

Abstract
INTRODUCTION
We investigated longitudinal associations between self‐reported exercise and Alzheimer's disease (AD)‐related biomarkers in individuals with autosomal dominant AD (ADAD) mutations.
METHODS
Participants were 308 ADAD mutation carriers aged 39.7 ± 10.8 years from the Dominantly Inherited Alzheimer's Network. Weekly exercise volume was measured via questionnaire and associations with brain volume (magnetic resonance imaging), cerebrospinal fluid biomarkers, and brain amyloid beta (Aβ) measured by positron emission tomography were investigated.
RESULTS
Greater volume of weekly exercise at baseline was associated with slower accumulation of brain Aβ at preclinical disease stages β = –0.16 [–0.23 to –0.08], and a slower decline in multiple brain regions including hippocampal volume β = 0.06 [0.03 to 0.08].
DISCUSSION
Exercise is associated with more favorable profiles of AD‐related biomarkers in individuals with ADAD mutations. Exercise may have therapeutic potential for delaying the onset of AD; however, randomized controlled trials are vital to determine a causal relationship before a clinical recommendation of exercise is implemented.
Highlights
Greater self‐reported weekly exercise predicts slower declines in brain volume in autosomal dominant Alzheimer's disease (ADAD).
Greater self‐reported weekly exercise predicts slower accumulation of brain amyloid beta in ADAD.
Associations varied depending on closeness to estimated symptom onset.
Keywords: Alzheimer's disease, exercise, magnetic resonance imaging, positron emission tomography, physical activity
1. BACKGROUND
Physical inactivity is an important modifiable risk factor for Alzheimer's disease (AD). 1 Greater physical activity is associated with reduced risk for all‐cause dementia and AD, 2 , 3 , 4 , 5 better cognition, 6 , 7 and greater brain volume in AD‐susceptible regions. 8 , 9 , 10 , 11 Physical activity is also associated with more favorable AD‐related biomarker profiles, including amyloid beta (Aβ) and tau, in the blood, cerebrospinal fluid (CSF), and brain. 12 , 13 Exercise (a structured or planned form of physical activity) is associated with better cognition across the AD trajectory. 14 , 15 However, it is currently unclear at which disease stage exercise is associated with the greatest benefit for AD‐related outcomes, and whether such benefits are sustained across the disease course.
Autosomal dominant AD (ADAD) is a rare form of AD attributable to mutations on either the amyloid precursor protein (APP), presenilin 1 (PSEN1), or presenilin 2 (PSEN2) genes. 16 Individuals with ADAD provide a unique opportunity to draw temporal conclusions about the disease course because they are destined to develop AD at a mutation‐specific age, meaning it is possible to examine at which disease stages intervention implementation might be most effective. Further, because individuals with ADAD are younger at disease onset, they are likely free of many age‐related comorbidities which make interpretation of the association between exercise and AD‐related biomarkers challenging in older adults.
Previous cross‐sectional studies have suggested that in individuals with ADAD, greater self‐reported exercise duration (≥ 150 minutes/week) was associated with better cognition at the expected age of symptom onset, lower levels of AD‐related biomarkers in CSF, and in those already accumulating brain Aβ (i.e., Aβ positive), lower brain Aβ levels. 17 , 18 However, to our knowledge, no studies have considered longitudinal associations between exercise and AD biomarkers in individuals with ADAD, and such research will help further our understanding of the potential of exercise as a therapeutic strategy to delay the onset of AD.
The current study used data from the Dominantly Inherited Alzheimer Network (DIAN), a multi‐site, international observational study of individuals from families who carry ADAD mutations. Our primary aim was to examine associations between self‐reported exercise and AD‐related markers (CSF biomarkers, brain Aβ, and brain structure) over time, and determine whether associations varied depending on closeness to estimated years from expected symptom onset (EYO). We therefore considered the following research questions: (1) are baseline and longitudinal self‐reported exercise levels associated with AD‐related markers over time? and (2) do baseline and longitudinal associations between self‐reported exercise and AD biomarkers differ depending on estimated years from expected symptom onset?
2. METHOD
2.1. Participants
From DIAN data freeze 14 (data collected from January 2009 to June 2019), there were a total of 534 participants with available data; after removing Dutch mutation carriers (APP E693Q mutation that causes cerebral amyloid angiopathy), mutation non‐carriers, outliers (defined below), and those with missing self‐reported exercise data, there remained 308 participants (see Figure 1). In the DIAN study the visit schedule for follow‐up assessments varies based on how close a participant is to their estimated year of symptom onset, with 3‐year intervals for asymptomatic individuals and 1‐year intervals for individuals within 3 years of the parent's age at onset. 19 Participants were included if they had data for ≥ 1 of our outcome variables of interest; thus, sample size varied depending on the outcome variable and ranged from n = 254 for brain Aβ to n = 307 for CSF (Figure 1). All assessment and imaging procedures for the DIAN study were approved by the Washington University Human Research Protection Office. Written informed consent was obtained from all individuals or their caregivers.
FIGURE 1.
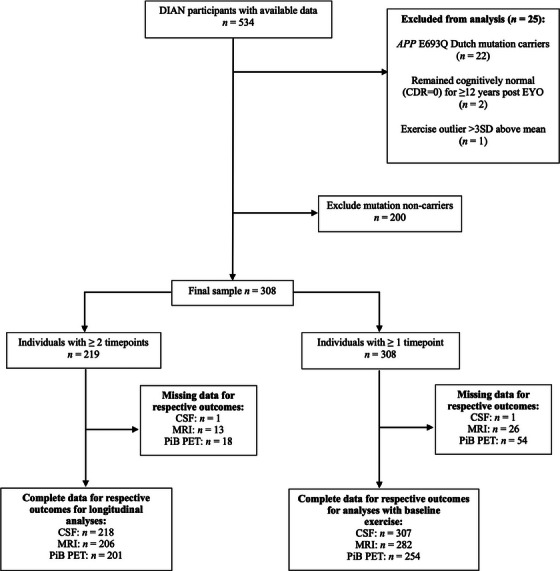
Flow diagram indicating number of participants with data available for inclusion for each analysis. Timepoints refer to number of visits at which both exercise and biomarker data were collected. APP, amyloid precursor protein; CDR, Clinical Dementia Rating; CSF, cerebrospinal fluid; DIAN, Dominantly Inherited Alzheimer Network; EYO, estimated years from expected symptom onset; MRI, magnetic resonance imaging; PiB PET, Pittsburgh compound B positron emission tomography; SD, standard deviation
RESEARCH IN CONTEXT
Systematic review: The authors reviewed the literature using traditional database searches. Previous studies have examined the association between physical activity and Alzheimer's disease (AD) biomarkers; however, only two previous cross‐sectional studies have examined this association in individuals with autosomal dominant AD mutations.
Interpretation: Our findings demonstrate that greater weekly exercise is associated with slower accumulation of brain amyloid beta and slower decline in brain volume in individuals who are destined to develop AD.
Future directions: Exercise may have therapeutic potential for delaying the onset of AD; however, randomized controlled trials are vital to determine a causal relationship before a clinical recommendation of exercise is implemented. These associations should be further studied in individuals with late‐onset AD.
2.2. Estimated years from symptom onset
The age at onset (AAO) for ADAD cognitive symptoms can be estimated based on parental AAO (the first progressive change in cognition or behavior of the parent) or mean mutation AAO (used if available; 20 otherwise, parental onset age is used). Thus, the disease stage can be projected based on how far away an individual is from their EYO. 21 If parental AAO is used, this is determined after careful discussion with a reliable collateral source, the participant, and any other sources of information that are useful for determination (e.g., medical records, other family members). EYO is calculated by subtracting the estimated AAO (from mutation data or parental age of onset) from the participant's age at the time of assessment, resulting in negative values indicating years until estimated conversion, and positive values indicating years since expected symptom onset. Efforts were made to account for differences between estimated and actual symptom onset, resulting in the removal of two individuals who remained cognitively normal (Clinical Dementia Rating [CDR] = 0) ≥ 12 years post EYO (Figure 1).
2.3. Exercise level evaluation
At baseline and each subsequent follow‐up visit, participants self‐reported their average minutes per week spent partaking in 10 leisure‐time exercise activities (e.g., walking, jogging, cycling, swimming, tennis, aerobics, yoga, and weight training) over the past 12 months. In the questionnaire instructions, participants were encouraged to have their responses corroborated by their collateral source (e.g., family member or friend). Research from our group has previously shown internal consistency for this questionnaire. 17 Consistent with our previous work, individual item responses were truncated to a maximum of 600 minutes per week (an adaptation of guidelines regarding maximum reports of daily activities to those recommended for the International Physical Activity Questionnaire; 22 a total of 38 responses were truncated). A continuous score was calculated from all items by combining minutes per week spent exercising in each activity. One outlier with 5160 exercise minutes/week was removed. The continuous exercise variable (i.e., total exercise minutes/week) was used for all analyses with baseline exercise. To create a categorical exercise change variable (i.e., change over time), a binary variable was created with a cut‐off of ≥ 365 minutes/week, derived from a two‐component mixture distribution analysis. 23 This methodology was selected because of a bimodal distribution in the physical activity data (Figure S1 in supporting information). Participants with ≥ 2 study visits were then classified into one of four categories, based on their activity levels at first and last follow‐up visits: “decreasers” (moved from ≥ 365 minutes/week to < 365 minutes/week from first to last visit); “stable low” (remained < 365 minutes/week at first and last visits); “stable high” (remained ≥ 365 minutes/week at first and last visits); or “increasers” (moved from < 365 minutes/week to ≥ 365 minutes/week from first to last visit). Quality control was run on these data to ensure participants did not show negligible exercise change (i.e., < 10 minutes/week) that resulted in a category change. Thus, all participants who changed categories did so with an exercise increase or decrease of at least 10 minutes/week. A limitation of this approach is that individuals have varying follow‐up lengths, thus one exercise change score may be calculated over 2 years, and one may be over 6 years, depending on how long participants remained in the study. This is a general limitation of dropouts in longitudinal research, and the mean length of follow‐up in the current sample was 1.9 ± 2 years.
2.4. Genotyping, CSF collection, and neuroimaging
Genotyping was performed on DNA extracted from blood samples to identify ADAD genetic mutations on the APP, PSEN1, or PSEN2 genes (see Supplementary Methods in supporting information).
Fasting CSF was collected in the morning via lumbar puncture, using previously described methods (Supplementary Methods). 24 Concentrations of CSF Aβ40, Aβ42, total tau (t‐tau), and total phosphorylated tau (p‐tau181) were measured by chemiluminescent enzyme immunoassay using an automated platform (LUMIPULSE G1200, Fujirebio) according to the manufacturer's specifications.
Brain volume measures were derived from T1‐weighted images using a magnetization‐prepared rapid acquisition gradient echo (MPRAGE) sequence on a 3T scanner 25 and were matched to the Alzheimer's Disease Neuroimaging Initiative (ADNI) protocol. 25 Images were processed using FreeSurfer (Supplementary Methods). A standardized uptake value ratio (SUVR) was calculated for brain Aβ measured using Pittsburgh compound B positron emission tomography (PiB PET) with regional spread function partial volume correction (Supplementary Methods). 26
2.5. Statistical analyses
Analyses were conducted using R statistical computing packages version 4.3.1 (R Core Team, 2021). To examine associations between exercise and AD‐related biomarkers, a series of linear mixed‐effects models (LMMs) were conducted (Supplementary Methods). We examined the interaction between exercise measured at baseline and time (in years) on the dependent variables of CSF Aβ42, CSF Aβ42/Aβ40, CSF p‐tau/Aβ42, CSF p‐tau181, and CSF t‐tau; hippocampal volume, total cortical volume, total gray matter volume, subcortical gray matter volume, and white matter hyperintensities; and PiB PET SUVR measured across multiple follow‐up visits. To examine longitudinal change in exercise and AD markers, these models were re‐run using our categorical exercise change variable instead of the baseline exercise variable. We then re‐ran all models with an added interaction term for baseline EYO (time x exercise x EYO) to examine whether associations differed based on disease stage. Baseline EYO was treated as a continuous variable in all analyses except in the case of brain Aβ because there was a non‐linear association between EYO and Aβ (Figure S2 in supporting information), thus EYO was separated into categories (≤ –15 EYO; n = 80; > –15 EYO to 0; n = 109; EYO > 0; n = 94, Supplementary Methods). All models included a random intercept for participant ID. A random slope was not estimated because many participants in DIAN have only two time points, meaning a random slope could not be accurately identified. Post hoc investigations for our three‐way interaction were conducted by creating 5‐year “bins” of the baseline EYO variable (i.e., –15 to –10, −9 to –5 years, etc.) and examining at which point the slope of exercise on the respective AD biomarker first became significant. The aim of this split was to balance adequate sample size within each bin while still improving our understanding of these associations. All analyses, except for post hoc, were corrected for multiple comparisons using the false discovery rate for interaction terms within each biomarker. 27 ,
TABLE 1.
Descriptive statistics for our subsample of the DIAN cohort, mutation carriers only.
| Subset with ≥ 1 visit (n = 308) | Subset with ≥ 2 visits (n = 219) | Test statistic | |
|---|---|---|---|
| Age, years | 39.7 (10.8) | 40.0 (10.8) | t = −0.49 |
| Sex, % female (n) | 56.5 (174) | 56.2 (123) | χ2 = 0.00 |
| APOE ε4 allele carriers, % (n) | 30.5 (94) | 32.0 (70) | χ2 = 0.04 |
| MMSE score | 26.6 (5.4) | 27.0 (4.5) | t = 1.10 |
| EYO at baseline, years | −7.0 (11.0) | −6.85 (10.9) | t = −0.25 |
| Education, years | 14.2 (3.0) | 14.2 (2.9) | t = 0.35 |
| Family mutation | χ2 = 0.36 | ||
| PSEN1, % (n) | 77.9 (240) | 78.1 (171) | |
| PSEN2, % (n) | 7.1 (22) | 5.5 (12) | |
| APP, % (n) | 14.9 (46) | 16.4 (36) | |
| Weekly exercise, min | 393.7 (342.9) | 393.8 (345.2) | t = 0.001 |
| Follow‐up, number of visits | 2.3 (1.2) | 2.8 (1.06) | t = −5.29 ** |
| CSF Aβ42, pg/mL | 610.8 (358.58) | 619.6 (380.6) | t = 0.24 |
| CSF Aβ42/40 | 0.07 (0.03) | 0.07 (0.03) | t = 0.13 |
| CSF p‐tau181, pg/mL | 76.5 (66.56) | 80.3 (68.9) | t = 0.56 |
| CSF t‐tau, pg/mL | 509.2 (350.22) | 540.0 (368.9) | t = 0.86 |
Note: If not otherwise described, data are presented as mean (standard deviation). CSF, MMSE, and weekly exercise means are at baseline.
Abbreviations: Aβ, amyloid beta; APOE, apolipoprotein E; APP, amyloid precursor protein; CSF, cerebrospinal fluid; DIAN, Dominantly Inherited Alzheimer's Network; EYO, estimated years from symptom onset; MMSE, Mini‐Mental State Examination; PSEN, presenilin gene; p‐tau, phosphorylated tau; t‐tau, total tau.
P < 0.001.
Studies using DIAN data often include the gene on which a participants’ mutation occurs (i.e., APP, PSEN1, or PSEN2) as a covariate, because although all mutations impact Aβ processing, the biological processes are distinct. 28 Additionally, phenotypic expression of cortical Aβ may be related to the specific location of the affected codon. 28 We aimed to statistically account for these differences by ranking specific mutations (e.g., Glu280Gly, Ala79Val, etc.) based on their average parental age of onset within the sample (regardless of kindred). We assigned each mutation a number, with 1 being assigned to the mutation with the youngest average AAO, and 80 having the oldest average AAO. We subsequently split this variable into quantiles (herein referred to as mutation rank) and tested it for inclusion as a covariate in our analyses.
We considered accounting for the decline in exercise levels across the disease course by controlling for CDR score. However, CDR was excluded as a covariate because it has an underlying association with AD outcome variables; its inclusion would result in incorrect model estimation. We have instead included supplementary analyses with CDR score in place of EYO for outcome variables that showed significant interactions (Supplemental Methods and Table S1 in supporting information).
Covariates differed depending on the outcome variable and were selected to maximize model fit. The following covariates were tested for each dependent variable: revisualized age, sex, apolipoprotein E (APOE) ε4 status (coded as carriage of at least one ε4 allele = 1, otherwise = 0), the gene on which the mutation exists (family mutation), and our mutation rank variable. The resultant models included different covariates for each outcome variable, namely: CSF variables included revisualized age, APOE ε4 status, mutation rank, and family mutation; magnetic resonance imaging (MRI) included revisualized age, sex, mutation rank, family mutation, and intracranial volume; PiB PET (brain Aβ) included only APOE ε4 status.
3. RESULTS
Descriptive data for participants are presented in Table 1. The sample had a mean age of 39.7 ± 10.8 years, 56.5% were female, had a mean weekly exercise level of 382 ± 330.6 minutes, and had a mean of 2.3 ± 1.2 follow‐up visits.
3.1. CSF markers
3.1.1. Baseline exercise and CSF biomarkers across time and EYO
Higher self‐reported exercise was associated with lower p‐tau/Aβ42, p‐tau, and t‐tau cross‐sectionally; however, these associations did not persist over time (Table 2; Model 1). There were no interactions between baseline exercise, time, and EYO on any CSF marker (Table 2; Model 2).
TABLE 2.
Partially standardized beta coefficients from linear mixed models examining exercise on Alzheimer's disease‐related biomarkers at baseline, over time, and across EYO.
| Outcome variable | Baseline exercise Model 1 β (SE) |
Baseline exercise x time Model 1 β (SE) |
Baseline exercise x time x EYO Model 2 β (SE) |
Exercise change category x time Model 3 β (SE) |
Exercise change category x time x EYO Model 4 β (SE) |
|---|---|---|---|---|---|
| CSF Aβ42 | 0.08 (0.06) | 0.02 (0.02) | 0.00 (0.02) | −0.03 (0.02) | 0.02 (0.02) |
| CSF Aβ42/Aβ40 | 0.05 (0.06) | −0.01 (0.01) | 0.01 (0.02) | −0.03 (0.01) | 0.02 (0.02) |
| CSF p‐tau/Aβ42 | −0.14 (0.06) * | −0.02 (0.01) | −0.01 (0.02) | 0.01 (0.01) | −0.02 (0.01) |
| CSF p‐tau | −0.16 (0.06) ** | −0.01 (0.01) | −0.01 (0.02) | −0.00 (0.01) | −0.01 (0.02) |
| CSF t‐tau | −0.21 (0.06) *** | 0.01 (0.02) | −0.00 (0.02) | −0.02 (0.02) | −0.03 (0.02) |
| Right hippocampal volume | 0.12 (0.06) | 0.04 (0.01) * | 0.06 (0.01) *** | 0.04 (0.02) * | 0.00 (0.01) |
| Left hippocampal volume | 0.14 (0.06) | 0.04 (0.02) * | 0.06 (0.02) * | 0.04 (0.02) | −0.00 (0.02) |
| Total cortical volume | 0.11 (0.04) * | 0.01 (0.01) | 0.03 (0.01) * | 0.01 (0.01) | 0.03 (0.01) * |
| Total subcortical gray matter volume | 0.12 (0.05) * | 0.01 (0.01) | 0.03 (0.01) * | 0.01 (0.01) | 0.01 (0.01) |
| Total gray matter volume | 0.12 (0.04) * | 0.01 (0.01) | 0.03 (0.01) * | 0.01 (0.01) | 0.03 (0.01) * |
| White matter hyperintensities | −0.10 (0.06) | −0.00 (0.02) | 0.01 (0.02) | −0.05 (0.02) | −0.01 (0.02) |
| Aβ (PiB) | −0.18 (0.06) ** | −0.02 (0.01) * | 0.04 (0.01) *** | −0.00 (0.01) | 0.01 (0.01) |
Note. All results presented as partially standardized beta coefficient (standard error). Predictor variables are standardized; outcome variables are not. Separate models run for each outcome variable. Significant results reported here have been corrected for multiple comparisons using FDR, applied within each outcome variable (i.e., for CSF 5 outcomes x 4 models = 20 P values corrected). FDR correction was applied to the highest order interaction term only (i.e., FDR was not applied to main effects from the same model for Models 1 and 3). Data for the main effect of exercise and exercise x time taken from the same model. All CSF and PiB outcome variables were log transformed. Covariates are as follows CSF: age, APOE ε4 status, mutation rank, family mutation; MRI: age, sex, intracranial volume, mutation rank, family mutation; PiB: APOE ε4 status; Exercise categories were created based on a 365 minute/week cut‐off and classified as 1 = decreased, 2 = remained low, 3 = remained high, 4 = increased, based on follow‐up data.
Abbreviations: Aβ, amyloid beta; APOE, apolipoprotein E; CSF, cerebrospinal fluid; EYO, estimated years from expected symptom onset; FDR, false discovery rate; MRI, magnetic resonance imaging; PiB, Pittsburgh compound B; p‐tau, phosphorylated tau; SE, standard error; t‐tau, total tau; β, partially standardized beta coefficient.
P < 0.05.
P < 0.01.
P < 0.001 (q values, i.e., FDR corrected, for interaction terms).
3.1.2. Change in exercise and CSF biomarkers across time and EYO
Exercise change category was not associated with CSF Aβ42, Aβ42/40, p‐tau/Aβ42, p‐tau, and t‐tau rates of change (Table 2; Model 3). There was no significant interaction for change in exercise, time, and EYO on any CSF marker (Table 2; Model 4).
3.2. MRI
3.2.1. Baseline exercise and MRI variables across time and EYO
Greater baseline exercise was associated with greater total brain volume, subcortical volume, and total gray matter volume cross‐sectionally, but the slopes of brain volume change over time were not associated with baseline exercise (Table 2; Model 1). For hippocampal volume, there was no cross‐sectional association with exercise; however, greater baseline exercise was associated with a slower decline in right and left hippocampal volume over time (not considering EYO; Table 2; Model 1).
There were significant interactions between baseline exercise, time, and EYO for every MRI outcome except white matter hyperintensities (Table 2; Model 2). These associations show that higher baseline exercise relates to slower decline in brain volume and that this becomes increasingly more pronounced as individuals move closer to, and beyond, their predicted EYO (Figure 2; only right hippocampal volume and total gray matter volume included here, other outcomes demonstrate a similar pattern and are included in Figure S3 in supporting information). Additional analyses indicated that the associations between exercise and structural brain outcomes were first observed at –5 EYO (i.e., 5 years before predicted onset), and were maintained up to 15 years post estimated symptom onset (P value range from < 0.001 – 0.023). Figure S4 in supporting information shows scatterplots for the raw uncorrected values by exercise group across EYO. There were no significant interactions between baseline exercise x time for any volumetric outcome within CDR groups when replacing EYO with CDR (Table S1).
FIGURE 2.
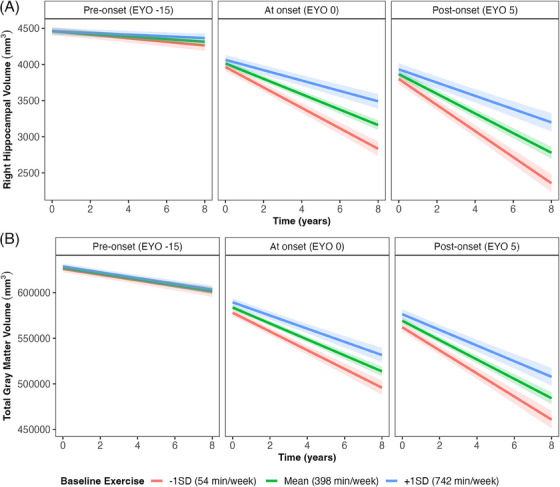
Baseline exercise x time interaction on structural brain outcomes at baseline EYO. Baseline EYO was treated as a continuous variable in all models and is only separated here for visual aid (i.e., EYO groups were not created); a negative EYO represents years before estimated symptom onset, positive EYO represents years post estimated symptom onset. The data presented here are fitted values from analyses modeling associations between exercise and brain outcomes given a particular EYO. Scatterplots of raw data are presented in Figure S4 in supporting information. The left‐hand panel shows the fitted values for change in brain volume over time if EYO is −15, the middle demonstrates the association if EYO is 0, and the right shows this association if EYO is 5. Gray bands show 95% confidence intervals. A, Right hippocampal volume. B, Total gray matter volume. EYO, estimated years from expected symptom onset; SD, standard deviation
3.2.2. Change in exercise and MRI variables across time and EYO
Those who maintained high exercise retained greater hippocampal volume over time compared to those who decreased their exercise (Table 2, Figure 3; decreases as a reference, 𝛽 = 0.13, standard error [SE] = 0.05, P = 0.006). There were significant interactions between changes in exercise, time, and EYO for total cortical volume and total gray matter volume (Table 2; Model 4; Figure 4). Those who maintained stable high exercise or increased exercise had slower decline in total cortical volume compared to those who decreased their exercise level (“stable high”: 𝛽 = 0.08, SE = 0.04, P = 0.028; “increasers”: 𝛽 = 0.08, SE = 0.04, P = 0.041), an effect that became more pronounced across EYO (Figure 4). A similar pattern was shown for total gray matter volume; however, individual comparisons between exercise categories were only trending toward significance (“decreasers” vs. “stable high”: 𝛽 = 0.06, SE = 0.03, P = 0.065; “decreasers” vs. “increasers”: 𝛽 = 0.07, SE = 0.04, P = 0.060; Figure 4).
FIGURE 3.
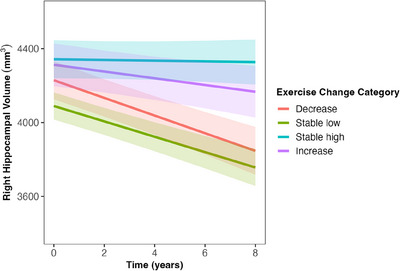
Association between change in exercise and change in right hippocampal volume over time. Exercise categories were created based on a 365 minutes/week cut off and classified as: 1 = “decreasers,” 2 = “stable low,” 3 = “stable high,” 4 = “increasers,” based on follow‐up data. Differences exist between “decreasers” versus “stable high category,” β = 0.13, SE = 0.05, P = .006 and “stable low”’ versus “stable high” category, β = 0.11, SE = 0.04, P = = 0.006. Gray bands represent 95% confidence intervals. EYO, estimated years from expected symptom onset; SE, standard error
FIGURE 4.
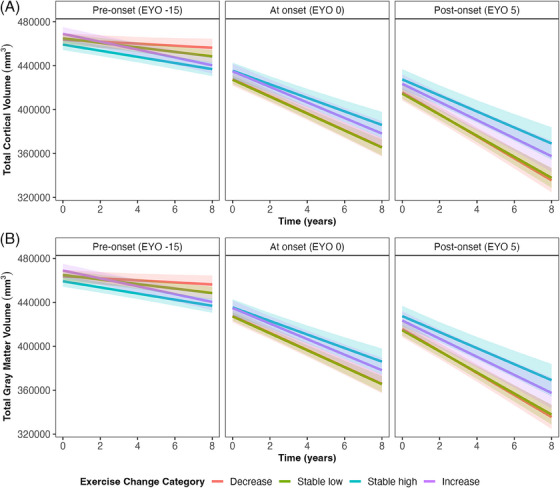
Change in exercise on structural brain outcomes over time and across EYO. Exercise categories were created based on a 365 minute/week cut off and classified as: 1 = “decreasers,” 2 = “stable low,” 3 = “stable high,” 4 = “increasers,” based on follow‐up data. EYO was treated as a continuous variable in all models and is only separated here for visual aid. Gray bands represent 95% confidence intervals. Total cortical volume, differences exist between decreasers versus stable high category, β = 0.08, SE = 0.04, P = .028; and decreasers versus increasers, β = 0.08, SE = 0.04, P = .041. Total gray matter volume, a trend exists for differences between decreasers versus stable high: β = 0.06, SE = 0.03, P = .065, and decreasers versus increasers: β = 0.07, SE = 0.04, P = .060. EYO, estimated years to symptom onset; SE, standard error
3.3. Brain Aβ
3.3.1. Baseline exercise and brain Aβ across time and EYO
Higher self‐reported baseline exercise was associated with lower brain Aβ as measured by PiB PET, both cross‐sectionally and over time (Table 2; Model 1). There was an interaction between baseline exercise, time, and EYO (Table 2; Model 2). Results showed that in the pre‐onset group (see Statistical Methods), greater exercise was associated with slower accumulation of Aβ (𝛽 = −0.16, SE = 0.04, P < 0.001; Figure 5). However, there was a trend for the opposite pattern in those nearing predicted onset, that is, those with high baseline exercise showed greater Aβ accumulation (𝛽 = 0.06, SE = 0.03, P = 0.067; Figure 5). In those past their predicted onset (i.e., EYO > 0), there was no interaction between exercise and time on brain Aβ (𝛽 = −0.01, SE = 0.02, P = 0.582; Figure 5). We examined demographic differences between our EYO groups to further understand these findings (Table S2 in supporting information). There were differences between EYO groups only for age and family mutation (Table S2); however, when adding these variables to the baseline exercise x time on PiB model for our nearing predicted onset group, results remained similar (𝛽 = 0.06, SE = 0.03, Pp = 0.070). Figure S4 (panel f) shows scatterplots for the raw uncorrected and untransformed brain Aβ values by exercise group across EYO; Table S1 shows these analyses separated by CDR score instead of EYO (i.e., baseline exercise x time within CDR groups), none of which were significant.
FIGURE 5.
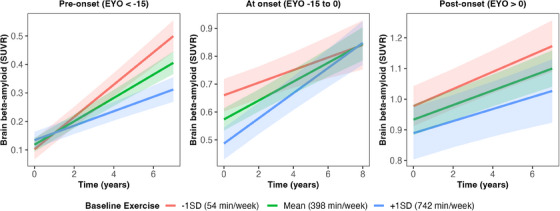
Baseline exercise x time on brain Aβ (log) in subsets of EYO stages (i.e., EYO groups were created). Data were subset based on EYO because of a non‐linear association between EYO and brain Aβ (Figure S2 in supporting information). The left panel demonstrates the change in Aβ over time separated by exercise level within the pre‐onset group (n = 80), the middle panel demonstrates change over time in those nearing EYO (n = 109), the right panel demonstrates those post EYO (n = 94). Gray bands represent 95% confidence intervals. Aβ, amyloid beta; EYO, estimated years from expected symptom onset; SD, standard deviation
3.3.2. Change in exercise and brain Aβ across time and EYO
Change in exercise was not associated with brain Aβ over time, and there was no interaction with EYO (Table 2; Models 3 and 4).
4. DISCUSSION
The primary aim of the current study was to examine associations between exercise levels and AD‐related biomarkers in ADAD mutation carriers. We report cross‐sectional associations between greater exercise participation and lower CSF measures of p‐tau, t‐tau, p‐tau/Aβ42, greater brain volume, and lower brain Aβ burden; and longitudinal associations between greater exercise and slower brain atrophy and Aβ accumulation, which varied dependent upon disease stage. These results illustrate consistent associations between exercise and AD‐related biomarkers, and although a causal relationship cannot be determined, they support the notion of additional randomized controlled trials investigating the therapeutic potential for exercise to delay the onset of AD.
Cross‐sectional analyses revealed associations between exercise and CSF markers, brain volume, and brain Aβ, all in the expected directions. These results are consistent with previous cross‐sectional research in the DIAN cohort, which showed associations between greater exercise and lower brain Aβ, 17 , less AD‐like CSF pathology, and better cognitive function. 18 In the current study, CSF markers were the only outcome that did not demonstrate longitudinal associations with exercise levels. Previous research within the DIAN cohort showed the greatest change in CSF markers > 5 to 10 years before EYO, and a slowing of these changes (or reversal in the case of p‐tau, which shows a significant decline) as individuals moved toward EYO. 24 , 29 This pattern was not observed in prior DIAN studies for other biomarkers such as brain Aβ and hippocampal volume, which steadily increased or decreased (respectively) over the disease course. 26 In the current study, the average EYO at baseline was 7.0 years before symptom onset, which is just before changes in CSF markers may begin to plateau. 30 Thus, exercise may have a greater influence on CSF markers earlier in the disease stage when they are more variable and sensitive to change.
Greater exercise was associated with a slower decline in brain volume over time; this association was evident as individuals moved toward, and beyond, their EYO; however, the effect size was relatively small. Associations between higher exercise and greater brain volume were first observed 5 years before EYO and were maintained up to 15 years post symptom onset. This is consistent with previous research in the DIAN cohort showing hippocampal atrophy begins ≈ 5 years before diagnosis. 29 Similarly, previous research in older adults at risk for late‐onset AD (LOAD; which may be comparable to ADAD because of similarities in pathophysiology 31 , 32 , 33 ) has demonstrated beneficial effects of exercise on the hippocampus, 8 , 34 although recent meta‐analyses have reported inconsistent findings for this association. 11 , 35 One reason for this inconsistency may be, as demonstrated in the current results, that exercise is only associated with AD biomarkers during times of greatest pathology‐related change (i.e., ≤ 5 years before symptom onset for MRI outcomes, and > 5 years before symptom onset for CSF), and pathological change, or lack thereof, is often difficult to determine in individuals at risk for LOAD. Thus, to better understand the utility of exercise as a therapeutic strategy, future studies in LOAD should consider enriching for individuals likely on the AD trajectory (e.g., those accumulating brain Aβ), identified using proxy measures such as CSF or plasma markers. 36 Such studies will improve our understanding of the efficacy of exercise in influencing biomarker changes in preclinical AD.
Baseline exercise was associated with slower brain Aβ accumulation ≥ 15 years before estimated symptom onset; however, there was no association between exercise and brain Aβ nearing, and post‐, estimated symptom onset. This is consistent with previous research from both LOAD and ADAD studies indicating that brain Aβ begins to accumulate up to 20 years before dementia diagnosis 29 , 37 and the notion that lifestyle interventions should be implemented at early disease stages, before Aβ‐associated inflammation and neuronal damage become irreversible. 38 It is noteworthy, however, that Aβ accumulation in ADAD individuals likely cannot be prevented due to mutations with near 100% penetrance, but may be delayed via exercise. Although the current results are derived from younger individuals with ADAD mutations, they may be generalizable to individuals with LOAD due to strong pathological similarities. 33 The current study supports the notion that lifestyle interventions to delay the onset of LOAD may be best implemented in midlife and early aging (i.e., in the sixth decade of life).
The current study used a self‐report measure of exercise. Although this measure has been previously validated, 17 it may be subject to bias and fallible memory. The American Heart Association recommends 150 minutes of moderate‐intensity exercise per week; the thresholds in the current study were high compared to those recommendations. This is likely attributed to either an overreporting of actual physical activity levels (on average 44% in other populations) 39 or increased activity levels because those in the DIAN cohort may be acutely aware of AD risk factors and attempt to mitigate them. The current study did, however, use high‐quality measures of brain structure, function, and underlying pathology, and used a comprehensive approach to statistical analyses. Due to the observational design, we are not able to determine causality from our results, and indeed, declines in functioning or increases in pathology may contribute to decreased exercise levels. We attempted to better understand these associations by examining exercise and AD biomarkers at different CDR levels (Table S1); however, this analysis is limited by group sample sizes and restricted granularity because the CDR has a limited range (five possible scores). Finally, the effect sizes (standardized betas) of our results are relatively small, and lifetime exercise habits which we were not able to account for may have influenced current findings. Thus, randomized controlled trials are required to confirm our results and determine the magnitude of potential clinical benefit.
Here, we demonstrate that greater exercise is associated with benefits for multiple AD‐related outcomes including CSF biomarkers, brain structure, and brain Aβ (only at preclinical stages) in those who carry ADAD mutations. Exercise is worthy of further investigation (using randomized controlled trials) as a potential strategy to alter AD trajectory, whether it be ADAD or sporadic AD, and preserve brain structure and function after AD onset.
CONFLICT OF INTEREST STATEMENT
R.J.B. reports grants from Eli Lilly, Roche, Pharma Consortium (AbbVie, AstraZeneca, Biogen, Eisai, Eli Lilly and Company, Hoffmann La‐Roche Inc, Janssen, Pfizer, Sanofi‐Aventis), and Tau SILK/PET Consortium (Biogen/AbbVie/Lilly); non‐financial support from Avid Radiopharmaceuticals; personal fees and other from Washington University, outside the submitted work. J.C.M. is currently participating in clinical trials of antidementia drugs from Eli Lilly and Company, Biogen, and Janssen. J.C.M. serves as a consultant for Lilly USA and receives research support from Eli Lilly/Avid Radiopharmaceuticals. T.B. receives grant funding from Avid Radiopharmaceuticals/Eli Lilly and participates in clinical trials sponsored by Eli Lilly, Avid Radiopharmaceuticals, Roche, and Pfizer. F.L. holds grants from NIH, NIA, Alzheimer Association, DIAN, Tau‐Consortium, Large PD, Biogen, and Roche. J.L. reports speaker fees from Bayer Vital, Biogen, EISAI, TEVA, Zambon, Merck, and Roche; consulting fees from Axon Neuroscience, EISAI, and Biogen; author fees from Thieme medical publishers and W. Kohlhammer GmbH medical publishers; and is an inventor in a patent “Oral Phenylbutyrate for Treatment of Human 4‐Repeat Tauopathies” (EP 23 156 122.6) filed by LMU Munich. In addition, J.L. reports compensation for serving as chief medical officer for MODAG GmbH, is beneficiary of the phantom share program of MODAG GmbH, and is an inventor in a patent “Pharmaceutical Composition and Methods of Use” (EP 22 159 408.8) filed by MODAG GmbH, all activities outside the submitted work. R.J.P.’s laboratory receives cost recovery funding from Biogen for tissue procurement and processing services related to ALS clinical trials, outside the submitted work. All other authors have declared no conflicts of interest. Author disclosures are available in the supporting information.
CONSENT STATEMENT
All participants and collateral sources provided written informed consent prior to any study‐related procedures.
Supporting information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
This manuscript has been reviewed by DIAN Study investigators for scientific content and consistency of data interpretation with previous DIAN Study publications. We acknowledge the altruism of the participants and their families and the contributions of the DIAN research and support staff at each of the participating sites for their contributions to this study. Data collection and sharing for this project was supported by The Dominantly Inherited Alzheimer Network (DIAN, U19AG032438) funded by the National Institute on Aging (NIA), the Alzheimer's Association (SG‐20‐690363‐DIAN, AARFD‐21‐851415), the German Center for Neurodegenerative Diseases (DZNE), Raul Carrea Institute for Neurological Research (FLENI), Partial support by the Research and Development Grants for Dementia from Japan Agency for Medical Research and Development, AMED, and the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), Spanish Institute of Health Carlos III (ISCIII), Canadian Institutes of Health Research (CIHR), Canadian Consortium of Neurodegeneration and Aging, Brain Canada Foundation, Fonds de Recherche du Québec – Santé, and the Korea Dementia Research Project through the Korea Dementia Research Center (KDRC), funded by the Ministry of Health & Welfare and the Ministry of Science and ICT, Republic of Korea.
Open access publishing facilitated by Murdoch University, as part of the Wiley ‐ Murdoch University agreement via the Council of Australian University Librarians.
APPENDIX A. DIAN COLLABORATORS
A.1.
| Last Name | First Name | Institution | Affiliation |
|---|---|---|---|
| Bateman | Randall | Washington University | Washington University School of Medicine in St. Louis |
| Daniels | Alisha J. | Washington University | Washington University in St. Louis |
| Courtney | Laura | Washington University | Washington University School of Medicine in St. Louis |
| McDade | Eric | Washington University | Washington University School of Medicine in St. Louis, Department of Neurology |
| Llibre‐Guerra | Jorge J. | Washington University |
Dominantly Inherited Alzheimer's Network Department of Neurology, Washington University School of Medicine in St. Louis |
| Supnet‐Bell | Charlene | Washington University | Washington University in St. Louis, School of Medicine, Department of Neurology |
| Xiong | Chengie | Washington University | Washington University in St. Louis, School of Medicine |
| Xu | Xiong | Washington University | Washington University in St. Louis, School of Medicine |
| Lu | Ruijin | Washington University | Washington University in St. Louis, School of Medicine |
| Wang | Guoqiao | Washington University | Washington University in St. Louis, School of Medicine |
| Li | Yan | Washington University | Washington University in St. Louis, School of Medicine |
| Gremminger | Emily | Washington University | Washington University in St. Louis, School of Medicine |
| Perrin | Richard J. | Washington University | Department of Pathology and Immunology, Department of Neurology, Knight Alzheimer Disease Research Center, Washington University School of Medicine, Saint Louis, MO, USA, |
| Franklin | Erin | Washington University | Department of Pathology and Immunology, Washington University in St. Louis |
| Ibanez | Laura | Washington University | Department of Psychiatry, Department of Neurology, and NeuroGenomics and Informatics Center |
| Jerome | Gina | Washington University | Washington University in St. Louis, School of Medicine, Department of Neurology |
| Herries | Elizabeth | Washington University | Washington University in St. Louis, School of Medicine, Department of Neurology |
| Stauber | Jennifer | Washington University | Washington University in St. Louis, School of Medicine, Department of Neurology |
| Baker | Bryce | Washington University | Washington University in St. Louis, School of Medicine, Department of Neurology |
| Minton | Matthew | Washington University | Washington University in St. Louis, School of Medicine, Department of Neurology |
| Cruchaga | Carlos | Washington University |
(1) Department of Psychiatry, Washington University School of Medicine, St. Louis, MO, USA (2) NeuroGenomics and Informatics Center, Washington University School of Medicine, St. Louis, MO, USA |
| Goate | Alison M. | Mount Sinai | Dept. of Genetics & Genomic Sciences, Dept. of Neuroscience, Ronald M. Loeb Center for Alzheimer's Disease, Icahn School of Medicine at Mount Sinai, NY, NY |
| Renton | Alan E. | Mount Sinai | Ronald M. Loeb Center for Alzheimer's Disease, Dept of Genetics and Genomic Sciences and Nash Family Dept of Neuroscience, Icahn School of Medicine at Mount Sinai |
| Picarello | Danielle M. | Mount Sinai | Ronald M. Loeb Center for Alzheimer's Disease, Dept of Genetics and Genomic Sciences and Nash Family Dept of Neuroscience, Icahn School of Medicine at Mount Sinai |
| Benzinger | Tammie | Washington University | Washington University in St. Louis, Department Radiology |
| Gordon | Brian A. | Washington University | Washington University in St. Louis, Department Radiology |
| Hornbeck | Russ | Washington University | Washington University in St. Louis, Department Radiology |
| Chen | Allison | Washington University | Washington University School of Medicine, St. Louis |
| Chen | Charles | Washington University | Washington University School of Medicine, St. Louis |
| Flores | Shaney | Washington University | Washington University School of Medicine, St. Louis |
| Joseph‐Mathurin | Nelly | Washington University | Washington University School of Medicine, St. Louis |
| Jarman | Steve | Washington University | Washington University School of Medicine, St. Louis |
| Jackson | Kelley | Washington University | Washington University School of Medicine, St. Louis |
| Keefe | Sarah | Washington University | Washington University School of Medicine, St. Louis |
| Koudelis | Deborah | Washington University | Washington University School of Medicine, St. Louis |
| Massoumzadeh | Parinaz | Washington University | Washington University School of Medicine, St. Louis |
| McCullough | Austin | Washington University | Washington University School of Medicine, St. Louis |
| McKay | Nicole | Washington University | Washington University School of Medicine, St. Louis |
| Nicklaus | Joyce | Washington University | Washington University School of Medicine, St. Louis |
| Pulizos | Christine | Washington University | Washington University School of Medicine, St. Louis |
| Wang | Qing | Washington University | Washington University School of Medicine, St. Louis |
| Sabaredzovic | Edita | Washington University | Washington University School of Medicine, St. Louis |
| Smith | Hunter | Washington University | Washington University School of Medicine, St. Louis |
| Scott | Jalen | Washington University | Washington University School of Medicine, St. Louis |
| Simmons | Ashlee | Washington University | Washington University School of Medicine, St. Louis |
| Rizzo | Jacqueline | Washington University | Washington University School of Medicine, St. Louis |
| Hassenstab | Jason | Washington University |
Associate Professor of Neurology and of Psychological & Brain Sciences Washington University in St. Louis |
| Smith | Jennifer | Washington University | Department of Neurology, Washington University in St. Louis |
| Stout | Sarah | Washington University | Department of Neurology, Washington University in St. Louis |
| Aschenbrenner | Andrew J. | Washington University |
Assistant Professor of Neurology Washington University in St. Louis |
| Karch | Celeste M. | Washington University | Department of Psychiatry, Washington University in St Louis |
| Marsh | Jacob | Washington University | DIAN Fibroblast and Stem Cell Bank, Washington University in St. Louis |
| Morris | John C. | Washington University | Washington University in St. Louis, Department of Neurology and the Knight Alzheimer's Disease Research Center |
| Holtzman | David M. | Washington University | Department of Neurology, Hope Center for Neurological Disorders, Knight Alzheimer's Disease Research Center, Washington University in St. Louis |
| Barthelemy | Nicolas | Washington University | Washington University in St. Louis, School of Medicine, Department of Neurology |
| Xu | Jinbin | Washington University | Department of Radiology, Washington University in St. Louis |
| Noble | James M. | Columbia University | Taub Institute for Research on Alzheimer's Disease and the Aging Brain, G.H. Sergievsky Center, Department of Neurology, Columbia University Irving Medical Center |
| Berman | Sarah B. | University of Pittsburgh | University of Pittsburgh Departments of Neurology and Clinical & Translational Science |
| Ikonomovic | Snezana | University of Pittsburgh | University of Pittsburgh, Department of Neurology |
| Nadkarni | Neelesh K. | University of Pittsburgh | University of Pittsburgh, Departments of Medicine (Geriatric Medicine) and Neurology |
| Day | Gregory | Mayo | Department of Neurology, Mayo Clinic in Florida; Jacksonville, FL |
| Graff‐Radford | Neill R. | Mayo | Department of Neurology, Mayo Clinic in Florida; Jacksonville, FL |
| Farlow | Martin | Indiana University | Indiana University School of Medicine |
| Chhatwal | Jasmeer P. | BWH | Massachusetts General Hospital, Brigham and Women's Hospital, Harvard Medical School |
| Ikeuchi | Takeshi | Niigata | Brain Research Institute, Niigata University |
| Kasuga | Kensaku | Niigata | Brain Research Institute, Niigata University |
| Niimi | Yoshiki | Tokyo | Specially appointed lecturer, Unit for Early and Exploratory Clinical Development, The University of Tokyo |
| Huey | Edward D. | Butler | Memory and Aging Program, Butler Hospital, Professor, Department of Psychiatry and Human Behavior, Alpert Medical School, Brown University |
| Salloway | Stephen | Butler | Memory and Aging Program, Butler Hospital, Departments of Psychiatry and Human Behavior and Neurology, Alpert Medical School, Brown University |
| Schofield | Peter R. | Sydney |
(1) Neuroscience Research Australia, Sydney NSW 2031 Australia (2) School of Medical Sciences, University of New South Wales, Sydney NSW 2052 Australia |
| Brooks | William S. | Sydney |
(1) Neuroscience Research Australia, Sydney NSW 2031 Australia; and (2) School of Medical Sciences, University of New South Wales, Sydney NSW 2052 Australia |
| Bechara | Jacob A. | Sydney | Neuroscience Research Australia, Sydney NSW 2031 Australia |
| Martins | Ralph | Perth | Edith Cowan University |
| Fox | Nick C. | UCL |
(1) Dementia Research Centre, UCL Queen Square Institute of Neurology, London, United Kingdom (2) UK Dementia Research Institute at UCL, London, United Kingdom |
| Cash | David M. | UCL |
(1) Dementia Research Centre, UCL Queen Square Institute of Neurology, London, United Kingdom (2) UK Dementia Research Institute at UCL, London, United Kingdom |
| Ryan | Natalie S. | UCL |
(1) Dementia Research Centre, UCL Queen Square Institute of Neurology, London, United Kingdom (2) UK Dementia Research Institute at UCL, London, United Kingdom |
| Jucker | Mathias | Tubingen |
(1) German Center for Neurodegenerative Diseases (DZNE) Tübingen, Tübingen, Germany (2) Hertie‐Institute for Clinical Brain Research, University of Tübingen, Tübingen, Germany |
| Laske | Christoph | Tubingen |
(1) German Center for Neurodegenerative Diseases (DZNE) Tübingen, Tübingen, Germany (2) Hertie‐Institute for Clinical Brain Research, University of Tübingen, Tübingen, Germany |
| Hofmann | Anna | Tubingen |
(1) German Center for Neurodegenerative Diseases (DZNE) Tübingen, Tübingen, Germany (2) Hertie‐Institute for Clinical Brain Research, University of Tübingen, Tübingen, Germany |
| Kuder‐Buletta | Elke | Tubingen | German Center for Neurodegenerative Diseases (DZNE) Tübingen, Tübingen, Germany |
| Graber‐Sultan | Susanne | Tubingen | German Center for Neurodegenerative Diseases (DZNE) Tübingen, Tübingen, Germany |
| Obermueller | Ulrike | Tubingen |
(1) German Center for Neurodegenerative Diseases (DZNE) Tübingen, Tübingen, Germany (2) Hertie‐Institute for Clinical Brain Research, University of Tübingen, Tübingen, Germany |
| Levin | Johannes | Munich | (1) German Center for Neurodegenerative Diseases, site Munich; (2) Department of Neurology, Ludwig‐Maximilians‐Universität München, Munich, Germany; (3) Munich Cluster for Systems Neurology (SyNergy), Munich, Germany |
| Roedenbeck | Yvonne | Munich | (1) German Center for Neurodegenerative Diseases, site Munich; (2) Department of Neurology, Ludwig‐Maximilians‐Universität München, Munich, Germany; (3) Munich Cluster for Systems Neurology (SyNergy), Munich, Germany |
| Vöglein | Jonathan | Munich |
(1) Department of Neurology, LMU University Hospital, LMU Munich, Munich, Germany (2) German Center for Neurodegenerative Diseases (DZNE), Munich, Germany |
| Lee | Jae‐Hong | Seoul | Asian Medical Center, Seoul, South Korea |
| Roh | Jee Hoon | Seoul | Korea University College of Medicine, Seoul, South Korea |
| Sanchez‐Valle | Raquel | Barcelona | Alzheimer's disease and other cognitive disorders group. Neurology Service. Hospital Clínic de Barcelona. FRCB‐IDIBAPS. University of Barcelona, Barcelona (Spain) |
| Rosa‐Neto | Pedro | McGill | Translational Neuroimaging Laboratory, McGill University Research Centre for Studies in Aging, Department of Neurology and Neurosurgery, Psychiatry and Pharmacology and Therapeutics, McGill University, Montreal, Canada |
| Allegri | Ricardo F. | FLENI/Salta | Department of Cognitive Neurology, Instituto Neurológico Fleni, Buenos Aires, Argentina |
| Chrem Mendez | Patricio | FLENI | Department of Cognitive Neurology, Institute for Neurological Research Fleni, Buenos Aires, Argentina |
| Surace | Ezequiel | FLENI | Department of Molecular Biology and Neuropathology, Institute for Neurological Research Fleni, Buenos Aires, Argentina |
| Vazquez | Silvia | FLENI | Center of Molecular Imaging, Institute for Neurological Research Fleni, Buenos Aires, Argentina |
| Lopera | Francisco | Medellin | Grupo de Neurociencias de Antioquia (GNA), Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia. |
| Leon | Yudy Milena | Medellin | Grupo de Neurociencias de Antioquia (GNA), Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia. |
| Ramirez | Laura | Medellin | Grupo de Neurociencias de Antioquia (GNA), Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia. |
| Aguillon | David | Medellin | Grupo de Neurociencias de Antioquia (GNA), Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia. |
| Levey | Allan I. | Emory | Goizueta Alzheimer's Disease Research Center, Emory University, Atlanta, GA 30329 |
| Johnson | Erik C.B | Emory | Goizueta Alzheimer's Disease Research Center, Emory University, Atlanta, GA 30329 |
| Seyfried | Nicholas T. | Emory | Goizueta Alzheimer's Disease Research Center, Emory University, Atlanta, GA 30329 |
| Ringman | John | University of Southern California | Department of Neurology, Keck School of Medicine of USC, University of Southern California |
| Fagan | Anne M. | Washington University | Department of Neurology, Washington University in St. Louis |
| Mori | Hiroshi | Osaka Metropolitan University | |
| Masters | Colin | University of Melbourne | Florey Institute, The University of Melbourne |
Sewell KR, Doecke JD, Xiong C, et al. Longitudinal associations between exercise and biomarkers in autosomal dominant Alzheimer's disease. Alzheimer's Dement. 2024;20:7923–7939. 10.1002/alz.14270
DIAN consortium author list in appendix
REFERENCES
- 1. Livingston G, Huntley J, Sommerlad A, et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet North Am Ed. 2020;396(10248):413‐446. doi: 10.1016/s0140-6736(20)30367-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Blondell SJ, Hammersley‐Mather R, Veerman JL. Does physical activity prevent cognitive decline and dementia? A systematic review and meta‐analysis of longitudinal studies. BMC Public Health. 2014;14(1):510. doi: 10.1186/1471-2458-14-510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stephen R, Hongisto K, Solomon A, Lönnroos E. Physical activity and Alzheimer's disease: a systematic review. J Gerontol A Biol Sci Med Sci. 2017;72(6):733‐739. [DOI] [PubMed] [Google Scholar]
- 4. Beckett MW, Ardern CI, Rotondi MA. A meta‐analysis of prospective studies on the role of physical activity and the prevention of Alzheimer's disease in older adults. BMC Geriatrics. 2015;15(1):9. doi: 10.1186/s12877-015-0007-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ogino E, Manly JJ, Schupf N, Mayeux R, Gu Y. Current and past leisure time physical activity in relation to risk of Alzheimer's disease in older adults. Alzheimers Dement. 2019;15(12):1603‐1611. doi: 10.1016/j.jalz.2019.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sewell KR, Rainey‐Smith SR, Peiffer J, et al. The relationship between objective physical activity and change in cognitive function. Alzheimers Dement. 2023;19(7):2984‐2993. doi: 10.1002/alz.12950 [DOI] [PubMed] [Google Scholar]
- 7. Erickson KI, Donofry SD, Sewell KR, Brown BM, Stillman CM. Cognitive aging and the promise of physical activity. Annu Rev Clin Psychol. 2022;18. doi: 10.1146/annurev-clinpsy-072720-014213 [DOI] [PubMed] [Google Scholar]
- 8. Erickson KI, Voss MW, Prakash RS, et al. Exercise training increases size of hippocampus and improves memory. Proc Natl Acad Sci. 2011;108(7):3017‐3022. doi: 10.1073/pnas.1015950108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tan ZS, Spartano NL, Beiser AS, et al. Physical activity, brain volume, and dementia risk: the Framingham study. J Gerontol A Biol Sci Med Sci. 2016;72(6):789‐795. doi: 10.1093/gerona/glw130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rovio S, Spulber G, Nieminen LJ, et al. The effect of midlife physical activity on structural brain changes in the elderly. Neurobiol Aging. 2010;31(11):1927‐1936. doi: 10.1016/j.neurobiolaging.2008.10.007 [DOI] [PubMed] [Google Scholar]
- 11. Wilckens KA, Stillman CM, Waiwood AM, et al. Exercise interventions preserve hippocampal volume: a meta‐analysis. Hippocampus. 2021;31(3):335‐347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brown BM, Peiffer J, SR R‐S. Exploring the relationship between physical activity, beta‐amyloid and tau: a narrative review. Ageing Res Rev. 2019;50:9‐18. doi: 10.1016/j.arr.2019.01.003 [DOI] [PubMed] [Google Scholar]
- 13. Law LL, Rol RN, Schultz SA, et al. Moderate intensity physical activity associates with CSF biomarkers in a cohort at risk for Alzheimer's disease. Alzheimers Dement. 2018;10:188‐195. doi: 10.1016/j.dadm.2018.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Northey JM, Cherbuin N, Pumpa KL, Smee DJ, Rattray B. Exercise interventions for cognitive function in adults older than 50: a systematic review with meta‐analysis. Br J Sports Med. 2018;52(3):154‐160. doi: 10.1136/bjsports-2016-096587 [DOI] [PubMed] [Google Scholar]
- 15. Huang X, Zhao X, Li B, et al. Comparative efficacy of various exercise interventions on cognitive function in patients with mild cognitive impairment or dementia: a systematic review and network meta‐analysis. J Sport Health Sci. 2022;11(2):212‐223. doi: 10.1016/j.jshs.2021.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bertram L, Lill CM, Tanzi RE. The genetics of Alzheimer disease: back to the future. Neuron. 2010;68(2):270‐281. [DOI] [PubMed] [Google Scholar]
- 17. Brown BM, Sohrabi HR, Taddei K, et al. Habitual exercise levels are associated with cerebral amyloid load in presymptomatic autosomal dominant Alzheimer's disease. Alzheimers Dement. 2017;13(11):1197‐1206. doi: 10.1016/j.jalz.2017.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Müller S, Preische O, Sohrabi HR, et al. Relationship between physical activity, cognition, and Alzheimer pathology in autosomal dominant Alzheimer's disease. Alzheimers Dement. 2018;14(11):1427‐1437. doi: 10.1016/j.jalz.2018.06.3059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Morris JC, Aisen PS, Bateman RJ, et al. Developing an international network for Alzheimer research: the Dominantly Inherited Alzheimer Network. Clin Investig. 2012;2(10):975‐984. doi: 10.4155/cli.12.93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ryman DC, Acosta‐Baena N, Aisen PS, et al. Symptom onset in autosomal dominant Alzheimer disease: a systematic review and meta‐analysis. Neurology. 2014;83(3):253‐260. doi: 10.1212/wnl.0000000000000596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bateman RJ, Xiong C, Benzinger TL, et al. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med. 2012;367(9):795‐804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Craig CL, Marshall AL, SJÖSTRÖM M, et al. International Physical Activity Questionnaire: 12‐Country reliability and validity. Med Sci Sports Exercise. 2003;35(8):1381‐1395. doi: 10.1249/01.mss.0000078924.61453.fb [DOI] [PubMed] [Google Scholar]
- 23. Tatiana B, Didier C, David R, Derek Y. mixtools: an R package for analyzing finite mixture models. J Stat Softw. 2009;32(6):1‐29. [Google Scholar]
- 24. Fagan AM, Xiong C, Jasielec MS, et al. Longitudinal Change in CSF Biomarkers in Autosomal‐Dominant Alzheimer's Disease. Sci Transl Med. 2014;6(226):226ra30‐226ra30. doi: 10.1126/scitranslmed.3007901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jack CR Jr, MA B, NC F, et al. The Alzheimer's disease neuroimaging initiative (ADNI): mRI methods. J Magn Reson Imaging. 2008;27(4):685‐691. doi: 10.1002/jmri.21049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McKay NS, Gordon BA, Hornbeck RC, et al. Positron emission tomography and magnetic resonance imaging methods and datasets within the Dominantly Inherited Alzheimer Network (DIAN). Nat Neurosci. 2023;26(8):1449‐1460. doi: 10.1038/s41593-023-01359-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Benjamini Y, Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Annals of statistics. 2001:1165‐1188. https://www.jstor.org/stable/2674075 [Google Scholar]
- 28. Chhatwal JP, Schultz SA, McDade E, et al. Variant‐dependent heterogeneity in amyloid β burden in autosomal dominant Alzheimer's disease: cross‐sectional and longitudinal analyses of an observational study. Lancet Neurol. 2022;21(2):140‐152. doi: 10.1016/S1474-4422(21)00375-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McDade E, Wang G, Gordon BA, et al. Longitudinal cognitive and biomarker changes in dominantly inherited Alzheimer disease. Neurology. 2018;91(14):e1295‐e1306. doi: 10.1212/wnl.0000000000006277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Llibre‐Guerra JJ, Li Y, Schindler SE, et al. Association of longitudinal changes in cerebrospinal fluid total tau and phosphorylated tau 181 and brain atrophy with disease progression in patients with Alzheimer disease. JAMA Netw Open. 2019;2(12):e1917126‐e1917126. doi: 10.1001/jamanetworkopen.2019.17126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bateman RJ, Aisen PS, De Strooper B, et al. Autosomal‐dominant Alzheimer's disease: a review and proposal for the prevention of Alzheimer's disease. Alzheimers Res Ther 2011;3(1):1. doi: 10.1186/alzrt59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xiong C, McCue LM, Buckles V, et al. Cross‐sectional and longitudinal comparisons of biomarkers and cognition among asymptomatic middle‐aged individuals with a parental history of either autosomal dominant or late‐onset Alzheimer's disease. Alzheimers Dement. 2023;19(7):2923‐2932. doi: 10.1002/alz.12912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Morris JC, Weiner M, Xiong C, et al. Autosomal dominant and sporadic late onset Alzheimer's disease share a common in vivo pathophysiology. Brain. 2022;145(10):3594‐3607. doi: 10.1093/brain/awac181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ten Brinke LF, Bolandzadeh N, Nagamatsu LS, et al. Aerobic exercise increases hippocampal volume in older women with probable mild cognitive impairment: a 6‐month randomised controlled trial. Br J Sports Med. 2015;49(4):248‐254. doi: 10.1136/bjsports-2013-093184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gogniat MA, Robinson TL, Miller LS. Exercise interventions do not impact brain volume change in older adults: a systematic review and meta‐analysis. Neurobiol Aging. 2021;101:230‐246. doi: 10.1016/j.neurobiolaging.2021.01.025 [DOI] [PubMed] [Google Scholar]
- 36. Winer JR, Mander BA, Helfrich RF, et al. Sleep as a potential biomarker of tau and β‐amyloid burden in the human brain. J Neurosci. 2019;39(32):6315‐6324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Villemagne VL, Burnham S, Bourgeat P, et al. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer's disease: a prospective cohort study. Lancet Neurol. 2013;12(4):357‐367. doi: 10.1016/s1474-4422(13)70044-9 [DOI] [PubMed] [Google Scholar]
- 38. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353‐356. [DOI] [PubMed] [Google Scholar]
- 39. Prince SA, Adamo KB, Hamel ME, Hardt J, Gorber SC, Tremblay M. A comparison of direct versus self‐report measures for assessing physical activity in adults: a systematic review. Int J Behav Nutr Phys Act. 2008;5(1):56. doi: 10.1186/1479-5868-5-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information