

Abstract
Background
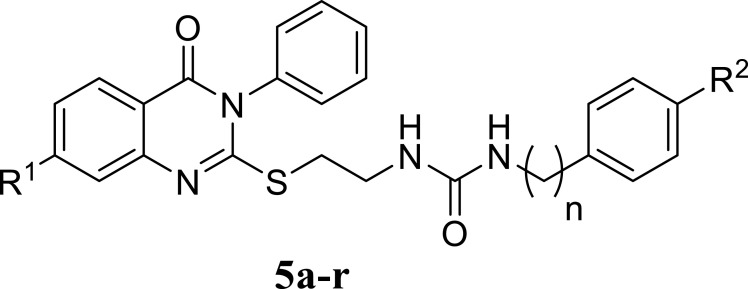
In response to the urgent need for continuous discovery of new anti-proliferative agents, a new series of quinazoline compounds 5a-r was prepared.
Methods
As a reference, four cancer cell lines—HCT116, HePG2, Hela, and MCF-7—and sorafenib (SOR) were used to assess the novel motifs’ in vitro anticancer efficacy. The most cytotoxic compounds were tested in a VEGFR-2 suppressive test and flow cytometric test. Docking analysis was done to the three novel motifs.
Results
Compound 5d showed the best anti-tumor activity of the tested compounds with IC50 6.09, 2.39, 8.94 and 4.81 μM in succession. In addition, compound 5h revealed a potent anticancer effect against HCT116 and HePG2 with IC50 5.89 and 6.74 μM, respectively. Also, compound 5p exhibited very strong activity against HCT116, HePG2 & MCF7 with IC50 8.32, 9.72 and 7.99, respectively. Compound 5p had the highest inhibition against VEGFR-2 with an IC50 of 0.117 μM, in contrast to 0.069 μM for SOR. According to flow cytometric testing, the most effective VEGFR-2 inhibitory agent, 5p, was shown to suppress the G1/S cell population in MCF-7 cells. Docking analysis confirmed that the three novel motifs could bind to the VEGFR-2 enzyme’s binding region like the co-crystallized ligand SOR did.
Conclusion
The enzyme inhibitory test of compound 5p showed that it is the most potent hybrid that caused MCF-7 cells to undergo apoptosis and generated a G1/S cell cycle arrest. Confirmation of the obtained results was done with the aid of the docking study, which showed that the three motifs might adhere to the enzyme’s major active sites, and the results were in good accordance with the experimental VEGFR-2 inhibitory results. We can conclude that the new quinazoline compounds 5a-r could be used as candidates for development of more efficient anticancer inhibitors.
Keywords: quinazolines, VEGFR-2 inhibitors, molecular docking, cell cycle analysis, apoptosis
Graphical Abstract

Introduction
Nowadays, cancer is considered the chief cause of mortality that overwhelms healthcare systems worldwide.1,2 Consequently, there is a pressing need to discover and develop novel and effective anti-cancer agents.3,4 Angiogenesis, a critical cancer hallmark, involves the creation of new blood vessels from the adjacent ones, which is vital for tumor growth and progression.5 Without this functional vascularity, cancer cells remain latent and lose their ability to metastasize.6,7 Therefore, angio-suppressive strategies have evolved as outstanding therapeutic approaches to overcome malignancies.6,8,9
One of the foremost essential regulators of angiogenesis is vascular endothelial growth factor, often known as VEGF.10,11 The binding of VEGF to Vascular endothelial growth factor receptors (VEGFR) stimulates endothelial cell proliferation, migration, and subsequent angiogenesis.12 VEGFR-2 is the principal receptor among the three VEGF receptors that regulate VEGF-induced cell proliferation and angiogenesis.5,13 Considering it, inhibiting the VEGF/VEGFR-2 pathway presents a potentially worthwhile strategy for anti-angiogenic therapy in the case of cancer treatment.7,13,14 To date, several small molecule VEGFR-2 inhibitors include SOR (I), Regorafenib (II), Sunitinib (III), Linifanib (IV), Lenvatinib (V), Tivozanib (VI) and Vandetanib (VII) have been approved for the treatment of angiogenesis dependent malignancies (Figure 1).7,15–20 Despite their efficacy in cancer treatment, FDA-approved VEGFR-2 inhibitors still encounter serious resistance that limits their use,21,22 so it is crucial to develop novel VEGFR-2 inhibitors with minimal toxicity and to combat cancer cell drug resistance.22,23 In general, the VEGFR-2 inhibitors displayed wide diversity in their structure, where they possessed various scaffolds that served as hinge binder motifs that occupied the ATP binding pocket, including picolinamide (eg, SOR (I) and Regorafenib (II)), indole (eg, Sunitinib (III)), indazole (eg, Linifanib (IV)), quinolone (eg, Lenvatinib (V) and Tivozanib (VI)), and quinazoline (eg, Vandetanib (VII)).7,14,24 However, most of them share the presence of urea of the diaryl urea moiety in their structure, which served as the essential pharmacophore in the design of VEGFR-2 inhibitors (Figure 1).25,26 Binding with the DFG motif of VEGFR-2 was greatly facilitated by the urea moiety, which acts as a donor-acceptor of hydrogen bonds. In particular, the urea group’s oxygen atom formed a hydrogen bond with VEGFR-2’s Asp1046 residue, and the NH groups coordinated with the Glu885 residue.27
Figure 1.

Chemical structures of VEGFR-2 inhibitors that the FDA clinically authorized.
The quinazoline nucleus is a common heterocycle in several well-known and commercially available anticancer medications.28–30 Several quinazoline derivatives were reported in the literature as effective anti-angiogenic VEGFR-2 inhibitors (Figure 1).31–33 For instance, Vandetanib (VII), an anilinoquinazoline derivative, is introduced by AstraZeneca as a multi-target VEGFR-2, EGFR, and Ret tyrosine kinases inhibitors.20 Cediranib (AZD2171, VIII) and AZD2932 (IX) are other quinazoline ether-containing compounds with powerful VEGFR-2 inhibitory activity.34–36 Cediranib (VIII) was reported to inhibit the VEGF-induced proliferation in a sub-nanomolar IC50 (IC50 < 1 nmol/L).35 AZD2932 (IX) was found to be a potential inhibitor of angiogenesis with an IC50 = 8 nM against VEGFR-2.36 SKLB1002 (X), a quinazoline containing 1,3,4-thiadiazole ring, has been reported as a potent VEGFR-2 inhibitor with an IC50 of 32 nmol/L.37 It also effectively inhibited a new microvasculature in zebrafish embryos.37 These findings highlighted the significance of the quinazoline scaffold in inhibiting VEGFR-2.31,38 Thus, several structural modifications on the quinazoline ring have been made during the last years to develop different quinazoline-based candidates showing anti-angiogenic agent effects through VEGFR-2 inhibition.39,40 In this context, quinazoline-4 (3H)-ones attracted outstanding attention as a very effective scaffold for VEGFR-2 inhibitors because it is an excellent hinge-binding moiety that occupies the ATP binding domain of the VEGFR-2 enzyme.41–44 For example, quinazoline-4 (3H)-one bearing thiadiazole-urea XI was reported to exhibit significant in vitro anti-cancer activity against prostate cancer PC3 cell line (IC50 = 17.7 μM) compared to SOR (IC50 = 17.3 μM) and displayed potent VEGFR inhibitory activity.45 Moreover, 3-phenylquinazolinone derivatives XII showed significant anticancer activity against many cell lines via their ability to effectively inhibit VEGFR-2 with IC50 of 0.34 μM superior to that of SOR (IC50 = 0.588 μM).42 Significant anticancer action and very potent suppression of VEGFR-2 were revealed by the 3-ethyl-6-nitroquinazoline-4-one derivative XIII. The IC50 values of 4.6 μM, which are in the micromolar range, are more significant than the IC50 value of 4.8 μM associated with the reference medicine pazopanib.44
Inspired by the information above, and in continuation of our previous work to design and develop new anticancer agents targeting VEGFR-2 inhibition,24,33 it was decided to synthesize new quinazoline-4 (3H)-one/urea hybrids that have similar pharmacophoric attributes as previously described VEGFR-2 blockers in an attempt to obtain more potent anti-cancers where substituted quinazoline-4 (3H)-one moiety was used to fit in ATP binding region (Figure 2). It was reported that the large size space of the ATP binding domain enables the bicyclic quinazoline ring to work on it effectively.43,46 An ethylthio bridge was introduced to connect quinazoline-4 (3H)-one ring with urea moiety, which comprises the essential HBA/HBD characteristics to act as the pharmacophore, forming the necessary hydrogen bonding with conserved DFG motif. Finally, a terminal hydrophobic system, either phenyl or benzyl moiety, is substituted with electron-withdrawing or electron-donating groups, which form various hydrophobic interactions with the back allosteric site, affecting the potency of the resulting hybrids. Furthermore, the SAR of these candidates was allowed to be investigated as effective anti-tumor surrogates and potent VEGFR-2 inhibitors.
Figure 2.

A selection of illustrative examples of Quinazolin-4(3H)-ones that have been demonstrated to inhibit VEGFR-2 and the discovery of the desired compounds.
Results and Discussion
Chemistry
The synthetic pathway of the new series of quinazoline compounds 5a-r is demonstrated in scheme 1, upon starting with the appropriate phenyl alkyl amine derivative 1a-i, which was reacted with 2-chloroethyl isocyanate, afforded the chloroethyl urea analogs 2a-i.47,48 On the other hand, modification of the Niementowski reaction was applied to form the quinazoline-2-thiol derivatives 4a, b. The title intermediates were furnished in good yield upon the reaction of anthranilic acid or its 4-chloro derivative with phenyl isothiocyanate.49,50 Our final target analogs were produced by refluxing the chloroethyl urea derivatives 2a-i with the quinazolines 4a,b in DMF in the presence of potassium carbonate as a base.50,51
Scheme 1.

Synthetic pathway for the target quinazoline compounds 5a-r. Reagents and Conditions: (a) THF, rt, overnight; (b) TEA, ethanol, reflux, 2 hr; (c) K2CO3, DMF, 70 °C, 24 hr.
The structures of the newly furnished quinazoline hybrids 5a-r were confirmed via different spectroscopic techniques using IR, 1HNMR, 13CNMR and MS. See Figures S1–S27 represent the NMR spectra (1H NMR or 13C-NMR) for final compounds. The IR spectra of all the title compounds showed two significant peaks in the range of 1620–1696 cm−1 corresponding to two carbonyl groups instead of one in all the starting compounds verifying the formation of our compounds. 1HNMR charts of the new analogs revealed two signals as triplet equivalent to the ethylthio bridge in the region of 3.2–3.4 ppm. Moreover, the appearance of the two mobile NH protons of urea moiety in the chart around 6.5 and 8.7 or 6.2 and 6.4 revealed the success of the S-substitution of quinazolines 4a, b. 13CNMR spectra of the new analogs showed two significant peaks at 157–161 ppm equivalent to the two carbonyl carbons in addition to two peaks in the aliphatic region around 33–39 ppm corresponding to SCH2CH2N in all the produced compounds. All the remaining data were in perfect accordance with the proposed structures. All the synthetic techniques and procedures, in addition to yields, are displayed in the experimental section.
Biological Activity
In vitro Analyzing the Cytotoxicity and Anti-Tumor Activity Using HCT-116, HeLa, HePG-2 and MCF-7
Tests were conducted using the standard MTT tests versus the colorectal (HCT116), hepatocellular (HePG2), human cervical (Hela) carcinoma and breast cancer (MCF-7) cell lines for determining the cytotoxic impact of the quinazoline derivatives 5a-r. The reference cell line was SOR.52 Table 1 displays the IC50 values that were computed.
Table 1.
The Activities of the Novel Compounds 5a-r Over Normal Cells (WI-38) and 4 Cancer Cell Lines (Using SOR as a Reference) and the Values of Their IC50 (μM) are Provided
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Comp. no.* | R1 | R2 | n | IC50 (µM)** | ||||
| HCT-116 | HePG-2 | Hela | MCF-7 | WI-38 | ||||
| 5a | H | OCH3 | 0 | 62.70±3.6 | 81.29±4.3 | 83.41±4.4 | 79.47±3.9 | 29.42±2.1 |
| 5b | H | Cl | 0 | 77.26±4.2 | 88.46±4.8 | >100 | 92.19±4.7 | 64.31±3.7 |
| 5c | H | Br | 0 | 48.22±3.0 | 57.82±3.6 | 67.67±3.9 | 55.15±3.2 | >100 |
| 5d | H | CF3 | 0 | 6.09±2.2 | 2.39±0.1 | 8.94±0.7 | 4.81±0.2 | 36.29±2.4 |
| 5e | H | H | 1 | 54.91±3.3 | 65.68±3.8 | 75.34±4.2 | 72.77±3.6 | >100 |
| 5f | H | OCH3 | 1 | 14.78±1.3 | 34.24±2.4 | 31.51±2.3 | 18.66±1.5 | 91.40±5.1 |
| 5g | H | Cl | 1 | 35.29±2.6 | 47.19±3.1 | 53.68±3.1 | 39.58±2.6 | 85.98±4.8 |
| 5h | H | H | 2 | 5.89±0.3 | 6.74±0.5 | 11.61±0.9 | 13.88±1.1 | 41.06±2.6 |
| 5i | H | Cl | 2 | 39.06±2.7 | 49.83±3.2 | 58.33±3.3 | 43.15±2.8 | >100 |
| 5j | Cl | OCH3 | 0 | 9.51±0.9 | 12.34±1.0 | 19.10±1.4 | 16.01±1.3 | 38.52±2.5 |
| 5k | Cl | Cl | 0 | 23.43±2.1 | 40.85±2.6 | 38.02±2.5 | 32.91±2.3 | >100 |
| 5l | Cl | Br | 0 | 28.64±2.3 | 44.74±2.9 | 49.41±2.9 | 37.25±2.5 | >100 |
| 5m | Cl | CF3 | 0 | 18.14±1.5 | 30.68±2.2 | 27.88±2.1 | 21.45±1.8 | 76.27±4.3 |
| 5n | Cl | H | 1 | 44.21±2.8 | 52.39±3.4 | 61.48±3.7 | 48.68±3.0 | 73.64±3.9 |
| 5o | Cl | OCH3 | 1 | 69.30±3.9 | 84.33±4.6 | 90.23±4.8 | 86.52±4.2 | 35.87±2.3 |
| 5p | Cl | Cl | 1 | 8.32±0.7 | 9.72±0.7 | 14.27±1.2 | 7.99±0.5 | 56.87±3.3 |
| 5q | Cl | H | 2 | 32.53±2.5 | 41.58±2.8 | 45.22±2.7 | 24.58±2.0 | 53.01±3.1 |
| 5r | Cl | Cl | 2 | 13.20±1.1 | 17.67±1.4 | 22.36±1.7 | 10.74±0.9 | 74.17±4.1 |
| SOR*** | - | - | - | 5.47±0.3 | 9.18±0.6 | 7.26±0.3 | 4.17±0.2 | 10.65±0.8 |
Note: *Symbols represent synthesized compounds. **The IC50 value is the dose at which 50% of tumor cell growth is inhibited. The data is shown as the mean ± SD from the dose-response graphs in triplicate. IC50 (mg/mL): 1–10 (very strong), 11–20 (strong), 21–50 (moderate), 51–100 (weak), 100–200 (very weak), above 200 (non-cytotoxic). ***SOR represents Sorafenib.
Based on the results, compounds 5d, 5h and 5p were the most potent candidates and exhibited intense anticancer inhibition effects versus the tested human tumor cell lines. In comparison to SOR, which had an IC50 value of 5.47 µM towards the HCT-116 cells, compounds 5d, 5h, 5j and 5p exhibited substantial anti-tumor activities have IC50 of 6.09, 5.89, 9.51, 8.32 µM, respectively. Moreover, compounds 5f, 5m and 5r conveyed intense activities with IC50 values of 14.78, 18.14 and 13.20 µM. Furthermore, compounds 5k and 5l had a modest cytotoxic effect on HCT-116 compared to SOR, with IC50 ratings of 23.43 and 28.64 µM. Compared to SOR (IC50 of 9.18), candidates 5d and 5h with IC50 of 2.39 and 6.74 µM exhibited more significant inhibitory activity against the HEPG2 cells. Additionally, the activity of compound 5p (IC50 of 9.72 µM) was nearly the same as that of the reference drug SOR. Compounds 5j and 5r demonstrated cytotoxic solid activity with IC50 of 12.34 and 17.67 µM, respectively. Compounds 5d, 5h, 5j and 5p evinced significant anti-tumor potency versus Hela cells have IC50 ratings of 8.94, 11.61, 19.10 and 14.27 µM in succession. Compared to the positive control (IC50 of 7.26 µM), compounds 5m and 5r exhibited moderate anticancer activity against Hela with IC50 of 27.88 and 22.36 in succession. Compound 5d showed cytotoxic solid action against the MCF-7 cell line, as indicated by the IC50 values (4.81 µM), which were roughly comparable to those of the gold standard medicine SOR (4.17 µM). Furthermore, the IC50 of 7.99 µM, compound 5p, had an incredibly potent action against MCF-7 cells. Intense activities were demonstrated by compounds 5h and 5j, which have IC50 ratings of 13.88 and 16.01 µM, respectively. Moreover, with an IC50 of 7.99 µM, compound 5p exhibited extremely potent action against MCF-7 cells. Compounds 5h and 5j were highly active and had IC50 ratings of 13.88 and 16.01 µM. Furthermore, substances 5m and 5q manifested a moderate anti-tumors effect towards MCF-7 with IC50 of 21.45 and 24.58 µM.
Structure-Activity Correlation
SAR analysis for the newly formed compounds as anti-proliferative agents against HCT-116, HePG-2, HeLa and MCF-7 cells was studied and illustrated in (Figure 3). The tested molecules can be classified into (i) 7-chloro-quinazoline derivatives 5a-i and (ii) 7-unsubstituted analogs 5j-r. Generally, it was found that the 7-chloro analogs exerted an overall better cytotoxic effect than the 7-unsubstituted analogs. The only exception was for unsubstituted analogs 5d, 5f and 5h, which exhibited anti-proliferative solid efficacy towards the 4 tested cell lines with IC50 range of 2.39–18.66 μM, respectively. Concerning the 7-unsubstituted analogs, the SAR analysis hinted that the anticancer activity is impacted by different substituted groups introduced to the aromatic ring attached to the urea group, where the appending of trifluoromethyl substituent to the aryl part had a potential impact on anticancer activity affording the most potent candidate within these series (5d with IC50 of 2.39–8.94 μM). The inclusion of EWD groups like 4-chloro (5b, IC50 = 77.26–100 μM), 4-bromo (5c, IC50 = 48.22–67.67 μM), or EDG like 4-methoxy (5a, IC50 = 62.70–83.41) dramatically declined the anticancer activity. Strikingly, it was deduced that a spacer, either one or two carbon atoms between the aromatic ring and urea moiety, affected the anticancer potency. For instance, the incorporation of one carbon atom spacer between 4-methoxyphenyl and urea moiety as in compound 5f (IC50 = 14.78–34.24 μM) highly enhanced the anticancer effect compared to its analog with no spacer 5a (IC50 of 62.70–83.41 μM). In a similar behavior, the inclusion of one carbon spacer between 4-chlorophenyl and urea moiety as in 5g (IC50 = 35.29–53.68 μM) improved the activity rather than its analog with no spacer 5b (IC50 = 77.26–100 μM) or with two carbon spacer 5i (IC50 = 39.06–58.33 μM). On the other hand, regarding 7-chloro analogs 5j-r, 4-methoxy-grafted analog 5j with no spacer between urea and aromatic ring revealed enhanced inhibitory action with IC50 of 9.51–16.01 μM. The replacement of 4-methoxy either with 4-chloro 5k (IC50 = 23.43–40.85 μM), 4-bromo 5l (IC50 = 28.64–49.41 μM) or 4-trifluoromethyl 5m (IC50 = 18.14–30.68 μM) negatively influenced the inhibitory activity. Moreover, adding a one-carbon spacer between 4-methoxyphenyl and urea reduces markedly the anticancer activity as in 5o with an IC50 range of 69.30–90.23 μM. Similarly, among 4-chloro substituted hybrids, analog with one carbon linker 5p (IC50 = 7.99–14.27 μM) is considered the most efficient anti-tumor inhibitor within these series and showed better activity than the two-carbon linker containing analog 5r (IC50 = 10.74–22.36 μM) and the no spacer one 5k (IC50 = 23.43–40.85 μM), which possessed the minor activity. Furthermore, the hybrids with unsubstituted aromatic groups connected to the urea motif either by one carbon 5n or two carbon linkers 5q exerted a pattern of weak activity. The IC50 curves are illustrated in Table S1.
Figure 3.

Summary of structure–activity correlation of target quinazoline-4(3H)-ones 5a-r..
In vitro Cytotoxicity Towards Normal Human Cells
All the new hybrids were tested for their safety profile by examining their cytotoxicity on standard Caucasian fibroblast-like fetus pulmonary WI-38 cell lines. As depicted from the results in Table 1, all the tested compounds revealed moderate to weak cytotoxicity against normal cells. Our most active anticancer hybrids, 5d, 5h, 5j, 5p and 5r showed moderate to weak activity against normal cells with IC50 values of 36.29, 41.06, 38.52, 56.87 and74.17 μM, respectively, revealing comparable selectivity of the new motifs when compared to the reference SOR (IC50 = 10.65 μM), indicating good safety profile of the new compounds.
VEGFR-2 Enzyme Inhibition Assay
The most active motifs, revealing the highest anti-tumor potencies 5d, 5h and 5p, were further assessed for their dose-dependent VEGFR-2 inhibitory activity at five different concentrations (0.01, 0.1, 1, 10, 100 μM) to find out their IC50 values. The results shown in Table 2 reveal that at doses ranging from 1 to 100 μM, all the substances tested exhibited a significant percentage of inhibition towards me against the tested enzyme at concentrations 1–100 μM. Compound 5p exhibited 70.49%, 88.29% and 93.52% inhibition against VEGFR-2 enzyme at 1, 10, and 100 μM, respectively. 5h showed 93.07%, 86.14% and 65.78% at the three concentrations, whereas compound 5d revealed 60.42%, 83.51% and 90.97% inhibition at the three concentrations above. IC50 of the three most active compounds against VEGFR-2 enzyme showed that compound 5p is the most active against the enzyme with IC50 of 0.117 μM, 5h exhibited IC50 of 0.215 μM and the weakest was 5d with IC50 0.274 μM, in comparison with 0.069 μM for SOR.
Table 2.
Inhibitory Effect of Compounds 5d, 5h and 5p Against VEGFR-2 Enzyme
| Comp.* | % Inhibition | IC50 (μM) | ||||
|---|---|---|---|---|---|---|
| 0.01 μM | 0.1 μM | 1 μM | 10 μM | 100 μM | ||
| 5d | 25.47 | 38.92 | 60.42 | 83.51 | 90.97 | 0.274 |
| 5h | 25.29 | 40.14 | 65.78 | 86.14 | 93.07 | 0.215 |
| 5p | 31.44 | 44.39 | 70.49 | 88.29 | 93.52 | 0.117 |
| SOR** | 35.43 | 47.62 | 75.44 | 90.15 | 94.63 | 0.069 |
Note: *Symbols represent synthesized compounds.**SOR represents Sorafenib.
Cell Cycle Analysis
Further inspection for the mechanism of action of the best active compound 5p against VEGFR-2 inhibiting the growth of cancer cells and how it might attain its effect. The propidium iodide staining test was utilized to examine the cell cycle assessment and its ability to induce apoptosis in MCF-7 cells.52,53 After a 24-hour incubation period alongside the IC50 of the test compound, MCF-7 cells were stained by PI, and their DNA content was assessed using flow cytometry. DMSO was used as a control.
Results (Table 3, Figure 4) indicated that compound 5p halted MCF-7 cells at G0-G1, where the overall percentage increased from 54.09% in the untreated cells to 57.16% in those treated with 5p. The test compound also caused an increase in the population in the S phase by 35.02% compared to 29.16% in the control. It inhibited cell growth in G2/M by 7.82% compared to 16.75% in the untreated cells. These findings prove that our target compound seized the cell growth at the G1/S phase.
Table 3.
The Impact of the Substance 5p on the Distribution of Cell Cycles in MCF-7 Cells, Including DMSO as a Control
| Comp. no | Cell Cycle Distribution (%) | ||
|---|---|---|---|
| G0-G1 | S | G2-M | |
| 5p | 57.16 | 35.02 | 7.82 |
| Control (DMSO) | 54.09 | 29.16 | 16.75 |
Figure 4.

The DNA ploidy in MCF-7 cells was examined using flow cytometry following treatment with compound 5p..
Detection of Apoptosis
Inducing apoptosis is a powerful and attractive way to develop new anti-proliferative candidates. Compound 5p’s apoptosis-inducing capabilities were thus assessed using a flow cytometry experiment that combined the Annexin V-FITC and propidium iodide staining techniques.54 The results show that compound 5p triggered early apoptosis in MCF-7 cells by 26.11% after 24 hours of incubation, compared to the untreated cells by 0.61% (Figure 5). Furthermore, it enhanced late apoptosis by 12.51% compared to the control (0.27%). Moreover, the test compound prompted necrosis by 4.23%. Compound 5p enhanced total apoptosis by 42.85% compared to the control (2.29%). To sum up, we can say that our target motif could inhibit the growth of cells via apoptotic induction.
Figure 5.

Impact of compound 5p on the percentage of Annexin V-FITC positive staining in MCF-7 cells after 24 hours of incubation, with DMSO used as a control. The four stages of cell death are Q1, necrotic cells; Q2, late apoptosis; Q3, living cells; and Q4, early apoptosis.
Molecular Docking
Results and Discussion
Compounds 5p, 5h and 5d 5p, 5h and 5d achieved the best cytotoxicity among the prepared compounds and were found to inhibit VEGFR-2 in enzyme inhibition assay. Molecular docking investigations have been used extensively to identify bioactive compounds and their binding mode.55–63 Hence, we utilized it to identify molecular features in the most active compounds responsible for the observed experimental enzyme inhibition; the three compounds achieved lower but good binding affinity comparable to the standard inhibitor SOR, as found in Table 4. The post-docking analysis highlighted specific significant interactions that are correlated with good inhibitory activity, such as the formation of hydrogen bonding with Asp1044, Glu883 and Cys919 and hydrophobic interactions through the interaction with amino acid in the ATP active site such as Ala866, Glu917, Leu1035, and Leu840 or in the linker area such as Cys1045, Phe1047, Val848, Val916 and Lys868 or in the extra hydrophobic pocket area such as Ile1044, Val898, Leu1019. In this context, compounds 5p, 5h, and 5d could reproduce the previously mentioned interactions to a great extent.
Table 4.
The Top Three Molecules Docked to VEGFR-2 (PDB: 4ASD) Have Lower Binding Energies Than the Co-Crystallized Ligand SOR
| Compound | Venna Score |
|---|---|
| SOR* | −12.3 |
| 5p | −10.5 |
| 5h | −10.2 |
| 5d | −9.8 |
Note: *SOR represents Sorafenib.
In the case of compound 5p, a hydrogen bond was established with Asp1046 and Glu885 through the ureido linker and hydrophobic interaction with Ala881, Leu889, Leu1019, Cys1024, His1026 and Leu1049 through the quinazoline moiety. Furthermore, the benzyl moiety formed extensive hydrophobic with Val848, Ala866, Val899, Val916, Leu1035, Cys1045, and Phe1047. Still, it did not interact with Cys919 or Glu917, a significant interaction required to achieve a potent inhibitory effect. It could explain the superior inhibitory effect of SOR over the compounds under investigation.64
Compound 5h retained the ability to form hydrogen bonds with Glu885 and Asp1046 through the ureido linker and Arg1027 through the carbonyl in the quinazoline moiety. Hydrophobic interactions with Asp814, Ile888, Cys1024 and Asp1046 were also recognized. Also, the benzyl moiety interacted with Val848, Ala866, Val916 and Cys1045 in the linker area. Nevertheless, fewer hydrophobic interactions were observed in the case of 5h compared to 5p, highlighting the importance of chloro substitution in both aryl quinazoline and benzyl moiety, justifying the lower inhibitory activity of compound 5h.
Finally, compound 5d exhibited a different binding mode rather than those compounds where the aryl moiety of the quinazoline extruded out the hydrophobic pocket, limiting the hydrophobic interaction to Ile888 and Cys1024 was compensated by maintaining hydrogen bond with Asp1046, Glu885 through the ureido spacer and hydrophobic interactions with Val848, Ala866, Val916, Leu1035, Cys1045 and Phe1047 through the benzyl moiety and its trifluoromethyl substitution. This follows the experimental enzyme inhibition assay where compound 5d achieved the lowest inhibitory activity compared to 5h, 5p and SOR. Figure 6 depicts how compounds 5p, 5h, and 5d interact through the VEGFR-2 active site.
Figure 6.

Molecular docking of the best active compounds in the active site of VEGFR-2 PDB: 4ASD. (a) 3D interaction of compound 5p with the active site of VEFR2 (b) 2D presentation of the interaction of compound 5p. (c) 3D interaction of compound 5h with the active site of VEFR2 (d) 2D presentation of the interaction of compound 5h. (e) 3D interaction of compound 5d with the active site of VEFR2 (f) 2D presentation of the interaction of compound 5d..
Conclusion
To summarize, the new quinazoline-2-thiol derivatives 5a-r were synthesized. The structure of the novel hybrids was confirmed using different spectroscopic techniques. They were assessed for their cytotoxic effect vs four cancer cell lines: HCT116, HePG2, Hela & MCF7. The most active ones were 5d, 5h and 5p. Compound 5d showed significant activity towards the four tested cell lines with IC50 6.09, 2.39, 8.94 and 4.81 μM in succession. 5h exhibited potent effect against HCT116 and HePG2 with IC50 5.89 and 6.74 μM, respectively. Also, compound 5p showed potent activity against HCT116, HePG2 & MCF7 with IC50 8.32, 9.72 and 7.99, respectively. Compound 5p showed the most effective activity towards the VEGFR-2 enzyme, with an IC50 of 0.117 μM, whereas SOR had an IC50 of 0.069 μM; in subsequent testing, the activities of the three compounds towards the VEGFR-2 enzyme. The enzyme inhibitory test of compound 5p showed that it is the most potent hybrid that caused MCF-7 cells to undergo apoptosis and generated a G1/S cell cycle arrest. Confirmation of the obtained results was done with the aid of the docking study, which showed that the three motifs might adhere to the enzyme’s major active sites, and the results were in good accordance with the experimental VEGFR-2 inhibitory results.
Experimental
Chemistry
The Stuart apparatus (SMP 30) measured melting points (°C). The FT-IR 200 spectrophotometer (ύ cm−1) at the Faculty of Pharmacy, Mansoura University, Egypt, was used to obtain IR spectra (KBr). In the NMR Unit of the Faculty of Pharmacy at Mansoura University in Egypt, 1H-NMR and 13C-NMR spectra were acquired in (DMSO-d6) at 1H-NMR 400 MHz and 13C-NMR 100 MHz, with TMS serving as an internal standard. Mass spectrometry was performed using the Thermo Scientific GCMS model ISQ at the Regional Center for Mycology and Biotechnology (RCMB), Al-Azhar University, Egypt. Chemicals and reagents were purchased from Aldrich Chemicals Co, USA. Reaction times were determined by thin-layer chromatography (TLC) on silica gel plates 60F245 E. Merk, using an eluting solution of (hexane: EtOAc; 1:1) and visualization under UV light (366–245nm). The essential precursors, chloroethyl ureas (2a-i) and mercaptoquinazolin-4(3H)-one derivative (4a-b), could be easily prepared as in literature.47–50
General Procedure for the Synthesis of 3.4-Dihydroquinazolin-2-Yl)thio)ethyl)ureas (5a-r)
A mixture of 2.3-dihydroquinazolin-4(1H)-one derivatives 4a-b (0.2 mmol), chloride 2a-i (1.2 eq), and K2CO3 in DMF (2 mL) was stirred at 70°C for 24 h. Then, the reaction mixture was poured into water/ice, and the formed precipitates were filtered, washed with cold water, and purified and recrystallized using ethyl acetate to give target compounds 5a-r.
1-(4-Methoxyphenyl)-3-(2-((4-Oxo-3-Phenyl-3,4-Dihydroquinazolin-2-Yl)thio)ethyl)urea (5a)
White solid (0.07 g, 79%). M.p. 199–201°C. IR (νmax/cm−1): 3320 (NH), 3068, 2994 (CH), 1691, 1632 (C=O), 1606, 1550, 1225.1H NMR (400 MHz, DMSO-d6) δ 8.32 (s, 1H), 8.10 (dd, J = 7.9, 1.1 Hz, 1H), 7.87–7.81 (m, 1H), 7.64 (d, J = 7.9 Hz, 1H), 7.59–7.53 (m, 3H), 7.50 (d, J = 7.2 Hz, 1H), 7.48–7.42 (m, 2H), 7.27 (d, J = 9.0 Hz, 2H), 6.82 (d, J = 9.0 Hz, 2H), 6.22 (t, J = 5.7 Hz, 1H), 3.70 (s, 3H), 3.46 (t, J = 6.3 Hz, 2H), 3.27 (t, J = 6.3 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 161.3, 157.5, 155.9, 154.5, 147.8, 136.4, 135.4, 133.9, 130.3, 130.0, 129.9, 129.9, 127.0, 126.6, 126.4, 120.0, 114.3, 55.6, 38.2, 33.2. MS m/z (%): 446.81 (M+, 25.88).
1-(4-Chlorophenyl)-3-(2-((4-Oxo-3-Phenyl-3,4-Dihydroquinazolin-2-Yl)thio)ethyl)urea (5b)
White solid (0.068 g, 75%). M.p. 220–221°C. IR (νmax/cm−1): 3327 (NH), 3060, 2927 (CH), 1692, 1640 (C=O), 1599, 1551, 1206. 1H NMR (400 MHz, DMSO-d6) δ 8.70 (s, 1H), 8.12 (d, J = 7.2 Hz, 1H), 7.86 (s, 1H), 7.65 (d, J = 7.2 Hz, 1H), 7.55 (s, 3H), 7.55–7.40 (m, 5H), 7.28 (s, 2H), 6.48 (s, 1H), 3.46 (t, J = 6.3 Hz, 2H), 3.28 (t, J = 6.3 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 161.3, 157.5, 155.6, 147.9, 139.9, 136.5, 135.3, 131.3, 129.9, 128.9, 127.9, 127.0, 126.8, 126.4, 125.2, 120.3, 119.9, 38.5, 33.0. MS m/z (%): 450.33 (M+, 30.47).
1-(4-Bromophenyl)-3-(2-((4-Oxo-3-Phenyl-3,4-Dihydroquinazolin-2-Yl)thio)ethyl)urea (5c)
White solid (0.076 g, 77%). M.p. 229–231°C. IR (νmax/cm−1): 3325 (NH), 3065, 2927 (CH), 1688, 1642 (C=O), 1599, 1551, 1206. 1H NMR (400 MHz, DMSO-d6) δ 8.96 (s, 1H), 8.10 (s, 1H), 7.83 (s, 1H), 7.64 (d, J = 7.2 Hz, 1H), 7.51–7.43 (m, 6H), 7.42–7.31 (s, 4H), 6.66 (s, 1H), 3.46 (t, J = 6.3 Hz, 2H), 3.28 (t, J = 6.3 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 161.3, 157.5, 155.6, 147.8, 140.4, 136.5, 135.3, 131.8, 130.3, 129.9, 128.3, 127.0, 126.6, 126.4, 120.1, 120.0, 112.7, 38.4, 33.0. MS m/z (%): 495.41(M+, 35.98).
1-(2-((4-Oxo-3-Phenyl-3,4-Dihydroquinazolin-2-Yl)thio)ethyl)-3-(4-(Trifluoromethyl)phenyl)urea (5d)
White solid; (0.082 g, 81%). M.p. 240–242°C. IR (νmax/cm−1): 3327 (NH), 3068, 2920 (CH), 1690, 1640 (C=O), 1599, 1551, 1245. 1H NMR (400 MHz, DMSO-d6) δ 8.79 (s, 1H), 8.10 (d, J = 7.2 Hz, 1H), 7.83 (s, 1H), 7.63 (d, J = 7.2 Hz, 1H), 7.55 (s, 3H), 7.51–7.38 (m, 5H), 7.28 (s, 2H), 6.47 (s, 1H), 3.46 (t, J = 6.3 Hz, 2H), 3.28 (t, J = 6.3 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 161.3, 157.5, 155.5, 147.8, 139.9, 136.5, 135.3, 130.3, 129.9, 128.9, 127.9, 127.0, 126.6, 126.5, 126.4, 125.0, 120.0, 119.6, 38.5, 33.0. MS m/z (%): 484.8 (M+, 25.12).
1-Benzyl-3-(2-((4-Oxo-3-Phenyl-3,4-Dihydroquinazolin-2-Yl)thio)ethyl)urea (5e)
White solid (0.067 g, 78%). M.p. 200–202°C. IR (νmax/cm−1): 3450, 3329 (NH), 3066, 2917 (CH), 1693, 1621 (C=O), 1581, 1546, 1256. 1H NMR (400 MHz, DMSO-d6) δ 8.09 (d, J = 6.8 Hz, 1H), 7.88–7.84 (m, 1H), 7.64 (d, J = 8.1 Hz, 1H), 7.61–7.54 (m, 3H), 7.53–7.41 (m, 3H), 7.35–7.26 (m, 2H), 7.25–7.17 (m, 3H), 6.44 (t, J = 5.7 Hz, 1H), 6.18 (t, J = 5.7 Hz, 1H), 4.19 (d, J = 5.8 Hz, 2H), 3.46 (t, J = 6.3 Hz, 2H), 3.23 (t, J = 6.3 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 161.3, 158.3, 157.4, 147.5, 140.5, 139.0, 135.2, 130.3, 130.0, 129.9, 129.3, 127.3, 127.0, 126.5, 126.4, 125.8, 120.0, 42.7, 38.7, 33.2. MS m/z (%): 431.2 (M+, 43.18).
1-(4-Methoxybenzyl)-3-(2-((4-Oxo-3-Phenyl-3,4-Dihydroquinazolin-2-Yl)thio)ethyl)urea (5f)
White solid (0.078 g, 85%). M.p. 203–205°C. IR (νmax/cm−1): 3448, 3331 (NH), 3069, 2932 (CH), 1686, 1624 (C=O), 1581, 1547, 1255. 1H NMR (400 MHz, DMSO-d6) δ 8.09 (dd, J = 7.9, 1.2 Hz, 1H), 7.89–7.82 (m, 1H), 7.64 (d, J = 8.0 Hz, 1H), 7.61–7.55 (m, 3H), 7.53–7.48 (m, 1H), 7.48–7.43 (m, 2H), 7.14 (d, J = 8.6 Hz, 2H), 6.85 (d, J = 8.6 Hz, 2H), 6.34 (t, J = 5.8 Hz, 1H), 6.13 (t, J = 5.7 Hz, 1H), 4.11 (d, J = 5.9 Hz, 2H), 3.73 (s, 3H), 3.34 (t, J = 6.2 Hz, 2H), 3.22 (t, J = 6.2 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 161.3, 158.5, 158.3, 157.6, 147.8, 136.5, 135.4, 133.1, 130.3, 129.9, 129.9, 128.8, 127.0, 126.6, 126.4, 120.0, 114.1, 55.5, 42.8, 38.7, 33.3. MS m/z (%): 460.91 (M+, 20.22).
1-(4-Chlorobenzyl)-3-(2-((4-Oxo-3-Phenyl-3,4-Dihydroquinazolin-2-Yl)thio)ethyl)urea (5g)
White solid (0.066 g, 71%). M.p. 207–209°C. IR (νmax/cm−1): 3447, 3328 (NH), 3067, 2925 (CH), 1695, 1624 (C=O), 1581, 1546, 1206. 1H NMR (400 MHz, DMSO-d6) δ 8.09 (d, J = 7.0 Hz, 1H), 7.91–7.80 (m, 1H), 7.63 (d, J = 8.1 Hz, 1H), 7.60–7.53 (m, 3H), 7.50 (d, J = 8.1 Hz, 1H), 7.48–7.43 (m, 2H), 7.35 (d, J = 8.3 Hz, 2H), 7.24 (d, J = 8.3 Hz, 2H), 6.49 (t, J = 5.9 Hz, 1H), 6.23 (t, J = 5.6 Hz, 1H), 4.18 (d, J = 6.0 Hz, 2H), 3.44 (t, J = 6.3 Hz, 2H), 3.22 (t, J = 6.3 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 161.3, 158.3, 157.6, 147.8, 140.5, 136.5, 135.4, 131.5, 130.3, 130.0, 129.9, 129.3, 128.6, 127.0, 126.6, 126.4, 120.0, 42.7, 38.7, 33.2. MS m/z (%): 465.37 (M+, 11.58).
1-(2-((4-Oxo-3-Phenyl-3,4-Dihydroquinazolin-2-Yl)thio)ethyl)-3-Phenethylurea (5h)
White solid (0.069 g, 78%). M.p. 162–164°C. IR (νmax/cm−1): 3354 (NH), 3064, 2969 (CH), 1694, 1632 (C=O), 1584, 1547, 1206. 1H NMR (400 MHz, DMSO-d6) δ 8.10 (d, J = 7.7 Hz, 1H), 7.86 (t, J = 7.5 Hz, 1H), 7.64 (d, J = 8.1 Hz, 1H), 7.62–7.53 (m, 3H), 7.50 (d, J = 8.1 Hz, 1H), 7.49–7.43 (m, 2H), 7.28 (t, J = 7.4 Hz, 2H), 7.23–7.15 (m, 3H), 6.14 (t, J = 5.5 Hz, 1H), 5.95 (t, J = 5.5 Hz, 1H), 3.32 (t, J = 6.2 Hz, 2H), 3.23–3.15 (m, 4H), 2.66 (t, J = 7.2 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 161.3, 158.3, 157.6, 147.8, 140.2, 136.5, 135.4, 130.3, 129.9, 129.1, 128.8, 127.0, 126.6, 126.5, 126.4, 120.0, 49.1, 41.4, 38.6, 36.6, 33.3. MS m/z (%): 444.61 (M+, 40.45).
1-(4-Chlorophenethyl)-3-(2-((4-Oxo-3-Phenyl-3,4-Dihydroquinazolin-2-Yl)thio)ethyl)urea (5i)
White solid (0.07 g, 73%). M.p. 172–174°C. IR (νmax/cm−1): 3323 (NH), 3065, 2941 (CH), 1693, 1621 (C=O), 1585, 1547, 1206. 1H NMR (400 MHz, DMSO-d6) δ 8.09 (d, J = 7.0 Hz, 1H), 7.91–7.79 (m, 1H), 7.64 (d, J = 8.1 Hz, 1H), 7.62–7.53 (m, 3H), 7.51 (d, J = 8.1 Hz, 1H), 7.48–7.39 (m, 2H), 7.32 (d, J = 8.3 Hz, 2H), 7.20 (d, J = 8.3 Hz, 2H), 6.17 (s, 1H), 5.98 (s, 1H), 3.31 (t, J = 6.2 Hz, 2H), 3.25–3.12 (m, 4H), 2.65 (t, J = 7.0 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 161.3, 158.3, 157.6, 147.9, 137.5, 136.4, 135.4, 131.5, 130.3, 129.9, 129.1, 129.0, 128.5, 126.6, 126.4, 120.0, 49.1, 41.4, 38.6, 36.6, 33.3. MS m/z (%): 480.31 (M+, 28.44).
1-(2-((7-Chloro-4-Oxo-3-Phenyl-3,4-Dihydroquinazolin-2-Yl)thio)ethyl)-3-(4-Methoxyphenyl)urea (5j)
White solid (0.081 g, 84%). M.p. 139–141°C. IR (νmax/cm−1): 3323 (NH), 3067, 2932 (CH), 1691, 1642 (C=O), 1603, 1547, 1241. 1H NMR (400 MHz, DMSO-d6) δ 8.44 (s, 1H), 8.08 (d, J = 8.5 Hz, 1H), 7.69 (s, 1H), 7.56–7.45 (m, 7H), 7.27 (d, J = 8.6 Hz, 2H), 6.81 (d, J = 8.6 Hz, 2H), 6.36 (s, 1H), 3.70 (s, 3H), 3.33 (t, J = 6.2 Hz, 2H), 3.26 (t, J = 6.2 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 160.7, 159.6, 155.9, 154.4, 148.7, 139.9, 136.3, 130.4, 130.0, 129.8, 129.1, 126.5, 125.8, 120.2, 120.0, 118.9, 114.3, 55.6, 38.5, 33.2. MS m/z (%): 480.32 (M+, 25.22).
1-(2-((7-Chloro-4-Oxo-3-Phenyl-3,4-Dihydroquinazolin-2-Yl)thio)ethyl)-3-(4-Chlorophenyl)urea (5k)
White solid (0.071 g, 73%). M.p. 165–167°C. IR (νmax/cm−1): 3384, 3336 (NH), 3066, 2907 (CH), 1688, 1657 (C=O), 1598, 1544, 1270. 1H NMR (400 MHz, DMSO-d6) δ 8.69 (s, 1H), 8.07 (d, J = 8.4 Hz, 1H), 7.65 (s, 1H), 7.56 (s, 3H), 7.52–7.43 (m, 3H), 7.40 (d, J = 8.3 Hz, 2H), 7.26 (d, J = 8.2 Hz, 2H), 6.39 (s, 1H), 3.43 (t, J = 6.2 Hz, 2H), 3.27 (t, J = 6.2 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 160.7, 159.5, 155.5, 148.7, 139.9, 139.8, 136.3, 130.4, 130.0, 129.8, 129.1, 128.9, 126.5, 125.8, 125.1, 119.6, 118.9, 38.6, 33.0. MS m/z (%): 485.42 (M+, 35.40).
1-(4-Bromophenyl)-3-(2-((7-Chloro-4-Oxo-3-Phenyl-3,4-Dihydroquinazolin-2-Yl)thio)ethyl)urea (5l)
White solid (0.08 g, 75%). M.p. 170–172°C. IR (νmax/cm−1): 3382, 3336 (NH), 3062, 2909 (CH), 1685, 1655 (C=O), 1598, 1544, 1270. 1H NMR (400 MHz, DMSO-d6) δ 8.68 (s, 1H), 8.07 (d, J = 8.5 Hz, 1H), 7.97 (s, 1H), 7.66 (d, J = 7.9 Hz, 1H), 7.56 (s, 2H), 7.50 (d, J = 7.9 Hz, 2H), 7.46 (s, 2H), 7.37 (d, J = 7.7 Hz, 2H), 6.38 (s, 1H), 6.17 (s, 1H), 3.43 (t, J = 6.2 Hz, 2H), 3.28 (t, J = 6.2 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 160.7, 159.5, 155.6, 148.5, 139.9, 139.6, 136.3, 131.5, 130.0, 129.6, 128.3, 127.0, 126.5, 125.8, 125.1, 119.6, 112.7, 38.6, 33.0. MS m/z (%): 530.21 (M+, 29.15).
1-(2-((7-Chloro-4-Oxo-3-Phenyl-3,4-Dihydroquinazolin-2-Yl)thio)ethyl)-3-(4-(Trifluoromethyl)phenyl)urea (5m)
White solid (0.062 g, 60%). M.p. 238–240°C. IR (νmax/cm−1): 3385, 3336 (NH), 3064, 2909 (CH), 1685, 1658 (C=O), 1598, 1544, 1255. 1H NMR (400 MHz, DMSO-d6) δ 8.95 (s, 1H), 8.07 (d, J = 8.2 Hz, 1H), 7.64 (s, 1H), 7.62–7.49 (m, 7H), 7.48 (s, 2H), 6.47 (s, 1H), 3.43 (t, J = 6.2 Hz, 2H), 3.29 (t, J = 6.2 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 160.7, 159.5, 155.3, 148.7, 144.6, 139.9, 136.3, 130.4, 129.9, 129.8, 129.1, 126.5, 126.5, 126.4, 125.8, 118.9, 117.7, 38.7, 32.9. MS m/z (%): 519.53 (M+, 20.71).
1-Benzyl-3-(2-((7-Chloro-4-Oxo-3-Phenyl-3,4-Dihydroquinazolin-2-Yl)thio)ethyl)urea (5n)
White solid (0.064 g, 69%). M.p. 169–171°C. IR (νmax/cm−1): 3559, 3324 (NH), 3069, 2941 (CH), 1683, 1623 (C=O), 1571, 1544, 1257. 1H NMR (400 MHz, DMSO-d6) δ 8.08 (s, 1H), 7.80–7.41 (m, 7H), 7.40–6.98 (m, 5H), 6.45 (s, 1H), 6.19 (s, 1H), 4.17 (d, J = 5.4 Hz, 2H), 3.40 (t, J = 6.2 Hz, 2H), 3.19 (t, J = 6.2 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 160.7, 159.7, 158.4, 148.7, 141.2, 139.9, 136.3, 130.4, 130.0, 129.8, 129.1, 128.7, 127.5, 127.0, 126.5, 125.8, 118.9, 43.4, 38.8, 33.3. MS m/z (%): 465.12 (M+, 25.33).
1-(2-((7-Chloro-4-Oxo-3-Phenyl-3,4-Dihydroquinazolin-2-Yl)thio)ethyl)-3-(4-Methoxybenzyl)urea (5o)
White solid (0.079 g, 81%). M.p. 175–177°C. IR (νmax/cm−1): 3520, 3320 (NH), 3065, 2945 (CH), 1690, 1623 (C=O), 1571, 1548, 1257. 1H NMR (400 MHz, DMSO-d6) δ 8.08 (d, J = 8.5 Hz, 1H), 7.70 (s, 1H), 7.57 (s, 3H), 7.52 (d, J = 8.6 Hz, 1H), 7.51–7.43 (m, 2H), 7.14 (d, J = 8.2 Hz, 2H), 6.86 (d, J = 8.2 Hz, 2H), 6.36 (s, 1H), 6.14 (s, 1H), 4.11 (d, J = 5.4 Hz, 2H), 3.73 (s, 3H), 3.40 (t, J = 6.2 Hz, 2H), 3.21 (t, J = 6.2 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 160.7, 159.7, 158.4, 154.5, 148.7, 141.3, 139.9, 136.3, 130.6, 130.4, 129.8, 129.5, 128.7, 127.0, 114.5, 55.6, 125.8, 118.9, 43.4, 38.8, 33.3. MS m/z (%): 494.61 (M+, 25.45).
1-(2-((7-Chloro-4-Oxo-3-Phenyl-3,4-Dihydroquinazolin-2-Yl)thio)ethyl)-3-(4-Chlorobenzyl)urea (5p)
White solid (0.074 g, 74%). M.p. 180–182°C. IR (νmax/cm−1): 3346, 3316 (NH), 3074, 2945 (CH), 1686, 1623 (C=O), 1569, 1546, 1257. 1H NMR (400 MHz, DMSO-d6) δ 8.08 (d, J = 8.4 Hz, 1H), 7.69 (s, 1H), 7.62–7.54 (m, 3H), 7.52 (d, J = 8.6 Hz, 1H), 7.51–7.42 (m, 2H), 7.35 (d, J = 7.9 Hz, 2H), 7.24 (d, J = 7.9 Hz, 2H), 6.51 (s, 1H), 6.24 (s, 1H), 4.18 (d, J = 5.5 Hz, 2H), 3.42 (t, J = 6.2 Hz, 2H), 3.22 (t, J = 6.2 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 160.7, 159.7, 158.3, 148.7, 140.4, 139.9, 136.3, 131.5, 130.4, 130.0, 129.8, 129.3, 129.1, 128.6, 126.5, 125.8, 118.9, 42.7, 38.8, 33.3. MS m/z (%): 499.75 (M+, 30.02).
1-(2-((7-Chloro-4-Oxo-3-Phenyl-3,4-Dihydroquinazolin-2-Yl)thio)ethyl)-3-Phenethylurea (5q)
White solid (0.077 g, 80%). M.p. 200–202°C. IR (νmax/cm−1): 3350 (NH), 3067, 2933 (CH), 1690, 1628 (C=O), 1603, 1547, 1258. 1H NMR (400 MHz, DMSO-d6) δ 8.08 (d, J = 8.5 Hz, 1H), 7.70 (s, 1H), 7.62–7.55 (m, 3H), 7.52 (d, J = 8.6 Hz, 1H), 7.50–7.43 (m, 2H), 7.28 (t, J = 7.3 Hz, 2H), 7.20 (t, J = 7.2 Hz, 3H), 6.18 (t, J = 5.5 Hz, 1H), 6.00 (t, J = 5.4 Hz, 1H), 3.30 (t, J = 6.2 Hz, 2H), 3.25–3.14 (m, 4H), 2.66 (t, J = 7.2 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 160.7, 159.7, 158.3, 148.7, 140.2, 139.9, 136.3, 130.4, 130.0, 129.8, 129.1, 128.8, 126.5, 126.4, 125.8, 118.9, 41.4, 38.6, 36.5, 33.4. MS m/z (%): 478.79 (M+, 19.92).
1-(2-((7-Chloro-4-Oxo-3-Phenyl-3,4-Dihydroquinazolin-2-Yl)thio)ethyl)-3-(4-Chlorophenethyl)urea (5r)
White solid (0.071 g, 69%). M.p. 253–255°C. IR (νmax/cm−1): 3331 (NH), 3071, 2938 (CH), 1696, 1626 (C=O), 1575, 1545, 1259. 1H NMR (400 MHz, DMSO-d6) δ 8.08 (d, J = 8.5 Hz, 1H), 7.69 (s, 1H), 7.63–7.54 (m, 3H), 7.52 (d, J = 8.5 Hz, 1H), 7.49–7.43 (m, 2H), 7.32 (d, J = 8.2 Hz, 2H), 7.21 (d, J = 8.1 Hz, 2H), 6.18 (s, 1H), 5.99 (t, J = 5.2 Hz, 1H), 3.30 (d, J = 6.2 Hz, 2H), 3.23–3.15 (m, 4H), 2.65 (t, J = 7.0 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 160.7, 159.7, 158.3, 148.7, 139.9, 139.2, 136.3, 131.1, 131.0, 130.4, 130.0, 129.8, 129.1, 128.6, 126.5, 125.8, 118.9, 41.2, 38.6, 35.8, 33.4. MS m/z (%): 514.19 (M+, 27.30).
Biological Evaluation
Anti-Proliferative Screening
The HCT-116, HeLa, HePG-2 and MCF-7 cancer cell lines and WI-38 normal fibroblast cells from American Type Culture Collection (ATCC) via the Holding company for biological products and vaccines (VACSERA) (Cairo, Egypt) were screened using RPMI-1640 medium (Sigma Co., St. Louis, USA) supplemented with 10% fetal bovine serum (GIBCO, UK), 100 units/mL penicillin, 100µg/mL streptomycin and maintained at 37°C in a 5% Co2 incubator. The cell lines were seeded in a 96-well plate at a density of 1.0×104 cells/well at 37°C for 48 h under 5% Co2. After incubation, the cells were treated with different concentration of tested compounds and SOR (1.56, 3.125, 6.25, 12.5, 25, 50, 100 µm) then incubated for 24 h. After drug treatment, 20 µL of MTT solution at 5mg/mL was added and incubated for 4 h. Dimethyl sulfoxide (DMSO) (Sigma Co., St. Louis, USA) in volume of 100 µL is added into each well to dissolve the purple formazan formed. The colorimetric assay is measured and recorded at absorbance of 570 nm using a plate reader (EXL 800, USA). The relative cell viability in percentage was calculated as (A570 of treated samples/A570 of untreated sample) X 100. The equation of Boltzmann sigmoidal concentration– response curve was used for calculating the IC50 by using Graph Pad Prism 6 and compared to the reference drug.33,65,66
In vitro VEGFR-2 Kinase Inhibitory Assay
The detecting reagent Kinase-Glo® MAX (Promega) was used to measure VEGFR-2 kinase activity. The VEGFR2 (KDR) Kinase assay kit (BPS Bioscience, Catalog # 40325) was added to 96-well plates with purified recombinant VEGFR-2 enzyme, VEGFR-2 substrate, ATP and kinase assay buffer according to manufacturer’s instructions. In brief, the master mixture was prepared (25 μL per well) and poured into each well. 5 μL of inhibitor solution was added to each well and designated as “Test Inhibitor”. The “Positive Control” and “Blank” groups received 5 μL of the same solution without the inhibitor (Inhibitor buffer). Prepare 3 mL of kinase buffer by combining 600 μL of kinase buffer with 2400 μL of water. The blank wells received 20 μL of kinase buffer. The amount of VEGFR-2 required for the test was measured and the enzyme was diluted to 1 ng/μL with kinase buffer. 20 μL of diluted VEGFR-2 enzyme was added to the wells designated as “Test Inhibitor Control” and “Positive Control” to initiate the reaction and the mixtures were incubated at 30°C for 45 minutes. After that, 50 μL of Kinase-Glo Max reagent was added to each well and the plate was incubated at room temperature for 15 minutes. The luminescence was measured with a microplate reader.
Flow Cytometry Analysis of the Cell Cycle Distribution
Cell-cycle analysis was performed by DNA staining with propidium iodide (PI). Briefly, MCF-7 cells were exposed to 5p at its IC50 at 37°C for 24 h under 5% CO2. Cells were washed twice with phosphate buffer saline (PBS), then collected by centrifugation. After that, the cells were mixed with ice-cold 70% (v/v) ethanol cells, resuspended with PBS buffer and incubated with 1 mL of PI staining reagent (50 mg/mL PI, 0.1 mg/mL RNaseA and 0.05% Triton X-100) for one hour at room temperature. Cells were evaluated by flow cytometry using FACSCalibur (Becton Dickinson) and the percentage of cells at each phase of the cell cycle was calculated using Cell-Questsoftware (Becton Dickinson).
Cell Apoptosis Analysis
The extent of apoptosis was measured by staining with annexin V-fluorescein isothiocyanate (FITC) (an apoptotic cell marker) and PI (a necrotic cell marker) using the Annexin V-FITC/PI apoptosis detection kit (BD Biosciences, San Diego, CA) according to the manufacturer’s instructions. Briefly, MCF-7 cells were treated with compound 5p and incubated for 24 h. Then, 1–5×105 cells were collected by centrifugation and suspended in 500 µL of binding buffer. After that, cells stained with 5 μL Annexin V-FITC and 5 μL PI and incubated for 5 min at room temperature in the dark. Analysis of Annexin-V-FITC binding was performed using FACS calibur flow cytometer (BD Biosciences, San Jose. CA).54,66,67
Docking Methods
Since compounds 5p, 5h and 5d significantly affected VEGFR-2, molecular docking was utilized to gain insights into their interaction with the active site. In brief, the VEGFR-2 3D structure was downloaded from the Protein Data Bank (PDB) using the code (4ASD) and then processed using the default settings on the Cb-dock-2 server (https://cadd.labshare.cn/cb-dock2/php/index.php). Also, the ligand preparation module of the same server was used to obtain the 3D chemical structure of the most active derivatives, as previously reported.68 Template-based docking using Auto Dock vina as the docking engine and the active site was determined as a grid box size X:20, Y:20, Z:20 using the following coordinates: X: −23.744, Y: −4.022, Z: −9.684. The docking software was validated by redocking the co-crystallized, and the RMSD was found to be 0.5. vina score was calculated for each compound. Finally, the Discovery Studio visualizer evaluated the binding of the Docked Pose with the active site.24,69
Funding Statement
This work was funded by the Deanship of Graduate Studies and Scientific Research at Jouf University under grant No. (DGSSR-2024-01-02062).
Data Sharing Statement
The data supporting this study’s findings are available from the corresponding author, M. M. A., upon reasonable request.
Disclosure
The authors declare no conflicts of interest in this work.
References
- 1.Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics 2023. Ca Cancer J Clin. 2023;73(1):17–48. doi: 10.3322/caac.21763 [DOI] [PubMed] [Google Scholar]
- 2.Hamdi A, Said E, Farahat AA, El-Bialy SAA, Massoud MAM. Synthesis and in vivo antifibrotic activity of novel leflunomide analogues. Lett Drug Des Discov. 2016;13(9):912–920. doi: 10.2174/1570180813666160630125624 [DOI] [Google Scholar]
- 3.Eckhardt S. Recent progress in the development of anticancer agents. Curr Med Chem Anticancer Agents. 2002;2(3):419–439. doi: 10.2174/1568011024606389 [DOI] [PubMed] [Google Scholar]
- 4.Ali R, Mirza Z, Ashraf GM, et al. New anticancer agents: recent developments in tumor therapy. Anticancer Res. 2012;32(7):2999–3005. [PubMed] [Google Scholar]
- 5.Shibuya M. Vascular endothelial growth factor (VEGF) and its receptor (VEGFR) signaling in angiogenesis: a crucial target for anti- and pro-angiogenic therapies. Genes Cancer. 2011;2:1097–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hall AP. The role of angiogenesis in cancer. Comp Clin Path. 2005;13(3):95–99. doi: 10.1007/s00580-004-0533-3 [DOI] [Google Scholar]
- 7.Modi SJ, Kulkarni VM. Vascular endothelial growth factor receptor (VEGFR-2)/KDR inhibitors: medicinal chemistry perspective. Med Drug Discov. 2019;2:100009. doi: 10.1016/j.medidd.2019.100009 [DOI] [Google Scholar]
- 8.Gasparini G, Longo R, Toi M, Ferrara N. Angiogenic inhibitors: a new therapeutic strategy in oncology. Nat Clin Pract Oncol. 2005;2(11):562–577. doi: 10.1038/ncponc0342 [DOI] [PubMed] [Google Scholar]
- 9.Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature. 2005;438(7070):967–974. doi: 10.1038/nature04483 [DOI] [PubMed] [Google Scholar]
- 10.Ann H, Bart L, Martin SH, Hans W, Allan TVO, Ernst ADB. Vascular endothelial growth factor and angiogenesis. Pharmacol Rev. 2004;56(4):549. doi: 10.1124/pr.56.4.3 [DOI] [PubMed] [Google Scholar]
- 11.Olsson A-K, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling? in control of vascular function. Nat Rev Mol Cell Biol. 2006;7(5):359–371. doi: 10.1038/nrm1911 [DOI] [PubMed] [Google Scholar]
- 12.Ferrara N. Vascular endothelial growth factor and the regulation of angiogenesis. Recent Prog Horm Res. 2000;55:15–35. [PubMed] [Google Scholar]
- 13.Shah AA, Kamal MA, Akhtar S. Tumor angiogenesis and VEGFR-2: mechanism, pathways and current biological therapeutic interventions. Curr Drug Metab. 2021;22(1):50–59. doi: 10.2174/1389200221666201019143252 [DOI] [PubMed] [Google Scholar]
- 14.Liu X-J, Zhao H-C, Hou S-J, et al. Recent development of multi-target VEGFR-2 inhibitors for the cancer therapy. Bioorg Chem. 2023;133:106425. doi: 10.1016/j.bioorg.2023.106425 [DOI] [PubMed] [Google Scholar]
- 15.Ziogas IA, Tsoulfas G. Evolving role of Sorafenib in the management of hepatocellular carcinoma. World J Clin Oncol. 2017;8(3):203. doi: 10.5306/wjco.v8.i3.203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roskoski R Jr. Sunitinib: a VEGF and PDGF receptor protein kinase and angiogenesis inhibitor. Biochem Biophys Res Commun. 2007;356(2):323–328. doi: 10.1016/j.bbrc.2007.02.156 [DOI] [PubMed] [Google Scholar]
- 17.Aversa C, Leone F, Zucchini G, et al. Linifanib: current status and future potential in cancer therapy. Expert Rev Anticancer Ther. 2015;15(6):677–687. doi: 10.1586/14737140.2015.1042369 [DOI] [PubMed] [Google Scholar]
- 18.Hao Z, Wang P. Lenvatinib in management of solid tumors. Oncologist. 2020;25(2):e302–e310. doi: 10.1634/theoncologist.2019-0407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jamil MO, Hathaway A, Mehta A. Tivozanib: status of development. Curr Oncol Rep. 2015;17(6):1–7. doi: 10.1007/s11912-015-0451-3 [DOI] [PubMed] [Google Scholar]
- 20.Chau NG, Haddad RI. Vandetanib for the treatment of medullary thyroid cancer. Clin Cancer Res. 2013;19(3):524–529. doi: 10.1158/1078-0432.CCR-12-2353 [DOI] [PubMed] [Google Scholar]
- 21.Khanna P, Soh HJ, Chen C-H, et al. ACE2 abrogates tumor resistance to VEGFR inhibitors suggesting angiotensin-(1-7) as a therapy for clear cell renal cell carcinoma. Sci Transl Med. 2021;13(577):eabc0170. doi: 10.1126/scitranslmed.abc0170 [DOI] [PubMed] [Google Scholar]
- 22.Liu Y, Li Y, Wang Y, et al. Recent progress on vascular endothelial growth factor receptor inhibitors with dual targeting capabilities for tumor therapy. J Hematol Oncol. 2022;15(1):89. doi: 10.1186/s13045-022-01310-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.S V, Kajal K, Mondal S, et al. Novel VEGFR-2 kinase inhibitors as anticancer agents: a review focusing on SAR and molecular docking studies (2016–2021). Chem Biodivers. 2023;20(2):e202200847. doi: 10.1002/cbdv.202200847 [DOI] [PubMed] [Google Scholar]
- 24.Al-Sanea MM, Hamdi A, Mohamed AAB, et al. New benzothiazole hybrids as potential VEGFR-2 inhibitors: design, synthesis, anticancer evaluation, and in silico study. J Enzyme Inhib Med Chem. 2023;38(1):2166036. doi: 10.1080/14756366.2023.2166036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Catalano A, Iacopetta D, Sinicropi MS, Franchini C. Diarylureas as antitumor agents. Appl Sci. 2021;11(1):374. doi: 10.3390/app11010374 [DOI] [Google Scholar]
- 26.Cheng K, Liu C-F, Rao G-W. Anti-angiogenic agents: a review on vascular endothelial growth factor receptor-2 (VEGFR-2) inhibitors. Curr Med Chem. 2021;28(13):2540–2564. doi: 10.2174/0929867327666200514082425 [DOI] [PubMed] [Google Scholar]
- 27.Lee K, Jeong K-W, Lee Y, et al. Pharmacophore modeling and virtual screening studies for new VEGFR-2 kinase inhibitors. Eur J Med Chem. 2010;45(11):5420–5427. doi: 10.1016/j.ejmech.2010.09.002 [DOI] [PubMed] [Google Scholar]
- 28.Ahmad I, Ahmad I. An insight into the therapeutic potential of quinazoline derivatives as anticancer agents. MedChemComm. 2017;8(5):871–885. doi: 10.1039/C7MD00097A [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hameed A, Al-Rashida M, Uroos M, Ali SA, Arshia MI, Khan KM. Quinazoline and quinazolinone as important medicinal scaffolds: a comparative patent review (2011–2016. Expert Opin Ther Pat. 2018;28(4):281–297. doi: 10.1080/13543776.2018.1432596 [DOI] [PubMed] [Google Scholar]
- 30.El-Shafey HW, Gomaa RM, El-Messery SM, Goda FE. Quinazoline based HSP90 inhibitors: synthesis, modeling study and ADME calculations towards breast cancer targeting. Bioorg Med Chem Lett. 2020;30(15):127281. doi: 10.1016/j.bmcl.2020.127281 [DOI] [PubMed] [Google Scholar]
- 31.Moradi M, Mousavi A, Emamgholipour Z, et al. Quinazoline-based VEGFR-2 inhibitors as potential anti-angiogenic agents: a contemporary perspective of SAR and molecular docking studies. Eur J Med Chem. 2023;259:115626. doi: 10.1016/j.ejmech.2023.115626 [DOI] [PubMed] [Google Scholar]
- 32.Zayed MF. Medicinal chemistry of quinazolines as anticancer agents targeting tyrosine kinases. Sci Pharm. 2023;91(2):18. doi: 10.3390/scipharm91020018 [DOI] [Google Scholar]
- 33.Hamdi A, El-Shafey HW, Othman DI, El-Azab AS, AlSaif NA, Alaa A-M. Design, synthesis, antitumor, and VEGFR-2 inhibition activities of novel 4-anilino-2-vinyl-quinazolines: molecular modeling studies. Bioorg Chem. 2022;122:105710. doi: 10.1016/j.bioorg.2022.105710 [DOI] [PubMed] [Google Scholar]
- 34.Alberts SR, Fitch TR, Kim GP, et al. Cediranib (AZD2171) in patients with advanced hepatocellular carcinoma: a Phase II north central cancer treatment group (NCCTG) clinical trial. Am J Clin Oncol. 2012;35(4):329. doi: 10.1097/COC.0b013e3182118cdf [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wedge SR, Kendrew J, Hennequin LF, et al. AZD2171: a highly potent, orally bioavailable, vascular endothelial growth factor receptor-2 tyrosine kinase inhibitor for the treatment of cancer. Cancer Res. 2005;65(10):4389–4400. doi: 10.1158/0008-5472.CAN-04-4409 [DOI] [PubMed] [Google Scholar]
- 36.Plé PA, Jung F, Ashton S, et al. Discovery of AZD2932, a new quinazoline ether inhibitor with high affinity for VEGFR-2 and PDGFR tyrosine kinases. Bioorg Med Chem Lett. 2012;22(1):262–266. doi: 10.1016/j.bmcl.2011.11.019 [DOI] [PubMed] [Google Scholar]
- 37.Zhang S, Cao Z, Tian H, et al. SKLB1002, a novel potent inhibitor of VEGF receptor 2 signaling, inhibits angiogenesis and tumor growth in vivo. Clin Cancer Res. 2011;17(13):4439–4450. doi: 10.1158/1078-0432.CCR-10-3109 [DOI] [PubMed] [Google Scholar]
- 38.Wu P, Nielsen TE, Clausen MH. Small-molecule kinase inhibitors: an analysis of FDA-approved drugs. Drug Discov Today. 2016;21(1):5–10. doi: 10.1016/j.drudis.2015.07.008 [DOI] [PubMed] [Google Scholar]
- 39.Haider K, Das S, Joseph A, Yar MS. An appraisal of anticancer activity with structure–activity relationship of quinazoline and quinazolinone analogues through EGFR and VEGFR inhibition: a review. Drug Dev Res. 2022;83:859–890. [DOI] [PubMed] [Google Scholar]
- 40.Hadi SRAE, Lasheen DS, Soliman DH, Elrazaz EZ, Abouzid KAM. Scaffold hopping and redesign approaches for quinazoline based urea derivatives as potent VEGFR-2 inhibitors. Bioorg Chem. 2020;101:103961. doi: 10.1016/j.bioorg.2020.103961 [DOI] [PubMed] [Google Scholar]
- 41.Mahdy HA, Ibrahim MK, Metwaly AM, et al. Design, synthesis, molecular modeling, in vivo studies and anticancer evaluation of quinazolin-4(3H)-one derivatives as potential VEGFR-2 inhibitors and apoptosis inducers. Bioorg Chem. 2020;94:103422. doi: 10.1016/j.bioorg.2019.103422 [DOI] [PubMed] [Google Scholar]
- 42.Eissa IH, El-Helby A-GA, Mahdy HA, et al. Discovery of new quinazolin-4(3H)-ones as VEGFR-2 inhibitors: design, synthesis, and anti-proliferative evaluation. Bioorg Chem. 2020;105:104380. doi: 10.1016/j.bioorg.2020.104380 [DOI] [PubMed] [Google Scholar]
- 43.El-Adl K, El-Helby A-GA, Ayyad RR, et al. Design, synthesis, and anti-proliferative evaluation of new quinazolin-4(3H)-ones as potential VEGFR-2 inhibitors. Bioorg Med Chem. 2021;29:115872. doi: 10.1016/j.bmc.2020.115872 [DOI] [PubMed] [Google Scholar]
- 44.Abdallah AE, Eissa SI, Al Ward MMS, Mabrouk RR, Mehany ABM, El-Zahabi MA. Design, synthesis and molecular modeling of new quinazolin-4(3H)-one based VEGFR-2 kinase inhibitors for potential anticancer evaluation. Bioorg Chem. 2021;109:104695. doi: 10.1016/j.bioorg.2021.104695 [DOI] [PubMed] [Google Scholar]
- 45.Faraji A, Motahari R, Hasanvand Z, et al. Quinazolin-4(3H)-one based agents bearing thiadiazole-urea: synthesis and evaluation of anti-proliferative and antiangiogenic activity. Bioorg Chem. 2021;108:104553. doi: 10.1016/j.bioorg.2020.104553 [DOI] [PubMed] [Google Scholar]
- 46.Aziz MA, Serya RAT, Lasheen DS, et al. Discovery of potent VEGFR-2 inhibitors based on furopyrimidine and thienopyrimidne scaffolds as cancer targeting agents. Sci Rep. 2016;6(1):24460. doi: 10.1038/srep24460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fortin JS, Lacroix J, Desjardins M, Patenaude A, Petitclerc É, René C. Alkylation potency and protein specificity of aromatic urea derivatives and bioisosteres as potential irreversible antagonists of the colchicine-binding site. Bioorg Med Chem. 2007;15(13):4456–4469. doi: 10.1016/j.bmc.2007.04.028 [DOI] [PubMed] [Google Scholar]
- 48.Mounetou E, Legault J, Lacroix J, C-Gaudreault R. Antimitotic antitumor agents: synthesis, structure−activity relationships, and biological characterization of N -Aryl- N ‘-(2-chloroethyl) ureas as new selective alkylating agents. J Med Chem. 2001;44(5):694–702. doi: 10.1021/jm0010264 [DOI] [PubMed] [Google Scholar]
- 49.Yan G, Zekarias BL, Li X, Jaffett VA, Guzei IA, Golden JE. Divergent 2-Chloroquinazolin-4(3 H)-one rearrangement: twisted-cyclic guanidine formation or ring-fused N -acylguanidines via a domino process. Chem Eur J. 2020;26(11):2486–2492. doi: 10.1002/chem.201905219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Elfeky SM, Almehmadi SJ, Tawfik SS. Synthesis, in-silico, and in-vitro study of novel chloro methylquinazolinones as PI3K-δ inhibitors, cytotoxic agents. Arab J Chem. 2022;15(2):103614. doi: 10.1016/j.arabjc.2021.103614 [DOI] [Google Scholar]
- 51.Sepehri N, Mohammadi-Khanaposhtani M, Asemanipoor N, et al. Novel quinazolin–sulfonamid derivatives: synthesis, characterization, biological evaluation, and molecular docking studies. J Biomol Struct Dyn. 2022;40(8):3359–3370. doi: 10.1080/07391102.2020.1847193 [DOI] [PubMed] [Google Scholar]
- 52.Ferrari M, Fornasiero MC, Isetta AM. MTT colorimetric assay for testing macrophage cytotoxic activity in vitro. J Immunol Methods. 1990;131(2):165–172. doi: 10.1016/0022-1759(90)90187-Z [DOI] [PubMed] [Google Scholar]
- 53.Tolba MF, Esmat A, Al‐Abd AM, et al. Caffeic acid phenethyl ester synergistically enhances docetaxel and paclitaxel cytotoxicity in prostate cancer cells. IUBMB Life. 2013;65(8):716–729. doi: 10.1002/iub.1188 [DOI] [PubMed] [Google Scholar]
- 54.Kumar R, Saneja A, Panda AK. An annexin V-FITC—propidium iodide-based method for detecting apoptosis in a non-small cell lung cancer cell line. Lung Cancer. 2021;213–223. [DOI] [PubMed] [Google Scholar]
- 55.Elgazar AA, Knany HR, Ali MS. Insights on the molecular mechanism of anti-inflammatory effect of formula from Islamic traditional medicine: an in-silico study. J Tradit Complement Med. 2019;9(4):353–363. doi: 10.1016/j.jtcme.2018.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Elsbaey M, Ibrahim MAA, Bar FA, Elgazar AA. Chemical constituents from coconut waste and their in silico evaluation as potential antiviral agents against SARS-CoV-2. S Afr J Bot. 2021;141:278–289. doi: 10.1016/j.sajb.2021.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Al-Sanea MM, Hamdi A, Brogi S, et al. Design, synthesis, and biological investigation of oxadiazolyl, thiadiazolyl, and pyrimidinyl linked antipyrine derivatives as potential non-acidic anti-inflammatory agents. J Enzyme Inhib Med Chem. 2023;38(1):2162511. doi: 10.1080/14756366.2022.2162511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Al-Sanea MM, Chilingaryan G, Abelyan N, et al. Combination of ligand and structure based virtual screening approaches for the discovery of potential PARP1 inhibitors. PLoS One. 2022;17(9):e0272065. doi: 10.1371/journal.pone.0272065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Al-Sanea MM, Abdel-Maksoud MS, El-Behairy MF, et al. Anti-inflammatory effect of 3-fluorophenyl pyrimidinylimidazo [2, 1-b] thiazole derivatives as p38α inhibitors. Bioorg Chem. 2023;139:106716. doi: 10.1016/j.bioorg.2023.106716 [DOI] [PubMed] [Google Scholar]
- 60.Hamdi A, Yaseen M, Ewes WA, et al. Development of new thiazolidine-2,4-dione hybrids as aldose reductase inhibitors endowed with antihyperglycaemic activity: design, synthesis, biological investigations, and in silico insights. J Enzyme Inhib Med Chem. 2023;38(1):2231170. doi: 10.1080/14756366.2023.2231170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Othman DI, Hamdi A, Abdel-Aziz MM, Elfeky SM. Novel 2-arylthiazolidin-4-one-thiazole hybrids with potent activity against mycobacterium tuberculosis. Bioorg Chem. 2022;124:105809. doi: 10.1016/j.bioorg.2022.105809 [DOI] [PubMed] [Google Scholar]
- 62.Hamdi A, Elhusseiny WM, Othman DI, et al. Synthesis, antitumor, and apoptosis-inducing activities of novel 5-arylidenethiazolidine-2, 4-dione derivatives: histone deacetylases inhibitory activity and molecular docking study. Eur J Med Chem. 2022;244:114827. doi: 10.1016/j.ejmech.2022.114827 [DOI] [PubMed] [Google Scholar]
- 63.Abass SA, Elgazar AA, El-Kholy SS, et al. Unraveling the nephroprotective potential of papaverine against cisplatin toxicity through mitigating oxidative stress and inflammation: insights from in silico, in vitro, and in vivo investigations. Molecules. 2024;29(9):1927. doi: 10.3390/molecules29091927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Othman DI, Hamdi A, Tawfik SS, Elgazar AA, Mostafa AS. Identification of new benzimidazole-triazole hybrids as anticancer agents: multi-target recognition, in vitro and in silico studies. J Enzyme Inhib Med Chem. 2023;38(1):2166037. doi: 10.1080/14756366.2023.2166037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1–2):55–63. doi: 10.1016/0022-1759(83)90303-4 [DOI] [PubMed] [Google Scholar]
- 66.El Hamaky NF, Hamdi A, Bayoumi WA, Elgazar AA, Nasr MN. Novel quinazolin-2-yl 1, 2, 3-triazole hybrids as promising multi-target anticancer agents: design, synthesis, and molecular docking study. Bioorg Chem. 2024;148:107437. doi: 10.1016/j.bioorg.2024.107437 [DOI] [PubMed] [Google Scholar]
- 67.Othman DI, Hamdi A, Elhusseiny WM, et al. Synthesis of novel spirochromane incorporating Schiff’s bases, potential antiproliferative activity, and dual EGFR/HER2 inhibition: cell cycle analysis and in silico study. Saudi Pharm J. 2023;31(11):101803. doi: 10.1016/j.jsps.2023.101803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.El-Senduny FF, Elgazar AA, Alwasify HA, et al. Bio-evaluation of untapped alkaloids from vinca minor enriched by methyl-jasmonate-induced stress: an integrated approach. Planta Med. 2023. 89 964-–978. [DOI] [PubMed] [Google Scholar]
- 69.Ewes WA, Tawfik SS, Almatary AM, et al. Identification of benzothiazoles bearing 1, 3, 4-thiadiazole as antiproliferative hybrids targeting VEGFR-2 and BRAF kinase: design, synthesis, BIO evaluation and in silico study. Molecules. 2024;29(13):3186. doi: 10.3390/molecules29133186 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data supporting this study’s findings are available from the corresponding author, M. M. A., upon reasonable request.