

Abstract
The tauopathies are defined by pathological tau protein aggregates within a spectrum of clinically heterogeneous neurodegenerative diseases. The primary tauopathies meet the definition of rare diseases in the United States. There is no approved treatment for primary tauopathies. In this context, designing the most efficient development programs to translate promising targets and treatments from preclinical studies to early‐phase clinical trials is vital. In September 2022, the Rainwater Charitable Foundation convened an international expert workshop focused on the translation of tauopathy therapeutics through early‐phase trials. Our report on the workshop recommends a framework for principled drug development and a companion lexicon to facilitate communication focusing on reproducibility and achieving common elements. Topics include the selection of targets, drugs, biomarkers, participants, and study designs. The maturation of pharmacodynamic biomarkers to demonstrate target engagement and surrogate disease biomarkers is a crucial unmet need.
Highlights
Experts provided a framework to translate therapeutics (discovery to clinical trials).
Experts focused on the “5 Rights” (target, drug, biomarker, participants, trial).
Current research on frontotemporal degeneration, progressive supranuclear palsy, and corticobasal syndrome therapeutics includes 32 trials (37% on biologics)
Tau therapeutics are being tested in Alzheimer's disease; primary tauopathies have a large unmet need.
Keywords: biomarkers, development, early‐phase clinical trials, preclinical, tauopathy, therapeutics
1. INTRODUCTION
1.1. Tau and tauopathies
Research in the mid‐1980s demonstrated that tau is the major protein in neurofibrillary tangles, one of the pathological hallmarks of Alzheimer's disease (AD). 1 , 2 Subsequent research determined that tau aggregates are the primary pathological feature of a spectrum of clinically heterogeneous neurodegenerative diseases collectively referred to as tauopathies. 3 Primary tauopathies, with tau as the predominant pathological component, include progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), Pick's disease, frontotemporal lobar degeneration (FTLD) due to mutations in the gene encoding microtubule‐associated protein tau (MAPT), globular glial tauopathy (GGT), age‐related tau astrogliopathy (ARTAG), primary age‐related tauopathy (PART), and argyrophilic grain disease (AGD). 4 Secondary tauopathies in which tau aggregation is believed to occur in response to other pathological proteins or events include AD, Parkinson's disease, dementia with Lewy bodies (DLB), and chronic traumatic encephalopathy (CTE). 4 , 5
Globally, > 55.2 million people have dementia; the annual incidence is ≈ 7 million new cases. By 2050, prevalence will increase to 139 million people worldwide. 6 AD accounts for 60% to 70% of all cases of dementia. 7 In the United States, ≈ 11% of people aged ≥ 65 years have AD dementia; 33% of those aged ≥ 85 years have AD dementia. 8 Prevalence estimates of the primary tauopathies are complicated by widespread misdiagnosis and a lack of specific diagnostic biomarkers. FTLD has been estimated to account for 2.6% of all‐cause dementia. 7 , 9 In epidemiological studies in Olmsted County, Minnesota, the prevalence rates of PSP and corticobasal syndrome (CBS) were estimated to be comparable (≈ 11/100,000). 10 Owing to the small number of cases studied to date, the incidence of CTE is currently unknown. 11 These estimates of the primary tauopathies conform to the definition of rare diseases with < 200,000 people affected with any of these disorders in the United States. 12
In recent years, important advances have been made in the development of tools and identification of cellular pathways for diagnostic and therapeutic targeting of tau pathology. The mediators of pathogenic tau‐induced neuronal dysfunction and death are being elucidated through studies of transcriptomics, including the disruption of nuclear and genomic architecture. The development of positron emission tomography (PET)‐based tau imaging has transformed in vivo visualization of tau pathology in AD, and the tracking of its pathological spread in this disease. Important advances in biologics include early‐phase clinical trials testing tau‐targeting antisense oligonucleotides (ASOs) to reduce tau production 13 as well as tau‐based immunotherapy with monoclonal antibodies (mAbs) targeting the extracellular spread of tau. 14 The development of small molecules for tauopathy has been directed at reducing its production, decreasing its deposition, and enhancing its clearance through autophagy and proteostasis as well as inhibiting the posttranslational modification of tau that is associated with a pro‐aggregating state.
In September 2022, the Rainwater Charitable Foundation, one of the largest independent primary tauopathy research funders with > $140 M invested since 2009, convened and sponsored a workshop focused on a framework for translating tauopathy therapeutics from drug discovery to preclinical development to early‐phase clinical trials. This initiative brought together a group of 35 leading preclinical and clinical research experts across neurodegenerative diseases, and more specifically in the tauopathies. Participants included 16 academic faculty, nine industry, and 10 charitable non‐profit organizations. The workshop had four objectives: (1) to develop a framework and companion lexicon for the translation of therapeutics from late preclinical development studies to early, proof‐of‐concept (PoC) clinical trials in primary tauopathies; (2) to share knowledge to foster an environment for capturing the state of the art across different phases of therapeutic development; (3) to identify key questions to be addressed for progress in future therapeutic development of the tauopathies; and (4) to publish the workshop proceedings to share the framework and findings with the broader research community. These workshop objectives align with those from the Alzheimer's Disease‐Related Dementias Summit in 2022. 15
1.2. The lexicon for tauopathy therapeutic development
Across the spectrum of neurodegenerative diseases, there have been several initiatives to develop a common lexicon to facilitate clinical drug development. 16 , 17 , 18 , 19 , 20 At this workshop, participants considered these existing frameworks across the neurodegenerative disease spectrum and applied them to tauopathy research. Table 1 provides the lexicon for tauopathies that was developed and advanced through this workshop. It refines study‐related terms and differentiates types of biomarkers that are critical to the successful translation of medicines from preclinical development through early‐phase clinical trials to the goal of achieving PoC.
TABLE 1.
Proposed lexicon.
| Term | Definition |
|---|---|
| Disease‐monitoring biomarker | Can be applied serially and used to detect a change in the severity of disease; may be used for PoP or PoC |
| Disease‐prognosis biomarker | Can forecast the rate of change associated with disease pathophysiology or natural history |
| Downstream biomarker | Response indicating an indirect impact on disease pathophysiology downstream to its initial site of action and target engagement |
| Drug repositioning | Developing a therapeutic for an indication other than for what it was originally intended, with prioritization during development and before approval |
| Drug repurposing | Application of established drug compounds to new therapeutic indications |
| In silico | Use of informatics (e.g., epidemiology, data mining) to identify novel compounds or for repurposing an existing compound |
| Patient selection | Defines the inclusion criteria that can use biomarkers to define or enrich a study population |
| Pharmacodynamic pathway biomarker | Pharmacodynamic biological response in a person linked to a pathway that is directly influenced by an intervention |
| Pharmacodynamic response biomarker | Biological response in a person linked to an intervention or exposure |
| Preclinical development | Phase of development that includes model systems in multiple species, including transgenics, knock‐in models, and human‐derived iPSCs |
| Preclinical disease stage | A clinical designation for human staging of disease that includes cognitively and behaviorally unimpaired individuals at risk of developing a tauopathy based on the presence of a known causal genetic mutation or disease‐state biomarkers. Alternatively referred to as asymptomatic, at risk, or presymptomatic |
| Proof of principle | Achieved when a biomarker response indicates that a directed intervention has modified the known or suspected pathology of a tauopathy |
| Proof of concept | Achieved when an intervention has produced a clinical response that may be predictive of efficacy in patients with a tauopathy |
| Proof of mechanism | Evidence that a molecular target has been engaged and has affected the biology of target cells in a non‐clinical model (aka TE) |
| Safety biomarker | Biomarker which can monitor the toxicity and/or safety of a drug through evaluation of risk |
| Stratification | Defines the approach to allocation of participants in a study population using biomarkers or other features (e.g., disease stage, severity, site, country) and allowing for a balance of factors identified as critical for a treatment response |
| Target engagement | A biomarker response that indicates the intervention reached its site of action and has sufficiently engaged its intended target |
| Target product profile | Describes the intended use, patient population, and other distinctive characteristics including efficacy and safety |
| TE biomarker | Biological response directly reflective of the intended target |
| Translational biomarker | Can be deployed in preclinical studies and then advanced to clinical trials, serving as a PD or TE measure |
| Treatment‐predictive biomarker | Can predict the nature and/or extent of a response to treatment |
Abbreviations: iPSCs, induced pluripotent stem cells; PD, pharmacodynamic; PoC, proof of concept; PoP, proof of principle; TE, target engagement.
1.3. Proposed framework to clinical PoC
The workshop focused its framework from drug discovery to the milestone of PoC. It leveraged the Rights of Precision Drug Development of Cummings, Feldman, and Scheltens, with adaptation and application to tauopathy therapeutics. 21 Milestones for PoC within this framework include establishing a favorable profile of pharmaceutical properties including safety and tolerability, pharmacokinetics (PK)/pharmacodynamics (PD), blood–brain barrier (BBB) penetration, maximum tolerated dose (or equivalent), and dose(s) to be tested. Early phase 2a studies include safety and tolerability, dose range, and biomarker measures. Preliminary clinical effects, including directional trajectories, effect sizes, and consistency across measures, are ascertained without anticipating statistically significant drug–placebo differences at this stage in these small preliminary trials. Phase 2b studies provide fundamental data for clinical PoC, as they provide a broader assessment of safety, tolerability, impact on target engagement biomarkers; biological effects on other disease biomarkers; and estimates of clinical effect sizes, including those that would be potentially clinically important and likely to be pursued in later‐stage registrational trials. Clinical PoC can be considered having been reached when there is a weight of converging evidence of dose, target engagement, PK/PD, biological effects, safety, and tolerability, as well as sufficient clinical evidence to support a decision to proceed to larger scale phase 3 confirmatory registrational trials. The demonstration of clinical efficacy in later stage trials is a prerequisite for full target validation.
2. LANDSCAPE OVERVIEW
With the clinical workshop goal to focus on primary tauopathies, a search in Global Data on January 12, 2024, identified 30 planned or ongoing phase 1 through 3 interventional studies of either frontotemporal degeneration (FTD), PSP, and/or CBD. An abbreviated list of trials is provided in Table 2, including 15 studies in PSP, 17 studies in FTD, and three studies in CBD. Across these diseases, the therapeutic targets and treatments are broad and diverse.
TABLE 2.
Current therapeutic trials for PSP, CBD, and FTD (as of January 12, 2024) a .
| Drug name | Company name | Development stage | Disease | Molecule type | Mechanism of action | Clinical trial details |
|---|---|---|---|---|---|---|
| Latozinemab | Alector Inc | Phase 3 | FTD | Monoclonal antibody | Sortilin inhibitor | NCT06111014 |
| Sodium phenylbutyrate + taurursodiol (AMX0035, Relyvrio, Albrioza) | Amylyx Pharmaceuticals Inc | Phase 3 | PSP | Small molecule | Apoptosis regulator BAX inhibitor | NCT06122662 |
| AZP‐2006 | AlzProtect SAS | Phase 2 | PSP | Small molecule | Prosaposin‐progranulin complex regulation | NCT04008355 |
| Apilimod mesylate (LAM‐002A) | OrphAI Therapeutics Inc | Phase 2 | FTD | Small molecule | 1‐phosphatidylinositol‐3‐phosphate‐5‐kinase (PIKFYVE) inhibitor | NCT05483322 |
| ASN‐90 | Grupo Ferrer Internacional SA | Phase 2 | PSP | Small molecule | Protein O‐GlcNAcase inhibitor | No active PSP trial found (to be initiated) |
| AVB‐101 | AviadoBio Ltd | Phase 2 | FTD | Gene therapy | Progranulin activator | NCT06064890 |
| Censavudine | Transposon Therapeutics Inc | Phase 2 | FTD, PSP | Small molecule | Reverse transcriptase inhibitor | NCT04993755 |
| Deulinoleate ethyl (R001) | Retrotope Inc | Phase 2 | PSP | Small molecule | Downregulates oxidative stress. It protects cells from damage, mediated through lipid peroxidation | NCT04937530 |
| Fasudil (BRAVYL) | Woolsey Pharmaceuticals Inc | Phase 2 | PSP, CBD | Small molecule | Rho kinase inhibitor | NCT04734379 |
| Hydromethylthionine mesylate (LMTX) | TauRx Therapeutics Ltd | Phase 2 | PSP | Small molecule | MAPT inhibitor; TAR DNA‐binding protein‐43 inhibitor | No active PSP trial found (to be planned based on results in AD) |
| Oxytocin (Syntocinon) | Phoenixus AG | Phase 2 | FTD | Synthetic peptide | Oxytocin receptor agonist | NCT03260920 |
| PBFT‐02 | Passage Bio Inc | Phase 2 | FTD | Gene therapy | Progranulin activator | NCT04747431 |
| Rotigotine ER | UCB SA | Phase 2 | FTD | Small molecule | Dopamine receptor agonist | NCT04937452 |
| Tertomotide (GV1001, Riavax) | GemVax & KAEL Co Ltd | Phase 2 | PSP | Subunit vaccine | Telomerase reverse transcriptase added MOA: Gonadotropin releasing hormone receptor (GNRHR) agonist | NCT05819658 |
| DNL593 | Denali Therapeutics Inc | Phase 2 | FTD | Fusion protein | Progranulin replacement | NCT05262023 |
| PR‐006 | Prevail Therapeutics Inc | Phase 2 | FTD | Gene therapy | Non‐replicating recombinant adeno‐associated virus serotype 9 (AAV9) to deliver codon‐optimized DNA encoding wild‐type progranulin | NCT04408625 |
| Bepranemab (UCB0107) | UCB SA | Phase 1 Phase 1 | PSP | Monoclonal antibody | Microtubule associated protein tau inhibitor | NCT04658199 |
| TPI‐287 | Cortice Biosciences Inc | Phase 1 ( b see comment under clinical trials) | PSP, CBD | Small molecule | Tubulin inhibitor | NCT02133846 (completed in 2019) |
| AHT‐434 | Alterity Therapeutics Ltd | Phase 1 | PSP, CBD | Small molecule | Α‐syn inhibitor | In phase 2 for multiple system atrophy (NCT05109091). No specific phase 1 trial found for PSP in Australia |
| AL‐101 | Alector Inc | Phase 1 | FTD | Monoclonal antibody | Sortilin inhibitor | NCT04111666 (obtained fast track designation for FTD in 2020) |
| ANAVEX‐371 | Anavex Life Sciences Corp | Phase 1 | FTD | Small molecule | Muscarinic acetylcholine receptor M1 agonist; Sigma non‐opioid intracellular receptor‐1 agonist | NCT04442945 (obtained orphan drug designation for FTD in 2016) |
| APNmAb‐005 | Aprinoia Therapeutics Inc | Phase 1 | FTD, PSP | Monoclonal antibody | Microtubule associated protein tau inhibitor | NCT05344989 |
| Buntanetap tartrate (Posiphen) | Annovis Bio Inc | Phase 1 | FTD | Small molecule | Α‐syn inhibitor; Aβ A4 protein inhibitor; MAPT inhibitor | No specific clinical trial found for FTD (except in pipeline on company website) |
| NAS‐150 | New Amsterdam Sciences Inc | Phase 1 | PSP | Small molecule | NADPH oxidase 4 inhibitor | No active phase 1 trial found (in planning on company website) |
| NIO‐752 | Novartis AG | Phase 1 | PSP | Antisense oligonucleotide | MAPT inhibitor | NCT04539041 |
| NP‐001 | Neuvivo Inc | Phase 1 | FTD | Small molecule | Purified form of sodium chlorite, which targets immune system macrophages | Listed by company as being in phase 1 but no clinical trial was found |
| ET‐STEM | Samsung Medical Center | Phase 1 | FTD | Cell therapy | NCT05315661 | |
| TQS‐168 | Tranquis Therapeutics Inc | Phase 1 | FTD | Small molecule | Peroxisome proliferator activated receptor gamma coactivator 1 alpha modulator | The company announced completion of phase 1 in 2022. Exploratory development ongoing in FTD |
| VES‐001 | Vesper Bio ApS | Phase 1 | FTD | Small molecule | Sortilin inhibitor | No clinical trial found but company announced dose to first volunteer in phase 1 study in Dec 2023 |
| OLX‐07010 | Oligomerix inc | IND/CTA filed | PSP | Small molecule | Microtubule associated protein tau inhibitor | In phase 1 for PSP |
Abbreviations: Aβ, amyloid beta; AD, Alzheimer's disease; CBD, corticobasal degeneration; FTD, frontotemporal degeneration; MAPT, microtubule‐associated protein tau; NADPH, nicotinamide adenine dinucleotide phosphate; PSP, progressive supranuclear palsy.
Source: Main source Global Data.
See comment under clinical trials.
RESEARCH IN CONTEXT
Systematic review: There has not been an approved drug treatment for primary tauopathies. As they are rare diseases, it is vitally important to design studies to move with optimum efficiency from preclinical studies to early‐phase clinical trials.
Interpretation: A principled approach to drug development for the tauopathies is recommended using the framework of the “5 Rights” including target, drug, biomarker, participant, and design. A companion lexicon can facilitate communication, with a focus on reproducibility and common elements.
Future directions: Academic and industry partnerships are fundamental to moving through bottlenecks and accelerating the translation of discoveries from academic labs (bench or basic research) into drug development programs for tauopathies. More timely data and sample sharing will support the necessary development of more robust disease modeling and simulation as well as back‐translation from failed trials to inform future corrections. The maturation of pharmacodynamic biomarkers that can demonstrate target engagement and act as surrogate disease biomarkers is a crucial unmet need.
Various approaches are being investigated with 20 small molecules across a diverse range of targets. There are four mAb programs, three gene therapy programs, and one program each of an ASO, cell therapy, fusion protein, synthetic peptide, and vaccine. Three of the programs include participants with PSP and CBD, and three different programs include both PSP and FTD participants. Of the PSP trials, only one is in phase 3, Amylyx's Relyvrio, 22 which is approved for usage in amyotrophic lateral sclerosis (ALS) patients. For the PSP trials, safety and tolerability is the most common primary clinical outcome of interest. There is a phase 3 trial for FTD testing an mAb from Alector called latozinemab. The majority of the agents being evaluated in FTD trials do not directly target tau biology. Instead, their mechanisms of action include targeting progranulin delivery or activation and sortilin inhibition, among others. The Clinical Dementia Rating Scale plus National Alzheimer's Coordinating Center's Frontotemporal Lobar Degeneration Module Sum of Boxes 23 is the most common composite clinical outcome. Of the studies that include biomarker outcomes, volumetric magnetic resonance imaging (MRI) and neurofilament light chains (NfL) in plasma or cerebrospinal fluid (CSF) are the most common biomarkers collected. Within the FTD trials, three trials are investigating agents directed at behavioral symptoms of FTD and are applicable to tauopathies. The Neuropsychiatric Inventory is the most common outcome in behavioral trials. 24 , ,
2.1. Applying lessons learned from related fields of neurodegenerative research
Many of the research challenges of tauopathies are similar to the other neurodegenerative diseases, particularly those diseases that share underlying tau pathophysiology in AD, Parkinson's disease, and ALS. Lessons learned from translational and later stage failed or negative trials in these diseases are relevant to planning future early‐phase tauopathy trials.
In AD, the therapeutic road to the successful development of amyloid‐lowering therapeutics has been long, complex, and informative. The regulatory approval of anti‐amyloid‐lowering mAbs, including aducanumab 25 and lecanemab, 26 and the promising phase 3 study results of donanemab 27 validate amyloid beta (Aβ) as a therapeutic target for AD drug development with the convincing demonstration that anti‐amyloid mAbs remove Aβ plaque, an effect that is associated with slowing of clinical decline. The slowing (25%–35%) across clinical outcome measures over 18 months in these phase 3 trials supports the amyloid hypothesis, as does definitive evidence of amyloid clearance on PET and reductions of phosphorylated tau (p‐tau) and other fluid biomarkers. The potential for augmenting treatment response in AD with combinatorial therapies directed at tauopathy and neuroinflammation and their interaction is an appealing strategy.
At the same time, the failure or negative results of multiple preceding Aβ‐targeted late‐stage AD trials are likely due to several factors that could be addressed in earlier phases of development, 19 , 28 including inconclusive definition of the target, insufficient evidence of target engagement, 29 , 30 inclusion of patients who did not have Aβ pathology, having non‐Aβ comorbid pathologies driving their clinical progression, and testing at an overly advanced stage of disease. For these failed Aβ‐targeted therapies, setting clinical PoC in phase 2 as a checkpoint before advancing to phase 3 trials would have required a better understanding of the range of doses for target engagement, the extent of pharmacodynamic response with Aβ lowering, and the risk–benefit of the higher doses that were eventually required for clinical efficacy.
Although Parkinson's disease is not considered a primary tauopathy, there is pathological tau aggregation and deposition in ≈ 50% of brains in affected patients, with evidence for cell‐to‐cell spread and interaction with α‐synucleinopathy. 5 The α‐synuclein (α‐syn) hypothesis of Parkinson's disease is based on the discovery that Lewy body inclusions contain aggregates of α‐syn in patients with familial Parkinson's disease. 31 In a consensus white paper proposing a roadmap for Parkinson's disease to achieve PoC in clinical trials targeting α‐syn, Merchant et al. applied lessons from both AD clinical trials and previous Parkinson's disease trials. 19 This Alpha‐Synuclein Working Group focused on the need to target early disease stages, establish target engagement and other biological measures of therapeutic response that are critical for the interpretation of study results, enrich trials with participants with demonstrated evidence of target pathology, identify clinical endpoints with greater sensitivity to demonstrate slowing of disease progression, and target multiple mechanisms for meaningful impact. The authors followed the five types of biomarker‐based evidence (i.e., diagnostic, monitoring, response, predictive, prognostic) 32 and, by doing so, developed a lexicon of terms to be used as part of their roadmap. To date, in clinical trials with α‐syn–targeted mAbs in Parkinson's disease, including prasinezumab 33 and cinpanemab, 34 α‐syn has been negative. The informative results of the failed amyloid antibody trials in AD may help clinicians understand where the hurdles exist in dosing, target engagement, or biological effects in Parkinson's disease. Indeed, recent biomarker data released from the cinpanemab trial may offer insight into some of these hurdles. 35
Among neurodegenerative diseases, ALS has recently seen significant therapeutic progress resulting from innovative drug development approaches. In a recent review, some of the above‐mentioned AD and Parkinson's disease therapeutic development challenges were addressed in the context of ALS, including how recent molecular discoveries, progress in the development of therapeutics (i.e., biomarkers, drug repurposing strategies, and high‐throughput drug screening), and new trial designs have been facilitated by improvements in patient‐reported outcome measures. 36 Existing obstacles in ALS in moving promising therapeutics to the clinic have included the lack of large‐scale research infrastructure to conduct clinical trials with the close follow‐up needed. 37 The extent of disease heterogeneity and the lack of proven biomarkers and clinical outcomes have been hampering; however, genetic signatures have helped inform individual course and treatment response, with precision medicine focus and with programs around disease mutations. Within the goal of rapidly identifying novel treatments, biomarkers, and trial endpoints, new approaches to early‐phase clinical trials have been developed. 38 The Healey ALS Platform Trial is a novel and informative approach using an adaptive platform design, with a master protocol allowing for the concurrent testing of multiple investigational products. 38 Platform trials like Healey ALS hold the potential to accelerate trials and drug development and are now being planned for the assessment of tau therapeutics. 39 They also create an opportunity for many patients to participate in clinical trials more easily. 40
The recent regulatory approvals of the ASO Tofersen (QALSODY, Biogen) for persons with autosomal dominantly inherited superoxide dismutase 1 mutation causing ALS provides an example of a targeted success for tailored, precision‐based RNA treatment approaches that might also aptly inform trials for those with tau mutations. 41 , 42 The approval pathway for Tofersen was accelerated based on biomarker effects on NfL as a surrogate biomarker that was considered reasonably likely to predict clinical benefit. This approach might be available for use in tau therapeutic programs if key biomarker changes can be convincingly linked to clinical outcomes.
Other disorders with tau pathology are emerging and may gain more attention as their tau pathophysiology is better understood. For instance, the results of myotonic dystrophy (DM) studies may inform the development of tau therapeutics. DM is characterized by a toxic RNA gain‐of‐function mechanism that disrupts RNA processing, localization, and translation. 43 Adult‐onset forms of DM have a congenital delay and progressive decline. 44 Some patients with DM types 1 and 2 show intracellular tau aggregates in the brain. 45 , 46 Another disorder displaying tau pathology includes tuberous sclerosis complex (TSC). TSC is an amyloid‐independent tauopathy associated with elevated phosphorylated 3R/4R tau aggregation. 47 TSC is a neurodevelopmental disorder associated with mutations in the TSC1 and TSC2 genes, leading to the growth of benign masses in several organ systems and, often, to behavioral, cognitive, and psychiatric difficulties. 48 , 49 ARTAG is a recently described 4R tauopathy. 50 ARTAG presents with astrocytic tau pathology and predominantly affects the elderly. ARTAG is believed to be distinct from PART, which involves mostly neuronal tau pathology. Finally, there are copathologies, including transactivation response DNA‐binding protein 43 kDa (TDP‐43) and tau. 51 The comorbid tau and TDP‐43 pathology may be synergistic with their neurotoxicity, and mixed pathology may be associated with severe AD outcomes. 52 Many of the tau therapeutics in development or the clinic today may be useful for these disorders in addition to the more traditionally known tauopathies. 53
3. THE 5 RIGHTS OF AD DRUG DEVELOPMENT: APPLIED TO TAUOPATHIES
Given the low success rates of clinical trials across neurodegenerative diseases including AD, Parkinson's disease, and ALS, coupled with the rare incidence of primary tauopathies, particular attention at this workshop focused on considerations around the most efficient and adaptive approaches to early‐phase drug development. To this end, this workshop adapted the five “rights” of precision drug development for AD 21 with application to the other tauopathies. The framework includes the right target, right drug, right biomarker, right participants, and right trials (Figure 1).
FIGURE 1.
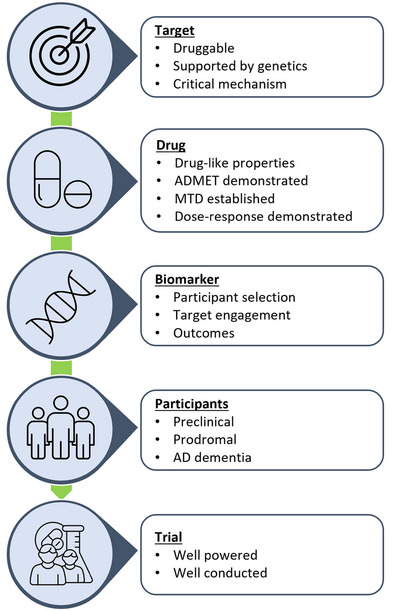
Five “rights” of precision drug development for Alzheimer's disease. 18 , 21 AD, Alzheimer's disease; ADMET, absorption, distribution, metabolism, excretion, toxicology; MTD, maximum tolerated dose.
3.1. The right target
The right “target” for tauopathy ideally includes those: (1) with a direct linkage to dysfunction in the disease, (2) are genetically causative, or (3) are associated with biomarker changes through the disease course. For tauopathies, this can include the production of tau species through effects on transcription and translation; cell‐to‐cell pathological spread via transsynaptic propagation, microtubule stabilization, and clearance, either intracellularly through autophagy and proteostasis; or through active or passive immunomodulation. 54 Furthermore, there are identifiable targets that can prevent tau's pathological posttranslational modification by acetylation, phosphorylation, O‐GlcNAcylation, nitration, ubiquitination, and SUMOylation, leading to its misfolding and aggregation. 55 This approach in development and target validation in drug discovery follows the therapeutic hypothesis based on genetic, human pathologic, and model data. Denali Therapeutics refers to genetically verified drug targets as degenogenes, which by analogy are similar to oncogenes that have served as targets for oncology drug development. 56 An alternative approach is to use proteomic, lipidomic, or metabolomic approaches to identify new targets, relying on CSF and other biospecimen samples being drawn from natural history studies.
To identify the right target, the criteria and process for preclinical studies in preparation for early clinical drug nomination must be established. The target identification process can follow genetic causes of disease, and the biochemical pathways altered as a result (e.g., from post‐mortem tissue). The genes and proteins in these pathways, as well as the consequential pathophysiology resulting from these alterations (e.g., neurofibrillary tangles), become the targets. Model systems have been valuable for studying these altered disease processes and identifying tractable targets with the potential for modulation provided that a suitable agent (e.g., the right drug) can be identified. Major caveats to identifying the right target for tauopathies include limited availability of patient data from genetically affected pedigrees, access to ante‐mortem and post‐mortem tissues, and the lack of well‐validated model systems that recapitulate the underlying biology and heterogeneity of disease for each specific tauopathy.
The workshop identified challenges and still unresolved issues in preclinical development and modeling of tauopathies. There is a clear need for better translational models that recapitulate human disease with which to advance target validation. This recommendation was born of the widespread recognition that all the tauopathy models to date have strengths and limitations, and it is unlikely that any one of the currently available models can capture all aspects of the clinical diseases. Rather, each model is used with appropriate awareness of its limitations and is selected based on the hypothesis that is being evaluated, and that is best matched to the mechanism of action of the drug being tested. Given these limitations, the workshop recognized that it is important to design a “fit‐for‐purpose” study with selected models to appropriately evaluate aspects of the disease that the model can represent. 57 , 58 The currently available murine tauopathy disease models do not generate human‐relevant neurodegeneration, nor do they capture disease heterogeneity. 59 To increase the relevance, such models can be humanized by “knocking‐in” a human gene to replace the mouse homologue rather than depending on protein‐overproducing transgenic models. For tauopathies in particular, there is an unmet need presently for mouse models that are specific to unique tauopathies, and that can facilitate mechanistic studies for testing compound activity.
The major challenge of recapitulating human tauopathies is finding a suitable model system that fully outlines the complex tau splicing in human brains. 60 , 61 For instance, rodent brains express neither the six isoforms of tau proteins found in adult humans nor the distinct patterns of 4‐repeat (4R tau) in a number of the human primary tauopathies (CBD, PSP, AGD, GGT, and many of the MAPT mutations), nor the 3‐repeat (3R) in others (Pick's disease), nor the 4R/3R (AD tauopathy, PART, CTE). 62 , 63 , 64 Studies also showed that the presence of murine tau protein interferes with human tau protein aggregation and toxicity, whereas forced expression of wild‐type human 4R tau is highly toxic to murine neurons. 65 , 66
Human neuronal cell models could provide a more relevant in vitro system to recapitulate tauopathy in human brain‐like conditions, including tau splicing. With recent advances in the inducible pluripotent stem cell (iPSC) and direct conversion technologies, human neurons with AD and tauopathy‐inducing mutations can be generated and maintained in standard cell culture conditions. 67 , 68 , 69 , 70 These cellular models enable the characterization of the early stages of tauopathy in human neurons and the influence of AD and human MAPT mutations. 67 , 68 , 69 , 70 , 71 , 72 However, most human iPSC‐derived neural culture models display the fetal tau‐splicing pattern with predominantly 3R isoforms but lacking 4R, which is critical for recapitulating robust tau pathology. 61 , 73 , 74 Intronic mutations altering 3R/4R tau splicing also induce tauopathy in humans. 60 , 74 Missense mutations causing tauopathy are concentrated in exon 10 of the tau gene, included only in the 4R tau isoform. 60 Therefore, insufficient expression of 4R tau in the iPSC‐derived neuronal models makes it challenging to use these mutations for disease modeling. Furthermore, due to the in vitro nature of iPSCs, they also lack the micro‐environment and cell–cell interactions that are critical to recapitulating human tauopathies. 61 , 75 , 76 Indeed, iPSC‐derived neurons have failed to produce robust tauopathy, including aggregation of hyperphosphorylated tau species and tau‐induced neurodegeneration.
Multiple technologies are being developed to address the shortcomings of insufficient 4R tau expression and to recapitulate robust tauopathy in human cellular models. Three‐dimensional (3D) culture conditions accelerate 4R tau generation and, thereby, tauopathy in human neural cell cultures. 77 , 78 , 79 , 80 , 81 , 82 Choi et al. have shown that Aβ accumulation in a 3D extracellular matrix is sufficient to induce robust tauopathy in human neural cells. 77 In this study, 3D‐differentiated human fetal neural progenitor cells expressed an equimolar ratio of 4R and 3R tau isoforms, similar to those found in human adult brains. 77 Human iPSC‐derived neurons, differentiated in the 3D matrix, also showed elevated 4R tau expression, although it took longer to display adult brain‐like tau pathology. 79 , 80 Finally, brain organoid models required even more extended maturation (9–18 months) to exhibit elevated 4R tau expression, and the expression level is lower than in the iPSC‐derived neurons in 3D culture systems. 83 , 84 The issue of slow maturation can be addressed to some degree through induced/transdifferentiated human neurons, which display more mature neuronal phenotypes and preserve disease‐associated phenotypes in standard two‐dimensional (2D) culture conditions, especially compared to iPSC‐derived neurons. 68 When induced by mitochondrial RNAs, transdifferentiated human neurons consistently expressed high levels of 4R‐related tau pathology comparable to human brains. 85 Adding a tauopathy mutation in the same model increases the 4R‐to‐3R tau ratio and promotes seed‐competent insoluble tau species. 85 Exogenous tau seeds, recombinant or patient derived, have also been used to accelerate tau pathology in iPSC‐derived human neurons. 86 , 87 , 88 , 89
Although progress has been made, triggering in vitro full‐blown tau pathology model systems remains challenging, as they lack the aging component observed in human brains. Direct conversion/trans‐differentiation could mimic patient cells’ age or stress conditions. Also, the biology in play is driven by the genetics of the donor. Furthermore, this technology depends on a supply of primary cells with low proliferative capacity, limiting the technology's generalized use for drug screening and validation. Recently, studies have demonstrated the critical role of microglia and peripheral immune cells in regulating brain tauopathy, 90 , 91 , 92 which has not yet been fully integrated into most human cellular tauopathy models but represents an emerging model area. Despite the challenges, new human cellular models of tauopathy provide an attractive platform to screen and validate drug candidates in human brain‐like conditions and can complement current rodent and non‐human primate tauopathy models, potentially bridging the rodent‐to‐human translational gap. 93
Beyond the models themselves, the workshop addressed the need for rigorous, reproducible non‐clinical studies and initiatives to confirm findings and create a commonality of approach with replication. The Model Organism Development and Evaluation for Late‐Onset Alzheimer's Disease (MODEL‐AD), 94 a consortium funded by the National Institute on Aging in 2016, represents such an effort to improve translation from animal models to humans. 94 MODEL‐AD has generated > 50 new mouse models that recapitulate genetic risk for late‐onset AD (LOAD) that are available to both non‐profit and for‐profit institutions, without any licensing restrictions. 95 The program has completed basic phenotypic analysis on > 30 of these models with comprehensive phenotyping including disease trajectories at the pathological, transcriptomic, proteomic, and functional levels on > 15 models. 95 Its aims include to (1) develop, characterize, and distribute the next generation of animal models of AD—with a focus on LOAD—based on human data; (2) establish and implement guidelines for rigorous preclinical testing in LOAD models with standards comparable to human clinical trials; and (3) provide a resource for standardized therapeutic efficacy testing of preclinical drug candidates that prioritizes translational biochemical and physiological endpoints over behavioral measures (e.g., mouse cognitive tests) using best practices. 96
As part of the MODEL‐AD consortium, the Preclinical Testing Core established a rigorous screening strategy with “Go/No‐Go” decision points that permit unbiased assessments of potential therapeutic agents in the mouse models characterized by the consortium. 96 Based on the available funds, the Preclinical Testing Core can evaluate up to two compounds per year, through a program that selects compounds nominated by investigators from the greater research community: Screening the Optimal Pharmaceutical for Alzheimer's Disease (STOP‐AD). 97 Upon selection, each compound is matched to an appropriate mouse model using a precision medicine–like approach (e.g., Right Patient) to ensure the target is expressed in the animal model within the stage of disease that is analogous to the interventional strategy of the anticipated clinical trial. For these studies, the selected outcome measures in the non‐clinical trials are prioritized for those that are most translational from mouse models to humans. More specifically, the initial screen evaluates drug stability, formulation, and PK to confirm appreciable brain exposure and in vivo target engagement in the disease model at the disease‐relevant ages. The findings from these initial steps allow for early identification and correction of drug formulation issues before advancing the drug to longer term, resource‐intensive studies. Then, PD and predictive PK/PD modeling determine the dose regimen for long‐term studies. The secondary screen evaluates target engagement and disease‐modifying activity using non‐invasive PET/computed tomography. If the compound meets the “Go” criteria for these endpoints, functional activity on behavioral endpoints is evaluated with a focus on identifying a therapeutic window and de‐risking for the potential for side effects. The post‐treatment analysis includes the evaluation of changes in proteomic and transcriptomic signatures after drug treatment. Importantly, the STOP‐AD program has developed an evaluation framework that can allow side‐by‐side comparisons of therapeutic candidates offering a common measure of their potential to be successful. 97 Studies have protocols that adhere to clear quantification within “Go/No‐Go” decision guidelines and are conducted according to the Animal Research: Reporting of In Vivo Experiments guidelines. 98 , 99 The standards and measures are defined, including an a priori inclusion and exclusion criterion, and the clarity with which the proof of principle (PoP) and/or PoC are described facilitates the decision‐making process. This resource is open to the greater research community, including for the screening of tauopathy‐targeted compounds.
3.2. The right drug
The right “drug” for tauopathy can be defined by a selectivity profile that includes a high affinity for its intended target, with low affinity for other off‐target effects; a demonstrated ability to modify the intended disease biology; with good adsorption and bioavailability achieving a potentially effective dose in the central nervous system (CNS), including BBB permeability and a favorable therapeutic index (i.e., the predicted therapeutic dose is well below the no‐observed‐adverse‐effect level). The important properties of drugs being selected for testing in tauopathies include having activity in the in vivo brain exposure that is relevant to in vitro data. This includes relevant free, unbound exposure in the brain after administration of acceptable doses. As drugs are selected and tested in early‐phase trials, a well‐characterized dose response in vivo, as reflected by robust PK/PD relationships, preferably with a clinically translatable biomarker, comprises key elements to be established. Characterizing the relationships among exposures in the brain, CSF, and plasma facilitates human studies and dose selection. If these relationships are 1:1:1, the move to the clinic can be made with greater confidence. By contrast, if there is a “brain penalty,” it must be anticipated in the dosing regimen. Establishing a sufficiently understood PK across the range of modalities of tauopathy drugs in development represents one of the complexities of finding the right drug. To identify the right drug, the workshop endorsed prespecifying criteria within the preclinical development for moving a compound into clinical development.
Tau may theoretically be a better AD therapeutic target than Aβ because, unlike the weaker relationship of brain Aβ plaque burden and cognition, the extent of insoluble tau pathology predicts both the onset of clinical symptoms of AD as well as the pattern of clinical decline. 100 , 101 , 102 , 103 , 104 Insoluble tau deposition measured by tau PET strongly correlates with the onset of clinical symptoms in autosomal dominant AD, 102 and temporal lobe tau PET uptake predicts the cortical spread of tau pathology, 105 brain atrophy, 106 and subsequent clinical decline in sporadic AD. 103 Moreover, the severity and type of symptoms in AD closely reflect the distribution of insoluble tau in the brain, suggesting that interventions reducing tau have the potential to produce large clinical effects. 107 , 108 , 109 , 110
Clinical trials of early tau therapies, including biologics with first‐generation N‐terminal anti‐tau mAbs, tilavonemab (ABBV‐8E12), 111 , 112 , 113 gosuranemab, 114 , 115 zagotenemab, and semorinemab 116 , 117 , 118 have failed to demonstrate consistent clinical benefits in AD and/or other tauopathies. 119 , 120 , 121 A trial of semorinemab in mild‐to‐moderate AD stands apart, having reported a ≈ 40% reduction in the rate of decline on the Alzheimer's Disease Assessment Scale‐Cognitive Composite co‐primary endpoint, but not on other endpoints, resulting in a lack of converging evidence and questions around the potential reproducibility of the findings. 122 By contrast, in prodromal‐mild AD, semorinemab did not demonstrate clinical benefit, with modestly reduced CSF p‐tau181 levels by ≈ 15% (mean of all doses). This effect was less than the ≈ 25% reductions reported for patients who responded to high‐dose aducanumab or lecanemab, possibly explaining the lack of clinical effect in this population. 25 , 123 Although all four of these first‐generation anti‐tau mAbs had evidence of target engagement of N‐terminal tau fragments in CSF, 116 , 120 , 124 , 125 none reported PD effects on insoluble aggregated tau. 119 , 123 Their lack of efficacy may have been due to the targeting of the N‐terminal tau fragments, which may be non‐pathogenic and may have diverted the necessary target engagement of mid‐ and C‐terminal regions. Second‐generation anti‐tau mAbs and an active vaccine 126 that target the mid‐domain microtubule‐binding region (MTBR) and C‐terminal regions have now entered clinical trials. 127 , 128 For current immunotherapies, including anti‐tau mAbs, only a very low proportion (≈ 0.1%) crosses the BBB and has brain uptake. 129 Research efforts are actively studying how to improve brain bioavailability through the enhancement of endogenous transport systems including receptor‐mediated transcytosis, with ligand‐receptor complexes, examples of which include transferrin receptors, low‐density lipoprotein 1, or nanoparticles. 130 Alternative approaches include molecules with heavy‐chain‐only antibodies 129 to improve delivery.
A range of tau‐targeted small‐molecule therapeutic programs span phases 1 and 2 in trials of both primary and secondary tauopathies. None have reached full clinical PoC to date. 131 These include compounds that alter posttranslational modification of tau with acetylation inhibition (i.e., salsalate 132 ), aggregation inhibition with methylene blue, 133 , 134 methylthionium, leuco‐methylthioninium (LMTM; i.e., Trx0237), 135 microtubule stabilization with davenutide, epothilone D, a macrolide, as well as taxane derivative abeteotaxane (i.e., TPI 287). 121 , 131 An array of glycoside hydrolase O‐GlcNAcase inhibitors, which block the formation of neurotoxic tau aggregates by increasing the glycosylation of tau and lowering propensity to aggregate, are currently being tested in clinical trials, including ASN 120290 in PSP, LY3372689 in AD, and ASN 51 in AD and Parkinson's disease. 136 , 137 , 138 Kinase inhibitors, including glycogen synthase kinase 3 beta, a serine‐threonine enzyme with the medications tideglusib, valproate, lithium, Fyn kinase of the Src family with saracatinib, and Abl tyrosine kinase with nilotinib have been through early‐phase trials. 139 , 140
The recent phase 1 results with the intrathecally delivered anti‐MAPT ASO MAPTRx (ISIS 814907/BIIB080) in AD achieved a dose‐responsive reduction in CSF total tau, p‐tau 181, and the ratio of total tau/Aβ42. 13 For the highest dose group of 60 mg every 4 weeks, a mean change of ≈ 60% in total tau was achieved supporting sufficient target engagement and advancement to phase 2. 13 Also, in a small number of patients, tau PET demonstrated a drug–placebo difference with less insoluble tau in patients on active therapy by Week 25 and a reduction of insoluble tau below baseline after 100 weeks of exposure. 141 The molecule is now being tested in a phase 2 trial. A similar mechanism of action by NIO752, an intrathecal anti‐MAPT ASO is being tested in phase 1 trials for AD and PSP.
Outcomes like these, combined with the learnings from failed anti‐Aβ and anti‐tau mAb clinical trials and a growing tauopathy therapeutic landscape, 53 directed a consensus in the workshop that any new therapies being developed for tauopathies should demonstrate PD effects on validated tau biomarkers (tau PET and/or mid‐domain MTBR sites for aggregated tau) or tau‐related targets including neuroinflammation or neurodegeneration before moving forward to large clinical efficacy studies.
The number of ongoing clinical trials of disease‐modifying therapies for AD has not increased significantly since 2012. 27 In 2018, the number of ongoing AD clinical trials was 40 times less than the number of ongoing trials in oncology. 28 In a recent evidence‐based review and Delphi consensus, Ballard et al. proposed alternative, lower‐risk approaches for new drug development in AD using drug repositioning and repurposing. 28 These approaches can expand drug development opportunities and accelerate the timelines for new treatments for AD and primary tauopathies. An advantage of drug repurposing is the known safety and tolerability profile of the candidate compound. Treatment‐related safety risks for tauopathies in early‐phase human trials can benefit from data and safety monitoring board oversight to identify and mitigate any safety and tolerability signals that arise. The designation of any potential adverse events of special interest that follow preclinical toxicology or phase 1 trials can help provide a focused evaluation of new compounds for early significant signals. With repositioned and repurposed drugs, the time and cost involved in progressing the candidate compound to clinical trials are reduced significantly. Additionally, costs associated with formulation optimization, manufacturing development, and drug–drug interaction studies have already been absorbed by the originating pharmaceutical companies. Thus, this could become a complementary and accessible route to drug development for tauopathy therapeutics by academic institutions, government agencies, and not‐for‐profit organizations. Such a pipeline can be further sourced from 3D human neural glial triculture drug screening, 142 in silico predictive modeling with the use of transcriptomic databases and computational modeling, 143 and through systematic reviews and Delphi panels, which rank potential candidates according to defined desirable features. 144 , 145
However, repurposing and repositioning medicines is not without its challenges. Repurposing can involve medicines that were not optimized for CNS diseases and for which little is known about the CNS properties of the medicines, including the dosing needed to achieve target engagement. 143 If their CNS properties are poor or not optimized, reformulation of prodrugs or even new chemical entities may be needed. 146 Phase 1 dose‐finding studies in healthy volunteers may be needed if the dose or age of the proposed population differs substantially from those of the original indication. Some current efforts use the process of “back‐translating,” which validates selected models by testing compounds that already have clinical data (e.g., levetiracetam) or for which there are large pharmacoepidemiology databases to investigate the effects of long‐term exposure on AD risk and course. 147 , 148 Back‐translation will become more feasible as clinically validated treatments such as the amyloid‐lowering mAbs set benchmarks for comparisons. For repurposing a molecule, there must be a demonstration that the desired effect is mediated by a cognate target of the molecule, effective exposure in the brain, and exposures for efficacy that are within the limits of the investigational new drug (IND) application toxicology studies for the original indication. Some molecules have metabolites, and some of these may be active. Thus, it is important to account for these when conducting clinical trials for a new indication. Additionally, there can be regulatory complexities to address. For example, repurposed or repositioned drugs may have less (or non‐existent) intellectual property–limiting patent life, which can then make it difficult to incentivize investment for registrational trials.
3.3. The right biomarker
The right “biomarkers” for tauopathy trials are selected across a range of uses that make each “fit for purpose” in development, including diagnosis and inclusion, PD evaluation of target engagement, monitoring of treatment response, and predictive and prognostic modeling. 32 Currently, the tau biomarkers available for use in primary tauopathies and those for secondary tauopathy associated with AD differ significantly. Whereas excellent progress has been realized in AD‐related tau biomarkers, there remains a significant unmet need in the tau canonical biomarkers for the primary tauopathies. The development of tau biomarkers specific to primary tauopathies was recognized in the workshop as being one of the most pressing gaps in this area of therapeutic research.
Through the anti‐amyloid mAb development in AD, much has been learned about the utility of amyloid and tau biomarkers. 149 Figure 2 is a schematic showing a mapping of fluid biomarkers of potential relevance in tauopathy therapeutic trials. Diagnostically, amyloid PET and CSF measures of Aβ42, ratio of Aβ42/40, p‐tau181, and p‐tau217, in various permutations have reached broad acceptance and validation as identifying in vivo amyloid pathology. 150 After a substudy within the bapineuzumab phase 3 clinical trials, it was recognized that 21.4% of participants for these anti‐amyloid mAb trials were amyloid‐negative with PET standard uptake value ratios below the diagnostic threshold. Thus, there was a trial paradigm shift to require biomarker confirmation for anti‐amyloid mAb clinical trials. 151 This improved diagnostic accuracy of participants with target pathology addressed a critical factor in the progress of anti‐amyloid mAbs in clinical trials by eliminating this significant trial design liability.
FIGURE 2.

Biomarkers of potential interest in tauopathies. Aβ, amyloid beta; APP, amyloid precursor protein; BDNF, brain‐derived neurotrophic factor; CSF, cerebrospinal fluid; GFAP, glial fibrillary acidic protein; MBP, myelin basic protein; MCP‐1, monocyte chemoattractant protein‐1; NF‐L, neurofilament light; NSE, neuron‐specific enolase; PDGFRβ, platelet‐derived growth factor receptor β; S100B, S100 calcium‐binding protein B; SBDP, spectrin breakdown products; SNAP 25, synaptosomal‐associated protein 25 kDa; SV2A, synaptic vesicle glycoprotein 2A; SYT 1, synaptotagmin 1; TDP‐43, transactive response DNA‐binding protein 43 kDa; TREM2, triggering receptor expressed on myeloid cells 2; UCHL1, ubiquitin C‐terminal hydrolase L1; VILIP, visinin‐like protein; YKL‐40, chitinase‐like protein based on 3 N‐terminal amino acids tyrosine (Y), lysine (K) and leucine (L) + molecular mass of 40 kDa 14.
The PD effects of aducanumab and lecanemab decisively lowering aggregated amyloid on PET scanning served to support the initial US Food and Drug Administration (FDA) accelerated approval because the medications were successfully targeting the fundamental pathophysiology of the disease with a reasonable likelihood of predicting clinical benefit for patients. 25 , 152 Subsequently, for lecanemab, full regulatory approval followed based on converging clinical evidence. 26 Other biomarker effects with lecanemab treatment included significant changes of plasma measures of glial fibrillary acid protein, p‐tau181, and Aβ42/40 at 6 months, providing the potential for early readout of biomarkers that might predict later clinical response.
Increased p‐tau concentrations in biofluids largely reflect Aβ pathology‐induced tau phosphorylation and secretion. 153 By contrast, there are no ideal or clinically validated tau fluid biomarkers or imaging tracers for diagnosis, PD monitoring, or disease monitoring in the primary tauopathies. Fluorodeoxyglucose PET is currently the functional imaging biomarker modality that is widely available for use as an outcome measure in primary tauopathies. Fluorodeoxyglucose PET findings of hypometabolism differ across the 4R tauopathy disorders, with some phenotypic patterns being appreciable. 154 Tau PET biomarkers have the potential to inform target engagement and infer drug effects; however, ligands are needed for 4R tauopathies and use outside of AD. The patterns of tau PET with 18F‐flortaucipir may also have some phenotypic utility; however, the degree of uptake does not correlate well with the extent of tauopathy and is unlikely to be sufficient for pharmacodynamic effects in the primary tauopathies. Second‐generation tau PET ligands, including [18F]PI‐2620, and [18F]PM‐PBB3 (also known as [18F]APN‐1607 and [18F]Florzolotau) primary tauopathies are being actively explored as 4R tau biomarkers with variable results in non‐AD tauopathies. 155 , 156 Overall, current data indicate that better 3R‐ and 4R‐tau PET ligands are needed to support clinical development. For other imaging biomarkers, structural MRI is a commonly used and robust modality to measure longitudinal changes in brain atrophy. Its measurement can run in parallel with clinical effects; however, its signal‐to‐noise ratio can be low, and the directionality of treatment effects is often uncertain. Similarly, across types of biomarkers, treatment effects may not be concordant or follow expected directions. Thus, predictions made in designing trials with different treatments may not always be correct, and development plans are best updated with preliminary data.
Recently, there has been potentially transformative tau biomarker discovery research that has identified tau MTBR isoforms that can distinguish primary tauopathies as well as tangle‐specific sites in AD. 157 , 158 Using an immunoprecipitation and mass spectrometry approach, Horie et al. reported on 4R isoform‐specific tau species from MTBR‐tau275 and MTBR‐tau282 that provide the first biomarkers that may significantly aid in the diagnosis of primary tauopathies and, in turn, in the facilitation of clinical trials and monitoring treatment response for these disorders. 157 Another tau fragment, ending at amino acid 368, has a similar potential. 159 Furthermore, abnormalities in the MTBR residue 243 (MTBR‐tau243) have been reported to be specific for insoluble tau aggregate pathology, reflecting cortical tangle pathology in AD. 158 When measured in CSF, MTBR‐tau243 is identified as a potential surrogate measure that can track the extent of this pathology. 158 When coupled with an assay for p‐tau205, the combination improves the prediction of both tau PET positivity and Mini‐Mental State Examination scores. 158 These MTBR tau biomarkers differ from the soluble p‐tau measures of 181, 231, and 217, which reflect the presence of amyloid pathology and amyloid PET correlations more so than insoluble tau tangle pathology and tau PET. 158 Confirmatory and further validation studies are anticipated. New insights from cryo‐electron microscopy studies may also lead to the development of potential new CSF tau biomarkers derived from microtubule‐binding domain peptides. 160 , 161
Outside tau biomarkers, NfL may be applicable for specific contexts of use, as it is sensitive to neuronal axonal damage/degeneration and has the potential to track disease progress and response to treatment. 41 Plasma levels of NfL have been reported to correlate with the severity of post‐mortem tangle pathology and neurodegeneration. 162 However, NfL lacks specificity across the tauopathies 163 and is elevated across a spectrum of neurologic and neurodegenerative diseases. 164 , 165 NfL may perform best as a biomarker of disease course in natural history studies and for prognostic segregation of treatment response as an enrollment enrichment tool in clinical trials, but may also have utility as a PD biomarker, as seen in clinical trials in ALS, multiple sclerosis, and spinal muscular atrophy. 166
The availability of more and better biomarkers will continue to advance drug development at all stages, including preclinical/non‐clinical and clinical stages of development. There is a need to carefully consider the context of use for biomarkers, as the evidentiary burden of supportive data will be different for safety, diagnostic, risk, prognostic, predictive, PD, and monitoring biomarkers. 167 Importantly, a biomarker need not be fully qualified by regulators to be used in a clinical trial and generate high‐value data. One goal in biomarker development is to have real‐world evidence to be sufficiently qualified to be included in the evidentiary package.
A useful resource for biomarker development is the FDA's BEST (Biomarkers, EndpointS, and other Tools) Resource. 32 The BEST Resource was developed by the FDA–National Institutes of Health Joint Leadership Council to provide harmonization of terms used in translational science and medical product development. There is an emphasis on endpoints and aims, to capture distinctions between biomarkers and clinical assessments and to describe their specific roles in biomedical research, clinical practice, medical product development, and in the regulation of products by the FDA.
3.4. The right participants
The phenotypic spectrum of tauopathies crosses diverse clinical features, presentations, and variable symptomatic progression rates through the disease course. This heterogeneity complicates the identification of the right participants at the most appropriate stage of the disease to evaluate novel tauopathy treatments. Through genetic testing for highly penetrant autosomal dominant MAPT mutations, it is possible to identify disease carriers in their preclinical phase of the disease. Staged development is needed to eventually design preventative clinical trials in such individuals. Initial efficacy on target PD and biological biomarkers can be followed by longer term studies of clinical efficacy and comparisons to modeled synthetic control groups or historical comparison groups such that trials can then be designed to test prevention/delay of disease onset in carriers in the preclinical phase of the disease.
Following the informative example of the Dominantly Inherited Alzheimer's Network Trials Unit (DIAN‐TU), interventions can be undertaken within a platform trial design that compares multiple early interventions with a prespecified biomarker interim analysis (stage gate) to proceed to longer term trials. 168 The ALLFTD 169 and GENFI 170 studies have successfully enrolled the largest numbers of families affected by genetic forms of MAPT mutation‐FTD as well as sporadic disease and have followed them in the largest international longitudinal observational natural history studies. 171 , 172 They have also come together in the FTD Prevention Initiative, 173 in which they are combining data for the organization of prevention clinical trials, and developing disease progression models and simulations. 174 For rare neurogenetic diseases, the FDA has indicated its interest in such innovative approaches to create disease progression models that could support clinical endpoints, enrollment criteria, and evaluation of changes in natural history from experimental treatment intervention. 175
For sporadic disease, there has been considerable interest in characterizing distinct phenotypes of 4R tauopathies including PSP with subtypes of Richardson's syndrome 176 and subtyping of CBS. 177 However, there remain inherent limitations. In PSP, the confirmation of diagnosis can take many years as the multisystem manifestations develop, and in usual care settings, misdiagnosis with Parkinson's disease is frequent. This delay leads to difficulties in identifying the right patient to enroll in clinical trials at an early stage. The sequence and speed at which symptoms progress can be highly variable, and the point of intervention may be very important. With the recent example of the anti‐amyloid mAb donanemab, it is evident that intervention in sporadic AD associated with less tau burden is more clinically efficacious than intervention at later stages of disease when greater tau pathology is present. 27 , 178 This sheds important light on the right patient tied to the right stage of disease and the right intervention from preclinical symptoms through the dementia stages.
There are also important patient factors for clinical trials that span dementia stages. The provisions for obtaining patient consent and advance planning for research are important, given that capacity will typically be lost in the course of the tauopathies, and intentions must be made clear to legally authorized representatives. Best practices for patient and caregiver engagement and communication can help consolidate interest in research and clinical trial participation and encourage brain donation on passing. Even at the early stages of the disease, anosognosia and metacognitive impairment in self‐awareness and self‐monitoring can occur in FTD, particularly behavioral variant FTD. The effects of being unaware of symptoms or impairment can limit directly reported symptoms and treatment effects. 179 , 180 Questionnaires and patient‐reported assessments need to be interpreted accordingly. It is important to shape inclusion criteria with consideration given to patient comorbidities and their concomitant medications, to increase enrollment and representativeness of patient populations.
Trial participation and retention is a key challenge for the rare disease primary tauopathies. FTD Disorders Registry data showed travel burden as the single largest factor discouraging individuals from participating in clinical trials. Workshop participants discussed the importance of reducing the costs of study‐related travel and costs by remote participation using digital health technologies (DHTs). DHTs, which are more widely available post‐COVID, can be further leveraged to better understand the patient experience, particularly as they can be administered and monitored at home, potentially improving the feasibility of trial participation and retention by reducing the number of required trips to the clinic. 181 At‐home use of DHTs also provides an opportunity to parse between good‐ and bad‐day assessments, some of which can be disease related. An elevated level of variability may reflect a primary mechanism of a patient's disease progression. In theory, increasing the frequency of at‐home assessments could increase the power of the study and reduce the number of needed participants.
Historically, the patient's perspective has been included only in later‐stage clinical development in relationship to marketing strategies, yet these perspectives can be addressed more inclusively earlier and diversely throughout the clinical drug development process. A participant's and family's risk tolerance can be surveyed in advance of a research program involving CSF‐administered biologic or gene therapy, particularly to consider the acceptability of potential drug delivery mechanisms and toxicity. This would be useful in evaluating any assumptions about the therapeutic intervention with a more formal understanding of the end user's perspective. Including patient feedback early in the process can help with the optimization of consent forms; increase patient retention, especially for pathologies such as PSP, which can progress quickly during a trial; and reduce study‐related travel burden. From the FDA's perspective, patient and caregiver engagement should be conducted systematically (i.e., via Patient‐Focused Drug Development program 182 ) to ensure the prespecified primary and secondary endpoints of a clinical trial are clearly and formally correlated with quality‐of‐life measures. Many of these points are yet to be elucidated in rare neurological disorders.
The Association for Frontotemporal Degeneration generated a Voice of the Patient Report 183 as part of the FDA's Externally Led Patient‐Focused Drug Development initiative. Using a combination of testimonials, polls, and a large survey executed in collaboration with the FTD Disorders Registry, the initiative documented insights from people living with FTD disorders, their care partners, and caregivers. 183 The meeting was designed to help the FDA understand the experiences and priorities of those affected by all FTD disorders, including PSP and CBS, and to use this knowledge to inform risk–benefit analyses of drug candidates and guide regulatory decisions throughout the drug development process. Meeting participants described their lived experience with FTD disorders, including their experiences seeking and receiving treatment as well as their priorities for the development of new therapies. 183 Overall, participants mentioned a broad spectrum of FTD symptoms and described the devasting impact of these symptoms on every aspect of their day‐to‐day lives. Participants expressed frustration and concern about the lack of effective therapies for symptom control and/or disease modification. Among those on medications, side effects were noted to be common and often exacerbated by inappropriate or off‐label prescriptions. The vast majority of participants expressed an overwhelming desire to participate in research studies of new treatment options. 183 Further insights from people with lived experience of FTD disorders can be accessed through the FTD Disorders Registry, a direct‐to‐participant registry available to researchers for data, collaboration, and recruitment support.
In a study conducted by UCB Biopharma SRL, patient interviews were conducted to obtain a better understanding of their “journey” with PSP. 184 Conceptually, interview questions addressed motor, non‐motor, and constitutional symptoms and dysfunction as well as the effects of PSP on daily life. The results showed that the current clinical journey involves cycling through the health‐care system, a delayed and terminal diagnosis, a lack of treatment, and rapid disease progression. The emotional journey was dominated by negative feelings, especially around diagnosis, with brief moments of positivity. To effectively address this, it was appreciated that patients, caregivers, and health‐care providers should be mindful of taking the diagnostic journey together and that health‐care providers communicate clearly what is known and unknown at specific time points as well as provide prognostic expectations, even if they are not optimistic. Education should be customized for all involved parties, and a coordinated effort is required from all stakeholders toward a common goal for better, more supportive care of people living with PSP and their caregivers. Recommendations included longer consultation times, closer collaboration among health‐care providers and patient organizations, and more diversified support and education for patients and caregivers.
3.5. The right trial
To identify the right trial, workshop participants discussed the options and considerations to optimize trial design for the rare disease primary tauopathies to advance to PoC. The workshop members agreed that fundamentally the right trial should demonstrate target engagement with a sensitive, validated biomarker that tracks to the mechanism of action and related biological impact on the disease. Confidence in dosing using sound PK and maximum tolerated dose and PD, where possible, serves to help bracket the dose range in early‐phase trials with allometric dose scaling as applicable from preclinical/non‐clinical models. Depending on the target population and the trial's primary and secondary outcomes, distinctive and different biomarkers may be required. Early‐phase trials, while serving as learning opportunities, still benefit from having a predefined “Go/No‐Go” matrix. The right trial should have a reliable algorithm for decision making, target engagement, and safety/tolerability, which may differ by tauopathy.
For the rare disease primary tauopathies, enrollment and sample sizes to achieve definitive clinical efficacy are likely to be a continuous challenge. Traditional designs, therefore, are not ideally suited to adapt to and learn from incoming trial data and will, in turn, be less than optimal. Fortunately, innovative design options with potential applicability for tauopathies are gaining familiarity and experience with researchers. Figure 3 shows the types of seamless designs with stage gates, basket and umbrella designs, and platform trials. Many of these have had their genesis in oncology trials. All of these have the potential to increase efficiency in early‐phase trials.
FIGURE 3.
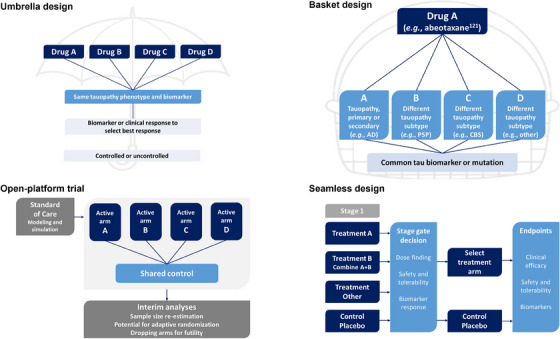
Trial schema designs. AD, Alzheimer's disease; CBS, corticobasal syndrome; PSP, progressive supranuclear palsy.
Several platform trial examples exist across neurodegenerative diseases, which serve as important points of reference. The Healey ALS Platform Trial is testing multiple compounds (regimens) concurrently in a perpetual open manner in ALS. 38 It has provisions for adding new treatments (regimens) as they are identified for testing on the platform as well as for dropping regimens when futility criteria are met. There is a shared control arm for all the active regimens enabling a higher randomization rate to active treatment of 3:1 active versus control within a given regimen. To achieve this type of open platform, there is an agreed‐to master protocol and common rules for all regimens and a shared single trial infrastructure and resources. 185 There are provisions for outside borrowing of historical or synthetic data, allowing multiple sources of information to be used within the platform. This design allows for the incorporation of all information, adaptation to accumulating evidence, and use of powerful analytic methods, even in the absence of clear PD biomarkers for agents that may be studied. All of the adaptations are planned before the trial starts with well‐defined criteria for adaptation clearly explained and key trial parameters specified (Figure 4). 186 Directed by Bayesian analyses, more patients can be adaptively randomized to better‐performing interventions. By “playing the winners,” there can be seamless transformation to confirmatory later‐stage trials. The strengths of this design come from the extensive modeling and simulation conducted before the trial launches around the natural history and disease progression, including the best endpoints and biomarkers for the phase of the disease being tested. The availability of natural history and patient‐level data for clinical trial simulation is key to this design, as is the development of disease progression models. 174 , 187 One advantage of characterizing progression rates is the ability to identify and differentiate rapid and slow disease progressors; a better understanding of rapid progression would help build confidence in pursuing PoP and PoC studies.
FIGURE 4.
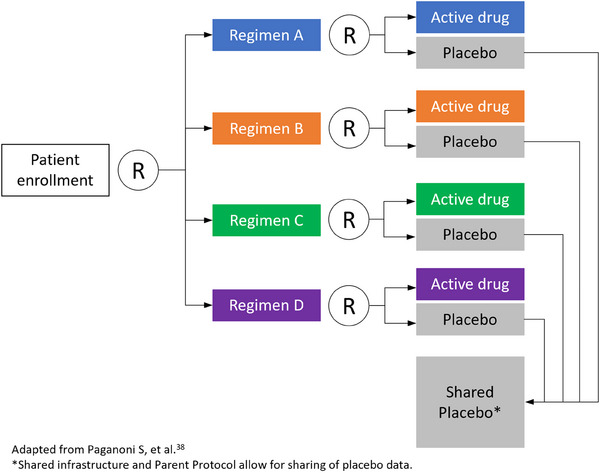
Participant flow in the Healey ALS Platform Trial. ALS, amyotrophic lateral sclerosis; R, randomization.
The challenges of this type of innovative design include the modeling and simulation needed upfront before the trial launches; the need for buy‐in from diverse stakeholders, including the health authorities (FDA) and those who contribute their compounds; the understanding that it may not be conducive to all treatment modalities (e.g., gene therapies); and that power and type 1 error of a trial do not apply. The DIAN‐TU provides another example of a platform trial that includes adaptive design and a disease progression model. 188 Within the context of autosomal dominant AD, the DIAN‐TU has successfully investigated the anti‐amyloid mAbs gantenerumab and solanezumab in those who were asymptomatic and mildly symptomatic, incorporating a seamless design in which the initial phase of biomarker outcome was used as the stage gate to the longer term clinical endpoints. 189 The DIAN‐TU Tau NexGen, 190 a secondary prevention trial in asymptomatic mutation carriers, is now poised to demonstrate biological engagement of tau and/or combination‐directed therapeutic drugs to significantly decrease insoluble tau as measured by tau PET, or soluble tau as measured in CSF and plasma, to reach PoC of potential clinical and cognitive benefit to support the transition to seamless phase 3 validation studies.
The urgency to develop more efficient clinical trial designs is growing for sporadic AD. With the approval of lecanemab, and possibly donanemab soon, it will be necessary to investigate the effects of other therapeutic modalities in combination with anti‐amyloid therapies. The number of potential combinations and control conditions suggests that traditional parallel trial designs will be prohibitively expensive and time consuming. The new National Institutes of Health‐funded AD Tau Platform Trial will be a phase 2, factorial design platform that will test at least two therapies alone or in combination with anti‐amyloid therapies in early sporadic AD with a focus on tau biomarkers for demonstrating biological PoC 191 scheduled to begin in late 2024 or 2025.
Given the poor translatability of non‐clinical models to human disease, it is uncertain which pathogenic mechanisms (and candidate therapies) may be related to which (if any) human disease. Increasingly, it is recognized that tau metabolism and likely pathogenic mechanisms vary in different tauopathies (e.g., AD and PSP). One approach to identifying the right participants (therapeutic indication) is to assess a tau therapy in multiple different tauopathies in parallel or successive clinical trials. In this paradigm, basket and umbrella trial designs (Figure 3) also offer some unique considerations for tauopathy therapeutic development. 185 Within a basket trial design, multiple tauopathy phenotypes, supported by biomarker evidence as likely being caused by underlying tau pathology, can be simultaneously treated with the same treatment. Outcomes of an early‐phase basket trial can, in turn, then include safety, tolerability, and biomarkers with comparison across the clinical phenotypes to determine which phenotype represents the best one to pursue into further development. 192 In the example of such a tauopathy basket trial, the microtubule stabilizer abeotaxane was tested and showed differential safety, tolerability, and biomarker profiles when tested in AD, PSP, and CBS. 121 These results enabled important comparative information to be identified early in development. 121 Alternatively, in an umbrella design, 185 multiple drugs can be used to treat the same tauopathy with biomarkers being used to identify which of the drugs emerges as the best drug to advance. For the primary tauopathies, one of the important rate‐limiting factors with basket designs is the dependency on robust and specific tauopathy biomarkers that are fit for purpose. By analogy to ALS, much can be learned from rapid, inexpensive trials, even in the absence of specific biomarkers, if there is sufficient power to measure an effect on a clinical endpoint. In PSP‐Richardson's syndrome, experience in mild–moderate disease with new, more powerful versions of the PSP rating scale suggests that such trials may now be feasible in this primary tauopathy.
Within‐subject or single‐case experimental designs provide an alternative design option that investigates change in individual patients as the unit of analysis rather than an aggregate change in a group of patients. This approach addresses the challenge of inherent heterogeneity by using the same individual during the pretreatment phase as the control condition. As applied in tauopathies, this n‐of‐1–based approach acquires a run‐in period (with or without placebo) with frequent repeated biomarker measures to create an optimal pretreatment profile followed by crossover to an active period of treatment. Determinations are made on the treatment response of each participant with a defined response rate of individuals defined within the trial. It also serves as an initial evaluation of PK and target engagement. This approach provides for efficient and economical screening (i.e., “rapid fail”) and early‐stage “Go/No‐Go” decisions, and establishes foundational human data to formulate intellectual property and refine the design of subsequent phase 2 and 3 trials. This approach has been incorporated into more complex designs that have been used previously for behavioral interventions within the Sequential Multiple Assignment Randomized Trial (SMART). 193 Statistical approaches include repeated measures of visual, time series, or Bayesian change analyses. Generalizability is evaluated using an analytic or metanalytic combination of single case trials.
4. PARTNERSHIPS AND ACCELERANTS
In this workshop, bottlenecks were discussed, and considerations were then proposed for accelerating the translation of discoveries made in academic laboratories into the clinical pipeline. These academic laboratories may have a promising new compound but may lack the resources or drug discovery knowledge to move it forward. Academic, not‐for‐profit foundations, lay support organizations, and industry partnerships are seen as fundamental vehicles to accelerate the transition of discoveries from academic labs (i.e., bench or basic research) into industry‐based drug development programs and successful early‐phase clinical trials (i.e., clinical trials). The end goal for academic and industry researchers is the same: to increase the capacity for good, rigorous drug discovery and development, leading eventually to new treatments for patients. Once this is established, different stakeholders can use their strengths to optimize the development process. Academic researchers excel at understanding the target profile, clinical and biomarker measures, and clinical adoption. Pharmaceutical and biotech companies rigorously test and effectively commercialize the product. It is important to understand the bottlenecks and to partner with agencies and organizations (e.g., drug discovery institutes) that can provide resources to overcome these hurdles. Academic researchers should develop their intellectual property strategy as early as possible, and academic technology transfer offices should seek support from external consultants and attorneys to ensure protection that provides the basis for proprietary development and market readiness. Finally, as a shared priority for the research community, funders could consider supporting efforts to create more and better compound libraries and to improve immediate access to these valuable resources.
5. CONCLUSION
The workshop identified important unmet needs in translational tau therapeutic research, including the need for high‐fidelity models, model‐to‐human translation biomarkers, more target mechanisms, more validated biomarkers (e.g., PD, diagnostic, predictive, response monitoring, safety), and more rigorous definitions of acceptable outcomes for “Go/No‐Go” decisions. More sensitive and tailored clinical outcomes, including digital biomarkers, are also tagged as important priority areas. When achieved and synthesized into comprehensive discovery and development programs, these advances will enable accelerated identification of new therapeutic options for patients with tauopathies.
CONFLICT OF INTEREST STATEMENT
Steven E. Arnold declares lecture honoraria from: Abbvie, Biogen, and Eisai; serving on advisory boards for: Allyx Therapeutics, Bob's Last Marathon, Cassava, Cortexyme, Sage Therapeutics, and vTv Therapeutics; consulting for: Athira, Boyle‐Shaugnessy Law, Cognito Therapeutics, M3 Biotech, Orthogonal Neuroscience, and Risen Pharmaceutical Technology; and institutional research grant support from: Abbvie, Alzheimer's Association, Alzheimer's Drug Discovery Foundation, Amylyx, Athira, Chromadex, Cyclerion Therapeutics, EIP Pharma, Janssen/Johnson & Johnson, NIH, Novartis, Seer Biosciences, and vTv Therapeutics. Clive Ballard declares grants from Synexus, reMYND, and Novo Nordisk; consultancy fees from Tau Therapeutics, Acadia Pharmaceuticals, Johnson & Johnson, Suven Life Sciences, Sunovion, Exciva, Roche, AbbVie, Orion Pharma, BioExcel, AARP, and Lilly; and honoraria from Bristol Myers Squibb, Axome Therapeutics, Tau Therapeutics, and Biogen. Dirk Beher is a co‐founder, employee, and shareholder of Asceneuron SA. Bradley F. Boeve received honorarium for SAB activities for the Tau Consortium; institutional research grant support from: Alector, Biogen, Transposon, Cognition Therapeutics, EIP Pharma, GE Healthcare; institutional NIH grant support from: P30 AG062677, U19 AG063911, R01 AG038791, U01 NS100620, U19 AG071754, U24 AG056270; institutional foundation support from: Lewy Body Dementia Association, American Brain Foundation; and institutional philanthropic support from: Mayo Clinic Dorothy and Harry T. Mangurian Jr. Lewy Body Dementia Program, the Little Family Foundation, Ted Turner Family Foundation. Adam L. Boxer received grant support from NIH U19AG063911, R01AG073482, R56AG075744, R01AG038791, RF1AG077557, R01AG071756, U01AG045390, Rainwater Charitable Foundation, Bluefield Project to Cure FTD, GHR Foundation, Alzheimer's Association, Association for Frontotemporal Degeneration, Alzheimer's Drug Discovery Foundation, UCSF Parkinson's Spectrum Disorders Center, University of California Cures AD Program; industry research support from: Biogen, Eisai, Regeneron; and did industry consulting for: AGTC, Alchemab, Alector, Amylyx, Arkuda, Arvinas, Arrowhead, Eli Lilly, Merck, Muna, Oscotec, Roche, Switch, Transposon, Wave. Jeffrey L. Cummings reports consulting for: Acadia, Alkahest, AlphaCognition, AriBio, Biogen, Cassava, Cerecin, Cortexyme, Diadem, EIP Pharma, Eisai, GemVax, Genentech, Green Valley, GAP Innovations, Grifols, Janssen, Karuna, Lilly, Lundbeck, LSP, Merck, NervGen, Novo Nordisk, Oligomerix, Ono, Otsuka, PRODEO, Prothena, ReMYND, Resverlogix, Roche, Sage Therapeutics, SignantHealth, Suven, TrueBinding, and Vaxxinity pharmaceutical, assessment, and investment companies; and grant funding from: NIGMS grant P20GM109025, NINDS grant U01NS093334, NIA grant R01AG053798, NIA grant P20AG068053, NIA grant P30AG072959, NIA grant R35AG71476, Alzheimer's Disease Drug Discovery Foundation (ADDF), Ted and Maria Quirk Endowment, and the Joy Chambers‐Grundy Endowment. Penny A. Dacks is an employee of AFTD. Kristophe Diaz is an employee of CurePSP. Colin Ewen is an employee of UCB Pharma. Howard H. Feldman reports UCSD service agreements for consulting from: Axon Neuroscience, Samus Therapeutics, SAB of the Tau Consortium (a program of the Rainwater Charitable Foundation), Novo Nordisk, Genentech (DSMB), Roche/Banner Institute (DMC), and Janssen LLC (DSMB); ADCS clinical trials grant funding from: Annovis (Posiphen), Biohaven (Troriluzole), Vivoryon (Varoglutamstat), AC Immune (ACI‐24‐1301), LuMind (LIFE‐DSR); research funding from: NIA/NIH (U19 AG010483, P30 AG062429, R01 AG061146‐01, 1R56 AG069130‐01, R01 AG051618, 1R01AG070353‐01), CIHR (CAN‐163902), Alzheimer's Association (SG‐20‐690388‐PEACE AD), UC Cures (BRD‐16‐501530), Brain Canada (4469); patent royalties from: Detecting and Treating Dementia (PGRN) Serial Number 12/3‐2691 U.S. Patent No. PCT/US2007/07008. Washington, DC: U.S. Patent and Trademark Office; and funding support from: The USC UCSD Epstein Family Alzheimer Research Collaboration. Brian Fiske works for The Michael J. Fox Foundation for Parkinson's Research and has no conflicts of interest to disclose. M. Isabel Gonzalez has no conflicts of interest to disclose. Glenn A. Harris works for the Rainwater Charitable Foundation, a sponsor of the workshop. Beth J. Hoffman is a full‐time employee of Origami Therapeutics, Inc.; receives grant support from: SBIR Phase I Award No. NINDS R43NS127716; is affiliated with: Huntington's Disease Society of America Board of Trustees; and receives honoraria from: Rainwater Charitable Foundation Tau Consortium, Biofrontera (NASDAQ: BFRI) Board of Directors. Doo Yeon Kim receives grant support from: NIH 1R01AG071496‐01, R01AG062547‐01, 1R01AG057635‐01A1, 1R01AG061891‐01A1, 1R01AG062547‐01, Cure Alzheimer's Fund, Coins for Alzheimer's Research Trust; is a co‐inventor of 3D Alzheimer's in‐a‐dish model; the technology and cell lines are licensed by Millipore/Sigma; and received patent loyalty from Acta Pharmaceutical for the drug candidates detected in cellular model screening. David S. Knopman serves on a data safety monitoring board for the Dominantly Inherited Alzheimer Network Treatment Unit study; was an investigator in Alzheimer's disease clinical trials sponsored by Biogen, Lilly Pharmaceuticals, and the University of Southern California, both of which have ended, and is currently an investigator in a trial in frontotemporal degeneration with Alector; has served as a consultant for Roche, AriBio, Linus Health, Biovie, and Alzeca Biosciences but receives no personal compensation; and receives funding from the NIH. Terina N. Martinez is an employee of the Critical Path Institute and has no conflicts of interest to disclose. Patrick C. May received honoraria for SAB activities for the Tau Consortium (a program of the Rainwater Charitable Foundation) and Cure Alzheimer's Fund. Eric McDade received research funding from: NIA (K23AG046363; U01AG059798), Anonymous Foundation, and GHR; institutional support from: DIAN‐TU Pharma Consortium, Eli Lilly, and Hoffmann La‐Roche; did speaker engagements for: Eli Lilly and Esai; served on an advisory board and DSMB for: Alector, Alzamend, Hoffmann La‐Roche, Merck, and Eli Lilly and is a co‐inventor of the “Methods of diagnosing AD with phosphorylation changes” technology licensed by Washington University to C2N Diagnostics (Washington University holds 5% equity in C2N; Through these relationships, Washington University and Dr. McDade are entitled to receive royalties from the license agreement with C2N). Laura K. Nisenbaum is an employee of the Alzheimer's Drug Discovery Foundation. Jose‐Alberto Palma is an employee of Novartis. Melanie Quintana is an employee of Berry Consultants. Gil D. Rabinovici received grant support from: NIH/NIA P30 AG062422, R35 AG072362, U01 AG057195, R56‐AG075744, U01‐AG082350, Alzheimer's Association, American College of Radiology (contributions from Eli Lilly, GE Healthcare, Life Molecular Imaging), Rainwater Charitable Foundation, Genentech; is a paid consultant for Alector, Eli Lilly, Merck; served on a DSMB for Johnson & Johnson; and is an associate editor for JAMA Neurology. Jonathan D. Rohrer has provided consultancy or been on an advisory board for Novartis, Wave Life Sciences, Prevail, Alector, Aviado Bio, Takeda, Arkuda Therapeutics, and Denali Therapeutics. Amy Rommel works for the Rainwater Charitable Foundation, a sponsor of the workshop. Howard J. Rosen is a consultant for Alchemab, Prevail, Genentech, Eisai; received grant support from: NIH U19 AG063911, P30 AG062422, P01 AG019724, R01 AG056749, CA Department of Public Health 18‐10612, 09‐11410. Adam M. Staffaroni reports consulting for Alector, Passage Bio, Prevail Therapeutics/Lilly, and Takeda; recent/current funding from: Association for Frontotemporal Dementia, ALS Association, Bluefield Project to Cure FTD, Larry L. Hillblom Foundation, and NIH‐NIA (K23 AG061253; RF1AG077557); and serves on ADDF Scientific Review Board. Stacey J. Sukoff Rizzo reports research funding from: NIA/NIH R01AG067289, U54AG054345, U19AG074866, P30AG024827, R24AG073190, U54AG065181, U54AG065187, R13 AG060708, RF1NS117486, U01AG079828, WoodNext Foundation, Hevolution; consulting for: Hager Biosciences, GenPrex Inc., Sage Therapeutics, Momentum Biosciences (shareholder); and travel/conference support from: Rainwater Charitable Foundation, Alzheimer's Association. Rudolph E. Tanzi has no conflicts of interest to disclose. Paul R. Territo reports research funding from: NIA/NIH U54AG054345, R01AG067289, R21AG078575, R21AG083791, R21AG083891, R13AG060708, Alzheimer's Association ALZDISCOVERY‐1051148. Matthew D. Troyer is an employee of Denali Therapeutics. Henrik Zetterberg serves on scientific advisory boards and/or as a consultant for Abbvie, Acumen, Alector, Alzinova, ALZPath, Amylyx, Annexon, Apellis, Artery Therapeutics, AZTherapies, Cognito Therapeutics, CogRx, Denali, Eisai, Merry Life, Nervgen, Novo Nordisk, Optoceutics, Passage Bio, Pinteon Therapeutics, Prothena, Red Abbey Labs, reMYND, Roche, Samumed, Siemens Healthineers, Triplet Therapeutics, and Wave; has given lectures in symposia sponsored by Alzecure, Biogen, Cellectricon, Fujirebio, Lilly, Novo Nordisk, and Roche; and is a co‐founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program (outside submitted work). Nick K. Ziogas is a prior employee and owned stock at Biogen and is a current employee and owns stock at Bristol Myers Squibb. Author disclosures are available in the supporting information.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
The Rainwater Charitable Foundation provided sponsorship and financial support for this standalone workshop. The authors would like to thank Linda Goldstein, PhD, ISMPP CMPP,™ from The Write Source MSC, for attending the workshop, collating notes from presenters and discussions, and providing medical writing assistance in development of the manuscript. Medical writing assistance was supported by the Rainwater Charitable Foundation.
Feldman HH, Cummings JL, Boxer AL, et al. A framework for translating tauopathy therapeutics: Drug discovery to clinical trials. Alzheimer's Dement. 2024;20:8129–8152. 10.1002/alz.14250
Dr. Ziogas was at Biogen through September 22, 2023; he is now at Bristol Myers Squibb.
REFERENCES
- 1. Goedert M, Wischik CM, Crowther RA, Walker JE, Klug A. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule‐associated protein tau. Proc Natl Acad Sci U S A. 1988;85(11):4051‐4055. doi: 10.1073/pnas.85.11.4051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wischik CM, Novak M, Thogersen HC, et al. Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci U S A. 1988;85(12):4506‐4510. doi: 10.1073/pnas.85.12.4506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Orr ME, Sullivan AC, Frost B. A brief overview of tauopathy: causes, consequences, and therapeutic strategies. Trends Pharmacol Sci. 2017;38(7):637‐648. doi: 10.1016/j.tips.2017.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boeve BF, Boxer AL, Kumfor F, Pijnenburg Y, Rohrer JD. Advances and controversies in frontotemporal dementia: diagnosis, biomarkers, and therapeutic considerations. Lancet Neurol. 2022;21(3):258‐272. doi: 10.1016/S1474-4422(21)00341-0 [DOI] [PubMed] [Google Scholar]
- 5. Zhang X, Gao F, Wang D, et al. Tau pathology in Parkinson's disease. Front Neurol. 2018;9:809. doi: 10.3389/fneur.2018.00809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. World Health Organization . Dementia. WHO. https://www.who.int/health‐topics/dementia#tab=tab_1 [Google Scholar]
- 7. Leroy M, Bertoux M, Skrobala E, et al. Characteristics and progression of patients with frontotemporal dementia in a regional memory clinic network. Alzheimers Res Ther. 2021;13(1):19. doi: 10.1186/s13195-020-00753-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Alzheimer's Association . 2022 Alzheimer's Disease Facts and Figures. https://www.alz.org/alzheimers‐dementia/facts‐figures
- 9. Turcano P, Stang CD, Mielke MM, et al. Incidence of frontotemporal disorders in Olmsted County: a population‐based study. Alzheimers Dement. 2020;16(3):482‐490. doi: 10.1016/j.jalz.2019.08.199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Coyle‐Gilchrist IT, Dick KM, Patterson K, et al. Prevalence, characteristics, and survival of frontotemporal lobar degeneration syndromes. Neurology. 2016;86(18):1736‐1743. doi: 10.1212/WNL.0000000000002638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Smith DH, Johnson VE, Trojanowski JQ, Stewart W. Chronic traumatic encephalopathy—confusion and controversies. Nat Rev Neurol. 2019;15(3):179‐183. doi: 10.1038/s41582-018-0114-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. U.S. Food and Drug Administration . Rare Diseases: Common Issues in Drug Development Guidance for Industry. Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research; 2019. https://www.fda.gov/media/119739/download [Google Scholar]
- 13. Mummery CJ, Borjesson‐Hanson A, Blackburn DJ, et al. Tau‐targeting antisense oligonucleotide MAPT(Rx) in mild Alzheimer's disease: a phase 1b, randomized, placebo‐controlled trial. Nat Med. 2023;29(6):1437‐1447. doi: 10.1038/s41591-023-02326-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Plotkin SS, Cashman NR. Passive immunotherapies targeting Abeta and tau in Alzheimer's disease. Neurobiol Dis. 2020;144:105010. doi: 10.1016/j.nbd.2020.105010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. National Institute of Neurological Disorders and Stroke . ADRD Summit 2022 Report to the National Advisory Neurological Disorders and Stroke Council. National Institutes of Health; 2022. https://www.ninds.nih.gov/news-events/events/adrd-summit-2022 [Google Scholar]
- 16. Boxer AL, Gold M, Feldman H, et al. New directions in clinical trials for frontotemporal lobar degeneration: methods and outcome measures. Alzheimers Dement. 2020;16(1):131‐143. doi: 10.1016/j.jalz.2019.06.4956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dubois B, Feldman HH, Jacova C, et al. Revising the definition of Alzheimer's disease: a new lexicon. Lancet Neurol. 2010;9(11):1118‐1127. doi: 10.1016/S1474-4422(10)70223-4 [DOI] [PubMed] [Google Scholar]
- 18. Friedman LG, McKeehan N, Hara Y, et al. Value‐generating exploratory trials in neurodegenerative dementias. Neurology. 2021;96(20):944‐954. doi: 10.1212/WNL.0000000000011774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Merchant KM, Cedarbaum JM, Brundin P, et al. A proposed roadmap for parkinson's disease proof of concept clinical trials investigating compounds targeting alpha‐synuclein. J Parkinsons Dis. 2019;9(1):31‐61. doi: 10.3233/JPD-181471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nicholson KA, Cudkowicz ME, Berry JD. Clinical trial designs in amyotrophic lateral sclerosis: does one design fit all? Neurotherapeutics. 2015;12(2):376‐383. doi: 10.1007/s13311-015-0341-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cummings J, Feldman HH, Scheltens P. The “rights” of precision drug development for Alzheimer's disease. Alzheimers Res Ther. 2019;11(1):76. doi: 10.1186/s13195-019-0529-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Paganoni S, Hendrix S, Dickson SP, et al. Effect of sodium phenylbutyrate/taurursodiol on tracheostomy/ventilation‐free survival and hospitalisation in amyotrophic lateral sclerosis: long‐term results from the CENTAUR trial. J Neurol Neurosurg Psychiatry. 2022;93(8):871‐875. doi: 10.1136/jnnp-2022-329024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Miyagawa T, Brushaber D, Syrjanen J, et al. Use of the CDR(R) plus NACC FTLD in mild FTLD: data from the ARTFL/LEFFTDS consortium. Alzheimers Dement. 2020;16(1):79‐90. doi: 10.1016/j.jalz.2019.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cummings JL, Mega M, Gray K, Rosenberg‐Thompson S, Carusi DA, Gornbein J. The Neuropsychiatric Inventory: comprehensive assessment of psychopathology in dementia. Neurology. 1994;44(12):2308‐2314. doi: 10.1212/wnl.44.12.2308 [DOI] [PubMed] [Google Scholar]
- 25. Budd Haeberlein S, Aisen PS, Barkhof F, et al. Two randomized phase 3 studies of aducanumab in early Alzheimer's disease. J Prev Alzheimers Dis. 2022;9(2):197‐210. doi: 10.14283/jpad.2022.30 [DOI] [PubMed] [Google Scholar]
- 26. van Dyck CH, Swanson CJ, Aisen P, et al. Lecanemab in early Alzheimer's disease. N Engl J Med. 2023;388(1):9‐21. doi: 10.1056/NEJMoa2212948 [DOI] [PubMed] [Google Scholar]
- 27. Sims JR, Zimmer JA, Evans CD, et al. Donanemab in Early Symptomatic Alzheimer Disease: the TRAILBLAZER‐ALZ 2 Randomized Clinical Trial. JAMA. 2023;330(6):512‐527. doi: 10.1001/jama.2023.13239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ballard C, Aarsland D, Cummings J, et al. Drug repositioning and repurposing for Alzheimer disease. Nat Rev Neurol. 2020;16(12):661‐673. doi: 10.1038/s41582-020-0397-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Karran E, Hardy J. A critique of the drug discovery and phase 3 clinical programs targeting the amyloid hypothesis for Alzheimer disease. Ann Neurol. 2014;76(2):185‐205. doi: 10.1002/ana.24188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Toyn JH, Ahlijanian MK. Interpreting Alzheimer's disease clinical trials in light of the effects on amyloid‐beta. Alzheimers Res Ther. 2014;6(2):14. doi: 10.1186/alzrt244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation in the alpha‐synuclein gene identified in families with Parkinson's disease. Science. 1997;276(5321):2045‐2047. doi: 10.1126/science.276.5321.2045 [DOI] [PubMed] [Google Scholar]
- 32. FDA‐NIH Biomarker Working Group . BEST (Biomarkers, EndpointS, and other Tools) Resource. U.S. Food and Drug Administration and National Institutes of Health; 2016. https://pubmed.ncbi.nlm.nih.gov/27010052/ [PubMed] [Google Scholar]
- 33. Pagano G, Taylor KI, Anzures‐Cabrera J, et al. Trial of Prasinezumab in Early‐stage Parkinson's disease. N Engl J Med. 2022;387(5):421‐432. doi: 10.1056/NEJMoa2202867 [DOI] [PubMed] [Google Scholar]
- 34. Lang AE, Siderowf AD, Macklin EA, et al. Trial of Cinpanemab in Early Parkinson's disease. N Engl J Med. 2022;387(5):408‐420. doi: 10.1056/NEJMoa2203395 [DOI] [PubMed] [Google Scholar]
- 35. Hutchison RM, Fraser K, Yang M, et al. Cinpanemab in Early Parkinson Disease: evaluation of biomarker results from the phase 2 SPARK clinical trial. Neurology. 2024;102(5):e209137. doi: 10.1212/WNL.0000000000209137 [DOI] [PubMed] [Google Scholar]
- 36. Kiernan MC, Vucic S, Talbot K, et al. Improving clinical trial outcomes in amyotrophic lateral sclerosis. Nat Rev Neurol. 2021;17(2):104‐118. doi: 10.1038/s41582-020-00434-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. van den Berg LH, Sorenson E, Gronseth G, et al. Revised Airlie House consensus guidelines for design and implementation of ALS clinical trials. Neurology. 2019;92(14):e1610‐e1623. doi: 10.1212/WNL.0000000000007242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Paganoni S, Berry JD, Quintana M, et al. Adaptive platform trials to transform amyotrophic lateral sclerosis therapy development. Ann Neurol. 2022;91(2):165‐175. doi: 10.1002/ana.26285 [DOI] [PubMed] [Google Scholar]
- 39. Aisen PS, Bateman RJ, Carrillo M, et al. Platform trials to expedite drug development in Alzheimer's disease: a report from the EU/US CTAD Task Force. J Prev Alzheimers Dis. 2021;8(3):306‐312. doi: 10.14283/jpad.2021.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bedlack RS, Pastula DM, Welsh E, Pulley D, Cudkowicz ME. Scrutinizing enrollment in ALS clinical trials: room for improvement? Amyotroph Lateral Scler. 2008;9(5):257‐265. doi: 10.1080/17482960802195913 [DOI] [PubMed] [Google Scholar]
- 41. Miller TM, Cudkowicz ME, Genge A, et al. Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N Engl J Med. 2022;387(12):1099‐1110. doi: 10.1056/NEJMoa2204705 [DOI] [PubMed] [Google Scholar]
- 42. Paganoni S, Macklin EA, Hendrix S, et al. Trial of sodium phenylbutyrate‐taurursodiol for amyotrophic lateral sclerosis. N Engl J Med. 2020;383(10):919‐930. doi: 10.1056/NEJMoa1916945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Thomas JD, Oliveira R, Sznajder LJ, Swanson MS. Myotonic dystrophy and developmental regulation of RNA processing. Compr Physiol. 2018;8(2):509‐553. doi: 10.1002/cphy.c170002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. De Serres‐Berard T, Pierre M, Chahine M, Puymirat J. Deciphering the mechanisms underlying brain alterations and cognitive impairment in congenital myotonic dystrophy. Neurobiol Dis. 2021;160:105532. doi: 10.1016/j.nbd.2021.105532 [DOI] [PubMed] [Google Scholar]
- 45. Caillet‐Boudin ML, Fernandez‐Gomez FJ, Tran H, Dhaenens CM, Buee L, Sergeant N. Brain pathology in myotonic dystrophy: when tauopathy meets spliceopathy and RNAopathy. Front Mol Neurosci. 2014;6:57. doi: 10.3389/fnmol.2013.00057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Laforce RJ, Dallaire‐Theroux C, Racine AM, et al. Tau positron emission tomography, cerebrospinal fluid and plasma biomarkers of neurodegeneration, and neurocognitive testing: an exploratory study of participants with myotonic dystrophy type 1. J Neurol. 2022;269(7):3579‐3587. doi: 10.1007/s00415-022-10970-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liu AJ, Lusk JB, Ervin J, Burke J, O'Brien R, Wang SJ. Tuberous sclerosis complex is a novel, amyloid‐independent tauopathy associated with elevated phosphorylated 3R/4R tau aggregation. Acta Neuropathol Commun. 2022;10(1):27. doi: 10.1186/s40478-022-01330-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. de Vries PJ, Whittemore VH, Leclezio L, et al. Tuberous sclerosis associated neuropsychiatric disorders (TAND) and the TAND Checklist. Pediatr Neurol. 2015;52(1):25‐35. doi: 10.1016/j.pediatrneurol.2014.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Northrup H, Krueger DA, International Tuberous Sclerosis Complex Consensus G. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 Iinternational Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49(4):243‐254. doi: 10.1016/j.pediatrneurol.2013.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kovacs GG, Ferrer I, Grinberg LT, et al. Aging‐related tau astrogliopathy (ARTAG): harmonized evaluation strategy. Acta Neuropathol. 2016;131(1):87‐102. doi: 10.1007/s00401-015-1509-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Riku Y, Yoshida M, Iwasaki Y, Sobue G, Katsuno M, Ishigaki S. TDP‐43 proteinopathy and tauopathy: do they have pathomechanistic links? Int J Mol Sci. 2022;23(24):15755. doi: 10.3390/ijms232415755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tome SO, Tsaka G, Ronisz A, et al. TDP‐43 pathology is associated with increased tau burdens and seeding. Mol Neurodegener. 2023;18(1):71. doi: 10.1186/s13024-023-00653-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cummings JL, Gonzalez MI, Pritchard MC, May PC, Toledo‐Sherman LM, Harris GA. The therapeutic landscape of tauopathies: challenges and prospects. Alzheimers Res Ther. 2023;15(1):168. doi: 10.1186/s13195-023-01321-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li C, Gotz J. Tau‐based therapies in neurodegeneration: opportunities and challenges. Nat Rev Drug Discov. 2017;16(12):863‐883. doi: 10.1038/nrd.2017.155 [DOI] [PubMed] [Google Scholar]
- 55. Gerson JE, Castillo‐Carranza DL, Kayed R. Advances in therapeutics for neurodegenerative tauopathies: moving toward the specific targeting of the most toxic tau species. ACS Chem Neurosci. 2014;5(9):752‐769. doi: 10.1021/cn500143n [DOI] [PubMed] [Google Scholar]
- 56. Denali Therapeutics . Genetic pathway potential. https://www.denalitherapeutics.com/science/pathway
- 57. ALZFORUM . Research Models. Accessed November 4, 2022. https://www.alzforum.org/research‐models/search?species=&diseases=&genes%5B%5D=33511&types=&keywords‐entry=&keywords=tau#results
- 58. Sahara N, Yanai R. Limitations of human tau‐expressing mouse models and novel approaches of mouse modeling for tauopathy. Front Neurosci. 2023;17:1149761. doi: 10.3389/fnins.2023.1149761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chung DC, Roemer S, Petrucelli L, Dickson DW. Cellular and pathological heterogeneity of primary tauopathies. Mol Neurodegener. 2021;16(1):57. doi: 10.1186/s13024-021-00476-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Liu F, Gong CX. Tau exon 10 alternative splicing and tauopathies. Mol Neurodegener. 2008;3:8. doi: 10.1186/1750-1326-3-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wray S. Modeling tau pathology in human stem cell derived neurons. Brain Pathol. 2017;27(4):525‐529. doi: 10.1111/bpa.12521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gotz J, Ittner LM. Animal models of Alzheimer's disease and frontotemporal dementia. Nat Rev Neurosci. 2008;9(7):532‐544. doi: 10.1038/nrn2420 [DOI] [PubMed] [Google Scholar]
- 63. Irwin DJ. Tauopathies as clinicopathological entities. Parkinsonism Relat Disord. 2016;22(Suppl 1):S29‐S33. doi: 10.1016/j.parkreldis.2015.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lee VM, Kenyon TK, Trojanowski JQ. Transgenic animal models of tauopathies. Biochim Biophys Acta. 2005;1739(2‐3):251‐259. doi: 10.1016/j.bbadis.2004.06.014 [DOI] [PubMed] [Google Scholar]
- 65. Ando K, Leroy K, Heraud C, et al. Accelerated human mutant tau aggregation by knocking out murine tau in a transgenic mouse model. Am J Pathol. 2011;178(2):803‐816. doi: 10.1016/j.ajpath.2010.10.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wegmann S, Maury EA, Kirk MJ, et al. Removing endogenous tau does not prevent tau propagation yet reduces its neurotoxicity. EMBO J. 2015;34(24):3028‐3041. doi: 10.15252/embj.201592748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Karch CM, Kao AW, Karydas A, et al. A comprehensive resource for induced pluripotent stem cells from patients with primary tauopathies. Stem Cell Reports. 2019;13(5):939‐955. doi: 10.1016/j.stemcr.2019.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Mertens J, Paquola ACM, Ku M, et al. Directly reprogrammed human neurons retain aging‐associated transcriptomic signatures and reveal age‐related nucleocytoplasmic defects. Cell Stem Cell. 2015;17(6):705‐718. doi: 10.1016/j.stem.2015.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Penney J, Ralvenius WT, Tsai LH. Modeling Alzheimer's disease with iPSC‐derived brain cells. Mol Psychiatry. 2020;25(1):148‐167. doi: 10.1038/s41380-019-0468-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663‐676. doi: 10.1016/j.cell.2006.07.024 [DOI] [PubMed] [Google Scholar]
- 71. Kuhn R, Mahajan A, Canoll P, Hargus G. Human induced pluripotent stem cell models of frontotemporal dementia with tau pathology. Front Cell Dev Biol. 2021;9:766773. doi: 10.3389/fcell.2021.766773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Schoch KM, DeVos SL, Miller RL, et al. Increased 4R‐tau induces pathological changes in a human‐tau mouse model. Neuron. 2016;90(5):941‐947. doi: 10.1016/j.neuron.2016.04.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Iovino M, Agathou S, Gonzalez‐Rueda A, et al. Early maturation and distinct tau pathology in induced pluripotent stem cell‐derived neurons from patients with MAPT mutations. Brain. 2015;138(11):3345‐3359. doi: 10.1093/brain/awv222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sposito T, Preza E, Mahoney CJ, et al. Developmental regulation of tau splicing is disrupted in stem cell‐derived neurons from frontotemporal dementia patients with the 10 + 16 splice‐site mutation in MAPT. Hum Mol Genet. 2015;24(18):5260‐5269. doi: 10.1093/hmg/ddv246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Israel MA, Yuan SH, Bardy C, et al. Probing sporadic and familial Alzheimer's disease using induced pluripotent stem cells. Nature. 2012;482(7384):216‐220. doi: 10.1038/nature10821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Moore S, Evans LD, Andersson T, et al. APP metabolism regulates tau proteostasis in human cerebral cortex neurons. Cell Rep. 2015;11(5):689‐696. doi: 10.1016/j.celrep.2015.03.068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Choi SH, Kim YH, Hebisch M, et al. A three‐dimensional human neural cell culture model of Alzheimer's disease. Nature. 2014;515(7526):274‐278. doi: 10.1038/nature13800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lin YT, Seo J, Gao F, et al. APOE4 causes widespread molecular and cellular alterations associated with alzheimer's disease phenotypes in human iPSC‐Derived brain cell types. Neuron. 2018;98(6):1141‐1154.e7. doi: 10.1016/j.neuron.2018.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Lomoio S, Pandey RS, Rouleau N, et al. 3D bioengineered neural tissue generated from patient‐derived iPSCs mimics time‐dependent phenotypes and transcriptional features of Alzheimer's disease. Mol Psychiatry. 2023;28(12):5390‐5401. doi: 10.1038/s41380-023-02147-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Miguel L, Rovelet‐Lecrux A, Feyeux M, et al. Detection of all adult Tau isoforms in a 3D culture model of iPSC‐derived neurons. Stem Cell Res. 2019;40:101541. doi: 10.1016/j.scr.2019.101541 [DOI] [PubMed] [Google Scholar]
- 81. Raja WK, Mungenast AE, Lin YT, et al. Self‐organizing 3D human neural tissue derived from induced pluripotent stem cells recapitulate Alzheimer's disease phenotypes. PLoS One. 2016;11(9):e0161969. doi: 10.1371/journal.pone.0161969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Seidel D, Krinke D, Jahnke HG, et al. Induced tauopathy in a novel 3D‐culture model mediates neurodegenerative processes: a real‐time study on biochips. PLoS One. 2012;7(11):e49150. doi: 10.1371/journal.pone.0049150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Bowles KR, Silva MC, Whitney K, et al. ELAVL4, splicing, and glutamatergic dysfunction precede neuron loss in MAPT mutation cerebral organoids. Cell. 2021;184(17):4547‐4563.e17 doi: 10.1016/j.cell.2021.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lovejoy C, Alatza A, Arber C, et al. Engineered cerebral organoids recapitulate adult Tau expression and disease‐relevant changes in Tau splicing. Research Square. 2020. doi: 10.21203/rs.3.rs-37620/v1 [DOI] [Google Scholar]
- 85. Capano LS, Sato C, Ficulle E, et al. Recapitulation of endogenous 4R tau expression and formation of insoluble tau in directly reprogrammed human neurons. Cell Stem Cell. 2022;29(6):918‐932.e8. doi: 10.1016/j.stem.2022.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Oakley DH, Klickstein N, Commins C, et al. Continuous monitoring of tau‐induced neurotoxicity in patient‐derived iPSC‐neurons. J Neurosci. 2021;41(19):4335‐4348. doi: 10.1523/JNEUROSCI.2590-20.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Usenovic M, Niroomand S, Drolet RE, et al. Internalized tau oligomers cause neurodegeneration by inducing accumulation of pathogenic tau in human neurons derived from induced pluripotent stem cells. J Neurosci. 2015;35(42):14234‐14250. doi: 10.1523/JNEUROSCI.1523-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Verheyen A, Diels A, Dijkmans J, et al. Using human iPSC‐derived neurons to model TAU aggregation. PLoS One. 2015;10(12):e0146127. doi: 10.1371/journal.pone.0146127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Verheyen A, Diels A, Reumers J, et al. Genetically engineered iPSC‐Derived FTDP‐17 MAPT neurons display mutation‐specific neurodegenerative and neurodevelopmental phenotypes. Stem Cell Reports. 2018;11(2):363‐379. doi: 10.1016/j.stemcr.2018.06.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Chen X, Firulyova M, Manis M, et al. Microglia‐mediated T cell infiltration drives neurodegeneration in tauopathy. Nature. 2023;615(7953):668‐677. doi: 10.1038/s41586-023-05788-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Perea JR, Llorens‐Martin M, Avila J, Bolos M. The role of microglia in the spread of tau: relevance for tauopathies. Front Cell Neurosci. 2018;12:172. doi: 10.3389/fncel.2018.00172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Shi Y, Manis M, Long J, et al. Microglia drive APOE‐dependent neurodegeneration in a tauopathy mouse model. J Exp Med. 2019;216(11):2546‐2561. doi: 10.1084/jem.20190980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Sukoff Rizzo SJ, Homanics G, Schaeffer DJ, et al. Bridging the rodent to human translational gap: marmosets as model systems for the study of Alzheimer's disease. Alzheimers Dement (N Y). 2023;9(3):e12417. doi: 10.1002/trc2.12417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. MODEL‐AD: Model Organism Development for Late Onset Alzheimer's Disease. Accessed November 4, 2022. https://model‐ad.org
- 95. Oblak AL, Forner S, Territo PR, et al. Model organism development and evaluation for late‐onset Alzheimer's disease: MODEL‐AD. Alzheimers Dement (N Y). 2020;6(1):e12110. doi: 10.1002/trc2.12110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Sukoff Rizzo SJ, Masters A, Onos KD, et al. Improving preclinical to clinical translation in Alzheimer's disease research. Alzheimers Dement (N Y). 2020;6(1):e12038. doi: 10.1002/trc2.12038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Quinney SK, Murugesh K, Oblak A, et al. STOP‐AD portal: selecting the optimal pharmaceutical for preclinical drug testing in Alzheimer's disease. Alzheimers Dement. 2023;19(11):5289‐5295. doi: 10.1002/alz.13108 [DOI] [PubMed] [Google Scholar]
- 98.Percie du Sert N, Ahluwalia A, Alam S, et al. Reporting animal research: explanation and elaboration for the ARRIVE guidelines 2.0. PLoS Biol. 2020;18(7):e3000411. doi: 10.1371/journal.pbio.3000411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Percie du Sert N, Hurst V, Ahluwalia A, et al. The ARRIVE guidelines 2.0: updated guidelines for reporting animal research. PLoS Biol. 2020;18(7):e3000410. doi: 10.1371/journal.pbio.3000410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Braak H, Braak E. Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol. 1991;82(4):239‐259. doi: 10.1007/BF00308809 [DOI] [PubMed] [Google Scholar]
- 101. Chang CW, Shao E, Mucke L. Tau: enabler of diverse brain disorders and target of rapidly evolving therapeutic strategies. Science. 2021;371(6532):eabb8255. doi: 10.1126/science.abb8255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Gordon BA, Blazey TM, Christensen J, et al. Tau PET in autosomal dominant Alzheimer's disease: relationship with cognition, dementia and other biomarkers. Brain. 2019;142(4):1063‐1076. doi: 10.1093/brain/awz019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Ossenkoppele R, Smith R, Mattsson‐Carlgren N, et al. Accuracy of tau positron emission tomography as a prognostic marker in preclinical and prodromal Alzheimer disease: a head‐to‐head comparison against amyloid positron emission tomography and magnetic resonance imaging. JAMA Neurol. 2021;78(8):961‐971. doi: 10.1001/jamaneurol.2021.1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Vogel JW, Young AL, Oxtoby NP, et al. Four distinct trajectories of tau deposition identified in Alzheimer's disease. Nat Med. 2021;27(5):871‐881. doi: 10.1038/s41591-021-01309-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Sanchez JS, Becker JA, Jacobs HIL, et al. The cortical origin and initial spread of medial temporal tauopathy in Alzheimer's disease assessed with positron emission tomography. Sci Transl Med. 2021;13(577):eabc0655. doi: 10.1126/scitranslmed.abc0655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. La Joie R, Visani AV, Baker SL, et al. Prospective longitudinal atrophy in Alzheimer's disease correlates with the intensity and topography of baseline tau‐PET. Sci Transl Med. 2020;12(524):eaau5732. doi: 10.1126/scitranslmed.aau5732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Boxer AL, Yu JT, Golbe LI, Litvan I, Lang AE, Hoglinger GU. Advances in progressive supranuclear palsy: new diagnostic criteria, biomarkers, and therapeutic approaches. Lancet Neurol. 2017;16(7):552‐563. doi: 10.1016/S1474-4422(17)30157-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Jack CR Jr, Bennett DA, Blennow K, et al. NIA‐AA Research Framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14(4):535‐562. doi: 10.1016/j.jalz.2018.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Scheltens P, De Strooper B, Kivipelto M, et al. Alzheimer's disease. Lancet. 2021;397(10284):1577‐1590. doi: 10.1016/S0140-6736(20)32205-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Wang Y, Mandelkow E. Tau in physiology and pathology. Nat Rev Neurosci. 2016;17(1):5‐21. doi: 10.1038/nrn.2015.1 [DOI] [PubMed] [Google Scholar]
- 111. Yanamandra K, Jiang H, Mahan TE, et al. Anti‐tau antibody reduces insoluble tau and decreases brain atrophy. Ann Clin Transl Neurol. 2015;2(3):278‐288. doi: 10.1002/acn3.176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Yanamandra K, Kfoury N, Jiang H, et al. Anti‐tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron. 2013;80(2):402‐414. doi: 10.1016/j.neuron.2013.07.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Florian H, Wang D, Arnold SE, et al. Tilavonemab in early Alzheimer's disease: results from a phase 2, randomized, double‐blind study. Brain. 2023;146(6):2275‐2284. doi: 10.1093/brain/awad024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Bright J, Hussain S, Dang V, et al. Human secreted tau increases amyloid‐beta production. Neurobiol Aging. 2015;36(2):693‐709. doi: 10.1016/j.neurobiolaging.2014.09.007 [DOI] [PubMed] [Google Scholar]
- 115. Sopko R, Golonzhka O, Arndt J, et al. Characterization of tau binding by gosuranemab. Neurobiol Dis. 2020;146:105120. doi: 10.1016/j.nbd.2020.105120 [DOI] [PubMed] [Google Scholar]
- 116. Ayalon G, Lee SH, Adolfsson O, et al. Antibody semorinemab reduces tau pathology in a transgenic mouse model and engages tau in patients with Alzheimer's disease. Sci Transl Med. 2021;13(593):eabb2639. doi: 10.1126/scitranslmed.abb2639 [DOI] [PubMed] [Google Scholar]
- 117. Lee SH, Le Pichon CE, Adolfsson O, et al. Antibody‐mediated targeting of tau in vivo does not require effector function and microglial engagement. Cell Rep. 2016;16(6):1690‐1700. doi: 10.1016/j.celrep.2016.06.099 [DOI] [PubMed] [Google Scholar]
- 118. Teng E, Manser PT, Shah M, et al. The use of episodic memory tests for screening in clinical trials for early Alzheimer's disease: a comparison of the free and cued selective reminding test (FCSRT) and the repeatable battery for the assessment of neuropsychological status (RBANS). J Prev Alzheimers Dis. 2023;10(1):41‐49. doi: 10.14283/jpad.2022.101 [DOI] [PubMed] [Google Scholar]
- 119. Dam T, Boxer AL, Golbe LI, et al. Safety and efficacy of anti‐tau monoclonal antibody gosuranemab in progressive supranuclear palsy: a phase 2, randomized, placebo‐controlled trial. Nat Med. 2021;27(8):1451‐1457. doi: 10.1038/s41591-021-01455-x [DOI] [PubMed] [Google Scholar]
- 120. Hoglinger GU, Litvan I, Mendonca N, et al. Safety and efficacy of tilavonemab in progressive supranuclear palsy: a phase 2, randomised, placebo‐controlled trial. Lancet Neurol. 2021;20(3):182‐192. doi: 10.1016/S1474-4422(20)30489-0 [DOI] [PubMed] [Google Scholar]
- 121. Tsai RM, Miller Z, Koestler M, et al. Reactions to multiple ascending doses of the microtubule stabilizer TPI‐287 in patients with Alzheimer disease, progressive supranuclear palsy, and corticobasal syndrome: a randomized clinical trial. JAMA Neurol. 2020;77(2):215‐224. doi: 10.1001/jamaneurol.2019.3812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Shugart J. First cognitive signal that tau immunotherapy works? ALZFORUM. 2021. Accessed September 2, 2021. https://www.alzforum.org/news/research‐news/first‐cognitive‐signal‐tau‐immunotherapy‐works [Google Scholar]
- 123. Teng E, Manser PT, Pickthorn K, et al. Safety and efficacy of semorinemab in individuals with prodromal to mild Alzheimer disease: a randomized clinical trial. JAMA Neurol. 2022;79(8):758‐767. doi: 10.1001/jamaneurol.2022.1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Boxer AL, Qureshi I, Ahlijanian M, et al. Safety of the tau‐directed monoclonal antibody BIIB092 in progressive supranuclear palsy: a randomised, placebo‐controlled, multiple ascending dose phase 1b trial. Lancet Neurol. 2019;18(6):549‐558. doi: 10.1016/S1474-4422(19)30139-5 [DOI] [PubMed] [Google Scholar]
- 125. Kim B, Mikytuck B, Suh E, et al. Tau immunotherapy is associated with glial responses in FTLD‐tau. Acta Neuropathol. 2021;142(2):243‐257. doi: 10.1007/s00401-021-02318-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Novak P, Kovacech B, Katina S, et al. ADAMANT: a placebo‐controlled randomized phase 2 study of AADvac1, an active immunotherapy against pathological tau in Alzheimer's disease. Nat Aging. 2021;1:521‐534. [DOI] [PubMed] [Google Scholar]
- 127. Courade JP, Angers R, Mairet‐Coello G, et al. Epitope determines efficacy of therapeutic anti‐Tau antibodies in a functional assay with human Alzheimer Tau. Acta Neuropathol. 2018;136(5):729‐745. doi: 10.1007/s00401-018-1911-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Ji C, Sigurdsson EM. Current status of clinical trials on tau immunotherapies. Drugs. 2021;81(10):1135‐1152. doi: 10.1007/s40265-021-01546-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Bhatt R, Ramos O. Blood‐brain barrier permeable antibodies for Alzheimer's potential therapeutic and diagnostic applications. Alzheimers Dement. 2023;19(S7)(Drug Development):e061328. doi: 10.1002/alz.061328 [DOI] [Google Scholar]
- 130. Zhao P, Zhang N, An Z. Engineering antibody and protein therapeutics to cross the blood‐brain barrier. Antib Ther. 2022;5(4):311‐331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. VandeVrede L, Boxer AL, Polydoro M. Targeting tau: clinical trials and novel therapeutic approaches. Neurosci Lett. 2020;731:134919. doi: 10.1016/j.neulet.2020.134919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Soeda Y, Takashima A. New insights into drug discovery targeting tau protein. Front Mol Neurosci. 2020;13:590896. doi: 10.3389/fnmol.2020.590896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Schirmer RH, Adler H, Pickhardt M, Mandelkow E. Lest we forget you – methylene blue…. Neurobiol Aging. 2011;32(12):2325.e7‐16. doi: 10.1016/j.neurobiolaging.2010.12.012 [DOI] [PubMed] [Google Scholar]
- 134. Wischik CM, Edwards PC, Lai RY, Roth M, Harrington CR. Selective inhibition of Alzheimer disease‐like tau aggregation by phenothiazines. Proc Natl Acad Sci U S A. 1996;93(20):11213‐11218. doi: 10.1073/pnas.93.20.11213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Wischik CM, Bentham P, Gauthier S, Miller S, Kook K, Schelter BO. Oral tau aggregation inhibitor for Alzheimer's disease: design, progress and basis for selection of the 16 mg/day dose in a phase 3, randomized, placebo‐controlled trial of hydromethylthionine mesylate. J Prev Alzheimers Dis. 2022;9(4):780‐790. doi: 10.14283/jpad.2022.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Ryan JM, Quattropani A, Abd‐Elaziz K, et al. O1‐12‐05: phase 1 study in healthy volunteers of the O‐GlcNAcase inhibitor ASN120290 as a novel therapy for progressive supranuclear palsy and related tauopathies. Alzheimers Dement. 2018;14(7S_Part_4):251. [Google Scholar]
- 137. Wang X, Li W, Marcus J, et al. MK‐8719, a novel and selective O‐GlcNAcase inhibitor that reduces the formation of pathological tau and ameliorates neurodegeneration in a mouse model of tauopathy. J Pharmacol Exp Ther. 2020;374(2):252‐263. doi: 10.1124/jpet.120.266122 [DOI] [PubMed] [Google Scholar]
- 138. Park J, Kim DY, Hwang GS, Han IO. Repeated sleep deprivation decreases the flux into hexosamine biosynthetic pathway/O‐GlcNAc cycling and aggravates Alzheimer's disease neuropathology in adult zebrafish. J Neuroinflammation. 2023;20(1):257. doi: 10.1186/s12974-023-02944-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Piscopo P, Crestini A, Carbone E, et al. A systematic review on drugs for synaptic plasticity in the treatment of dementia. Ageing Res Rev. 2022;81:101726. doi: 10.1016/j.arr.2022.101726 [DOI] [PubMed] [Google Scholar]
- 140. Yadikar H, Torres I, Aiello G, et al. Screening of tau protein kinase inhibitors in a tauopathy‐relevant cell‐based model of tau hyperphosphorylation and oligomerization. PLoS One. 2020;15(7):e0224952. doi: 10.1371/journal.pone.0224952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Edwards AL, Collins JA, Junge C, et al. Exploratory tau biomarker results from a multiple ascending‐dose study of BIIB080 in Alzheimer disease: a randomized clinical trial. JAMA Neurol. 2023;80(12):1344‐1352. doi: 10.1001/jamaneurol.2023.3861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Park J, Wetzel I, Marriott I, et al. A 3D human triculture system modeling neurodegeneration and neuroinflammation in Alzheimer's disease. Nat Neurosci. 2018;21(7):941‐951. doi: 10.1038/s41593-018-0175-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Taubes A, Nova P, Zalocusky KA, et al. Experimental and real‐world evidence supporting the computational repurposing of bumetanide for APOE4‐related Alzheimer's disease. Nat Aging. 2021;1(10):932‐947. doi: 10.1038/s43587-021-00122-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Cunniffe N, Vuong KA, Ainslie D, et al. Systematic approach to selecting licensed drugs for repurposing in the treatment of progressive multiple sclerosis. J Neurol Neurosurg Psychiatry. 2021;92(3):295‐302. doi: 10.1136/jnnp-2020-324286 [DOI] [PubMed] [Google Scholar]
- 145. O'Brien JT, Chouliaras L, Sultana J, Taylor JP, Ballard C, Group RS. RENEWAL: REpurposing study to find NEW compounds with Activity for Lewy body dementia‐an international Delphi consensus. Alzheimers Res Ther. 2022;14(1):169. doi: 10.1186/s13195-022-01103-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Boyarko B, Podvin S, Greenberg B, et al. Evaluation of bumetanide as a potential therapeutic agent for Alzheimer's disease. Front Pharmacol. 2023;14:1190402. doi: 10.3389/fphar.2023.1190402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Desai RJ, Varma VR, Gerhard T, et al. Comparative risk of Alzheimer Disease and related dementia among medicare beneficiaries with rheumatoid arthritis treated with targeted disease‐modifying antirheumatic agents. JAMA Netw Open. 2022;5(4):e226567. doi: 10.1001/jamanetworkopen.2022.6567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Varma VR, Desai RJ, Navakkode S, et al. Hydroxychloroquine lowers Alzheimer's disease and related dementias risk and rescues molecular phenotypes related to Alzheimer's disease. Mol Psychiatry. 2023;28(3):1312‐1326. doi: 10.1038/s41380-022-01912-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. FDA Converts Novel Alzheimer's Disease Treatment to Traditional Approach. 2023. Accessed July 6, 2023. https://www.fda.gov/news‐events/press‐announcements/fda‐converts‐novel‐alzheimers‐disease‐treatment‐traditional‐approval
- 150. Dumas A, Destrebecq F, Esposito G, Suchonova D, Steen Frederiksen K. Rethinking the detection and diagnosis of Alzheimer's disease: outcomes of a European Brain Council project. Aging Brain. 2023;4:100093. doi: 10.1016/j.nbas.2023.100093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Salloway S, Sperling R, Fox NC, et al. Two phase 3 trials of bapineuzumab in mild‐to‐moderate Alzheimer's disease. N Engl J Med. 2014;370(4):322‐333. doi: 10.1056/NEJMoa1304839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. FDA's Decision to Approve New Treatment for Alzheimer's Disease. U.S. Food & Drug Administration; 2021. Accessed June 7, 2021. https://www.fda.gov/drugs/our‐perspective/fdas‐decision‐approve‐new‐treatment‐alzheimers‐disease [Google Scholar]
- 153. Karikari TK, Ashton NJ, Brinkmalm G, et al. Blood phospho‐tau in Alzheimer disease: analysis, interpretation, and clinical utility. Nat Rev Neurol. 2022;18(7):400‐418. doi: 10.1038/s41582-022-00665-2 [DOI] [PubMed] [Google Scholar]
- 154. Stamelou M, Respondek G, Giagkou N, Whitwell JL, Kovacs GG, Hoglinger GU. Evolving concepts in progressive supranuclear palsy and other 4‐repeat tauopathies. Nat Rev Neurol. 2021;17(10):601‐620. doi: 10.1038/s41582-021-00541-5 [DOI] [PubMed] [Google Scholar]
- 155. Bischof GN, Brendel M, Barthel H, et al. Improved tau PET SUVR quantification in 4‐repeat tau phenotypes with [(18)F]PI‐2620. J Nucl Med. 2024;65(6):952‐955. doi: 10.2967/jnumed.123.265930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Volter F, Beyer L, Eckenweber F, et al. Assessment of perfusion deficit with early phases of [(18)F]PI‐2620 tau‐PET versus [(18)F]flutemetamol‐amyloid‐PET recordings. Eur J Nucl Med Mol Imaging. 2023;50(5):1384‐1394. doi: 10.1007/s00259-022-06087-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Horie K, Barthelemy NR, Spina S, et al. CSF tau microtubule‐binding region identifies pathological changes in primary tauopathies. Nat Med. 2022;28(12):2547‐2554. doi: 10.1038/s41591-022-02075-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Horie K, Salvado G, Barthelemy NR, et al. CSF MTBR‐tau243 is a specific biomarker of tau tangle pathology in Alzheimer's disease. Nat Med. 2023;29(8):1954‐1963. doi: 10.1038/s41591-023-02443-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Blennow K, Chen C, Cicognola C, et al. Cerebrospinal fluid tau fragment correlates with tau PET: a candidate biomarker for tangle pathology. Brain. 2020;143(2):650‐660. doi: 10.1093/brain/awz346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Fitzpatrick AWP, Falcon B, He S, et al. Cryo‐EM structures of tau filaments from Alzheimer's disease. Nature. 2017;547(7662):185‐190. doi: 10.1038/nature23002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Shi Y, Zhang W, Yang Y, et al. Structure‐based classification of tauopathies. Nature. 2021;598(7880):359‐363. doi: 10.1038/s41586-021-03911-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Ashton NJ, Leuzy A, Lim YM, et al. Increased plasma neurofilament light chain concentration correlates with severity of post‐mortem neurofibrillary tangle pathology and neurodegeneration. Acta Neuropathol Commun. 2019;7(1):5. doi: 10.1186/s40478-018-0649-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Bacioglu M, Maia LF, Preische O, et al. Neurofilament light chain in blood and CSF as marker of disease progression in mouse models and in neurodegenerative diseases. Neuron. 2016;91(2):494‐496. doi: 10.1016/j.neuron.2016.07.007 [DOI] [PubMed] [Google Scholar]
- 164. Bridel C, van Wieringen WN, Zetterberg H, et al. Diagnostic value of cerebrospinal fluid neurofilament light protein in neurology: a systematic review and meta‐analysis. JAMA Neurol. 2019;76(9):1035‐1048. doi: 10.1001/jamaneurol.2019.1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Meeter LHH, Vijverberg EG, Del Campo M, et al. Clinical value of neurofilament and phospho‐tau/tau ratio in the frontotemporal dementia spectrum. Neurology. 2018;90(14):e1231‐e1239. doi: 10.1212/WNL.0000000000005261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Zetterberg H, Teunissen C, van Swieten J, et al. The role of neurofilament light in genetic frontotemporal lobar degeneration. Brain Commun. 2023;5(1):fcac310. doi: 10.1093/braincomms/fcac310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. U.S. Food and Drug Administration . Biomarker Qualification Program. Context of Use. 2021. https://www.fda.gov/drugs/biomarker‐qualification‐program/context‐use
- 168. Mills SM, Mallmann J, Santacruz AM, et al. Preclinical trials in autosomal dominant AD: implementation of the DIAN‐TU trial. Rev Neurol (Paris). 2013;169(10):737‐743. doi: 10.1016/j.neurol.2013.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. The ALLFTD Research Study . The ARTFL‐LEFFTDS Longitudinal Frontotemporal Lobar Degeneration Study. https://www.allftd.org/
- 170. GENFI: Genetic FTD Initiative. https://www.genfi.org/
- 171. Boeve B, Bove J, Brannelly P, et al. The longitudinal evaluation of familial frontotemporal dementia subjects protocol: framework and methodology. Alzheimers Dement. 2020;16(1):22‐36. doi: 10.1016/j.jalz.2019.06.4947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Rohrer JD, Nicholas JM, Cash DM, et al. Presymptomatic cognitive and neuroanatomical changes in genetic frontotemporal dementia in the Genetic Frontotemporal dementia Initiative (GENFI) study: a cross‐sectional analysis. Lancet Neurol. 2015;14(3):253‐262. doi: 10.1016/S1474-4422(14)70324-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. FTD Preventive Initiative . Boxer Lab, University of California San Francisco. 2023. https://boxerlab.ucsf.edu/study/fpi [Google Scholar]
- 174. Staffaroni AM, Quintana M, Wendelberger B, et al. Temporal order of clinical and biomarker changes in familial frontotemporal dementia. Nat Med. 2022;28(10):2194‐2206. doi: 10.1038/s41591-022-01942-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. U.S. Food and Drug Administration . Human Gene Therapy for Neurodegenerative Diseases: Guidance for Industry. Center for Biologics Evaluation and Research; 2022. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/human-gene-therapy-neurodegenerative-diseases [Google Scholar]
- 176. Kowalska A, Jamrozik Z, Kwiecinski H. Progressive supranuclear palsy – parkinsonian disorder with tau pathology. Folia Neuropathol. 2004;42(2):119‐123. [PubMed] [Google Scholar]
- 177. Jellinger KA. Pathomechanisms of cognitive and behavioral impairment in corticobasal degeneration. J Neural Transm (Vienna). 2023. doi: 10.1007/s00702-023-02691-w [DOI] [PubMed] [Google Scholar]
- 178. Mintun MA, Lo AC, Duggan Evans C, et al. Donanemab in Early Alzheimer's Disease. N Engl J Med. 2021;384(18):1691‐1704. doi: 10.1056/NEJMoa2100708 [DOI] [PubMed] [Google Scholar]
- 179. Eslinger PJ, Dennis K, Moore P, Antani S, Hauck R, Grossman M. Metacognitive deficits in frontotemporal dementia. J Neurol Neurosurg Psychiatry. 2005;76(12):1630‐1635. doi: 10.1136/jnnp.2004.053157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Rosen HJ. Anosognosia in neurodegenerative disease. Neurocase. 2011;17(3):231‐241. doi: 10.1080/13554794.2010.522588 [DOI] [PubMed] [Google Scholar]
- 181. Taylor JC, Heuer HW, Clark AL, et al. Feasibility and acceptability of remote smartphone cognitive testing in frontotemporal dementia research. Alzheimers Dement (Amst). 2023;15(2):e12423. doi: 10.1002/dad2.12423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. U.S. Food and Drug Administration . Patient‐Focused Drug Development: Selecting, Developing, or Modifying Fit‐for‐Purpose Clinical Outcome Assessments. Center for Drug Evaluation and Research; 2022. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/patient-focused-drug-development-selecting-developing-or-modifying-fit-purpose-clinical-outcome [Google Scholar]
- 183. Dodge S, Dacks P, Niehoff D, et al. Frontotemporal Degeneration (FTD): A Voice of the Patient Report. The Association for Frontotemporal Degeneration; 2021:1‐75. [Google Scholar]
- 184. Respondek G, Breslow D, Amirghiasvand C, et al. The lived experiences of people with progressive supranuclear palsy and their caregivers. Neurol Ther. 2023;12(1):229‐247. doi: 10.1007/s40120-022-00420-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Woodcock J, LaVange LM. Master protocols to study multiple therapies, multiple diseases, or both. N Engl J Med. 2017;377(1):62‐70. doi: 10.1056/NEJMra1510062 [DOI] [PubMed] [Google Scholar]
- 186. U.S. Food and Drug Administration. Center for Biologics Evaluation and Research, Center for Drug Evaluation Research . Adaptive Design Clinical Trials for Drugs and Biologics Guidance for Industry. Center for Biologics Evaluation and Research, Center for Drug Evaluation and Research; 2019. FDA‐2018‐D‐3124. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/adaptive-design-clinical-trials-drugs-and-biologics-guidance-industry [Google Scholar]
- 187. Wang G, Berry S, Xiong C, et al. A novel cognitive disease progression model for clinical trials in autosomal‐dominant Alzheimer's disease. Stat Med. 2018;37(21):3047‐3055. doi: 10.1002/sim.7811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Bateman RJ, Benzinger TL, Berry S, et al. The DIAN‐TU Next Generation Alzheimer's prevention trial: adaptive design and disease progression model. Alzheimers Dement. 2017;13(1):8‐19. doi: 10.1016/j.jalz.2016.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Salloway S, Farlow M, McDade E, et al. A trial of gantenerumab or solanezumab in dominantly inherited Alzheimer's disease. Nat Med. 2021;27(7):1187‐1196. doi: 10.1038/s41591-021-01369-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Dominantly Inherited Alzheimer Network Trial: An Opportunity to Prevent Dementia . A Study of Potential Disease Modifying Treatments in Individuals With a Type of Early Onset Alzheimer's Disease Caused by a Genetic Mutation (DIAN‐TU) (DIAN‐TU). National Library of Medicine, National Center for Biotechnology Information. https://clinicaltrials.gov/study/NCT05269394 [Google Scholar]
- 191. Boxer AL, Sperling R. Accelerating Alzheimer's therapeutic development: the past and future of clinical trials. Cell. 2023;186(22):4757‐4772. doi: 10.1016/j.cell.2023.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Cummings J, Montes A, Kamboj S, Cacho JF. The role of basket trials in drug development for neurodegenerative disorders. Alzheimers Res Ther. 2022;14(1):73. doi: 10.1186/s13195-022-01015-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Almirall D, Nahum‐Shani I, Sherwood NE, Murphy SA. Introduction to SMART designs for the development of adaptive interventions: with application to weight loss research. Transl Behav Med. 2014;4(3):260‐274. doi: 10.1007/s13142-014-0265-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information