

Abstract
Background
Approximately half of hypertrophic cardiomyopathy (HCM) patients lack a precise genetic diagnosis. The likelihood of identifying clinically relevant variants increased over time.
Methods
In this study, we conducted a gene-centric reanalysis of exome data of 200 HCM cases 5 years after the initial analysis. This reanalysis prioritized genes with a matched HCM entry in the OMIM database and recently emerging HCM-associated genes gathered using a text mining-based literature review. Further classification of the identified genes and variants was performed using the Clinical Genome Resource (ClinGen) resource and American College of Medical Genetics and Genomics (ACMG) guidelines to assess the robustness of gene–disease association and the clinical actionability of the prioritized variants.
Results
As expected, the majority of patients carried variants in MYBPC3 and MYH7 genes, 26% (n = 51) and 8% (n = 16), respectively, in accordance with the initial analysis. The vast majority of pathogenic (P) and likely pathogenic (LP) variants were found in MYBPC3 (22 out of 40 variants) and MYH7 (8 out of 16 variants) genes. Three genes—not included in the initial analysis—were identified: SVIL, FHOD3, and TRIM63. Considering only patients with unique variants in the last three genes, there was a 9% enhancement in variant identification. Importantly, SVIL variant carriers presented apical and septal HCM, aortopathies, and severe scoliosis for one patient. Ten patients (5%) carried variants in the FHOD3 gene, six in hotspot regions (exons 12 and 15). We identified seven variants within the TRIM63 gene in 12 patients (6%). Homozygous variants were detected in 2.5% of the cohort in MYBPC3 (n = 1), MYL3 (n = 1), and TRIM63 (n = 3) genes.
Conclusion
Our study revealed that no variants were found in the ACTC1, TPM1, and TNNI3 genes in the HYPERGEN cohort. However, we identified variants in five out of the eight HCM core genes, with a high prevalence in young patients. We identified variants in three recent HCM-associated genes (SVIL, FHOD3, and TRIM63) in 35 patients, with 18 patients carrying unique variants (9%). Our results further emphasize the usefulness of exome data reanalysis, particularly in genotype-negative patients.
Keywords: HCM, exome reanalysis, sarcomeric genes, non-sarcomeric genes, novel-associated genes, FHOD3, TRIM63, SVIL
Introduction
Hypertrophic cardiomyopathy (HCM) is an inherited cardiac disease, defined by left ventricular (LV) wall thickness greater than 15 mm, in the absence of other loading conditions that could explain the hypertrophy (1). The degree, localization, and distribution of the hypertrophy are variable (2). The LV systolic function can be preserved, increased, or reduced (2). Consequently, HCM is characterized by a phenotypic heterogeneity that could be partly explained by the heterogeneity of the genetic underlying etiology.
The estimated prevalence of HCM is 1:500 in the general population based on the recognition of the disease phenotype (3–5). However, considering familial transmission, genotype-positive cases for sarcomeric genes, and subclinical cases, a higher prevalence (1:200) is reported (6).
The clinical manifestations of HCM typically occur between the ages of 20 and 40, although they can develop at any age (7). It is noteworthy that 50–60% of cases are diagnosed after the age of 30, reflecting the variable penetrance and expressivity of the disease (3). The average life expectancy for HCM patients is typically favorable, approaching 70 years in effectively managed cases (6), although outcomes are greatly dependent on the presence of risk factors for complications such as early onset of the disease, arrhythmias, and heart failure. The annual risk of sudden cardiac death (SCD) in high-risk patients is estimated to range from 0.5 to 1%, with the highest risk observed in young cases under 30 years of age (8).
HCM is widely recognized as a sarcomeric disease since the vast majority of patients carry variants within the eight core genes encoding sarcomeric proteins (9). Indeed, the myosin-binding protein C (MYBPC3) and the β-myosin heavy chain (MYH7) genes account for 50–70% of HCM cases that undergo genetic testing (1–3, 10, 11). The remaining patients harbor variants in other sarcomeric genes, such as myosin light chain (MLC) genes (MYL2 and MYL3), troponin encoding genes (TNNT2 and TNNI3), tropomyosin 1 (TPM1), and actin α-cardiac muscle 1 (ACTC1) (4, 11, 12). Collectively, actionable variants in sarcomeric-positive (sarc+) patients account for >90% of the totality of pathogenic (P) variants in HCM patients (12). Of note, these percentages vary widely depending on the studied populations, the clinical profiling, the sequencing method used (panel, whole-exome sequencing [WES], etc.), and variant prioritization criteria.
Several additional genes have been linked to HCM encoding non-sarcomeric proteins, including, but not restricted to, proteins of the Z-disk (ACTN2, TCAP, and VCL), calcium handling genes (TNNC1, RYR2, JPH2, PLN), and the proteasome (TRIM63) (4). Similarly to the sarcomeric genes, both inheritance models are reported in these genes with a predominance of the autosomal dominant pattern (12, 13). However, the contribution of these genes is minor, ranging from 0.06 to 8.7% based on combined published data involving genes with strong, moderate, and weak evidence of causality and HCM-associated genes supported only by functional data (13). These findings reinforce the fact that sarcomeric genes predominantly cause HCM and emphasize the significant proportion of the missing heritability in HCM (13). Therefore, up to 50% of HCM patients do not have an identifiable pathogenic disease-causing variant (14, 15). So far, this missing heritability is increasingly explained by the fact that sarcomeric-negative (Sarc-) and genotype-negative patients have a non-Mendelian or near-Mendelian disease caused by a joint effect of genetic and non-genetic factors (13, 16). Thus, the additive effect of allelic heterogeneity and additional genetic variants, along with variants in cis causing allelic imbalance, could also partially explain the missing heritability, the phenotypic variability, and the incomplete penetrance. In this line, many studies have suggested the oligogenic inheritance model in HCM patients lacking an identifiable highly penetrant variant (16–20). Furthermore, rare variants are broadly acknowledged among the plausible sources of missing heritability and the likely contributors to the oligogenic inheritance rather than common variants. Indeed, variants with strong effects are expected to be kept under selective pressure and remain at low to extremely low frequencies in the population (21, 22). Altogether, incomplete penetrance, variable expressivity, and missing heritability in HCM make the genetic diagnosis and clinical prognosis challenging.
The efficiency of WES reanalysis has been demonstrated to improve genetic testing yield, especially by adding the newly associated genes previously unrecognized as clinically relevant. It is estimated that the genetic diagnostic yield could be enhanced by approximately 15% when including new disease-gene associations, up-to-date software, variant frequency databases, and text mining for genetic and clinical re-evaluation (23). Moreover, it is recommended that the dataset initially analyzed over 2 years ago should be prioritized for reanalysis (24).
In this study, we sought to refine the initial Hypertension Genetic Epidemiology Network (HYPERGEN) analysis by focusing on HCM-causing genes (that are known and recently associated). Additionally, we assessed the usefulness of our reanalysis regarding the clinical actionability of the identified variants by applying the American College of Medical Genetics and Genomics/Association of Molecular Pathologists (ACMG/AMP) criteria.
Methods
This study was approved by an institutional review committee. All subjects gave informed consent for genetic studies. WES was performed on the NGS Illumina HiSeq2500 platform using Agilent SureSelect V6 technology (Genwiz, United States) (25).
The HYPERGEN cohort
The HYPERGEN cohort included 132 men and 68 women (mean age, 55 years; range, 19–91 years; 86.5% above 40; 34% familial cases). The patients were enrolled in five French centers: Marseille, Bordeaux, Paris, Dijon, and Rennes.
A definite echocardiographic evidence of HCM was considered based on the measurements of maximal wall thickness ≥ 15 mm in sporadic and > 13 mm in familial cases without dilatation or any cardiovascular comorbidities or systemic disease.
None of the patients was reported by the clinicians as syndromic or likely syndromic. However, the first HYPERGEN study identified one novel variant in the GLA gene (p.Leu311Profs*4), and the clinical re-evaluation of the patient confirmed a Fabry disease diagnosis. A second case with the disease-causing variant TTR: p.Val50Met has been reported with transthyretin cardiac amyloidosis (25). Of note, this study’s variant prioritization strategy is different from the first HYPERGEN analysis. Thus, an important caveat to our reanalysis is that an accurate comparison of the genetic diagnosis yield cannot be assessed as the initial analysis pipeline differs from this reanalysis. Additionally, since we do not have access to the variant coordinates identified in the first analysis, it is impossible to determine whether any variants have been downgraded or upgraded. The significant differences are detailed in the following.
Initial HYPERGEN analysis vs. the 5 years interval analysis
In the initial HYPERGEN study, bioinformatic analyses were performed by retaining exonic and intronic variants with a minor allele frequency (MAF) lower than 1% and predicted as P or likely pathogenic LP mainly according to the Universal Mutation Database (UMD) predictor. The first step of WES data analysis consisted of searching for variants in a virtual panel of 167 genes involved in cardiomyopathies and other various cardiac hereditary diseases (25).
In contrast, in this reanalysis, priority was given to genes with a matched HCM entry in Mendelian disease in the Online Mendelian Inheritance in Man (OMIM) database and the ClinGen1 as well as recently emerging HCM-associated genes gathered using text mining-based literature review from 2016 to 2023 (Figure 1).
Figure 1.

Venn diagram visualization of genes across initial and 5-year analyses. The “previously analyzed genes” section includes genes from the 167-gene panel used in the original HYPERGEN study; the “overlapping genes” section contains genes identified in both the initial and current analyses; the “newly identified genes” section consists of genes associated with HCM between 2019 and 2024.
More importantly, variant pathogenicity was assessed according to ACMG/AMP guidelines, and the consistency of a predicted deleterious effect by at least 3 prediction tools and a high combined annotation-dependent depletion (CADD) score.
As aforementioned, the initial HYPERGEN analysis showed two HCM phenocopies. In this study, a preanalysis included the significant syndromic genes and genes yielding known HCM phenocopies. None of the variants identified within these genes were classified as P or LP according to ACMG or reported in ClinVar database (data not shown). Moreover, all these variants were found along with other HCM-relevant variants. Thus, in this reanalysis, variants in genes causing syndromic HCM and phenocopies are not included. The TTN gene is not included as well.
The Venn diagram shown in Figure 1 illustrates the relationship between the genes previously prioritized in the first analysis and those reanalyzed in this study. The overlapping section represents genes shared between both analyses.
Variant filtering, prioritization, and sanger validation
The first criterion of this reanalysis is variant rarity. Thus, a filtering allele frequency of <1% in gnomAD database was applied. Subsequently, intergenic, intronic, and synonymous variants were removed. The remaining variants were prioritized based on their in silico predicted impact on protein function. This analysis’s primary scoring tool is the CADD (26). Indeed, variants predicted as damaging or probably damaging, deleterious, disease-causing by PolyPhen-2 and SIFT, and/or MutationTaster while also having a CADD score > 15 were considered as variants of high confidence of pathogenicity (27–29). Of note, up to two-thirds of the variants of uncertain significance (VUS) in confirmed sarcomeric genes are considered causal of HCM (30). Thus, suspicious VUS with some evidence of causality (a rarity, VUS within well-established HCM genes, mutational hotspots…) is reported in this reanalysis.
Finally, to gauge the clinical actionability of the detected variants, a subsequent classification according to ACMG criteria was performed (31).
All the prioritized variants within the core and minor HCM genes were validated by Sanger sequencing when DNA samples were available (Supplementary File 1).
Biological pathway and gene ontology analyses
Given a large number of variants, even after the filtering steps, variants within HCM-minor genes could be missed. To facilitate and ensure the identification of these variants, a biological pathway analysis including Reactome and KEGG databases as well as gene (GO) and human phenotype (HPO) ontologies terms was carried out using VarAFT software (32–36). For example, querying the HPO and GO databases with the keywords “cardiomyopathy phenotype” and “cardiac hypertrophy” results in 3 HPO term identifiers (HP:0001639, HP:0005157, HP:0031992), 27 disease terms (matching all the OMIM phenotypes) and 83 additional genes. VarAFT software allowed us to query each variant call format (VCF) file for all the possible combinations.
French control cohort
French healthy controls (n = 960) were queried to compare variants allele frequencies. Exons coordinate on GRCh37 were extracted for the genes of interest and extended by 50 bases. Those regions were called using GATK (4.3.0.0) with a set of Binary Alignment Map (BAM) files using a nextflow-based pipeline for joint-genotyping.2 Resulting VCF was annotated with snpEff (37) and jvarkit/GnomAD.3 Variants known to be filtered in gnomAD or having an allele frequency greater than 1% in the non-finish population were excluded. Protein truncating and splice site variants were kept using jvarkit/vcffilterso.
Results
Variant identification among the definitive HCM genes
MYBPC3 and MYH7 genes: the permanently significant HCM genes
Definitive HCM genes with a well-established disease association are mainly the eight sarcomeric genes with strong evidence of causality (MYBPC3, MYH7, TNNT2, TNNI3, TPM1, ACTC1, MYL3, and MYL2) (38, 39). In our analysis, variants were found in five out of these eight well-established genes.
Not surprisingly, the most prevalent genes in our case cohort are MYBPC3 and MYH7 genes, which is in accordance with the initial HYPERGEN study (Figure 2).
Figure 2.

Variant distribution within HCM well-established genes. The number of patients harboring variants in each HCM core gene is dark purple. The number of variants within each gene is indicated in light purple.
Variants coordinates are detailed in Tables 1–4.
Table 1.
Variants identified in the MYBPC3 gene (transcript NM_000256, MANE select).
| Patient ID | Genotype | Variant type | AA change | rs ID | CADD | ACMG/ClinVar | GnomAD-AF | Additional variant |
|---|---|---|---|---|---|---|---|---|
| HCM-1 | Het | Splicing | Ex4:c.505 + 2 T > A | NA | 23.5 | LP/NA | NA | Unique variant |
| HCM-2 | Het | Stopgain | Ex4:p.(Ser139*) | rs730880704 | 35 | LP/LP | NA | Unique variant |
| HCM-3 | Het | Missense | Ex5:p.(Arg177His) | rs201012766 | 23.5 | B/B | 0.001201 | Unique variant |
| HCM-4 | Het | Stopgain | Ex5:p.(Gln205*) | rs397516061 | 36 | LP/P | NA | Unique variant |
| HCM-5 | Het | Missense | Ex5:p.(Leu183Ile) | NA | 24.3 | VUS/NA | NA | MYBPC3:c.1928-2A > G |
| HCM-6 | Het | Missense | Ex5:p.(Ser217Gly) | rs138753870 | 14.62 | B/Conflicting | 0.001693 | MYBPC3:p.(Arg943*) |
| HCM-7 | Het | Missense | Ex5:p.(Val189Ile) | rs11570052 | 20.0 | B/B | 0.002459 | Unique variant |
| HCM-8 | Het | Missense | Ex5:p.(Val189Ile) | rs11570052 | 20.0 | B/B | 0.002459 | Unique variant |
| HCM-9 | Het | Missense | Ex6:p.(Arg238Cys) | rs771143409 | 33 | VUS/VUS | 0.00001615 | ACTN2: p.(Thr347Met) |
| HCM-10 | Het | Missense | Ex6:p.(Glu258Lys) | rs397516074 | 28.8 | P/P | 0.00002199 | Unique variant |
| HCM-11 | Het | Missense | Ex8:p.(Arg281Gly) | rs371711564 | 23.4 | VUS/VUS | NA | Unique variant |
| HCM-12 | Het | Frameshift deletion | Ex11:p.(Phe305ProfsTer27) | rs397516080 | NA | LP/P | NA | Unique variant |
| HCM-13 | Het | Missense | Ex12:p.(Arg326Gln) | rs34580776 | 24.0 | B/B | 0.004359 | Unique variant |
| HCM-14 | Het | Missense | Ex12:p.(Gly341Ser) | rs397515881 | 34 | VUS/VUS | 0.00004999 | MYBPC3: p.(Pro955ArgfsTer95) |
| HCM-15 | Het | Missense | Ex12:p.(Val321Met) | rs200119454 | 26.5 | VUS/Conflicting | 0.0003297 | Unique variant |
| HCM-16 | Het | Missense | Ex13:p.(Arg382Trp) | rs11570076 | 34 | B/B | 0.004239 | Unique variant |
| HCM-17 | Het | Splicing | Ex14:c.1351 + 2 T > C | rs397515897 | 23.1 | LP/P | NA | Unique variant |
| HCM-18 | Het | Frameshift deletion | Ex14:p.(Val437GlyfsTer13) | rs397515896 | NA | LP/P | NA | Unique variant |
| HCM-19 | Het | Splicing | Ex16:c.1624 + 4A > T | rs397515916 | NA | LP/P | 0.00001334 | Unique variant |
| HCM-20 | Het | Missense | Ex16:p.(Arg495Gln) | rs200411226 | 30 | LP/P | 0.00002408 | Unique variant |
| HCM-21 | Het | Missense | Ex16:p.(Arg495Gln) | rs200411226 | 30 | LP/P | 0.00002408 | Unique variant |
| HCM-22 | Het | Missense | Ex16:p.(Arg495Gly) | rs397515905 | 25.4 | P/P | 0.000004013 | Unique variant |
| HCM-23 | Het | Missense | Ex16:p.(Arg502Trp) | rs375882485 | 34 | P/P | 0.00004632 | Unique variant |
| HCM-24 | Het | Missense | Ex16:p.(Arg502Trp) | rs375882485 | 34 | P/P | 0.00004632 | Unique variant |
| HCM-25 | Het | Missense | Ex16:p.(Arg502Trp) | rs375882485 | 34 | P/P | 0.00004632 | Unique variant |
| HCM-26 | Hom | Missense | Ex17:p.(Ala562Thr) | rs397515919 | 31 | VUS/VUS | NA | Unique variant |
| HCM-27 | Het | Missense | Ex17:p.(Arg597Gln) | rs727503195 | 35 | P/P | 0.00002996 | FHOD3: p.(Arg637Gln) |
| HCM-28 | Het | Missense | Ex18:p.(Glu611Lys) | rs730880555 | 35 | VUS/Conflicting | 0.00002350 | Unique variant |
| HCM-29 | Het | Splicing | Ex20:c.1928-2A > G | rs397515937 | 23.8 | P/P | NA | Unique variant |
| HCM-30 | Het | Splicing | Ex20:c.1928-2A > G | rs397515937 | 23.8 | P/P | NA | Unique variant |
| HCM-31 | Het | Splicing | Ex20:c.1928-2A > G | rs397515937 | 23.8 | P/P | NA | Unique variant |
| HCM-32 | Het | Splicing | Ex20:c.1928-2A > G | rs397515937 | 23.8 | P/P | NA | Unique variant |
| HCM-5 | Het | Splicing | Ex20:c.1928-2A > G | rs397515937 | 23.8 | P/P | NA | MYBPC3: p.(Leu183Ile) |
| HCM-33 | Het | Splicing | Ex20:c.1928-2A > G | rs397515937 | 23.8 | P/P | NA | Unique variant |
| HCM-34 | Het | Splicing | Ex20:c.1928-2A > G | rs397515937 | 23.8 | P/P | NA | Unique variant |
| HCM-35 | Het | Missense | Ex22:p.(Asp770Asn) | rs36211723 | 34 | LP/Conflicting | 0.00001606 | Unique variant |
| HCM-36 | Het | Frameshift insertion | Ex22:p.(Lys754GlufsTer79) | rs774521272 | NA | LP/P | 0.000008025 | Unique variant |
| HCM-37 | Het | Frameshift insertion | Ex22:p.(Lys754GlufsTer79) | rs774521272 | NA | LP/P | 0.000008025 | Unique variant |
| HCM-38 | Het | Frameshift insertion | Ex23:p.(Trp792ValfsTer41) | rs397515963 | NA | P/P | NA | Unique variant |
| HCM-39 | Het | Frameshift insertion | Ex23:p.(Trp792ValfsTer41) | rs397515963 | NA | P/P | NA | SVIL: p.(Ala1496Asp) |
| HCM-40 | Het | Missense | Ex24:p.(Ala833Val) | rs3729952 | 26.9 | B/B | 0.002538 |
MYH6: p.(Arg1532Leu) SVIL: p.(Thr1630Ser) |
| HCM-41 | Het | Missense | Ex24:p.(Arg810His) | rs375675796 | 35 | LP/Conflicting | 0.00004816 | Unique variant |
| HCM-6 | Het | Stopgain | Ex26:p.(Arg943*) | rs387907267 | 43 | P/P | 0.00001214 | MYBPC3: p.(Ser217Gly) |
| HCM-42 | Het | Stopgain | Ex26:p.(Gln969*) | rs397515992 | 38 | LP/P | NA | Unique variant |
| HCM-43 | Het | frameshift deletion | Ex26:p.(Pro955ArgfsTer95) | rs397515990 | NA | P/P | 0.00003187 | Unique variant |
| HCM-14 | Het | frameshift deletion | Ex26:p.(Pro955ArgfsTer95) | rs397515990 | NA | P/P | 0.00003187 | MYBPC3: p.(Gly341Ser) |
| HCM-44 | Het | Missense | Ex28:p.(Arg1022Pro) | rs397516000 | 34 | VUS/Conflicting | 0.00002517 |
MYH7: p.(His1185Gln) TRIM63: p.(Cys23Tyr) |
| HCM-45 | Het | Stopgain | Ex28:p.(Gln1061*) | rs397516005 | 39 | LP/P | 0.00001477 | Unique variant |
| HCM-46 | Het | Missense | Ex30:p.(Arg1138His) | rs187705120 | 35 | VUS/B | 0.001139 | Unique variant |
| HCM-47 | Het | Missense | Ex30:p.(Ile1131Thr) | rs370890951 | 22.5 | B/Conflicting | 0.0008403 | Unique variant |
| HCM-48 | Het | Frameshift deletion | Ex31:p.(Cys1202Leufs*35) | NA | NA | NA | NA | FHOD3: p.(Arg637Gln) |
| HCM-49 | Het | stopgain | Ex32:p.(Cys1244*) | rs730880600 | 38 | LP/P | NA | Unique variant |
| HCM-50 | Het | Stopgain | Ex32:p.(Cys1244*) | rs730880600 | 38 | LP/P | NA | Unique variant |
| HCM-51 | Het | Missense | Ex32:p.(Ser1213Pro) | NA | 25.3 | VUS/NA | NA | Unique variant |
AA, amino acid; ACMG, American College of Medical Genetics and Genomics; Het, heterozygous; Hom, homozygous; B, benign; P, pathogenic; LP, likely pathogenic; VUS, variant of uncertain significance; NA, no data available; Conflicting, conflicting interpretations of pathogenicity; CADD, combined annotation-dependent depletion scores according to CADD model v1.3; GnomAD-AF, allele frequency according to gnomAD V2.1.
Table 4.
Variants identified in myosin light chain (MLC) genes.
| Patient ID | Gene | Transcript | Genotype | Variant type | AA change | rs ID | CADD | ACMG/ClinVar | GnomAD-AF | Additional variant |
|---|---|---|---|---|---|---|---|---|---|---|
| HCM-80 | MYL2 | NM_000432 | Het | Missense | Ex5:p.(Gly92Ala) | NA | 25.2 | VUS/NA | NA | Unique variant |
| HCM-77 | MYL3 | NM_000258 | Hom | Missense | Ex3:p.(Ala57Asp) | rs139794067 | 29.8 | VUS/Conflicting | 0.0001627 | Unique variant |
| HCM-78 | MYL3 | NM_000258 | Het | Missense | Ex3:p.(Arg94Cys) | rs730880961 | 33 | VUS/VUS | 0.00002388 | Unique variant |
| HCM-79 | MYL3 | NM_000258 | Het | Missense | Ex5:p.(Arg163Thr) | rs752165383 | 25.0 | VUS/VUS | 0.000003998 | Unique variant |
AA, amino acid; ACMG, American College of Medical Genetics and Genomics; Het, heterozygous; Hom, homozygous; VUS, variant of uncertain significance; NA, no data available; Conflicting, conflicting interpretations of pathogenicity; CADD, combined annotation-dependent depletion scores according to CADD model v1.3; GnomAD-AF, allele frequency according to gnomAD V2.1.
We identified 24 missense variants and 16 protein-truncating variants (PTV) in MYBPC3 gene (Figure 2). The vast majority of the identified MYBPC3 variants are unique (42 out of 51 patients; 82%), 3 patients are double heterozygous (HCM-5, HCM-6, and HCM-14) harboring each a missense variant classified as a VUS and a second PTV or splice-site variant reported in ClinVar database and classified as P according to ACMG. Six patients with variants in the MYBPC3 carrying one or more additional variants in minor HCM genes (ACTN2 and MYH6) and in recent HCM-associated genes (FHOD3, TRIM63, and SVIL) (Table 1; see Figure 3).
Figure 3.

Variant type distribution of MYBPC3 gene.
We identified 16 distinct variants in the MYH7 gene, suggesting that these variants are more likely private. Importantly, in our case cohort, no PTV MYH7 variant has been identified, and 8 out of the 16 missense variants are classified P and reported P or LP according to ACMG rules and ClinVar Database, respectively (Table 2). It should be noted that MYBPC3 and MYH7 are not only the most prevalent genes but also the most genes with P/LP classified variants following ACMG/AMP criteria (Figure 4). The remaining variants in this study are predicted to be deleterious through several tools. Each variant’s detailed in silico prediction is available in Supplementary File 2.
Table 2.
Variants identified in the MYH7 gene (transcript NM_000257, MANE select).
| Patient ID | Genotype | Variant type | AA change | rs ID | CADD | ACMG/ClinVar | GnomAD-AF | Additional variant |
|---|---|---|---|---|---|---|---|---|
| HCM-52 | Het | Missense | Ex8:p.(Asn232Ser) | NA | 24.8 | VUS/NA | NA |
MYH6: p.(His1526Arg) CAV3: p.(Thr78Met) |
| HCM-53 | Het | Missense | Ex9:p.(Ile250Phe) | rs397516268 | 25.4 | VUS/Conflicting | NA | Unique variant |
| HCM-54 | Het | Missense | Ex14:p.(Arg453Cys) | rs121913625 | 33 | P/P | NA | Unique variant |
| HCM-55 | Het | Missense | Ex18:p.(Arg663His) | rs371898076 | 27.8 | P/P | 0.00001414 | Unique variant |
| HCM-56 | Het | Missense | Ex19:p.(Arg694Cys) | rs727504240 | 34 | VUS/Conflicting | 0.00001989 | Unique variant |
| HCM-57 | Het | Missense | Ex19:p.(Arg719Gln) | rs121913641 | 34 | P/P | NA | SVIL: p.(Val1438Met) |
| HCM-58 | Het | Missense | Ex20:p.(Gly741Arg) | rs121913632 | 33 | P/P | 0.00003185 | Unique variant |
| HCM-59 | Het | Missense | Ex20:p.(Ile736Thr) | rs727503261 | 24.7 | P/P | NA | Unique variant |
| HCM-60 | Het | Missense | Ex21:p.(Ala797Thr) | rs3218716 | 20.5 | P/P | 0.00002386 | Unique variant |
| HCM-61 | Het | Missense | Ex21:p.(Arg787Cys) | rs145677314 | 22.7 | VUS/Conflicting | 0.00006364 |
VCL: p.(His636Arg) MYPN: p.(Asp1208Gly) |
| HCM-62 | Het | Missense | Ex21:p.(Gly768Arg) | rs727503260 | 35 | P/P | NA | SVIL: p.(Ser1414Thr) |
| HCM-63 | Het | Missense | Ex22:p.(Arg869His) | rs202141173 | 34 | VUS/LP | 0.00002386 |
MYLK2: p.(Ser510Leu) TRIM63: p.(Cys75Tyr) |
| HCM-44 | Het | Missense | Ex27:p.(His1185Gln) | NA | 24.0 | VUS/NA | NA |
MYBPC3:p.(Arg1022Pro) TRIM63: p.(Cys23Tyr) |
| HCM-64 | Het | Missense | Ex33:p.(Glu1546Gly) | NA | 29.3 | VUS/NA | NA | Unique variant |
| HCM-65 | Het | Missense | Ex35:p.(Arg1712Gln) | rs193922390 | 28.3 | LP/P | 0.00002125 | Unique variant |
| HCM-66 | Het | Missense | Ex37:p.(Glu1829Lys) | rs143562243 | 27.9 | VUS/VUS | 0.00001594 | FLNC: p.(Arg1719Cys) |
AA, amino acid; ACMG, American College of Medical Genetics and Genomics; Het, heterozygous; P, pathogenic; LP, likely pathogenic; VUS, variant of uncertain significance; NA, no data available; Conflicting: Conflicting interpretations of pathogenicity; CADD, combined annotation-dependent depletion scores according to CADD model v1.3; GnomAD-AF: allele frequency according to gnomAD V2.1.1.
Figure 4.
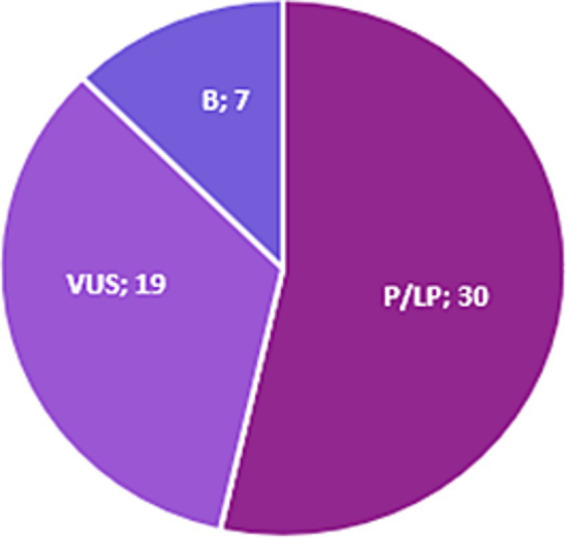
Proportion of MYBPC3 and MYH7 variants with P/LP ACMG classification. B, Benign; P, Pathogenic; LP, Likely Pathogenic; VUS, Variant of Uncertain Significance.
Troponin T2 and MLC genes: the minor sarcomeric genes
We identified a small proportion of patients carrying variants in TNNT2, MYL2, and MYL3 genes (n = 6; 3%) (Table 3). Two patients carried missense variants in TNNT2 (p.Val95Met and p.Lys283Glu), reported in ClinVar as LP and P, respectively. Furthermore, we identified four variants in MYL2 (n = 1) and MYL3 (n = 3) genes, one of which is at a homozygous state (MYL3 p.Ala57Asp) with the conflicting interpretation of pathogenicity in ClinVar (Table 4).
Table 3.
Variants identified in the TNNT2 gene (transcript NM_001276345, MANE select).
| Patient ID | Genotype | Variant type | AA change | rs ID | CADD | ACMG/ClinVar | GnomAD-AF | Additional variant |
|---|---|---|---|---|---|---|---|---|
| HCM-73 | Het | Missense | Ex9:p.(Val95Met) | NA | 30 | VUS/LP | NA | Unique variant |
| HCM-74 | Het | Missense | Ex16:p.(Lys283Glu) | rs1553279294 | 28.4 | VUS/P | NA | Unique variant |
AA, amino acid; ACMG, American College of Medical Genetics and Genomics; Het, heterozygous; P, pathogenic; LP, likely pathogenic; VUS, variant of uncertain significance; NA, no data available; CADD, combined annotation-dependent depletion scores according to CADD model v1.3; GnomAD-AF, Aalele frequency according to gnomAD V2.1.1.
All the variants in troponin T2 and MLC genes are unique.
Variant identification among minor HCM genes
Several additional genes are consistently reported as HCM-causing genes encoding sarcomeric and non-sarcomeric proteins and contributing to a small proportion of HCM genetic etiology (13). In our reanalysis, we identified variants in 12 HCM minor genes. Rare and deleterious variants in FLNC, MYH6, MYPN, and ACTN2 genes accounted for the majority of the prioritized variants among these genes (Table 5).
Table 5.
Variants identified in minor HCM genes.
| Patient ID | Genotype | Variant type | AA change | rs ID | CADD | ACMG/ClinVar | GnomAD-AF | Additional variant |
|---|---|---|---|---|---|---|---|---|
| Variants identified in the FLNC gene (transcript NM_001458, MANE select) | ||||||||
| HCM-92 | Het | Missense | Ex4:p.(Ala280Thr) | rs778751919 | 34 | VUS/VUS | 0.00002004 | Unique variant |
| HCM-93 | Het | Missense | Ex13:p.(Asp693Ala) | rs34972246 | 28.9 | VUS/LB | 0.003442 | Unique variant |
| HCM-67 | Het | Missense | Ex23:p.(Arg1341Gln) | rs149641783 | 27.5 | VUS/Conflicting | 0.0009425 | MYH6: p.(Gln277His) |
| HCM-94 | Het | Missense | Ex28:p.(Ala1588Gly) | rs148545460 | 27.5 | VUS/Conflicting | 0.0001603 | Unique variant |
| HCM-95 | Het | Missense | Ex28:p.(Ser1624Leu) | rs879255639 | 34 | VUS/Conflicting | 0.00003189 | Unique variant |
| HCM-66 | Het | Missense | Ex30:p.(Arg1719Cys) | rs773260834 | 34 | VUS/VUS | 0.000008022 | MYH7: p.(Glu1829Lys) |
| HCM-96 | Het | Missense | Ex34:p.(Arg1860Cys) | rs181067717 | 34 | VUS/LB | 0.005209 | Unique variant |
| HCM-97 | Het | Missense | Ex38:p.(Gly2106Val) | NA | 28.6 | NA | NA | Unique variant |
| HCM-98 | Het | Missense | Ex38:p.(Tyr2108Asn) | NA | 28.6 | NA | NA | Unique variant |
| HCM-99 | Het | Missense | Ex40:p.(Gly2199Arg) | rs368977589 | 23.3 | VUS/Conflicting | 0.00003239 | Unique variant |
| HCM-100 | Het | Missense | Ex42:p.(Arg2364His) | rs201672146 | 25.7 | VUS/LB | 0.001724 | MYPN: p.(Pro1112Leu) |
| HCM-101 | Het | Missense | Ex42:p.(Arg2364His) | rs201672146 | 25.7 | VUS/LB | 0.001724 | Unique variant |
| HCM-102 | Het | Missense | Ex47:p.(Lys2637Gln) | rs767794768 | 20.9 | VUS/VUS | 0.00006479 | Unique variant |
| Variants identified in the MYH6 gene (Transcript NM_002471, MANE select) | ||||||||
| HCM-67 | Het | Missense | Ex10:p.(Gln277His) | rs140660481 | 21.9 | VUS/Conflicting | 0.0003677 | FLNC: p.(Arg1341Gln) |
| HCM-68 | Het | Missense | Ex21:p.(Arg860His) | rs115845031 | 24.9 | B /Conflicting | 0.001778 | Unique variant |
| HCM-69 | Het | Missense | Ex22:p.(Glu932Lys) | NA | 34 | VUS/NA | NA | Unique variant |
| HCM-70 | Het | Missense | Ex23:p.(Ala1004Ser) | rs143978652 | 22.9 | LP/Conflicting | 0.0009651 | Unique variant |
| HCM-71 | Het | Missense | Ex28:p.(Glu1295Gln) | rs34935550 | 29.1 | LB/B | 0.003167 | Unique variant |
| HCM-72 | Het | Missense | Ex28:p.(Glu1295Gln) | rs34935550 | 29.1 | LB/B | 0.003167 | Unique variant |
| HCM-40 | Het | Missense | Ex32:p.(Arg1532Leu) | rs34330111 | 34 | VUS/Conflicting | 0.0002794 |
MYBPC3: p.(Ala833Val) SVIL: p.(Thr1630Ser) |
| HCM-52 | Het | Missense | Ex32:p.(His1526Arg) | NA | 25.5 | NA | NA |
MYH7: p.(Asn232Ser) CAV3: p.(Thr78Met) |
| Variants identified in the MYPN gene (transcript NM_032578, MANE select) | ||||||||
| HCM-124 | Het | Missense | Ex2:p.(Gln193Arg) | rs573684358 | 23.0 | LB/VUS | 0.0002188 | Unique variant |
| HCM-100 | Het | Missense | Ex17:p.(Pro1112Leu) | rs71534278 | 27.3 | VUS/VUS | 0.003060 | FLNC: p.(Arg2364His) |
| HCM-125 | Het | Missense | Ex17:p.(Pro1112Leu) | rs71534278 | 27.3 | VUS/VUS | 0.003060 | Unique variant |
| HCM-126 | Het | Missense | Ex17:p.(Pro1112Leu) | rs71534278 | 27.3 | VUS/VUS | 0.003060 | Unique variant |
| HCM-127 | Het | Missense | Ex17:p.(Ala1141Thr) | rs150404143 | 27.7 | B/B | 0.001085 | JPH2: p.(Phe221Leu) |
| HCM-61 | Het | Missense | Ex18:p.(Asp1208Gly) | NA | 29.7 | NA | NA |
MYH7: p.(Arg787Cys) VCL: p.(His636Arg) |
| Variants identified in the ACTN2 gene (transcript NM_001103, MANE select) | ||||||||
| HCM-9 | Het | Missense | Ex10:p.(Thr347Met) | rs727504590 | 33 | B/Conflicting | 0.0001343 | MYBPC3: p.(Arg238Cys) |
| HCM-121 | Het | Missense | Ex12:p.(Arg438Trp) | rs563171274 | 34 | VUS/Conflicting | 0.00001194 | Unique variant |
| HCM-116 | Het | Missense | Ex12:p.(Val458Met) | rs895000018 | 33 | LB/LB | 0.000003985 | TRIM63: p.(Gln247*) |
| HCM-122 | Het | Missense | Ex17:p.(Lys694Asn) | rs748034053 | 24.7 | LB/Conflicting | 0.00003182 | Unique variant |
| HCM-123 | Het | Missense | Ex21:p.(Arg851His) | rs201335965 | 34 | B/Conflicting | 0.00004601 | Unique variant |
| Variants identified in the ALPK3 gene (transcript NM_020778, MANE select) | ||||||||
| HCM-86 | Het | Missense | Ex5:p.(Tyr497Cys) | rs116077141 | 25.6 | B/B | 0.001299 | Unique variant |
| HCM-87 | Het | Missense | Ex7:p.(Arg1483Trp) | rs370816513 | 34 | VUS/VUS | 0.00002784 | Unique variant |
| HCM-88 | Het | Missense | Ex7:p.(Gly1522Asp) | NA | 24.4 | NA | NA | Unique variant |
| HCM-76 | Het | Missense | Ex14:p.(Arg1907Gln) | rs116585466 | 32 | VUS/VUS | 0.0001469 | MYLK2: p.(Gly89Asp) |
| Variants identified in the CSRP3 gene (transcript NM_003476, MANE select) | ||||||||
| HCM-81 | Het | Missense | Ex3:p.(Trp4Arg) | rs45550635 | 26.3 | B/Conflicting | 0.002285 | Unique variant |
| HCM-82 | Het | Missense | Ex3:p.(Trp4Arg) | rs45550635 | 26.3 | B/Conflicting | 0.002285 | SVIL: p.(Thr1630Ser) |
| HCM-83 | Het | Missense | Ex3:p.(Phe30Leu) | NA | 32 | VUS/VUS | NA | Unique variant |
| HCM-84 | Het | Missense | Ex5:p.(Arg100His) | rs138218523 | 24.9 | B/Conflicting | 0.001323 | Unique variant |
| Variants identified in the MYLK2 gene (transcript NM_033118, MANE select) | ||||||||
| HCM-75 | Het | Missense | Ex3:p.(Ala87Thr) | rs753089175 | 22.8 | VUS/VUS | 0.00003595 | Unique variant |
| HCM-76 | Het | Missense | Ex3:p.(Gly89Asp) | rs115398036 | 23 | B/B | 0.004242 | ALPK3: p.(Arg1907Gln) |
| HCM-63 | Het | Missense | Ex11:p.(Ser510Leu) | rs907738836 | 27.9 | NA | NA |
MYH7: p.(Arg869His) TRIM63: p.(Cys75Tyr) |
| Variants identified in the NEXN gene (transcript NM_144573, MANE select) | ||||||||
| HCM-89 | Het | Missense | Ex9:p.(Glu332Ala) | rs201763096 | 23.2 | B/B | 0.004068 | Unique variant |
| HCM-90 | Het | Missense | Ex9:p.(Glu332Ala) | rs201763096 | 23.2 | B/B | 0.004068 | Unique variant |
| Variants identified in the CAV3 gene (transcript NM_033337, MANE select) | ||||||||
| HCM-52 | Het | Missense | Ex2:p.(Thr78Met) | rs72546668 | 23.7 | NA | 0.002667 |
MYH7: p.(Asn232Ser) MYH6: p.(His1526Arg) |
| HCM-91 | Het | Missense | Ex2:p.(Thr78Met) | rs72546668 | 23.7 | NA | 0.002667 | VCL: p.(Arg285Cys) |
| Variants identified in the VCL gene (transcript NM_014000, MANE select) | ||||||||
| HCM-91 | Het | Missense | Ex7:p.(Arg285Cys) | rs757517552 | 34 | VUS/VUS | 0.00001591 | CAV3: p.(Thr78Met) |
| HCM-61 | Het | Missense | Ex14:p.(His636Arg) | rs71579374 | 23.9 | VUS/Conflicting | 0.001485 |
MYH7: p.(Arg787Cys) MYPN: p.(Asp1208Gly) |
| Variants identified in the TCAP gene (transcript NM_003673, MANE select) | ||||||||
| HCM-85 | Het | Missense | Ex2:p.(Met71Thr) | rs143465226 | 23 | VUS/NA | 0.00001669 | Unique variant |
| Variants identified in the JPH2 gene (transcript NM_020433, MANE select) | ||||||||
| HCM-127 | Het | Missense | Ex2:p.(Phe221Leu) | rs558770240 | 23.2 | B/Conflicting | 0.001931 | MYPN: p.(Ala1141Thr) |
AA, amino acid; ACMG, American College of Medical Genetics and Genomics; Het, heterozygous; B, benign; P, pathogenic; LP, likely pathogenic; VUS, variant of uncertain significance; NA, no data available; Conflicting, conflicting interpretations of pathogenicity; CADD, combined annotation-dependent depletion scores according to CADD model v1.3; GnomAD-AF, allele frequency according to gnomAD V2.1.
Ten patients were found to carry unique variants in the FLNC gene, and three were digenic (HCM-67, HCM-66, and HCM-100) harboring an additional variant in the MYH6, MYH7, and MYPN genes, respectively. Unique variants in the MYH6 gene were identified in five patients. Three out of six patients with MYPN variants carried the same p.Pro1112Leu variant.
Less unique variants were detected in the remaining minor genes ALPK3, CSRP3, MYLK2, CAV3, VCL, and JPH2 genes. Of note, variants in NEXN (p.Glu332Ala) and TCAP (p.Met71Thr) are unique.
Emerging HCM genes: TRIM63, FHOD3, and SVIL
The main goal of this reanalysis was to identify variants within the recently associated genes not included in the initial HYPERGEN analysis. Three genes were identified toward this goalTRIM63, FHOD3, and SVIL.
TRIM63 gene
The TRIM63 gene was associated with an autosomal recessive form of HCM (40, 41). In this study, we identified variants within the TRIM63 gene in 12 patients. Three patients presented homozygous variants for TRIM63 gene, and 6 variants were unique (Table 6). Indeed, the missense variants (p.Cys23Tyr and p.Cys75Tyr) were identified in patients at the heterozygous and homozygous states. Similarly, the stop variant (p.Gln247*) was found at the homozygous state in one patient and heterozygous for two other patients. According to ACMG/AMP classification, the missense variants Cys23Tyr and Cys75Tyr are LP. The TRIM63 p.Gln247* stop variant is reported in ClinVar with conflicting interpretations of pathogenicity. A second stopgain variant (p.Glu261*) was found in a single patient (Figure 5). Of note, PTVs were identified only in MYBPC3 and TRIM63 genes.
Table 6.
Variants identified in the TRIM63 gene (transcript NM_032588, MANE select).
| Patient ID | Genotype | Variant type | AA change | rs ID | CADD | ACMG/ClinVar | GnomAD-AF | Additional variant |
|---|---|---|---|---|---|---|---|---|
| HCM-44 | Het | Missense | Ex1:p.(Cys23Tyr) | rs754874432 | 31 | LP/NA | 0.00003182 |
MYBPC3:p.(Arg1022Pro) MYH7: p.(His1185Gln) |
| HCM-111 | Hom | Missense | Ex1:p.(Cys23Tyr) | rs754874432 | 31 | LP/NA | 0.00003182 | SVIL: p.(Ser1414Thr) |
| HCM-112 | Hom | Missense | Ex2:p.(Cys75Tyr) | rs200811483 | 31 | LP/Conflicting | 0.00009952 | Unique variant |
| HCM-113 | Het | Missense | Ex2:p.(Cys75Tyr) | rs200811483 | 31 | LP/Conflicting | 0.00009952 | Unique variant |
| HCM-63 | Het | Missense | Ex2:p.(Cys75Tyr) | rs200811483 | 31 | LP/Conflicting | 0.00009952 |
MYH7: p.(Arg869His) MYLK2: p.(Ser510Leu) |
| HCM-114 | Hom | Stopgain | Ex5:p.(Gln247*) | rs148395034 | 38 | VUS/Conflicting | 0.0006787 | Unique variant |
| HCM-115 | Het | Stopgain | Ex5:p.(Gln247*) | rs148395034 | 38 | VUS/Conflicting | 0.0006787 | Unique variant |
| HCM-116 | Het | Stopgain | Ex5:p.(Gln247*) | rs148395034 | 38 | VUS/Conflicting | 0.0006787 | ACTN2: p.(Val458Met) |
| HCM-117 | Het | Stopgain | Ex5:p.(Glu261*) | rs149312738 | 40 | VUS/NA | 0.00001416 | Unique variant |
| HCM-118 | Het | Missense | Ex5:p.(Glu269Lys) | rs61749355 | 26.4 | LB/NA | 0.003159 | Unique variant |
| HCM-119 | Het | Missense | Ex5:p.(Thr262Ile) | rs889710255 | 25.6 | VUS/NA | 0.000003982 | SVIL: p.(Glu1286Lys) |
| HCM-120 | Het | Missense | Ex7:p.(Ile322Thr) | rs368532655 | 23.6 | VUS/VUS | 0.00001989 | SVIL: p.(Val1490Ala) |
AA, amino acid; ACMG, American College of Medical Genetics and Genomics; Het, heterozygous; Hom, homozygous; LB, likely benign; LP, likely pathogenic; VUS, variant of uncertain significance; NA, no data available; Conflicting, conflicting interpretations of pathogenicity; CADD, combined annotation-dependent depletion scores according to CADD model v1.3; GnomAD-AF, allele frequency according to gnomAD V2.1.
Figure 5.

Schematic representation of the TRIM63 gene. Upside: Rare variants identified in HYPERGEN cohort: Green: B/LB; Yellow: VUS; Red: LP/P; Downside: Variants reported in Clinvar database (clinical significance at last reviewed): Grey: VUS and conflicting interpretations of pathogenicity; Black: LP/P; Protein domains: Red = RING-finger domain (Zn-finger of 40 to 60 residue); Blue = B-Box-type zinc finger; Green = Zn2+ binding site; Yellow = Inter-Src homology 2 (iSH2) helical domain of Class IA Phosphoinositide 3-kinase Regulatory subunits. Figure created with ProteinPaint (https://proteinpaint.stjude.org/).
Clinical data were gathered for some patients with TRIM63 variants. Two patients have septal hypertrophy and normal left ventricular ejection fraction (LVEF) (HCM-44 and HCM-113). Two patients with homozygous TRIM63 variants (HCM-112 and HCM-114) have apical hypertrophy. The patient (HCM-117) with the stop variant p.Glu261* has a severe biventricular HCM with LVEF = 49% and mild aortic regurgitation.
FHOD3 gene
The FHOD3 locus is one of the most vital signals for HCM in genome-wide association study (GWAS) studies (42). Pathogenic variants are mainly located in two regions in the FHOD3 diaphanous inhibitory domain (exon 12) and the coiled–coil domain (exons 15 and 16). Moreover, it has been demonstrated that exons 11 and 12 are crucial for MybpC-mediated localization of the FHOD3 protein to the sarcomeric C-zone (12, 43–45). Our reanalysis further strengthens these associations by the identification of 3 missense variants in exon 12 and 15 in 6 patients of the HYPERGEN cohort. In total, 7 rare variants were prioritized in the FHOD3 gene in 10 patients (Figure 6). The majority of variants are unique except for two patients (HCM-48 and HCM-27) with the recurrent FHOD3 p.Arg637Gln variant. Both patients carried MYBPC3 variants, p.(Cys1202Leufs*35) and p.(Arg597Gln), respectively (Table 7).
Figure 6.

Schematic representation of the FHOD3 gene. Upside: Rare variants identified in HYPERGEN cohort: Green = B/LB; Yellow = VUS; Red = LP/P; Downside: Variants reported in ClinVar database (clinical significance at last reviewed): Gray = VUS and conflicting interpretations of pathogenicity; Black = LP/P; protein domain: Green = Formin Homology 2 Domain. Figure was created with ProteinPaint (https://proteinpaint.stjude.org/).
Table 7.
Variants identified in the FHOD3 gene (transcript NM_001281740, MANE select).
| Patient ID | Genotype | Variant type | AA change | rs ID | CADD | ACMG/ClinVar | GnomAD-AF | Additional variant |
|---|---|---|---|---|---|---|---|---|
| HCM-103 | Het | Missense | Ex1:p.(Arg27Pro) | rs755501978 | 33 | VUS/VUS | 0.0001619 | Unique variant |
| HCM-104 | Het | Missense | Ex6:p.(Leu177Phe) | rs781645381 | 26.8 | VUS/NA | NA | Unique variant |
| HCM-105 | Het | Missense | Ex12:p.(Ser549Asn) | NA | 21.1 | NA | NA | Unique variant |
| HCM-106 | Het | Missense | Ex15:p.(Arg637Gln) | rs151313792 | 31 | VUS/LB | 0.001728 | Unique variant |
| HCM-48 | Het | Missense | Ex15:p.(Arg637Gln) | rs151313792 | 31 | VUS/LB | 0.001728 | MYBPC3: p.(Cys1202Leufs*35) |
| HCM-27 | Het | Missense | Ex15:p.(Arg637Gln) | rs151313792 | 31 | VUS/LB | 0.001728 | MYBPC3: p.(Arg597Gln) |
| HCM-107 | Het | Missense | Ex15:p.(Arg637Gln) | rs151313792 | 31 | VUS/LB | 0.001728 | Unique variant |
| HCM-108 | Het | Missense | Ex15:p.(Arg638Trp) | rs141148037 | 35 | VUS/NA | 0.0004252 | SVIL: p.(Glu291Lys) |
| HCM-109 | Het | Missense | Ex17:p.(Glu687Lys) | rs778872098 | 24.8 | VUS/NA | 0.000007986 | Unique variant |
| HCM-110 | Het | Missense | Ex28:p.(Val1570Ile) | rs201824593 | 33 | VUS/NA | 0.0007425 | Unique variant |
AA, amino acid; ACMG, American College of Medical Genetics and Genomics; Het, heterozygous; LB, likely benign; VUS, variant of uncertain significance; NA, no data available; CADD, combined annotation-dependent depletion scores according to CADD model v1.3; GnomAD-AF, allele frequency according to gnomAD V2.1.
We had access to the clinical description of one patient (HCM-104) with FHOD3 variant (p.Leu177Phe). A definite HCM diagnosis was made at the age of 13 years. The patient had a concentric HCM with biventricular dilation. At the age of 31 years, his LVEF = 71% and RVEF = 53%.
SVIL gene
Recently, the SVIL gene was associated with HCM (46, 47). Thus, we performed a gene-targeted analysis for the SVIL gene. No PTV or homozygous variants were found in patients of the HYPERGEN cohort. Nevertheless, we identified 10 missense SVIL variants in 13 patients (Table 8; Figure 7). The prioritized variants are absent in 1920 control alleles. Five variants are unique. To better characterize the clinical presentation of SVIL variant carriers, cardiac and extracardiac features were gathered for 7 patients, two women and 5 men (Table 9). The age at onset of women patients was 29 and 27 years, one of them in the postpartum period. Moreover, the patient with SVIL: p.(Arg215Trp) had severe scoliosis with permanent bracing and muscle fasciculations. Three patients had aortopathies including a bicuspid aortic valve with severe regurgitation, isolated ectasia of the Valsalva sinus, and degenerative aortic insufficiency. A consistent pattern of fibrosis localization was noted in these patients in the septum and LV apex. Magnetic resonance imaging (MRI) findings showed significant myocardial fibrosis for the majority of patients with intramyocardial delayed contrast in the inferior and lateral walls (Table 9).
Table 8.
Variants identified in the SVIL gene (transcript NM_021738, MANE select).
| Patient ID | Genotype | Variant type | AA change | rs ID | CADD | ACMG/ClinVar | GnomAD-AF | Additional variant |
|---|---|---|---|---|---|---|---|---|
| HCM-129 | Het | Missense | Ex6:p.(Arg215Trp) | rs74132329 | 26.2 | B/B | 0.003420 | Unique variant |
| HCM-108 | Het | Missense | Ex7:p.(Glu291Lys) | rs372012664 | 30 | B/NA | 0.00004242 | FHOD3: p.(Arg638Trp) |
| HCM-130 | Het | Missense | Ex8:p.(Glu313Gly) | rs138539716 | 25.7 | B/NA | 0.004283 | Unique variant |
| HCM-119 | Het | Missense | Ex21:p.(Glu1286Lys) | rs145704372 | 27.6 | LB/NA | 0.0003 | TRIM63: p.(Thr262Ile) |
| HCM-111 | Het | Missense | Ex23:p.(Ser1414Thr) | rs144661370 | 29.6 | B/LB | 0.001681 | TRIM63: p.(Cys23Tyr) |
| HCM-134 | Het | Missense | Ex23:p.(Ser1414Thr) | rs144661370 | 29.6 | B/LB | 0.001681 | Unique variant |
| HCM-62 | Het | Missense | Ex23:p.(Ser1414Thr) | rs144661370 | 29.6 | B/LB | 0.001681 | MYH7: p.(Gly768Arg) |
| HCM-120 | Het | Missense | Ex24:p.(Val1490Ala) | rs899379947 | 29.2 | B/NA | 0.00002477 | TRIM63: p.(Ile322Thr) |
| HCM-39 | Het | Missense | Ex25:p.(Ala1496Asp) | 32 | MYBPC3: p.(Trp792ValfsTer41) | |||
| HCM-40 | Het | Missense | Ex27:p.(Thr1630Ser) | rs28451028 | 22.7 | VUS/NA | 0.004201 |
MYBPC3: p.(Ala833Val) MYH6: p.(Arg1532Leu) |
| HCM-82 | Het | Missense | Ex27:p.(Thr1630Ser) | rs28451028 | 22.7 | VUS/NA | 0.004201 | CSRP3: p.(Trp4Arg) |
| HCM-138 | Het | Missense | Ex31:p.(Arg1870Gln) | rs762138005 | 35 | VUS/NA | 0.00001065 | Unique variant |
| HCM-139 | Het | Missense | Ex33:p.(Lys1960Arg) | 19 | 0.000003976 | Unique variant |
AA, amino acid; ACMG, American College of Medical Genetics and Genomics; Het, heterozygous; B, benign; VUS, variant of uncertain significance; NA, no data available; CADD, combined annotation-dependent depletion scores according to CADD model v1.3; GnomAD-AF, allele frequency according to gnomAD V2.1.1.
Figure 7.

Schematic representation of the SVIL gene. Upside: Rare variants identified in HYPERGEN cohort: Green = B/LB; Yellow = VUS; Downside: Variants reported in ClinVar database (clinical significance at last reviewed): Gray = VUS and conflicting interpretations of pathogenicity; Black = LP/P; Protein domains: Green, blue, yellow, gray, and brown = gelsolin-like domains. Figure was created with ProteinPaint (https://proteinpaint.stjude.org/).
Table 9.
Clinical findings of SVIL variant carriers.
| Patient ID | HCM-129 | HCM-130 | HCM-108 | HCM-134 | HCM-119 | HCM-138 | HCM-139 |
|---|---|---|---|---|---|---|---|
| Gender | Female | Female | Male | Male | Male | Male | Male |
| Age at onset | 29 years (postpartum period) | 27 years | 40 years | 29 years | 48 years | 24 years | 60 years |
| Initial symptoms | NA | Asthenia and dyspnea since childhood | SCV (VF) | Asthenia | A symptomatic | Asymptomatic | Syncopes during effort |
| Muscular abnormalities/deformities | Severe scoliosis with permanent bracing, muscle fasciculations | Isolated back pain | Not checked | None | Not reported by the patient/not checked | Not reported by the patient/not checked | Not reported by the patient/not checked |
| Arrhythmias | AF | NSVT | AF and VT/VF | AF | None | None | None |
| LV thickness TTE/MRI | 26/19 mm | 32/22 mm | 31/30 mm | 23/25 mm | 19/19 mm | 22/22 mm | 16/15 mm |
| Fibrosis localization on MRI | LGE intramyocardial anteroseptal and apical | Severe intramyocardial LGE Apical and on the RV (rare) | Severe intramyocardial LGE inferoseptal and lateral | Severe intramyocardial LGE Septal | LGE intramyocardial Septal and apical | LGE Not significant | LGE intramyocardial anteroseptal |
| LVEF (%) TTE/MRI | 60/30 | 50/64 | 50/63 | 70/67 | 65/70 | 75/79 | 70/74 |
| Aortic phenotype | Normal | Normal | Normal | Bicuspid aortic valve associated with dilation of Valsalva’s sinus (41 mm) and severe aortic regurgitation Aortic valve replacement |
Isolated ectasia of Valsalva’s sinus (39 mm) | Normal | Degenerative Aortic insufficiency (mild) without aortic dilatation |
| CK (0–170) | 150–200 | 100–200 | 315 | 150–900 | 95 | CPK (39–308): 79 | CPK (39–308): 161 |
| Tropo T (<14) | 29–34 | 15–21 | 32–35 | Tropo Ic US (0,000 – 0,050): 0,197 | 13 | Tropo Ic US (0,000 – 0,050): 0,023 | Tropo Ic US (0,000 – 0,050): <0,006 |
| NT-proBNP | 1,100–3,000 | 2,350 | 300–900 | BNP (0–100): 713 | 379 | BNP (0–100): 70 | BNP (0–100): 66 |
| SVIL variant | SVIL: p.(Arg215Trp) | SVIL: p.(Glu313Gly) | SVIL: p.(Glu291Lys) | SVIL: p.(Ser1414Thr) | SVIL: p.(Glu1286Lys) | SVIL: p.(Arg1870Gln) | SVIL: p.(Lys1960Arg) |
| Additional variant | None | None | FHOD3: p.(Arg638Trp) | None | TRIM63: p.(Thr262Ile) | None | None |
| Extracardiac features | Sickle cell disease; Bilateral hips prothesis; Appendicitis Osteoporosis Type-2 diabetes Hyperuricemia Arterial hypertension Tuberculosis (15 years) Non-secreting adrenal benign tumor Glaucoma |
Sleep apnea syndrome | NA | NA | NA | NA | NA |
| Neurological exam/findings | Muscular fasciculations | Mild paresthesia (nocturnal) | Normal | Not performed | Not performed | Not performed | Not performed |
AF, atrial fibrillation; CK, creatine kinase; HCM, hypertrophic cardiomyopathy; LGE, late gadolinium enhancement; LVEF, left ventricular ejection fraction; RV, right ventricular; VF, Ventricular fibrillation; VT, ventricular tachycardia; MRI, magnetic resonance imaging; NSVT, non-sustained ventricular tachycardia; NA, not available; SCD, sudden cardiac death; TTE, transthoracic echocardiography.
Only seven variants of the HYPERGEN cohort were present in the control cohort with low allele frequencies (Table 10). Moreover, four out of these seven variants were found to have the highest allele frequencies in the European non-Finnish population in gnomAD.
Table 10.
HYPERGEN variants found in the control cohort.
| Variant | Allele count | Number of homozygotes | Allele number | AF in a French control cohort | MAF Highest population in gnomAD |
|---|---|---|---|---|---|
| TCAP: p.Met71Thr | 2 | 0 | 1920 | 0.001 | African/African American: 6.582e-5 |
| ALPK3: p.Arg1483Trp | 1 | 0 | 1920 | 5.2083e-4 | European non-Finnish: 6.155e-5 |
| VCL: p.His636Arg | 1 | 0 | 1920 | 5.2083e-4 | European Finnish: 0.0063 |
| FLNC: p.Arg1860Cys | 28 | 2 | 1920 | 0.0145 | European non-Finnish: 0.0082 |
| TRIM63: p.Glu269Lys | 6 | 0 | 1920 | 0.0031 | European non-Finnish: 0.0058 |
| TRIM63: p.Ile322Thr | 6 | 0 | 1920 | 0.0031 | European non-Finnish: 4.397e-5 |
| MYPN: p.Pro1112Leu | 3 | 0 | 1920 | 0.0015 | Ashkenazi Jewish: 0.0097 |
AF, allele frequency; MAF, minor allele frequency.
The genetic landscape of young HYPERGEN patients
The HYPERGEN cohort included 27 patients with HCM occurring at a very young age or in early adulthood (13.5%). The HCM clinical diagnosis of the patients was definite before the age of 40 years. We sought to determine the genetic architecture of this young proportion. Indeed, sarcomeric genes were the most involved genes, as 50% of the identified variants are within the MYBPC3 gene. Interestingly, variants in SVIL and FHOD3 genes accounted for 10 and 7% of young HYPERGEN patients, respectively (Figure 8).
Figure 8.

Percentages of gene variants distribution among young patients.
In summary, a total of 20 genes have been identified in the HYPERGEN cohort, with a significant implication of the MYBPC3 gene, followed by MYH7 and SVIL genes (Figure 9). According to ClinGen, 8 out of the 20 identified genes are classified with a robust association with HCM, 7 with disputed/limited evidence, and the JPH2 gene with a moderate gene–disease validity. The FLNC, CAV, and SVIL are not curated for HCM, and the FHOD3 gene is under curation (Table 11). However, those genes are reported in the literature and in the OMIM database in association with HCM.
Figure 9.

Genes implicated in the HYPERGEN cohort. The number of variants is indicated in dark purple, and the number of patients is indicated in light purple.
Table 11.
List of the identified genes in the HYPERGEN cohort and their matched ClinGen classification.
| Gene symbol | Gene name | Gene-HCM validity classification | Inheritance | Gene–disease validity classification | Inheritance | |
|---|---|---|---|---|---|---|
| MYBPC3 | Myosin binding protein C3 | Definitive (MONDO:0005045) | AD | Dilated cardiomyopathy (MONDO:0005021) | Limited | AD |
| Arrhythmogenic right ventricular cardiomyopathy (MONDO:0016587) | Limited | |||||
| MYH7 | Myosin heavy chain 7 | Definitive (MONDO:0005045) | AD | Dilated cardiomyopathy (MONDO:0005021) | Definitive | AD |
| MYH7-related skeletal myopathy (MONDO:0008050) | Definitive | |||||
| Arrhythmogenic right ventricular cardiomyopathy (MONDO:0016587) | Limited | |||||
| Congenital heart disease (MONDO:0005453) | Limited | |||||
| MYL2 | MLC 2 | Definitive (MONDO:0005045) Definitive (MONDO:0012112) |
AD | Dilated cardiomyopathy (MONDO:0005021) | Limited | AD |
| Arrhythmogenic right ventricular cardiomyopathy (MONDO:0016587) | No known disease relationship | |||||
| MYL3 | MLC 3 | Definitive (MONDO:0005045) | AD | Arrhythmogenic right ventricular cardiomyopathy (MONDO:0016587) | Limited | AD |
| Dilated cardiomyopathy (MONDO:0005021) | Disputed | |||||
| TNNT2 | Troponin T2, Cardiac Type | Definitive (MONDO:0005045) | AD | Dilated cardiomyopathy (MONDO:0005021) | Definitive | AD |
| Arrhythmogenic right ventricular cardiomyopathy (MONDO:0016587) | No known disease relationship | |||||
| MYLK2 | MLC Kinase 2 | Disputed (MONDO:0005045) | AD | NA | NA | NA |
| CSRP3 | Cysteine and glycine-rich protein 3 | Definitive (MONDO:0005045) | SD | Dilated cardiomyopathy (MONDO:0005021) | Limited | AD |
| Moderate (MONDO:0005045) | AD | |||||
| MYH6 | Myosin heavy chain 6 | Limited (MONDO:0005045) | AD | Dilated cardiomyopathy (MONDO:0005021) | Limited | AD |
| ACTN2 | Actinin α2 | Definitive (MONDO:0000591) | AD | NA | NA | NA |
| ALPK3 | α-Kinase 3 | Definitive (MONDO:0005045) | AR | NA | NA | NA |
| VCL | Vinculin | Disputed (MONDO:0005045) | AD | Dilated cardiomyopathy (MONDO:0005021) | Moderate | NA |
| JPH2 | Junctophilin 2 | Moderate (MONDO:0005045) | AD | Dilated cardiomyopathy (MONDO:0005021) | Moderate | SD |
| NEXN | Nexilin F-actin binding protein | Limited (MONDO:0005045) | AD | Dilated Ccrdiomyopathy (MONDO:0005021) | Moderate | AD |
| MYPN | Myopalladin | Disputed (MONDO:0005045) | AD | MYPN-related Myopathy (MONDO:0015023) | Definitive | AR |
| Dilated cardiomyopathy (MONDO:0005021) | Limited | AD | ||||
| TCAP | Titin-Cap | Disputed (MONDO:0005045) | AD | Limb–girdle muscular dystrophy (MONDO:0015152) | Definitive | AR |
| Dilated cardiomyopathy (MONDO:0005021) | Limited | AD | ||||
| FLNC | Filamin C | NA | NA | Dilated cardiomyopathy (MONDO:0005021) | Definitive | AD |
| Myofibrillar myopathy 5 (MONDO:0012289) | Definitive | |||||
| FHOD3 | Formin homology 2 domain containing 3 | Under active curation | NA | NA | NA | |
| CAV3 | Caveolin 3 | NA | NA | Caveolinopathy (MONDO:0016146) | Definitive | AD |
| Long QT syndrome (MONDO:0002442) | Limited | |||||
| TRIM63 | Tripartite motif containing 63 | Moderate (MONDO:0005045) | AR | NA | NA | NA |
| Disputed (MONDO:0005045) | AD | |||||
| SVIL | Supervillin | NA | NA | NA | NA | NA |
AD: autosomal dominant; AR: autosomal recessive; HCM, hypertrophic cardiomyopathy; MLC, myosin light chain; NA, not available; SD: semidominant.
Our reanalysis yielded 16 novel variants, including four in the MYBPC3 gene, three in MYH7, two variants were found in each of MYH6 and FLNC genes, and one novel variant in TNNT2, MYL2, MYPN, ALPK3, and CSRP3, respectively.
Of note, considering only patients with unique variants in TRIM63, FHOD3, and SVIL genes, there was a 9% enhancement in variant identification following this reanalysis.
All the identified genes and their matched phenotypes in the OMIM database are summarized in Supplementary File 3.
Discussion
Despite the advent of next-generation sequencing, approximately 50% of cases remain genotype-elusive. In this study, a reanalysis of exome data from 200 HCM patients was carried out 5 years after the initial analysis with the goal of refining the initial analysis, reporting all the relevant, prioritized variants, and particularly identifying rare variants within the novel HCM-associated genes.
In the present reanalysis, nearly 34% of patients were found to carry variants in MYBPC3 and MYH7 genes. This yield of sarc+ patients reached 37% when including patients with TNNT2, MYL2, and MYL3 variants.
Haploinsufficiency is the primary mechanism driving HCM linked to MYBPC3 (myosin binding protein C3) gene (38, 48). Thus, non-truncating variants (missense, in-frame indels, PTV predicted to escape non-sense mediated decay [NMD], and stop-loss) are prioritized whether they are predicted as pathogenic with high or low confidence. Our reanalysis yielded a total of 40 MYBPC3 variants, including 24 missense, 4 splice-site, 6 stopgain, and 6 frameshift variants. The most recurrent and common HCM causal variant MYBPC3, p.Arg502Trp, was identified in only three patients (1.5%). This variant was found in 2.4% of the European-descent patients (15, 49–51). However, the pathogenic MYBPC3: c1928-2A > G variant was the most commonly identified variant in the HYPERGEN cohort (n = 7; 3.5%). Notably, these variants are unique except for one patient (HCM-5) harboring a second MYBPC3 variant (p.Leu183Ile) (Table 1). Contrary to the MYBPC3 gene, mostly known with haploinsufficiency and allelic imbalance as the underlying mechanism of the disease, the pathophysiology mechanism of MYH7 (myosin heavy chain 7) missense variants is the dominant negative effect, which implies that the MYH7 gene is tolerant to loss of function (LoF) variants (10, 38, 39, 52, 53). Thus, prioritizing the MYH7 missense variant with a predicted deleterious impact according to different in silico algorithms may increase the identification rate of actionable variants. Moreover, gene regions in which rare and likely causal variants are significantly clustered; therefore, a new etiological fraction-based ACMG rule on rare missense MYH7 variants was proposed to improve genetic testing yield in HCM (30). All the identified MYH7 variants in this study are missense (n = 16) with 8 P/LP variants. Of note, variants in the MYH7 gene were associated with significant LV hypertrophy, which is the hallmark of HCM and an unfavorable prognosis compared to patients carrying variants in the other HCM genes (54).
We identified a small proportion of patients (3%) carrying variants in TNNT2, MYL2, and MYL3 genes. Only two variants in the TNNT2 gene were identified. The TNNT2 gene encodes the cardiac troponin T2 which is a regulatory protein of the thin filament troponin complex in the sarcomere playing a crucial role in contractility function (39, 55). More than 30 TNNT2 P/LP variants have been linked to HCM, with individual variants being unique and private to distinct families (55). Moreover, phenotypic variability was reported among patients carrying the same TNNT2 variant (55). The MLCs are composed of a regulatory light chain (MYL2) and an essential light chain (MYL3). These genes contribute to the stability of myosin head and the regulation of cardiac contraction by phosphorylation and Ca2+ binding (52). Despite these genes being considered well-established HCM genes, their contribution to HCM etiology is limited. A study by Borrelli et al. estimated that the contribution of genes encoding sarcomere thin filaments does not exceed 5% (11). In our cohort, no high or moderate confidence variants were found in ACTC1, TPM1, TNNC1, or TNNI3 genes. A recent study by Allouba et al. (56) showed that homozygous variants are more prevalent within MYL2 and MYL3 genes than within major sarcomeric HCM genes. In the HYPERGEN cohort, only one homozygous variant was identified (MYL3: p.Ala57Asp) in a patient with a family history of HCM.
Although HCM has been recognized as a monogenic disease for a long time, the wide utilization of high-throughput sequencing has demonstrated that it may be caused by the occurrence of more than one variant, particularly for sarc−patients. In this reanalysis, considering sarc+patients, we identified three patients with two variants within the MYBPC3 gene, seven cases carrying digenic variants, and five patients carrying more than two variants (Tables 1, 2). In some sarc−cases, unique variants were found in genes encoding proteins located in the Z-disc, namely, FLNC, ACTN2, CSRP3, TRIM63, and SVIL (Tables 5–8). Of note, variants within CAV3, VCL, and JPH2 genes were found along with additional sarcomeric and non-sarcomeric variants.
We identified three CSRP3 variants, including the known p.Trp4Arg variant. The CSRP3 gene, encoding cysteine and glycine-rich protein 3, is one of the non-sarcomeric HCM-associated genes with strong evidence for a primary pathogenic role in HCM (13). It plays different roles in mechanosensory functions and actin cytoskeleton assembly (12, 57). Functional analysis of Csrp3 knock-in animals (Csrp3Trp4Arg/+) and (Csrp3Trp4Arg/Trp4Arg) showed an age-and gene dosage-dependent HCM and heart failure, characterized by a nearly complete loss of contractile function under catecholamine-induced stress. Moreover, Cspr3 mRNA and protein levels were significantly decreased in the hearts of heterozygous and homozygous Cspr3Trp4Arg knock-in animals (57).
We identified five variants in the ACTN2 gene, three of which were unique, and two patients were digenic and were carrying additional variants in MYBPC3 and TRIM63 genes. The ACTN2 gene (major Z-disc cross-linking protein) is of particular interest as variants within this gene have been linked to diverse cardiac phenotypes such as HCM, dilated cardiomyopathy (DCM), LV non-compaction (LVNC), and SCD (Supplementary File 3) (58–61). Patients with ACTN2 pathogenic variants showed no specific hypertrophy pattern, as septal, apical, concentric, and biventricular hypertrophy were reported. More importantly, patients with mild hypertrophy had severe complications such as resuscitated cardiac arrest and heart failure (12, 61–64).
A small number of variants was identified within the ALPK3 gene (n = 4), 3 out of the 4 variants were unique. Of note, the ALPK3 gene (α-protein kinase 3) was initially linked to an autosomal recessive form of severe pediatric mixed cardiomyopathy (HCM/DCM phenotype) (65–67). In 2020, the ALPK3 gene reached a definitive classification of strong evidence for an autosomal recessive form of HCM with an infant onset (46). However, cases harboring heterozygous LoF variants were reported with mild-to-moderate phenotypes, and an autosomal dominant pattern of inheritance was subsequently associated with an adult-onset HCM (12, 46, 68, 69). Recently, rare missense ALPK3 variants have been identified in Asian HCM patients (46, 70).
Three genes were recently associated with HCM and identified in this reanalysis: TRIM63, FHOD3, and SVIL. TRIM63 is one of the rare genes recently described as a cause of HCM with autosomal-recessive inheritance. TRIM63 encodes muscle-specific RING-finger protein 1 (MuRF1), a member of the ubiquitin ligases subfamily, such as MuRF-2 and MuRF-3 (71, 72). It is an E3 ubiquitin−protein that regulates the degradation of sarcomeric proteins such as Mybpc3 and Myh6 through ubiquitylation (12, 72). Homozygous and compound heterozygous rare variants in the TRIM63 gene were linked to HCM. TRIM63 variant carriers showed concentric LV hypertrophy, significant cardiac fibrosis, LV systolic dysfunction, and arrhythmias (12, 40, 41). Furthermore, systolic dysfunction and late gadolinium enhancement have been reported as characteristic features of TRIM63-associated cardiomyopathies (41). Rare missense variants in TRIM63 at the heterozygous state were reported as genetic modifiers of HCM. Although the TRIM63 missense variants had limited evidence of disease causality, TRIM63 knockouts are likely to be associated with HCM, given their enrichment in HCM patients and their absence in gnomAD (12, 41). In our reanalysis, the two LP missense variants (p.Cys23Tyr and p.Cys75Tyr) were identified in 5 patients of the HYPERGEN cohort. The TRIM63: (p.Gln247*) stop variant was identified in three patients, one at the homozygous state and two patients were heterozygous. The p.Gln247* variant is reported in the ClinVar database with a conflicting interpretation of pathogenicity. Nevertheless, in vitro and in vivo functional studies showed near complete loss of auto-ubiquitination in cells transduced with the TRIM63Q247* lentiviral construct (12, 41). Several other TRIM63 variants impair the ubiquitination of Trim63 substrates in adult cardiomyocytes. These findings implicate the impaired protein degradation as a pathophysiology mechanism of HCM (40, 41).
Of note, in the HYPERGEN cohort, five patients (2.5%) carried homozygous variants in MYBPC3 (n = 1), MYL3 (n = 1), and TRIM63 (n = 3) genes.
The second recent gene identified in this reanalysis is the FHOD3 gene. FHOD3 encodes the cardiac formin homology 2 domain containing three proteins that localize in the thin filament of the sarcomere and promote actin filament polymerization in cardiomyocytes (44).
Rare pathogenic FHOD3 variants were linked to HCM etiology through association analysis of 3,189 HCM patients and familial segregation studies (43). FHOD3 variants associated with HCM are mostly non-truncating and disturb the diaphanous inhibitory domain of the protein. Patients carrying likely causative FHOD3 variants showed mild-to-moderate LV hypertrophy and a 1% annual incidence of cardiovascular mortality (43). Additionally, FHOD3-HCM patients were diagnosed in adulthood (mean age 46.1 years), and two-thirds (66%) were men. The majority of patients (82%) had asymmetric septal hypertrophy (mean 18.8 ± 5 mm). LV ejection fraction <50% was present in 14% of the cohort and hypertrabeculation in 16% (43). Moreover, the cardiomyopathic phenotype of cMyBP-C null mice was aggravated by Fhod3 overexpression with a sarcomere integrity disruption. This phenotype was partially improved by a reduction in the Fhod3 protein levels, suggesting that Fhod3 has a damaging impact on cardiac function under cMyBP-C null conditions where Fhod3 is mis-localized (44). These findings suggest the likely contribution of Fhod3 to the pathogenesis of cMyBP-C-related cardiomyopathy and that Fhod3 is implicated in cardiac cMyBP-C-mediated regulation via direct interaction (44). In this study, rare missense FHOD3 variants were identified in the hotspot exons (12 and 15), and two patients with the recurrent FHOD3 p.Arg637Gln variant were digenic, carrying additional variants in the MYBPC3 gene (p.Cys1202Leufs*35 and p.Arg597Gln).
More recently, the SVIL gene was associated with HCM (46, 47). The SVIL (Supervillin) encodes a multidomain actin and myosin-binding protein in the Z-disc (73, 74). Biallelic LoF SVIL variants were identified in patients with skeletal myopathy, mild LV hypertrophy, and smaller descending aortic diameter (75). Indeed, a 10.5-fold excess burden of SVIL rare PTV variants in HCM cases has been demonstrated in a recent GWAS (47). Patients harboring rare truncating SVIL variants were found to have an increased LV contractility in both obstructive and non-obstructive forms of HCM, as demonstrated by Mendelian randomization analyses (47). In one family, the SVIL: p.(Gln255*) variant was found in two affected cousins, providing some evidence of familial segregation (47). In this reanalysis, clinical data were gathered for 7 out of the 13 patients with SVIL variants (Table 9). Three patients harboring the SVIL: p.(Ser1414Thr), p.(Glu1286Lys), and p.(Lys1960Arg) variants presented an aortic phenotype including bicuspid aortic valve disease, isolated ectasia of Valsalva sinus, and mild aortic insufficiency. Overall, the majority of SVIL patients showed severe intramyocardial late gadolinium enhancement. One patient (HCM-108) experienced SCD, and another patient (HCM-139) had a history of syncopes during physical effort. Of note, the patient (HCM-108) carried an additional variant in the FHOD3 gene (p.Arg638Trp). A severe scoliosis was diagnosed in one patient (HCM-129).
Interestingly, variants in SVIL and FHOD3 genes accounted for 10 and 7%, respectively, of patients with early HCM onset. Patients with sarcomere mutations have been reported to show earlier adverse complications and a worse prognosis (76). Several published HCM cohorts, studies, and data provided by the Sarcomeric Human Cardiomyopathy Registry (SHaRe) have shown that cases with sarcomeric variants had an early onset of the disease and a more severe phenotype with malignant complications. Thus, patients diagnosed in early adulthood (<40 years) had more severe outcomes with many adverse complications compared to patients with late-onset HCM. Moreover, HCM young patients (20–29 years) had a 4-fold increase in the risk of death compared to the general population (76–78).
All the identified variants in this reanalysis were searched in a French control cohort, gathering 960 healthy individuals. Only seven variants were present (Table 10). Of note, TCAP: p.(Met71Thr), ALPK3: p.(Arg1483Trp), FLNC: p.(Arg1860Cys) and MYPN: p.(Pro1112Leu) variants were unique in HYPERGEN carriers. The VCL: p.(His636Arg) and MYPN: p.P (ro1112Leu) variants were found in Finnish and Ashkenazi Jewish populations, respectively, suggesting that these variants could be more prevalent in these bottleneck populations.
Although the identification of variants in genes lacking strong evidence for disease causality and/or VUS in minor HCM genes does not increase the clinically actionable genes/variants discovery, it sheds light on the possibility of a combinatorial joint effect where VUS variants may act as a small risk increasing genetic factor.
Gathering strong proof of disease causality remains challenging for many reasons, such as the unavailability of family members to undergo co-segregation and funding constraints for functional studies. Indeed, genes such as ALPK3 and FHOD3 have been considered disease-causing by large gene-centric case–control studies. This strategy is an effective approach to reaching the needed statistical power supporting gene–disease association (46). Furthermore, the publication of cases with variants of low to moderate evidence was recommended, as the identical variants may be detected in extended families with similar phenotypes by researchers interested in functional validation (46).
Presently, we aimed to reanalyze a cohort of 200 HCM patients by focusing on genes strongly associated with HCM while also investigating newly linked genes with limited or insufficient evidence of causality to determine their potential involvement in our cohort. Overall, our results strengthen the implication of FHOD3, TRIM63, and SVIL genes in HCM as minor genes. Variants in the SVIL gene, recently linked to HCM, were found in 13 HYPERGEN patients. More importantly, patients harboring SVIL variants presented a similar hypertrophic pattern (significant myocardial fibrosis for six out of the seven patients/apical for three patients), and aortopathy was reported for three patients. The findings from our study extend the clinical and genetic spectrum of SVIL gene carriers. While Tadros et al. (47) identified SVIL variants in HCM patients, our study reveals additional cases, underscoring the need for expanded genetic screening. This broader understanding can facilitate future investigations into the pathophysiological mechanisms associated with SVIL variants, potentially leading to improved risk stratification and management strategies for patients.
The assessing and reporting of the identified variants—particularly within the novel associated genes—may provide additional strength to the previously reported variants and identify novel variants that help an accurate classification and interpretation. This study contributes to defining the genetic landscape of French HCM patients, which facilitates cascade genetic testing in familial cases and ultimately could guide personalized treatment.
The main goal of this study was to assess and report actionable and high-confidence variants in known and emerging HCM genes. However, there are several limitations in our study, making an accurate estimation of the increased diagnostic yield challenging. This is mainly due to the absence of detailed genetic results from the initial analysis. In addition, detailed patient phenotyping and clinical data are lacking. Thus, genotype–phenotype correlations and risk stratification were difficult to achieve.
Funding Statement
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by SANOFI GENZYME SA (contract number C01550), the "Association Française contre les Myopathies" (NMH-Decrypt Project), and the "Institut National de la Santé et de la Recherche Médicale" to SZ. HJ received a postdoctoral fellowship from the "Fondation Lefoulon Delalande".
Footnotes
Data availability statement
The data presented in the study are deposited in the ClinVar database under the following link: https://www.ncbi.nlm.nih.gov/clinvar/submitters/508840/.
Ethics statement
The studies involving humans were approved by Comité d’éthique de la Recherche d’Aix-Marseille Université. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
HJ: Conceptualization, Data curation, Formal analysis, Investigation, Validation, Writing – original draft, Writing – review & editing. VM: Data curation, Formal analysis, Writing – review & editing. HM: Data curation, Investigation, Writing – review & editing. PL: Data curation, Formal analysis, Methodology, Writing – review & editing. LC: Formal analysis, Investigation, Writing – review & editing. MH: Formal analysis, Investigation, Writing – review & editing. CL: Data curation, Writing – review & editing. FM: Resources, Supervision, Validation, Writing – review & editing. HG: Data curation, Investigation, Resources, Supervision, Validation, Writing – review & editing. J-JS: Data curation, Resources, Validation, Writing – review & editing. SZ: Conceptualization, Data curation, Investigation, Resources, Supervision, Validation, Writing – review & editing. KN: Data curation, Formal analysis, Investigation, Resources, Validation, Writing – review & editing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2024.1480947/full#supplementary-material
References
- 1.Richard P, Charron P, Carrier L, Ledeuil C, Cheav T, Pichereau C, et al. Hypertrophic cardiomyopathy: distribution of disease genes, Spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation. (2003) 107:2227–32. doi: 10.1161/01.CIR.0000066323.15244.54 [DOI] [PubMed] [Google Scholar]
- 2.Muresan ID, Agoston-Coldea L. Phenotypes of hypertrophic cardiomyopathy: genetics, clinics, and modular imaging. Heart Fail Rev. (2021) 26:1023–36. doi: 10.1007/s10741-020-09931-1, PMID: [DOI] [PubMed] [Google Scholar]
- 3.Maron BJ, Desai MY, Nishimura RA, Spirito P, Rakowski H, Towbin JA, et al. Diagnosis and evaluation of hypertrophic cardiomyopathy: JACC state-of-the-art review. J Am Coll Cardiol. (2022) 79:372–89. doi: 10.1016/j.jacc.2021.12.002 [DOI] [PubMed] [Google Scholar]
- 4.Litt MJ, Ali A, Reza N. Familial hypertrophic cardiomyopathy: diagnosis and management. Vasc Health Risk Manag. (2023) 19:211–21. doi: 10.2147/VHRM.S365001, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bourfiss M, van Vugt M, Alasiri AI, Ruijsink B, van Setten J, Schmidt AF, et al. Prevalence and disease expression of pathogenic and likely pathogenic variants associated with inherited cardiomyopathies in the general population. Circ Genomic Precis Med. (2022) 15:e003704. doi: 10.1161/CIRCGEN.122.003704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maron BJ, Desai MY, Nishimura RA, Spirito P, Rakowski H, Towbin JA, et al. Management of Hypertrophic Cardiomyopathy: JACC state-of-the-art review. J Am Coll Cardiol. (2022) 79:390–414. doi: 10.1016/j.jacc.2021.11.021 [DOI] [PubMed] [Google Scholar]
- 7.Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol. (2015) 65:1249–54. doi: 10.1016/j.jacc.2015.01.019, PMID: [DOI] [PubMed] [Google Scholar]
- 8.Members WC, Ommen SR, Mital S, Burke MA, Day SM, Deswal A, et al. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: Executive summary: A report of the American College of Cardiology/American Heart Association joint committee on clinical practice guidelines. Circulation. (2020) 142:e533–57. doi: 10.1161/CIR.0000000000000938 [DOI] [PubMed] [Google Scholar]
- 9.Morimoto S. Sarcomeric proteins and inherited cardiomyopathies. Cardiovasc Res. (2008) 77:659–66. doi: 10.1093/cvr/cvm084, PMID: [DOI] [PubMed] [Google Scholar]
- 10.Sabater-Molina M, Pérez-Sánchez I, JPH R, Gimeno JR. Genetics of hypertrophic cardiomyopathy: a review of current state. Clin Genet. (2018) 93:3–14. doi: 10.1111/cge.13027 [DOI] [PubMed] [Google Scholar]
- 11.Borrelli F, Losi MA, Canciello G, Todde G, Perillo EF, Ordine L, et al. Sarcomeric versus non-Sarcomeric HCM. Cardiogenetics. (2023) 13:92–105. doi: 10.3390/cardiogenetics13020009 [DOI] [Google Scholar]
- 12.Walsh R, Offerhaus JA, Tadros R, Bezzina CR. Minor hypertrophic cardiomyopathy genes, major insights into the genetics of cardiomyopathies. Nat Rev Cardiol. (2022) 19:151–67. doi: 10.1038/s41569-021-00608-2, PMID: [DOI] [PubMed] [Google Scholar]
- 13.Walsh R, Buchan R, Wilk A, John S, Felkin LE, Thomson KL, et al. Defining the genetic architecture of hypertrophic cardiomyopathy: re-evaluating the role of non-sarcomeric genes. Eur Heart J. (2017) 38:3461–8. doi: 10.1093/eurheartj/ehw603, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lopes LR, Syrris P, Guttmann OP, O’Mahony C, Tang HC, Dalageorgou C, et al. Novel genotype–phenotype associations demonstrated by high-throughput sequencing in patients with hypertrophic cardiomyopathy. Heart. (2015) 101:294–301. doi: 10.1136/heartjnl-2014-306387, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lopes LR, Zekavati A, Syrris P, Hubank M, Giambartolomei C, Dalageorgou C, et al. Genetic complexity in hypertrophic cardiomyopathy revealed by high-throughput sequencing. J Med Genet. (2013) 50:228–39. doi: 10.1136/jmedgenet-2012-101270, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maron BJ, Maron MS, Maron BA, Loscalzo J. Moving beyond the sarcomere to explain heterogeneity in hypertrophic cardiomyopathy. J Am Coll Cardiol. (2019) 73:1978–86. doi: 10.1016/j.jacc.2019.01.061, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marian AJ. Oligogenic cardiomyopathy. J Cardiovasc. Aging. (2022) 2:3. doi: 10.20517/jca.2021.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baulina NM, Kiselev IS, Chumakova OS, Favorova OO. Hypertrophic cardiomyopathy as an Oligogenic disease: transcriptomic arguments. Mol Biol. (2020) 54:840–50. doi: 10.1134/S0026893320060023 [DOI] [PubMed] [Google Scholar]
- 19.Helms AS, Day SM. Other side of the coin: the missing heritability in hypertrophic cardiomyopathy. Eur Heart J. (2017) 38:3469–71. doi: 10.1093/eurheartj/ehx024, PMID: [DOI] [PubMed] [Google Scholar]
- 20.Li L, Bainbridge MN, Tan Y, Willerson JT, Marian AJ. A potential Oligogenic etiology of hypertrophic cardiomyopathy novelty and significance: a classic single-gene disorder. Circ Res. (2017) 120:1084–90. doi: 10.1161/CIRCRESAHA.116.310559, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gorlov I, Gorlova O, Frazier M, Spitz M, Amos C. Evolutionary evidence of the effect of rare variants on disease etiology. Clin Genet. (2011) 79:199–206. doi: 10.1111/j.1399-0004.2010.01535.x, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zuk O, Schaffner SF, Samocha K, Do R, Hechter E, Kathiresan S, et al. Searching for missing heritability: designing rare variant association studies. Proc Natl Acad Sci. (2014) 111:E455–64. doi: 10.1073/pnas.1322563111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schobers G, Schieving JH, Yntema HG, Pennings M, Pfundt R, Derks R, et al. Reanalysis of exome negative patients with rare disease: a pragmatic workflow for diagnostic applications. Genome Med. (2022) 14:66. doi: 10.1186/s13073-022-01069-z, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hiatt SM, Amaral MD, Bowling KM, Finnila CR, Thompson ML, Gray DE, et al. Systematic reanalysis of genomic data improves quality of variant interpretation. Clin Genet. (2018) 94:174–8. doi: 10.1111/cge.13259, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nguyen K, Roche S, Donal E, Odent S, Eicher J-C, Faivre L, et al. Whole exome sequencing reveals a large genetic heterogeneity and revisits the causes of hypertrophic cardiomyopathy. Circ Genomic Precis Med. (2019) 12:e002500. doi: 10.1161/CIRCGEN.119.002500, PMID: [DOI] [PubMed] [Google Scholar]
- 26.Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. (2019) 47:D886–94. doi: 10.1093/nar/gky1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using poly Phen-2. Curr Protoc Hum Genet. (2013) Chapter 7: Unit 7.20 doi: 10.1002/0471142905.hg0720s76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. (2003) 31:3812–4. doi: 10.1093/nar/gkg509, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schwarz JM, Cooper DN, Schuelke M, Seelow D. Mutation taster 2: mutation prediction for the deep-sequencing age. Nat Methods. (2014) 11:361–2. doi: 10.1038/nmeth.2890, PMID: [DOI] [PubMed] [Google Scholar]
- 30.Walsh R, Mazzarotto F, Whiffin N, Buchan R, Midwinter W, Wilk A, et al. Quantitative approaches to variant classification increase the yield and precision of genetic testing in Mendelian diseases: the case of hypertrophic cardiomyopathy. Genome Med. (2019) 11:5. doi: 10.1186/s13073-019-0616-z, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med Off J Am Coll Med Genet. (2015) 17:405–24. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. (2016) 44:D457–62. doi: 10.1093/nar/gkv1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jassal B, Matthews L, Viteri G, Gong C, Lorente P, Fabregat A, et al. The reactome pathway knowledgebase. Nucleic Acids Res. (2020) 48:D498–503. doi: 10.1093/nar/gkz1031, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Köhler S, Gargano M, Matentzoglu N, Carmody LC, Lewis-Smith D, Vasilevsky NA, et al. The human phenotype ontology in 2021. Nucleic Acids Res. (2021) 49:D1207–17. doi: 10.1093/nar/gkaa1043, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gene Ontology Consortium . Gene ontology consortium: going forward. Nucleic Acids Res. (2015) 43:D1049–56. doi: 10.1093/nar/gku1179, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Desvignes J-P, Bartoli M, Delague V, Krahn M, Miltgen M, Béroud C, et al. Var AFT: a variant annotation and filtration system for human next generation sequencing data. Nucleic Acids Res. (2018) 46:W545–53. doi: 10.1093/nar/gky471, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). (2012) 6:80–92. doi: 10.4161/fly.19695, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park J, Packard EA, Levin MG, Judy RL, Damrauer SM, Day SM, et al. A genome-first approach to rare variants in hypertrophic cardiomyopathy genes MYBPC3 and MYH7 in a medical biobank. Hum Mol Genet. (2021) 31:827–37. doi: 10.1093/hmg/ddab249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ingles J, Goldstein J, Thaxton C, Caleshu C, Corty EW, Crowley SB, et al. Evaluating the clinical validity of hypertrophic cardiomyopathy genes. Circ Genomic Precis Med. (2019) 12:e002460. doi: 10.1161/CIRCGEN.119.002460, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Andreeva S, Chumakova O, Karelkina E, Lebedeva V, Lubimtseva T, Semenov A, et al. Case report: two new cases of autosomal-recessive hypertrophic cardiomyopathy associated with TRIM63-compound heterozygous variant. Front Genet. (2022) 13:743472. doi: 10.3389/fgene.2022.743472, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Salazar-Mendiguchía J, Ochoa JP, Palomino-Doza J, Domínguez F, Díez-López C, Akhtar M, et al. Mutations in TRIM63 cause an autosomal-recessive form of hypertrophic cardiomyopathy. Heart. (2020) 106:1342–8. doi: 10.1136/heartjnl-2020-316913, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harper AR, Goel A, Grace C, Thomson KL, Petersen SE, Xu X, et al. Common genetic variants and modifiable risk factors underpin hypertrophic cardiomyopathy susceptibility and expressivity. Nat Genet. (2021) 53:135–42. doi: 10.1038/s41588-020-00764-0, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ochoa JP, Sabater-Molina M, García-Pinilla JM, Mogensen J, Restrepo-Córdoba A, Palomino-Doza J, et al. Formin homology 2 domain containing 3 (FHOD3) is a genetic basis for hypertrophic cardiomyopathy. J Am Coll Cardiol. (2018) 72:2457–67. doi: 10.1016/j.jacc.2018.10.001, PMID: [DOI] [PubMed] [Google Scholar]
- 44.Matsuyama S, Kage Y, Fujimoto N, Ushijima T, Tsuruda T, Kitamura K, et al. Interaction between cardiac myosin-binding protein C and formin Fhod 3. Proc Natl Acad Sci USA. (2018) 115:E4386–95. doi: 10.1073/pnas.1716498115, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wooten EC, Hebl VB, Wolf MJ, Greytak SR, Orr NM, Draper I, et al. Formin homology 2 domain containing 3 variants associated with hypertrophic cardiomyopathy. Circ Cardiovasc Genet. (2013) 6:10–8. doi: 10.1161/CIRCGENETICS.112.965277, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chumakova OS, Baulina NM. Advanced searching for hypertrophic cardiomyopathy heritability in real practice tomorrow. Front Cardiovasc Med. (2023) 10:1236539. doi: 10.3389/fcvm.2023.1236539, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tadros R, Zheng SL, Grace C, Jordà P, Francis C, Jurgens SJ, et al. Large scale genome-wide association analyses identify novel genetic loci and mechanisms in hypertrophic cardiomyopathy. med Rxiv. [Preprint] (2023); 2023.01.28.23285147:44) 44. doi: 10.1093/eurheartj/ehad655.3197 [DOI] [Google Scholar]
- 48.Josephs KS, Roberts AM, Theotokis P, Walsh R, Ostrowski PJ, Edwards M, et al. Beyond gene-disease validity: capturing structured data on inheritance, allelic-requirement, disease-relevant variant classes, and disease mechanism for inherited cardiac conditions. Med Rxiv Prepr Serv Health Sci. (2023) doi: 10.1186/s13073-023-01246-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang XL, De S, McIntosh LP, Paetzel M. Structural characterization of the C3 domain of cardiac myosin binding protein C and its hypertrophic cardiomyopathy-related R502W mutant. Biochemistry. (2014) 53:5332–42. doi: 10.1021/bi500784g [DOI] [PubMed] [Google Scholar]
- 50.Saltzman AJ, Mancini-DiNardo D, Li C, Chung WK, Ho CY, Hurst S, et al. Short communication: the cardiac myosin binding protein C Arg502Trp mutation: a common cause of hypertrophic cardiomyopathy. Circ Res. (2010) 106:1549–52. doi: 10.1161/CIRCRESAHA.109.216291, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Walsh R, Thomson KL, Ware JS, Funke BH, Woodley J, McGuire KJ, et al. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet Med. (2017) 19:192–203. doi: 10.1038/gim.2016.90, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Andersen PS, Havndrup O, Bundgaard H, Moolman-Smook JC, Larsen LA, Mogensen J, et al. Myosin light chain mutations in familial hypertrophic cardiomyopathy: phenotypic presentation and frequency in Danish and south African populations. J Med Genet. (2001) 38:43e–443e. doi: 10.1136/jmg.38.12.e43, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marian AJ. Molecular genetic basis of hypertrophic cardiomyopathy. Circ Res. (2021) 128:1533. doi: 10.1161/CIRCRESAHA.121.318346, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mathew J, Zahavich L, Lafreniere-Roula M, Wilson J, George K, Benson L, et al. Utility of genetics for risk stratification in pediatric hypertrophic cardiomyopathy. Clin Genet. (2018) 93:310–9. doi: 10.1111/cge.13157, PMID: [DOI] [PubMed] [Google Scholar]
- 55.Pham JH, Giudicessi JR, Tweet MS, Boucher L, Newman DB, Geske JB. Tale of two hearts: a TNNT2 hypertrophic cardiomyopathy case report. Front Cardiovasc Med. (2023) 10:1167256. doi: 10.3389/fcvm.2023.1167256, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Allouba M, Walsh R, Afify A, Hosny M, Halawa S, Galal A, et al. Ethnicity, consanguinity, and genetic architecture of hypertrophic cardiomyopathy. Eur Heart J. (2023) 44:5146–58. doi: 10.1093/eurheartj/ehad372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Knöll R, Kostin S, Klede S, Savvatis K, Klinge L, Stehle I, et al. A common MLP (muscle LIM protein) variant is associated with cardiomyopathy. Circ Res. (2010) 106:695–704. doi: 10.1161/CIRCRESAHA.109.206243, PMID: [DOI] [PubMed] [Google Scholar]
- 58.Bagnall RD, Molloy LK, Kalman JM, Semsarian C. Exome sequencing identifies a mutation in the ACTN2 gene in a family with idiopathic ventricular fibrillation, left ventricular noncompaction, and sudden death. BMC Med Genet. (2014) 15:99. doi: 10.1186/s12881-014-0099-0, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fan L-L, Huang H, Jin J-Y, Li J-J, Chen Y-Q, Xiang R. Whole-exome sequencing identifies a novel mutation (p.L320R) of alpha-Actinin 2 in a Chinese family with dilated cardiomyopathy and ventricular tachycardia. Cytogenet Genome Res. (2019) 157:148–52. doi: 10.1159/000496077 [DOI] [PubMed] [Google Scholar]
- 60.Ranta-aho J, Olive M, Vandroux M, Roticiani G, Dominguez C, Johari M, et al. Mutation update for the ACTN2 gene. Hum Mutat. (2022) 43:1745–56. doi: 10.1002/humu.24470, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kraoua L, Jaouadi H, Allouche M, Achour A, Kaouther H, Ahmed HB, et al. Molecular autopsy and clinical family screening in a case of sudden cardiac death reveals ACTN2 mutation related to hypertrophic/dilated cardiomyopathy and a novel LZTR1 variant associated with Noonan syndrome. Mol Genet Genomic Med. (2022) 10:e1954. doi: 10.1002/mgg3.1954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Girolami F, Iascone M, Tomberli B, Bardi S, Benelli M, Marseglia G, et al. Novel α-actinin 2 variant associated with familial hypertrophic cardiomyopathy and juvenile atrial arrhythmias: a massively parallel sequencing study. Circ Cardiovasc Genet. (2014) 7:741–50. doi: 10.1161/CIRCGENETICS.113.000486, PMID: [DOI] [PubMed] [Google Scholar]
- 63.Arvanitis M, Tampakakis E, Zhang Y, Wang W, Auton A, 23andMe Research Team et al. Genome-wide association and multi-omic analyses reveal ACTN2 as a gene linked to heart failure. Nat Commun. (2020) 11:1122. doi: 10.1038/s41467-020-14843-7, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chiu C, Bagnall RD, Ingles J, Yeates L, Kennerson M, Donald JA, et al. Mutations in alpha-actinin-2 cause hypertrophic cardiomyopathy: a genome-wide analysis. J Am Coll Cardiol. (2010) 55:1127–35. doi: 10.1016/j.jacc.2009.11.016, PMID: [DOI] [PubMed] [Google Scholar]
- 65.Agarwal R, Wakimoto H, Paulo JA, Zhang Q, Reichart D, Toepfer C, et al. Pathogenesis of cardiomyopathy caused by variants in ALPK3, an essential Pseudokinase in the cardiomyocyte nucleus and sarcomere. Circulation. (2022) 146:1674–93. doi: 10.1161/CIRCULATIONAHA.122.059688, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Çağlayan AO, Sezer RG, Kaymakçalan H, Ulgen E, Yavuz T, Baranoski JF, et al. ALPK3 gene mutation in a patient with congenital cardiomyopathy and dysmorphic features. Cold Spring Harb Mol Case Stud. (2017) 3:a001859. doi: 10.1101/mcs.a001859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jaouadi H, Kraoua L, Chaker L, Atkinson A, Delague V, Levy N, et al. Novel ALPK3 mutation in a Tunisian patient with pediatric cardiomyopathy and facio-thoraco-skeletal features. J Hum Genet. (2018) 63:1077–82. doi: 10.1038/s10038-018-0492-1, PMID: [DOI] [PubMed] [Google Scholar]
- 68.Lopes LR, Garcia-Hernández S, Lorenzini M, Futema M, Chumakova O, Zateyshchikov D, et al. Alpha-protein kinase 3 (ALPK3) truncating variants are a cause of autosomal dominant hypertrophic cardiomyopathy. Eur Heart J. (2021) 42:3063–73. doi: 10.1093/eurheartj/ehab424, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Walsh R, Bezzina CR. ALPK3: a full spectrum cardiomyopathy gene? Eur Heart J. (2021) 42:3074–7. doi: 10.1093/eurheartj/ehab415, PMID: [DOI] [PubMed] [Google Scholar]
- 70.Dai J, Li K, Huang M, Sun Y, Liu H, Li Z, et al. The involvement of ALPK3 in hypertrophic cardiomyopathy in East Asia. Front Med. (2022) 9:915649. doi: 10.3389/fmed.2022.915649, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Su M, Wang J, Kang L, Wang Y, Zou Y, Feng X, et al. Rare variants in genes encoding MuRF1 and MuRF2 are modifiers of hypertrophic cardiomyopathy. Int J Mol Sci. (2014) 15:9302–13. doi: 10.3390/ijms15069302, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen SN, Czernuszewicz G, Tan Y, Lombardi R, Jin J, Willerson JT, et al. Human molecular genetic and functional studies identify TRIM63, encoding muscle RING finger protein 1, as a novel Gene for human hypertrophic cardiomyopathy. Circ Res. (2012) 111:907–19. doi: 10.1161/CIRCRESAHA.112.270207, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Smith TC, Fridy PC, Li Y, Basil S, Arjun S, Friesen RM, et al. Supervillin binding to myosin II and synergism with anillin are required for cytokinesis. Mol Biol Cell. (2013) 24:3603–19. doi: 10.1091/mbc.e12-10-0714, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pope RK, Pestonjamasp KN, Smith KP, Wulfkuhle JD, Strassel CP, Lawrence JB, et al. Cloning, characterization, and chromosomal localization of human superillin (SVIL). Genomics. (1998) 52:342–51. doi: 10.1006/geno.1998.5466, PMID: [DOI] [PubMed] [Google Scholar]
- 75.Hedberg-Oldfors C, Meyer R, Nolte K, Abdul Rahim Y, Lindberg C, Karason K, et al. Loss of supervillin causes myopathy with myofibrillar disorganization and autophagic vacuoles. Brain J Neurol. (2020) 143:2406–20. doi: 10.1093/brain/awaa206, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Olivotto I, Girolami F, Ackerman MJ, Nistri S, Bos JM, Zachara E, et al. Myofilament protein gene mutation screening and outcome of patients with hypertrophic cardiomyopathy. Mayo Clin Proc. (2008) 83:630–8. doi: 10.1016/S0025-6196(11)60890-2, PMID: [DOI] [PubMed] [Google Scholar]
- 77.Canepa M, Fumagalli C, Tini G, Vincent-Tompkins J, Day SM, Ashley EA, et al. Temporal trend of age at diagnosis in hypertrophic cardiomyopathy: an analysis of the international Sarcomeric human cardiomyopathy registry. Circ Heart Fail. (2020) 13:e007230. doi: 10.1161/CIRCHEARTFAILURE.120.007230, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Marstrand P, Han L, Day SM, Olivotto I, Ashley EA, Michels M, et al. Hypertrophic cardiomyopathy with left ventricular systolic dysfunction: insights from the SHaRe registry. Circulation. (2020) 141:1371–83. doi: 10.1161/CIRCULATIONAHA.119.044366, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data presented in the study are deposited in the ClinVar database under the following link: https://www.ncbi.nlm.nih.gov/clinvar/submitters/508840/.