

Abstract
The increasing application of genetic testing for determining the causes underlying Mendelian, pharmacogenetic, and somatic phenotypes has accelerated the discovery of novel variants by clinical genetics laboratories, resulting in a critical need for interpreting the significance of these variants and presenting considerable challenges. Launched in 2013 at the National Center for Biotechnology Information, National Institutes of Health, ClinVar is a public database for clinical laboratories, researchers, expert panels, and others to share their interpretations of variants with their evidence. The database holds 600,000 submitted records from 1,000 submitters, representing 430,000 unique variants. ClinVar encourages submissions of variants reviewed by expert panels, as expert consensus confers a high standard. Aggregating data from many groups in a single database allows comparison of interpretations, providing transparency into the concordance or discordance of interpretations. In its first five years, ClinVar has successfully provided a gateway for the submission of medically relevant variants and interpretations of their significance to disease. It has become an invaluable resource for the clinical genetics community seeking guidance from consensus interpretations. Building on the platform of providing transparency and leveraging aggregation of variant interpretations, ClinVar is now well positioned to help the clinical genetics community improve interpretations.
Keywords: Variant interpretation, ClinVar, Bioinformatics, Clinical genetics, Database, Genetic testing, Medical genetics
Introduction
ClinVar is an archive of human genetic variants and their relationships to human health and disease. It was created at the National Center for Biotechnology Information (NCBI) at the National Institutes of Health (NIH) to provide a centralized, public open-access database for data needed to aid users in interpreting variants. Other centralized databases of variation related to human disease existed before ClinVar; databases like the Human Gene Mutation Database (HGMD®; Stenson et al., 2017) and OMIM (www.omim.org/) focused on variants found in the literature but not variants identified in clinical testing laboratories. Other centralized databases focused on certain types of variants, such as COSMIC (Forbes et al., 2017) for somatic variation. Gene-specific databases also existed, most notably those created with the Leiden Open Variation Database (LOVD; Fokkema et al., 2011) framework. The existence of many databases provides alternative options in how data are collected, stored, and presented, but it also presents a challenge to the user who must become aware of each database that is relevant to clinical testing or research results, and then learn to navigate and query each database in a specific way to find relevant available data. Additionally, some resources are not accessible by the public, whether they be subscription-based, like HGMD®, or proprietary. Housed and operated in a non-profit entity, ClinVar is a centralized resource that aggregates data from many sources, facilitating searches for variant information.
Data generated by individual research laboratories and consortia have traditionally been shared through publication in journals. The same has not been true for data generated from clinical genetic testing. Thus a core part of ClinVar’s mission is to provide a repository for clinical genetic testing laboratories to share interpretations of variants and their evidence for making such interpretations. Before phrases such as “open data” and “precision medicine” became a part of mainstream discussions, there was little precedent for clinical genetics laboratories to share their variant data. In fact, a small number of laboratories rushed to patent and license newly identified gene-to-disease correlations incorporated into new tests, staking sole ownership of data as an income generator. Each laboratory maintained its own (likely proprietary) data, and this approach was sufficient at that time.
A confluence of factors led up to ClinVar: (1) changes in policy on genetic data sharing (GDS); (2) the use of genetic data in healthcare and employment, such as GINA [The Genetic Information Nondiscrimination Act of 2008;https://www.eeoc.gov/laws/statutes/gina.cfm]; (3) the decision of the U.S. Supreme Court that human genes are not patentable (Association for Molecular Pathology v. Myriad Genetics, Inc., 569 U. S; 2013); (4) rapid advancements in genetic measurement, assessment, and storage technologies; and (5) an overall shift in opinions on collaboratively sharing data. The advent and adoption of Next-Generation Sequencing (NGS) technologies into clinical genetic test protocols has resulted in the identification of many novel variants that must be classified for the first time. Many of these variants are rare and an individual laboratory may only ever see a rare variant once.
In the past, laboratories researched variants in PubMed and online databases such as HGMD®, but it was difficult to know if other testing laboratories had observed the variant as well, because contacting other laboratories to find out was logistically impractical and unscalable with respect to time, growing numbers of variants, and laboratory-specific data-sharing rules. Other approaches to delineate pathogenicity are available to clinical laboratories, including sequencing family members, comparison to large numbers of controls, and running translational/functional studies. However, the results of these studies are not always publicly available, and these studies are often prohibitively costly and time-consuming. ClinVar scaled these hurdles by being the first centralized database allowing clinical testing laboratories to share their interpretations of variants along with their evidence and making the data freely available, thereby enabling aggregation of interpretations for identical variants submitted by different laboratories to better substantiate interpretations. The need for such a database is supported by statements by the American Medical Association (AMA, 2013), the American College of Medical Genetics and Genomics (ACMG Board of Directors, 2017), and the National Society of Genetic Counselors (NSGC, 2015), all of which recommend sharing genomic variant data and interpretations of that data in a publicly accessible repository, like ClinVar. In the five years since the first public release of the database in April 2013 (Landrum et al., 2014), submission to ClinVar has now become routine for some clinical testing laboratories. As of July 11, 2018 (the date on which all data in this paper were compiled), a total of 1,000 organizations share data through ClinVar, including 430 organizations that share interpretations from clinical testing, and the database holds more than 600,000 submitted interpretations representing 430,000 unique variants. The aggregation of data for the same variant or variant-disease pair allows ClinVar to report when different submitters agree or disagree on the interpretation of the same variant. There has been substantial interest in the number of variants in ClinVar with conflicts in interpretation (Balmaña et al., 2016; Bland et al., 2018; Gradishar et al., 2017; Lincoln et al., 2017; Pepin et al., 2016; Rehm et al., 2017; Shah et al., 2018; Vail et al., 2015; Yang et al., 2017a) and in efforts to minimize these discrepancies, particularly by investigators in the Clinical Genome Resource (ClinGen) [Harrison et al., 2017].
From its inception, an ancillary goal for ClinVar was to amass interpretations from expert groups, including “penultimate” consensus interpretations. A few expert panels for variant interpretation existed prior to ClinVar (InSiGHT [Thompson et al., 2014], CFTR2 [http://cftr2.org], ENIGMA [Spurdle et al., 2012], and PharmGKB [Whirl-Carrillo et al., 2012]). Shortly after ClinVar’s public debut, the Clinical Genome Resource (ClinGen) was awarded funding by the National Institutes of Health (NIH), including support for the creation of new expert panels (Rehm et al., 2015).
Variant curation by expert panels is a valuable element of ClinVar, and this importance is reflected in a field called the “review status”. The review status indicates the level of review that supports a submitted or an aggregate interpretation (https://www.ncbi.nlm.nih.gov/clinvar/docs/review_status/). For submitted records, the review status considers whether the interpretation is from a practice guideline or from expert panel review; otherwise, it considers whether the submitter’s rules of classification were provided to ClinVar. ClinVar aggregates submitted data by variant (web displays for the Variation ID) and variant-disease (ClinVar records with an RCV prefix for “reference ClinVar” record). Aggregate records also have a review status based on the considerations mentioned above, plus whether there is consensus among interpretations from submitters who are not formally recognized in ClinVar as expert groups. The review status for aggregate records is represented in ClinVar’s web displays with a number of stars. ClinVar uses the review status of an aggregate record to help calculate the aggregate interpretation. If a variant has an interpretation from an expert panel, only that interpretation is used as the aggregate interpretation, regardless of how other submitters interpreted the variant. However, the submitted interpretations from other organizations remain in the database and available to users. Thus, an interpretation from an expert panel overrides any other conflicts in the interpretation. Several of the expert panels developed through ClinGen, including a number of new expert panels, are described in other articles within this issue.
Note on Terminology
Several different terms have been used in ClinVar for the field that represents the relationship between the variant and a condition. These terms include “clinical significance”, “assertion”, and “interpretation”, and were used based on feedback from ClinVar’s community of users. There does not appear to be a standard that is used uniformly across all of clinical genetics. In this paper, the term “interpretation” is used to refer to the submitter’s interpretation of the relationship of a variant to a condition. This relationship is considered a variant-level interpretation, not a patient-specific interpretation. It may represent the relationship between a variant and Mendelian disease, cancer, drug response, or other condition. As the relationship may be asserted through clinical testing, research, or curation, an “interpretation” is neither limited to a clinical context, nor to classifications of pathogenicity for disease.
Scope of the Database
ClinVar includes variants from any region of the genome — genes, intergenic regions, or the mitochondrial genome. The database includes variants of any size, from single-nucleotide variants to small insertions and deletions, and large copy-number variants (CNVs). Note that while small variants typically have a precise genomic location, copy number variants that are detected by microarray are generally described with an imprecise genomic location. This imprecision is represented in ClinVar using inner and outer start and stop locations (https://www.ncbi.nlm.nih.gov/dbvar/content/overview/), as provided by the submitter. ClinVar constructs a name for each CNV including the genome assembly, the cytogenetic band(s), the chromosome number, the outermost genomic coordinates, and the copy number. For example, the name for Variation 60214, a copy number loss variant on chromosome 2, is GRCh38/hg38 2q22.3(chr2:143988786–144558029)x1.
Growth of the Database
ClinVar’s first public release contained 30,000 submitted records representing 27,000 unique variants (Figure 1). Within its first five years, ClinVar has become an international resource, with 1000 submitting organizations from 65 countries (www.ncbi.nlm.nih.gov/clinvar/docs/map/). Much of the early growth of the database can be attributed to the efforts of several clinical laboratories that have been members of ClinGen, notably: EGL Genetic Diagnostics, Eurofins Clinical Diagnostics; GeneDx; Invitae; and Laboratory for Molecular Medicine, Partners HealthCare Personalized Medicine. These laboratories were early to submit their own data to ClinVar and they encouraged other clinical laboratories to share as well (Bean et al., 2013; Rehm et al., 2017; Yang et al., 2017b). As of July 11, 2018, ClinVar holds more than 680,000 submitted records representing 430,000 unique variants. The database continues to grow steadily. Each month, an average of 20 new organizations submit to ClinVar and NCBI staff process an average of 19,000 submitted records representing 11,000 variants. Most data in ClinVar are from 451 organizations providing results from clinical testing, and from 461 organizations providing results from research; note that some organizations submit results from both clinical testing and research. Data are also provided by locus-specific databases (LSDBs), expert panels, and resources such as OMIM®, GeneReviews® (https://www.ncbi.nlm.nih.gov/books/NBK1116/), and UniProt (The UniProt Consortium, 2017). More recently, ClinVar has invited clinicians and patient registries to submit phenotypic data for patients who have had genetic testing (Landrum et al., 2018), since clinical testing laboratories rarely receive this information. Data in ClinVar are freely available; therefore, it is important that each submitter has obtained appropriate consent to make the data public, and for submissions to exclude any identifiable data such as a patient name or a recognizable identifier.
Figure 1.
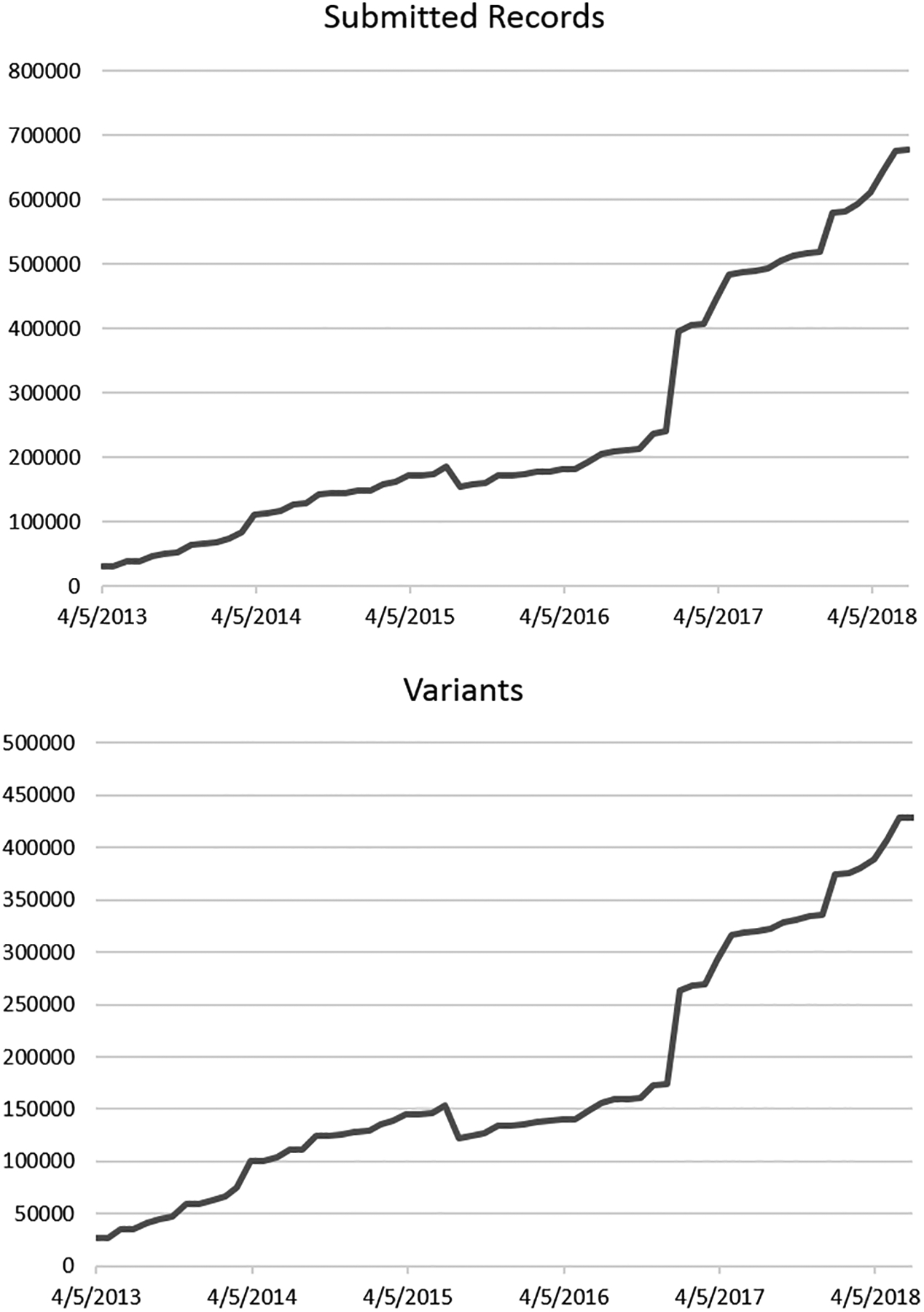
Growth of the number of unique variants and submitted records in ClinVar since its inception.
As noted above, variants of all sizes are found in ClinVar. Considering all variants, including large CNVs, 30,000 genes are affected by variants in ClinVar. Smaller variants typically lie in a single gene. These variants are present in 6,010 genes with TTN, BRCA1, and BRCA2 having the most variants in the database (Table 1). ClinVar includes variants in all of the 59 genes recommended by the American College of Medical Genetics and Genomics (ACMG) for the reporting of incidental findings (ACMG SF v2.0; Kalia et al., 2017). Among these 59 genes in ACMG SF v2.0, the top ten genes for total number of variants and number of variants reported as pathogenic or likely pathogenic in ClinVar are BRCA1, BRCA2, APC, TSC2, MSH6, MSH2, LDLR, MLH1, FBN1, and RYR1 (Table 2).
Table 1.
Top ten genes for number of variants in ClinVar.
| Gene | Number of variants |
|---|---|
| TTN | 10615 |
| BRCA2 | 9372 |
| BRCA1 | 6614 |
| ATM | 5304 |
| APC | 4774 |
| TSC2 | 3607 |
| NF1 | 3398 |
| MSH6 | 3297 |
| MSH2 | 2771 |
| LDLR | 2435 |
Table 2.
Top ten genes from ACMG SF v2.0 for total number of variants and number of pathogenic or likely pathogenic variants in ClinVar.
| Gene | Total variants | P*/LP** variants |
|---|---|---|
| BRCA2 | 9372 | 2961 |
| BRCA1 | 6614 | 2525 |
| APC | 4774 | 711 |
| TSC2 | 3607 | 355 |
| MSH6 | 3297 | 619 |
| MSH2 | 2771 | 837 |
| LDLR | 2435 | 1721 |
| MLH1 | 2263 | 800 |
| FBN1 | 2081 | 1034 |
| RYR1 | 1690 | 227 |
P, pathogenic
LP, likely pathogenic
Submitted records in ClinVar include varying degrees of evidence. Much of the evidence is structured as observations of the variant. An observation may represent an aggregate of individuals in whom the variant was observed; cases or single individuals in whom the variant was observed; or experimental observations. Only three fields related to each observation are required for submission: (1) the “allele origin”, i.e., whether the variant was found to be in the germline or was somatic; (2) the “affected status”, which describes whether or not individuals with the variant were affected by the interpreted condition; and (3) the method used to collect the data for the interpretation (the “collection method”), such as clinical testing or research. However, it is possible to submit many other types of evidence in support of an interpretation. Approximately 60% of submitted records include one of these optional forms of evidence. These types of evidence include citations identified while researching the variant and/or a free text comment that explains the rationale for the variant’s classification which are frequently provided; number of individuals and number of families with the variant are less frequently entered. The guidelines published by ACMG and the Association of Molecular Pathologists (AMP; Richards et al., 2015) name several other types of evidence that should be considered when classifying a variant, including functional evidence. ClinVar welcomes submissions based partially or entirely on functional evidence, including submissions from research laboratories that describe a functional assay and the functional consequence of the variant on the protein, but do not interpret the variant for a disease or other condition. ClinVar currently has 459 variants that include data about the experimental method and result; 1,212 variants include the functional significance of the variant provided by a submitter. To date, these variants represent 46 genes; only 7 genes have more than 10 variants with functional data (Table 3). Researchers who are generating functional data for variants are highly encouraged to share their data through ClinVar, which will make it easily accessible to clinical testing laboratories and expert panels.
Table 3.
Genes in ClinVar with the most variants having functional evidence.
| Gene | # variants with experimental results |
|---|---|
| LDLR | 313 |
| AGXT | 102 |
| APOB | 28 |
| ABCA4 | 23 |
| GATM | 15 |
| GRHPR | 15 |
| PCSK9 | 12 |
Concordance of Interpretations
The primary function of ClinVar is to archive the interpretations of variants that are submitted to the database. A secondary function is to aggregate the submitted data by variant and by variant-disease pairs. Aggregation allows users to pool evidence from multiple laboratories, which can improve their ability to interpret rare variants. Importantly, aggregation also exposes the concordance or discordance in the interpretation of the variant among different submitters. The level of discordance among laboratories that submit data to ClinVar has been a surprise and a source of concern for some in the clinical genetics community, as evidenced by several published comparisons using data from ClinVar (Balmaña et al., 2016; Bland et al., 2018; Gradishar et al., 2017; Harrison et al., 2017; Lincoln et al., 2017; Pepin et al., 2016; Rehm et al., 2017; Shah et al., 2018; Vail et al., 2015; Yang et al., 2017a). These studies used various approaches to analyze discordance within ClinVar. Some focused on certain genes or disease areas, a subset of records submitted by specific laboratories, or only medically actionable differences (Pathogenic or Likely pathogenic versus Benign, Likely benign, or Uncertain significance). One study (Harrison et al., 2017) was designed to coordinate clinical laboratories that had conflicting interpretations in ClinVar, analyze the discrepancies between those laboratories, and reach a consensus to resolve those discrepancies where possible. Notably, some studies included comparisons to private datasets. As a consequence, there are striking differences in the conclusions of these studies. At one end of the spectrum, a very high level of discordance led Gradishar et al. (2017) to “call into question the practicality of checking all test results against a database”. At the other end of the spectrum, Lincoln et al. (2017) concluded that “significant classification disagreements among the professional clinical laboratories represented in ClinVar are infrequent yet important”.
Despite the lack of a consensus opinion on the concordance level in ClinVar, such evaluations are an indication of ClinVar’s fundamental success. Five years ago, such inquiry was not feasible. Several initiatives to organize and implement a central resource for cataloging medically relevant variation information in a freely accessible, transparent model did not reach completion nor were they successfully adopted. The detailed analysis and appraisal that is given to variant data in ClinVar underscores the essential role ClinVar plays in clinical contexts and the success of its core mission — to provide transparency into how variants have been interpreted. Transparency indicates where there are conflicts in the interpretation, which underscores the need for consistent approaches to interpretation in addition to standardization of associated data and terms. It helps researchers prioritize variants that need more experimental data to inform the interpretation. Expert panels can prioritize both variants of uncertain significance and those with conflicting interpretations for in-depth review. Individual laboratories gain the opportunity to share evidence and come to a consensus on the interpretation (Harrison et al., 2017). Investigating conflicts allows individual laboratories to discuss differences in how they assign weight to certain pieces of evidence and use their professional judgement, even within the framework of a standard approach to interpretation. The ClinVar staff does not directly resolve conflicts in interpretation. However, ClinVar supports efforts to resolve conflicts in interpretation by providing a report of all pairwise interpretations with conflicts so that these data are easy to access and prioritize. A submitted record may be updated by its submitter at any time. Submitters who have changed a classification for any reason, including conflict resolution, are encouraged to update their data in ClinVar. Updates to submitted records are tracked by providing a version number for each record.
ClinVar calculates conflicts only for the terms recommended by ACMG and AMP for the interpretation of Mendelian diseases. ClinVar does not calculate a conflict for other types of interpretations. For example, a variant that is reported as pathogenic for a disease and also involved in a drug response is not considered to have a conflict in ClinVar. Instead, the aggregate interpretation is reported as “Pathogenic; drug response”. Additionally, ClinVar only reports conflicts for the variant among three levels: Pathogenic and/or Likely pathogenic, Uncertain significance, and Benign and/or Likely benign. In other words, differences in likelihood are not considered conflicts. As of July 11, 2018, there were 22,819 variants with conflicts in the interpretation. This corresponds to 5.2% of all variants in ClinVar, and 20% of variants that have more than one submission and thus are more likely to have a conflict. Note that there is also a small number of variants (217) with submission only from a single submitter that is a consortium, where the consortium reports a conflict by submitting “conflicting data from submitters” as the interpretation.
It is important to note that the number of conflicts reported above includes variants that have been reported with different interpretations for multiple disorders. The number of conflicting interpretations for variant-disease pairs (ClinVar RCV records) is 7083, or 1.1% of all RCV records. Note that some conflicts are not apparent in RCV records because different submitters may use slightly different terms for the same disease (e.g., hereditary breast cancer versus familial breast cancer), and those submissions aggregate into different RCV records. Some users prefer to consider only “medically significant” conflicts, namely Pathogenic or Likely pathogenic versus Benign, Likely benign, or Uncertain significance. Currently, there are 757 variants in ClinVar (0.2%) with a “medically significant” conflict.
Expert Panels
Interpretations from expert panels are an important subset of data in ClinVar. Members of ClinGen worked closely with ClinVar staff to develop guidelines (https://www.ncbi.nlm.nih.gov/clinvar/docs/review_guidelines/) for the composition of an expert panel, namely:
It is recommended that the expert panel include healthcare professionals caring for patients relevant to the disease gene in question: medical geneticists, clinical laboratory diagnosticians, and/or molecular pathologists who report such findings; and appropriate researchers relevant to the disease, gene, functional assays, and statistical analyses.
It is expected that the individuals comprising the expert panel represent multiple institutions.
It is expected that the individuals comprising the expert panel should be international in scope and are experts in the field based on publications and long-standing scope of work.
The goal is to establish only one expert panel per gene that is inclusive of known experts in the field. The ClinGen Steering Committee (https://www.clinicalgenome.org/about/clingen-leadership/) provides approval of expert panels, so that users can have high confidence in the expert-curated interpretations in ClinVar. As mentioned in the Introduction, the interpretation of a variant from an expert panel supersedes other submitted interpretations in ClinVar. Thus, interpretations from expert panels can be considered a form of conflict resolution. It is important to note that expert panels are not considered infallible or unchangeable; a submitted record from an expert panel may also be updated by that expert panel if new information becomes available and its interpretation changes.
Currently, there are six expert panels with data available in ClinVar. InSiGHT (Thompson et al., 2014), CFTR2 (http://cftr2.org), ENIGMA (Spurdle et al., 2012), and PharmGKB (Whirl-Carrillo et al., 2012) were the first four expert panels to submit to ClinVar. These four groups pre-date ClinGen and were used as models for recommendations for the composition of an expert panel. More recently, two expert panels from ClinGen have shared data through ClinVar: the ClinGen RASopathy Expert Panel (Gelb et al., 2018) and the ClinGen Inherited Cardiomyopathy Expert Panel (Kelly et al., 2018). To date, there are a total of 9,324 variants in ClinVar with a submitted interpretation from an expert panel (Table 4). This includes variants that were interpreted for their relationship to Mendelian diseases, cancer, and/or response to a drug. Currently there are 737 variants in ClinVar that would have a conflict in the aggregate interpretation without a submission from an expert panel.
Table 4.
Number of variants in ClinVar with a submitted record from an expert panel.*
| Expert panel | Curated genes | Total variants | Pathogenic or Likely pathogenic variants |
|---|---|---|---|
| ENIGMA | BRCA1, BRCA2 | 6156 | 4829 |
| InSiGHT | MSH2, PMS2, MSH6, MLH1 | 2389 | 1371 |
| PharmGKB | DPYD, VKORC1, CFTR, and 99 more | 196 | None – classified for drug response |
| CFTR2 | CFTR | 291 | 291 |
| ClinGen RASopathy Expert Panel | BRAF, HRAS, KRAS, MAP2K1, MAP2K2, PTPN11, RAF1, SHOC2, SOS1 | 233 | 58 |
| ClinGen Inherited Cardiomyopathy Expert Panel | MYH7 | 102 | 39 |
Some variants may have more than one expert submission, if they were interpreted both for a disease and for response to a drug.
Limitations of the Database
While ClinVar provides an invaluable resource for interpretations of variants, it is important for users to be aware of certain caveats. First, ClinVar does not include records for all variants that have been identified in a human genome. Also, no effort has been made to represent all possible variants in the human genome. The scope of the database is limited to variants that have been interpreted for their relationship to a condition, not variants that were merely observed in a patient. ClinVar only includes variants that have been submitted to the database. As submission to ClinVar is voluntary, the database is not comprehensive for all variants that are in scope for the database.
Second, some interpretations in ClinVar are more recent than others. ClinVar encourages submitters to update records when the data have changed, particularly if there is a change to the interpretation. However, updates are made at the submitter’s discretion. Some organizations update their ClinVar records on a regular schedule or as needed, while other organizations do not update their records. As an example of the latter, a research group may not follow up on a particular variant that they originally reported to ClinVar as pathogenic, or a clinical laboratory may close. ClinVar does not address the issue of out-of-date interpretations at this time; all submitted records are considered current, regardless of the date of submission or the date that the variant was interpreted by the submitter (“date last evaluated”). One alternative approach is to designate a time period for currency of submitted records. After that time period has passed, the submitted record could be moved to a “legacy” category, such that the submitted record is available to users but it does not contribute to the aggregate data for the variant. Feedback on this suggested approach is welcome.
Third, ClinVar accepts submissions of interpretations from a wide variety of organizations. Some organizations collect the data and interpret variants in a clinical context, while others interpret variants as part of research or a curation project; this context is represented by the “collection method” in ClinVar. Some submitting organizations may have more experience or expertise in a particular gene or disease. ClinVar staff members do not try to determine if an interpretation is correct, nor do they determine how qualified a submitting organization may be. ClinVar does provide factual information to help the user evaluate submitted data. For example, calculation of the review status considers whether the submitting organization provided documentation of the criteria that it uses to interpret variants (“assertion criteria”). The review status does not indicate the appropriateness of those criteria; the user must make that judgement. PubMed (NCBI Resource Coordinators, 2018) may be considered as an analogy for ClinVar. PubMed includes citations for many papers, of varying quality. Tagging a citation as a review article suggests a higher level of expertise of the authors, similar to a higher review status in ClinVar. However, a user of PubMed must use their own judgement to determine which citations are of high quality and valuable to their work. In the same way, database users should look to ClinVar not as a single source of truth, but rather as a guide for access to interpretations provided by members of the clinical genetics community, and combine that data with their own judgement to make their own interpretation of a variant. A practical approach to apply such judgement is to download data from ClinVar and filter the data by review status to exclude any records for which assertion criteria have not been provided. A similar filter can be applied to search results on the ClinVar web site (Landrum et al., 2014).
Finally, the ClinVar database is not a finished product. Projecting that ClinVar will continue to accumulate data, the ClinVar team has identified two major priorities to improve the utility of the database. One is to implement an easier, more intuitive search for data on the ClinVar website. Those who are interested in volunteering for usability testing of ClinVar’s search function should email clinvar@ncbi.nlm.nih.gov. The other is to improve the turnaround time for submissions of data to ClinVar. It is anticipated that new features to aid submitters to ClinVar will be made available within the next two years.
Conclusion
ClinVar has changed the way information is shared within the clinical genetics community. It has now become routine for clinical genetics testing laboratories to share their interpretations of variants publicly. Laboratories can more easily identify when another laboratory has seen a rare variant; they can also potentially learn when a variant has been observed in a patient with a disease or phenotype that was not previously associated with the gene. Laboratories are now aware of differences in interpretations of a variant, resulting in opportunities to review evidence from different sources and reclassify the variant, if necessary. Researchers easily identify variants that would benefit from additional experimental data to bolster the evidence for the interpretation. Expert panels focus their curation efforts on the difficult variants, including variants with uncertain significance and variants with conflicting interpretations. Genetic counselors, clinical geneticists, and other clinical providers consult ClinVar to gather more information about variants reported by a testing laboratory.
In ClinVar’s first five years, its vision has been realized. A free, publicly available resource now stands to archive and provide full access to vital information that forms the bridge between variation and clinical interpretation. Transparency has allowed for variant conflict interrogation and resolution. Analysis of such data can now be initiated, which is further pushing the field of clinical genetics and ClinVar forward. It is anticipated that the next five years will bring sustained growth of ClinVar submissions, including an increase in somatic and structural variants plus a wealth of associated phenotypic data. ClinVar will continue to strive for ease of access and submission, plus gains in interoperability. In addition to promoting standards, ClinVar aims to foster validation, consistency, and collaboration among participants. With its commitment to advancing human health through the growth and development of the database, ClinVar is expected to cement its place of importance in the clinical genetics community.
Acknowledgments
The authors thank Donna Maglott and Shanmuga Chitipiralla for their database expertise, Marilu Hoeppner for her meticulous copy-editing skills, and the entire ClinVar team for their tireless efforts to maintain and improve ClinVar. The authors also acknowledge collaborators in the ClinGen project. This work was supported by the Intramural Research Program of the National Library of Medicine, National Institutes of Health.
Footnotes
Conflict of interest
None declared.
References
- ACMG Board of Directors. (2017). Laboratory and clinical genomic data sharing is crucial to improving genetic health care: a position statement of the American College of Medical Genetics and Genomics. Genetics in Medicine, 19(7):721–722. doi: 10.1038/gim.2016.196. [DOI] [PubMed] [Google Scholar]
- American Medical Association. (2013). Genome analysis and variant identification. Policy D-460.971.
- Balmaña J, Digiovanni L, Gaddam P, Walsh MF, Joseph V, Stadler ZK, Nathanson KL, Garber JE, Couch FJ, Offit K, Robson ME, & Domchek SM (2016). Conflicting Interpretation of Genetic Variants and Cancer Risk by Commercial Laboratories as Assessed by the Prospective Registry of Multiplex Testing. Journal of Clinical Oncology, 34(34), 4071–4078. doi: 10.1200/JCO.2016.68.4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean LJ, Tinker SW, da Silva C, & Hegde MR (2013). Free the data: one laboratory’s approach to knowledge-based genomic variant classification and preparation for EMR integration of genomic data. Hum Mutat. 34(9), 1183–8. doi: 10.1002/humu.22364. Epub 2013 Aug 5. [DOI] [PubMed] [Google Scholar]
- Bland A, Harrington EA, Dunn K, Pariani M, Platt JCK, Grove ME, & Caleshu C (2018). Clinically impactful differences in variant interpretation between clinicians and testing laboratories: a single-center experience. Genetics in Medicine, 20(3), 369–373. doi: 10.1038/gim.2017.212. [DOI] [PubMed] [Google Scholar]
- Fokkema IF, Taschner PE, Schaafsma GC, Celli J, Laros JF, & den Dunnen JT (2011). LOVD v.2.0: the next generation in gene variant databases. Human Mutation, 32(5), 557–63. doi: 10.1002/humu.21438. [DOI] [PubMed] [Google Scholar]
- Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, Cole CG, Ward S, Dawson E, Ponting L, Stefancsik R, Harsha B, Kok CY, Jia M, Jubb H, Sondka Z, Thompson S, De T, & Campbell PJ (2017). COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Research, 45 (D1), D777–D783. 10.1093/nar/gkw1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelb BD, Cavé H, Dillon MW, Gripp KW, Lee JA, Mason-Suares H, Rauen KA, Williams B, Zenker M, Vincent LM. (2018). ClinGen’s RASopathy Expert Panel consensus methods for variant interpretation. Genetics in Medicine. Mar 1. doi: 10.1038/gim.2018.3. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradishar W, Johnson K, Brown K, Mundt E, & Manley S (2017). Clinical Variant Classification: A Comparison of Public Databases and a Commercial Testing Laboratory. Oncologist, 22(7), 797–803. doi: 10.1634/theoncologist.2016-0431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison SM, Dolinsky JS, Knight Johnson AE, Pesaran T, Azzariti DR, Bale S, Chao EC, Das S, Vincent L, & Rehm HL (2017). Clinical laboratories collaborate to resolve differences in variant interpretations submitted to ClinVar. Genetics in Medicine, 19(10), 1096–1104. doi: 10.1038/gim.2017.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia SS, Adelman K, Bale SJ, Chung WK, Eng C, Evans JP, Herman GE, Hufnagel SB, Klein TE, Korf BR, McKelvey KD, Ormond KE, Richards CS, Vlangos CN, Watson M, Martin CL, & Miller DT (2017). Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genetics in Medicine, 19(2), 249–255. doi: 10.1038/gim.2016.190. [DOI] [PubMed] [Google Scholar]
- Kelly MA, Caleshu C, Morales A, Buchan J, Wolf Z, Harrison SM, Cook S, Dillon MW, Garcia J, Haverfield E, Jongbloed JDH, Macaya D, Manrai A, Orland K, Richard G, Spoonamore K, Thomas M, Thomson K, Vincent LM, Walsh R, Watkins H, Whiffin N, Ingles J, van Tintelen JP, Semsarian C, Ware JS, Hershberger R, Funke B (2018). Adaptation and validation of the ACMG/AMP variant classification framework for MYH7-associated inherited cardiomyopathies: recommendations by ClinGen’s Inherited Cardiomyopathy Expert Panel. Genet Med. 20(3):351–359. doi: 10.1038/gim.2017.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, & Maglott DR. (2014). ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Research, 42, D980–5. doi: 10.1093/nar/gkt1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Jang W, Karapetyan K, Katz K, Liu C, Maddipatla Z, Malheiro A, McDaniel K, Ovetsky M, Riley G, Zhou G, Holmes JB, Kattman BL, & Maglott DR (2018). ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Research, 46(D1), D1062–D1067. doi: 10.1093/nar/gkx1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lincoln SE, Yang S, Cline MS, Kobayashi Y, Zhang C, Topper S, Haussler D, Paten B, & Nussbaum RL (2017). Consistency of BRCA1 and BRCA2 Variant Classifications Among Clinical Diagnostic Laboratories. JCO Precision Oncology. 1. doi: 10.1200/PO.16.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Society of Genetic Counselors (NSGC). (2015, April). Clinical Data Sharing (Position Statement) [Blog post]. Retrieved from: https://www.nsgc.org/p/bl/et/blogaid=330
- NCBI Resource Coordinators. (2018). Database resources of the National Center for Biotechnology Information. Nucleic Acids Research, 4;46(D1):D8–D13. doi: 10.1093/nar/gkx1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepin MG, Murray ML, Bailey S, Leistritz-Kessler D, Schwarze U, & Byers PH. (2016). The challenge of comprehensive and consistent sequence variant interpretation between clinical laboratories. Genetics in Medicine, 18(1), 20–4. doi: 10.1038/gim.2015.31. [DOI] [PubMed] [Google Scholar]
- Rehm HL, Berg JS, Brooks LD, Bustamante CD, Evans JP, Landrum MJ, Ledbetter DH, Maglott DR, Martin CL, Nussbaum RL, Plon SE, Ramos EM, Sherry ST, Watson MS; & ClinGen. (2015). ClinGen—the Clinical Genome Resource. New England Journal of Medicine, 372(23):2235–42. doi: 10.1056/NEJMsr1406261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehm HL, Harrison SM, & Martin CL (2017). ClinVar Is a Critical Resource to Advance Variant Interpretation. Oncologist, 22(12), 1562. doi: 10.1634/theoncologist.2017-0246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, & Rehm HL; ACMG Laboratory Quality Assurance Committee. (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–24. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah N, Hou YC, Yu HC, Sainger R, Caskey CT, Venter JC, & Telenti A (2018). Identification of Misclassified ClinVar Variants via Disease Population Prevalence. American Journal of Human Genetics, 102(4), 609–619. doi: 10.1016/j.ajhg.2018.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spurdle AB, Healey S, Devereau A, Hogervorst FB, Monteiro AN, Nathanson KL, Radice P, Stoppa-Lyonnet D, Tavtigian S, Wappenschmidt B, Couch FJ, Goldgar DE; & ENIGMA. (2012). ENIGMA—evidence-based network for the interpretation of germline mutant alleles: an international initiative to evaluate risk and clinical significance associated with sequence variation in BRCA1 and BRCA2 genes. Human Mutation, 33(1), 2–7. doi: 10.1002/humu.21628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, Hussain M, Phillips AD, & Cooper DN (2017). The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Human Genetics, 136(6), 665–677. doi: 10.1007/s00439-017-1779-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson BA, Spurdle AB, Plazzer JP, Greenblatt MS, Akagi K, Al-Mulla F, Bapat B, Bernstein I, Capellá G, den Dunnen JT, du Sart D, Fabre A, Farrell MP, Farrington SM, Frayling IM, Frebourg T, Goldgar DE, Heinen CD, Holinski-Feder E, Kohonen-Corish M, Robinson KL, Leung SY, Martins A, Moller P, Morak M, Nystrom M, Peltomaki P, Pineda M, Qi M, Ramesar R, Rasmussen LJ, Royer-Pokora B, Scott RJ, Sijmons R, Tavtigian SV, Tops CM, Weber T, Wijnen J, Woods MO, Macrae F, & Genuardi M (2014). Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nature Genetics, 46(2), 107–115. doi: 10.1038/ng.2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The UniProt Consortium. (2017). UniProt: the universal protein knowledgebase. Nucleic Acids Res. 45(D1):D158–D169. doi: 10.1093/nar/gkw1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vail PJ, Morris B, van Kan A, Burdett BC, Moyes K, Theisen A, Kerr ID, Wenstrup RJ, & Eggington JM (2015). Comparison of locus-specific databases for BRCA1 and BRCA2 variants reveals disparity in variant classification within and among databases. Journal of Community Genetics, 6(4), 351–9. doi: 10.1007/s12687-015-0220-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whirl-Carrillo M, McDonagh EM, Hebert JM, Gong L, Sangkuhl K, Thorn CF, Altman RB, & Klein TE (2012). Pharmacogenomics knowledge for personalized medicine. Clinical Pharmacology & Therapeutics, 92(4), 414–7. doi: 10.1038/clpt.2012.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Lincoln SE, Kobayashi Y, Nykamp K, Nussbaum RL, & Topper S (2017a). Sources of discordance among germ-line variant classifications in ClinVar. Genetics in Medicine, 19(10), 1118–1126. doi: 10.1038/gim.2017.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Cline M, Zhang C, Paten B, & Lincoln SE (2017b). Data sharing and reproducible clinical genetic testing: Successes and challenges. Pac Symp Biocomput. 22, 166–176. doi: 10.1142/9789813207813_0017. [DOI] [PMC free article] [PubMed] [Google Scholar]