

Abstract
INTRODUCTION
Small molecules and antibodies are being developed to lower amyloid beta (Aβ) peptides.
METHODS
We describe MEDI1814, a fully human high‐affinity monoclonal antibody selective for Aβ42, the pathogenic self‐aggregating species of Aβ.
RESULTS
MEDI1814 reduces free Aβ42 without impacting Aβ40 in the cerebrospinal fluid of rats and cynomolgus monkeys after systemic administration. MEDI1814 administration to patients with Alzheimer's disease (AD; n = 57) in single or repeat doses up to 1800 mg intravenously or 200 mg subcutaneously was associated with a favorable safety and tolerability profile. No cases of amyloid‐related imaging abnormalities were observed. Predictable dose‐proportional changes in serum exposures for MEDI1814 were observed across cohorts. Cerebrospinal fluid (CSF) analysis demonstrated central nervous system penetration of MEDI1814. Pharmacodynamic data showed dose‐dependent suppression of free Aβ42, increases in total (bound and free) Aβ42, but no change in total Aβ40 in CSF across doses.
DISCUSSION
MEDI1814 offers a differentiated approach to impacting Aβ in AD via selective reduction of free Aβ42.
Keywords: amyloid beta 42, drug development, pharmacodynamics, pharmacokinetics, preclinical, safety, tolerability
1. BACKGROUND
Alzheimer's disease (AD) is the most common type of dementia and is characterized by cognitive deterioration and impaired activities of daily living. 1 The predominant pathologies associated with AD in the brain are aggregates of amyloid beta (Aβ) deposited in the extracellular space, and intraneuronal neurofibrillary tangles of the microtubule‐associated protein tau. 2 The amyloid cascade hypothesis 3 , 4 , 5 , 6 supported by preclinical work on oligomers 7 , 8 , 9 and recent clinical evaluation of plaque lowering mean that these remain a dominant model hypothesized for the progression of AD. As a consequence, Aβ has been a primary target for therapeutic interventions, with most experimental drugs in clinical trials over the last two decades aiming to either reduce its production (with small molecule γ‐secretase and β‐site amyloid precursor protein cleaving enzyme inhibitors) or promoting its clearance (with immunotherapies). 10 Of the forms of Aβ targeted, Aβ42 and Aβ43 are highly hydrophobic and self‐aggregating, whereas Aβ40 is less so. Indeed, Aβ40 has been postulated to have anti‐amyloidogenic and neuroprotective effects in the brain, although this has not been demonstrated in clinical trials. 11 , 12 Presenilin 1 (PSEN1, the catalytic subunit of the γ‐secretase complex) is considered to play an important role in the generation of Aβ, with most mutations not increasing total Aβ generation, but instead increasing the release and ratio of longer (less trimmed), amyloidogenic species of Aβ (≥ Aβ42). 13 , 14 In addition to full‐length Aβ42, other highly amyloidogenic and neurotoxic forms of Aβ are also abundant in the brains of AD patients, including N‐terminal truncated versions, such as pyroglutamate‐modified Aβ3‐42 (pGlu‐Aβ3‐42). 15 , 16 , 17
MEDI1814 is a fully human high‐affinity monoclonal antibody selective for amyloid beta (Aβ)42.
MEDI1814 reduces free Aβ42 without impacting Aβ40 after systemic administration.
Predictable dose‐proportional changes in serum exposures for MEDI1814 were observed.
MEDI1814 was associated with a favorable safety and tolerability profile.
Developments relating to the identification of cerebrospinal fluid (CSF) and positron emission tomography (PET) imaging biomarkers for AD have enabled earlier diagnosis, and their application in clinical research has led to successful Phase 2 and 3 clinical trials of immunotherapies. 18 , 19 , 20 As a result of some of these trials, the US Food and Drug Administration (FDA) has granted accelerated approval for, and approved, two passive immunotherapy treatments, aducanumab‐avwa 21 and lecanemab‐irmb, 22 which both demonstrate significant dose‐ and time‐dependent reduction of amyloid plaques.
However, passive immunotherapies targeting aggregated forms of Aβ, including those that have been approved for therapeutic use, are associated with increased amyloid‐related imaging abnormalities (ARIAs) and immunotherapies targeting soluble forms of Aβ in completed late‐stage trials have proven unsuccessful despite strong binding to total Aβ in periphery and reduction of free Aβ. 23 , 24 One other critical factor to consider is that all the passive immunotherapeutic approaches have targeted both the Aβ40 and Aβ42 forms of the peptide, thus reducing levels of Aβ40, which has been suggested to have a neuroprotective effect. 25
Here, we describe the discovery, non‐clinical, and early clinical development of MEDI1814, a fully human, effector‐null monoclonal antibody that has high affinity and selectivity for full‐length and N‐terminal truncated forms of the amyloidogenic species Aβ42/43 versus the neuroprotective species Aβ40. Sequestering the free Aβ42 monomer should prevent the further formation and accumulation of toxic oligomeric forms of Aβ42, thereby slowing disease progression. We also report early data on the downstream biomarker neurofilament light chain (NfL). Clinical data are presented from single ascending dose (SAD) and multiple ascending dose (MAD) studies undertaken to evaluate the safety and pharmacokinetics (PK) of MEDI1814 in patients with AD. Based on these data and the observed PK/pharmacodynamic (PD) profile for MEDI1814‐mediated Aβ42 suppression in the CSF, this study identifies a potential dosing regimen for further clinical study in patients with AD.
2. METHODS
2.1. Clinical study design
The Phase 1 clinical study was a randomized, double‐blind, placebo‐controlled, two‐part investigation to evaluate the safety, tolerability, PK, PD, and immunogenicity of single (Part 1) and multiple doses (Part 2) of MEDI1814 administered to patients with mild to moderate AD (Figure S1 in supporting information). The protocol was executed in the United States under an investigational new drug application in accordance with the principles of Good Clinical Practice, having received the relevant institutional review board and FDA approvals (ClinicalTrials.gov Identifier NCT02036645; AstraZeneca Protocol D4750C00001). Written informed consent was obtained from patients or their legal representative prior to any study procedures. Part 1 of the study was undertaken as a dose‐ascending study of single intravenous (IV) MEDI1814 doses (25, 100, 300, 90, and 1800 mg) or a single subcutaneous (SC) dose (100 mg) administered across six cohorts of patients with AD (n = 5 for 25 mg IV; n = 8 for other cohorts; and n = 2 placebo per cohort). In Part 2 of the study, multiple ascending doses were evaluated after IV (300, 900, and 1800 mg) or SC (200 mg) administration across four cohorts of patients with AD (n = 8 for all multiple dose cohorts; n = 2 placebo and n = 6 MEDI1814 per cohort). Three doses were administered, each dose separated by 29 days. For both study parts, eligibility criteria were for male and female (non‐childbearing potential) patients, aged 55 to 85 years, with mild to moderate AD (National Institute on Aging–Alzheimer's Association criteria 26 ), with cognitive and functional symptoms of probable AD present for ≥ 6 months prior to randomization (Mini‐Mental State Examination [MMSE] score ≥ 16 to 26; magnetic resonance imaging [MRI] scan at screening consistent with a diagnosis of dementia; Rosen Modified Hachinski Ischemic score £4 and body mass index 17–32 kg/m2). Patients enrolled in Part 2 were also required to have a CSF Aβ1‐42 concentration of < 550 pg/mL.
Study participants were assessed at follow‐up visits for ≈ 13 weeks after the single dose (Part 1) and after the last repeat dose (Part 2). Safety and tolerability were monitored throughout both parts of the study by adverse events, clinical assessments (vital signs, electrocardiograms, physical and neurological examinations, MMSE, and Columbia‐Suicide Severity Rating Scale) and laboratory tests (clinical chemistry, hematology, and urinalysis). Data were reviewed by a safety review committee between successive cohorts of patients. A follow‐up MRI scan was undertaken at day 36 after the single dose (Part 1) and after the last repeat dose (Part 2). Blood was sampled for MEDI1814 PK and PD biomarkers (total Aβ1‐42) and immunogenicity throughout the respective follow‐up periods. Lumbar puncture was performed at day 29 after the single dose (Part 1) and after the last repeat dose (Part 2) for analysis of CSF biomarkers (total and free Aβ1‐42, total Aβ1‐40).
RESEARCH IN CONTEXT
Systematic review: The authors reviewed the scientific literature using PubMed. Small molecules and antibodies are being developed to lower amyloid beta (Aβ) peptides and have been evaluated in clinical studies. However, these typically lower both Aβ40 and Aβ42, and targeting the key toxic building blocks of Aβ while sparing Aβ40 may provide advantages in terms of potential safety and efficacy.
Interpretation: We describe the discovery and early development of MEDI1814, which selectively lowers free Aβ42, but not Aβ40, in the cerebrospinal fluid of patients with Alzheimer's disease (AD). Overall, MEDI1814 was well tolerated in human subjects after both single‐ and multiple‐dose intravenous administration.
Future directions: The question remains to be addressed whether selective reduction of free Aβ42 can provide efficacy for patients with AD. Further clinical evaluation of MEDI1814 may provide greater insight into the involvement of Aβ in the initiation of AD.
2.2. Materials
Aβ peptides were purchased from Anaspec, rPeptide, or Bachem and were reconstituted to 1 mg/mL in 1% ammonium hydroxide (v/v). Batches of MEDI1814 and other immunoglobulin G (IgG) antibodies were produced through Chinese hamster ovary (CHO) cell expression and subsequent protein A purification (as described by Daramola et al. 27 ).
2.3. Discovery of MEDI1814
Fab phage display libraries 28 were used to isolate Aβ1‐42 specific antibodies via a series of selection cycles on synthetic human biotinylated Aβ1‐42 (rPeptide), essentially as previously described. 29 , 30 Soluble Fab fragments were generated 28 and tested for binding to human Aβ1‐42 and Aβ1‐40 in a homogenous time‐resolved fluorescence assay (HTRF, Cisbio Bioassays). Abet0007 was converted to scFv format and cloned into a modified pCantab6 vector. 30 Large scFv‐phage libraries were derived by oligonucleotide‐directed mutagenesis of the VH and VL complementarity determining regions (CDR)3s using standard molecular biology techniques as described by Clarkson and Lowman. 31 These were subjected to three rounds of selection on decreasing concentrations of Aβ1‐42. ScFv fragments were screened as above for improved binding over Abet0007, with binding kinetics measured by surface plasmon resonance (SPR; see methods below). Abet0144 underwent further affinity maturation by targeting all six CDRs as above and by selection using ribosome display (essentially as described by Lewis and Lloyd 32 ). Briefly, each individual randomized CDR was subjected to several rounds of affinity selection and then recombined by polymerase chain reaction in a pair‐wise fashion to produce new libraries with 2/6 CDRs mutated. This proceeded until 4/6 CDRs were recombined and a total of 11 rounds of selection had been performed.
2.4. BIAcore affinity measurements and peptide competition assay
The binding kinetics of anti‐Aβ42 antibodies were measured by SPR using the BIAcore T‐100 (essentially as described by Karlsson et al. 33 ). Antibodies were covalently coupled by amine linkage to a CM5 chip surface and Aβ peptides flowed over a range of concentrations (depending on KD of antibody tested). The peptide competition was performed by testing the ability of unlabeled Aβ peptides to compete for binding of Abet0144 or Abet0380 IgG to biotinylated Aβ1‐42 using HTRF, with europium cryptate labelled anti‐human Fc IgG and XL665‐labelled streptavidin from CisBio.
2.5. Animal care and use
All procedures were performed in accordance with the AstraZeneca Care and Use of Animals policy, complied with the Animals (Scientific Procedures) Act 1986, and were approved by a local ethics committee. The cynomolgus monkey study was performed at WIL Research (Lyon, France), study number AB141816, and was approved by their local ethics committee. The study was conducted in compliance with International Council for Harmonisation (ICH) and European Medicines Agency guidelines on repeated dose toxicity (CPMP/SWP/1042/99). In accordance with recommendations of the Weatherall report, the cynomolgus monkeys used in the study were housed in socialized groups of up to three of the same dose group in a communal housing compartment (dimensions: 1.80 × 1.20 × 2.10 m) with sawdust as bedding and separated into single units for the time necessary to complete the treatment procedures and the individual observations/examinations. The partitions between the units and main compartment were then removed to allow socialization. They were fed with expanded complete commercial primate diet (Special Diets Services: OWM [E] BANANA SQC SHORT), as well as receiving fruit or vegetables daily. The animals were fasted for at least 15 hours before clinical laboratory blood sampling, during urine collection, and before necropsy. All animals were also fasted before all procedures requiring anesthesia.
2.6. In vivo pharmacodynamic effect of MEDI1814
MEDI1814 was formulated in a histidine buffer for IV and SC administration. Control animals were treated with the appropriate vehicle. The rat in vivo pharmacology study was performed in Sprague–Dawley rats (8–12 males per group), with MEDI1814 administered at 0.25, 1, 5, and 10 mg/kg IV weekly over 2 weeks.
In vivo animal toxicity studies were performed in Sprague–Dawley rats (14 animals per sex per group) and cynomolgus monkeys (4 animals per sex per group) with weekly doses of 0, 10, and 100 mg/kg IV or 75 mg/kg SC (or IV/SC vehicle) administered over 13 weeks, followed by an 8‐week (rats) or 9‐week (monkeys) treatment‐free period. Vehicle was administered by both routes to the same animals. At the end of the dosing period, CSF samples were collected from ten rats and six monkeys per group and, at the end of the treatment free period, from the five rats per group that continued on study (10 mg/kg IV not included), and the additional two monkeys included in the vehicle and 100 mg/kg IV groups. Free and total Aβ42, and total Aβ40 were measured from CSF, while total Aβ42 was measured in plasma.
2.7. Quantitation of free and total Aβ42 and Aβ40 in rat and non‐human primate samples
Aβ42 and Aβ40 were assayed using commercially available enzyme‐linked immunosorbent assay (ELISA) kits validated at AstraZeneca. Methods were derived from Bogstedt et al. 5 Briefly, to assess free peptide, Aβ42 bound to MEDI1814 was removed by immunoprecipitation (IP) prior to ELISA analysis of the free Aβ42. To assess total peptide, samples were either heat treated to remove interfering MEDI1814 prior to analysis of total Aβ42 by ELISA, or an excess of MEDI1814 was applied to the samples with a subsequent detection of the Aβ42–MEDI1814 complex. Aβ40 was measured directly in CSF by ELISA.
2.7.1. Free Aβ42 in CSF
Free Aβ42 was analyzed in CSF from non‐human primates (NHPs) according to Bogstedt et al. 5 Briefly, Aβ42 bound to MEDI1814 was removed by IP prior to ELISA analysis of the free Aβ42 using a commercial ELISA kit (Innogenetics: cat no 80177). Free Aβ42 was analyzed in CSF from rats according to Bogstedt et al. 5 with some modifications. Further details can be found in the supporting information.
2.7.2. Total Aβ42 in CSF
Total Aβ42 was analyzed in CSF from NHPs according to Bogstedt et al. 5 Briefly, the CSF was heat treated to remove interfering MEDI1814 prior to analysis using a commercial ELISA kit (Innogenetics: cat no 80177). Total Aβ42 was analyzed in CSF from rats according to Bogstedt et al. 5 either by using an excess of MEDI1814 and subsequent detection of the Aβ42‐MEDI1814 complex by a secondary anti‐hIgG antibody, or by heat treatment of the CSF to remove interfering MEDI1814 prior to analysis using a commercial ELISA kit. Further details can be found in the supporting information.
2.7.3. Aβ40 in CSF
Aβ40 was analyzed in CSF from NHPs using the High Sensitivity Human Amyloid β40 ELISA kit (MilliporeSigma; EZHS40). The ELISA analysis was performed according to the manufacturer's instructions and Bogstedt et al. 5
Analytical methods for quantification of total Aβ42 in plasma and the brain, and Aβ40 in the brain can be found in the supporting information.
2.8. Quantitation of free and total Aβ42 and Aβ40 in human samples
Aβ42 and Aβ40 were assayed using commercially available electrochemiluminescence immunoassay (ECLIA) kits validated at AstraZeneca (Meso Scale Diagnostics [MSD]). Briefly, to assess free peptide, Aβ42 bound to MEDI1814 was removed by IP using peptide A and peptide G beads (Dynabeads, Novex by Life Technologies) prior to analysis, while unbound free Aβ1‐42 remained in the supernatant. To assess total Aβ42, samples were heat treated to remove interfering MEDI1814. Total Aβ1‐40 was measured directly in CSF by ECLIA. Further details can be found in the supporting information.
2.9. PK analysis and PK/PD modeling
MEDI1814 PK concentrations in rat and cynomolgus monkey serum and CSF were measured using a quantitative MSD electrochemiluminescent (ECL) assay. Anti‐drug antibody (ADA) evaluations in rat and cynomolgus monkey serum were performed using a solution phase, ECL bridging immunoassay developed and validated at AstraZeneca. A validated ECL assay using an MSD platform was used for quantitative determination of MEDI1814 in human serum and CSF and the detection of ADA to MEDI1814 in human serum. Non‐compartmental PK analysis was performed using Phoenix WinNonlin Professional (version 6.3, Certera).
A two‐compartment model with IV/SC input and first‐order elimination was used to describe the PK data in cynomolgus monkeys. The relationship between serum concentration of MEDI1814 and plasma total Aβ42 was described using a PK/PD model in which MEDI1814 binds with free Aβ42 to form a MEDI1814‐Aβ42 complex. Population PK/PD modeling was conducted using non‐linear mixed‐effects modeling (NONMEM version 7.2, ICON plc).
The PK/PD model based on cynomolgus monkeys was used to simulate the PK/PD profiles of MEDI1814 concentration profiles in humans with scaled human PK and PD parameter estimates and assumptions. The total and free Aβ42 in CSF were simulated using a similar structural PK/PD model (as in blood) with a MEDI1814 CSF‐to‐serum concentration of ≈ 0.1%, and used to determine the starting and target doses of MEDI1814 for the Phase 1 clinical study.
2.10. Quantitation of NfL and P‐tau217 in human samples
The Quanterix NfL assay (Catalog number: 103186) was run in accordance with the manufacturer's protocol on the Quanterix HD‐1 instrument. Briefly, human samples were thawed on wet ice and vortexed prior to centrifugation at 2000 × g for 10 minutes at 4°C. Plasma samples were then diluted 1:4 in the provided sample diluent, CSF samples were diluted 1:100 in the provided sample diluent from the kit, and each sample was run on the Quanterix HD‐1.
Eli Lilly and Company provided the measurements of the previously published in‐house immunoassay for phosphorylated tau (p‐tau)217 using the MSD platform and a streptavidin small spot plate (MSD, L45SA). A p‐tau217–specific biotinylated monoclonal antibody was used as the capture antibody (Lilly, biotinylated‐IBA493) and sulfo‐tagged amino‐terminal tau antibody as the detector (Lilly, SULFO‐TAG‐Ru‐4G10‐E2). Antibodies were conjugated with NHS‐PEG4‐Biotin (Thermo Scientific, catalog number: A39259) or MSD GOLD SULFO‐TAG NHS‐Ester (MSD, catalog number: R91AO‐1) according to the manufacturer's protocol. The assay was run as previously described. 34
2.11. Brain MRI
MRI, with central radiological read, was used during the course of this study to help assess safety such as the occurrence of ARIAs. These include hemosiderin deposition (ARIA‐H) and signs potentially indicative of inflammation or vasogenic edema (ARIA‐E). 35 MRI examination was performed on three occasions: screening, Day 36 (after a single dose), and Day 92 (after multiple doses). The MRI sequences to assess safety included 3D T1‐weighted gradient echo scans, T2*‐weighted gradient echo scans, T2‐weighted spin echo scans, and T2‐weighted fluid attenuated inversion recovery scans.
2.12. Statistical analysis
For the non‐clinical in vivo profile of MEDI1814, decreases in free and total Aβ42 in CSF and total Aβ40 and Aβ42 in the brain were assessed relative to isotype control using one‐way analysis of variance and Tukey multiple comparison (GraphPad Prism version 8.3).
For the clinical study, the safety analysis set included all subjects who received at least one dose of MEDI1814 or placebo, and for whom any post‐dose data were available. The PK analysis set included all evaluable PK data appropriate for the evaluation of interest from all subjects who received at least one dose of MEDI1814. Subjects who received placebo were not part of the PK analysis set. The PD (biomarker) analysis set included all evaluable biomarker data appropriate for the evaluation of interest. Statistical analyses were performed using SAS Enterprise Guide 4.3 supported by SAS version 9.2 on UNIX.
The main presentation period for adverse events (AEs) was the time from first dose to follow‐up. In addition, to capture AEs related to CSF sampling, AEs are also presented separately for each CSF sampling time period. Project‐specific reference limits were used to flag potentially clinically significant values for each subject. When project‐specific reference limits were not available, the reference limits of the local laboratory were used.
Dose proportionality was evaluated using a power model applied to each of the following PK parameters derived from MEDI1814 serum concentrations: maximum serum concentration (Cmax), area under the serum concentration‐time curve (AUC) from zero to the last measurable concentration (AUC0‐t), AUC from zero to infinity (AUC0‐∞), and AUC over the dosing interval (AUC0 τ). For Part 2 (MAD), separate analyses were conducted for Day 1 and Day 57. For CSF free Aβ1‐42, total Aβ1‐42, and total Aβ1‐40, an analysis of covariance linear model was used to estimate the difference between MEDI1814 and placebo in percent change from baseline response, with baseline value included in the model as a covariate.
Due to the exploratory nature of the study, the sample size was not based on formal statistical considerations. The sample size was based on experience from previous similar Phase 1 studies with other compounds to obtain adequate safety, tolerability, and PK data to achieve the objectives of the study while exposing as few subjects as possible to study medication and procedures.
3. RESULTS
3.1. Discovery of MEDI1814
Abet0007, the parental precursor of MEDI1814, was derived by phage display panning of antibody libraries against Aβ1‐42. This precursor had the desired Aβ42 selectivity, but only a moderate affinity (KD) of 473 nM (Table S1 in supporting information). Abet0007 was then subjected to in vitro affinity maturation by targeting the variable heavy (VH) and variable light (VL) CDRs through the generation of randomized libraries. First, libraries just targeting the CDR3 domains underwent further phage display panning and screening for higher affinity variants. This resulted in a variant, Abet0144, with a moderate improvement in affinity at a KD of 9.5 nM (Table S1), with retained selectivity for Aβ42 over Aβ40. Thereafter, further affinity optimization was undertaken (as above), but this time targeting all six CDRs of Abet0144, using ribosome display for the panning 32 , 36 and iteratively recombining beneficial mutations across the different CDR regions. This resulted in a large panel of scFv variants with improved KD versus Abet0144, while maintaining selectivity for Aβ42. The highest affinity clones were reformatted to human IgG1‐triple mutation (TM) 37 and profiled further. The lead Abet0380 had a measured KD of 322 pM (Table S1), with an improved off‐rate over Abet0144. The selectivity profile of Abet0380 for different Aβ peptides was reconfirmed in competition binding experiments (Figure 1). Inhibition was observed with full‐length Aβ1‐42, full‐length Aβ1‐43, and all Aβ1‐42 peptide forms truncated only at the N‐terminus, with half‐maximal inhibitory concentration (IC50) values within 3‐fold of each other. No inhibition was observed with the truncated peptides Aβ1‐16 and Aβ12‐28, and only relatively weak and incomplete inhibition was observed with Aβ1‐40 (≈ 2000‐fold weaker than Aβ1‐42). This is consistent with MEDI1814 having a C‐terminal epitope, with high selectivity for full‐length Aβ1‐42 and Aβ1‐43 (versus Aβ1‐40) and high affinity for both full and N‐terminal–only truncated forms of Aβ1‐42.
FIGURE 1.
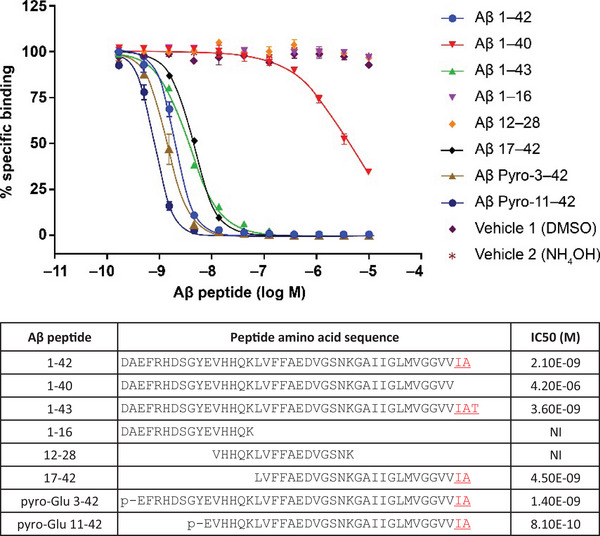
Aβ peptide selectivity of MEDI1814. Aβ peptide competition of MEDI1814 to biotinylated Aβ1‐42 using homogeneous time‐resolved fluorescence, with europium cryptate labelled anti‐human Fc immunoglobulin and XL665‐labelled streptavidin from CisBio. Peptide sequences and half‐maximal inhibitory concentration values are depicted. MEDI1814 has high selectivity for full length Aβ1‐42 and Aβ1‐43 (versus Aβ1‐40) and high affinity for both full and N‐terminal‐only truncated forms of Aβ1‐42. Aβ, amyloid beta.
3.2. Non‐clinical in vivo profile of MEDI1814
In a 14‐day rat study (Figure 2A‐D), two IV administrations of MEDI1814 (0.25–10 mg/kg) at days 0 and 7 gave a dose‐dependent decrease of free Aβ42 in CSF measured at trough (7 days post second treatment). Maximal suppression of free Aβ42 was observed at 5 and 10 mg/kg down to the limit of quantification (LOQ) in the assay (38pg/mL). Significant effects were observed at 0.5 to 10 mg/kg relative to isotype control (0.5 mg/kg, p < 0.01; 1.0 mg/kg, p < 0.001; 5 and 10 mg/kg, p < 0.0001). Due to sequestration of Aβ42 by MEDI1814, a dose‐dependent increase of total (bound and free) Aβ42 was demonstrated in both CSF and brain (both p < 0.0001 vs. isotype control at 5 and 10 mg/kg). Total Aβ40 in the brain was unaffected by either isotype control or MEDI1814, thus demonstrating the selectivity of MEDI1814 for Aβ42 in the brain. Plasma and CSF exposure to MEDI1814 were measured only at the end of the experiment, with 0.1% to 0.15% of the plasma concentration being present in the CSF (Figure S2 in supporting information).
FIGURE 2.
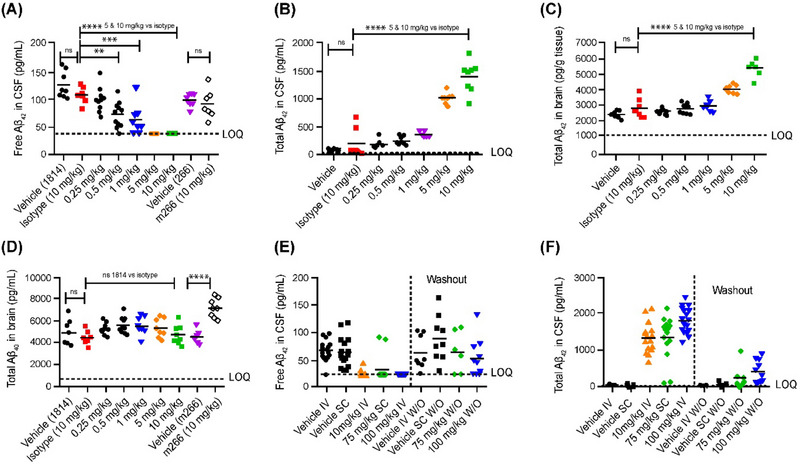
Pharmacodynamics of MEDI1814 in Sprague–Dawley rats. Intravenous administration of MEDI1814 and m266 twice over 14 days with samples collected 7 days post second dose (A–D). Maximal suppression of free Aβ42 was observed at 5 and 10 mg/kg, with significant effects being observed at 0.5–10 mg/kg relative to isotype control. Dose‐dependent increase of total (bound and free) Aβ42 is observed both CSF and brain. Total Aβ40 in the brain was unaffected by either isotype control or MEDI1814, showing selectivity of MEDI1814 for Aβ42 in the brain. MEDI1814 dosed 14 times over 13 weeks, with samples collected 8 weeks after the final dose (E and F). Free Aβ42 in CSF decreased in all the MEDI1814 treatment groups after 14 weekly doses (E). Total CSF Aβ42 increased dose‐dependently in the 10 mg/kg and 100 mg/kg IV and 75 mg/kg SC groups compared to the appropriate vehicle group after 14 doses (F). At the end of the treatment‐free period, 8‐ and 3.5‐fold higher total Aβ42 levels in the CSF were observed in the 100 mg/kg IV and 75 mg/kg SC groups, respectively. Concentrations shown are absolute in pg/mL as determined by ELISA. One‐way analysis of variance and Tukey multiple comparison: ns p ≥ 0.05, *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. Aβ, amyloid beta; CSF, cerebrospinal fluid; ELISA, enzyme‐linked immunosorbent assay; IV, intravenous; SC, subcutaneous.
A 13‐week, repeat‐dose study was conducted to assess the toxicological profile of MEDI1814 (10 mg/kg IV, 100 mg/kg IV, or 75 mg/kg SC) in Sprague–Dawley rats and cynomolgus monkeys. In rats, free Aβ42 in CSF was reduced to below the LOQ in all the MEDI1814 treatment groups after 14 weekly doses (p < 0.0001; Figure 2E). After an 8‐week treatment‐free period, there was a 17.8% and 28.8% reduction in free Aβ42 in the CSF remaining in the 100 mg/kg IV and 75 mg/kg SC groups, respectively, relative to the appropriate vehicle (p > 0.05; Figure 2E). Total CSF Aβ42 increased dose‐dependently by 27‐, 36‐, and 26‐fold in the 10 mg/kg and 100 mg/kg IV and 75 mg/kg SC groups compared to the appropriate vehicle group after 14 doses (Figure 2F). At the end of the treatment‐free period, 8‐ and 3.5‐fold higher total Aβ42 levels in the CSF were observed in the 100 mg/kg IV and 75 mg/kg SC groups, respectively, compared to the appropriate vehicle group (p > 0.05 for both; Figure 2F). MEDI1814 serum concentrations over the course of the 13‐week treatment period in rats are shown in Figure S3 in supporting information.
In cynomolgus monkeys, a dose‐dependent increase in total Aβ42 in both plasma (1.1‐fold [p = 0.0002], and 2.1‐ and 1.5‐fold [both p < 0.0001]) and CSF (3.7‐fold [p < 0.01], and 7.7‐ and 6.1‐fold [both p < 0.0001]) was observed at the end of the treatment phase in the 10 mg/kg IV, 100 mg/kg IV, or 75 mg/kg SC groups, respectively (Figure 3). Greater than 95% suppression of free Aβ42 in CSF was achieved at all dose levels at the end of the treatment phase (p < 0.0001; Figure 3A). At the end of a 9‐week treatment‐free period, total Aβ42 levels in plasma were 1.8‐fold higher in the 100 mg/kg IV dose group compared to the vehicle group (Figure 3B); total CSF Aβ42 was 3.5‐fold higher (not significant vs. vehicle group, Figure 3C), and free CSF Aβ42 was suppressed by 87.5% compared to the vehicle group (p < 0.0001). The specificity of MEDI1814 binding to Aβ42 over Aβ40 was confirmed in cynomolgus monkeys by lack of effect on total CSF Aβ40 at all doses tested (Figure 3D, p > 0.05 between all groups). MEDI1814 serum concentrations over the course of the 13‐week treatment period in cynomolgus monkeys are shown in Figure S4 in supporting information.
FIGURE 3.
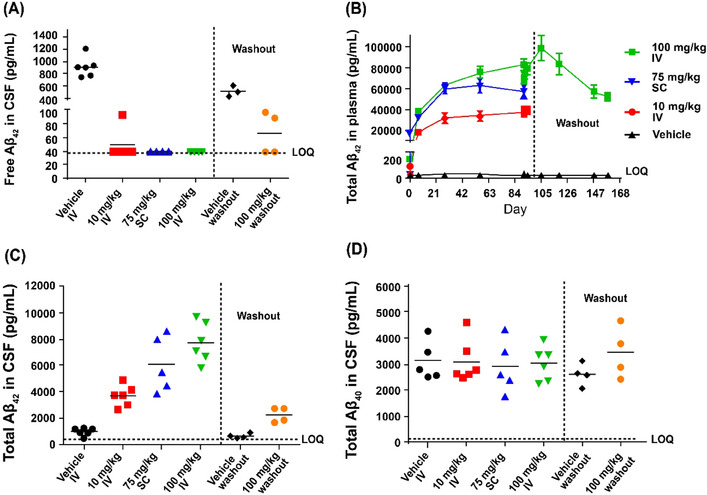
Pharmacodynamics of MEDI1814 in cynomolgus monkeys. Intravenous weekly administration of MEDI1814 over 13 weeks in cynomolgus monkey: (A), free CSF Aβ42; (B) total plasma Aβ42; (C) total CSF Aβ42; (D) total CSF Aβ40. A dose‐dependent increase in total Aβ42 in both plasma and CSF was observed at the end of the treatment phase in the 10 mg/kg IV, 100 mg/kg IV, and 75 mg/kg SC groups. The majority of free Aβ42 (> 95%) was suppressed in CSF at all dose levels at the end of the treatment phase (A). At the end of a 9‐week treatment‐free period, total Aβ42 levels in plasma increased in the 100 mg/kg IV dose group compared to the vehicle group (B); compared to the vehicle group, total CSF Aβ42 was higher, though not significant (C), and free CSF Aβ42 was suppressed. The specificity of MEDI1814 binding to Aβ42 over Aβ40 was confirmed by lack of effect on total CSF Aβ40 at all tested doses (D). Washout denotes a period of 9 weeks after dosing had stopped. Aβ, amyloid beta; CSF, cerebrospinal fluid; ELISA, enzyme‐linked immunosorbent assay; IV, intravenous; LOQ, limit of quantification; SC, subcutaneous.
3.3. Chronic non‐clinical safety of MEDI1814
The 13‐week repeat‐dose toxicity study described above was ICH S6‐compliant and conducted in two pharmacologically relevant animal species as part of the MEDI1814 non‐clinical development program. There were no MEDI1814‐related adverse effects or target organ toxicity identified in young adult male and female Sprague–Dawley rats or cynomolgus monkeys after 14 weekly administrations of 10 or 100 mg/kg IV, or 75 mg/kg SC MEDI1814.
3.4. Projection of MEDI1814 non‐clinical data for human dose
The no‐observed‐adverse‐effect level was determined as 100 mg/kg IV, based on 14 weekly doses of MEDI1814 in the 13‐week repeat‐dose study described above. The safety margins based on rat and cynomolgus monkeys were similar, but the safety margins for human dose were based on cynomolgus monkeys because (1) no AEs were observed in either species after complete target (Aβ42) suppression; (2) ≈ 33% of rats were ADA‐positive in treatment groups, impacting toxicity exposure; (3) rat toxicokinetic exposure in satellite animals might not reflect toxicity exposure in main study animals because of ADA; and (4) more PK and PD data were available from cynomolgus monkeys.
The PK/PD profiles of MEDI1814 in humans were simulated from a PK/PD model based on cynomolgus monkeys (with appropriately scaled parameters; Figure S5 in supporting information). The cynomolgus monkey model adequately described the MEDI1814 and total Aβ42 concentration–time profiles after both IV and SC dosing regimens, with estimates of clearance (7.68 mL/day/kg) and apparent volume of distribution at steady state (118.4 mL/kg), very similar to values for a typical IgG1 in cynomolgus monkeys. The plasma half‐life of free Aβ42, binding affinity of MEDI1814 to free Aβ42, and half‐life of MEDI1814‐Aβ42 complex were estimated at ≈ 5 minutes, ≈ 10 pM, and ≈ 6.73 days, respectively, with the complex half‐life similar to that of a typical monoclonal antibody in cynomolgus monkeys. Mean baseline‐free Aβ42 in CSF of ≈ 527 pg/mL (preliminary recovery data from control animals) and first‐order degradation constant (Kdeg) of ≈ 1.386 days−1 (or half‐life of 12 hours) were assumed (see Methods for details on these parameters). Other PD parameters including KD, off dissociation constant (Koff), and Kdeg for the CSF compartment were assumed to be the same as for the blood compartment.
Using a version of this model with scaled human PK/PD parameters, a starting dose of 25 mg IV MEDI1814 (fixed dosing) was chosen for the initial Phase 1 clinical study based on expected suppression of free Aβ42 in both plasma and CSF. At this dose level, a maximum suppression of ≈ 97% and 76% in free Aβ42 in plasma and CSF, with Aβ42 returning to baseline in ≈ 28 and 7 days, respectively, was expected. After IV doses of 300 mg every 4 weeks (Q4W; fixed dosing), ≥ 50% suppression of free Aβ42 in CSF was expected throughout the dosing interval in the clinic. Hence, 300 mg IV Q4W was identified as the initial target efficacious dose in patients with AD.
3.5. MEDI1814 selectively reduces CSF free Aβ42 in patients with AD
The favorable non‐clinical profile described provided a basis for the evaluation of MEDI1814 in a first‐in‐human randomized, double‐blind, placebo‐controlled clinical trial in patients with mild‐to‐moderate AD receiving single doses (Part 1; n = 45) and multiple doses (Part 2; n = 32) of the antibody (see Table 1 for demographic information and Figure S1 for study design schematic). Eleven subjects participated in both Part 1 and Part 2. In total 57 patients received single or repeat doses of MEDI1814 up to 1800 mg IV or 200 mg SC and 20 patients received placebo.
TABLE 1.
Demographics and baseline characteristics.
| A. Single ascending dose (safety analysis set) | |||||||
|---|---|---|---|---|---|---|---|
| MEDI1814 | |||||||
|
Placebo n = 12 |
25 mg IV n = 3 |
100 mg IV n = 6 |
300 mg IV n = 6 |
900 mg IV n = 6 |
1800 mg IV n = 6 |
100 mg SC n = 6 |
|
| Age (years), mean (SD) | 66.3 (7.22) | 74.0 (8.89) | 66.8 (1.94) | 69.0 (6.10) | 71.8 (5.15) | 64.8 (5.91) | 69.3 (4.84) |
| Female, n (%) | 7 (58.3) | 0 (0.0) | 6 (100.0) | 5 (83.3) | 4 (66.7) | 1 (16.7) | 3 (50.0) |
| White, n (%) | 10 (83.3) | 1 (33.0) | 6 (100.0) | 4 (66.7) | 5 (83.3) | 6 (100.0) | 5 (83.3) |
| BMI (kg/m2), mean (SD) | 26.8 (2.90) | 27.1 (3.40) | 28.2 (1.67) | 27.2 (2.74) | 24.7 (1.68) | 27.6 (3.70) | 27.8 (2.29) |
| Baseline MMSE score, mean (SD) | 21.8 (3.02) | 22.0 (2.65) | 22.3 (3.27) | 22.5 (3.73) | 22.2 (2.99) | 21.0 (2.00) | 23.2 (2.04) |
| B. Multiple ascending dose (safety analysis set) | |||||
|---|---|---|---|---|---|
| MEDI1814 | |||||
|
Placebo n = 8 |
300 mg IV n = 6 |
900 mg IV n = 6 |
1800 mg IV n = 6 |
200 mg SC n = 6 |
|
| Age (years), mean (SD) | 70.0 (5.90) | 71.7 (6.62) | 70.8 (6.62) | 62.5 (6.16) | 69.3 (8.71) |
| Female, n (%) | 5 (62.5) | 3 (50.0) | 2 (33.3) | 3 (50.0) | 5 (83.3) |
| White, n (%) | 8 (100.0) | 5 (83.3) | 6 (100.0) | 6 (100.0) | 5 (83.3) |
| BMI (kg/m2), mean (SD) | 27.9 (2.90) | 31.2 (0.73) | 26.2 (4.45) | 28.0 (3.93) | 30.1 (1.52) |
| Baseline MMSE score, mean (SD) | 22.1 (2.48) | 19.8 (2.04) | 20.2 (1.72) | 20.0 (2.45) | 22.5 (2.59) |
Abbreviations: BMI, body mass index; IV, intravenous; MMSE, Mini‐Mental State Examination; SC, subcutaneous; SD, standard deviation.
3.5.1. Pharmacodynamics
Relative to baseline, a dose‐dependent reduction of free Aβ42 in CSF and increase in total (bound and free) Aβ42 was observed at day 29 after single MEDI1814 doses; a profile consistent with antibody‐mediated target engagement of Aβ42 in the central compartment (Figure 4A). Free Aβ42 in CSF was reduced by ≈ 90% (median) in the highest dose cohorts (300–1800 mg IV), but lower levels of suppression were observed for the earlier doses: –72% (100 mg IV), –11% (100 mg SC), and –34% (25 mg IV) compared to –8% for placebo (see Figure 5 and Table S2 in supporting information). The CSF profile for free Aβ42 over the MEDI1814 dose range was largely consistent with that predicted using the PK/PD model based on cynomolgus monkey data. Although the levels of free Aβ42 suppression were greater than expected over the dose range 25 to 300 mg IV, near maximal levels of suppression were observed after the 900 and 1800 mg IV doses, as predicted (Figure 4A). The same trend was observed in repeat dosing of MEDI1814 (Figure 4B). At the higher dose levels, as expected, substantial increases in total (bound and free) Aβ42 were observed: 273% (median, 1800 mg IV), 323% (900 mg IV), and 135% (300 mg IV), compared to 0.2% for placebo (Table S2). MEDI1814–placebo differences (least squares [LS] mean) for percentage change from baseline in free Aβ42 in CSF ranged from –27% to –124%, and from 13% to 332% for total Aβ42 over the dose range (Table S2). However, in keeping with the known selectivity of MEDI1814 for Aβ42, no significant changes from baseline in total Aβ40 levels in CSF or in MEDI1814–placebo differences for Aβ40 were observed (Table S2).
FIGURE 4.
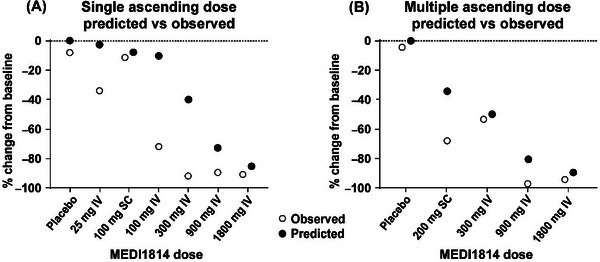
Comparison of observed and predicted pharmacodynamic responses to MEDI1814 after administration to patients with AD. Data show median % change from baseline in CSF free Aβ42 after administration of placebo and MEDI1814 single doses (25, 100, 300, 900, and 1800 mg IV or 100 mg SC in the single ascending dose study [A]) or repeat doses (300, 900, and 1800 mg IV or 200 mg SC, administered monthly in the multiple ascending dose study [B]). Relative to baseline, a dose‐dependent reduction of free Aβ42 in CSF and increase in total (bound and free) Aβ42 was observed at day 29 after single MEDI1814 doses (A). Near maximal levels of suppression were observed after the 900 and 1800 mg IV repeat doses (B). Aβ, amyloid beta; AD, Alzheimer's disease; CSF, cerebrospinal fluid; assay; IV, intravenous; SC, subcutaneous.
FIGURE 5.
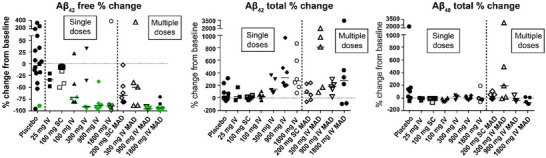
Effect of MEDI1814 on CSF Aβ biomarkers after single and multiple dose administration to patients with AD. Data show percentage change from baseline to follow‐up day 29 in CSF levels of (A), free Aβ42, (B) total Aβ42, and (C) total Aβ40 after single doses of placebo or MEDI1814 (25, 100, 300, 900, and 1800 mg IV or 100 mg SC). Median and individual data points are shown for % change from baseline. Free Aβ42 in CSF was greatly reduced in the highest dose cohorts (300–1800 mg IV), but lower levels of suppression were observed for the lower doses (100 mg IV, 100 mg SC, and 25 mg IV), compared to placebo. All individual data with median values shown where baseline and post‐dose sample available. Data are pooled for the placebo group after single dosing across the dose range. Data are pooled for the placebo groups after multiple dosing according to administration (IV or SC). Green symbol – Aβ42 free data at LLOQ (single ascending dose = 32 pg/mL; multiple ascending dose = 12 pg/mL). Aβ, amyloid beta; AD, Alzheimer's disease; CSF, cerebrospinal fluid; IV, intravenous; LLOQ, lower limit of quantification; MAD, multiple ascending dose; SC, subcutaneous.
A comparable dose‐related CSF biomarker response was observed at day 85 for multiple doses of MEDI1814. Free Aβ42 in CSF was reduced by 4% (median, placebo), 50% (300 mg IV), 67% (200 mg SC), and by ≈ 95% for the 900 and 1800 mg MEDI1814 IV doses (Figure 5 and Table S3 in supporting information). The observed profile for suppression of free Aβ42 in the CSF was entirely consistent with the PK/PD profile predicted using cynomolgus monkey data (Figure 4). In contrast, increases in total Aβ42 in the CSF of ≈ 70% to 800% (median) were observed over the MEDI1814 dose range, compared to ≈ –30% for placebo (Figure 5 and Table S3). The profile was again reflected in the MEDI1814–placebo differences (LS mean) for change from baseline in free Aβ42 (≈ –90% at the 900 and 1800 mg IV doses) and in total Aβ42 (≈ 860% for the 1800 mg IV dose; Table S3). The multiple‐dose regimen further confirmed the absence of any significant change in the total Aβ40 profile in the CSF after dosing with MEDI1814 (Figure 5 and Table S3).
In addition to Aβ biomarkers, CSF and plasma samples from the multiple‐dose part of the study were evaluated for downstream biomarkers of AD including NfL and p‐tau217 as exploratory endpoints. CSF NfL levels were reduced by a median 53% compared to baseline levels after three monthly IV doses of MEDI1814 1800 mg compared to a median increase in placebo‐treated patients of 4% over the same time duration (Figure 6A). Plasma NfL levels were reduced by a median 21% compared to baseline levels after three monthly IV doses of MEDI1814 1800 mg compared to a median increase in placebo‐treated patients of 2% over the same time duration (Figure 6B). Significant correlation was observed across the CSF and plasma NfL measurements (Spearman r: 0.47 [p‐value < 0.05] at baseline and 0.74 [p‐value < 0.05] at the day 85 timepoint). These correlations were consistent with previously observed human age‐related profiles for NfL in serum and CSF. 38 In addition, a median reduction of plasma p‐tau217 of 36% was observed compared to baseline levels after three monthly IV doses of MEDI1814 1800 mg compared to a median increase in placebo treated patients of 3% over the same time duration (Figure 6C).
FIGURE 6.
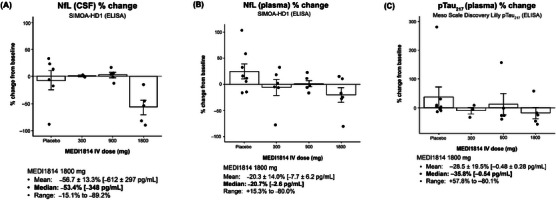
Effect of MEDI1814 on CSF (A), plasma NfL (B), and p‐tau217 (C) levels after administration of three monthly IV doses to patients with AD. Compared to baseline, CSF NfL levels were reduced after three monthly IV doses of MEDI1814 1800 mg compared to a median increase in placebo‐treated patients (A). Similarly, plasma NfL levels were reduced after three monthly IV doses of MEDI1814 1800 mg compared to a median increase in placebo‐treated patients (B). Data are mean ± standard error. Aβ, amyloid beta; AD, Alzheimer's disease; CSF, cerebrospinal fluid; IV, intravenous; NfL, neurofilament light chain; p‐tau, phosphorylated tau.
3.5.2. Pharmacokinetics
The PK properties of MEDI1814 were consistent across single‐ and multiple‐dosing paradigms. Serum exposures were observed to be dose‐proportion, and concentrations declined in a biphasic manner with similar rates of elimination (effective mean serum half‐life ≈ 14–20 days; Figure 7 and Table S4 in supporting information). Mean clearance for MEDI1814 at steady state (day 57 after multiple doses) ranged from 145 to 223 mL/day−1. Serum accumulation of MEDI1814 over the period was moderate (0.75‐ to 1.15‐fold for Cmax and 0.83‐ to 1.62‐fold for AUC; mean across all doses; Table S5 in supporting information). Median tmax at steady state after multiple SC dosing was 14 days. MEDI1814 bioavailability after a single 100 mg SC dose was 33% (based on AUC0‐∞ using 100 mg IV dose as reference). MEDI1814 was quantifiable in CSF at doses ≥ 300 mg after both single and repeat dose administration. CSF:serum concentration ratios ranged from 0.09% to 0.30% after single doses and from 0.08% to 0.59% after multiple doses.
FIGURE 7.
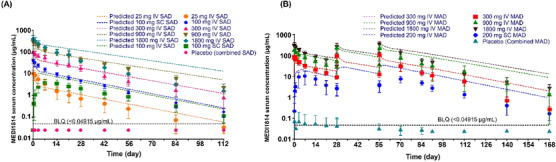
Serum concentration profiles of MEDI1814 in patients with AD. (A) Single ascending dose. (B) Multiple ascending dose. Pharmacokinetic properties of MEDI1814 were consistent across single‐ and multiple‐dosing paradigms. Serum exposures were dose‐proportional, and concentrations declined in a biphasic manner with similar rates of elimination (effective mean serum half‐life ≈ 14–20 days). AD, Alzheimer's disease; IV, intravenous; MAD, multiple ascending dose; SAD, single ascending dose; SC, subcutaneous.
3.5.3. Tolerability and immunogenicity
MEDI1814 was generally well tolerated after single‐ and multiple‐dose administration in patients with AD. The incidence of AEs was comparable between MEDI1814 and placebo‐treated subjects (Tables S6 and S7 in supporting information). There were no apparent dose‐related trends in the occurrence of AEs. Most events were assessed as mild, with few of moderate intensity; none were severe. No serious AEs, discontinuations due to AEs, or deaths were reported. There were no clinically significant changes in vital signs, electrocardiogram parameters, laboratory results, or on follow‐up physical and neurological examinations. There was no indication of cognitive deterioration (MMSE, data not shown), or of suicidal ideation or behavior (Columbia‐Suicide Severity Rating Scale, data not shown) after treatment.
Importantly, MRI assessments did not reveal any occurrences of ARIAs, either with respect to the formation of edema (ARIA‐E) or to hemosiderin deposition (ARIA‐H; Tables S8, S9, S10 in supporting information). No ADA, otherwise suggestive of immunogenicity, were detected for any subject in the study (all titers < 50).
4. DISCUSSION
We hypothesized that Aβ42 was the principal driver of amyloid toxicity in AD, and thus set out to selectively target Aβ42 and not Aβ40 using the monoclonal antibody MEDI1814. The approach was considered to offer potential benefits over existing therapeutic approaches as it would avoid a reduction in Aβ40, which may be neuroprotective, while also enabling robust suppression of the target Aβ42. We therefore prioritized target engagement throughout the development process, rather than using mouse models of cognitive function, for example Morris Water Maze, to guide decision making. In addition, it is recognized that these models do not typically enable demonstration of an effect size sufficient to allow for efficient PK/PD modeling and have generally not shown good clinical translation in the context of AD. We set about generating a high affinity (pM range) Aβ42 selective antibody by panning human antibody phage display libraries to the peptide, and subsequent affinity maturation using directed evolution, which was achieved to generate MEDI1814.
Selectivity of MEDI1814 for Aβ42 over Aβ40 was demonstrated using in vitro peptide competition studies, which showed a > 3 log unit difference in displacement of Aβ1‐42 binding versus Aβ1‐40. This is remarkable, as Aβ42 only differs from Aβ40 by the additional two amino acids at the C‐terminus. However, Aβ42 is reported to have more hairpin‐like structure at the C‐terminus compared to Aβ40, 39 which likely will have facilitated the observed selectivity. Peptide binding data also show that MEDI1814 can engage Aβ1‐43, 14 and the pyroglutamate modified Aβp3‐42 and Aβp11‐42. 40 , 41 This target engagement profile is considered potentially advantageous, as these are all clinically relevant forms of Aβ, enriched in the plaque of patients with AD and having similar propensity to form toxic oligomers.
In vivo, MEDI1814 reduced free Aβ42 in CSF to the LOQ in rats and cynomolgus monkeys, with a concomitant increase in total Aβ42 as the peptide assumes a longer half‐life due to high affinity binding to the antibody (an Aβ half‐life of hours 42 vs. 7 and 13 days for MEDI1814 in rats and cynomolgus monkey, respectively; Figures 2 and 3). These effects agree with the in vitro peptide binding data showing selectivity for Aβ42, as no change in Aβ40 was observed in either rat (brain or CSF) or cynomolgus monkey (CSF) after in vivo administration, even at high (100 mg/kg IV) doses. The rat data presented here clearly demonstrate a different profile from m266 (rodent version of solanezumab), which showed no change in free Aβ42 in CSF but did increase total Aβ40 in brain tissue. The fluid biomarker data presented here clearly demonstrate a different clinical profile from solanezumab, which showed increased total Aβ42 and NfL. 43 , 44 Upon cessation of MEDI1814 dosing, the levels of free and total Aβ42 were observed to return toward baseline, indicating reversibility in both non‐clinical species. Furthermore, the plasma data for total Aβ42 showed that an equilibrium of antibody–Aβ42 complex is reached after three to four doses, which would also be expected to occur in CSF and brain compartments as an equilibrium of antibody–peptide complex formation and clearance is reached.
The non‐clinical target engagement data enabled the development of a translational PK/PD model that effectively predicted CSF Aβ42 suppression in patients with AD after both single and multiple dosing. In clinical evaluation, MEDI1814 was observed to have similar Aβ pharmacodynamic characteristics to those observed in rats and cynomolgus monkeys; single and multiple dosing at the 900 and 1800 mg IV dose levels led to near maximal suppression of free Aβ42 in CSF, together with a concomitant increase in total Aβ42 and no change in Aβ40. The potential for MEDI1814 to almost fully suppress monomeric CSF Aβ42 while sparing Aβ40 provides an opportunity to test an aspect of the amyloid hypothesis that has not hitherto been possible. Interestingly, a reduction of plasma p‐tau217 and of CSF and plasma NfL levels from baseline was also observed after three monthly IV doses of MEDI1814 1800 mg suggesting an impact on downstream neurodegeneration process. This is the first observation of NfL lowering from baseline by an amyloid‐targeting immunotherapy, and the data would benefit from confirmation in another clinical study that included a larger sample.
Overall, MEDI1814 was well tolerated by the study subjects after both single‐ and multiple‐dose IV administration. There were no clinically important findings across any of the safety endpoints over the MEDI1814 dose range explored. There was no evidence of an immunogenic response to the antibody. Importantly, based on MRI evaluation, there was no evidence for provocation of either ARIA‐E or ARIA‐H after MEDI1814 exposure. It should be noted that PET with amyloid tracers was not included in this particular study as MEDI1814 was not designed to actively clear amyloid plaques. However, quantitative amyloid plaque estimation via PET should be included in future studies involving more patients and over a longer duration. This favorable safety profile is thought to be due to the mechanism‐of‐action of MEDI1814 by virtue of its capacity to sequester soluble Aβ42, rather than to bind and dissolute amyloid plaques, therefore precluding the need for effector function which may otherwise contribute to the increased ARIA observed with other amyloid targeting antibodies.
Herein, we have described the discovery, non‐clinical, and early clinical development, together with early data on downstream biomarkers for AD, of a fully human, effector‐null monoclonal antibody (MEDI1814) that has high affinity and selectivity for full‐length and N‐terminal truncated forms of Aβ42/43 versus Aβ40. We believe that this could have advantages over other potential Aβ therapeutics evaluated to date, in terms of potential safety and efficacy, as there is preferential targeting of the key toxic building blocks of Aβ while sparing Aβ40. The question remains to be addressed as to whether selective reduction of free Aβ42 can provide efficacy for patients with AD. In our opinion, late‐stage clinical evaluation of MEDI1814 may allow the most rigorous test of the involvement of Aβ in the initiation of AD to date, providing MEDI1814 is dosed in patients at a clinically relevant time frame for therapeutic intervention (e.g., in the pre‐symptomatic, prodromal, or early phases of the disease).
AUTHOR CONTRIBUTIONS
Conceptualization: Christopher Lloyd, Maria T. Groves, Craig Shering, Keith Tan, Thor Ostenfeld, Iain P. Chessell, Andy Billinton, Jeffrey L. Dage. Methodology: Christopher Lloyd, Adrian Nickson, Maria T. Groves, Philip Newton, David Lowne, Anna Bogstedt, Michael Pomfret, Andy Billinton, Susanna Eketjäll, Maria T. Groves, Fraser Welsh, Susanne Gustavsson, Mary McFarlane, Richard Turner, Zulma Santisteban Valencia, Eva Lindqvist, Keith Tan, Tharani Chessell, Amanda D. Dudley, Thor Ostenfeld, Jeffrey L. Dage, Nicholas Kyle Proctor. Investigation: Susanne Gustavsson, Keith Tan, Michael Pomfret, Tharani Chessell, Amanda D. Dudley, Thor Ostenfeld. Visualization: Christopher Lloyd, Craig Shering, Keith Tan, Thor Ostenfeld, Anna Bogstedt. Funding acquisition: N/A. Project administration: Amanda D. Dudley. Supervision: Amanda D. Dudley. Writing—original draft: Christopher Lloyd, Craig Shering, Keith Tan, Thor Ostenfeld, Anna Bogstedt. Writing—review & editing: Christopher Lloyd, Per‐Ola Freskgård, Philip Newton, David Lowne, Adrian Nickson, Anna Bogstedt, Susanna Eketjäll, Kina Höglund, Susanne Gustavsson, Fraser Welsh, Tharani Chessell, Mary McFarlane, Ratan V. Bhat, Richard Turner, Michael S. Perkinton, Zulma Santisteban Valencia, Eva Lindqvist, Michael Pomfret, Amanda D. Dudley, Tristan J. Vaughan, Maria T. Groves, Fanni Natanegara, Yingdong Feng, John R. Sims, Nicholas Kyle Proctor, Jeffrey L. Dage, Craig Shering, Keith Tan, Thor Ostenfeld, Iain P. Chessell, Andy Billinton. All authors contributed to study design and data interpretation, drafting the manuscript or revising it critically for important intellectual content, gave final approval of the version to be published, and agree to be accountable for all aspects of the work. Christopher Lloyd, Adrian Nickson, and Maria T. Groves designed and performed antibody engineering. Philip Newton performed peptide competition studies. David Lowne performed BIAcore studies. Anna Bogstedt, Michael Pomfret, Andy Billinton, Susanna Eketjäll, Maria T. Groves, Fraser Welsh, and Susanne Gustavsson designed and interpreted the rodent pharmacology studies. Mary McFarlane designed the 13‐week toxicology studies. Richard Turner, Zulma Santisteban Valencia, Eva Lindqvist, and Andy Billinton contributed to PK and PD analysis and PK‐PD modeling. Keith Tan, Michael Pomfret, Tharani Chessell, Amanda D. Dudley, and Thor Ostenfeld contributed to the design and running of the Phase 1 trial. Jeffrey L. Dage designed and interpreted the human plasma and CSF NfL and p‐tau217 studies. Nicholas Kyle Proctor performed human plasma P‐tau217 studies. Christopher Lloyd, Anna Bogstedt, Thor Ostenfeld, Mary McFarlane, Keith Tan wrote the manuscript with contributions and review from all authors.
CONFLICT OF INTEREST STATEMENT
CL, PN, DL, AN, ABo, SE, SG, FW, TC, MMcF, RVB, RT, MSP, ZSV, EL, MP, ADD, TJV, MTG, CS, KT, TO, IPC, and ABi are either current or former employees of AstraZeneca, and either own or owned shares/options in AstraZeneca. P‐OF is an inventor on patents or patent applications. FN, YF, JRS, NKP, and JLD are current or former employees of Eli Lilly. JLD is an inventor on patents or patent applications of Eli Lilly and Company relating to the assays, methods, reagents and / or compositions of matter for p‐tau assays. JLD has served as a consultant or on advisory boards for Eisai, Abbvie, Genotix Biotechnologies Inc, Gates Ventures, Karuna Therapeutics, AlzPath Inc., Cognito Therapeutics, Inc., and received research support from ADx Neurosciences, Fujirebio, AlzPath Inc., Roche Diagnostics, and Eli Lilly and Company in the past 2 years. JLD has received speaker fees from Eli Lilly and Company. JLD is a founder and advisor for Monument Biosciences. JLD has stock or stock options in Eli Lilly and Company, Genotix Biotechnologies, AlzPath Inc., and Monument Biosciences. MEDI1814 is covered by PCT patent application WO2014060444. KH has nothing to declare. Author disclosures are available in the supporting information.
CONSENT STATEMENT
For studies involving humans, written informed consent was obtained from patients or their legal representative prior to any study procedures.
Supporting information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
The authors thank Alison Scott for project management of the 13‐week toxicology studies. The authors thank all participating patients, their families, and the study investigators. The authors also thank Vicky Hinstridge and Mark Davies of inScience Communications, Springer Healthcare Ltd, UK, for providing medical writing support, and Sophie Robinson of inScience Communications, Springer Healthcare Ltd, UK, for providing project management support, which was funded by AstraZeneca. AstraZeneca funded the study and had a role in study design, data collection, data analysis, data interpretation, and writing of the report. The corresponding author had full access to all the data and had final responsibility to submit for publication.
Lloyd C, Freskgård P‐O, Newton P, et al. MEDI1814 selectively reduces free Aβ42 in cerebrospinal fluid of non‐clinical species and Alzheimer's disease patients. Alzheimer's Dement. 2024;20:7762–7776. 10.1002/alz.14238
DATA AVAILABILITY STATEMENT
The MEDI1814 Phase 1 clinical trial described here is registered on ClinicalTrials.gov with the reference NCT02036645.
REFERENCES
- 1. World Health Organization . Dementia. 2022.
- 2. Braak H, Del Tredici K. The pathological process underlying Alzheimer's disease in individuals under thirty. Acta Neuropathol. 2011;121:171‐181. [DOI] [PubMed] [Google Scholar]
- 3. Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256:184‐185. [DOI] [PubMed] [Google Scholar]
- 4. Hardy J. Testing times for the “amyloid cascade hypothesis”. Neurobiol Aging. 2002;23:1073‐1074. [DOI] [PubMed] [Google Scholar]
- 5. Bogstedt A, Groves M, Tan K, Narwal R, McFarlane M, Hoglund K. Development of immunoassays for the quantitative assessment of amyloid‐beta in the presence of therapeutic antibody: application to pre‐clinical studies. J Alzheimers Dis. 2015;46:1091‐1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med. 2016;8:595‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Walsh DM, Klyubin I, Fadeeva JV, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long‐term potentiation in vivo. Nature. 2002;416:535‐539. [DOI] [PubMed] [Google Scholar]
- 8. Kayed R, Lasagna‐Reeves CA. Molecular mechanisms of amyloid oligomers toxicity. J Alzheimers Dis. 2013;33(1):S67‐S78. Suppl. [DOI] [PubMed] [Google Scholar]
- 9. Cline EN, Bicca MA, Viola KL, Klein WL. The amyloid‐beta oligomer hypothesis: beginning of the third decade. J Alzheimers Dis. 2018;64:S567‐S610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Panza F, Lozupone M, Watling M, Imbimbo BP. Do BACE inhibitor failures in Alzheimer patients challenge the amyloid hypothesis of the disease? Expert Rev Neurother. 2019;19:599‐602. [DOI] [PubMed] [Google Scholar]
- 11. El‐Agnaf OM, Irvine GB. Review: formation and properties of amyloid‐like fibrils derived from alpha‐synuclein and related proteins. J Struct Biol. 2000;130:300‐309. [DOI] [PubMed] [Google Scholar]
- 12. Kim J, Onstead L, Randle S, Price R, Smithson L, Zwizinski C, et al. Abeta40 inhibits amyloid deposition in vivo. J Neurosci. 2007;27:627‐633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kuperstein I, Broersen K, Benilova I, et al. Neurotoxicity of Alzheimer's disease Abeta peptides is induced by small changes in the Abeta42 to Abeta40 ratio. EMBO J. 2010;29:3408‐3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Saito T, Suemoto T, Brouwers N, et al. Potent amyloidogenicity and pathogenicity of Abeta43. Nat Neurosci. 2011;14:1023‐1032. [DOI] [PubMed] [Google Scholar]
- 15. Jawhar S, Wirths O, Schilling S, Graubner S, Demuth HU, Bayer TA. Overexpression of glutaminyl cyclase, the enzyme responsible for pyroglutamate A{beta} formation, induces behavioral deficits, and glutaminyl cyclase knock‐out rescues the behavioral phenotype in 5XFAD mice. J Biol Chem. 2011;286:4454‐4460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Perez‐Garmendia R, Gevorkian G. Pyroglutamate‐modified amyloid beta peptides: emerging targets for Alzheimer s disease immunotherapy. Curr Neuropharmacol. 2013;11:491‐498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Meissner JN, Bouter Y, Bayer TA. Neuron loss and behavioral deficits in the TBA42 mouse model expressing N‐truncated pyroglutamate amyloid‐beta3‐42. J Alzheimers Dis. 2015;45:471‐482. [DOI] [PubMed] [Google Scholar]
- 18. Mintun MA, Lo AC, Duggan Evans C, Wessels AM, et al. Donanemab in early Alzheimer's disease. N Engl J Med. 2021;384:1691‐1704. [DOI] [PubMed] [Google Scholar]
- 19. van Dyck CH, Swanson CJ, Aisen P, et al. Lecanemab in early Alzheimer's disease. N Engl J Med. 2022;388:9‐21. [DOI] [PubMed] [Google Scholar]
- 20. Budd Haeberlein S, O'Gorman J, Chiao P, et al. Clinical development of aducanumab, an anti‐aβ human monoclonal antibody being investigated for the treatment of early Alzheimer's disease. J Prev Alzheimers Dis. 2017;4:255‐263. [DOI] [PubMed] [Google Scholar]
- 21. US Food & Drug Administration . Drug Approval Package: Aduhelm (aducanumab‐avwa). 2021.
- 22. US Food & Drug Administration . Drugs@FDA: FDA‐Approved Drugs—Lecanemab irmb. 2023.
- 23. Willis BA, Sundell K, Lachno DR, et al. Central pharmacodynamic activity of solanezumab in mild Alzheimer's disease dementia. Alzheimers Dement (N Y). 2018;4:652‐660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Siemers ER, Sundell KL, Carlson C, et al. Phase 3 solanezumab trials: secondary outcomes in mild Alzheimer's disease patients. Alzheimers Dement. 2016;12:110‐120. [DOI] [PubMed] [Google Scholar]
- 25. Plotkin SS, Cashman NR. Passive immunotherapies targeting Aβ and tau in Alzheimer's disease. Neurobiol Dis. 2020;144:105010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:263‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Daramola O, Stevenson J, Dean G, et al. A high‐yielding CHO transient system: coexpression of genes encoding EBNA‐1 and GS enhances transient protein expression. Biotechnol Prog. 2014;30:132‐141. [DOI] [PubMed] [Google Scholar]
- 28. Hoet RM, Cohen EH, Kent RB, et al. Generation of high‐affinity human antibodies by combining donor‐derived and synthetic complementarity‐determining‐region diversity. Nat Biotechnol. 2005;23:344‐348. [DOI] [PubMed] [Google Scholar]
- 29. Hawkins RE, Russell SJ, Winter G. Selection of phage antibodies by binding affinity. Mimicking affinity maturation. J Mol Biol. 1992;226:889‐896. [DOI] [PubMed] [Google Scholar]
- 30. Vaughan TJ, Williams AJ, Pritchard K, et al. Human antibodies with sub‐nanomolar affinities isolated from a large non‐immunized phage display library. Nat Biotechnol. 1996;14:309‐314. [DOI] [PubMed] [Google Scholar]
- 31. Clarkson T, Lowman HB. Phage Display: A Practical Approach. Oxford University Press, 2004; 2004. [Google Scholar]
- 32. Lewis L, Lloyd C. Optimisation of antibody affinity by ribosome display using error‐prone or site‐directed mutagenesis. Methods Mol Biol (Clifton, NJ). 2012;805:139‐161. [DOI] [PubMed] [Google Scholar]
- 33. Karlsson R, Michaelsson A, Mattsson L. Kinetic analysis of monoclonal antibody‐antigen interactions with a new biosensor based analytical system. J Immunol Methods. 1991;145:229‐240. [DOI] [PubMed] [Google Scholar]
- 34. Ashton NJ, Puig‐Pijoan A, Mila‐Aloma M, et al. Plasma and CSF biomarkers in a memory clinic: head‐to‐head comparison of phosphorylated tau immunoassays. Alzheimers Dement. 2023;19:1913‐1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:280‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Groves MA, Nickson AA. Affinity maturation of phage display antibody populations using ribosome display. Methods Mol Biol (Clifton, NJ). 2012;805:163‐190. [DOI] [PubMed] [Google Scholar]
- 37. Oganesyan V, Damschroder MM, Leach W, Wu H, Dall'Acqua WF. Structural characterization of a mutated, ADCC‐enhanced human Fc fragment. Mol Immunol. 2008;45:1872‐1882. [DOI] [PubMed] [Google Scholar]
- 38. Khalil M, Pirpamer L, Hofer E, et al. Serum neurofilament light levels in normal aging and their association with morphologic brain changes. Nat Commun. 2020;11:812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sgourakis NG, Yan Y, McCallum SA, Wang C, Garcia AE. The Alzheimer's peptides Abeta40 and 42 adopt distinct conformations in water: a combined MD /NMR study. J Mol Biol. 2007;368:1448‐1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schilling LP, Pascoal TA, Zimmer ER, et al. Regional amyloid‐beta load and white matter abnormalities contribute to hypometabolism in Alzheimer's dementia. Mol Neurobiol. 2019;56:4916‐4924. [DOI] [PubMed] [Google Scholar]
- 41. Schlenzig D, Manhart S, Cinar Y, et al. Pyroglutamate formation influences solubility and amyloidogenicity of amyloid peptides. Biochemistry. 2009;48:7072‐7078. [DOI] [PubMed] [Google Scholar]
- 42. Patterson BW, Elbert DL, Mawuenyega KG, et al. Age and amyloid effects on human central nervous system amyloid‐beta kinetics. Ann Neurol. 2015;78:439‐453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Farlow M, Arnold SE, van Dyck CH, et al. Safety and biomarker effects of solanezumab in patients with Alzheimer's disease. Alzheimers Dement. 2012;8:261‐271. [DOI] [PubMed] [Google Scholar]
- 44. Salloway S, Farlow M, McDade E, et al. A trial of gantenerumab or solanezumab in dominantly inherited Alzheimer's disease. Nat Med. 2021;27:1187‐1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Data Availability Statement
The MEDI1814 Phase 1 clinical trial described here is registered on ClinicalTrials.gov with the reference NCT02036645.