

The epigenome consists of nuclear information, heritable during cell division, that controls development, tissue differentiation, and cellular responsiveness. Epigenetic information is controlled by genome sequence, environmental exposure, and stochasticity, or random chance. As such, epigenetics stands at the interface of the genome, development, and environmental exposure.
All cells of the body have essentially the same DNA, yet different organs and tissues serve vastly different functions and also retain their identity as their cells divide. This cellular identity is epigenetic information, or information that is added onto the genes themselves. As originally defined in the 1950s by the embryologist Conrad Waddington, epigenetics is the branch of biology that studies the interactions between genes and their products that bring phenotype into being.1 Waddington’s definition was based on a highly deterministic view of the ultimate destiny of tissue development: although it might vary somewhat according to environmental exposure, the end point was inexorably determined by the genes, not the environment. Waddington described an “epigenetic landscape,” in which a pluripotent cell acquires differentiated properties as it rolls down “canals” to its eventual fate.
A major change in epigenetic thinking came from the realization that the environment has a profound effect on developmental plasticity, particularly with aging and susceptibility to common disease.2 The modern definition of epigenetics takes this plasticity into account: modifications of DNA or associated factors that have information content, other than the DNA sequence itself, are maintained during cell division, are influenced by the environment, and cause stable changes in gene expression. Thus, the epigenetic landscape is now viewed more dynamically than it was initially.3
FORMS OF EPIGENETIC INFORMATION
Epigenetic information takes three forms, the first of which is DNA methylation (see the Glossary), a covalent modification of the nucleotide cytosine at the 5′ position, which is generally associated with gene silencing (Fig. 1). DNA methylation is the best-understood epigenetic modification and the clearest example of epigenetic information for several reasons. First, the information can be copied, in this case by the enzyme DNA methyltransferase 1, which recognizes hemimethylated CpG sites (locations in DNA at which a cytosine precedes a guanosine in the 5′ to 3′ sequence) on newly replicated DNA and methylates the daughter-strand cytosine at the complementary CpG. Second, the information can be interpreted, in this case by differential binding of transcription factors and enhancers, depending on the methylation state. Third, new sites of DNA methylation can be introduced by de novo methyltransferases. Finally, the information can be erased, either passively during cell division or by means of an enzymatic process involving teneleven translocation (TET) methylcytosine dioxygenases, followed by glycosylation and replacement with an unmethylated cytosine.4 DNA methylation is the most useful epigenetic marker for human disease studies because it is stable over a period of decades and is present in archival specimens, including paraffin blocks.5
Figure 1. The Cellular Nature of Epigenetic information.
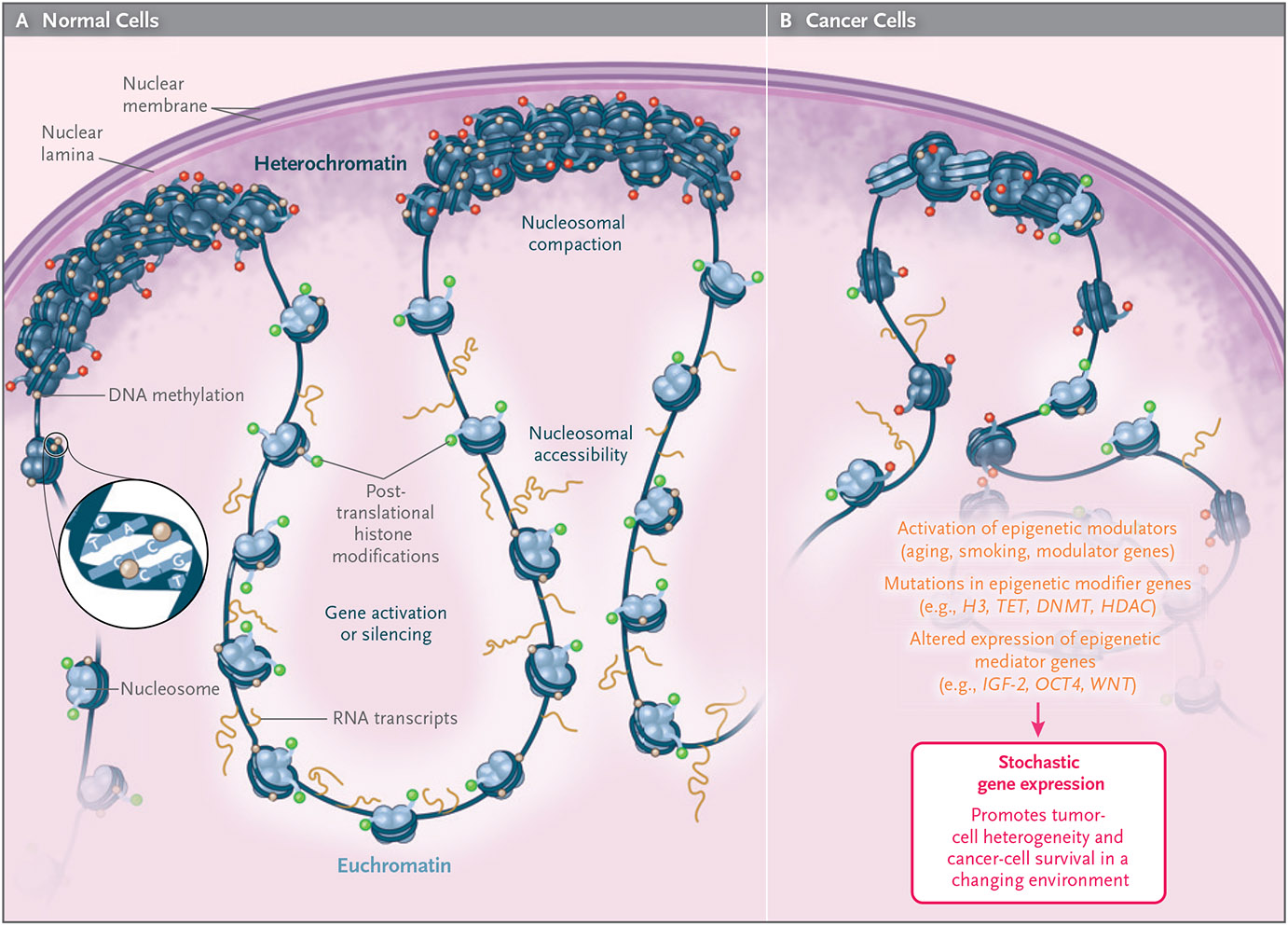
The DNA double helix is modified at the nucleotide cytosine by DNA methylation (brown dots). The nucleosomes around which the DNA is coiled undergo post-translational modifications of their component histones (green dots, depicting activation marks; red dots depict silencing marks), leading to gene activation (light-blue nucleosomes, with RNA transcripts originating nearby) or silencing (dark-blue nucleosomes). Higher-order chromatin structure involves nucleosomal compaction often near the nuclear membrane (heterochromatin) or nucleosomal accessibility (euchromatin). The nuclear periphery is primarily repressive but probably also contains transcriptionally permissive subcompartments. Higher-order large blocks of heterochromatin often involve large epigenomic domains termed lamina-associated domains (LADs) and large, organized chromatin lysine (K) modifications (LOCKs). In cancer, both large and smaller heterochromatic domains become euchromatic. In addition, epigenetic modulators such as environmental exposure and aging, as well as cancer mutations in epigenetic modifier genes, affect the expression of epigenetic mediators controlling pluripotency and cellular self-renewal. All these factors lead to increased stochastic gene expression in cancer, promoting tumor-cell heterogeneity and cancer-cell survival in a changing environment (e.g., as a result of metastasis or chemotherapy).
The second form of epigenetic information comprises more than 200 known post-translational modifications of nucleosomal histones about which the double helix is itself wound, in an ATP-independent process involving acetylation, methylation, phosphorylation, ubiquitylation, and sumoylation. Each modification is associated with gene activity, gene silencing, or insulation between active and inactive gene regions (Fig. 1). Post-translational modifications act through recruitment of transcription factors, activation of transcriptional enhancers, recruitment of repressive proteins, and interaction with the DNA methylation machinery. Just as DNA methylation can be erased, so too can post-translational modifications (e.g., by lysine demethylases and deacetylases and by replacement of histone 3 with histone 3.3), but how information is copied during cell division is less clear. A related form of epigenetic information is nucleosome remodeling by means of an ATP-dependent process that changes the density of nucleosomes, making them more or less available for transcription. The replication of this pattern during cell division is even more opaque. An outstanding review of chromatin modifications is available elsewhere.6
The third form of epigenetic information is higher-order chromatin structure, examples of which include loop organization revealed by chromosome-conformation-capture methods (i.e., techniques used to analyze the higher-order organization of chromatin in a cell); large, organized chromatin lysine (K) modifications (LOCKs) that condense a major fraction of the silent genome7; and nuclear lamina-associated domains (LADs)8 involved in nuclear compartmentation of multigene regions (Fig. 1). This higher-order chromatin structure also partitions the genome into regions of many tens of kilobases, which can co-associate in topologically associated domains (TADs) that allow for enhancer–promoter interaction.9,10 These domains have some tissue specificity, with relevant functional genomic elements juxtaposed for the purpose of a particular organ or set of cells.
MODULATION OF EPIGENETIC INFORMATION BY THE ENVIRONMENT
Human epidemiologic studies have long pointed to the role of diet in changing the genetic program over multiple generations. Men whose grandfathers were exposed to the Swedish famine in Överkalix before puberty tend to die at an earlier age from various common diseases than men whose grandfathers were not exposed to the famine.11 Both the Dutch Hunger Winter and the Great Leap Forward of China involved mass starvation of the population, and in both cases, fetal exposure to famine during the first trimester of gestation was associated with an incidence of schizophrenia in adulthood that was twice as high as the incidence among adults who had not been exposed during gestation.12
The first convincing example of intergenerational dietary epigenetic effects was an experiment involving mice with an insertional mutation in the Agouti locus that controls coat color and weight, termed Avy (Agouti viable yellow). These phenotypes are regulated by dietary methionine, the essential amino acid precursor for DNA methylation. When pregnant dams are exposed to a diet rich in methionine, Avy is variably silenced, with pups in the same litter having a range of phenotypes from brown and thin to yellow and obese.13 A great deal of epidemiologic evidence supports a relationship between dietary exposure in early life and long-term health,14 an idea first proposed by Barker and Osmond15 and supported by more recent studies.16
Moreover, diet can cause profound changes in the epigenome, leading to human disease. For example, deprivation of the essential amino acid methionine and folate deficiency are associated with liver and colon cancer in animals and humans.17,18 Folate deficiency impairs biosynthesis of the active precursor for DNA methylation, S-adenosylmethionine, and also impairs synthesis of thymidylate. A recent randomized trial also showed that dietary fat composition affects DNA methylation in adipocytes.19 Many studies have shown that the metabolic syndrome and related disorders are linked to epigenetic changes detected in blood DNA.20 Exposure to nicotine and other toxins causes substantial epigenetic changes in smokers, as well as in the cord blood and placenta of fetuses exposed prenatally, affecting genes involved in normal pulmonary function and cancer.21-25 Three epigenetic loci for IgE concentration, which is strongly linked to allergic response, account for 13% of variation in IgE levels.26 Exercise has mechanistically important effects on the skeletal-muscle epigenome,27 as may trauma in early life.28 Recently, post-traumatic stress disorder has been linked to epigenetic changes prospectively.29
CANCER AS THE PARADIGM OF COMMON EPIGENETIC DISEASE
It has been known since the 1980s that most or all tumors are associated with widespread losses and some gains of DNA methylation throughout the genome.30 The Beckwith–Wiedemann syndrome is an overgrowth disorder that causes Wilms’ tumors of the kidney and other so-called embryonal tumors that arise from fetal cells and persist after birth. The frequency of tumors is increased by a factor of more than 1000 among patients with the Beckwith–Wiedemann syndrome as compared with the general population. The syndrome is genetically heterogeneous, but the risk of cancer is associated specifically with loss of imprinting of the gene encoding insulin-like growth factor 2 (IGF-2), activating the normally silent maternal allele and leading to a double dose of IGF-2 protein.31,32 These observations prove that the epigenetic changes precede and increase the risk of cancer rather than arise after tumor formation.33
A common description of cancer is that it is many different diseases34 caused by differing mutational mechanisms, that each cancer type is distinct and requires particular therapies, and even that each tumor of a particular type is distinct and could be treated individually on the basis of genomic sequencing. However, the differences in tumor types are related to the tissue of origin and often to the spectrum of mutations associated with that organ, whereas the properties of tumor heterogeneity and therapeutic resistance are epigenetic and are shared among tumor types.
My colleagues and I, as well as others, have argued that cancers are in fact more alike than different and that the central feature of cancer is a disrupted and unstable epigenome, usually but not always caused by mutations and often preceded by epigenetic changes to the normal tissues themselves as a result of age and injury.2,35,36 These changes lead to epigenetic instability, erosion of defined chromatin regions, and variability of gene expression, resulting in tumor-cell heterogeneity. Moreover, mutations specifically driving metastasis have not been identified in cancer, yet epigenetic changes in large areas of the genome have been shown to drive metastasis. Comprehensive genome-scale analysis of DNA methylation shows that the methylation changes in cancer involve blocks of tens to hundreds of kilobases that overlap the large heterochromatin structures noted above, termed LOCKs and LADs.37,38 The transition to cancer occurs through regional loss of heterochromatin and loss of DNA methylation, with stochastic gene expression in these regions (Fig. 1). Changes in DNA methylation in these regions lead to enhanced variability in DNA methylation and expression of genes within the regions,37 which may be the mechanism for tumor-cell heterogeneity. Such heterogeneity is the defining feature of cancer that leads to chemoresistance, impaired DNA repair, metastasis, and death. A recent study has shown that large regions termed superenhancers have similar hypomethylation, leading to aberrant gene expression.39
There have been many reviews of epigenetic changes in cancer, including a recent review by my colleagues and me.40 As discussed in much greater detail there, the epigenetic changes in cancer can be grouped into three categories: epigenetic modifiers, epigenetic mediators, and epigenetic modulators (Fig. 1). Epigenetic modifiers are the easiest to understand and are the genes whose products modify the epigenome directly (e.g., through three forms of epigenetic information: DNA methylation, post-translational modification of chromatin, or higher-order chromatin structure). Most of the genes altered by mutation in cancer are in fact epigenetic modifiers, and thus both genetic and epigenetic changes are channeled through the epigenome. Examples of chromatin-remodeling genes that are mutated in cancer are SMARC in rhabdoid tumors, lung cancer, and Burkitt’s lymphoma; ARID in ovarian and hepatocellular cancers; IDH in glioblastoma41; and CHD in chronic lymphocytic leukemia and many solid tumors. DNA methyltransferases, TET demethylases, and the MBD (methyl-CpG-binding domain) family of methylation-recognition genes are mutated in lymphoma and colon cancer.
The epigenetic mediators in cancer, which are downstream of the epigenetic modifiers, are the targets of epigenetic modification by the modifiers, and this alteration contributes to a cell-state change toward stem-cell–like phenotypes. Epigenetic mediators include IGF-2 and several pluripotency factors such as NANOG, OCT4, and SOX2, which act either alone or in cooperation with signaling factors such as WNT in breast, skin, testicular, lung, colon, and esophageal cancers.
The epigenetic modulators, which are upstream of the modifiers, are the factors that infiuence the activity or localization of the epigenetic modifiers in order to destabilize differentiation-specific epigenetic states. They represent the bridge between the environment and the epigenome, whose disordered function confers a predisposition to and acceleration of cancer development. An example is nuclear factor κB (NF-κB)–mediated inflammatory responses, which trigger an epigenetic switch to a positive feedback loop with interleukin-6 and STAT3, transforming mammary epithelia. STAT3, in turn, helps maintain the expression of OCT4, NANOG, and SOX2 by binding to their enhancers. Aging is another epigenetic modulator.2 Genomewide hypomethylation in blood is associated with breast cancer years later.42 A recent study identified large-scale epigenomic blocks of methylation changes, similar to those seen in cancer, in photoaging skin.36 Cancer-specific methylation changes in squamous-cell cancer within these aged skin regions occurred only in those blocks.36
The idea that cancer is fundamentally an epigenetic disease is also reflected in the relationship between cancer and the epigenetic landscape. Since the epigenetic modifiers are the major targets of cancer mutations, the mutations can have widespread effects on the stability of the landscape. A key concept in understanding this change is entropy, defined in information theory as p × log p, where p is the probability of DNA methylation at a given site43; this use of the term entropy is distinct from its use in thermodynamics. My colleagues and I have observed that large-scale hypomethylated blocks increase gene-expression variability,44 corresponding to these regions of high entropy.43 Moreover, “hypermethylated CpG islands” and “hypomethylated shores,” the classic subcategories of DNA methylation changes, are in fact both often products of increased entropy within these large epigenomic structures43,45 (Fig. 1). These same domains may show higher entropy than the rest of the genome43 and may exist normally to allow the tissue-type switching, such as the transition from epithelial to mesenchymal tissue, that is necessary for normal embryogenesis and wound repair.46
Driver mutations (i.e., mutations that are present in all the cells of the primary cancer and that are thought to cause tumor growth) are almost universally present in primary cancers, with some exceptions, such as ependymomas, which appear to be entirely epigenetic.47 However, driver mutations for metastasis have not generally been found, even though it is the metastases that usually result in death. My colleagues and I recently identified large-scale changes in the epigenome, with loss of DNA methylation and heterochromatin including LOCKs, associated with distant metastases of pancreatic adenocarcinoma.48 These epigenetic alterations were also present in the particular regions of the primary tumors that gave rise to the metastases and thus were drivers of metastasis, but there were no mutated genetic drivers of the metastases. Moreover, this epigenetic disruption was linked to activation of the oxidative pentose phosphate pathway, which, when inhibited experimentally, led to partial reversal of the epigenetic changes and abrogation of tumor-cell growth in an in vitro model of invasion.48 Activation of this pathway has also been shown to promote the growth of other tumor types,49,50 and it will be interesting to look for the epigenetic link in these tumors as well. A potential mechanism could involve TET or KDM (lysine demethylase) dioxygenases, leading to loss of heterochromatin and loss of DNA methylation. Thus, defective metabolism may be an epigenetic modulator for metastasis, driving epigenomic changes that confer a survival advantage on cells that seed distant organ sites. If so, and if the reversibility that has been observed is confirmed, then one should be able to target primary tumors or micrometastases to abrogate or slow metastatic progression.
EPIGENETIC VARIABILITY AS A DRIVING FORCE FOR DISEASE
These recent findings on metastasis are consistent with a model in which loss of LOCKs and hypomethylated blocks in cancer underlies gene-expression variability within those domains and affects genes involved in tumor invasion and metastasis.45 Differentially methylated regions (DMRs) associated with cancer substantially correspond to tissue-specific DMRs.44,45 This hypervariability would increase the adaptability of tumor cells in an evolutionary sense as cells with a growth advantage at the expense of the normal cells in the host. The study of pancreatic cancer noted above would fit this model, in that epigenetic drivers evolving gradually within the primary tumor appear to cause distant metastasis.
This epigenetic change, and in particular the variability of epigenetic marks, may also be a valuable diagnostic and prognostic tool for cancer. For example, increased methylation variability is linked to more aggressive disease in leukemia and lymphoma.51,52 In addition, among biopsy samples that had been obtained because of a suspicion of cervical or breast cancer and that turned out to be negative, variability in DNA methylation was markedly increased in the samples from women in whom cancer developed years later, as compared with the samples from women in whom cancer did not develop.50 This work linking methylation entropy to cancer prediction has been extended to a clinically practical platform in the study of ovarian cancer.53 Similar observations have been made for outcome prediction in hematopoietic cancers, with chronic lymphocytic leukemia showing high intrasample methylation variability, which is associated with transcriptional variation and a poor outcome.54 The importance of epigenetic variability in disease risk is not limited to cancer and includes autoimmune disease55 and body-mass index.56 In addition, increased variability in DMRs has been found in monozygotic twins with type 1 diabetes, as compared with their unaffected twin siblings.57
THE NEW FIELD OF EPIGENETIC EPIDEMIOLOGY
It is now well established that common genetic variants in the population explain only a small fraction of the hereditary component of disease risk.58 This conundrum, termed the “missing heritability of common disease,” is being addressed by an exponentially growing effort to identify rare population variants that cumulatively explain most disease risk. The use of sequencing alone to assess disease risk is even more limited because sequencing studies cannot easily capture the role of the environment, which is thought to account for 80% of disease risk in humans. For example, a Western diet is the leading cause of type 2 diabetes and a major cause of cancer. Smoking is the leading cause of several cancers and also contributes to autoimmune and respiratory disease. Inflammation is caused by a variety of exposures and underlies autoimmune disease and cancer risk. Diseases related to aging, which constitute the foremost growing health problem, are in large part a result of long-term environmental damage.
A new field, epigenetic epidemiology, or the study of epigenetics in populations, has grown over the past decade to incorporate genetic variation with environmental exposure in explaining common diseases mediated by the epigenome. An understanding of gene–environment interaction is central to epidemiology generally. The relatively new idea is that epigenetics might in part mediate this interaction.59,60 This idea has gained considerable plausibility, since we now know that much of genetic variation is mediated through the epigenome.61,62
The principal tool for epigenetic epidemiology is the epigenome-wide association study (Fig. 2B). Such studies have focused almost entirely on DNA methylation, since this modification is stable over a period of decades and can be evaluated in genomic DNA samples from existing epidemiologic cohorts. In contrast, the genomewide association study relies on the statistical association with a genetic variant in the population with the inherited genotype (Fig. 2A). Normally, the detected variant is not in or even near what might be the causative gene. The epigenome-wide association study relies on the association between exposures and epigenetic changes, and their connection, in turn, with disease phenotypes. However, causality is more of a problem with this approach than with the genomewide approach, since one must determine whether the epigenetic changes were a cause or a consequence of disease, using statistical tools, animal models, or biochemical studies.
Figure 2. Epigenetic Approach to Epidemiology.
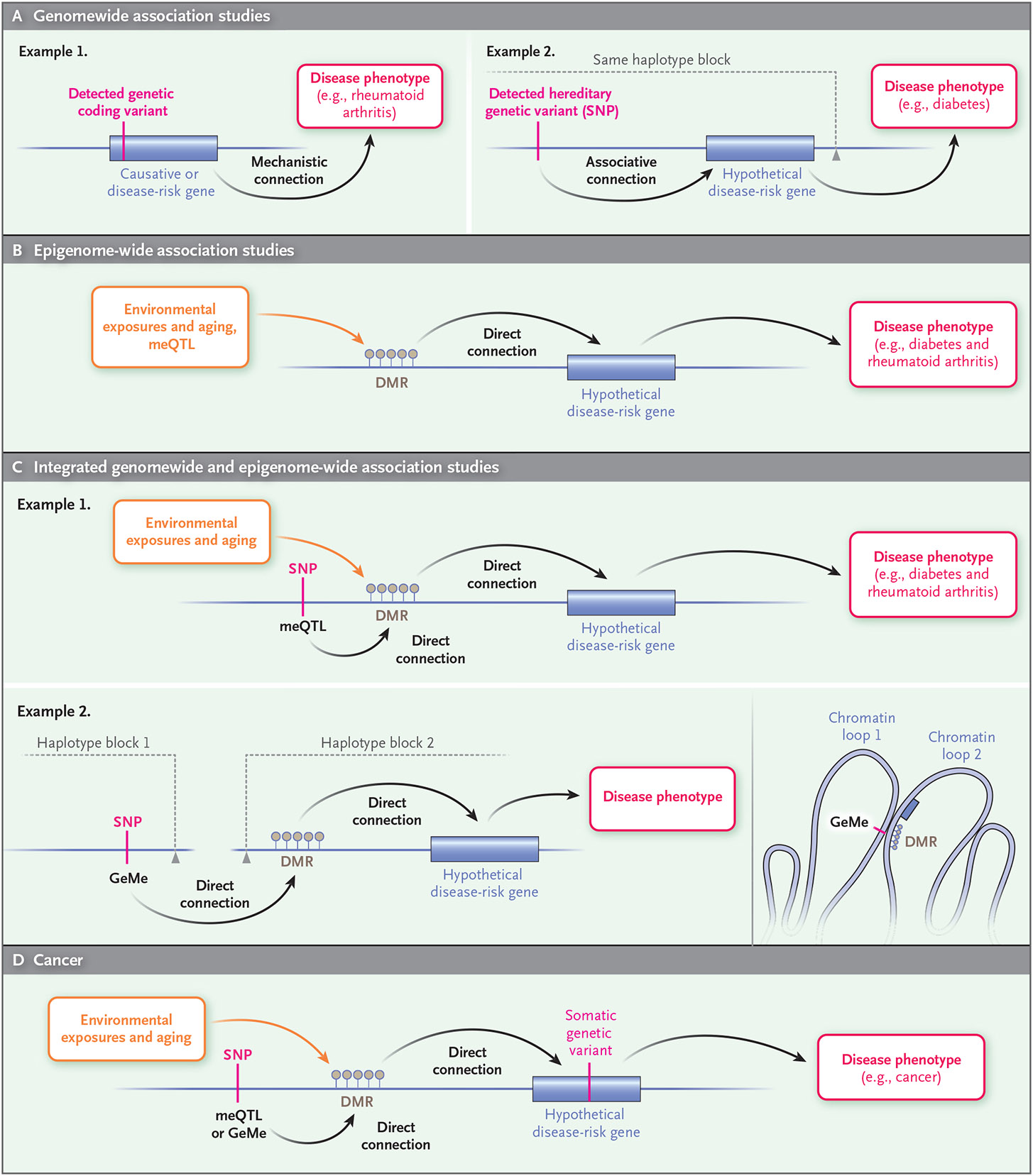
Common diseases in humans (e.g., cancer, diabetes, and rheumatoid arthritis) can be better understood through the combination of conventional genomewide association studies and epigenome-wide association studies. Conventional genomewide association studies (Panel A) link a hereditary DNA sequence variant or single-nucleotide polymorphism (SNP), through a presumed connection to a gene, to a disease phenotype (e.g., diabetes). Epigenome-wide association studies (Panel B) link environmental exposures (for which establishing causality requires statistical tools, animal models, or biochemical studies) and aging to a DNA methylation change and subsequently to a disease phenotype (e.g., diabetes or rheumatoid arthritis). An integrated approach (Panel C) incorporates both genetic and environmental exposure by relating genetic variants to epigenetic changes (methylation quantitative trait locus [meQTL]) in disease (e.g., diabetes and rheumatoid arthritis). Moreover, the combination of genomewide and epigenome-wide association studies can identify genetic variants regulating epigenetic marks (clusters of DNA methylation under genetic control [GeMes]) across linkage disequilibrium blocks that are normally penalized mathematically in conventional genomewide association studies (i.e., even though they are not in the same linkage disequilibrium block and thus not normally considered associated, they can be topologically associated through higher-order folding of chromatin in the nucleus, as shown in example 2). Similarly, cancer epigenetics (Panel D) enriches conventional cancer genetics by including environmental exposure and epigenetic changes together with hereditary genetic variants in risk assessment. DMR denotes differentially methylated region.
The integration of genetic and epigenetic studies can reinforce the strengths of both (Fig. 2C). For example, changes in DNA methylation might occur at a DMR that is in turn regulated by genetic variants identified in genome-wide association studies. Similarly, somatic mutations in cancer that are caused by the environment or chance are difficult to associate with disease unless they are within the coding sequence, since more than 99% of cancer mutations are “passenger mutations” of no mechanistic consequence because of the clonality of the disease. However, unlike mutations, epigenetic changes in cancer are also often found in the normal cells near the cancer, including age-related changes that are associated with abnormal regulation of tumor genes (Fig. 2C).
A problem specific to epigenome-wide association studies is the role of cell type. What one measures in blood may not be representative of what occurs in a target tissue such as brain. This surrogate tissue problem is a subject of new funding initiatives such as TaRGET II, from the National Institute of Environmental Health Sciences, which is designed to compare the epigenetic effects of toxins on target and surrogate tissues in mice, for eventual application to surrogate tissue measurements in humans. Moreover, most cell populations involve multiple cell types that may have varying DNA methylation, requiring either cellular fractionation or statistical correction. Several studies have identified epigenetic markers for schizophrenia in cord blood,63-65 but because of the issues described above, their mechanistic connection to the disease is still unclear.
Nevertheless, these problems can be overcome, and the markers can identify loci in genomewide association studies that are not apparent on the basis of purely genetic analyses. For example, in newly incident rheumatoid arthritis, DMRs could be identified at a locus not evident in conventional genomewide studies, in which the epigenome mediated genetic susceptibility to disease and was replicated in additional persons.55 Another example is type 1 diabetes, which was shown to be associated with specific DMRs in discordant monozygotic twins; a causal role was established by examination of cord blood from newborns in whom type 1 diabetes later developed.57 A recent study showed that many replicated DMRs are probably a consequence of increased body-mass index, but that was not true for all DMRs, and some were predictive of type 2 diabetes.66 Earlier, my colleagues and I used a species-comparative epigenomic approach to the study of obesity and diabetes, showing that some diet-induced DMRs in mouse adipocytes could be replicated in obese humans; were partially reversed by bariatric surgery; were themselves nearby known or previously unapparent single-nucleotide polymorphisms (SNPs), on the basis of genomewide association studies; and played a causal role in glucose uptake in vitro.67 Recent studies have identified dietary changes in the microbiome that can influence host methylation,68 providing another experimental tool to study the role of epigenetics in gene–environment interaction. In addition, genotype and in utero exposure to maternal smoking have been integrated in the analysis of neonates.69
Even if DNA methylation is at times a consequence rather than a cause of disease, there are data to suggest that it can serve as a presymptomatic marker (e.g., of Alzheimer’s disease),70,71 and developmental changes in DNA methylation in the prefrontal cortex identify SNPs that are associated with schizophrenia.72 SNPs affecting DNA methylation, known as methylation quantitative trait loci (meQTLs) (Fig. 2C), can thus be used for more precise genomewide association studies of large populations. For example, SNPs for a wide variety of common diseases are enriched for meQTLs, and these methylation sites could identify the most likely genes involved that were not obvious on examination of the SNPs themselves.73,74 Although meQTLs are in the same chromosomal region as the affected differentially methylated positions (DMPs), the SNPs and DMPs need not be immediately contiguous, or even within the same linkage disequilibrium block, yet may still regulate methylation, presumably because of topologic looping in the nucleus.75
INCORPORATING EPIGENETICS INTO RISK ASSESSMENT AND DISEASE PREVENTION
We now know that epigenetic changes play a causal role in cancer and occur long before cancer develops, and they appear to be the principal targets of genetic change and, at least in pancreatic cancer, the principal drivers of distant metastasis. Yet we do not have a mechanism for assessing the epigenetic risk of cancer or for discovering agents that could be used as epigenetic chemoprotection or epigenetic adjuvant therapy for primary cancer in order to abrogate or retard metastasis. We must bank frozen primary cancers for which there are matched outcome measures (recurrence or therapy response) so that we can identify the epigenetic field effects in normal tissue that predict progression and, at the same time, identify the epigenetic changes and genes that mediate progression.
Tamoxifen was discovered as an adjuvant treatment because it already had biologic activity against cancer. But our best chemopreventive agents may have no effect on tumors themselves, and we would never know they exist. We must therefore also fund animal research designed to identify mechanistically significant gene–environment interactions related to exposure and cancer prevention.
None of our current drug-screening tools are designed to test whether a drug reduces the variability of gene expression or associated epigenetic marks. In contrast, we assess both the risk and status of disease on the basis of measurements of mean values (genomic or epigenomic). However, recent studies of epigenetic variability and entropy suggest that we must also measure cell-to-cell variation in assessing disease risk. This will require collaboration among outstanding biologists, pharmacologists, and applied mathematicians to be successful.
It is important to combine genomewide and epigenome-wide association studies in order to uncover mechanisms in other common diseases. Attractive targets include autoimmune disease, the treatment of which is difficult after the cytokine storm occurs (which is the point at which patients usually seek care) but might be amenable to epigenetic intervention in the prodromal stages. We already know that the genome and epigenome conspire to cause rheumatoid arthritis55 and food allergy.76 Similarly, type 2 diabetes is difficult to treat once organ damage has occurred, including insulin resistance. Yet there are strong data supporting a genetic–epigenetic connection conferring a predisposition to the disease. Practical “precision epigenetic medicine” may already be possible. For example, metabolic pathological testing could use existing data showing that meQTLs are associated with body-mass index and metabolic phenotypes; causal inference testing shows that genetic variants often have an effect through DNA methylation.77,78
Finally, epigenetic analysis might be used in completely novel ways that have received almost no attention to date. For example, it could be used to predict therapeutic response in ways that purely genetic analysis cannot do, because epigenetic analysis measures the effect of the genome and the patient’s existing environmental load. Epigenetic analysis could also be used to assess in utero and transgenerational effects. For example, we already know that epigenetic changes are found in the offspring of women who smoke during pregnancy24 and that there are methylation changes in the sperm of fathers of children with autism who have subsequent children with autism.79 Epigenetics can lead us at last to an era of comprehensive medical understanding, unlocking the relationships among the patient’s genome, environment, prenatal exposure, and disease risk in time for us to prevent diseases or mitigate their effects before they take their toll on health.
Acknowledgments
I thank Michael Koldobskiy, Tomas Ekstrom, Anita Gondor, Yun Liu, Lindsay Rizzardi, and Xin Li for critical reading and suggestions.
Glossary
- Differentially methylated position (DMP)
A site of DNA methylation that is evaluated in epigenome-wide association studies.
- Differentially methylated region (DMR)
A region of DNA methylation that is evaluated in epigenome-wide association studies.
- DNA methylation
A covalent modification of the nucleotide cytosine, which is heritable during cell division and is associated generally with gene silencing.
- Entropy
A measure of disorder in a system; specifically, in information theory, a measure of unpredictability (known as Shannon entropy), defined as the sum of P(xi)logP(xi) of each state xi of a discrete random variable X.
- Epigenetic epidemiology
The study of the relationship between epigenetic variants and disease phenotype in the population.
- Epigenetic mediators
The gene targets of epigenetic modifiers that contribute to stem-cell–like phenotypes in cancer cells, including cellular reprogramming factors.
- Epigenetic modifiers
Genes whose products modify the epigenome directly through DNA methylation, post-translational modifications of chromatin, or higher-order chromatin structure; they are commonly mutated in cancer.
- Epigenetic modulators
Factors that influence the activity or localization of epigenetic modifiers, representing a bridge between the environment and the epigenome.
- Epigenetic stochasticity
A normal developmental, injury-response, or cancer-associated mechanism for increased variability of epigenetic marks at a given location. Cancer-associated epigenetic stochasticity leads to tumor-cell heterogeneity and increased survival of tumor cells in an environment undergoing change (e.g., as a result of metastasis or chemotherapy).
- Epigenome
The epigenetic information in a cell, comprising DNA methylation, post-translational modifications of histones, and higher-order chromatin structure.
- Epigenome-wide association studies (EWAS)
Studies of the relationship between epigenetic variants (differentially methylated regions [DMRs] or differentially methylated positions [DMPs]) in the population and disease phenotypes.
- Genetic methylation unit (GeMe)
A cluster of differentially methylated positions (DMPs) and the single-nucleotide polymorphisms (SNPs) regulating their methylation in the same chromosomal region; GeMes include methylation quantitative trait loci (MeQTLs) and also noncontiguous regions separating the DMPs and their controlling SNPs, even outside the same linkage disequilibrium block.
- Genomewide association studies (GWAS)
Studies of the relationship between DNA variants (generally single-nucleotide polymorphisms [SNPs], but also copy-number variants) in the population and disease phenotypes.
- Genomic imprinting
Parent-of-origin–specific epigenetic marks generally associated with comparative silencing of the allele transmitted to the offspring, regardless of the sex of the offspring.
- Large organized chromatin lysine (K) modifications (LOCKs)
Histone 3 (H3) lysine 9 (K9) dimethylation and trimethylation regions associated with gene silencing, lamina-associated domains (LADs), and large, DNA-hypomethylated blocks in cancer.
- Methylation quantitative trait loci (meQTLs)
Single-nucleotide polymorphisms (SNPs) associated with differentially methylated positions (DMPs), constituting a link between genomewide and epigenome-wide association studies.
Footnotes
Disclosure forms provided by the author are available with the full text of this article at NEJM.org.
References
- 1.Waddington CH. The strategy of the genes: a discussion of some aspects of theoretical biology. London: Allen & Unwin, 1957. [Google Scholar]
- 2.Teschendorff AE, West J, Beck S. Age-associated epigenetic drift: implications, and a case of epigenetic thrift? Hum Mol Genet 2013;22(R1):R7–R15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pujadas E, Feinberg AP. Regulated noise in the epigenetic landscape of development and disease. Cell 2012;148:1123–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen ZX, Riggs AD. DNA methylation and demethylation in mammals. J Biol Chem 2011;286:18347–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thirlwell C, Eymard M, Feber A, et al. Genome-wide DNA methylation analysis of archival formalin-fixed paraffin-embedded tissue using the Illumina Infinium Human-Methylation27 BeadChip. Methods 2010; 52:248–54. [DOI] [PubMed] [Google Scholar]
- 6.Soshnev AA, Josefowicz SZ, Allis CD. Greater than the sum of parts: complexity of the dynamic epigenome. Mol Cell 2016; 62:681–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wen B, Wu H, Shinkai Y, Irizarry RA, Feinberg AP. Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells. Nat Genet 2009;41:246–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harr JC, Luperchio TR, Wong X, Cohen E, Wheelan SJ, Reddy KL. Directed targeting of chromatin to the nuclear lamina is mediated by chromatin state and A-type lamins. J Cell Biol 2015;208:33–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lupiáñez DG, Kraft K, Heinrich V, et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 2015;161: 1012–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lieberman-Aiden E, van Berkum NL, Williams L, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009;326:289–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaati G, Bygren LO, Edvinsson S. Cardiovascular and diabetes mortality determined by nutrition during parents’ and grandparents’ slow growth period. Eur J Hum Genet 2002;10:682–8. [DOI] [PubMed] [Google Scholar]
- 12.Bygren LO. Intergenerational health responses to adverse and enriched environments. Annu Rev Public Health 2013; 34:49–60. [DOI] [PubMed] [Google Scholar]
- 13.Dolinoy DC, Weidman JR, Waterland RA, Jirtle RL. Maternal genistein alters coat color and protects Avy mouse offspring from obesity by modifying the fetal epigenome. Environ Health Perspect 2006;114:567–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Slomko H, Heo HJ, Einstein FH. Minireview: epigenetics of obesity and diabetes in humans. Endocrinology 2012;153: 1025–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barker DJ, Osmond C. Diet and coronary heart disease in England and Wales during and after the second world war. J Epidemiol Community Health 1986;40: 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seki Y, Williams L, Vuguin PM, Charron MJ. Minireview: epigenetic programming of diabetes and obesity: animal models. Endocrinology 2012;153:1031–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Poirier LA. The effects of diet, genetics and chemicals on toxicity and aberrant DNA methylation: an introduction. J Nutr 2002;132:Suppl:2336S–2339S. [DOI] [PubMed] [Google Scholar]
- 18.Giovannucci E. Epidemiologic studies of folate and colorectal neoplasia: a review. J Nutr 2002;132:Suppl:2350S–2355S. [DOI] [PubMed] [Google Scholar]
- 19.Perfilyev A, Dahlman I, Gillberg L, et al. Impact of polyunsaturated and saturated fat overfeeding on the DNA-methylation pattern in human adipose tissue: a randomized controlled trial. Am J Clin Nutr 2017;105:991–1000. [DOI] [PubMed] [Google Scholar]
- 20.Bianco-Miotto T, Craig JM, Gasser YP, van Dijk SJ, Ozanne SE. Epigenetics and DOHaD: from basics to birth and beyond. J Dev Orig Health Dis 2017;8:513–9. [DOI] [PubMed] [Google Scholar]
- 21.Meng W, Zhu Z, Jiang X, et al. DNA methylation mediates genotype and smoking interaction in the development of anti-citrullinated peptide antibody-positive rheumatoid arthritis. Arthritis Res Ther 2017;19:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bakulski KM, Lee H, Feinberg JI, et al. Prenatal mercury concentration is associated with changes in DNA methylation at TCEANC2 in newborns. Int J Epidemiol 2015;44:1249–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bellavia A, Urch B, Speck M, et al. DNA hypomethylation, ambient particulate matter, and increased blood pressure: findings from controlled human exposure experiments. J Am Heart Assoc 2013; 2(3):e000212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Joubert BR, Håberg SE, Nilsen RM, et al. 450K Epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environ Health Perspect 2012;120: 1425–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Richmond RC, Simpkin AJ, Woodward G, et al. Prenatal exposure to maternal smoking and offspring DNA methylation across the lifecourse: findings from the Avon Longitudinal Study of Parents and Children (ALSPAC). Hum Mol Genet 2015;24:2201–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liang L, Willis-Owen SAG, Laprise C, et al. An epigenome-wide association study of total serum immunoglobulin E concentration. Nature 2015;520:670–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barrès R, Yan J, Egan B, et al. Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab 2012;15:405–11. [DOI] [PubMed] [Google Scholar]
- 28.Szyf M. The early life environment and the epigenome. Biochim Biophys Acta 2009;1790:878–85. [DOI] [PubMed] [Google Scholar]
- 29.Rutten BPF, Vermetten E, Vinkers CH, et al. Longitudinal analyses of the DNA methylome in deployed military servicemen identify susceptibility loci for post-traumatic stress disorder. Mol Psychiatry 2017. June 20 (Epub ahead of print). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983;301:89–92. [DOI] [PubMed] [Google Scholar]
- 31.Steenman M, Westerveld A, Mannens M. Genetics of Beckwith-Wiedemann syndrome-associated tumors: common genetic pathways. Genes Chromosomes Cancer 2000;28:1–13. [DOI] [PubMed] [Google Scholar]
- 32.Maas SM, Vansenne F, Kadouch DJ, et al. Phenotype, cancer risk, and surveillance in Beckwith-Wiedemann syndrome depending on molecular genetic subgroups. Am J Med Genet A 2016;170:2248–60. [DOI] [PubMed] [Google Scholar]
- 33.DeBaun MR, Niemitz EL, McNeil DE, Brandenburg SA, Lee MP, Feinberg AP. Epigenetic alterations of H19 and LIT1 distinguish patients with Beckwith-Wiedemann syndrome with cancer and birth defects. Am J Hum Genet 2002;70:604–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.American Cancer Society. What is cancer? 2017. (https://www.cancer.org/cancer/cancer-basics/what-is-cancer.html).
- 35.Maegawa S, Gough SM, Watanabe-Okochi N, et al. Age-related epigenetic drift in the pathogenesis of MDS and AML. Genome Res 2014;24:580–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vandiver AR, Irizarry RA, Hansen KD, et al. Age and sun exposure-related widespread genomic blocks of hypomethylation in nonmalignant skin. Genome Biol 2015;16:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hansen KD, Timp W, Bravo HC, et al. Increased methylation variation in epigenetic domains across cancer types. Nat Genet 2011;43:768–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berman BP, Weisenberger DJ, Aman JF, et al. Regions of focal DNA hyper-methylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat Genet 2011;44:40–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heyn H, Vidal E, Ferreira HJ, et al. Epigenomic analysis detects aberrant superenhancer DNA methylation in human cancer. Genome Biol 2016;17:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feinberg AP, Koldobskiy MA, Göndör A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet 2016;17:284–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu C, Ward PS, Kapoor GS, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012;483:474–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Veldhoven K, Polidoro S, Baglietto L, et al. Epigenome-wide association study reveals decreased average methylation levels years before breast cancer diagnosis. Clin Epigenetics 2015;7:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jenkinson G, Pujadas E, Goutsias J, Feinberg AP. Potential energy landscapes identify the information-theoretic nature of the epigenome. Nat Genet 2017;49:719–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hansen KD, Sabunciyan S, Langmead B, et al. Large-scale hypomethylated blocks associated with Epstein-Barr virus-induced B-cell immortalization. Genome Res 2014;24:177–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Timp W, Feinberg AP. Cancer as a dys-regulated epigenome allowing cellular growth advantage at the expense of the host. Nat Rev Cancer 2013;13:497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McDonald OG, Wu H, Timp W, Doi A, Feinberg AP. Genome-scale epigenetic reprogramming during epithelial-to-mesenchymal transition. Nat Struct Mol Biol 2011;18:867–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mack SC, Witt H, Piro RM, et al. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature 2014;506:445–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McDonald OG, Li X, Saunders T, et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat Genet 2017;49:367–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lin Q, Wagner W. Epigenetic aging signatures are coherently modified in cancer. PLoS Genet 2015;11(6):e1005334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang ZZ, Lee EE, Sudderth J, et al. Glutathione depletion, pentose phosphate pathway activation, and hemolysis in erythrocytes protecting cancer cells from vitamin C-induced oxidative stress. J Biol Chem 2016;291:22861–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li S, Garrett-Bakelman FE, Chung SS, et al. Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nat Med 2016;22: 792–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pan H, Jiang Y, Boi M, et al. Epigenomic evolution in diffuse large B-cell lymphomas. Nat Commun 2015;6:6921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bartlett TE, Jones A, Goode EL, et al. Intra-gene DNA methylation variability is a clinically independent prognostic marker in women’s cancers. PLoS One 2015; 10(12):e0143178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Landau DA, Clement K, Ziller MJ, et al. Locally disordered methylation forms the basis of intratumor methylome variation in chronic lymphocytic leukemia. Cancer Cell 2014;26:813–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu Y, Aryee MJ, Padyukov L, et al. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat Biotechnol 2013;31:142–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang J, Loos RJ, Powell JE, et al. FTO genotype is associated with phenotypic variability of body mass index. Nature 2012;490:267–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Paul DS, Teschendorff AE, Dang MA, et al. Increased DNA methylation variability in type 1 diabetes across three immune effector cell types. Nat Commun 2016;7:13555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Blanco-Gómez A, Castillo-Lluva S, Del Mar Sáez-Freire M, et al. Missing heritability of complex diseases: enlightenment by genetic variants from intermediate phenotypes. Bioessays 2016;38:664–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rakyan VK, Down TA, Balding DJ, Beck S. Epigenome-wide association studies for common human diseases. Nat Rev Genet 2011;12:529–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bjornsson HT, Fallin MD, Feinberg AP. An integrated epigenetic and genetic approach to common human disease. Trends Genet 2004;20:350–8. [DOI] [PubMed] [Google Scholar]
- 61.Do C, Shearer A, Suzuki M, et al. Genetic-epigenetic interactions in cis: a major focus in the post-GWAS era. Genome Biol 2017;18:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.The Roadmap Epigenomics Consortium. Integrative analysis of 111 reference human epigenomes. Nature 2015;518:317–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hannon E, Dempster E, Viana J, et al. An integrated genetic-epigenetic analysis of schizophrenia: evidence for co-localization of genetic associations and differential DNA methylation. Genome Biol 2016; 17:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Aberg KA, McClay JL, Nerella S, et al. Methylome-wide association study of schizophrenia: identifying blood biomarker signatures of environmental insults. JAMA Psychiatry 2014;71:255–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Montaño CM, Irizarry RA, Kaufmann WE, et al. Measuring cell-type specific differential methylation in human brain tissue. Genome Biol 2013;14(8):R94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wahl S, Drong A, Lehne B, et al. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature 2017;541:81–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Multhaup ML, Seldin MM, Jaffe AE, et al. Mouse-human experimental epigenetic analysis unmasks dietary targets and genetic liability for diabetic phenotypes. Cell Metab 2015;21:138–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Krautkramer KA, Kreznar JH, Romano KA, et al. Diet-microbiota interactions mediate global epigenetic programming in multiple host tissues. Mol Cell 2016;64: 982–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Teh AL, Pan H, Chen L, et al. The effect of genotype and in utero environment on interindividual variation in neonate DNA methylomes. Genome Res 2014;24: 1064–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lunnon K, Smith R, Hannon E, et al. Methylomic profiling implicates cortical deregulation of ANK1 in Alzheimer’s disease. Nat Neurosci 2014;17:1164–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.De Jager PL, Srivastava G, Lunnon K, et al. Alzheimer’s disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat Neurosci 2014;17:1156–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jaffe AE, Gao Y, Deep-Soboslay A, et al. Mapping DNA methylation across development, genotype and schizophrenia in the human frontal cortex. Nat Neurosci 2016;19:40–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bonder MJ, Luijk R, Zhernakova DV, et al. Disease variants alter transcription factor levels and methylation of their binding sites. Nat Genet 2017;49:131–8. [DOI] [PubMed] [Google Scholar]
- 74.Liu Y, Li X, Aryee MJ, et al. GeMes, clusters of DNA methylation under genetic control, can inform genetic and epigenetic analysis of disease. Am J Hum Genet 2014;94:485–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen L, Ge B, Casale FP, et al. Genetic drivers of epigenetic and transcriptional variation in human immune cells. Cell 2016;167(5):1398–414. e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hong X, Hao K, Ladd-Acosta C, et al. Genome-wide association study identifies peanut allergy-specific loci and evidence of epigenetic mediation in US children. Nat Commun 2015;6:6304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Volkov P, Olsson AH, Gillberg L, et al. A genome-wide mQTL analysis in human adipose tissue identifies genetic variants associated with DNA methylation, gene expression and metabolic traits. PLoS One 2016;11(6):e0157776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hanson MA, Godfrey KM. Genetics: epigenetic mechanisms underlying type 2 diabetes mellitus. Nat Rev Endocrinol 2015;11:261–2. [DOI] [PubMed] [Google Scholar]
- 79.Feinberg JI, Bakulski KM, Jaffe AE, et al. Paternal sperm DNA methylation associated with early signs of autism risk in an autism-enriched cohort. Int J Epidemiol 2015;44:1199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]