obtained from human subjects by the Institutional Review Board of the University of Florida, in accordance with the guidelines and regulations of the Declaration of Helsinki. Signed informed consent to participate and consent to publish deidentified data was obtained from all subjects.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Grieneisen, L. et al. Gut microbiome heritability is nearly universal but environmentally contingent. Science. 373, 181–186. 10.1126/science.aba5483 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bang, C. et al. Metaorganisms in extreme environments: do microbes play a role in organismal adaptation? Zool. (Jena). 127, 1–19. 10.1016/j.zool.2018.02.004 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Runge, S. & Rosshart, S. P. The mammalian metaorganism: a holistic view on how microbes of all kingdoms and niches shape local and systemic immunity. Front. Immunol.12, 702378. 10.3389/fimmu.2021.702378 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nogales, J. & Garmendia, J. Bacterial metabolism and pathogenesis intimate intertwining: time for metabolic modelling to come into action. Microb. Biotechnol.15, 95–102. 10.1111/1751-7915.13942 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stevens, B. R., Pepine, C. J., Richards, E. M., Kim, S. & Raizada, M. K. Depressive hypertension: a proposed human endotype of brain/gut microbiome dysbiosis. Am. Heart J.239, 27–37. 10.1016/j.ahj.2021.05.002 (2021). [DOI] [PubMed] [Google Scholar]
- 6.Stevens, B. R. et al. Depression phenotype identified by using single nucleotide exact amplicon sequence variants of the human gut microbiome. Mol. Psychiatry. 26, 4277–4287. 10.1038/s41380-020-0652-5 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harker, M. et al. Functional characterisation of a SNP in the ABCC11 allele - effects on axillary skin metabolism, odour generation and associated behaviours. J. Dermatol. Sci.73, 23–30. 10.1016/j.jdermsci.2013.08.016 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Natsch, A. & Emter, R. The specific biochemistry of human axilla odour formation viewed in an evolutionary context. Philos. Trans. R Soc. Lond. B Biol. Sci.375, 20190269. 10.1098/rstb.2019.0269 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baumann, T. et al. Glutathione-conjugated sulfanylalkanols are substrates for ABCC11 and gamma-glutamyl transferase 1: a potential new pathway for the formation of odorant precursors in the apocrine sweat gland. Exp. Dermatol.23, 247–252. 10.1111/exd.12354 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rudden, M. et al. The molecular basis of thioalcohol production in human body odour. Sci. Rep.10, 12500. 10.1038/s41598-020-68860-z (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Minhas, G. S. et al. Structural basis of malodour precursor transport in the human axilla. Elife. 7. 10.7554/eLife.34995 (2018). [DOI] [PMC free article] [PubMed]
- 12.Bawdon, D., Cox, D. S., Ashford, D., James, A. G. & Thomas, G. H. Identification of axillary Staphylococcus sp. involved in the production of the malodorous thioalcohol 3-methyl-3-sufanylhexan-1-ol. FEMS Microbiol. Lett.36210.1093/femsle/fnv111 (2015). [DOI] [PubMed]
- 13.Troccaz, M. et al. Mapping axillary microbiota responsible for body odours using a culture-independent approach. Microbiome. 3, 3. 10.1186/s40168-014-0064-3 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hasegawa, Y., Yabuki, M. & Matsukane, M. Identification of new odoriferous compounds in human axillary sweat. Chem. Biodivers.1, 2042–2050. 10.1002/cbdv.200490157 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Martin, A. et al. A functional ABCC11 allele is essential in the biochemical formation of human axillary odor. J. Invest. Dermatol.130, 529–540. 10.1038/jid.2009.254 (2010). [DOI] [PubMed] [Google Scholar]
- 16.Kutsuwada, Y. et al. Association of HLA-DPB1, NLRP10, OVOL1, and ABCC11 with the axillary microbiome in a Japanese population. J. Dermatol. Sci.105, 98–104. 10.1016/j.jdermsci.2022.01.003 (2022). [DOI] [PubMed] [Google Scholar]
- 17.Di Cicco, F. et al. Intrinsic and extrinsic factors affecting axillary odor variation. A comprehensive review. Physiol. Behav.270, 114307. 10.1016/j.physbeh.2023.114307 (2023). [DOI] [PubMed] [Google Scholar]
- 18.Lam, T. H. et al. Understanding the microbial basis of body odor in pre-pubescent children and teenagers. Microbiome. 6, 213. 10.1186/s40168-018-0588-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Preti, G. & Leyden, J. J. Genetic influences on human body odor: from genes to the axillae. J. Invest. Dermatol.130, 344–346. 10.1038/jid.2009.396 (2010). [DOI] [PubMed] [Google Scholar]
- 20.The_UniProt_Consortium. https://www.uniprot.org/uniprotkb/Q96J66/entry. Nucleic Acids Research 51, D523-D531, doi:UniProt: the Universal Protein Knowledgebase in (2023). 10.1093/nar/gkac1052 (2022). [DOI] [PMC free article] [PubMed]
- 21.Council, S. E. et al. Diversity and evolution of the primate skin microbiome. Proc. Biol. Sci.28310.1098/rspb.2015.2586 (2016). [DOI] [PMC free article] [PubMed]
- 22.Li, H. et al. Axillary fossaa microbial dysbiosis and its relationship with axillary osmidrosis patients. Microb. Pathog.. 173, 105886. 10.1016/j.micpath.2022.105886 (2022). [DOI] [PubMed] [Google Scholar]
- 23.Ohashi, J., Naka, I. & Tsuchiya, N. The impact of natural selection on an ABCC11 SNP determining earwax type. Mol. Biol. Evol.28, 849–857. 10.1093/molbev/msq264 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Nicholas-Haizelden, K., Murphy, B., Hoptroff, M. & Horsburgh, M. J. Bioprospecting the skin microbiome: advances in therapeutics and Personal Care products. Microorganisms. 1110.3390/microorganisms11081899 (2023). [DOI] [PMC free article] [PubMed]
- 25.Joglekar, P. et al. Integrated genomic and functional analyses of human skin-associated Staphylococcus reveal extensive inter- and intra-species diversity. Proc. Natl. Acad. Sci. U.S.A.120 (e2310585120). 10.1073/pnas.2310585120 (2023). [DOI] [PMC free article] [PubMed]
- 26.Yamada, A. et al. High expression of ATP-binding cassette transporter ABCC11 in breast tumors is associated with aggressive subtypes and low disease-free survival. Breast Cancer Res. Treat.137, 773–782. 10.1007/s10549-012-2398-5 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Toyoda, Y. & Ishikawa, T. Pharmacogenomics of human ABC transporter ABCC11 (MRP8): potential risk of breast cancer and chemotherapy failure. Anticancer Agents Med. Chem.10, 617–624. 10.2174/187152010794473975 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Toyoda, Y. et al. Earwax, osmidrosis, and breast cancer: why does one SNP (538G > A) in the human ABC transporter ABCC11 gene determine earwax type? FASEB J.23, 2001–2013. 10.1096/fj.09-129098 (2009). [DOI] [PubMed] [Google Scholar]
- 29.Ishikawa, T., Toyoda, Y., Yoshiura, K. & Niikawa, N. Pharmacogenetics of human ABC transporter ABCC11: new insights into apocrine gland growth and metabolite secretion. Front. Genet.3, 306. 10.3389/fgene.2012.00306 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nielsen, M. et al. (ed K., M.) Current state of SLC and ABC transporters in the skin and their relation to sweat metabolites and skin diseases. Proteomes9. 10.3390/proteomes9020023 (2021). [DOI] [PMC free article] [PubMed]
- 31.Uhlen, M. et al. Proteomics. Tissue-based map of the human proteome. Data available at https://v23.proteinatlas.org. Science. 347, 1260419. 10.1126/science.1260419 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Karlsson, M. et al. A single-cell type transcriptomics map of human tissues. Data available from https://v23.proteinatlas.org. Sci Adv 7, 10.1126/sciadv.abh2169 (2021). [DOI] [PMC free article] [PubMed]
- 33.Toyoda, Y., Gomi, T., Nakagawa, H., Nagakura, M. & Ishikawa, T. Diagnosis of human axillary osmidrosis by genotyping of the human ABCC11 gene: clinical practice and basic scientific evidence. Biomed Res. Int. 7670483. 10.1155/2016/7670483 (2016). [DOI] [PMC free article] [PubMed]
- 34.Toyoda, Y. et al. Clinical and molecular evidence of ABCC11 protein expression in Axillary Apocrine glands of patients with Axillary Osmidrosis. Int. J. Mol. Sci.1810.3390/ijms18020417 (2017). [DOI] [PMC free article] [PubMed]
- 35.NCBI. dbSNP [Internet]. Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information; 1998 – [cited 2024 Jan 05]. https://www.ncbi.nlm.nih.gov/snp/rs17822931 (2024).
- 36.Yoshiura, K. et al. A SNP in the ABCC11 gene is the determinant of human earwax type. Nat. Genet.38, 324–330. 10.1038/ng1733 (2006). [DOI] [PubMed] [Google Scholar]
- 37.Nakano, M., Miwa, N., Hirano, A., Yoshiura, K. & Niikawa, N. A strong association of axillary osmidrosis with the wet earwax type determined by genotyping of the ABCC11 gene. BMC Genet.10. 10.1186/1471-2156-10-42 (2009). [DOI] [PMC free article] [PubMed]
- 38.Loos, H. M. et al. Past, Present, and Future of Human Chemical Communication Research. Perspect. Psychol. Sci.17456916231188147. 10.1177/17456916231188147 (2023). [DOI] [PMC free article] [PubMed]
- 39.Du, H., Ding, S., Gao, L., Zeng, J. & Lu, J. Microecological investigation and comparison of two clinical methods to evaluate axillary osmidrosis. Mol. Med. Rep.22, 4207–4212. 10.3892/mmr.2020.11528 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Onwuliri, V., Agbakoba, N. R. & Anukam, K. C. Topical cream containing live lactobacilli decreases malodor-producing bacteria and downregulates genes encoding PLP-dependent enzymes on the axillary skin microbiome of healthy adult nigerians. J. Cosmet. Dermatol.20, 2989–2998. 10.1111/jocd.13949 (2021). [DOI] [PubMed] [Google Scholar]
- 41.Fadrosh, D. W. et al. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome. 2, 6. 10.1186/2049-2618-2-6 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Prokop-Prigge, K. A. et al. Ethnic/racial and genetic influences on cerumen odorant profiles. J. Chem. Ecol.41, 67–74. 10.1007/s10886-014-0533-y (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.James, A. G., Austin, C. J., Cox, D. S., Taylor, D. & Calvert, R. Microbiological and biochemical origins of human axillary odour. FEMS Microbiol. Ecol.83, 527–540. 10.1111/1574-6941.12054 (2013). [DOI] [PubMed] [Google Scholar]
- 44.Ceccarani, C., Severgnini, M. A. & comparison between Greengenes, S. I. L. V. A. RDP, and NCBI reference databases in four published microbiota datasets. bioRxiv, 2023.2004.2012.535864, 10.1101/2023.04.12.535864. https://www.biorxiv.org/content/biorxiv/early/2023/04/13/2023.04.12.535864.full.pdf (2023).
- 45.Dixit, K. et al. Benchmarking of 16S rRNA gene databases using known strain sequences. Bioinformation. 17, 377–391. 10.6026/97320630017377 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Szpak, M. et al. FineMAV: prioritizing candidate genetic variants driving local adaptations in human populations. Genome Biol.19, 5. 10.1186/s13059-017-1380-2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Toyoda, Y. et al. MRP class of human ATP binding cassette (ABC) transporters: historical background and new research directions. Xenobiotica. 38, 833–862. 10.1080/00498250701883514 (2008). [DOI] [PubMed] [Google Scholar]
- 48.Gunther, T. et al. Population genomics of Mesolithic Scandinavia: investigating early postglacial migration routes and high-latitude adaptation. PLoS Biol.16, e2003703. 10.1371/journal.pbio.2003703 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Isshiki, M. et al. Admixture with indigenous people helps local adaptation: admixture-enabled selection in polynesians. BMC Ecol. Evol.21, 179. 10.1186/s12862-021-01900-y (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Severn, M. M. et al. The ubiquitous human skin Commensal Staphylococcus hominis protects against opportunistic pathogens. mBio. 13, e0093022. 10.1128/mbio.00930-22 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arlanov, R. et al. Functional characterization of common protein variants in the efflux transporter ABCC11 and identification of T546M as functionally damaging variant. Pharmacogenomics J.16, 193–201. 10.1038/tpj.2015.27 (2016). [DOI] [PubMed] [Google Scholar]
- 52.Mauro, M. et al. A scoping review of guidelines for the use of race, ethnicity, and ancestry reveals widespread consensus but also points of ongoing disagreement. Am. J. Hum. Genet.109, 2110–2125. 10.1016/j.ajhg.2022.11.001 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Callewaert, C. et al. Characterization of Staphylococcus and Corynebacterium clusters in the human axillary region. PLoS One. 8, e70538. 10.1371/journal.pone.0070538 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Callahan, B. J. et al. DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods. 13, 581–583. 10.1038/nmeth.3869 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cole, J. R. et al. Ribosomal database project: data and tools for high throughput rRNA analysis. Nucleic Acids Res.42, D633–642. 10.1093/nar/gkt1244 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Edgar, R. Taxonomy annotation and guide tree errors in 16S rRNA databases. PeerJ. 6, e5030. 10.7717/peerj.5030 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McMurdie, P. J. & Holmes, S. Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 8, e61217. 10.1371/journal.pone.0061217 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lemos, L. N., Fulthorpe, R. R., Triplett, E. W. & Roesch, L. F. Rethinking microbial diversity analysis in the high throughput sequencing era. J. Microbiol. Methods. 86, 42–51. 10.1016/j.mimet.2011.03.014 (2011). [DOI] [PubMed] [Google Scholar]
- 59.Douglas, G. M. et al. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol.38, 685–688. 10.1038/s41587-020-0548-6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Caspi, R. et al. The MetaCyc database of metabolic pathways and enzymes - a 2019 update. Nucleic Acids Res.48, D445–D453. 10.1093/nar/gkz862 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Oksanen, J. et al. vegan community ecology package version 2.6-2 April (2022). https://www.researchgate.net/publication/360782912_vegan_community_ecology_package_version_26-2_April_2022. (2022).
- 62.Friedman, J. & Alm, E. J. Inferring correlation networks from genomic survey data. PLoS Comput. Biol.8, e1002687. 10.1371/journal.pcbi.1002687 (2012). https://www.ncbi.nlm.nih.gov/pubmed/23028285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Broad_Institute. Morpheus Interactive heat maps using morpheus.js and htmlwidgets. https://software.broadinstitute.org/morpheus (2017).
- 64.Lu, Y. et al. MicrobiomeAnalyst 2.0: comprehensive statistical, functional and integrative analysis of microbiome data. Nucleic Acids Res.51, W310–W318. 10.1093/nar/gkad407 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim, S. et al. PubChem 2023 update. Nucleic Acids Res.51, D1373–D1380. 10.1093/nar/gkac956 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.R_Core_Team., R. A language and environment for statistical computing. Foundation for Statistical Computing, Vienna, Austria.. https://www.R-project.org/ (2024).
- 67.Petrone, J. R. et al. RESCUE: a validated Nanopore pipeline to classify bacteria through long-read, 16S-ITS-23S rRNA sequencing. Front. Microbiol.14, 1201064. 10.3389/fmicb.2023.1201064 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Langaee, T. & Ronaghi, M. Genetic variation analyses by Pyrosequencing. Mutat. Res.573, 96–102. 10.1016/j.mrfmmm.2004.07.023 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data availability. The datasets supporting the conclusions of this article are available in the NCBI Sequence Read Archive (SRA) repository under BioProject Accession RJNA1077601 (https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1077601).