

Abstract
Alveolar capillary barrier disruption induces local edema and inflammation that impairs pulmonary function and promotes alveolar destruction in COPD. This study aimed to determine how cigarette smoke modulated the serine-threonine phosphatase protein phosphatase 2 A (PP2A) to alter the barrier function of human lung microvascular endothelial cells (HLMVECs). Cigarette smoke exposure lowered overall PP2A activity and enhanced endothelial permeability in HLMVECs. However, directly decreasing PP2A activity with Fostriecin significantly reduced endothelial cell permeability. Protein fractionation studies determined that cigarette smoke diminished cytosolic PP2A activity but increased membrane and cytoskeletal activity. These changes coincided with the translocation of PP2A to the membrane, which reduced occludin phosphorylation in the membrane. Cigarette smoke decreased protein tyrosine phosphatase 1B (PTP1B) activity, a PP2A activator which also counters calcium intracellular influx. The decrease in PTP1B activity correlated with reduced calcium efflux in endothelial cells and these changes in calcium flux regulated PP2A activity. Indeed, culturing endothelial cells in low calcium medium prevented the decrease in cytosolic PP2A activity mediated by cigarette smoke. Together, these findings outline a mechanism whereby cigarette smoke acts via calcium to traffic PP2A from the cytosol to the membrane where it dephosphorylates occludin to increase endothelial cell permeability.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-77776-x.
Keywords: Chronic obstructive pulmonary disease, Inflammation, Protein phosphatase 2A, Endothelium, Permeability, And cigarette smoke
Subject terms: Cell biology, Molecular biology, Physiology, Respiratory tract diseases
Introduction
The lung is a multicellular structure of bifurcating airways that transmits air to the alveolus to deliver oxygen to and eliminate carbon dioxide from the circulation. The integrity of the alveolar unit is critical and disruption of intercellular connections within the alveolus can promote destructive inflammation or local flooding that impairs gas exchange1. The alveolar endothelium is a semipermeable barrier, which fuses with the alveolar epithelial basement membrane to facilitate gas transfer by minimizing the distance between the red blood cell in the capillary and the airspace within the alveolus2. The coordinated actions of the cytoskeleton and the adherens complex maintain endothelial function to orchestrate the proper flow of nutrients to preserve normal lung action and avert the burden of lung dysfunction3. It also prevents the deregulated flow of fluids and cells that would interfere with normal gas exchange4.
In response to cellular stressors, kinases phosphorylate proteins within the cytoskeleton and adherens complex to alter their activity5. COPD is one of the leading causes of death worldwide and several studies show that cigarette smoke promotes lung disease development by altering kinase activity and enhancing endothelial cell permeability6. Indeed, glycogen synthase kinase 3 beta (GSK-3β) stimulation by cigarette smoke activated histone deacetylase 6 (HDAC6) leading to α-tubulin deacetylation and microtubule disassembly7. Conversely, the oxidants present in cigarette smoke decreased the activity of RhoA and Focal adhesion kinase (FAK)8, which play a critical role in the coordinated assembly of focal adhesion complexes, adherens junctions (AJ), and cortical F-actin fibers9. Likewise, cigarette smoke-derived oxidants increase endothelial cell permeability by inducing the production of local ceramides10. These ceramides modulate cell permeability by blocking SET1, a protein phosphatase 2 A (PP2A) inhibitor or by altering the ceramide content and biophysical properties of the plasma membrane11.
Phosphatases counter kinases to return the system to homeostasis and prevent prolonged changes in cellular permeability. PP2A is the primary serine, threonine phosphatase of eukaryotic cells. It is ubiquitously expressed and accounts for 0.3–1% of the total cellular protein in the mammalian cell12. PP2A is a heterotrimeric protein comprised of an A structural, B regulatory and C catalytic subunit. On demand, B regulatory subunits can be interchanged to alter the substrate target of the protein complex13. PP2A exerts complex effects on lung endothelial cell permeability with studies showing both a positive and negative effect14,15. We previously determined that acute smoke exposure activates PP2A within human airway epithelial cells16 but chronic exposure decreased PP2A activity17. Here, we sought to determine how cigarette smoke influences PP2A activity within key intracellular compartments in human microvascular lung endothelial cells. We also assessed how cigarette smoke modulated PP2A activity to alter endothelial cell permeability and neutrophil adhesion. We found that exposure to cigarette smoke extract (CSE) acted via a calcium dependent mechanism to induce the translocation of PP2A to the cytoskeletal and cell membrane compartments. This translocation correlated with a decrease in occludin phosphorylation and an increase in cell permeability. Treating the endothelial cells with a PP2A inhibitor Fostriecin decreased cell permeability while neutrophil adhesion trended higher in PP2AC silenced endothelial cells. Together, these findings delineate a novel mechanism by which cigarette smoke alters endothelial cell permeability and inflammation by modulating the intracellular trafficking of PP2A.
Results
Cigarette smoke extract caused compartment-specific changes in PP2A activity. To determine if CSE administration altered overall PP2A activity within HLMVECs over 24-hours, PP2A-specific phosphatase activity assays were performed. These studies determined that overall PP2A activity decreased within these cells by 6 h and remained decreased for at least 24 h (Fig. 1A). PP2A is highly regulated and its activity can vary within intracellular compartments18. Given this, the effect of CSE on PP2A activity within specific cellular compartments was assessed. CSE significantly decreased PP2A activity within the cytosol at the 24-hour timepoint while no change in PP2A activity occurred within the nuclear compartment (Fig. 1B). In contrast, CSE significantly increased PP2A activity within both the membrane and cytoskeletal protein fractions of HLMVECs (Fig. 1B). Of interest, the increase in membrane PP2A activity was associated with a translocation of PP2A to the membrane compartment (Fig. 1C).
Fig. 1.

Cigarette smoke extract alters PP2A activity and distribution within endothelial cells. (A) Human microvascular lung endothelial cells (HLMVEC) were grown to 80% confluence in 6-well plates and treated with 5% CSE for 10 min, 30 min, 1, 6, 12 and 24 h. PP2A activity assays were conducted on lysate protein isolated from the cells at baseline and after 5% CSE treatment. (B) PP2A activity assays were conducted on cytosolic, nuclear, cytoskeletal and membrane protein fractions of HLMVECs treated with and without 5% CSE for 24 h. (C) Immunoblots for the catalytic subunit of PP2A (PP2AC) were conducted on membrane fractions of HLMVECs treated with control media or 5% CSE for 24 h. Actin immunoblots were conducted on membrane fractions as a loading control.
PTP1B regulates PP2A activity in endothelial cells and cigarette smoke extract reduces PTP1B activity. Our research group previously showed that PTP1B is a critical regulator of PP2A activity within airway epithelial cells19 and neutral sphingomyelinase (NSMase) has been reported to regulate endothelial PP2A activity20. Thus, we silenced NSMase and PTP1B and measured the effects on PP2A in HLMVECs. Silencing NSMase did not alter PP2A activity while silencing PTP1B significantly reduced PP2A activation (Fig. 2A). Since PP2A activity was PTP1B dependent, we assessed how CSE affected PTP1B activity in these cells. CSE treatment significantly reduced PTP1B activity within six hours of treatment and caused a trend reduction in PTP1B activity at 24 h (Fig. 2B) paralleling the CSE-mediated changes in PP2A activation.
Fig. 2.

.PTP1B regulates PP2A activity in endothelial cells and cigarette smoke extract reduces PTP1B activity (A) PP2A activity assays were conducted on human lung microvascular endothelial cells grown to 70% confluence in 6-well plates and then treated with control, NSMase or PTP1B siRNA for 24 h. (B) PTP1B activity assays were conducted on human microvascular lung endothelial cells (HLMVEC) grown to 80% confluence in 6-well plates and treated with 5% CSE for 6–24 h.
The effects of cigarette smoke extract on PP2A are calcium dependent
Nicotine stimulates nicotinic acetylcholine receptors on the cell surface to mediate an influx of calcium into cells21. Intracellular calcium levels regulate PP2A activation and intracellular distribution22,23. Thus, we sought to determine whether the changes in PP2A activity and distribution within CSE-treated HLMVECs were calcium-dependent. The administration of CSE to these cells significantly reduced calcium efflux (Fig. 3A), which is consistent with nicotine’s known effects on calcium influx24. This decrease in calcium efflux was reproducible with every concentration of CSE tested. CSE also required calcium to mediate the decrease in overall PP2A activity in these cells. When cells were cultured in low calcium media (< 0.05 mM) and then treated with CSE in low calcium media, no change in PP2A activity was noted (Fig. 3B). However, switching from low calcium media to CSE with high calcium media (1.0 mM) restored the effects of CSE on PP2A activity.
Fig. 3.

Cigarette smoke extract decreases calcium efflux and regulates PP2A activity in a calcium-dependent manner. (A) Human microvascular lung endothelial cells (HLMVEC) were grown to 80% confluence in 24-well plates and treated with control media or media with 1, 2, 5 or 10% CSE for 10 min. A calcium release assay was conducted on media collected from these cells. (B) PP2A activity assays were conducted on human microvascular lung endothelial cells (HLMVEC) grown to 80% confluence in 6-well plates and were treated with low calcium media that was switched to high calcium media or high calcium media that was switched to low calcium media after 5% CSE treatment.
The effects of CSE and PP2A on cell permeability.
In agreement with other studies, we found that CSE significantly increased endothelial cell permeability at all timepoints tested (Fig. 4A). This increase in permeability correlated with decreased PP2A activity in the cytosol and increased PP2A activity within the cell membrane. To determine the effects of PP2A on HLMVEC permeability, cells were treated with the PP2A inhibitor Fostriecin. Inhibiting PP2A significantly decreased endothelial cell permeability (Fig. 4B). The phosphorylation of threonine residues on the membrane protein occludin plays an important role in tight junction assembly25. Dephosphorylating occludin leads to the disassembly of these tight junctions leading to increased cell permeability26. PP2A is known to bind and dephosphorylate occludin. Coincident with PP2A’s translocation to the membrane, CSE treatment reduced occludin phosphorylation in these cells (Fig. 4C).
Fig. 4.

Cigarette smoke extract and PP2A inhibition alter endothelial cell permeability and occluding phosphorylation. (A) FITC permeability assays were conducted on human microvascular endothelial cells monolayers grown to 100% confluence on 3-µm pore collagen-coated PTFE membranes that were treated with 5% CSE for 2, 4, 6, 18 and 24 h. (B) FITC permeability assays were conducted on human microvascular endothelial cell monolayers grown to 100% confluence on 3-µm pore collagen-coated PTFE membranes that were treated with 1-µM Fostriecin for 24 h. (C) Occludin protein was immunoprecipitated from the endothelial cell lysates using a specific antibody. Immunoblots for p-threonine and total occludin were conducted on the immunoprecipitated protein.
PP2A alters neutrophil adhesion to HLMVECs
The trafficking of neutrophils to the alveolus during cigarette smoke exposure plays a central role in emphysema formation27. A key first step in this process is the adhesion of neutrophils to the endothelium which subsequently leads to their translocation to the alveolar compartment. As noted above, CSE treatment decreased overall PP2A activity within the endothelium. To test how this modulated neutrophil adhesion, we silenced PP2AC in these cells and then measured the effects on neutrophil adhesion. The loss of PP2AC expression, by itself, induced a trend increase in the adhesion of neutrophils to the endothelium (Fig. 5) though this did not reach statistical significance. The level of neutrophil adhesion in PP2AC silenced cells was comparable to the levels observed in CSE-treated cells.
Fig. 5.
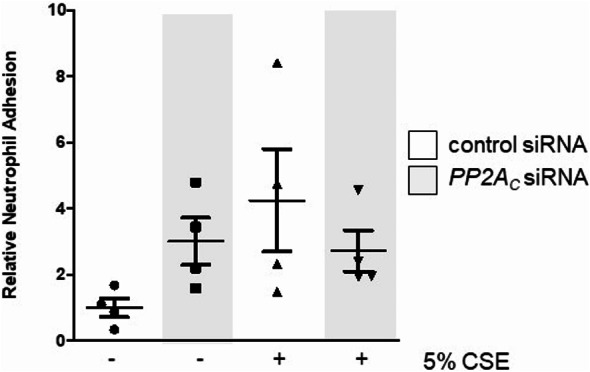
PP2ACsilencing increases neutrophil adhesion in lung endothelial cells. Human lung microvascular endothelial cells were grown to 70% confluence in 6-well plates and then treated with control or PP2AC siRNA for 24 h. The wells were incubated with Calcein-labeled neutrophils and the fluorescence intensity of each well was then measured.
Discussion
This study shows that CSE enhances intracellular calcium levels to effectuate changes in PP2A activity and distribution within human lung microvascular endothelial cells. CSE acted in a calcium dependent manner to induce the translocation of PP2A to the cell membrane resulting in the gain of activity within the cytoskeleton and membrane. The increase in PP2A activity within the membrane coincided with the dephosphorylation of the tight junction protein occludin and the increase in cell permeability (Fig. 6). Lastly, CSE decreased overall PP2A activity in these cells and silencing PP2AC induced a trend increase in neutrophil adhesion to the endothelium. Together, these findings suggest that the smoke-mediated modulation of PP2A activity and distribution triggers alveolar injury by altering endothelial permeability and inflammation.
Fig. 6.

Schema on the effects of cigarette smoke on PP2A translocation and occludin dephosphorylation.
Endothelial permeability is a key determinant of lung function and studies show that PP2A exerts critical effects on cellular permeability28,29. PP2A associates with microtubules and inhibiting PP2A with okadaic acid increased tau phosphorylation and disassembly of the microtubular network30. This impairs microtubular function in the endothelium leading to an increase in permeability31. Enhancing the expression of PP2A prevented microtubule dissolution and preserved vascular integrity in human pulmonary artery endothelial cells15. Similarly, in brain endothelial cells, the PP2A inhibitor Semaphorin3A increased VE-cadherin serine phosphorylation. This caused the internalization of VE-cadherin and the destabilization of intercellular junctions32. Thus, these results demonstrate that PP2A preserves the barrier function of the endothelium.
In contrast, several reports indicate that PP2A can increase cell permeability by affecting the phosphorylation status of the tight junction protein occludin. Phosphorylated occludin localizes to tight junctions but when occludin is dephosphorylated, it redistributes to the basolateral membrane and cytoplasmic vesicles33. Dephosphorylation of occludin in both epithelial and endothelial cells alters tight junctions at the cell surface to increase cell permeability34. In alveolar epithelial cells, hypoxia induces superoxide production which increases cell permeability by acting through PP2A to dephosphorylate and internalize occludin35. Likewise, cigarette smoke stimulates NADPH oxidase36 which activates PP2A in microvascular endothelial cells14. Inhibition of PP2A with Calyculin A prevented occludin dephosphorylation, the redistribution of tight junction proteins and the increase in epithelial permeability in these cells14. It is important to note that under basal conditions the PP2A inhibitor Fostriecin decreased endothelial cell permeability in our studies. Thus, the decrease in PP2A activity mediated by CSE cannot, by itself, explain the CSE-mediated increase in permeability. Instead, our findings indicate that CSE increased PP2A activity within the plasma membrane leading to occludin dephosphorylation and increased cellular permeability. This demonstrates the importance of site-specific changes in PP2A activity within the endothelium.
The results from these studies demonstrate that CSE required calcium to mediate intracellular changes in PP2A activity. Calcium binding plays an important role in PP2A holoenzyme assembly and substrate activity22. Moreover, calcium influx influences the activity of multiple kinases that phosphorylate PP2A subunits to alter the enzyme complex’s activity and intracellular distribution23. Endothelial cells express several nicotinic acetylcholine receptors (nAchR)37. However, α7nAchR plays the dominant role in nicotinic signaling in these cells and this receptor is more permeable to calcium than monovalent cations38,39. Indeed, silencing α7nAchR prevented nicotine from elevating intracellular calcium levels in the endothelium38. Conversely, stimulating α7nAchR inhibits PTP1B activity40 and this is important since PTP1B counters calcium influx into the cell41. Thus, it is conceivable that the nicotine present in CSE acted via α7nAchR to inhibit PTP1B thereby decreasing overall PP2A activity and enhancing calcium influx that redistributes PP2A to the cell membrane. Of note, our study examined overall calcium flux in response to CSE. Future studies will need to address the specific role of Sarco/endoplasmic reticulum calcium ATPase (SERCA) which regulates intracellular calcium stores and levels within the endothelium.
In agreement with our findings, researchers showed that hydrogen peroxide-mediated changes in PP2A activity in colonic epithelial cells were calcium-dependent42. Furthermore, this group showed that depleting calcium inhibited PP2A, augmented occludin phosphorylation, and accelerated the assembly of tight junctions to preserve cellular resistance43. We demonstrated that occludin was dephosphorylated on threonine residues upon CSE treatment. This is significant as the phosphorylation of occludin on threonine residues, but not on serine residues, is dramatically reduced during the disassembly of tight junctions43. The calcium influx mediated by CSE was associated with reduced overall PP2A activity but increased PP2A membrane localization, activity and occludin dephosphorylation. The dephosphorylation of occludin and endothelial permeability coincided with PP2A membrane translocation. Since PP2A regulates occludin phosphorylation, it is reasonable to assume that this translocation mediated the change in permeability. This could be demonstrated conclusively by inhibiting PP2A specifically within the membrane but there are no technical approaches that can do this at this time. Taken together, these findings indicate the smoke-mediated changes in intracellular calcium redistribute PP2A to the membrane where it dephosphorylates occludin to increase cell permeability.
The CSE induced changes in PP2A activity within the cellular compartments was most likely due to intracellular trafficking of this enzyme complex. The precise mechanisms by which CSE mediates these effects remain to be determined. Interestingly, Wnt3a induced the translocation of PP2A to the membrane and cytoskeleton and stimulated its binding to the phosphoprotein Disheveled 2 (Dvl2)44. Nicotine activates the Wnt3a pathway45. Therefore, nicotine present in CSE may act via Wnt signaling to mediate PP2A membrane shuttling. It is important to note that B regulatory subunits target PP2A holoenzymes to specific cellular compartments and determine the substrate specificity of the PP2A enzyme complex. The B55α, B55β, and B55δ target activity to the cytosol while B55γ is enriched in the cytoskeletal fraction46,47 and B56γ1 colocalizes to adhesion proteins on the cell membrane48. As discussed previously, B subunits are interchangeable and thus they can rapidly redistribute PP2A localization and activity within cells. Indeed, the dynamic nature of PP2A shuttling by B subunits plays a central role in regulating cellular division49. Calcium can regulate the activity and binding of specific B subunits22. Thus, it is conceivable that the changes in calcium concentration mediated by nicotine exposure in our study altered B subunit binding to promote PP2A trafficking to the cell membrane.
There are several limitations to this study. For one, we treated cells with CSE and we do not know the individual contributions of specific smoke components like nicotine, oxidants or acrolein on PP2A activity and cell permeability. However, since the changes in PP2A activity were calcium-dependent, we suspect that nicotine mediated these effects as it modulates intracellular calcium concentrations in these cells50. Secondly, endothelial cells express a broad range of nicotinic acetylcholine receptors so we do not know which receptor mediated the responses seen in this study. We speculate that it was most likely α7nAchR as it mediates calcium influx and is the predominant receptor in this cell51. Both the epithelium and endothelium contribute to permeability in vivo. Utilizing the in vitro system allowed us to examine the specific contribution of endothelial cells while an in vivo model would not. Future studies will need to address how these changes in PP2A translocation influence permeability in vivo. Lastly, we measured changes in enzymatic activity within distinct compartments; however, we did not assess whether these changes were associated with alteration in PP2A composition or post-translational modifications. Future studies are needed to address these questions to better understand the effects of cigarette smoke on endothelial cell integrity.
In summary, our findings show that CSE alters calcium concentrations within the endothelium to redistribute PP2A to the cell membrane where it increases cell permeability by dephosphorylating the adhesion protein occludin. These findings provide important new insights into the mechanisms by which cigarette smoke contributes to alveolar inflammation and lung dysfunction by impairing the barrier function of the endothelium. Future studies will need to address whether targeting PP2A activity within the endothelium could preserve endothelial resistance and prevent the exudation of fluid and cells that perpetuate alveolar injury and destruction.
Methods
Culture of human lung microvascular endothelial cells
Of note, all methods were carried out in accordance with relevant guidelines and regulations. Primary human lung microvascular endothelial cells (PromoCell Gmbh, Heidelberg, Germany) were grown to 70–80% confluence in endothelial cell growth media in 6-well plates. The human cells for research were obtained from tissue samples donated by informed consent from healthy volunteers or patients and/or their legal guardian. Cigarette smoke extract (CSE) was prepared as previously described52 and then added to the cells at concentrations of 1, 2, 5 and 10%. LDH assays (Cayman Chemical, Ann Arbor, MI) were conducted as per the manufacturer’s instructions to assess for cellular toxicity. PP2A activity was determined in the cells at time intervals following CSE administration using the Millipore PP2A activity assay (17–313; Millipore-Sigma, St. Louis, MO). To determine how CSE altered PP2A activity within specific cell compartments, endothelial cell protein was divided into cytosolic, nuclear, membrane and cytoskeletal fractions using the Millipore cell fractionation kit (Millipore-Sigma). PP2A activity was measured within each fraction after CSE treatment. PTP1B activity was assessed using the Millipore PTP1B activity assay (Millipore-Sigma). Cells were also transfected with PTP1B siRNA, neutral sphingomyelinase (NSMase) siRNA, PP2A siRNA or scrambled siRNA (Santa Cruz, Santa Cruz, CA, PTP1B- sc-36328, PP2A-C sc-43509, NSmase- sc-62655).
Calcium release assay and calcium switch studies. Primary human lung microvascular endothelial cells were grown to 80% confluence in 24-well plates and then treated with 0, 1, 2, 5 and 10% CSE for 10 min. The effects on intracellular calcium release were determined using a fluorometric calcium assay kit (Abcam, Cambridge, UK). To assess how changes in calcium mediated by CSE treatment influenced PP2A activity, human lung microvascular endothelial cells were treated with low calcium media that was switched to high calcium media or high calcium media that was switched to low calcium media after CSE treatment.
Cell permeability assays
Human microvascular endothelial cells monolayers were grown to 100% confluence on 3-µm pore collagen-coated PTFE membranes (Corning). 5% CSE was added to the basal media for 2, 4, 6, 18 and 24 h. FITC-dextran was added to the apical chamber and transcellular passage was determined by measuring the fluorescence intensity of the basal chamber with a fluorescence plate reader. To determine the effect of PP2A on cell permeability, 1-µM Fostriecin was added to the basal chamber and cell permeability was assessed as described above after 24 h.
Immunoblot analysis
Immunoblots for PP2A, SET1, occludin, and actin were conducted on protein fractions from the cytosol and membrane as per standard protocol52. To assess occludin threonine phosphorylation, occludin protein was immunoprecipitated from the endothelial cell lysates. After electrophoresis and transfer to a nitrocellulose membrane, immunoblots for total occludin and threonine phosphorylation of occludin were conducted using specific antibodies (Cell Signaling, Danvers, MA). (PP2AC 2038, Occludin 91131, SET1 97411, Actin 4967)
Neutrophil adhesion assay
15 ml of blood was obtained from a healthy volunteer via venipuncture. The protocol for blood isolation was approved by Downstate Health Sciences University’s IRB. The blood was mixed with 2.5 ml of acidified citrate to prevent clotting. 5 ml of a 5% dextran solution in PBS was added to the mixture and allowed to sit at room temperature for 45 min. The plasma was collected and the cells were pelleted at 600 x g for 10 min at 4 °C. The cellular pellet was suspended in 5 ml of HBSS and then layered over a cushion of 4 ml of Ficoll Paque (Sigma, Histopaque-1077) and centrifuged at 600 x g for 20 min at 4 °C. To eliminate red blood cells, the pellet was treated for twenty seconds with 5 ml of 0.2% NaCl. Then, 5 ml of 1.6% NaCl was immediately added to the mixture. The cells were pelleted and then suspended in DMEM media. CalceinAM (Molecular Probes; C3099) 5 µg/ml was added to the 5 ml suspension of neutrophils in DMEM media for 30 min at 37 °C. Neutrophils were then washed twice in PBS by centrifuging 300 x g for 8 min at 4 °C. Human lung microvascular endothelial cells were grown to 70% confluence in 6-well plates and then treated with control or PP2AC siRNA for 24 h. The HLMVEC monolayers were then washed three times with 3 ml of filter-sterilized (0.22-µm) RPMI 1640 (without phenol red) containing 3% BSA per well. 8 × 106 calcein-labeled neutrophils were added per well and then incubated for 20 min in a 37 °C, 5% CO2 incubator. The wells were then washed five times with 3 ml of PBS and then patted dry. 3 ml of RPMI 1640 media (without phenol) was added to each well and then the fluorescence intensity of each well was measured with an excitation wavelength of 485 nm and an emission wavelength of 520 nm.
Statistical analyses. The majority of the data are expressed as dot plots with the means ± S.E.M. highlighted. A comparison of groups was performed by Student’s t-test (two-tailed). Experiments with more than 2 groups were analyzed by 2-way ANOVA with Bonferroni posttests analysis. p values for significance were set at 0.05 All analyses were performed using GraphPad Prism Software (Version 9).
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Acknowledgements
This work was supported by grants made available to P.G. Flight Attendant Medical Research Institute (CIA160005) and the Alpha-1 Foundation (493373) and to R.F. Flight Attendant Medical Research Institute (CIA160028), Alpha One Foundation, and the National Institutes of Health 1 R01 HL162590-01A1.
Author contributions
Performed experiments: A.J.D., P.G., S.R., W.E., J.T. and R.F.F.; Study design: P.G. and R.F.F.; Analysis of data: A.J.D., P.G., and R.F.F.; Drafting the manuscript for important intellectual content: P.G. and R.F.F. Manuscript review and editing: A.J.D., S.M., O.E. P.G., and R.F.F.
Data availability
The original data that support the findings of this study are available from the corresponding author, [RF], upon reasonable request.
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Zhou, H., Fan, E. K. & Fan, J. <ArticleTitle Language=“En”>Cell-Cell Interaction mechanisms in Acute Lung Injury. Shock. 55, 167–176. 10.1097/SHK.0000000000001598 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weibel, E. R. On the tricks alveolar epithelial cells play to make a good lung. Am. J. Respir Crit. Care Med.191, 504–513. 10.1164/rccm.201409-1663OE (2015). [DOI] [PubMed] [Google Scholar]
- 3.Disease, G. B. D., Injury, I. & Prevalence, C. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 392, 1789–1858. 10.1016/S0140-6736(18)32279-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hopkins, A. M., Walsh, S. V., Verkade, P., Boquet, P. & Nusrat, A. Constitutive activation of Rho proteins by CNF-1 influences tight junction structure and epithelial barrier function. J. Cell Sci.116, 725–742. 10.1242/jcs.00300 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Ling, K. et al. Type I gamma phosphatidylinositol phosphate kinase modulates adherens junction and E-cadherin trafficking via a direct interaction with mu 1B adaptin. J. Cell Biol.176, 343–353. 10.1083/jcb.200606023 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rounds, S. & Lu, Q. Cigarette smoke alters lung vascular permeability and endothelial barrier function. Pulm Circ.8, 2045894018794000. 10.1177/2045894018794000 (2018). (2017 Grover Conference Series). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Borgas, D. et al. Cigarette Smoke Disrupted Lung Endothelial Barrier Integrity and Increased Susceptibility to Acute Lung Injury via Histone Deacetylase 6. Am. J. Respir Cell. Mol. Biol.54, 683–696. 10.1165/rcmb.2015-0149OC (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu, Q. et al. Cigarette smoke causes lung vascular barrier dysfunction via oxidative stress-mediated inhibition of RhoA and focal adhesion kinase. Am. J. Physiol. Lung Cell. Mol. Physiol.301, L847–857. 10.1152/ajplung.00178.2011 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Braga, V. M., Machesky, L. M., Hall, A. & Hotchin, N. A. The small GTPases Rho and Rac are required for the establishment of cadherin-dependent cell-cell contacts. J. Cell Biol.137, 1421–1431. 10.1083/jcb.137.6.1421 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schweitzer, K. S. et al. Mechanisms of lung endothelial barrier disruption induced by cigarette smoke: role of oxidative stress and ceramides. Am. J. Physiol. Lung Cell. Mol. Physiol.301, L836–846. 10.1152/ajplung.00385.2010 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Henry, B., Ziobro, R., Becker, K. A., Kolesnick, R. & Gulbins, E. Acid sphingomyelinase. Handb. Exp. Pharmacol.10.1007/978-3-7091-1368-4_4 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Ruediger, R., Van Wart Hood, J. E., Mumby, M. & Walter, G. Constant expression and activity of protein phosphatase 2A in synchronized cells. Mol. Cell. Biol.11, 4282–4285. 10.1128/mcb.11.8.4282-4285.1991 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Slupe, A. M., Merrill, R. A. & Strack, S. Determinants for Substrate Specificity of Protein Phosphatase 2A. Enzyme Res.10.4061/2011/398751 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu, F. & Wilson, J. X. Peroxynitrite-dependent activation of protein phosphatase type 2A mediates microvascular endothelial barrier dysfunction. Cardiovascular. Res.81, 38–45. 10.1093/cvr/cvn246 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tar, K. et al. Role of protein phosphatase 2A in the regulation of endothelial cell cytoskeleton structure. J. Cell. Biochem.98, 931–953. 10.1002/jcb.20829 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Wallace, A. M. et al. Protein phosphatase 2A regulates innate immune and proteolytic responses to cigarette smoke exposure in the lung. Toxicol. Sci.126, 589–599. 10.1093/toxsci/kfr351 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nath, S. et al. Chronic Cigarette Smoke Exposure Subdues PP2A Activity by Enhancing Expression of the Oncogene CIP2A. Am. J. Respir Cell. Mol. Biol.10.1165/rcmb.2018-0173OC (2018). [DOI] [PubMed] [Google Scholar]
- 18.Janssens, V. & Goris, J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem. J.353, 417–439. 10.1042/0264-6021:3530417 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Geraghty, P. et al. The glutathione peroxidase 1-protein tyrosine phosphatase 1B-protein phosphatase 2A axis. A key determinant of airway inflammation and alveolar destruction. Am. J. Respir Cell. Mol. Biol.49, 721–730. 10.1165/rcmb.2013-0026OC (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shamseddine, A. A., Airola, M. V. & Hannun, Y. A. Roles and regulation of neutral sphingomyelinase-2 in cellular and pathological processes. Adv. Biol. Regul.57, 24–41. 10.1016/j.jbior.2014.10.002 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dajas-Bailador, F. A., Mogg, A. J. & Wonnacott, S. Intracellular Ca2 + signals evoked by stimulation of nicotinic acetylcholine receptors in SH-SY5Y cells: contribution of voltage-operated Ca2 + channels and Ca2 + stores. J. Neurochem.81, 606–614. 10.1046/j.1471-4159.2002.00846.x (2002). [DOI] [PubMed] [Google Scholar]
- 22.Wlodarchak, N. et al. Structure of the Ca2+-dependent PP2A heterotrimer and insights into Cdc6 dephosphorylation. Cell Res.23, 931–946. 10.1038/cr.2013.77 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Colbran, R. J. Protein phosphatases and calcium/calmodulin-dependent protein kinase II-dependent synaptic plasticity. J. neuroscience: official J. Soc. Neurosci.24, 8404–8409. 10.1523/JNEUROSCI.3602-04.2004 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takahashi, T. et al. Component of nicotine-induced intracellular calcium elevation mediated through alpha3- and alpha5-containing nicotinic acetylcholine receptors are regulated by cyclic AMP in SH-SY 5Y cells. PLoS One. 15, e0242349. 10.1371/journal.pone.0242349 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rao, R. Occludin phosphorylation in regulation of epithelial tight junctions. Ann. N Y Acad. Sci.1165, 62–68. 10.1111/j.1749-6632.2009.04054.x (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wong, V. Phosphorylation of occludin correlates with occludin localization and function at the tight junction. Am. J. Physiol.273, C1859–1867. 10.1152/ajpcell.1997.273.6.C1859 (1997). [DOI] [PubMed] [Google Scholar]
- 27.Travis, J., Pike, R., Imamura, T. & Potempa, J. The role of proteolytic enzymes in the development of pulmonary emphysema and periodontal disease. Am. J. Respir Crit. Care Med.150, 143–146. 10.1164/ajrccm/150.6_Pt_2.S143 (1994). [DOI] [PubMed] [Google Scholar]
- 28.Patil, R. S. et al. Serine/Threonine Protein Phosphatases 1 and 2A in Lung Endothelial Barrier Regulation. Biomedicines 11 (2023). 10.3390/biomedicines11061638 [DOI] [PMC free article] [PubMed]
- 29.Li, Z. et al. Low-dose endothelial monocyte-activating polypeptide-II increases permeability of blood-tumor barrier via a PKC-zeta/PP2A-dependent signaling mechanism. Exp. Cell Res.331, 257–266. 10.1016/j.yexcr.2014.12.021 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Tar, K. et al. Phosphatase 2A is involved in endothelial cell microtubule remodeling and barrier regulation. J. Cell. Biochem.92, 534–546. 10.1002/jcb.20036 (2004). [DOI] [PubMed] [Google Scholar]
- 31.Karki, P. & Birukova, A. A. Microtubules as Major Regulators of Endothelial Function: Implication for Lung Injury. Front. Physiol.12, 758313. 10.3389/fphys.2021.758313 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Le Guelte, A. et al. Semaphorin 3A elevates endothelial cell permeability through PP2A inactivation. J. Cell Sci.125, 4137–4146. 10.1242/jcs.108282 (2012). [DOI] [PubMed] [Google Scholar]
- 33.Sakakibara, A., Furuse, M., Saitou, M., Ando-Akatsuka, Y. & Tsukita, S. Possible involvement of phosphorylation of occludin in tight junction formation. J. Cell Biol.137, 1393–1401. 10.1083/jcb.137.6.1393 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morgan, L. et al. Inflammation and dephosphorylation of the tight junction protein occludin in an experimental model of multiple sclerosis. Neuroscience. 147, 664–673. 10.1016/j.neuroscience.2007.04.051 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Caraballo, J. C. et al. Hypoxia increases transepithelial electrical conductance and reduces occludin at the plasma membrane in alveolar epithelial cells via PKC-zeta and PP2A pathway. Am. J. Physiol. Lung Cell. Mol. Physiol.300, L569–578. 10.1152/ajplung.00109.2010 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang, K. H. et al. NADPH oxidase (NOX) 1 mediates cigarette smoke-induced superoxide generation in rat vascular smooth muscle cells. Toxicol. Vitro. 38, 49–58. 10.1016/j.tiv.2016.10.013 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Macklin, K. D., Maus, A. D., Pereira, E. F., Albuquerque, E. X. & Conti-Fine, B. M. Human vascular endothelial cells express functional nicotinic acetylcholine receptors. J. Pharmacol. Exp. Ther.287, 435–439 (1998). [PubMed] [Google Scholar]
- 38.Wu, J. C. et al. Cholinergic modulation of angiogenesis: role of the 7 nicotinic acetylcholine receptor. J. Cell. Biochem.108, 433–446. 10.1002/jcb.22270 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pena, V. B., Bonini, I. C., Antollini, S. S., Kobayashi, T. & Barrantes, F. J. alpha 7-type acetylcholine receptor localization and its modulation by nicotine and cholesterol in vascular endothelial cells. J. Cell. Biochem.112, 3276–3288. 10.1002/jcb.23254 (2011). [DOI] [PubMed] [Google Scholar]
- 40.Sun, P. et al. Deficiency of alpha7 nicotinic acetylcholine receptor attenuates bleomycin-induced lung fibrosis in mice. Mol. Med.23, 34–39. 10.2119/molmed.2016.00083 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hsu, S. et al. Tyrosine phosphatase PTP1B modulates store-operated calcium influx. Cell. Signal.15, 1149–1156. 10.1016/s0898-6568(03)00088-3 (2003). [DOI] [PubMed] [Google Scholar]
- 42.Sheth, P., Samak, G., Shull, J. A., Seth, A. & Rao, R. Protein phosphatase 2A plays a role in hydrogen peroxide-induced disruption of tight junctions in Caco-2 cell monolayers. Biochem. J.421, 59–70. 10.1042/BJ20081951 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Seth, A., Sheth, P., Elias, B. C. & Rao, R. Protein phosphatases 2A and 1 interact with occludin and negatively regulate the assembly of tight junctions in the CACO-2 cell monolayer. J. Biol. Chem.282, 11487–11498. 10.1074/jbc.M610597200 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Yokoyama, N. & Malbon, C. C. Phosphoprotein phosphatase-2A docks to Dishevelled and counterregulates Wnt3a/beta-catenin signaling. J. Mol. Signal.2, 12. 10.1186/1750-2187-2-12 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zou, W., Zou, Y., Zhao, Z., Li, B. & Ran, P. Nicotine-induced epithelial-mesenchymal transition via Wnt/beta-catenin signaling in human airway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol.304, L199–209. 10.1152/ajplung.00094.2012 (2013). [DOI] [PubMed] [Google Scholar]
- 46.Strack, S., Zaucha, J. A., Ebner, F. F., Colbran, R. J. & Wadzinski, B. E. Brain protein phosphatase 2A: developmental regulation and distinct cellular and subcellular localization by B subunits. J. Comp. Neurol.392, 515–527 (1998). [PubMed] [Google Scholar]
- 47.Strack, S., Chang, D., Zaucha, J. A., Colbran, R. J. & Wadzinski, B. E. Cloning and characterization of B delta, a novel regulatory subunit of protein phosphatase 2A. FEBS Lett.460, 462–466. 10.1016/s0014-5793(99)01377-0 (1999). [DOI] [PubMed] [Google Scholar]
- 48.Ito, A. et al. A truncated isoform of the PP2A B56 subunit promotes cell motility through paxillin phosphorylation. EMBO J.19, 562–571. 10.1093/emboj/19.4.562 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee, T. Y. et al. The B56gamma3 regulatory subunit of protein phosphatase 2A (PP2A) regulates S phase-specific nuclear accumulation of PP2A and the G1 to S transition. J. Biol. Chem.285, 21567–21580. 10.1074/jbc.M109.094953 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang, Y. et al. Nicotine stimulates adhesion molecular expression via calcium influx and mitogen-activated protein kinases in human endothelial cells. Int. J. Biochem. Cell Biol.38, 170–182. 10.1016/j.biocel.2005.08.004 (2006). [DOI] [PubMed] [Google Scholar]
- 51.Cooke, J. P. & Ghebremariam, Y. T. Endothelial nicotinic acetylcholine receptors and angiogenesis. Trends Cardiovasc. Med.18, 247–253. 10.1016/j.tcm.2008.11.007 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Geraghty, P., Hardigan, A. & Foronjy, R. F. Cigarette smoke activates the proto-oncogene c-src to promote airway inflammation and lung tissue destruction. Am. J. Respir Cell. Mol. Biol.50, 559–570. 10.1165/rcmb.2013-0258OC (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The original data that support the findings of this study are available from the corresponding author, [RF], upon reasonable request.