

Abstract
INTRODUCTION
Transposable element (TE) dysregulation is associated with neuroinflammation in Alzheimer's disease (AD) brains. Yet, TE quantitative trait loci (teQTL) have not been well characterized in human aged brains with AD.
METHODS
We leveraged large‐scale bulk and single‐cell RNA sequencing, whole‐genome sequencing (WGS), and xQTL from three human AD brain biobanks to characterize TE expression dysregulation and experimentally validate AD‐associated TEs using CRISPR interference (CRISPRi) assays in human induced pluripotent stem cell (iPSC)–derived neurons.
RESULTS
We identified 26,188 genome‐wide significant TE expression QTLs (teQTLs) in human aged brains. Subsequent colocalization analysis of teQTLs with AD genetic loci identified AD‐associated teQTLs and linked locus TEs. Using CRISPRi assays, we pinpointed a neuron‐specific suppressive role of the activated short interspersed nuclear element (SINE; chr11:47608036–47608220) on expression of C1QTNF4 via reducing neuroinflammation in human iPSC‐derived neurons.
DISCUSSION
We identified widespread TE dysregulation in human AD brains and teQTLs offer a complementary analytic approach to identify likely AD risk genes.
Highlights
Widespread transposable element (TE) dysregulations are observed in human aging brains with degrees of neuropathology, apolipoprotein E (APOE) genotypes, and neuroinflammation in Alzheimer's disease (AD).
A catalog of TE quantitative trait loci (teQTLs) in human aging brains was created using matched RNA sequencing and whole‐genome sequencing data.
CRISPR interference assays reveal that an upregulated intergenic TE from the MIR family (chr11: 47608036–47608220) suppresses expression of its nearest anti‐inflammatory gene C1QTNF4 in human induced pluripotent stem cell–derived neurons.
Keywords: Alzheimer's disease, CRISPR interference, neuroinflammation, transposable elements, transposable element expression quantitative trait loci
1. BACKGROUND
Transposable elements (TEs), known as “jumping genes” or “viral elements,” constitute ~45% of the human genome. 1 TEs are transcriptionally silenced by epigenetic mechanisms, such as DNA methylation and histone modifications. 2 However, the effectiveness of this silencing declines with age and in neurodegenerative disorders, including Alzheimer's disease (AD). 2 In the brains of tau transgenic AD mice, TEs, especially from the endogenous retrovirus (ERV) class, can be activated at RNA, DNA, and protein levels in the context of brain aging and tauopathy. 3 By integrating studies of AD from human post mortem brain tissues and Drosophila melanogaster models, it has been demonstrated that tau is sufficient to induce TE activation and that this activation is associated with active chromatin signatures at multiple ERV genomic loci. 4 Loss of nuclear TAR DNA‐binding protein 43 (TDP‐43) is associated with chromatin de‐condensation around long interspersed nuclear elements (LINEs) and leads to increased retrotransposition of LINE‐1 in post mortem frontotemporal degeneration–amyotrophic lateral sclerosis (FTD‐ALS). 5 TE activation is also highly important during neurodevelopment, 6 indicating that TE regulation in the brain is a common feature across the human lifespan.
Elevated ERVs in neurons have been linked to activated microglia and inflammatory responses during mouse brain development. 7 De‐repression of a panel of LINE and long‐terminal repeat (LTR) families causes degenerative phenotypes in human TDP‐43 Drosophila neurons and glia, which can be rescued by genetically blocking expression of this TE. 8 Reactivated ERV can also promote protein aggregate spreading in a cell line model. 9 Thus, therapeutically targeting TEs might help treat aging‐related brain disorders. 10 It has also been demonstrated that genetic stabilization of heterochromatin suppresses aging‐associated TE activation and extends life span in Drosophila. 11 Likewise, downregulation of Tc1 (a superfamily of interspersed repeats and DNA transposons) extends lifespan in Caenorhabditis elegans. 12 In addition, nucleoside reverse‐transcriptase (RT) inhibitors and antiretroviral human immunodeficiency drugs rescue inflammation and cellular senescence triggered by TE activation, which extends lifespan. 13 Thus, understanding the mechanistic basis of TE dysregulation in the aging human brain could help identify therapeutic strategies for preserving brain health throughout aging.
RESEARCH IN CONTEXT
Systematic review: The authors reviewed the literature using traditional sources. Transposable elements (TEs), known as “jumping genes” or “viral elements”, constitute ~45% of the human genome. Although TE dysregulation has been identified in aged brains, TE‐mediated quantitative trait loci (teQTL) has not been well characterized in Alzheimer's disease (AD). We posit that the systematic identification of TE dysregulation and teQTLs in human aged brains will identify novel insights into non‐coding genetics and genome regulatory architecture and offer a promising avenue for understanding AD genetics and identifying novel targets for therapeutic development.
Interpretation: We identified widespread TE dysregulation in human aged brains through leveraging large‐scale RNA sequencing (RNA‐seq), whole‐genome sequencing (WGS), and various brain‐specific QTL data (xQTL) from three brain biobanks: (a) Mount Sinai Brain Bank (MSBB), (b) Mayo Clinic (Mayo), and (c) Religious Orders Study (ROS) or the Rush Memory and Aging Project (MAP) (ROS/MAP) brain biobanks. Colocalization analysis of teQTLs with large AD genome‐wide association study loci prioritized multiple AD likely causal genes (i.e., C1QTNF4 and FDFT1) regulated by teQTLs. Using CRISPR interference assays, we demonstrated that an upregulated intergenic TE from the MIR family (chr11: 47608036–47608220) suppresses expression of its nearest anti‐inflammatory gene C1QTNF4 in human induced pluripotent stem cell–derived neurons, highlighting the regulatory role of AD‐associated TE activation underlying the AD neuroinflammation.
Future directions: These findings demonstrate widespread TE dysregulation in human AD brains and teQTLs offer a powerful analytic approach to identifying AD risk genes. Further investigations using long‐read RNA‐seq data from ethnically diverse cohorts and experimental models are essential to establish a likely causal relationship of TE activation and disease etiologies and to identify TE‐targeted biomarkers and therapeutics for AD.
In this study, we sought to systematically characterize genetic control of TE expression and identify dysregulated TEs that could potentially contribute to disease pathogenesis in human aging brains with AD. First, we used two complementary approaches to investigate TE expression and dysregulation across diverse AD pathologies (including tau and amyloid beta [Aβ], apolipoprotein E [APOE] genotypes, and sex) using bulk RNA sequencing (RNA‐seq) data from three large‐scale human brain biobanks (Figure 1). To identify genetic control of expressed TEs, we integrated TE transcriptomic profiles with matched whole‐genome sequencing (WGS) data. This identified 26,188 genome‐wide significant TE‐mediated quantitative trait loci (teQTLs) in human brains. We then used colocalization analysis to prioritize risk loci associated with TE dysregulation by integrating AD genome‐wide association study (GWAS) datasets with xQTLs, including teQTLs, gene expression QTLs (eQTLs), DNA methylation QTLs (meQTLs), and H3K27 histone acetylation QTLs (haQTLs). The regulatory relationship between an upregulated TE and its potential target gene, such as a short interspersed nuclear element (SINE; chr11: 47608036–47608220) and C1QTNF4, were experimentally investigated using human brain cell type‐specific enhancer‐promoter interactome maps and CRISPR interference (CRISPRi) assays in human induced pluripotent stem cell (iPSC)‐derived excitatory neurons.
FIGURE 1.
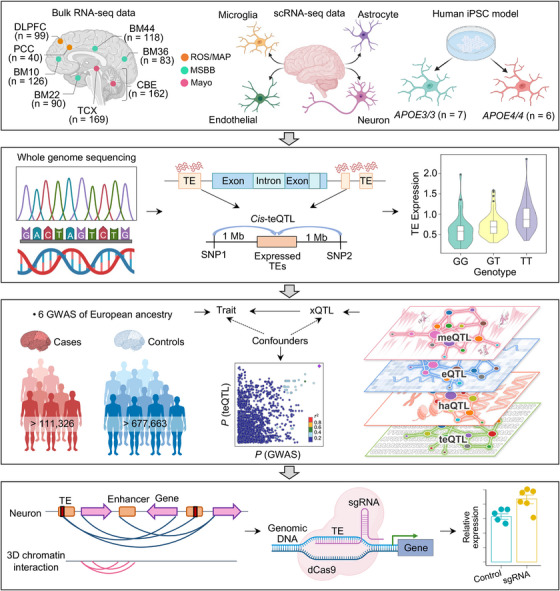
A diagram illustrating systematic characterization of TE dysregulation in human brains with AD. Bulk RNA‐seq data sets from three brain biobanks (MSBB, Mayo, and ROS/MAP) were downloaded from the AD knowledge portal. RNA‐seq data from the MSBB cohort was derived from four brain BM, including BM10, BM22, BM36, and BM44. RNA‐seq data from the Mayo cohort consisted of 156 AD patients and 175 cognitive HCs from cerebellum and TCX tissues. RNA‐seq data from the ROS/MAP cohort were collected from 98 AD and 41 cognitive HCs in two cortex regions. Results obtained from bulk RNA‐seq data were validated using three brain cell‐type RNA‐seq datasets: (1) the first FACS‐purified RNA‐seq data from four major brain cell populations from AD and control frozen cortex tissues, (2) the second snRNA‐seq datasets consisting of 482,472 nuclei from non‐demented control brains and AD brains with both Aβ and tau pathology, and (3) the third RNA‐seq data from human iPSC‐derived population APOE brain microglia cells. Family‐ and locus‐based TE expression levels were calculated using three tools (see Methods). We profiled Atlas of Human Brain teQTLs by integrating locus‐based expressed TEs with corresponding WGS data from the Mayo brain biobank. We then leveraged colocalization analysis of xQTLs with three AD GWAS summary statistics data to identify AD likely causal genes regulated by brain teQTLs. The underlying mechanisms between TE and AD causal genes were dissected using brain cell type–specific enhancer–promoter interactome maps. Finally, we used CRISPRi to confirm the regulatory relationship between locus TE and its target gene. Aβ, amyloid beta; AD, Alzheimer's disease; APOE, apolipoprotein E; BM, Brodmann areas; CRISPRi, CRISPR interference; DLPFC, dorsolateral prefrontal cortex; FACS, fluorescence‐activated cell sorting; GWAS, genome‐wide association study; HC, healthy controls; iPSC, induced pluripotent stem cell; Mayo, Mayo Clinic; MSBB, Mount Sinai brain bank; PCC, posterior cingulate cortex; RNA‐seq, RNA sequencing; ROS/MAP, Religious Orders Study/Rush Memory and Aging Project; TCX, temporal cortex; TE, transposable element; teQTLs, transposable element quantitative trait loci; WGS, whole‐genome sequencing.
2. METHODS
2.1. RNA‐seq data collection
We obtained fastq files for RNA‐seq datasets in similar brain regions from age‐ and sex‐matched subjects from the three primary Accelerating Medicines Partnership Alzheimer's Disease (AMP‐AD) cohort studies in the Synapse database (Table S1 in supporting information). The first dataset consists of RNA‐seq data generated as part of the Mount Sinai Brain Bank (MSBB) study (synapse ID: syn3159438). For this study, RNA was collected from 272 AD patients and 145 healthy controls (HCs) of European ancestry across four brain Brodmann areas (BM), including BM10 (n = 126), BM22 (n = 90), BM36 (n = 83), and BM44 (n = 118). The second dataset consists of RNA‐seq data generated as part of the Mayo RNA‐seq study (Mayo) study (synapse ID: syn5550404). For this study, RNA was collected from the cerebellum and temporal cortex (TCX) of 156 AD patients and 175 HCs of European ancestry. The third dataset consists of RNA‐seq data generated as part of the Religious Orders Study Rush Memory and Aging Project (ROS/MAP; synapse ID: syn3219045). For this study, RNA was collected from the posterior cingulate cortex and dorsolateral prefrontal cortex (DLPFC) of 98 AD patients and 41 HCs of European ancestry. We first used Plink 14 to confirm the ancestry information of subjects from the three brain biobanks based on reference samples of HapMap III (Figure S1A in supporting information). Clinical and pathological variables for the subjects, including RNA integrity number (RIN), sex, race, age at death, post mortem interval (PMI), disease status, APOE genotype, Braak staging, plaque density, and sequencing batch were also retrieved from the Synapse database (syn21241740).
2.2. Analysis of TE expression using bulk RNA‐seq data
We first used the SQuIRE software package for analyzing TE expression at both the subfamily and locus level. 15 The SQuIRE pipeline includes four tools: Fetch, Map, Count, and Call. By default, SQuIRE automatically downloads hg38 annotation files for genes and TEs from RefSeq and the University of California Santa Cruz (UCSC) Genome Browser RepeatMasker track with the fetch tool. The reference panel of the RepeatMasker database includes locus‐based TEs for each subfamily's members within the retrotransposon (SINE, LINE, LTR) and DNA transposon (DNA) families (Table S2 in supporting information). The Map tool uses parameters tailored to the alignment of TEs. By default, the reads were mapped to both the hg38 genome and the RepeatMasker annotation using STAR 16 with the parameters ‐outFilterScoreMinOverLread 0.4 ‐outFilterMatchNminOverLread 0.4 ‐chimSegmentMin 100 to allow for multi‐mapping and discordant alignments. The output BAM file was further processed by the Count tool using StringTie with default settings, which incorporates both unique‐ and multi‐mapped reads to calculate read counts and fragments per kilobase of transcript per million mapped reads (FPKM) for each TE locus. Briefly, Count first identifies reads that map to TEs; reads that only align to unique locations in the genome are termed unique‐mapped reads and reads that map to multiple genomic loci are termed multi‐mapped reads. Then Count assigns fractions of a read to each TE and further calculates the probability that the TE contributes to that read. The uniquely mapped reads have 100% probability to give rise to that read. TEs without uniquely mapped reads (n) receive fractions inversely proportional to the number of loci (N). Thus, TEs with unique reads obtain the remainder fraction (). Then Count normalizes each unique count () to the number of individual unique read start positions, or each TE's uniquely aligned length (). TEs with unique mapped reads (s ) are compared to each other. The fraction of a read is calculated as the ratio of normalized unique count () to the combined normalized unique count of all TEs (), as shown in the following Equation (1):
| (1) |
Count further refines this initial assignment using an expectation maximization (EM). Count normalizes a TE's total read count ( unique read counts multi‐aligned fractions from the previous step) by the effective transcript length (): Then the relative normalized total count is compared to the combined normalized total count of all of the TEs being compared (), as shown in the following Equation (2):
| (2) |
The output read count file was further processed for differential expression analysis among different biological comparison groups using a well‐established RNA‐seq harmonization approach released by the AMP‐AD consortium (syn21241740).
To test the replication rate of SQuIRE, we first used TEtranscripts 17 to assess the accuracy of family‐based TE expression. TEtranscripts first assigns unique‐mapped and multi‐mapped reads to each TE according to sequence similarity. Then, TEtranscripts combines those inserted locus TE RNA abundances into subfamilies according to definitions and nomenclature from RepeatMasker. 18 We then used the multi mode to assign weight to the contribution of the ambiguously mapped reads at each mapped locus. The EM algorithm was further used to determine the maximum likelihood of multi‐mapped reads to all TE transcripts. We then used Telescope 19 to confirm the accuracy of locus‐based TE expression. After the alignment stage, Telescope re‐assigns one or more possible alignments for each fragment, along with the respective alignment scores. Telescope then calculates penalties for each position in the alignment. Finally, Telescope iteratively optimizes the estimated penalties and reassigns fragments using a Bayesian statistical model. 19 Compared to other tools, Telescope has greater resolution and is sensitive to differences in sequencing platforms.
2.3. Differential expression analysis
For each brain biobank, we selected covariable factors from the RNA‐seq harmonization study released by the AMP‐AD consortium (syn21241740), including clinical variables (diagnosis, APOE genotype, sex, age at death, race, brain region, individual ID, and PMI) and sequencing variables (RIN, sequencing batch, and sequencing statistic results). To account for potential non‐linear dependence for RIN, squared terms (RIN2) were included. We first used the software variancePartition (version 1.21.6) 20 package to calculate percent variation in TE expression explained by each variable. It is apparent that variation of individuals is the strongest biological driver of variation followed by differences across sex and age at death (Figure S1B). The following model was fitted for each brain biobank: TE expression ∼ diagnosis + apoe4_allele + sex + age_death + brain region + sequencingBatch + pmi + RIN + RIN2 + race + ethnicity + individualID + alignment Summary Metrics_PCT_PF_READS_ALIGNED + RnaSeqMetrics_PCT_INTRONIC_BASES + RnaSeq Metrics_PCT_INTERGENIC_BASES + RnaSeq Metrics_PCT_CODING_BASES. We then merged the RNA‐seq data from different brain regions for each brain biobank. We further applied a well‐established data harmonization approach released by the AMP‐AD consortium (syn21241740) to identify both subfamily‐ and locus‐based differentially expressed TEs based on the above resultant TE count tables. Briefly, we removed poorly expressed TEs from the voom‐normalized TE expression matrix. TE expression was further filtered by comparing transcriptomic changes between TEs and the nearest gene. We retained TEs harboring oppositive log2FC value between the TE and its nearest gene. Then we used fixed/mixed effects modeling to adjust for the possible factors mentioned above on the trimmed mean of M–normalized count matrix table from the edgeR and voom package. 21 The filterByExpr function in the edgeR package provides an automatic way to filter expression of TEs, while keeping more highly expressed TEs for downstream analysis. P values were adjusted for multiple testing using the Benjamini–Hochberg method, with TEs considered differentially expressed at |log2FC| ≥ 1.0 and q < 0.05. Locus‐based differentially expressed TEs that overlapped with gene exons were filtered using bedtools. 22 The differentially expressed genes were retrieved from the RNA‐seq harmonization study (syn21241740). We applied this harmonization approach across different biological comparison groups, including AD versus HC, APOE ε4 AD versus HC, female AD versus female HC, and male AD versus male HC for each brain biobank.
2.4. Genotype data preprocessing
The WGS datasets from 349 subjects in variant call format (VCF) were downloaded from the Mayo study (synapse ID: syn11707308). Samples that overlapped with the RNA‐seq harmonization study (syn9702085) were selected for teQTL analysis. In total, 152 subjects were diagnosed with AD or were HCs and had matched WGS and RNA‐seq data in the TCX region of the Mayo study. Ancestry information was estimated based on reference samples of known ethnicities from HapMap III (Figure S1A). We use the following steps to improve genotype data quality, based on published protocols widely used for GWAS 23 and eQTL studies. 24 First, variants with a genotype missing rate ≥ 5% were excluded. In total, 2,020,217 variants were excluded in this step. We then checked the genotype missing rate at subject level. Subjects with genotype missing rate < 5% were left for further analysis. No subjects with excessive missing values were excluded from this step. We further filtered variants with the Hardy–Weinberg equilibrium (HWE) test using P value < 10−6. From the 199 subjects, 715,854 variants were removed in this step. We then used the Mishap test incorporated in PLINK 14 to predict the genotype missingness status of a single nucleotide polymorphism (SNP) by neighbor SNPs. Using 10−9 as the P value threshold in the Mishap test, we obtained 16,944,001 variants passing the filters. Due to limited sample size, 7,027,116 variants with minor allele frequency (MAF) ≤ 0.01 were excluded for subsequent analysis. 25 To improve sequencing data quality, we used independent SNPs to calculate heterozygosity rates. No samples with heterozygosity rates 4 standard deviations from the mean were excluded. 26 In total, 152 TCX brain region samples consisting of 8,575,054 SNPs passed the quality control in the preprocessing of genotype data.
2.5. TE expression data preprocessing
We selected TEs with > 1 FPKM in at least 50% samples for teQTL analysis. 15 We used the bedtools 22 intersect function to identify the nearest genes for each locus TE. Locus‐based differentially expressed TEs that overlapped with gene exons were filtered out. We retained TEs that showed oppositive log2FC compared to the nearest genes, based on differential expression analysis and RNA harmonization study (syn21241740). We further used three approaches to exclude sample outliers with problematic expression profiles, including relative log expression (RLE) analysis, pair‐wise correlation‐based hierarchical clustering, and D‐statistics analysis. 26 Samples with problematic expression profiles separated from normal samples were labeled as outliers. In total, we obtained 152 samples with 43,254 expressed TE loci for subsequent teQTL mapping analysis.
2.6. teQTL mapping
We used Matrix eQTL 27 to perform teQTL mapping using all the preprocessed genomic autosomal variants and TE expression data in the Mayo cohort. For cis‐teQTL mapping, we restricted our search to variants within 1 Mb upstream and downstream of each expressed TE. 28 This software used linear regression and analysis of variance models to test associations between gene expression levels and genotypes. We included covariates, including sex, age at death, APOE genotype, post mortem interval, Braak staging scores, and Thal amyloid stages when performing association testing. Multiple testing of Matrix eQTL was addressed by calculating the false discovery rate (FDR) for the TE–SNP pairs that passed a user‐defined significance threshold. This calculation was followed by the Bonferroni and Hochberg procedure, which is to let P (1) < P (2) < … < P (K) be the P values that passed the user‐defined significance threshold and N be the total number of tests performed. We used the FDR value of 0.05 as the threshold for cis‐teQTL.
We used RNA‐seq data and matched WGS data from the ROS/MAP cohort to replicate our teQTL findings (n = 45). We applied the same approach mentioned above to test local TE–SNP pairs. Replication rate of teQTL findings between the two cohorts was further assessed using the statistic; a detailed description of this estimation is described in section 2.7. All the TEs shown in this study have been annotated with assembly GRCh38. We converted genomic coordinates between different genome assemblies using CrossMap (version 0.6.4). 29
2.7. Estimation of teQTL SNP sharing with different molecular QTLs
We first obtained full summary statistic QTL data from three databases: (1) the eQTL dataset from the Mayo brain biobank 30 derived from cerebellum and TCX of European ancestry; (2) x‐QTL datasets from DLPFC from the ROS/MAP brain biobank (synapse ID: syn17015233), 31 including eQTL, cis DNA meQTL, and cis haQTL, all derived from European ancestry; (3) the Metabrain eQTL dataset from cortex of European ancestry. 32 Detailed information for QTL datasets used in this study are provided in Table S3 in supporting information. We then used a pairwise statistic 33 to test sharing and replication rate of teQTL with the three types of xQTL datasets using the qvalue R (version 4.2.0) package. Using sharing between teQTL and eQTL as an example, this analysis could perform FDR estimation with a given set of P values from the tested TE expression–SNP associations (test phenotype) that overlapped with meQTL SNPs (discovery phenotype). Thus, if the most SNPs could affect the two molecular phenotypes, then the corresponding would be high.
2.8. summary statistics in AD
We downloaded three AD GWAS summary statistics datasets: (1) late‐onset AD from GWAS catalog with accession number GCST007511 34 (63,926 participants of European ancestry); (2) late‐onset AD by Wightman et al. with accession number GCST90044699 35 (1,126,563 subjects of European ancestry); and (3) GWAS of AD from GWAS catalog with accession number GCST90027158 36 (788,989 participants). We extracted the MAF value from 1000 Genomes Phase 1 and selected SNPs with MAF > 1% for subsequent analysis. We defined genome‐wide significant loci with a threshold of GWAS P < 5.0 × 10−8. Loci with suggestive association were defined at a relaxed threshold of GWAS P < 1.0 × 10−5. Detailed information for the three GWAS datasets can be found in Table S4 in supporting information.
2.9. Colocalization analysis
To assess whether two molecular phenotypes were consistent with a shared causal variant, we performed Bayesian colocalization analysis using the coloc 37 package in R (version 4.2.0) for AD GWAS loci with different molecular phenotypes, including teQTL, eQTL, meQTL, and haQTL. We selected any SNPs passing genome‐wide significance (P < 5.0 × 10−8) in the three AD GWAS datasets. For xQTL datasets, we selected a relaxed threshold of xQTL P < 1.0 × 10−5 based on a previous study. 25 SNPs with MAF > 1% were selected for subsequent analysis. This method takes the two sets of summary statistics as input (for traits 1 and traits 2, which we refer to as “configuration”). Each possible colocalization pair can be assigned to one of five hypotheses:
H0: No association with either trait
H1: Association with trait 1, not with trait 2
H2: Association with trait 2, not with trait 1
H3: Association with trait 1 and trait 2, two independent SNPs
H4: Association with trait 1 and trait 2, one shared SNP
The colocalization framework can group the configurations into five sets: S0, S1, S2, S3, and S4. It can compute the posterior probabilities for each of the five hypotheses according to the following Equation (5):
| (5) |
where P(S) is the posterior probability of a configuration, P(D|S) is the observed posterior probability data D for a given configuration S, and the sum is over all configurations S which are consistent with a given hypothesis , where h = (1,2,3,4). Then it can reformulate the posterior probability as a ratio for each hypothesis. Using posterior probability under hypothesis 4 (PP4) as an example:
| (6) |
where the represents the posterior probability under each hypothesis, where = (0,1,2,3,4). The ratio in the numerator and denominator of the above equation is:
| (7) |
The coloc package can assess the posterior probabilities in hypothesis H4. For each genome‐wide significant and subthreshold GWAS loci, we first used PLINK 14 to select the top lead SNP and then extracted all SNPs within 500 Kb upstream or downstream of the top lead SNP (1 Mb sliding window size). In each QTL dataset, we extracted SNP–gene pairs within that range and then tested posterior probabilities of colocalization between those GWAS SNPs and genes using approximate Bayes factor computations. 37 For the TEs regulated by teQTL, CpGs regulated by meQTL, and peaks regulated by haQTL, we used bedtools 22 to assign the nearest genes according to the genomic region where the TEs, CpGs, and peaks are located. We assumed the two traits might be regulated by a single causal variant when the posterior probability of a colocalized signal (PP4) was > 0.5.
2.10. TE transcriptome‐wide association analysis
We leveraged FUSION 38 to perform transcriptome‐wide association analysis (TWAS) to identify significant TE expression–trait associations for the 64 control brains with available genotype and expression data from human TCX from the Mayo brain biobank (synapse ID: syn11707308). FUSION is an R package that implements the TWAS scheme. For genotype data, we first used PLINK 14 to convert the VCF files to a standard binary PLINK format. For TE expression data, TEs with expression levels < 1 in at least 50% of the samples were filtered for further analysis. We first estimated cis‐SNP heritability for those expressed TEs for SNPs in the 1 Mb flanking regions of the TE. Only TEs that were significant for heritability estimates at a Bonferroni‐corrected P < 0.05 were retained for subsequent analysis. We then constructed the reference panel by computing SNP–expression weights between genotype SNPs and TE expression for 64 samples using several regularized linear models, including best linear unbiased prediction (BLUP), least absolute shrinkage and selection operator, Elastic Net, and an additional Bayesian linear mixed model (BSLMM). The prediction accuracy of each model was measured by 5‐fold cross‐validation by a random sampling of 1000 highly heritable TEs. The computed Z score was used to assess the association strength between implicated TEs and disease. To account for multiple hypotheses, we applied a q < 0.05 for the TE expression reference panel. We further used FUSION's conditional analyses to validate the TWAS findings.
2.11. Genomic annotations of teQTL
We leveraged two approaches to annotate the teQTLs with default settings. We first leveraged genomic locations for each teQTL to different functional genomic elements according to the annotation database from the UCSC Genome Browser. Then, we applied the Genomic Regulatory Elements and GWAS Overlap algoRithm (GREGOR) 39 to estimate global enrichment patterns of teQTLs in epigenomic features using a permutation‐based approach. We downloaded 15 chromatin states from five chromatin marks in eight brain cell types from the Roadmap Epigenomics projects, 40 including brain angular gyrus (E067), brain anterior caudate (E068), brain cingulate gyrus (E069), brain germinal matrix (E070), brain hippocampus middle (E071), brain inferior temporal lobe (E072), brain DLPFC (E073), and brain substantia nigra (E074).
2.12. Brain cell type–specific analysis
We used four brain cell type RNA‐seq datasets to validate our findings. First, we used RNA‐seq data from four sorted brain cell populations, including microglia, astrocytes, endothelial cells, and neurons from frozen cerebral cortical tissues from AD and control brains 41 to identify cell type–specific TE activation in human aging brains. Briefly, FASTQ files were downloaded from the National Center for Biotechnology Information Gene Expression Omnibus (GEO) database under accession number GSE125050. 41 Clinical variables, including PMI, sex, and APOE genotype, were also downloaded from the original publication. 41 Next, differentially expressed TEs at the locus level between different disease status within the same cell types, such as between AD and HC, between heterozygous APOE ε4 and heterozygous APOE ε3, and between females and males, were identified by linear model analysis using the data harmonization approach described above. TEs with Benjamini–Hochberg corrected P < 0.05 after multiple testing were identified as locus‐based differential expressed TEs. The second brain cell type RNA‐seq dataset that was used was from human iPSC‐derived population APOE brain microglia cells, which could be downloaded from the GEO/Sequence Read Archive repository GSE190187. 42 This dataset was used to identify APOE ε4–driven lipid metabolic dysregulation in astrocytes and microglia. Those cell lines were selected from 43 Europeans, controlling for sex and disease status, and confirming APOE ε4/ε4 as the main AD contributor. Then, we used the same RNA harmonization approach mentioned above to identify locus‐based differentially expressed TEs solely contributed by APOE ε4/ε4 by comparing APOE ε4/ε4 to APOE ε3/ε3. The third brain cell type RNA‐seq dataset we used was that of 482,472 nuclei (GSE148822) from human non‐demented control brains and AD brains. 43 For each donor, two brain regions were included: the occipital cortex (OC) with Aβ pathology and the occipitotemporal cortex (OTC) with both Aβ and tau pathology. We further used scTE 44 to profile subfamily‐based differentially expressed TEs between AD brains (n = 10) and HCs (n = 8; Figure S2 in supporting information). The fourth brain cell type RNA‐seq dataset we used was human brain cell type–specific enhancer–promoter interactome maps for microglia, astrocytes, neurons, and oligodendrocytes. 45 Those cell types were subjected to the assay for transposase‐accessible chromatin sequencing (ATAC‐seq), H3K27ac and H3K4me3 chromatin immunoprecipitation sequencing (ChIP‐seq), and proximity ligation‐assisted ChIP‐seq (PLAC‐seq).
2.13. GWAS enrichment analysis
We used GWAS Analysis of Regulatory or Functional Information Enrichment with linkage disequilibrium (LD) correction (GARFIELD) 46 to test for enrichment of the three AD GWAS SNPs among teQTLs. GARFIELD performs greedy pruning of GWAS SNPs based on LD information (r2 > 0.1) and then annotates them based on functional information overlap. It quantifies enrichment using odds ratio (OR) at GWAS P < 1.0 × 10−5 and P < 5.0 × 10−8 significant cut‐offs and assesses significance by using generalized linear model testing, while accounting for MAF, distance to nearest transcription start site, and number of LD proxies (r2 > 0.8). Within this framework, GARFIELD accounts for major sources of confounding that current methods do not offer. In this case, we used the three GWAS summary statistics data as the annotation files, and then assessed the enrichment of teQTL signals in the three features extracted from the three GWAS studies (Table S4). We further used LD score regression analysis (LDSC) 47 to validate GARFIELD findings.
2.14. Functional enrichment analysis
AD‐related molecular signatures were obtained from the Molecular Signatures Database (MSigDB). We searched several terms in MSigDB, including “AD,” “amyloid,” “astrocyte,” “immune,” “microglia,” “mitochondria,” “myelin,” “neurofibrillary_tangle,” “neurogenesis,” “neuroinflammation,” “organic_acid,” “oxidation,” “protein_metab,” “synapses,” and “tau.” In total, we obtained 4904 AD‐related gene sets for enrichment analysis. Functional enrichment analysis of locus‐based differentially expressed TEs was performed using the enricher function in the clusterProfiler 48 package in R (version 4.2.0). Terms with Benjamini–Hochberg corrected P < 0.05 were defined as significantly enriched terms and pathways. For the SNPs located around locus‐based differentially expressed TEs, we leveraged FUMA 49 to perform functional mapping and annotation functional mapping of those SNPs. The SNP2GENE function in FUMA first classifies those SNPs as input and then provides extensive functional annotation for all SNPs in genomic areas identified by the lead SNP. Then, the GENE2FUNC function in FUMA takes a list of gene IDs as identified by the SNP2GENE function and annotates those genes in biological context. Significant enrichment at Bonferroni‐corrected P ≤ 0.05 were selected.
FIGURE 2.
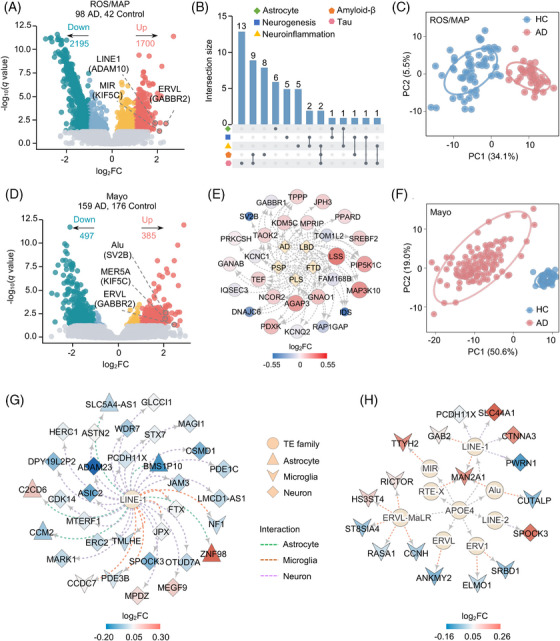
Locus‐based differential expression of TEs across two brain biobanks and AD with varying degree of neuropathology. A, D, Volcano plots of locus‐based differentially expressed TEs between AD subjects and cognitively HC across ROS/MAP (A) and Mayo (D) cohorts. TEs are colored by whether differentially expressed (q < 0.05; gray), differentially expressed but with modest effects (|log2FC| < 1.0; orange and light blue), or with stronger effects ((|log2FC| > 1.0; red and dark blue). Numbers of genes in each category are above the plot. TEs highlighted here represent the identified commonly differentially expressed TEs across the two brain biobanks. Genes in parentheses represent the nearest genes of differentially expressed TEs under consideration. B, Upset plot showing enriched AD‐related molecular signatures among the nearest genes of locus‐based differentially expressed TEs in ROS/MAP brain biobank. Dots with connecting lines indicate signature combinations. C, F, PCA based on the locus‐based differentially expressed TEs of the ROS/MAP (C) and Mayo (F) brain biobanks. Circles represent that AD and cognitive HC subjects could be separated at a confidence interval of 95%. E, Network depicting the nearest gene sets of locus‐based differentially expressed TEs in ROS/MAP brain biobank. Diamonds represent five neurodegenerative traits and circles represent the nearest genes of locus‐based differentially expressed TEs. The color represents log2FC of differentially expressed genes from the RNA‐seq Harmonization Study (syn21241740). G, H, TE–gene regulatory networks depicting cell type–specific differentially expressed TEs across tau and Aβ neuropathology (G) and heterozygous APOE ε4 genotyping (H). Circles in the inner layer represent TE families. Genes on the outer layer surface refer to the nearest genes of the locus‐based TEs. Colors on the outer layer panel refer to log2FC of differentially expressed genes from the RNA‐seq Harmonization Study (syn21241740). Genes differentially expressed in microglia are shown as V, neuron as diamond, and astrocytes as triangle. Aβ, amyloid beta; AD, Alzheimer's disease; APOE, apolipoprotein E; HC, healthy controls; Mayo, Mayo clinic; PCA, principal component analysis; RNA‐seq, RNA sequencing; ROS/MAP, Religious Orders Study/Rush Memory and Aging Project; TEs, transposable elements.
2.15. Functional validation of TEs by CRISPRi
i3N‐WTC11 neurons were differentiated with a two‐step protocol as previously described. 50 dCas9‐KRAB driven by CAG promoters was knocked‐in to a safe harbor locus in the intronic region of CLYBL to enable robust transgene expression through differentiation. sgRNAs targeting locus TEs and promoters of target genes were designed by CHOP‐CHOP with high efficiency. 51 To minimize the chance of off‐target effects, we can identify off‐target sites with 1 bp mismatch (MM1), 2 bp mismatches (MM2), or 3 bp mismatches (MM3). We selected sgRNAs with 0 MM1 and MM2, and < 5 MM3, to confirm the specificity of each sgRNA used in our study. Candidate TEs which have sgRNAs passing the above specificity criteria or have GC‐content between 40% and 70% were selected for CRISPRi validation. Two independent sgRNAs were used for each locus TE and listed in Table S5 in supporting information. sgRNA oligos were inserted into lentiGuide‐puro vector (Addgene 52963) and the plasmids were co‐transfected with lentivirus packaging plasmids, including pMD2.G (Addgene, 12259), and psPAX (Addgene, 12260), into 293T cells by PolyJet (SignaGen Laboratories SL100688). Virus‐containing media was collected for 48 hours, filtered through 0.45 μm filters (Millipore SLHV033RS), and concentrated with Amicon Ultra centrifugal filters (Millipore UFC801024). The virus was titrated in iPSCs by antibiotic selection. For CRISPRi experiments, iPSCs were first differentiated into 2‐week excitatory neurons. The neurons were then treated with lentivirus expressing sgRNAs (multiplicity of infection ≈ 3) and subjected to puromycin selection (0.5 ug/mL) for 4 days. Cells were collected for mRNA extraction 7 days post‐transfection. Gene expression was determined using both RNA‐seq and real‐time polymerase chain reaction (RT‐qPCR). DNase treated samples were sent to the genomics core of the Lerner Research Institute, Cleveland Clinic for RNA‐seq. Pair‐end 75 bp RNA‐seq libraries were constructed following Illumina's protocols. RNA‐seq analysis was conducted with two biologically independent replicates. Genes with Benjamini–Hochberg corrected P value < 0.1 were identified as differentially expressed. RT‐PCR was performed according to previously published protocol. 52 The relative expression values from two biological replicates were compared using Student t test.
2.16. Statistical analysis
Principal component analysis (PCA) was conducted using the ggbiplot package in R (version 4.2.0) based on the locus‐based differentially expressed TEs of the two RNA‐seq brain biobanks. We used the bedtools 22 intersect function to identify the nearest genes for each locus TE. Spearman correlation analysis between locus TE and its nearest gene was performed using the cor.test function in R. All plots created in this study were generated using the ggplot2 53 package in R.
3. RESULTS
3.1. TE expression atlas in human AD brains
We first quantified TE expression at the family level across the three brain biobanks using SQuIRE (adjusted P‐value [q] < 0.1, see Methods). We then performed differential expression analysis of subfamily TEs between AD subjects and cognitive HCs in each brain biobank, controlling for harmonized covariable factors from the AMP‐AD RNA‐seq Harmonization Study (syn9702085, see Methods).
Using SQuIRE (see Methods), we found 8, 2, and 9 upregulated TE subfamilies in the MSBB, Mayo clinic (Mayo), and the ROS/MAP cohorts, respectively (Figure S3A and Table S6 in supporting information). These differentially expressed subfamily TEs were further detected using TE transcripts (Figure S3B,C and Table S6). 17 As transcription factors (TFs) mediate sequence‐specific recognition and activation of TEs, 54 we investigated TF–TE regulatory relationships using enrichment analysis of known binding motifs for the top two upregulated subfamily TEs (Figure S3D). For example, we pinpointed that upregulated FOXO4 (log2FC = 0.31, q = 1.16 × 10−8) was significantly enriched within the upregulated ERV1 elements (log2FC = 0.46, q = 4.46 × 10−5) in AD brains from the ROS/MAP cohort (Figure S3D), suggesting potential regulatory roles between activated subfamily TE and FOXO4 expression in AD.
We next investigated locus‐based TE expression using SQuIRE. 15 After adjusting the harmonized covariable factors (see Methods), we identified locus‐based differentially expressed TEs between AD and HC brains for each brain biobank. In total, 10, 882, and 3895 TEs showed significant transcriptomic changes across MSBB, Mayo, and ROS/MAP brain biobanks, respectively (|log2FC| ≥ 1.0, q < 0.05, Figure 2A,D, and Table S7 in supporting information). The limited number of locus‐based differentially expressed TEs in the MSBB cohort could reflect low coverage and short single‐end sequencing reads (n = 100 bp). 55 We further analyzed differentially expressed TEs from the Mayo and ROS/MAP cohorts. Among the dysregulated TEs in both Mayo and ROS/MAP brain biobanks, we found that the class with the most significantly elevated TE expression was LINE (Figure S4A in supporting information), with LINE‐1 found to be the predominant type of elevated LINE in the two AD brain biobanks (Figure S4B). We further used Telescope 19 to evaluate robustness of locus‐based differentially expressed TEs identified by SQuIRE. 15 We found that the replication rate of Telescope ranged from 49.9% to 72.9% in the ROS/MAP biobank (Figure S5A and Table S7 in supporting information) and from 62.6% to 73.7% in the Mayo brain biobank (Figure S5B and Table S7). Cross‐biobank analysis of locus‐based differentially expressed TEs subsequently revealed a significant overlap across different biological comparison groups, including AD versus HC (Fisher exact test P < 2.2 × 10−16), heterozygous APOE ε4 AD versus HC (P = 1.9 × 10−6), female AD versus female HC (P < 2.2 × 10−16), and male AD versus male HC (P < 2.2 × 10−16; Figure S6A–D in supporting information).
We then examined genome regulatory sequences of TEs by mapping dysregulated TEs with human brain–specific regulatory sequences, including open chromatin regions, 56 TF binding sites, 57 and ROS/MAP enhancer and promoter sequences. 31 Here, we found that differentially expressed TEs were more enriched in open chromatin regions and promoter sequences in both Mayo and ROS/MAP brain biobanks (P = 0.0095, Figure S7 in supporting information), suggesting crucial gene regulatory roles of TEs. Next, we performed Spearman correlation tests of expression levels between dysregulated TEs and their nearest genes. The Spearman correlation coefficients (R) were 0.32 (P < 2.2 × 10−16) and 0.37 (P < 2.2 × 10−16) in the Mayo and ROS/MAP brain biobanks, respectively, indicating the expression of TEs is independent from the expression of nearby local genes. We then investigated pathway enrichment of nearest genes from the significantly dysregulated TEs. Of note, the ROS/MAP cohort was significantly enriched by AD‐related gene signatures, such as genes associated with tau (q = 4.36 × 10−10), Aβ (q = 0.016), and neuroinflammation (q = 0.028; Figure 2B). For the Mayo cohort, we identified significantly enriched gene signatures related to neurodegenerative disorders (Figure 2E), such as upregulated gene AGAP3 (log2FC = 0.24, q = 1.07 × 10−4) and MAP3K10 (log2FC = 0.26, q = 1.55 × 10−4). These significantly enriched AD‐related pathways revealed that TE dysregulation might be associated with potential AD pathobiological pathways. We then performed PCA for AD cases and cognitively HCs based on expression of locus‐based differentially expressed TEs and found that TE expression accurately predicted AD cases and cognitively HCs in both Mayo and ROS/MAP brain biobanks (Figure 2C,F). This indicates that monitoring differentially expressed TEs might aid clinical diagnosis of AD.
3.2. Cell type–specific TE dysfunction across degrees of AD neuropathology
We further analyzed RNA‐seq data from fluorescence‐activated cell sorting (FACS) cell types from frozen brains to investigate cell type–specific locus‐based TE dysregulation across different AD pathologies, including tau and Aβ neuropathology, APOE genotypes, and sex. In total, we pinpointed significantly upregulated TEs in AD patients’ frontal cortex compared to HCs across neurons (n = 3748), microglia (n = 6814), astrocytes (n = 6133), and endothelial cells (n = 1196; Table S8 in supporting information). Using gene set enrichment analysis (GSEA), we found that nearest genes of cell type–specific upregulated TEs were enriched in AD‐related pathways in cell type–specific manners: (1) nearest genes of upregulated TEs from astrocytes were enriched in pathways related to neurofibrillary tangles (q = 2.42 × 10−4), (2) nearest genes of upregulated TEs from microglia were enriched in amyloid fiber formation (q = 4.26 × 10−4) and immune system process (q = 1.51 × 10−5), and (3) nearest genes of upregulated TEs from neurons (q = 1.03 × 10−4) and endothelial cells (q = 0.018) were enriched in neuroinflammation and glutamatergic signaling (Table S9 in supporting information). We then tested whether upregulated TEs identified from the two brain biobanks exhibited cell type–specific patterns. As exemplified by the upregulated TEs from the ROS/MAP cohort, we observed 30, 14, and 10 elevated locus TEs in AD patients’ frontal cortex with a high score of tau‐ and Aβ neuropathology across neurons, microglia, and astrocytes, respectively (Table S8). Approximately 73.2% of those cell type–specific upregulated TEs were derived from the LINE‐1 family (Figure 2G). We also found a tau‐ and Aβ‐specific overexpressed TE from a LINE‐1 element (chr2: 206494817–206499652) in neurons; the nearest gene is ADAM23 (log2FC = −0.23, q = 0.0023), which promotes neuronal differentiation of human neural progenitor cells. 58 We additionally pinpointed a microglial‐specific upregulated TE from a LINE‐2 element (chr14: 75441408–75441632) that was affected by both tau and Aβ neuropathology (Table S8); the nearest gene here is JDP2, which regulates oxidative stress in human brains. 59
We next inspected APOE ε4 genotype–specific TE dysregulation. After adjusting for various confounding factors, we identified 706 and 1326 upregulated TEs in AD individuals with heterozygous APOE ε4 (log2FC ≥ 0.5, q < 0.05) compared to heterozygous APOE ε3 brains in the Mayo and ROS/MAP brain biobanks, respectively (Table S7). For the ROS/MAP cohort, we found that the closest genes to upregulated TEs were significantly enriched in gene signatures upregulated in early‐stage AD (q = 0.034). We further evaluated cell type–specific TE dysregulation of heterozygous APOE ε4 by leveraging the same brain cell‐type RNA‐seq datasets (see Methods, Table S8). This identified 16, 6, and 2 upregulated TEs in microglia, neuronal cells, and astrocytes, respectively, for AD individuals with heterozygous APOE ε4 (Table S8). We also constructed a TE–gene regulatory network by linking cell type–specific differentially expressed TEs to the nearest gene in the ROS/MAP cohort (Figure 2H). This pinpointed a SINE element (chr5: 109799806‐109800106) that was significantly overexpressed in microglia from heterozygous APOE ε4 AD patients’ frontal cortex, with MAN2A1 the nearest gene that correlates with AD progression. 60
It is well known that women are disproportionately affected by AD in terms of both disease prevalence and severity. 61 Thus, we further inspected TE dysregulation between females and males after adjusting for confounding factors. Compared to male subjects, we found 817 locus‐based differentially expressed TEs in female AD brains compared to male AD brains from the ROS/MAP brain biobank (log2FC ≥ 0.5, q < 0.05, Table S7). The nearest genes for female‐biased TEs were significantly enriched in gene signatures related to tau pathology (q = 0.012) and protein–protein interactions (PPI) at synapses (q = 0.001). The most female‐biased TE was from a SINE element (chr16: 24875912–24876225; log2FC = 2.55, q = 0.023), with SLC5A11 the nearest gene. We next examined female‐specific upregulated TEs using human brain cell type–specific RNA‐seq data (Table S8). Specifically, we identified 82, 6, and 3 female upregulated TEs from the ROS/MAP brain biobank for endothelial cells, microglia, and astrocytes, respectively (Table S8 and Figure S8A in supporting information). Of note, a LINE‐1 element (chr6: 35681337–35681679) was specifically overexpressed in female neurons and its nearest gene is FKBP5, which has been implicated in sex‐specific cognitive and emotional behavior. 62 We also highlighted 45 male‐biased upregulated TEs from the ROS/MAP brain biobank for endothelial cells (Table S8 and Figure S8B) and found that a SINE element (chr17: 44909153–44909360) was significantly upregulated in male endothelial cells (log2FC = 1.12, q = 1.04 × 10−5). The nearest gene GFAP was also upregulated in male AD brains in the ROS/MAP biobank (log2FC = 0.54, q = 0.0087). Interestingly, sex differences in GFAP expression have been studied in the brainstem of rats with neuropathic pain. 63 Taken together, our observations reveal distinct TE regulation underlying differences in AD pathobiology between male and female brains.
FIGURE 3.
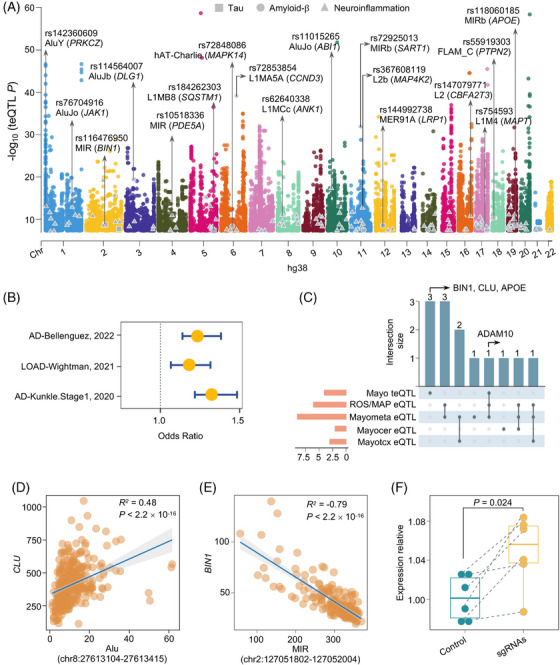
teQTL analysis in human brains and their enrichment with genetic loci of AD. A, Manhattan plot showing the ‐log10(P value) distribution of teQTLs identified in the Mayo brain biobank, of which 152 samples have both RNA‐seq and the matched WGS data. Different color denotes 22 human chromosomes. Genes (in the parentheses) highlighted represent AD‐related genes, including tau (square color coded in gray), A (circle color coded in gray), and neuroinflammation (triangle color coded in gray). teQTLs regulating known AD‐related genes are plotted in the first row. Subfamily TEs associated with those teQTLs are highlighted in the second row. All the TEs shown in this study are annotated with assembly GRCh38. B, teQTLs are enriched in AD‐associated variants across three AD GWAS studies. C, Upset plot showing AD risk genes identified across five types of xQTLs, including teQTL from the Mayo cohort, eQTLs from the Mayo cerebrum, eQTLs from the Mayotcx, eQTLs from the Mayo meta‐analysis, and eQTLs from the ROS/MAP DLPFC. The three genes highlighted are uniquely identified by teQTL findings. D, Expression correlation analysis between a SINE element (chr8: 27613104‐27613415) and its nearest gene CLU uniquely regulated by brain teQTL. E, Expression correlation analysis between a SINE element (chr2: 127051802‐127052004) and its nearest gene BIN1 uniquely regulated by brain teQTL. The x axis represents expression level of the TE and the y axis represents the expression level of its nearest gene. P values were calculated using the Spearman correlation test. R2 values showing the strength of expression correlation by Spearman correlation test. F, Box plot shows relative expression of BIN1 between control (green) and a SINE element (chr2: 127051802‐127052004) sgRNA (yellow) targeted iPSC‐derived neurons. Aβ, amyloid beta; AD, Alzheimer's disease; DLPFC, dorsolateral prefrontal cortex; eQTL, expression QTL; iPSC, induced pluripotent stem cell; Mayotcx, Mayo temporal cortex; RNA‐seq, RNA sequencing; ROS/MAP, Religious Orders Study/Rush Memory and Aging Project; SINE, short interspersed nuclear element; TE, transposable element; teQTL, transposable element quantitative trait loci; WGS, whole‐genome sequencing.
3.3. teQTLs in human aging brains
We next inspected associations between TE expression and common genetic variants (MAF > 1%) using matched RNA‐seq and WGS data for 152 human TCX brains in the Mayo cohort. We selected TEs with > 1 FPKM in at least 50% of brains and showing transcriptomic changes in the opposite direction with its nearest gene (see Methods). In total, we obtained 12,481 TE loci for our teQTL mapping analysis. We defined teQTL signals as variants located within 1 Mb upstream or downstream of the expressed TEs 28 and found that 1452 brain‐expressed TEs were significantly associated with 26,188 SNPs (teQTL P < 5 × 10−8; Figure 3A). Specifically, 36.11% of teQTLs were mapped to intergenic regions and 45.72% were mapped to intronic regions (Figure S9A in supporting information). We further found that the genes regulated by human brain teQTLs were significantly enriched in metabolic pathways of Aβ (Fisher exact test P = 0.036), tau (P = 0.019), and neuroinflammation (P = 2.1 × 10−4). For instance, teQTL rs754593 (P = 2.0 × 10−10) regulates expression of a LINE‐1 element (chr17: 46238321‐46238612) in the Mayo brain biobank, with MAPT the nearest gene. We found that LRP1 was also regulated by brain teQTL rs144992738 (P = 1.91 × 10−8). LRP1 is a major neuronal receptor that mediates brain homeostasis and controls Aβ metabolism. 64 We also found significant enrichment in 119 neuroinflammation‐related genes regulated by brain teQTLs (Fisher exact test P = 2.1 × 10−4). The most significant gene was SART1 (teQTL rs72925013, P = 1.22 × 10−32), for which upregulation leads to developmental defects in zebrafish central nervous system. 65 We next mapped teQTLs to the 15 chromatin states from eight human brain cell lines from the Roadmap Epigenomic Consortium project 40 and found teQTLs primarily enriched in the 5' and 3' transcribed regions, as well as in flanking bivalent transcription start sites/enhancers, indicating genome regulatory roles encoded by those human brain teQTLs (Figure S9B).
We next used both SNP‐based and gene‐based approaches to test common or unique regulatory roles between teQTL and eQTL. We first examined the enrichment between teQTLs with well‐documented human brain eQTLs, including: (1) eQTLs from Mayo cerebellum (Mayocer), Mayo temporal cortex (Mayotcx), and Mayo meta‐analysis (Mayometa); and (2) eQTLs from DLPFC from the ROS/MAP cohort (Table S3). This showed that only 47.3% (39.1%–58.4%) of teQTLs were identified as an eQTL across two eQTL datasets (Figure S9C,D), revealing the unique gene regulatory roles of brain teQTLs. We further assessed the replication rate of brain teQTLs with eQTLs from the two brain biobanks using the statistic approach, which estimates the proportion of eQTLs that are also significant in brain teQTLs. Using the overlap between teQTL and eQTL as an example, this analysis could perform FDR estimation with teQTL P values from the tested TE expression‐SNP associations (test phenotype) that overlapped with meQTL SNPs (discovery phenotype). We estimated that between teQTLs and eQTLs was 0.434 from the Mayo cerebellum, 0.416 from the Mayotcx, 0.485 from the Mayo metadata, and 0.445 from ROS/MAP. We then evaluated a gene‐based comparison between teQTLs and eQTLs in the Mayo cohort and identified 703 nearby genes at 26,188 unique genome‐wide significant teQTLs (P < 5 × 10−8). Among the 370 (52.6%) overlapping genes regulated by both teQTL and eQTL from Mayo meta‐analysis, 256 (69.2%) genes possessed teQTL signals distinct from the eQTL signals, as evidenced by the low LD value of the two SNPs (r2 < 0.1; Figure S10A in supporting information). Altogether, these observations reveal that teQTLs regulate distinct genes from traditional eQTLs, suggesting crucial roles of teQTLs in genome regulation.
TE expression can also be regulated by epigenetic mechanisms in brain health disorders. 66 Thus, we further investigated the co‐occurrence of teQTLs with variants affecting epigenetic marks, such as DNA methylation (meQTL) and histone H3 acetylation on lysine 9 (H3K9ac, haQTL) from the ROS/MAP cohort DLPFC, a region of the brain related to higher functioning. For the 26,188 genome‐wide significant teQTLs, 43.0% were meQTLs and 32.1% were haQTLs. Via statistical analysis, we found overlap between teQTLs and meQTLs ( = 0.440) and haQTLs (= 0.425, Figure S10B). Thus, epigenomic regulation may play a crucial role in regulating TE expression in AD, consistent with previous studies in other human tissues. 67
3.4. teQTLs tune likely causal genes in AD brains
We next investigated whether SNPs associated with teQTLs influence AD susceptibilities. Using three AD GWAS summary statistics data (Table S4, see Methods), we found significant enrichment of brain teQTLs across three AD GWAS datasets (Figures 3B and S10C). Functional enrichment analysis using the clusterProfiler 48 package (see Methods) revealed that genes regulated by human brain teQTLs were significantly enriched in multiple AD‐related pathways, such as pathways involved in microglia pathology (q = 1.39 × 10−15), delayed and abnormal myelination (q = 1.7 × 10−4), fatty acid beta oxidation (q = 0.043), and tau protein binding (q = 0.013; Figure S11 in supporting information). These findings suggest the potential roles of teQTLs in deciphering functional consequences of non‐coding GWAS loci in AD.
As eQTLs directly explain variation in mRNA expression, we examined whether teQTLs and eQTLs (Mayo cerebrum, Mayotcx, Mayo meta‐analysis, and ROS/MAP DLPFC) regulate consensus AD risk genes derived from GWAS studies. We thus focused our subsequent analysis on xQTLs with a genome‐wide significance threshold of P < 5.0 × 10−8. Of the 29 well‐defined AD risk genes identified in a previous study (Table S10 in supporting information), 68 ADAM10 was commonly regulated by three SNPs located in low LD regions (r2 < 0.1), including a teQTL (rs149055375) and two eQTLs (rs12437552 and rs12592302; Figure 3C). ADAM10 is involved in the generation of Aβ and tau pathology in AD. 69 In addition, three AD‐risk genes (BIN1, CLU, and APOE) were uniquely identified by teQTL analysis (Figure 3C). We found significant expression correlation between a SINE element (chr8: 27613104–27613415; teQTL rs143378944, P = 2.85 × 10−8) and its nearest gene CLU (R = 0.48, P < 2.2 × 10−16; Figure 3D). We found that expression of a SINE element (chr2: 127051802–127052004) exhibited significant negative correlation with BIN1 mRNA abundance (R = −0.79, Spearman correlation P < 2.2 × 10−16; Figure 3E). The corresponding teQTL was rs116476950 (P = 6.31 × 10−10). We also found an intronic SINE element (chr2: 127051802–127052004) harboring hallmarks of enhancer elements (including open chromatin status [from ATAC‐seq data], enrichment of H3K27ac, and chromatin looping) and promoters of BIN1 (Figure S12 in supporting information), suggesting functional roles of the SINE element in BIN1 expression.
In support of a likely regulatory relationship between the SINE element (chr2: 127051802–127052004) and BIN1 expression, we performed CRISPRi experimental assays in human iPSC‐derived neurons (see Methods). Here, we designed two single guide RNAs (sgRNAs) targeting the SINE element (chr2: 127051802–127052004) and BIN1 promoters (Table S5). The iPSC‐derived neurons were transduced with lentivirus expressing the two sgRNAs or non‐targeting sgRNA controls. After selecting the cells using puromycin, we used RT‐qPCR to quantify BIN1 gene expression. We found that BIN1 was significantly upregulated in human iPSC‐derived neurons upon CRISPRi in both sgRNA (P = 0.024, Figure 3F). Altogether, these observations revealed that teQTLs provide a complementary method to identify AD‐associated genes from non‐coding loci, in addition to traditional QTL approaches.
3.5. AD GWAS loci colocalize with human brain teQTLs
To identify AD‐associated variants and genes associated with TE dysregulation, we performed genome‐wide colocalization analysis through leveraging three AD GWAS summary statistics datasets (Table S4, see Methods) with genome‐wide significant teQTL findings (Figure 3A). We used three AD GWAS datasets: (1) a small but unique AD GWAS cohort with clinically diagnosed AD cases, 34 (2) the second AD GWAS cohort with late‐onset AD cases, 35 and (3) an AD GWAS cohort with the largest number of AD cases. 36 In total, we identified four genome‐wide significant AD GWAS loci with statistically significant colocalization by human brain teQTLs (PP4 > 0.5, see Methods, Table S11 in supporting information), including rs2906657 (P = 7.99 × 10−16), rs11605348 (P = 1.92 × 10−11), rs1065712 (P = 5.46 × 10−9), and rs55910656 (P = 1.01 × 10−8; Figure 4A). The nearest genes regulated by four genome‐wide significant teQTLs were ZCWPW1, C1QTNF4, FDFT1, and RABEP1.
FIGURE 4.
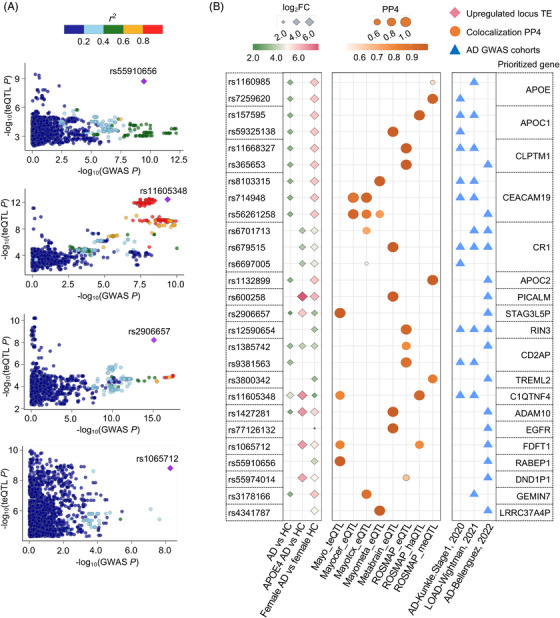
Colocalization analysis between teQTL and AD risk loci. A, LocusCompare plots for four high‐probability genome‐wide colocalized signals between teQTLs and AD GWAS loci. The colocalized SNPs are labeled with variant identifiers and annotated as squares. Plots are colored based on LD bins relative to the lead SNPs (red, 0.8; orange, 0.6–0.8; green, 0.4–0.6; light blue, 0.2–0.4; and dark blue, < 0.2). The SNP pairwise LD data were calculated based on the 1000 Genomes Phase 3 (ALL) reference panel. B, Colocalization analysis around 500 Kb flanking regions of locus‐based differentially expressed TEs between AD GWAS SNPs and xQTLs (annotated by y axis), including teQTL from Mayo; meQTL from ROS/MAP, haQTL from ROS/MAP; and eQTL from ROS/MAP, Mayo, and MetaBrain datasets. We showed SNPs with genome‐wide significant AD GWAS P < 5.0 × 10−8. Loci are named for their nearest protein‐coding genes or the regulated eGene. All genes and loci shown here with PP4 0.5 in at least one xQTL dataset. Circles size and color refer to PP4 value. Diamonds refer to locus‐based differentially expressed TEs from different biological comparison groups. Diamonds size and color refer to log2FC of locus‐based differentially expressed TEs under consideration. Triangles refer to the three AD GWAS datasets. AD, Alzheimer's disease; eQTL, expression QTLs; haQTL, histone acetylation QTLs; GWAS, genome‐wide association study; LD, linkage disequilibrium; meQTL, methylation QTLs; PP4, posterior probability of colocalization hypothesis 4; SNP, single nucleotide polymorphism; teQTL, transposable element quantitative trait loci; TEs, transposable elements.
We next extended colocalization analysis around flanking regions of locus‐based differentially expressed TEs. Here, we leveraged AD GWAS loci located in 500 Kb flanking regions of locus‐based differentially expressed TEs harboring brain teQTLs from Mayo; brain meQTLs from ROS/MAP; brain haQTLs from the ROS/MAP; and brain eQTLs from the ROS/MAP, Mayo, and MetaBrain datasets (Table S3). Using FUMA, 49 we identified that AD GWAS loci close to differentially expressed TEs were significantly enriched in multiple AD‐associated pathways (Figure S13 in supporting information), including cerebrospinal fluid phosphorylated tau levels (q = 3.1 × 10−17), hippocampal volume in AD dementia (q = 2.8 × 10−13), low‐density lipoprotein cholesterol levels (q = 7.4 × 10−5), and lipid metabolism phenotypes (q = 0.009). We further pinpointed 27 genome‐wide significant loci (GWAS P < 5.0 × 10−8) harboring significant xQTL colocalization with several well‐known AD risk genes around 500 Kb flanking regions of locus‐based differentially expressed TEs in the ROS/MAP cohort, including APOE, APOC1, CR1, PICALM, and ADAM10 (Figure 4B and Table S12 in supporting information). In total, 18 out of the 27 (66.7%) GWAS SNPs were replicated in the Mayo brain biobank (Figure S14 in supporting information). We found that two GWAS SNPs, including rs1065712 (P = 5.46 × 10−9) and rs11605348 (GWAS P = 1.92 × 10−11), were also indicative of colocalization with teQTLs and haQTLs. The locus TE regulated by rs1065712 (AD GWAS) was from a SINE element (chr8: 11840915–11841089) with its nearest gene being FDFT1, which is involved in cholesterol synthesis in cerebellar granule cells and pre‐cerebellar nuclei of mouse models. 70 We also pinpointed two locus‐based TEs regulated by teQTL rs11605348, including a SINE element (chr11: 47608036–47608220) and a LINE element (chr11: 47605296–47605575). The nearby genes are C1QTNF4 and NDUFS3, respectively. The anti‐inflammatory role of C1QTNF4 has been reported in inflammatory mouse models 71 and NDUFS3 is a central modulator of mitochondrial metabolism in aging brains. 72 In summary, teQTL colocalization analysis of AD genetic loci enabled prioritization of new AD‐associated genes that warrant further functional testing. ,
FIGURE 5.
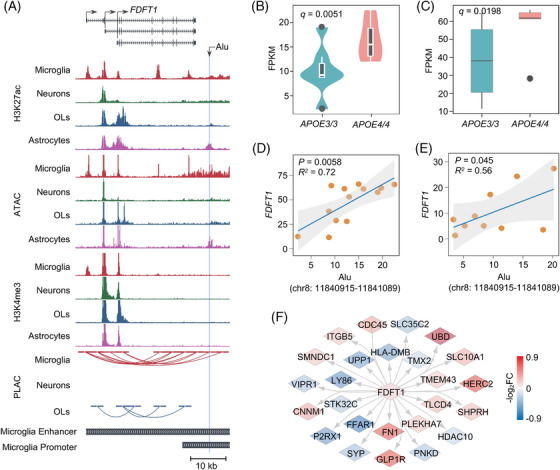
APOE ε4‐associated microglia‐specific TE activation and chromatin looping in AD. A, UCSC Genome Browser visualization of chromatin interactions between Alu family TE‐located microglia‐specific enhancer and promoter of FDFT1. B, C, Expression comparison between a SINE element (chr8: 11840915–11841089) (B) and FDFT1 (C) in APOE ε4/ε4 driven human microglia compared to APOE ε3/ε3. D, E, Expression correlation between the SINE element (chr8: 11840915–11841089) and its nearest gene FDFT1 in APOE ε4/ε4 microglia from human iPSC‐derived population APOE brain cell models (D) and in APOE ε4/ε4 and APOE ε3/ε4 SFG microglia from AD patients (E). P values are calculated using the Spearman correlation test. R2 values showing the strength of expression correlation by Spearman correlation test. F, Molecular networks containing 26 genes interacted with FDFT1. Edge color is proportional to corresponding log2FC between AD and HC in Mayo brain biobank. AD, Alzheimer's disease; APOE, apolipoprotein E; HC, healthy controls; iPSC, induced pluripotent stem cell; OLs, oligodendrocytes; SFG, superior frontal gyrus; SINE, short interspersed nuclear element; TE, transposable element; UCSC, University of California Santa Cruz.
FIGURE 6.
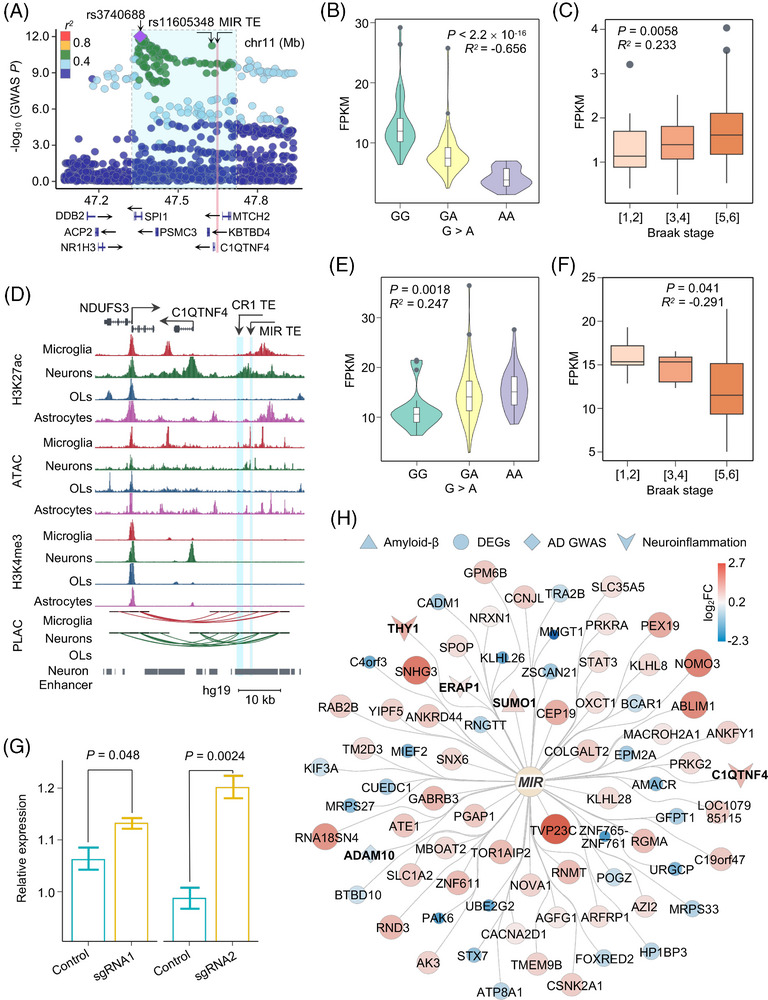
CRISPR interference reveals neuron‐specific suppressive role of upregulated MIR family TE on anti‐inflammatory response. A, Regional SNP association plots with TE from a SINE element (chr11: 47608036‐47608220) shown in LD blocks of rs3740688 in AD. Plots are colored based on LD bins relative to the lead SNPs rs3740688 (red, 0.8; orange, 0.6–0.8; green, 0.4–0.6; light blue, 0.2–0.4; and dark blue, < 0.2). The SNP pairwise LD data were calculated based on 1000 Genomes Phase 3 (ALL) reference panel. Gene annotations: the National Center for Biotechnology Information RefSeq Select database. Assembly GRCh37, scale in Mb. B, E, Expression level of the TE from a SINE element (chr11: 47608036‐47608220) (B) and C1QTNF4 (E) showed significant correlation with the presence of risk allele rs11605348. D, UCSC Genome Browser visualization of chromatin interactions between the SINE and LINE element TE‐located neuron‐specific enhancer and promoter regions of C1QTNF4 and NDUFS3, respectively. C, F, Expression of the TE from a SINE element (chr11: 47608036‐47608220) (C) and C1QTNF4 (F) showed significant correlation with the clinical Braak staging score. P values are calculated using the Spearman correlation test. R values showing the strength of expression correlation by Spearman correlation test. G, Box plot shows relative expression of C1QTNF4 between control (green) or SINE element (chr11: 47608036‐47608220) sgRNA (yellow) targeted iPSC‐derived neurons. H, Network depicting the significantly dysregulated genes from RNA‐seq analysis upon a SINE element (chr11: 47608036‐47608220) perturbation in iPSC‐derived neurons. Circles in the inner layer represent TE families. Genes on the outer layer surface refer to the dysregulated genes. Diamonds represent AD GWAS genes, triangles represent genes associated with Aβ, V represents neuroinflammation associated genes, and circles represent other differentially expressed genes. Colors and size on the outer layer panel refer to log2FC of differentially expressed genes from the RNA‐seq analysis. Genes related with Aβ, neuroinflammation, and GWAS traits are shown in bold fonts. Aβ, amyloid beta; AD, Alzheimer's disease; GWAS, genome‐wide association study; iPSC, induced pluripotent stem cell; LD, linkage disequilibrium; LINE, long interspersed nuclear element; OLs, oligodendrocytes; RNA‐seq, RNA‐sequencing; SINE, short interspersed nuclear element; SNP, single nucleotide polymorphism; TE, transposable element; UCSC, University of California Santa Cruz.
3.6. APOE ε4‐associated TE activation in microglia
Repeat polymorphisms in LD blocks with trait‐associated SNPs have genome regulatory roles. 73 We found that the TE from a SINE element (chr8: 11840915–11841089) was within the LD block region of the lead GWAS SNP rs1065712 (r 2 > 0.30; Figure S15 in supporting information), and then identified elevated expression of both the TE from the SINE element (chr8: 11840915–11841089) and its nearest gene FDFT1 in APOE ε4 AD brains from Mayo and ROS/MAP brain biobanks (Figure S16A,B in supporting information). Furthermore, this SINE element (chr8: 11840915–11841089; R = 0.288, P = 4.41 × 10−4) expression and FDFT1 (R = 0.256, P = 7.90 × 10−4) upregulation were positively correlated with the presence of the minor (risk) allele for rs1065712 (GWAS P = 5.46 × 10−9; Figure S16C,D) and with Braak staging score (Figure S16E,F). TE expression is regulated by complex patterns of epigenetic marks, 74 and we found that this SINE element (chr8: 11840915–11841089) resided within a microglia‐specific enhancer region defined by enrichment of H3K27ac and ATAC‐seq peaks from interactome maps in human microglia and publicly available datasets (Figure S17 in supporting information). The FDFT1 microglia‐specific enhancer frequently participated in long‐range chromatin interactions with multiple FDFT1 promoters, suggesting the regulatory roles of the SINE element (chr8: 11840915–11841089; Figure 5A).
FDFT1, a membrane‐associated enzyme involved in cholesterol biosynthesis, 75 is altered in microglia by APOE ε4 genotypes. 42 Notably, we observed elevated expression of FDFT1 in AD patients’ brains with heterozygous APOE ε4 compared to heterozygous APOE ε3 (log2FC = 0.25, q = 0.032) from the Mayo brain biobank (Figure S16G). Using RNA‐seq data from human iPSC‐derived APOE ε4/ε4 microglia cell models, we found that the SINE element (chr8: 11840915–11841089, log2FC = 0.60, q = 0.0051, Figure 5B) and FDFT1 (log2FC = 0.44, q = 0.0198, Figure 5C) expression were elevated in APOE ε4/ε4 versus APOE ε3/ε3 microglia. Moreover, the SINE element (chr8: 11840915–11841089) was significantly co‐expressed with its nearest gene FDFT1 (R = 0.72, P = 0.0058, Figure 5D). This co‐expression was also observed in a second cell type–specific RNA‐seq profile of superior frontal gyrus (SFG) microglia from AD patient brains 41 with heterozygous APOE ε4 genotypes (R = 0.56, P = 0.045, Figure 5E). We next reconstructed human PPI networks for FDFT1 using our GPSnet algorithm 76 (Figure 5F and Methods) and found that proteins interacted with FDFT1 involved in cholesterol‐related pathways (Figure 5F). Notably, reduced expression of SYP (log2FC = −0.30, q = 3.95 × 10−4) is associated with progressive cognitive decline and neurodegenerative illnesses. 77 Our results suggest that the activated SINE element (chr8: 11840915–11841089) is associated with dysregulation of FDFT1 involved in cholesterol‐related pathways in human aging AD brains.
3.7. CRISPRi assay identifies neuron‐specific suppressive role of activated MIR family TE via anti‐inflammatory responses
We pinpointed that a GWAS SNP rs11605348 (GWAS P = 1.92 × 10−11) showed a strong posterior probability for colocalization with both teQTL (PP4 = 0.84) and haQTL (PP4 = 0.97; Figure 4B). The two TEs regulated by rs11605348, including a SINE element (chr11: 47608036–47608220; log2FC = 2.22, q = 7.27 × 10−5) and a LINE element (chr11: 47605296–47605575; log2FC = 1.07, q = 0.048), showed consistent upregulation identified by SQuIRE 15 and Telescope 19 in human AD brains from the ROS/MAP biobank (Table S7). The SINE element (chr11: 47608036–47608220) also showed female‐specific upregulation in AD brains (log2FC = 5.1, q = 1.71 × 10−5). We found that the SINE element was within the lead AD GWAS SNP rs3740688 LD block region (r 2 > 0.30; Figure 6A). The SINE element expression was negatively correlated with the presence of the minor (risk) allele for rs11605348 (Spearman correlation R = −0.656, P < 2.2 × 10−16, Figure 6B) and positively correlated with the Braak staging score (R = 0.233, P = 0.0058, Figure 6C) in the ROS/MAP brain biobanks, supporting potential roles of teQTL rs11605348 with AD‐risk SNPs.
Using enhancer–promoter interactome maps across four major brain cell types, including neurons, microglia, oligodendrocytes, and astrocytes, we found that the SINE element (chr11: 47608036–47608220) may serve as a cis‐regulatory element by enrichment of open chromatin and H3K27ac signals (Figure 6D). We further confirmed this cis‐regulatory relationship using multi‐omics data from the UCSC Genome Brower database (Figure S17), suggesting that this SINE element may act as an alternative enhancer. 78 We detected frequent chromatin interactions from this SINE element to the promoter for C1QTNF4 in neurons, resulting in enrichment of H3K27ac and H3K4me3 signals in the promoter region of C1QTNF4 in neurons (Figure 6D). The prioritization of C1QTNF4 as an AD‐associated gene was also confirmed through TE transcriptome‐wide association analysis using the FUSION 38 package in the Mayo brain biobank (Figure S18 in supporting information). Functional significance of this chromatin looping was confirmed using human brain transcriptomic changes from the AMP‐AD RNA‐seq harmonization study (syn21241740), in which significant downregulation of C1QTNF4 in female AD brains was found in both Mayo (log2FC = −0.51, q = 8.97 × 10−5) and ROS/MAP (log2FC = −0.17, q = 0.041) brain biobanks. We further observed significant correlation between C1QTNF4 expression with the presence of the minor (risk) allele for SNP rs11605348 (Spearman correlation R = 0.247, P = 0.0018, Figure 6E) and with the Braak staging score (R = −0.291, P = 0.041, Figure 6F) in AD patient brains.
As we also detected frequent chromatin interactions between a LINE element (chr11: 47605296–47605575) and promoters of NDUFS3 (Figures 6D and S17), we next tested the regulatory roles of the SINE element and a LINE element via CRISPRi assays. We found significant downregulation of NDUFS3 at the LINE‐targeted element (Figure S19 in supporting information) and significant upregulation of C1QTNF4 at the SINE‐targeted element (Figure 6G). We further performed RNA‐seq to test transcriptomic changes of SINE‐targeted iPSC‐derived neurons. We identified 78 differentially expressed genes upon CRISPRi (Figure 6H and Table S13). In addition to Aβ‐associated SUMO1 (log2FC = 0.77, q = 0.035) and AD GWAS gene ADAM10 (log2FC = −0.37, q = 0.094), we also found that three upregulated anti‐inflammatory genes upon CRISPRi, including C1QTNF4 (log2FC = 1.16, q = 0.023), THY1 (log2FC = 1.39, q = 0.037), and ERAP1 (log2FC = 0.51, q = 0.070), suggests a repressive role for activated SINE element on inflammatory responses.
4. DISCUSSION
In this study, we comprehensively investigated TE dysregulation in human brains and dissected the genome regulatory effects of TEs in AD. We showed that TE dysregulation in AD was associated with tau pathology, amyloid neuropathology, and APOE ε4 genotypes, and acted in sex‐specific and cell type–specific manners. We further studied the relationship of DNA variants genome wide, particularly in AD‐associated loci, with TE expression. Through colocalization analysis between teQTLs with AD GWAS loci, brain cell type–specific 3D chromatin structure, and CRISPRi, we observed that upregulated TEs were associated with AD‐related gene expression changes in human iPSC‐derived neurons, including C1QTNF4. These findings show that teQTLs offer a powerful QTL analytic approach to identify TE‐related risk genes in AD and suggest that this could be broadly applied to investigate other neurodegenerative diseases as well.
We first applied the AMP‐AD RNA‐seq harmonization approach to account for clinical heterogeneity across samples, studies, experimental batch effects, and unwanted RNA‐seq technical variations and identified highly reproducible locus‐based (Figure S6) and region‐specific (Figure S20 in supporting information) differentially expressed TEs between both Mayo and ROS/MAP brain biobanks. We found ERV1 (a major TE family) was significantly dysregulated in both ROS/MAP and MSBB brain biobanks. Compared to human AD brains and tau transgenic Drosophila, we found an overexpression of LINE‐1 and ERVs among the classes of activated TE in the context of brain aging. 4 Elevated transcript levels of LINE‐1 and ERVs are also present in human neurodegenerative disorders including but not limited to ALS and ataxia. 79 , 80 , 81 The predominant type of activated LINE class was LINE‐1 in both Mayo and ROS/MAP brain biobanks. By analyzing chronological ordering of LINE‐1 subfamily TEs, we found that evolutionary younger elements, such as L1HS, L1PA ,and L1PB, are more vulnerable in human aging brains with AD, compared to ancient‐inserted subfamily L1ME TEs (Figure S21 in supporting information). This result is consistent with prior work that a panel of evolutionary young LINE‐1 possessed retrotransposition activity in the developing and adult human brain. 82 We further found that TEs are also decreased in human aging AD brains, consistent with previous studies in tau transgenic Drosophila brains 83 as well as in post mortem human brains with AD and progressive supranuclear palsy. 84 Impaired activation of TEs has been associated with weak interferon response in SARS‐CoV‐2 human cellular models, 85 suggesting that decreased TE expression could also affect host immune response. We also found that the number of locus‐based differentially expressed TEs was proportional to the sequencing read length (MSBB: single‐end 100 bp; Mayo: paired‐end 101 bp; ROS/MAP: paired‐end 151 bp). Although ROS/MAP has the smallest sample size across the three brain biobanks, we found the sequence length of locus‐based differentially expressed TEs specifically identified in the ROS/MAP brain biobank (n = 3738) was more than that of the Mayo brain biobank (n = 725). These results indicate that longer sequencing reads, such as PacBio HiFi reads and Oxford nanopore sequencing reads, may retrieve more TE transcripts. 86
Dynamic patterns of TE expression contribute to cell heterogeneity, 44 and by analyzing RNA‐seq data from FACS‐sorted cell types from frozen brains we pinpointed an APOE ε4‐specific TE activation from a SINE element (chr5: 109799806–109800106) in microglia. The nearest gene is a key TF MAN2A1, whose expression has been reported to correlate with AD progression. 60 We also profiled female‐specific TE dysregulation and found that the most perturbed cell type was endothelial cells. By incorporating sex‐specific locus‐based differentially expressed TEs with expression changes of nearby genes, we found that ≈ 33.8% of locus‐based differentially expressed TE can be explained by sex‐specific expression of nearby genes, suggesting that those TEs may play roles in sex‐specific gene regulatory networks. These results support the functional roles of TE dysregulation in AD pathogenesis by APOE‐, cell type‐, and sex‐specific manners.
Via analysis of WGS and matched RNA‐seq data from the Mayo brain biobank, we also illustrated the first catalog of teQTLs for human AD brains (Figure 3A), which demonstrated a 35.4% replication rate of teQTL findings between Mayo and ROS/MAP brain biobanks (Figure S22 in supporting information). We found that those teQTLs were significantly enriched across three AD GWAS datasets (Figure 3B), indicating potential roles of teQTLs for interpreting the results of GWAS. The other two GWAS datasets were derived from family history of AD (Proxy‐AD). 68 , 87 The overall low quality of AD phenotypes in those two Proxy‐AD datasets may be the potential reason for lack of significant enrichment. We also found significant enrichment of AD‐related genes regulated by brain teQTLs (Figure 3A). Specifically, we found that blocking activity of MIR family TE was associated with elevated expression of BIN1 using CRISPRi experiments in human iPSC‐derived neurons (Figure 3F), indicating a potential AD‐associated gene regulatory mechanism by a SINE element (chr2: 127051802–127052004).
We also identified potential AD risk genes and drug targets impacted by teQTLs via colocalization analysis. For example, a key teQTL rs1065712 (GWAS P = 5.46 × 10−9) is related with upregulated FDFT1, which encodes a key enzyme involved in cholesterol biosynthesis. 88 We found that FDFT1 and a nearby SINE element (chr8: 11840915–11841089) exhibited significantly elevated expression in APOE ε4/ε4 patient iPSC‐derived microglia (Figure 6B,C). Currently, several FDFT1 inhibitors, such as lapaquistat acetate, 89 are under investigation as an anti‐cholesterol agent. Notably, anti‐lipid and anti‐cholesterol agents have been identified as potential treatments for AD. 90 Thus, FDFT1 identified from teQTL analysis offers a potential risk gene and drug target for AD. More functional evaluation is warranted to test the causal relationship of teQTL on gene regulation underlying AD etiologies in the future.
We also identified another candidate teQTL, rs11605348 (GWAS P = 1.92 × 10−11), which is related to an upregulated SINE element (chr11: 47608036–47608220). We prioritized C1QTNF4 as an AD risk gene across three AD GWAS cohorts through additional TE transcriptome‐wide association analysis in the Mayo brain biobank (Figure S23 in supporting information). We additionally identified two additional teQTLs from TE transcriptome‐wide association analysis, including rs10838698 (AD GWAS P = 1.59 × 10−11) and rs896817 (AD GWAS P = 8.45 × 10−11), which was further supported by FUSION conditional analysis (Figure S18). We found that three AD GWAS loci were in the same high LD block region (LD r2 > 0.2, Figure S24 in supporting information), especially between rs10838698 and rs11605348 (r2 = 0.75), suggesting the co‐regulation of the three AD GWAS loci on C1QTNF4 expression. Furthermore, we found that upregulated MIR family TE was associated with repression of anti‐inflammatory genes upon CRISPRi, including C1QTNF4, THY1, and ERAP1. As inflammation is a central mechanism in AD and C1QTNF4 can exert an anti‐inflammatory activity, 71 these observations suggest novel anti‐inflammation associated AD risk genes associated with TE activation in AD and human aging brains. Thus, our results suggest that TE can not only serve as a potent source of diverse cis‐regulatory sequences in mammalian sequences, but it can be linked to gene expression changes associated with diseases. 91 While changes in chromatin marks may contribute to both transcriptomic changes and co‐regulation between TEs and nearby genes, we cannot conclude any causality of TEs on AD pathogenesis in the current study. We speculate that the activated TE might derive dsRNA and sRNAs, including short‐interfering RNAs (siRNAs) and PIWI‐interacting RNAs (piRNAs), which may affect expression of nearby genes involved in the inflammatory response. 92 The biological mechanisms across chromatin changes, TE activation, and target gene dysregulation require further functional evaluation in the future.
We acknowledge several potential limitations of our study. First, our teQTL findings might be influenced by the sample size of matched WGS and RNA‐seq data. Integration of large‐scale WGS and RNA‐seq data could identify more genome‐wide significant teQTLs in the future. Second, most donors for our WGS and RNA‐seq data were derived from European ancestry. In future studies, we will leverage more diverse ancestries (including African American) from the Alzheimer's Disease Sequencing Project. 93 Third, although we used multiple complementary bioinformatic tools to quantify TE expression, including SQuIRE, 15 Telescope, 19 and TEtranscripts, 17 those tools could not fully filter out co‐transcribed TEs within genes or remove the effects of pervasive transcription. Applying machine learning models may identify more accurate TE transcripts in the future. Finally, we only investigated cell type–specific locus‐based TE expression using bulk cell RNA‐seq data and one scRNA‐seq dataset. Incorporating large‐scale long‐read single‐cell/nucleus RNA‐seq data in the future may identify cell type–specific novel TE‐mediated risk genes in AD and other diseases if broadly applied.
AUTHOR CONTRIBUTIONS
Feixiong Cheng conceived the study. Yayan Feng performed experiments and data analyses. Yadi Zhou downloaded and managed the sequencing data from the AD Knowledge Portal. Xiaoyu Yang and Yin Shen performed CRISPRi experiments. Yuan Hou, James B. Leverenz, Andrew A. Pieper, Charis Eng, and Alison Goate discussed and interpreted results. Yayan Feng, Xiaoyu Yang, and Feixiong Cheng drafted the manuscript. Yayan Feng, Feixiong Cheng, Andrew A. Pieper, Alison Goate, and Yin Shen critically revised the manuscript. All authors gave final approval of the manuscript.
CONFLICT OF INTEREST STATEMENT
Dr. Leverenz has received consulting fees from Vaxxinity, grant support from GE Healthcare, and serves on a data safety monitoring board for Eisai. Dr. Goate serves on a data safety monitoring board for Genentech and Muna Therapeutics. The other authors have declared no competing interests. Author disclosures are available in the supporting information.
CONSENT STATEMENT
All human subjects used in this study have been adequately informed with consent by the ROS/MAP cohort, Mayo Clinic Cohort, and MSBB cohort. The consent was not necessary for this study.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
We thank members from The National Institute on Aging Genetics of Alzheimer's Disease Data Storage Site and The Alzheimer's Disease Knowledge portal to provide technical support for multi‐omics data access and analysis. This work was primarily supported by the National Institute on Aging (NIA) under Award Number R01AG084250, R56AG074001, U01AG073323, R01AG066707, R01AG076448, R01AG082118, RF1AG082211, and R21AG083003, and the National Institute of Neurological Disorders and Stroke (NINDS) under Award Number RF1NS133812, and the Alzheimer's Association award (ALZDISCOVERY‐1051936) to F.C. This work was supported in part by the Cleveland Alzheimer's Disease Research Center (NIH/NIA: P30AGO62428) to F.C., A.A.P., and J.B.L. A.A.P. was also supported by The Valour Foundation, the Rebecca E. Barchas MD Chair in Translational Psychiatry of Case Western Reserve University, the Morley‐Mather Chair in Neuropsychiatry of University Hospitals of Cleveland Medical Center, the American Heart Association and Paul Allen Foundation Initiative in Brain Health and Cognitive Impairment (19PABH134580006), Department of Veterans Affairs Merit Award I01BX005976, and the Louis Stokes VA Medical Center resources and facilities. ROS/MAP study data were provided by the Rush Alzheimer's Disease Center at Rush University Medical Center. This work was supported in part by the NIA under Award Number R01AG079291 and RF1AG079557 to Y.S. Data collection was supported through funding by National Institute on Aging (NIA) grants P30AG10161, R01AG15819, R01AG17917, R01AG30146, R01AG36836, U01AG32984, U01AG46152, and U01AG61356 and by the Illinois Department of Public Health. Mayo RNA‐seq study data were provided by the following sources: the Mayo Clinic Alzheimer's Disease Genetic Studies, led by N. Ertekin‐Taner and S. G. Younkin (Mayo Clinic, Jacksonville, Florida), using samples from the Mayo Clinic Study of Aging, the Mayo Clinic Alzheimer's Disease Research Center and the Mayo Clinic Brain Bank. Data collection was supported through funding by NIA grants P50 AG016574, R01 AG032990, U01 AG046139, R01 AG018023, U01 AG006576, U01 AG006786, R01 AG025711, R01 AG017216, and R01 AG003949; by National Institute of Neurological Disorders and Stroke (NINDS) grant R01 NS080820; MSBB data were generated from post mortem brain tissue collected through the Mount Sinai VA Medical Center Brain Bank and were provided by E. Schadt of the Mount Sinai School of Medicine through funding from NIA grant U01AG046170. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Feng Y, Yang X, Hou Y. Widespread transposable element dysregulation in human aging brains with Alzheimer's disease. Alzheimer's Dement. 2024;20:7495–7517. 10.1002/alz.14164
DATA AVAILABILITY STATEMENT
All data (including teQTL) and analytic code written in R are available in the GitHub repository: https://github.com/ChengF‐Lab/alzteQTL. CRISPRi‐targeted raw RNA‐seq data were deposited in the GEO database with the following accession number: GSE246052.
REFERENCES
- 1. Chuong EB, Elde NC, Feschotte C. Regulatory activities of transposable elements: from conflicts to benefits. Nat Rev Genet. 2017;18(2):71‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang K, Liu H, Hu Q, et al. Epigenetic regulation of aging: implications for interventions of aging and diseases. Signal Transduct Target Ther. 2022;7(1):374. doi: 10.1038/s41392-022-01211-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ramirez P, Zuniga G, Sun W, et al. Pathogenic tau accelerates aging‐associated activation of transposable elements in the mouse central nervous system. Prog Neurobiol. 2022;208:102181. doi: 10.1016/j.pneurobio.2021.102181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Guo C, Jeong HH, Hsieh YC, et al. Tau activates transposable elements in Alzheimer's disease. Cell Rep. 2018;23(10):2874‐2880. doi: 10.1016/j.celrep.2018.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liu EY, Russ J, Cali CP, Phan JM, Amlie‐Wolf A, Lee EB. Loss of nuclear TDP‐43 is associated with decondensation of LINE retrotransposons. Cell Rep. 2019;27(5):1409‐1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brattas PL, Jonsson ME, Fasching L, et al. TRIM28 controls a gene regulatory network based on endogenous retroviruses in human neural progenitor cells. Cell Rep. 2017;18(1):1‐11. doi: 10.1016/j.celrep.2016.12.010 [DOI] [PubMed] [Google Scholar]
- 7. Jonsson ME, Garza R, Sharma Y, et al. Activation of endogenous retroviruses during brain development causes an inflammatory response. EMBO J. 2021;40(9):e106423. doi: 10.15252/embj.2020106423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Krug L, Chatterjee N, Borges‐Monroy R, et al. Retrotransposon activation contributes to neurodegeneration in a Drosophila TDP‐43 model of ALS. PLoS Genet. 2017;13(3):e1006635. doi: 10.1371/journal.pgen.1006635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu S, Heumuller SE, Hossinger A, et al. Reactivated endogenous retroviruses promote protein aggregate spreading. Nat Commun. 2023;14(1):5034. doi: 10.1038/s41467-023-40632-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Evering TH, Marston JL, Gan L, Nixon DF. Transposable elements and Alzheimer's disease pathogenesis. Trends Neurosci. 2023;46(3):170‐172. doi: 10.1016/j.tins.2022.12.003 [DOI] [PubMed] [Google Scholar]
- 11. Wood JG, Jones BC, Jiang N, et al. Chromatin‐modifying genetic interventions suppress age‐associated transposable element activation and extend life span in Drosophila. Proc Natl Acad Sci. 2016;113(40):11277‐11282. doi: 10.1073/pnas.1604621113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sturm A, Saskoi E, Hotzi B, et al. Downregulation of transposable elements extends lifespan in Caenorhabditis elegans. Nat Commun. 2023;14(1):5278. doi: 10.1038/s41467-023-40957-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. De Cecco M, Ito T, Petrashen AP, et al. L1 drives IFN in senescent cells and promotes age‐associated inflammation. Nature. 2019;566(7742):73‐78. doi: 10.1038/s41586-018-0784-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Purcell S, Neale B, Todd‐Brown K, et al. PLINK: a tool set for whole‐genome association and population‐based linkage analyses. Am J Hum Genet. 2007;81(3):559‐575. doi: 10.1086/519795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang WR, Ardeljan D, Pacyna CN, Payer LM, Burns KH. SQuIRE reveals locus‐specific regulation of interspersed repeat expression. Nucleic Acids Res. 2019;47(5):e27. doi: 10.1093/nar/gky1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA‐seq aligner. Bioinformatics. 2013;29(1):15‐21. doi: 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jin Y, Tam OH, Paniagua E, Hammell M. TEtranscripts: a package for including transposable elements in differential expression analysis of RNA‐seq datasets. Bioinformatics. 2015;31(22):3593‐3599. doi: 10.1093/bioinformatics/btv422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wheeler TJ, Clements J, Eddy SR, et al. Dfam: a database of repetitive DNA based on profile hidden Markov models. Nucleic Acids Res. 2013;41(Database issue):D70‐D82. doi: 10.1093/nar/gks1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bendall ML, de Mulder M, Iniguez LP, et al. Telescope: characterization of the retrotranscriptome by accurate estimation of transposable element expression. PLoS Comput Biol. 2019;15(9):e1006453. doi: 10.1371/journal.pcbi.1006453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hoffman GE, Schadt EE. variancePartition: interpreting drivers of variation in complex gene expression studies. BMC Bioinf. 2016;17(1):483. doi: 10.1186/s12859-016-1323-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pimentel H, Bray NL, Puente S, Melsted P, Pachter L. Differential analysis of RNA‐seq incorporating quantification uncertainty. Nat Methods. 2017;14(7):687‐690. doi: 10.1038/nmeth.4324 [DOI] [PubMed] [Google Scholar]
- 22. Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841‐842. doi: 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chang D, Nalls MA, Hallgrimsdottir IB, et al. A meta‐analysis of genome‐wide association studies identifies 17 new Parkinson's disease risk loci. Nat Genet. 2017;49(10):1511‐1516. doi: 10.1038/ng.3955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Consortium GT. Genetic effects on gene expression across human tissues. Nature. 2017;550(7675):204‐213. doi: 10.1038/nature24277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. de Klein N, Tsai EA, Vochteloo M, et al. Brain expression quantitative trait locus and network analyses reveal downstream effects and putative drivers for brain‐related diseases. Nat Genet. 2023;55(3):377‐388. doi: 10.1038/s41588-023-01300-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang T, Liu Y, Ruan J, Dong X, Wang Y, Peng J. A pipeline for RNA‐seq based eQTL analysis with automated quality control procedures. BMC Bioinf. 2021;22(9):403. doi: 10.1186/s12859-021-04307-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shabalin AA. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics. 2012;28(10):1353‐1358. doi: 10.1093/bioinformatics/bts163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Consortium GT. Erratum: genetic effects on gene expression across human tissues. Nature. 2018;553(7689):530. doi: 10.1038/nature25160 [DOI] [PubMed] [Google Scholar]
- 29. Zhao H, Sun Z, Wang J, Huang H, Kocher J‐P, Wang L. CrossMap: a versatile tool for coordinate conversion between genome assemblies. Bioinformatics. 2014;30(7):1006‐1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sieberts SK, Perumal TM, Carrasquillo MM, et al. Large eQTL meta‐analysis reveals differing patterns between cerebral cortical and cerebellar brain regions. Sci Data. 2020;7(1):340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ng B, White CC, Klein HU, et al. An xQTL map integrates the genetic architecture of the human brain's transcriptome and epigenome. Nat Neurosci. 2017;20(10):1418‐1426. doi: 10.1038/nn.4632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. de Klein N, Tsai EA, Vochteloo M, et al. Brain expression quantitative trait locus and network analysis reveals downstream effects and putative drivers for brain‐related diseases. Nat Genet. 2023;55(3):377‐388. doi: 10.1038/s41588-023-01300-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci. 2003;100(16):9440‐9445. doi: 10.1073/pnas.1530509100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kunkle BW, Grenier‐Boley B, Sims R, et al. Genetic meta‐analysis of diagnosed Alzheimer's disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet. 2019;51(3):414‐430. doi: 10.1038/s41588-019-0358-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wightman DP, Jansen IE, Savage JE, et al. A genome‐wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer's disease. Nat Genet. 2021;53(9):1276‐1282. doi: 10.1038/s41588-021-00921-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bellenguez C, Küçükali F, Jansen IE, et al. New insights into the genetic etiology of Alzheimer's disease and related dementias. Nat Genet. 2022;54(4):412‐436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Giambartolomei C, Vukcevic D, Schadt EE, et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLos Genet. 2014;10(5):e1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gusev A, Ko A, Shi H, et al. Integrative approaches for large‐scale transcriptome‐wide association studies. Nat Genet. 2016;48(3):245‐252. doi: 10.1038/ng.3506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schmidt EM, Zhang J, Zhou W, et al. GREGOR: evaluating global enrichment of trait‐associated variants in epigenomic features using a systematic, data‐driven approach. Bioinformatics. 2015;31(16):2601‐2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kundaje A, Meuleman W, Ernst J, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518(7539):317‐330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Srinivasan K, Friedman BA, Etxeberria A, et al. Alzheimer's patient microglia exhibit enhanced aging and unique transcriptional activation. Cell Rep. 2020;31(13):107843. doi: 10.1016/j.celrep.2020.107843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tcw J, Qian L, Pipalia NH, et al. Cholesterol and matrisome pathways dysregulated in astrocytes and microglia. Cell. 2022;185(13):2213‐2233 e25. doi: 10.1016/j.cell.2022.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gerrits E, Brouwer N, Kooistra SM, et al. Distinct amyloid‐beta and tau‐associated microglia profiles in Alzheimer's disease. Acta Neuropathol. 2021;141(5):681‐696. doi: 10.1007/s00401-021-02263-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. He J, Babarinde IA, Sun L, et al. Identifying transposable element expression dynamics and heterogeneity during development at the single‐cell level with a processing pipeline scTE. Nat Commun. 2021;12(1):1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nott A, Holtman IR, Coufal NG, et al. Brain cell type‐specific enhancer‐promoter interactome maps and disease‐risk association. Science. 2019;366(6469):1134‐1139. doi: 10.1126/science.aay0793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Iotchkova V, Ritchie GR, Geihs M, et al. GARFIELD classifies disease‐relevant genomic features through integration of functional annotations with association signals. Nat Genet. 2019;51(2):343‐353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Finucane HK, Bulik‐Sullivan B, Gusev A, et al. Partitioning heritability by functional annotation using genome‐wide association summary statistics. Nat Genet. 2015;47(11):1228‐1235. doi: 10.1038/ng.3404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wu T, Hu E, Xu S, et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation. 2021;2(3):100141. doi: 10.1016/j.xinn.2021.100141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Watanabe K, Taskesen E, van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun. 2017;8(1):1826. doi: 10.1038/s41467-017-01261-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang C, Ward ME, Chen R, et al. Scalable production of iPSC‐derived human neurons to identify tau‐lowering compounds by high‐content screening. Stem Cell Rep. 2017;9(4):1221‐1233. doi: 10.1016/j.stemcr.2017.08.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Labun K, Montague TG, Krause M, Torres Cleuren YN, Tjeldnes H, Valen E. CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 2019;47(W1):W171‐W174. doi: 10.1093/nar/gkz365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yang X, Wen J, Yang H, et al. Functional characterization of Alzheimer's disease genetic variants in microglia. Nat Genet. 2023;55(10):1735‐1744. doi: 10.1038/s41588-023-01506-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wickham H, Chang W, Wickham MH. Package ‘ggplot2’. Create elegant data visualisations using the grammar of graphics version. 2016;2(1):1‐189. [Google Scholar]
- 54. Hermant C, Torres‐Padilla ME. TFs for TEs: the transcription factor repertoire of mammalian transposable elements. Genes Dev. 2021;35(1‐2):22‐39. doi: 10.1101/gad.344473.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang M, Beckmann ND, Roussos P, et al. The Mount Sinai cohort of large‐scale genomic, transcriptomic and proteomic data in Alzheimer's disease. Sci Data. 2018;5:180185. doi: 10.1038/sdata.2018.185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fullard JF, Hauberg ME, Bendl J, et al. An atlas of chromatin accessibility in the adult human brain. Genome Res. 2018;28(8):1243‐1252. doi: 10.1101/gr.232488.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Loupe JM, Anderson AG, Rizzardi LF, et al. Extensive profiling of transcription factors in postmortem brains defines genomic occupancy in disease‐relevant cell types and links TF activities to neuropsychiatric disorders. Biorxiv. 2023. doi: 10.1101/2023.06.21.545934 [DOI] [Google Scholar]
- 58. Markus‐Koch A, Schmitt O, Seemann S, et al. ADAM23 promotes neuronal differentiation of human neural progenitor cells. Cell Mol Biol Lett. 2017;22:16. doi: 10.1186/s11658-017-0045-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chiou SS, Wang SS, Wu DC, et al. Control of oxidative stress and generation of induced pluripotent stem cell‐like cells by jun dimerization protein 2. Cancers. 2013;5(3):959‐984. doi: 10.3390/cancers5030959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Arizaca Maquera KA, Welden JR, Margvelani G, et al. Alzheimer's disease pathogenetic progression is associated with changes in regulated retained introns and editing of circular RNAs. Front Mol Neurosci. 2023;16:1141079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Koran MEI, Wagener M, Hohman TJ. Alzheimer's Neuroimaging Initiative . Sex differences in the association between AD biomarkers and cognitive decline. Brain Imaging Behav. 2017;11:205‐213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. van Doeselaar L, Stark T, Mitra S, et al. Sex‐specific and opposed effects of FKBP51 in glutamatergic and GABAergic neurons: implications for stress susceptibility and resilience. Proc Natl Acad Sci USA. 2023;120(23):e2300722120. doi: 10.1073/pnas.2300722120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Boorman DC, Keay KA. Sex differences in morphine sensitivity are associated with differential glial expression in the brainstem of rats with neuropathic pain. J Neurosci Res. 2022;100(10):1890‐1907. doi: 10.1002/jnr.25103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kanekiyo T, Cirrito JR, Liu CC, et al. Neuronal clearance of amyloid‐beta by endocytic receptor LRP1. J Neurosci. 2013;33(49):19276‐19283. doi: 10.1523/JNEUROSCI.3487-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Henson HE, Taylor MR. A sart1 zebrafish mutant results in developmental defects in the central nervous system. Cells. 2020;9(11):2340. doi: 10.3390/cells9112340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Misiak B, Ricceri L, Sąsiadek MM. Transposable elements and their epigenetic regulation in mental disorders: current evidence in the field. Front Genet. 2019;10:580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Fedoroff NV. Transposable elements, epigenetics, and genome evolution. Science. 2012;338(6108):758‐767. doi: 10.1126/science.338.6108.758 [DOI] [PubMed] [Google Scholar]
- 68. Jansen IE, Savage JE, Watanabe K, et al. Genome‐wide meta‐analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nat Genet. 2019;51(3):404‐413. doi: 10.1038/s41588-018-0311-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yuan XZ, Sun S, Tan CC, Yu JT, Tan L. The role of ADAM10 in Alzheimer's disease. J Alzheimers Dis. 2017;58(2):303‐322. doi: 10.3233/JAD-170061 [DOI] [PubMed] [Google Scholar]
- 70. Funfschilling U, Saher G, Xiao L, Mobius W, Nave KA. Survival of adult neurons lacking cholesterol synthesis in vivo. BMC Neurosci. 2007;8:1. doi: 10.1186/1471-2202-8-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wang L. CTRP4: a new member of the adipocytokine family. Cell Mol Immunol. 2017;14(10):868‐870. doi: 10.1038/cmi.2017.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Giannos P, Prokopidis K, Raleigh SM, Kelaiditi E, Hill MW. Altered mitochondrial microenvironment at the spotlight of musculoskeletal aging and Alzheimer's disease. Sci Rep. 2022;12(1):11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. van Bree EJ, Guimaraes R, Lundberg M, et al. A hidden layer of structural variation in transposable elements reveals potential genetic modifiers in human disease‐risk loci. Genome Res. 2022;32(4):656‐670. doi: 10.1101/gr.275515.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. He J, Fu X, Zhang M, et al. Transposable elements are regulated by context‐specific patterns of chromatin marks in mouse embryonic stem cells. Nat Commun. 2019;10(1):34. doi: 10.1038/s41467-018-08006-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Saher G, Brügger B, Lappe‐Siefke C, et al. High cholesterol level is essential for myelin membrane growth. Nat Neurosci. 2005;8(4):468‐475. [DOI] [PubMed] [Google Scholar]
- 76. Cheng F, Lu W, Liu C, et al. A genome‐wide positioning systems network algorithm for in silico drug repurposing. Nat Commun. 2019;10(1):3476. doi: 10.1038/s41467-019-10744-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Rao JS, Kellom M, Kim HW, Rapoport SI, Reese EA. Neuroinflammation and synaptic loss. Neurochem Res. 2012;37(5):903‐910. doi: 10.1007/s11064-012-0708-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Todd CD, Deniz Ö, Taylor D, Branco MR. Functional evaluation of transposable elements as enhancers in mouse embryonic and trophoblast stem cells. Elife. 2019;8:e44344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Takahashi T, Stoiljkovic M, Song E, et al. LINE‐1 activation in the cerebellum drives ataxia. Neuron. 2022;110(20):3278‐3287. doi: 10.1016/j.neuron.2022.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Shpyleva S, Melnyk S, Pavliv O, Pogribny I, Jill James S. Overexpression of LINE‐1 retrotransposons in autism brain. Mol Neurobiol. 2018;55(2):1740‐1749. doi: 10.1007/s12035-017-0421-x [DOI] [PubMed] [Google Scholar]
- 81. Xiao‐Jie L, Hui‐Ying X, Qi X, Jiang X, Shi‐Jie M. LINE‐1 in cancer: multifaceted functions and potential clinical implications. Genet Med. 2016;18(5):431‐439. doi: 10.1038/gim.2015.119 [DOI] [PubMed] [Google Scholar]
- 82. Garza R, Atacho DAM, Adami A, et al. LINE‐1 retrotransposons drive human neuronal transcriptome complexity and functional diversification. Sci Adv. 2023;9(44):eadh9543. doi: 10.1126/sciadv.adh9543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ochoa E, Ramirez P, Gonzalez E, Mange JD, Ray JW, Bieniek KF. Bess frost B. Pathogenic tau‐induced transposable element‐derived dsRNA drives neuroinflammation. Sci Adv. 2023;9(1):eabq5423. doi: 10.1126/sciadv.abq5423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Sun W, Samimi H, Gamez M, Zare H, Frost B. Pathogenic tau‐induced piRNA depletion promotes neuronal death through transposable element dysregulation in neurodegenerative tauopathies. Nat Neurosci. 2018;21(8):1038‐1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Sorek M, Meshorer E, Schlesinger S. Impaired activation of transposable elements in SARS‐CoV‐2 infection. EMBO Rep. 2022;23(9):e55101. doi: 10.15252/embr.202255101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Rebollo R, Cumunel E, Mary A, et al. Detection and identification of transposable element transcripts using long read RNA‐seq in Drosophila germline tissues. Biorxiv. 2023. [Google Scholar]
- 87. Schwartzentruber J, Cooper S, Liu JZ, et al. Genome‐wide meta‐analysis, fine‐mapping and integrative prioritization implicate new Alzheimer's disease risk genes. Nat Genet. 2021;53(3):392‐402. doi: 10.1038/s41588-020-00776-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Do R, Kiss RS, Gaudet D, Engert JC. Squalene synthase: a critical enzyme in the cholesterol biosynthesis pathway. Clin Genet. 2009;75(1):19‐29. doi: 10.1111/j.1399-0004.2008.01099.x [DOI] [PubMed] [Google Scholar]
- 89. Liao JK. Squalene synthase inhibitor lapaquistat acetate: could anything be better than statins? Circulation. 2011;123(18):1925‐1928. doi: 10.1161/CIRCULATIONAHA.111.028571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Xu J, Mao C, Hou Y, et al. Interpretable deep learning translation of GWAS and multi‐omics findings to identify pathobiology and drug repurposing in Alzheimer's disease. Cell Rep. 2022;41(9):111717. doi: 10.1016/j.celrep.2022.111717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Gebrie A. Transposable elements as essential elements in the control of gene expression. Mobile DNA. 2023;14(1):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Lanciano S, Cristofari G. Measuring and interpreting transposable element expression. Nat Rev Genet. 2020;21(12):721‐736. doi: 10.1038/s41576-020-0251-y [DOI] [PubMed] [Google Scholar]
- 93. Beecham GW, Bis JC, Martin ER, et al. The Alzheimer's disease sequencing project: study design and sample selection. Neurol Genet. 2017;3(5):e194. doi: 10.1212/NXG.0000000000000194 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Data Availability Statement
All data (including teQTL) and analytic code written in R are available in the GitHub repository: https://github.com/ChengF‐Lab/alzteQTL. CRISPRi‐targeted raw RNA‐seq data were deposited in the GEO database with the following accession number: GSE246052.