

Abstract
Every cell must solve the problem of how to fold its genome. We describe how the folded state of chromosomes is the result of the combined activity of multiple conserved mechanisms. Homotypic affinity-driven interactions lead to spatial partitioning of active and inactive loci. Molecular motors fold chromosomes through loop extrusion. Topological features such as supercoiling and entanglements contribute to chromosome folding and its dynamics, and tethering loci to sub-nuclear structures adds additional constraints. Dramatically diverse chromosome conformations observed throughout the cell cycle, and across the tree of life can be explained through differential regulation and implementation of these basic mechanisms. We propose that the first functions of chromosome folding are to mediate genome replication, compaction and segregation, and that mechanisms of folding have subsequently been co-opted for other roles including long-range gene regulation in different conditions, cell types, and species.
In 1970 Ris and Kubai wrote1: “The analysis of chromosome structure seeks to describe the spatial relationships of the various molecular components of chromosomes and to relate changes in these configurations to chromosome functions such as replication, transcription, and genetic recombination”. Around the same time Thomas wrote2: “We don’t know how chromosomes are organized, but there are some tantalizing clues, and we may be on the edge of finding out”.
Ris and Kubai described the challenge of the field of Chromosome Biology, which holds true to this day. Thomas was correct that we were going to find out, though it took decades, the development of new methods, the contributions from different disciplines ranging from molecular to cell biology to evolutionary biology, and the sequencing of complete genomes. Parallel developments in physics continued changing our view of the nature of chromosomes, further pushing the envelope of Polymer Physics. Today, we know at least some of the structures, molecules, and mechanisms driving chromosome folding. Yet, how these processes and resulting chromosome structures relate to chromosome function continues to be a topic of intense study and debate. Still, there is the same sense of optimism that we may be on the edge of finding out the connections between folding and the working of the genome.
The view of chromosomes 50 years ago
Among the first articles in Cell covering chromatin structure, chromosome folding and nuclear organization were studies on fine-scale organization of eukaryotic chromatin fibers as “beads-on-a-string”3, formation of histone tetramers4, folded prokaryotic nucleoids5, and speculations on folding of DNA within the peculiar nuclei of dinoflagellates6, eukaryotes with apparent liquid crystalline chromosomes that have become to the topic of renewed interest recently7,8.
In the 1960s it was known that chromosomes were each composed of a single long strand of DNA and the question of how that DNA was spatially arranged to fulfill its role as genetic carrier was only just begun to be asked, and few answers were available. At first glance, it appeared that there were few commonalities between the structure of chromosomes from organisms from different kingdoms (e.g., eukaryotes vs. prokaryotes), between chromosomes from different tissues within a species (e.g., Drosophila polytene chromosomes in salivary glands, vs. more conventional eukaryotic chromatin in other tissues), or between chromosomes observed at different stages of the cell cycle for any one cell type (the classical X-shaped compacted mitotic chromosomes readily visible in the light microscope, vs. interphase chromosomes that essentially disappeared from view). Chromosomes could be linear, circular, regularly packed in proteinaceous capsids in phages, lightly packed in nucleoids in bacteria, wrapped around nucleosomes in (most) eukaryotes, supercoiled or not, and sometimes arranged in loops.
As for many other fields, the study of chromosome folding awaited the development of new technologies and assays that would ultimately allow the visualization of entire genomes at near base pair resolution in three dimensions within single cells. These developments were complemented by the joining together of scientists from a range of disciplines, including cell biology, molecular biology, structural biology on the one hand, and physics, computational biology, and bioinformatics on the other. This interdisciplinary effort has over the last few decades started to identify common principles of chromosome folding across the tree of life.
The need to fold chromosomes
In all organisms, the lengths of their genomes are large compared to the dimensions of the cell or nucleus. Stretched out, the genome of E. coli is ~ 1.7 millimeters long compared to a cell diameter of 2 micrometers. The length of the human genome in all chromosomes is ~2 meters, and the cell nucleus is only ~5–10 micrometers in diameter. However, given that DNA fibers are so thin (2 nm of DNA, or 11 nm of the chromatin fiber), the volume of the genome fits very comfortably in the cell or nucleus. So, is there really a need for cells to actively organize and fold chromosomes?
There are strong arguments for why cells need to actively fold their chromosomes. First, assuming that chromatin behaves as an unconstrained polymer (i.e. an ideal chain, resembling a random walk), and that it has a persistence length of a ~70nm containing ~3–5 kilobases 9,10, the diameter of an unconstrained chromosome of the length of just 1 copy of human chromosome 1 would be about 15 micrometers, exceeding that of the whole nucleus (; diameter of the coils is twice that: 16um). Clearly, extensive compaction is required to fit in all 46 chromosomes.
Second, we previously11 outlined that the polymer state of chromosomes has important implications for which loci can physically interact (e.g., genes and their regulatory elements), and the kinetics by which such interactions can form and in what fraction of cells. In the absence of any constraints or active processes folding chromatin, short range interactions between genomic loci, e.g., separated by up to tens of kilobases, will be frequent enough to occur in most cells in a reasonable amount of time (e.g., the duration of the cell cycle). However, longer-range interactions will be rare (~6% of the time for 0.5Mb separation 12), and will not be formed in most cells even at very large time scales. Finally, in the absence of active and controlled folding processes, it is hard to imagine how specificity in interactions can be obtained (below).
Third, in the absence of active management of chromosome organization, any genomic process would be compromised. For instance, replicating long DNA molecules leads to pairs of sister DNAs that are topologically intertwined, creating a significant challenge to the cell, as was realized by Delbruck already in 195613. To segregate these sister molecules to daughter cells, cells need to compact each so that they are short and rigid to facilitate segregation, while simultaneously also topologically unlinking them. This process is fundamental to life and is the intuitively most obvious case for the need for processes to actively fold chromosomes.
The chromosome folding problem
The (cell-type-dependent) folding of chromosomes is related to the (cell-type-dependent) linear epigenome patterning along the genome: the presence, location, and activity of cis regulatory elements such as enhancers, insulators and promoters, and (in eukaryotes) the presence of regions of specific combinations of histone modifications that define different chromatin states including euchromatin and heterochromatin. Like the protein folding problem, defined as the question how the primary amino acid sequence of a protein dictates its three-dimensional folding, the chromosome folding problem can be defined as the question how the linear epigenome is related to the spatial arrangement and folding of chromosomes in the cell.
However, and this is different from protein folding (see14 for review), chromosome folding is not only driven by affinities and interactions between genomic element, it also involves biological activities that directly fold and refold chromosomes, and these molecular processes can in some cases fold chromosomes in ways largely unrelated to the linear epigenome, e.g., in mitosis15–20. Chromosome conformation is also highly variable between individual cells, a result of the very large length of chromosomes combined with their stochastic dynamics of self-assembly21–23, and the action of highly dynamic folding processes such as loop extrusion that rearrange chromosomes at the scale of hundreds of kilobases over tens of minutes. Further, chromosomes can rapidly, in mere minutes, change their folding state, e.g., during entry and exit of mitosis16,24–27.
We propose a more expansive definition of the chromosome folding problem: the question how biophysical forces and molecular mechanisms, through the action of specific folding machineries, act on the linear epigenome to dynamically fold and refold chromosomes at different length and time scales, e.g., during the cell cycle, development, and other biological transitions.
Chromosome folding appears to differ in dramatic ways between species from different kingdoms (e.g., prokaryotes vs. eukaryotes), and as cells progress through the cell cycle. Such observations suggest that many different solutions to the chromosome folding problem may exist, and that species- and condition-specific folding mechanisms must have evolved. One of the most exciting discoveries of the last two decades has been that only a small number of universal biophysical and molecular processes drive chromosome folding. These major folding mechanisms are deeply conserved across the tree of life, but are directed, regulated, and deployed in different ways to produce different folded states of chromosomes to accommodate the many different functions of genomes.
Breakthroughs in determining chromosome folding
Chromosomes were first described using microscopic methods. Given that initially only large mitotic and meiotic chromosomes in plants and amphibians could be individually observed by microscopic means, initial studies focused on these chromosomes. First concepts of chromosome folding in mitosis developed the notion of the “folded fiber”, ranging from irregular fibers28, to radial loop structures29,30, to hierarchical models19,31–34. Initial physical models of interphase chromosomes started to arise when Fluorescence In Situ Hybridization (FISH) imaging established how the spatial distance between loci increases with the genomic separation between the probes. The first quantitative models considered an interphase chromosome as a random-walk polymer35; or a confined or tethered polymer36. Alternative models were proposed where chromosomes were folded into megabase-size loops along otherwise random-walk polymers37,38 (Figure 1).
Figure 1. Evolving physical models over the last decades.
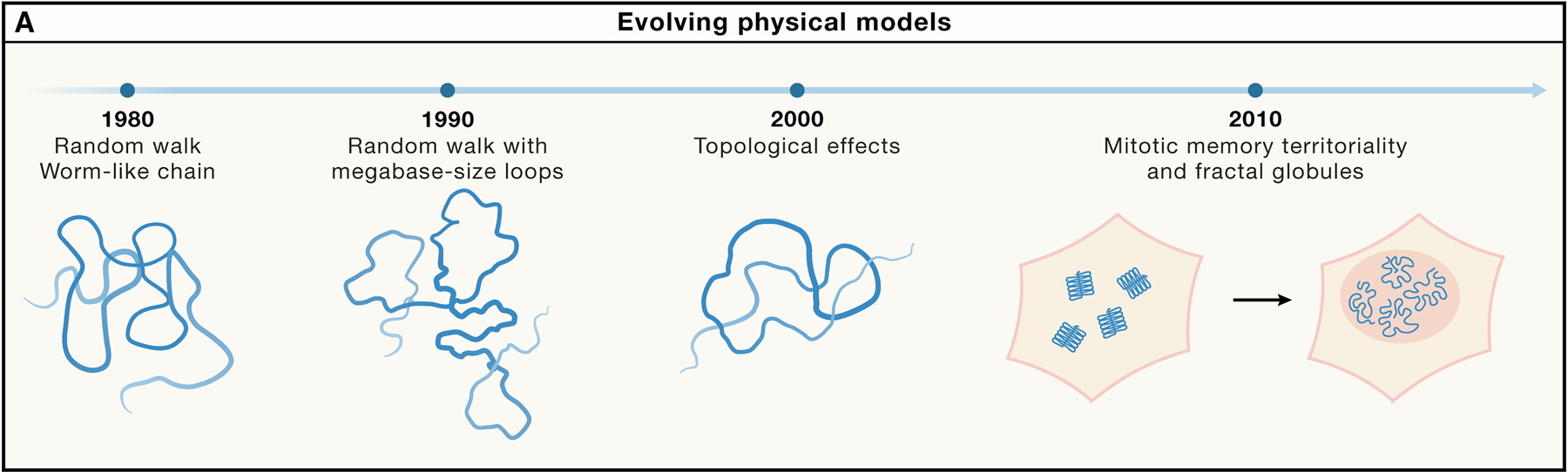
Over the last 50 years, Polymer Physics has put forth increasingly refined models of chromosome conformation. See text for details.
Over the last two decades, four important developments have greatly enhanced and transformed the study of chromatin, chromosomes, and entire genomes.
First, the ability to determine the sequence of complete genomes for many species. While initial genome sequencing efforts in the 1990s focused on smaller genomes of bacteria and some model organisms (e.g., budding yeast S. cerevisiae39 , the nematode C. elegans40, and the fruit fly D. melanogaster41, and large-scale international efforts were required to sequence the mouse and human genomes42–44, further increases in throughput and lowering of cost now enable the sequencing of any number of species and individuals. Now (near) full length genome sequences are available for thousands of species, ranging from bacteria, to protozoa, archaea, fungi, plants, birds, mammals etc. (e.g.,45–47). Notably, Hi-C can also be used to assemble the linear genome (first shown by Kaplan and Dekker48 and Burton and co-workers 49 in 2013) and is now routinely used to assemble genomes of new species (e.g., 50,51, and see 52 for an evaluation of these approaches).
Second, the development of genomic methods to probe the folding of chromosomes now allows mapping chromosome structure directly to genome sequence. Some of these methods are based on chromosome conformation capture (3C53, 4C54, 5C55, Hi-C56, Micro-C57, DNAse-Hi-C58, Chia-PET59, HiChIP60 , Plac-Seq61, etc.), while other methods rely on mapping DNA sequences near sub-nuclear structures such as DAM-ID62, TSA-seq63, or the identification of loci co-located in clusters or specific sections of the nucleus (GAM64; SPRITE65, etc.). In recent years, the resolution of these methods has increased so that 3D maps of genomes can be acquired at sub-kilobase resolution, as well as in single cells (e.g.,66–71).
Third, the development of imaging methods that can analyze the spatial locations of thousands of loci, so that the 3D structure of entire chromosomes, or even genomes can be traced in single cells. These include large scale locus tracing72,73; OligoStorm and OligoDNA Paint 74,75, and ORCA76. Those paralleled developments of live-cell tracking of chromosomal dynamics that sheds light on underlying folding processes 77, Gabriele, 2022 #1804.
Fourth, developments in the understanding of chromosome folding from the polymer physics point of view: from the random-walk35 and worm-like chain models in early 90s36, to early models of chains with loops37,38, to appreciation of topological effects78–80, to more recent studies of active polymers81 or polymer driven by motors (loop extrusion)82,83, and folded onto loops 84, to models of polymer dynamics and response to external forces85, Grosse-Holz, 2023 #1771.
The development and application of genomic and imaging-based methods to determine the structure of chromosomes has been extensively reviewed elsewhere86–90, and we refer the reader to those reviews and the primary literature cited therein. Here we focus on current views of what the structure of chromosomes is, under different conditions and in different species, the mechanisms by which these structures form, and how chromosome structure and function are related.
Four mechanisms for folding chromosomes
Studies in many species have shown that they share key mechanisms by which they fold chromosomes. Here we describe these mechanisms.
1. Compartmentalization
One of the first features described for the spatial organization of chromatin inside eukaryotic interphase nuclei is the spatial segregation of inactive heterochromatic chromatin from active euchromatin (Figure 2), as first described by Emil Heitz91. Classic microscopy studies showed dense, compacted chromatin clustered near the nuclear periphery, while decondensed, open chromatin was located more centrally . Later studies showed that more gene dense chromosomes tend to locate centrally, while gene poor chromosomes are more peripheral in the nucleus92–94. Studies on the timing of DNA replication also showed spatial segregation of early and late replicating chromatin, correlating with eu- and heterochromatin95.
Figure 2: Two processes for nuclear organization: compartmentalization through homotypic affinities and tethering to the nuclear periphery.
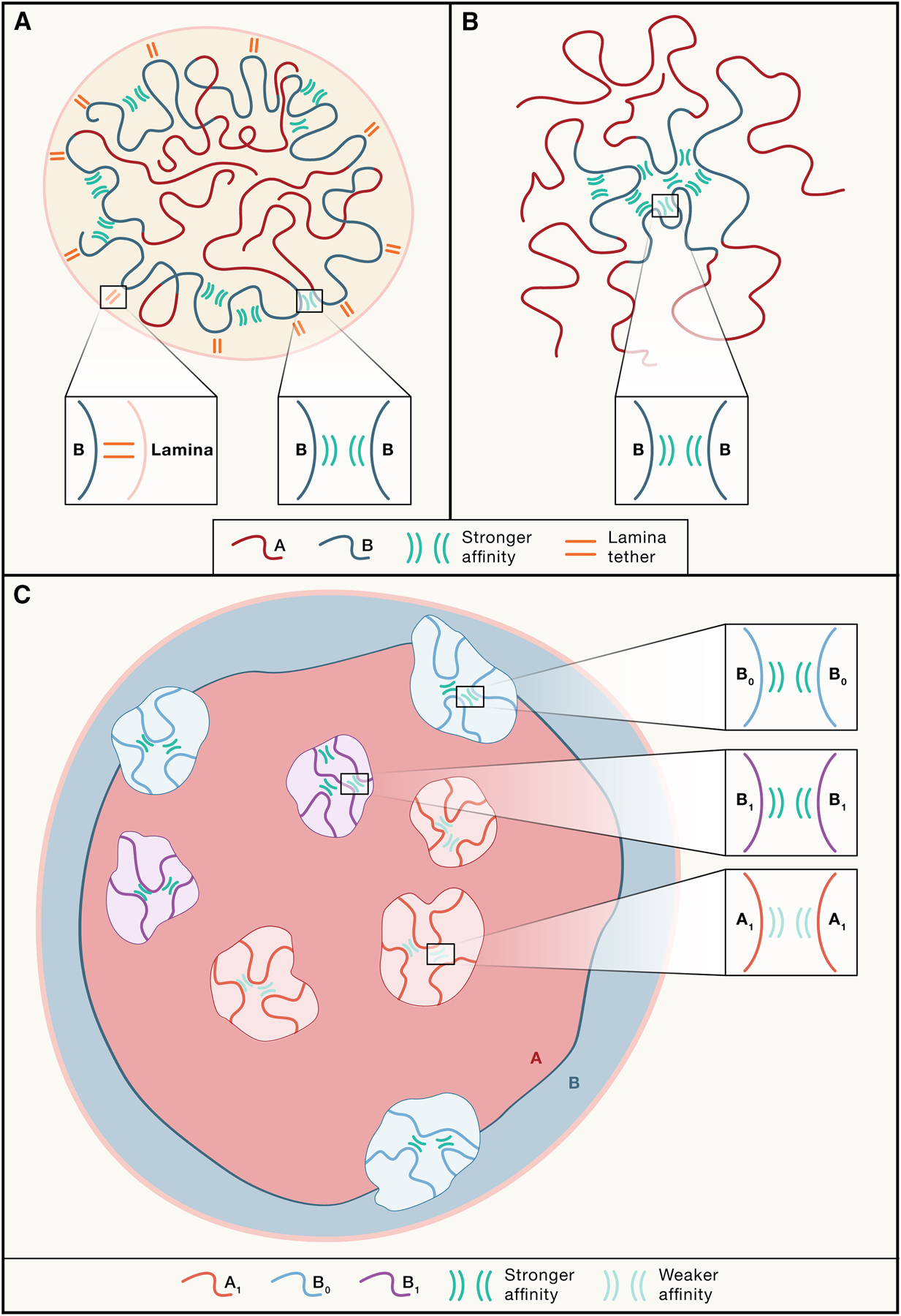
A. Eukaryotic chromosomes are composed of alternating A and B compartments. In conventional nuclear organization, strong B-B affinities lead to spatial separation of A and B compartments. A-A affinities are much weaker and contribute to a lesser extent. In addition, Some B compartments are tethered to the nuclear periphery, resulting in enrichment of heterochromatin at the nuclear periphery, leaving euchromatin located centrally. B. In the absence of tethering of B compartment domains to the nuclear periphery, A/B compartmentalization occurs normally, but the strong B-B affinities result in clustering of all B compartments in the center of the nucleus, with A compartments located at the periphery (inverted nucleus). C. A more complex picture when more than 2 compartment types are present. A and B compartments can be split in different subcompartments that can also display significant preferential homotypic affinities, leading to their spatial segregation.
Genomic assays such as DamID directly mapped sequences near the nuclear lamina, again identifying regions poor in genes, and mostly transcriptionally silent96,97. The very first 4C and Hi-C datasets showed spatially segregated eu- and heterochromatic domains at genome-wide scale54,56. In Hi-C, enriched interactions are readily detected between loci of similar chromatin and activity state: active and open chromatin interacts with other active and open loci, along the same chromosome (cis), and between chromosomes (trans). Similarly, inactive loci interact with each other. This phenomenon is referred to as compartmentalization.
Initial lower resolution Hi-C studies (Mb-scale) showed the presence of just two types of chromatin that self-interact, and these two types (A and B compartments) had all the hallmarks of euchromatin and heterochromatin respectively56. Subsequent higher resolution Hi-C maps showed that each of these two major types of compartments can be split in so-called subcompartments that differ in their precise chromatin composition, e.g., histone modification patterns, and can be as small as several kilobases98. Each of these sub-compartments display characteristic patterns of long-range interactions with other loci, but most display a preference to interact with other loci of the same sub-compartment type (Figure 2A). A major recent insight is that the number of types of subcompartments is larger than previously anticipated (Figure 2C), and that these are not necessarily universally present, i.e., in a given cell type not all subcompartment types may be observed99,100.
Compartmentalization is thought to be driven by homotypic affinities between loci 101–105 (Figure 2). The molecular nature of the factors that mediate these affinities are not known in detail. It is intriguing that compartmentalization has mostly been detected in eukaryotes that have nucleosomes, and not in eukaryotes without nucleosomal DNA, e.g., dinoflagellates7,8 and in some archaea106. Though other explanations can be proposed, it may indicate a key role for histones in this process. Compartmentalization is correlated with the presence of histone modifications: each sub-compartment has a characteristic combination of histone modifications99,100,107. In vitro, short chromatin fibers carrying H3K9Me3 can form condensates, indicating that the modified histones themselves can play a role in the clustering of heterochromatin108. Further, factors that recognize patterns of histone modification can act as bridging factors and, in that way, connect distal loci to stabilize compartmentalization. For instance, HP1 proteins can bind H3K9Me3, and can bind multiple histone tails simultaneously. Such bridging factors can also phase separate themselves, leading to aggregation of such proteins together with multiple loci109–113. While HP1 proteins can contribute to compartmentalization in that manner, loss of HP1 has surprisingly little effect on compartment formation114, pointing to roles for other yet to be identified factors. Other examples are polycomb complexes that mediate deposition of H3K27Me3 modifications and then associate with chromatin carrying this mark, stabilizing long-range interactions in cis and in trans between loci silenced by these factors (for reviews of the extensive literature on this topic see115,116). While many polycomb-bound loci reside in larger A compartment, and can be located in nuclei centers 117, they tend to engage in prominent long-range with other polycomb-bound sites, e.g., in Drosophila 118 and vertebrates, especially in embryonic stem cells119,120.
Compartmentalization constitutes a phenomenon of microphase separation i.e. a phenomenon where a polymer (chromosome) made of blocks of A and B monomers (or more types) forms spatially separated domains, sizes of which depend on the sizes of blocks in the sequence. Such a process can be driven by attractions between homotypic elements (A-A and/or B-B). Phase separation has also been seen in in in vitro chromatin reconstruction experiments108. Initial studies demonstrated that the characteristic pattern of A/B compartmentalization seen in Hi-C data can be reproduced by models with homotypic affinities105,121, yet the relative contributions of these affinities for A and B compartments remained unknown. A solution came from a rare biological system of the “inverted” nucleus in the rod photoreceptors122. Natural loss of attachment of heterochromatin to the nuclear periphery in such nuclei resulted in repositioning of heterochromatin to the center with the euchromatin taking peripheral locations. Both microscopy and Hi-C103 confirmed that despite their inversion such nuclei are perfectly compartmentalized, demonstrating that compartmentalization is driven by interactions within chromatin rather than anchoring of heterochromatic loci to the lamina. Polymer models further demonstrated that homotypic affinities of heterochromatic regions drive compartmentalization with affinities between euchromatic regions being much weaker (Figure 2B).
These predictions from models were confirmed with direct observation of dissociation kinetics of chromatin interactions by liquid chromatin Hi-C123. In liquid chromatin Hi-C, chromatin is fragmented in situ leading to progressive dissociation of chromatin interactions over time, which can be measured using Hi-C. It was found that chromosomes remain compartmentalized even when chromatin is fragmented to an average size of 10–20 kilobases. When chromatin is fragmented to a size of less than 6 kilobases, interacting chromatin segments dissociate within tens of minutes. Importantly, the kinetics of dissociation were related to the chromatin state, with heterochromatic loci dissociating slower than euchromatic loci pointing to higher affinities between heterochromatic loci. Together with observations obtained with very deeply sequenced Hi-C datasets98, these data show that subcompartments can be as small as several kilobases. Heterochromatic interactions between H3K9Me3-marked loci were found to be more stable and thus contribute most strongly to compartmentalization, as predicted by modeling103.
A mechanism of compartmentalization driven by largely relatively stable interactions between H3K9me3-marked heterochromatic loci does not rule out affinities between other regions, e.g., euchromatic loci (that among other factors can be direct or mediated by nuclear speckles) (Figure 2C). In fact, enrichment of contacts between H3K27ac regions is evident from Micro-C124,125 by averaging over thousands of regions in Hi-C126, but most distinctly observed with region-capture Micro-C127,128. While indicating that euchromatin regions also have some homotypic affinities, the need for averaging or exceedingly deep sequencing suggest that such contacts are rare, consistent with microscopy126. Although they can help to stabilize otherwise transient interactions between regulatory elements and promoters at sub-megabase separations, the rarity of such contacts at larger genomic distances suggests little functional roles they can play. Interactions between active loci can also be driven by bridging factors. For instance, Brd2/3/4 proteins can drive clustering of chromatin marked with H3K27Ac both in vivo and in vitro108. A particularly interesting case is an oncogenic BRD4-NUT protein fusion that can lead to the spreading of H3K27Ac through large regions, which in turn results in spatial clustering of such hyperacetylated “megadomains” 129. This may be relevant for normal cells as well, where even relatively small H3K27Ac-enriched loci such as enhancers and promoters can form small microcompartment domains127,130,131.
An important aspect of compartmentalization as an affinity-driven process is that such clustering and spatial segregation will occur both in cis and in trans. This makes this process fundamentally different from other mechanisms of chromosome folding such as loop extrusion that acts strictly in cis (see below). Furthermore, compartmentalization is driven by local interactions leading to stochastic assemblies at the scale of whole chromosomes or genomes. In other words, in each cell a different configuration is obtained, but in each cell active loci preferentially cluster with other active loci, but which specific sets of loci cluster together can differ. The process results in stochastic colocalization of loci of similar chromatin state but has otherwise limited specificity in terms of which specific DNA sequences interact in any given cell. This aspect is important for understanding potential functional roles for compartmentalization.
Although compartmentalization is mostly observed in eukaryotes, the biophysical process that underlies this phenomenon, affinity-driven clustering loci, likely also occurs in prokaryotes (below).
2. Loops, extrusion, and their control
A first description of chromosome loops in mitotic chromosomes appeared in this journal in 1977132, leading to the radial loop model of mitotic chromosomes30,133. Proposals about the presence of loops in interphase chromosomes have started to appear at about the same time based on sedimentation data134,135 and later as physical models fit to microscopy data37,38,136.
What is now known as the process of loop extrusion (Figure 3) has a rich history. The ideas of enzyme-mediated loop growth started to emerge in the literature as many unrelated hypothetical mechanisms underlying VDJ recombination137, compaction of interphase chromosomes 138, enhancer-promoter interactions138,139, supercoiling140, and mitotic compaction and segregation141. Many works attributed these processes to SMC complexes142–144, with indications that SMCs can function as chromatin compacting motors145. In the context of mitotic compaction, loop extrusion was first mathematically modeled 146. However, because of the lack of polymer models to make concrete predictions following from activity of these mechanisms, and experimental data to test and validate the models, these proposals remained largely hypothetical.
Figure 3: Two regimes of loop extrusion produce different conformations, consistent with interphase and mitosis.
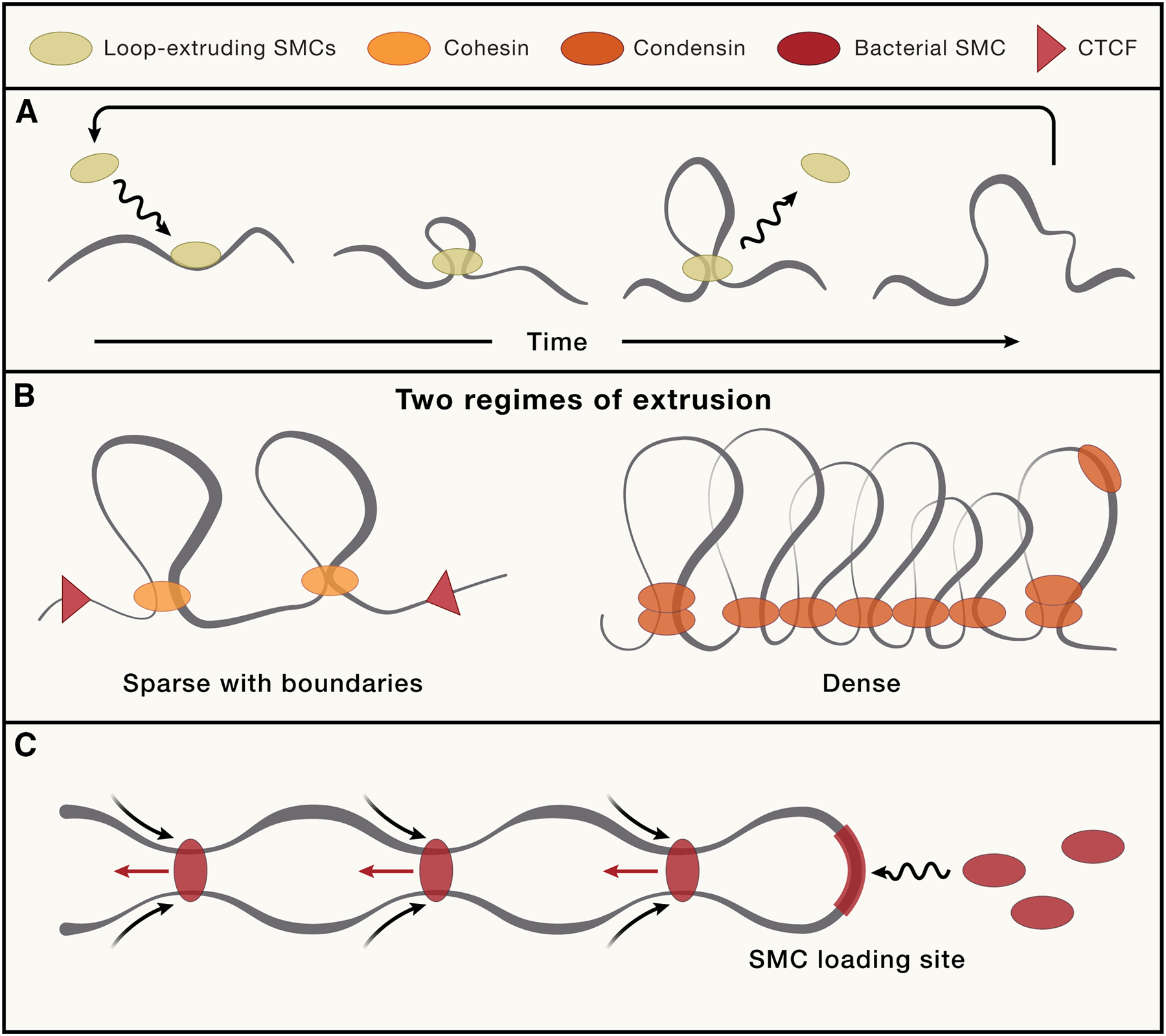
A. Activity of a loop extruder: the complex loads, extrudes some amount of time after which it may dissociate, or is actively unloaded. B. During interphase (in vertebrates), cohesin is the main loop extrusion complex. It has a short residence time, generating a low density of transient loops, and the chromosomes appear diffuse in shape. Cohesin can be blocked by CTCF-bound sites, generating enrichment of positioned loops at these elements. C. During mitosis, condensins are the main loop extrusion complexes. Condensin II has a long residence time, generating stable arrays of consecutive loops that lead to compaction into the rod-shaped mitotic chromosomes. Condensin is not blocked by CTCF, and the loop array is not positioned at reproducible loci in the cell population. C. In bacteria, repeated loading of loop extruding complexes at defined loading sites can lead to juxtaposition of the chromosome arms, sequences on either side of the loading site.
Emergence of chromosome conformation capture data provided rich grounds for developing and testing mechanisms of chromosome folding by loop extrusion. One prediction of the theory146 was that achieving mitotic compaction would lead to formation of a loop array where non-overlapping loops follow each other (if extruders cannot bypass each other). Indeed 5C and Hi-C data for mitotic cells were found to be consistent with such organization of loops15. Polymer simulations further indicated that loop extrusion can compact and segregate polymers of chromatids leading to morphologies of simulated chromosomes resembling those of early prophase chromosomes147. Loop extrusion during interphase148,149 was suggested as a mechanism underlying the then recently discovered Topologically Associating Domains (TADs150,151) and associated features such as stripes and dots observed in Hi-C interaction maps99. These features emerge naturally in polymer models of loop extrusion when extrusion is occluded by boundaries. Modeling studies also suggested that loop extruding motors are SMC complexes: cohesins in interphase and condensins during mitosis, while extrusion barriers are DNA-bound CTCF proteins. The loop extrusion hypothesis gained broad support by CTCF and cohesin depletion152,153 and modulation experiments, all generating model-predicted outcomes154. Direct visualization and characterization of loop extrusion by SMCs in single-molecule experiments (see Davidson and Peters155 and Hoencamp and Rowland156 for reviews) demonstrated that these complexes are indeed loop extrusion motors, as anticipated141,146, Fudenberg, 2016 #1292. The demonstration that the same mechanism in different regimes157 can lead to either interphase organization or to mitotic compaction (Figure 3B), suggesting that loop extrusion by SMCs can be a universal mechanism organizing chromosomes.
SMCs are ring-shaped and flexible protein complexes and include cohesin, condensin, SMC5/6, their bacterial counterparts, and possibly other complexes involved in DNA repair156. While condensins were known to be essential for mitotic chromosome compaction158–160, their mode of action was long unknown. Cohesin has been characterized as a complex that keeps two sister chromatids together141. Predicted loop extrusion activity of SMCs146, Fudenberg, 2016 #1292 was initially a surprise but single-molecule experiments have definitively demonstrated that SMC complexes can extrude loops in an ATP dependent manner161–165. In such in vitro experiments with naked DNA as template, as well as in live cells on endogenous chromatin12,24, loop extrusion is fast: ~1–3 Kb/sec. Different SMCs were found to be either one-sided or two-sided loop extruders161–164, with two-sided extrusion possibly resulting from rapid switching of one-sided extrusion activity166. These complexes have relatively low stall forces161, Golfier, 2020 #1726 (i.e., forces of 0.1-1pN suffice to stop extrusion) and some complexes such as cohesin are blocked by obstacles such as RNA polymerases167, CTCF, and MCM proteins (the latter two through a specific protein-protein interaction168,169). On the other hand, condensins display the surprising ability to bypass each other170 or obstacles much bigger than their size171. Yet the molecular mechanism of loop extrusion and force generation remains enigmatic and the area of active research172,173.
These developments paralleled studies of SMCs and their activity in bacteria. 5C and Hi-C in B.subtilis174 and C. crescentus175,176 revealed folding of the chromosomes “in half” with two juxtaposed, in an SMC-dependent manner. Further studies not only indicated loading of some SMC at specific sites (origin-proximal in many species, e.g., at ParS sites near the origin in B. subtilis177), but also directly visualized how such loading resulted in a progressive juxtaposition of the arms (Figure 3C). Strikingly, ahead of studies in eukaryotes, time-resolved Hi-C in bacteria allowed measuring the speed of loop extrusion in living cells at ~1 Kb/sec178.
Our understanding of the loop extrusion mechanism has significantly progressed in recent years. Practically every assumption and prediction of the original loop extrusion model has been challenged and mostly confirmed. As anticipated, cohesin depletion leads to increase in distances between all loci as seen by chromatin tracing 73, and ~50% of chromatin is located in extruded loops at any moment 12. Many processes and complexes on crowded DNA function as barriers to loop extrusion, including the process of transcription167, elements of the replicative machinery179, even in G1. CTCF remains the strongest known barrier that relies on a specific peptide that can halt extrusion99,152,168. Single CTCF sites, however, are permeable180 and loops that bridge two CTCF sites are rare and transient 77, Gabriele, 2022 #1804. Broadly these findings indicate that patterns of contacts formed by loop extrusion are transient, suggesting that it is the process of extrusion rather than specific patterns that can play functional roles181,182.
Several “rules of engagement” for SMCs, that determine how encounters between different complexes along the chromosome are resolved, are being discovered24,183 (Figure 4): they can block each other (likely cohesins183), one triggering unloading of the other (e.g., condensins triggering unloading of extruding cohesins24), or they can bypass each other and thus forming more complex overlapping loops (as seen in single-molecule experiments171 and in bacteria184). Interactions of loop extruding SMCs with SMCs holding sisters chromatids (“cohesive cohesins”) can vary as well, with yeast cohesins stopping at sites of sister cohesion183, while animal condensins bypassing such sites in mitosis24.
Figure 4: Rules of engagement for different SMCs results in different loop organization and structures of compacted chromosomes.
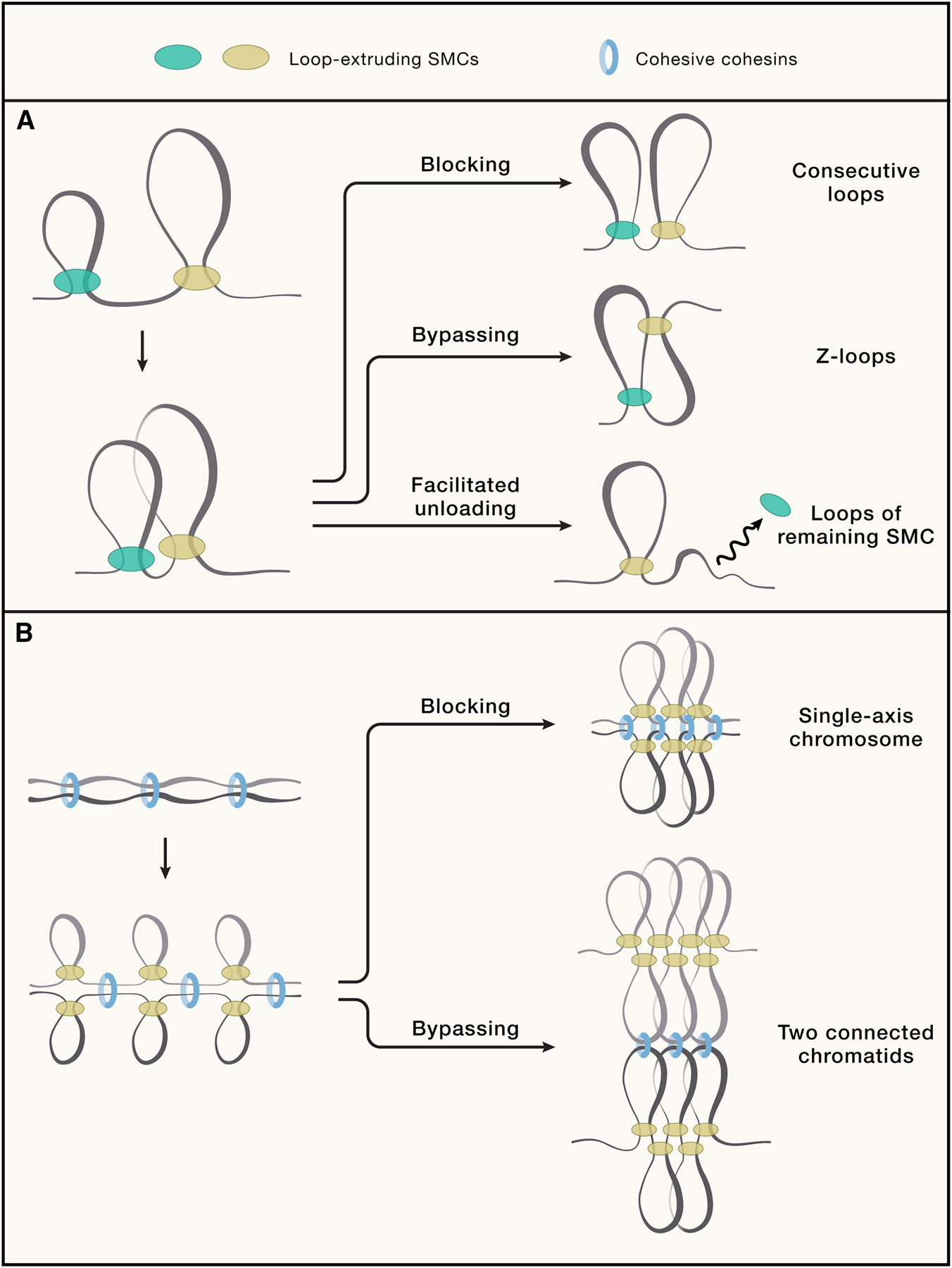
A. Three possible outcomes of an encounter between loop-extruding SMCs (orange and purple): they can block each other, leading to formation of consecutive loops; bypass each other forming so-called Z-loop170; or one can facilitate dissociation of another (other outcomes are also possible, e.g. one pushing the other back, etc). B. Two possible outcomes of interactions between cohesive cohesins (blue rings) and loop extruding SMCs (purple). Top: when extruders are blocked by cohesive complexes, sister chromatids are predicted to be connected at the bases of the loops, forming a single axis (as in meiotic prophase I and early mitotic prophase). When extruders can bypass cohesive complexes, sister chromatids are predicted to be connected through the tips of their loops (as in mitotic prometaphase).
Many of these mechanisms can be controlled by genomic elements and epigenetic context182,185. Examples known so far include methylation-dependent CTCF binding; targeted loading (at parS sites in bacteria186, and likely loading at active enhancers182,185,187, but not at promoters in animals); domains of localized extrusion activity (suggested in the silkworm188); sites of SMC unloading (e.g., 3’ ends of active genes in human182 and mouse189). The role of epigenetic context in regulating loop extrusion is less well-understood. While extrusion is active in both euchromatin and heterochromatin compartments100, heterochromatin is refractory to CTCF binding and hence devoid of boundaries.
Learning rules in one organism and extrapolating to others, we anticipate that (i) the speed of extrusion, loading and unloading can be controlled epigenetically in animals; (ii) replication forks and likely other genomic processes can halt/pause extrusion, thus breaking or establishing extrusion-mediated interactions; (iii) extrusion activity can be non-uniform along the genome. Broadly, changes in extrusion-mediated patterns through differentiation and development suggest that epigenetic marks can control extrusion and barriers. Extrusion may, in turn, play a role in the localization and spreading of epigenetic marks, as suggested by CTCF-demarcated domains of gammaH2AX spreading upon DSB repair190,191.
3. Associations with landmarks of the nucleus
In eukaryotes, loci can become tethered to the nuclear periphery, the nucleolus as well as other structures such as nuclear speckles that are enriched in RNA processing and splicing factors. In prokaryotes specific loci can be found tethered to the cell wall, e.g., the ParS sites in C. crescentus are tethered to the wall at one pole of the elongated cell.
The molecular mechanisms of tethering are becoming clearer only for a few of such associations. In vertebrates, tethering of heterochromatic domains to the nuclear periphery has been studied extensively. Large domains, referred to as Lamin-Associated Domains or LADs, are found associated with the nuclear periphery96,97. These domains are enriched in particular histone modifications such as H3K9Me3 and H3K9Me2 and are typically transcriptionally silent and compacted. Lamins may not be exclusively involved192,193 and other factors such as the Lamin B Receptor have been found to play roles194. However much less is known about the factors that determine clustering of loci around nucleoli or speckles, but a role for CTCF has been proposed195.
The functional relevance of tethering loci at sub-nuclear structures is largely unknown. While most cells have heterochromatic domains localized at the nuclear periphery, in specialized cell types such as rod cells, heterochromatin is not tethered and now is instead localized in the center of the nucleus122. This does not affect compartmentalization and does not appear to have dramatic effects on gene expression. It should also be noted that some active chromatin can be found localized at the nuclear periphery as well, especially around nuclear pores. The functional relevance of these associations is not established, but one possible role could be that this would facilitate rapid mRNA export (“gene gating” 196.
In other organisms or conditions, tethering of loci to the cell wall (bacteria197) or the nuclear envelope, is critical for chromosome segregation or chromosome pairing (e.g., meiosis I198). Although tethering is a straightforward way to facilitate spatial positioning of loci, much work is needed to explore the molecular players193, as well as roles of any cis-elements, that participate in these events, and to explore the functional relevance.
4. Topological constraints
The role of topological effects in the way cells manage their exceedingly long chromosomes, disentangling strands, and compacting long chains, have concerned biologists199 and physicists13,78 alike. Topoisomerase II was found to be essential for chromosome individualization200, and argued to be essential for fast mitotic compaction78. Effects of topological constraints and entanglements on polymer dynamics have been well known in physics201 and hypothesized to impact the way the genome is folded79,202.
Polymer theory suggested that the presence of topological constraints, i.e., when topoisomerase II activity is absent or very low, can lead to two phenomena: (i) exceedingly slow mixing between chromosomes after exit from mitosis, leading to formation of chromosomal territories; (ii) slow equilibration within each chromosome leading to the folding of the chain into a non-equilibrium and long-lived hierarchically organized and unknotted, state known as the fractal (or crumpled) globule79.
Polymer simulations and analysis of microscopy data for Drosophila suggested that polymers within each chromosome are folded into such a crumpled state203, and likely equilibrate exceedingly slowly. The first Hi-C data and polymer simulations provided compelling evidence that human interphase chromosomes are folded into the fractal globule state at the scale below ~10 Mb56. In this state a chromosome resembles a “space-filling” curve, i.e., continuous regions of the chromosome form compact spatial blobs, as was seen by microscopy204. The contact probability between genomic loci decays with genomic distance as with 205. This contrasts a polymer without topological constraints, i.e., strand passage can freely occur, which when compacted, resembles a random walk configuration in a confinement, where short continuous regions are expanded rather than compacted, leading to a rapid decay of the contact probability with genomic distance . Interestingly, chromosomes of yeast S.cerevisiae that are relatively short (up to 1.5 Mb), and don’t have a compact mitotic state, show this random walk folding 9. Multi-contact 3C data, and polymer simulation to test matches to that data, also demonstrate that each chromosome is largely unknotted206. It remains to be seen whether such unknotted and locally compact fractal globule folding and chromosomal territories have any specific functional roles or simply represent a memory of the unentangled telophase state207 preserved by topological constraints.
Recently, a more complex picture of how cells manage topological states of chromosomes started to emerge. Activity of topoisomerase II allows strand passage, turning the chain into a topologically unconstrained one, which can result in increased or decreased level of entanglement. Interestingly, loop extrusion can bias topoisomerase II activity towards unknotting an initially knotted chain208. Extruded loops can also buffer topological interactions making the chain constrained by topological interactions 84, e.g., in interphase chromosomes. Self-entanglement of mitotic chromosomes have long been anticipated, due to the critical role of topoisomerase II in mitotic compaction200,209,210, and as demonstrated in micromechanical experiments211. A recent study showed that mitotic and interphase chromosomes have very different topological states, with mitotic chromosomes being highly self-entangled while interphase is relatively free of knots, and suggested a pathway that allows cells to interconvert between them as cells exit mitosis207. To convert highly entangled mitotic chromosomes into an unentangled interphase state, cells require high activity of topoisomerase II during mitotic exit. To direct topoisomerase II activity toward unentanglement, and then preserve this unentangled interphase state a two-stage mitotic exit mechanism was proposed207. At the first stage decompaction while preserving mitotic loops biases topoisomerase II to disentangle the mitotic state, creating the unentangled compact state at telophase. During the second stage, chromosomes expand without much topoisomerase II activity thus forming chromosomal territories and fractal globule states in G1.
Sister chromatids are initially topologically intertwined during and after S-phase. Such topological connections will maintain connections between sister chromatids even in the absence of cohesive cohesin complexes. For segregation, these intertwines need to be removed. Loop extrusion in the presence of topoisomerase II activity has been shown by modeling to drive compaction of each sister chromatid, while unlinking them147. In effect, extrusion pulls the sisters away from each other which will drive the otherwise unbiased strand passage reaction by topoisomerase II towards decatenation.
The physical state of chromosomes
While Hi-C provides crucial information about the global state of chromatin in the scaling of the contact probability with genomic distance 205, microscopy measures a complementary characteristic: spatial separation . Interphase animal chromosomes typically yield and 73,212, for both consistent with the fractal (crumpled) globule folding, i.e. nearly space-filling organization where long continuous regions of the genome occupy continuous volumes in space. The fractal globule state is perturbed by extruded loops, and thus is best visible when cohesin is depleted and in synchronized cells, leading to scaling from 10 Kb to 1 0Mb (e.g. 213, Hsieh, 2022 #1793, Samejima, 2024 #1747). In S.cerevisiae, Hi-C and microscopy yield and 214–216 (for regions away from clustered centromere and telomeres) both characteristic of a largely unconstrained (random walk) chains, indicating that yeast chromosomal arms, away from clustered centromeres and anchored telomeres, are unconstrained polymers 214. In vertebrates, the fractal globule nature of folding is fully consistent with compartmentalization and loop extrusion 84,207. Yet the physical nature of this crumpled state, and thus the interphase vertebrate chromosomes remains enigmatic as evident from their dynamics and force-response.
Studies of chromosome dynamics provide a view complementary to that learned from Hi-C and microscopy. Moreover, timescales and frequencies of contacts measured by live-cell microscopy provide a foundation for understanding interactions between functional elements (see 217,218 for reviews).
Early works in chromosome dynamics in vertebrates used histone-fused photo-activateable GFP allowing to track changes in patterns of global chromosome organization in the nucleus and brought two key insights 219. First, dynamics during interphase is rather slow with a displacement of ~1 micron during ~24h interphase. Second, a great deal of randomization of positions of individual loci occurs after a cell division.
Tracking of individual (or pairs of) loci in live cells allowed quantifying mean-squared displacement (MSD) over an interval t, yielding MSD~tα with α=0.35-0.5 in bacteria, yeast, fly, and mammalian cells 12,77,220,221,222{Lucas, 2014 #1242,223. In S.cerevisiae, measured α=0.5 221,224 is in perfect agreement with Hi-C and microscopy, and is characteristic of a motion of a locus of a flexible but otherwise unconstrained polymer (so called Rouse model). Surprisingly, most of studies in animal cells, also reported α=0.5 12,77,225 that is hard to reconcile with and , and broadly the fractal globule that is expected to give α=0.2-0.4 (see 226 for review). Such inconsistency between “crumpled” R(s) and “unconstrained” MSD became most evident when both characteristics were measured using the same approach and in the same cells (see 223 and 218 for reviews). Some studies in mammalian cells yielded α=0.2 interpreting as a reflection of the properties of the nucleoplasm and suggesting a near-gel state of the chromatin 222. Live-cell measurements, however, and their analysis heavily rely on specifics of the experiment and correction for localization uncertainty (see 218 for review).
Live-cell tracking also estimates times it takes for a chromosomal region to sample its conformations. For example, in mammalian cells it takes about 40 minutes for a chromosomal region of 0.5 Mb to sample its conformations 12, only 5 minutes for two loci departed by 150 Mb to come sufficiently close (~100–200 nm) 77, while a larger (~2Mb) region didn’t equilibrate in 40 minutes 227. These times don’t simply imply same times for functional molecular interactions between chromosomal loci. For example, CTCF sites separated by 0.5 Mb form a stable interaction only about once per day and require cohesin-mediated extrusion 12. Broadly, it remains to be seen how proximity translates into functional interactions, which can critically depend on the radii over which such interactions can be established and molecular context, and how such molecular interactions result in transcriptional responses.
Studies of chromosome mechanics
Complementary to pictures obtained by microscopy and Hi-C are studies of chromosome mechanics. Loop formation and expansion and compaction/expansion of chromatin domains occur in the context of a crowded chromatin environment, leading to mechanical forces acting on chromosomes. Interplay between molecular processes that fold chromosomes and mechanical forces have been proposed to guide mitotic and meiotic chromosome compaction and chromosome segregation228–230.
Mechanical perturbation of the whole nucleus demonstrated that for displacements the nuclear response is driven by elastic properties of the polymer of chromatin attached to the nuclear lamina, while for larger displacements it is the stretching of the lamina that determines the response231. Inducing elevated levels of histone methylation makes chromatin stiffer, while elevated histone acetylation makes it softer232. This suggests that in some cell types, chromatin may play a role in providing optimal mechanical properties of the nucleus.
Micro-mechanical studies of isolated human mitotic chromosomes230 have provided important insights by demonstrating (i) chromosomes are extraordinarily elastic being able to extend more than 5 times their length233; (ii) the role of histone methylation in rigidifying chromatin, consistent with self-affinity of such heterochromatin regions234; (iii) the role of HP1alpha in mediating some of these interactions110; (iv) suggested key roles of condensins and topological entanglement in providing mechanical stability of mitotic chromosomes211. Broadly, these studies suggested that significant crosslinking turns a mitotic chromosome into a gel230. However, the nature of these crosslinks -- topological vs SMC vs non-SMC-based -- are yet to be understood.
A recent study of interphase chromosomes was able to perform a pull-release mechanical perturbation in live human cells85. These experiments showed that chromosomes responded as almost unconstrained (Rouse) polymers, consistent with their dynamics (see above). Surprisingly, chromosomal loci could travel micrometers across the nucleus in mere minutes. Models of a free polymer subject to weak affinities to the surrounding media can reproduce this behavior of chromatin, arguing that interphase chromosomes, unlike mitotic ones, are not gel-like or crosslinked. In summary, developing a physical model of interphase chromosomes that can unify Hi-C, microscopy, live-cell dynamics and mechanics is an important challenge218.
Folding chromosomes through the combined action of different folding mechanisms
The final folding state of a chromosome, or whole genome, is determined by the combined action of the several folding mechanisms described above22,235,236 (Figure 5). In addition to physical linkage, every locus is subject to the forces imposed by these mechanisms that combined determine its position with respect to other loci, its local dynamics, and its association with subnuclear structures such as the nuclear periphery or nuclear bodies including speckles and nucleoli. In addition, there is interplay between folding mechanisms, e.g., between loop extrusion and compartmentalization so that chromosome folding is not just the additive effect of each process in isolation. We discuss the folding of vertebrate interphase genome folding as an example given that this represents the best understood case but emphasize that we believe such combined action can explain chromosome folding more generally.
Figure 5: Current models of interphase chromosome organization through integrated activity of multiple mechanisms.
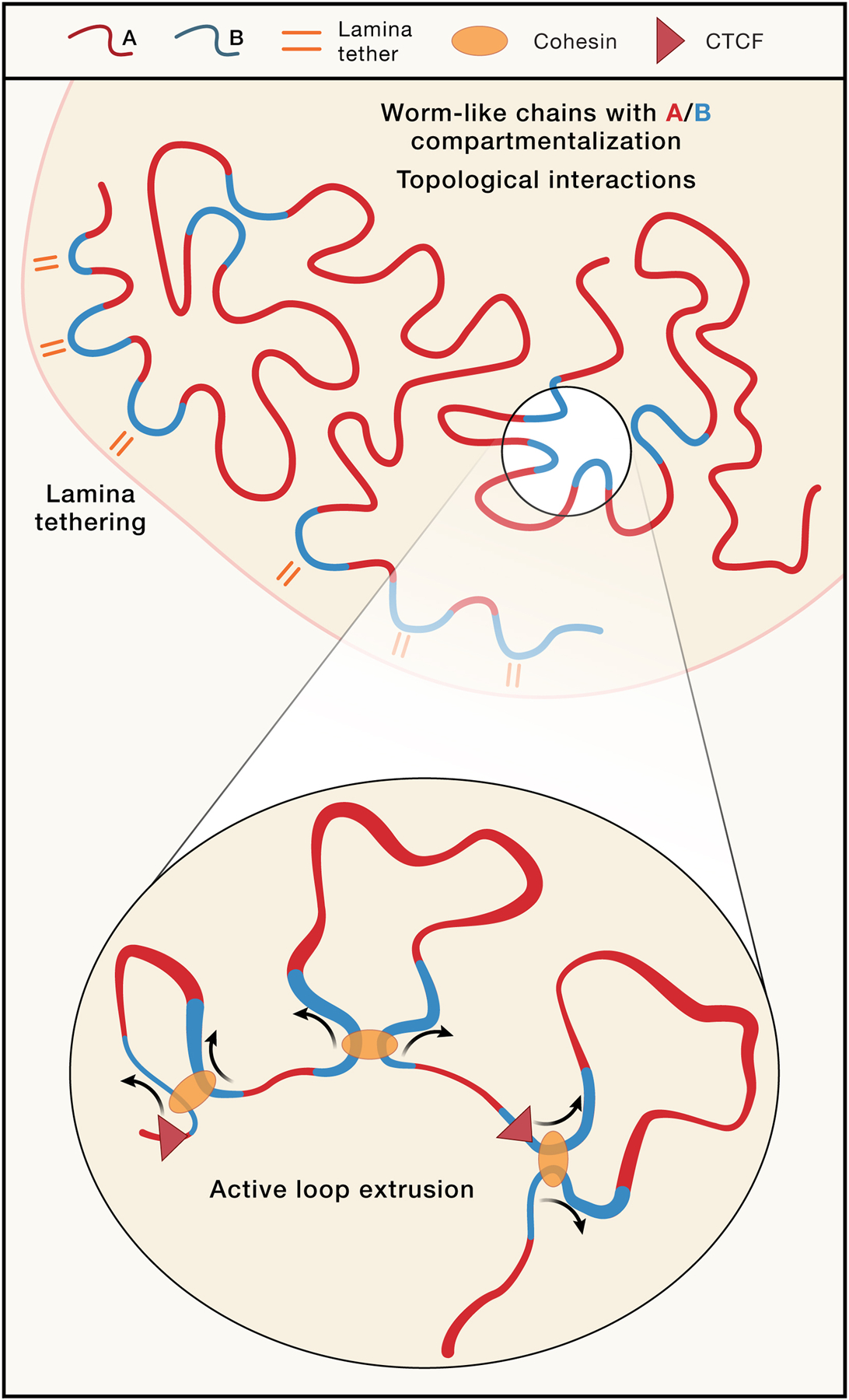
Schematic depiction of interphase chromosome conformation in eukaryotes as the combined and integrated result of multiple folding mechanisms. The chromosome is a worm-like chain that phase separates in distinct compartments (A/B compartments or finer subcompartments) driven by homotypic affinities. Tethering of domains to sub-nuclear structures such as the nuclear lamina, the nucleolus, or nuclear bodies including speckles, leads to positioning of loci and chromosomes at specific nuclear locations. Topological contraints prevent mixing in interphase but self-entanglements are formed in mitosis, facilitating full and fast compaction. At the scale of hundreds of Kb, loop extrusion, guided by cis elements that determine loading, unloading, and blocking (CTCF) of loop extruders, and with extensive interplay with other folding mechanisms, including compartmentalization, adds an additional layer of chromosome folding.
In interphase, along the length of each chromosome the epigenome alternates forming a sequence of chromatin domains of different types. The process of (sub-) compartmentalization will lead to spatial clustering of loci of similar types, through an affinity-driven process. This process naturally produces a stochastic assembly at the Mb-to-whole chromosome scale. Additional constraints are imposed through tethering loci to the nuclear periphery, the nucleolus, speckles etc. In addition, highly dynamic cohesin-mediated loop extrusion will bring loci together at the scale of up to hundreds of kilobases. All of these are acting on a chromatin fiber that is subject to topological constraints. As described above, cohesin extrusion patterns across the genome are guided by the presence of active enhancers that can recruit cohesin, CTCF-bound sites that can block extrusion, and sites where cohesin is unloaded (e.g., downstream of active genes). These cis elements determine a cohesin “traffic pattern”182,185 that produce over the cell population a range of structural features observed by Hi-C: formation of contiguous domains of enriched extrusion-dependent chromatin contacts (TADs) bounded by nearby CTCF sites; transient loops between convergent CTCF sites, enriched contacts between CTCF sites and flanking domains (stripes or flares in Hi-C maps); and some enhancer-promoter interactions facilitated by loop extrusion and also by affinity driven interactions (see below).
There is important interplay between different folding mechanisms. This is perhaps best exemplified by the interaction between compartmentalization and loop extrusion237. Loop extrusion can extend to hundreds of kilobases and can cross from one sub-compartment domain into another, thereby bringing together loci of different chromatin states that would otherwise tend to spatially segregate. This affects not just directly adjacent domains, the increased mixing of chromatin also appears to lead to mixing of domains at larger scale, e.g., interactions between compartment domains separated by large genomic distances, or even located on different chromosomes. In effect, loop extrusion makes different subcompartments segregate less than they otherwise would.
Simulations suggest that these are not only extruded loops but the whole active process of extrusion itself that weakens compartmentalization237. This effect is particularly clearly visible in experiments where loop extrusion is abolished through rapid depletion of cohesin, e.g., using degron approaches to inducibly degrade subunits of the cohesin complex213,238. In such experiments, compartmentalization is more pronounced, i.e., compartments segregate by type more strongly. In addition, smaller compartment domains emerged that in control cells appeared subsumed by the flanking domains in a cohesin-dependent manner. The compartment pattern seen in cohesin depleted cells correlates better with the epigenome profile, again showing that loop extrusion interferes with the natural tendency for domains to compartmentalize via intrinsic affinity-driven processes.
While tethering of loci from the B-compartment to the periphery is not directly driving compartmentalization itself103, it does determine which B-domains interact with which other B domains on other chromosomes, observed with Hi-C239. In the absence of such tethering, e.g., in the inverted nuclei of rod cells, the process of compartmentalization is unaffected but the pattern in interchromosomal interactions between B compartment domains is altered.
Finally, even though topological entanglements along and between chromosomes appear to be rare in interphase (in eukaryotes), this does not mean that topological transitions do not play a role in modulating interphase chromosome folding in eukaryotes. For instance, the increased compartmentalization observed upon acute depletion of cohesin is partly dependent on topoisomerase II activity207. Any real-time changes in compartmentalization may involve movement of loci, which may be facilitated by allowing topoisomerase II-dependent strand passage in general.
The same mechanisms can produce different folded states
In multicellular organisms, during interphase different cell types express different genes through differential activity of cis-regulatory elements, different patterns of histone modification and DNA methylation. Given that affinity-driven compartmentalization, as well as cohesin-mediated loop extrusion are directly guided and regulated by these features and cis-elements (above), the way the genome is folded in different cell types is different. However, although different loci will be clustered together or looped, the general folding principles are the same: affinities between subcompartments will drive their spatial clustering, and loop extrusion will occur throughout the genome with cohesin being recruited, unloaded and blocked at cis elements active in that cell type.
In contrast, chromosome organization can appear very different in different species and kingdoms (e.g., prokaryotes vs. eukaryotes), and across the mitotic and meiotic cell cycles, suggesting the possibility that in these cases very different folding principles and mechanisms may be at work. A key insight from extensive studies over the last decade on many different species, and with cells that synchronously progress through the cell cycle has been that in all cases folding is driven by the same small set of mechanisms described above. The reason this is possible is that these mechanisms, and especially the process of loop extrusion, are particularly malleable and can be regulated in many different ways resulting in a variety of chromosome architectures.
Below we provide examples how differential deployment of loop extrusion, compartmentalization, tethering, and topological entanglements can give rise to a large diversity of structures seen throughout the cell cycle, and even across kingdoms.
Interphase versus mitosis
The dramatic changes in chromosome morphology during the cell cycle serve as an excellent example of how cells can fold, unfold, and re-fold their genomes to accommodate gene expression in interphase, and accurate chromosome segregation during mitosis (Figure 3B, 4B). As originally proposed based on extensive microscopy studies30,132, biochemical and imaging experiments140,159,240, and later genomic (5C and Hi-C) studies and polymer modeling15,16,24, we now understand that by late prometaphase each sister chromatid is folded as a compressed array of consecutive loops. These loops are formed by condensin complexes: condensin II initially generates relatively large (400 kb - 1 Mb) loops in prophase, and during prometaphase condensin I then splits these in smaller (100 kb) loops16. This generates a nested arrangement of loops. In contrast to interphase, where many loops are positioned at reproducible sites (e.g., CTCF sites), positioned loops are not observed in mitosis. The array of loops then acquires a helical organization. This helical organization requires condensin II and is irregular. Perversions, where the handedness of the helical turns alternates every half turn, have been observed as well and these have been linked to the presence of connections between sister chromatids241. At the same time chromatin condenses through global reduction in histone acetylation, leading to general affinity-driven locus-locus interactions242. In Hi-C studies, no, or only very weak A or B compartments have been observed.
The organization of mitotic chromosomes appears very distinct from interphase chromosomes described above. However, both states are driven by mechanistically similar loop extrusion processes and affinity-driven locus-locus interactions. What makes the structures distinct is the way these mechanisms are implemented.
First, in interphase, cohesin is the main loop extruding complex , whereas in mitosis two types of condensin complexes act. This simple switch in extruder complexes explains much of the difference in interphase and mitotic chromosome folding. All complexes are estimated to extrude DNA at similar speeds (1–2 kb/sec). However, cohesin has a relatively short residence time on chromatin (10–20 minutes), and therefore interphase loops are relatively sparse, short-lived, and dynamic. In contrast, condensin II complexes appear to rarely or ever dissociate during mitosis243, and therefore can extrude larger and more stable loops. This also explains why in interphase only a fraction of DNA is extruded in loops at any given time, while by prometaphase almost the entire genome is extruded and contained within condensin loops (Figure 3B).
Second, cohesin and condensin differ in how they resolve encounters with other complexes and proteins while they extrude chromatin (above; Figure 4). Cohesin is blocked at CTCF-bound sites in a directional manner, leading to positioned and more stable loops between pairs of convergent CTCF sites that can be cell type specific. Condensins however, do not get blocked by CTCF244, or any other complex as far as we know, and therefore do not form positioned loops. Interestingly, during mitosis in living cells, condensins do not appear to bypass one another in vivo (Figure 4), leading to consecutive rather than overlapping loops15,16,24.
We note that the clear separation of action of cohesins and condensins during interphase and mitosis respectively described above for vertebrates, is not always so clear. In Drosophila condensin II plays roles in chromosome folding in interphase, and in C. elegans, condensin I contributes to folding the interphase genome245. In budding yeast, cohesin extrudes loops during mitosis246.
Third, cell type-specific affinity-driven compartmentalization is a major feature of interphase chromosomes but is absent within mitotic chromosomes. This has been puzzling because the patterns of histone modifications along chromosomes that correlate strongly with (sub-) compartments is largely preserved throughout the cell cycle. There are several possible explanations. First, it is possible that the factors that in interphase mediate the affinity between compartment domains are inactivated. A good example is the family of HP1 proteins. These proteins can bridge loci containing the H3K9Me3 modification. In mitosis, the residue immediately adjacent to K9 (H3S10) becomes phosphorylated. Histone tails carrying both modifications, H3K9Me3 and H3S10P, cannot be bound by HP1 proteins247. Given that histone tails become massively phosphorylated during mitosis, it is possible that many other bridging factors cannot bind, and thus affinity-driven compartmentalization will be prevented. An alternative, or additional explanation comes from a very recent study that showed that when condensins are depleted while cells are arrested in prometaphase, some form of compartmentalization is observed114. This result suggests that the factors and mechanisms for compartmentalization are active during mitosis but are somehow overruled by the condensin-driven loop array formation, similar to how in interphase cohesin-mediated loop extrusion counteracts compartmentalization.
Fourth, tethering of loci to the nuclear periphery, nucleoli, and speckles dominate interphase nuclear organization. During mitosis these structures are disassembled, and as a result the genome becomes untethered so that free rod-shaped mitotic chromosomes can form.
Fifth, topological entanglements within each chromosome are rare in interphase, but self-entanglements within individual sister chromatids are abundant in mitosis (above). This difference can at least in part be explained simply by the fact that topoisomerase IIa activity is high in mitosis, which together with a condensed and compacted chromatin state will drive the chromosomes towards becoming self-entangled. Polymer theory predicts that such change in topological state will facilitate rapid compaction, as would be required during prometaphase78. However, more active processes driving self-entanglements may also be at work.
Long versus short mitotic chromosomes
In vitro, condensin complexes can bypass rather larger objects 171, including other condensin complexes 170. In vivo, during mitosis it appears that such bypassing of condensins is rare, at least for condensin II in vertebrates 16,24. As a result, condensin II complexes extrude loops till they encounter each other and then stop so that a tightly spaced consecutive loop array is formed. This also strictly requires two-sided extrusion activity by condensin II248, as observed in single-molecule experiments 163,164. The size of these loops, assuming condensin II does not turn over, will be determined by how many condensins are recruited to chromatin 157. When many condensins are recruited, loops will tend to be small and mitotic chromatids will be relatively long and narrow. When fewer condensins are recruited, loops will on average be larger, and mitotic chromatids will be shorter and wider. Thus, in theory, the overall dimensions of mitotic chromosomes can be regulated simply by regulating condensin recruitment.
Interestingly, mitotic chromosomes can have very different dimensions when compared between species, or even within a species but at different stages of development. For instance, when mitotic chromosome dimensions are compared for human and mouse cells, it was observed that they differ in the amount of DNA that is packed per micron length of chromosome244. The difference was correlated with different loop sizes: in mouse cells the mitotic loops are considerably larger than in human cells (1 Mb vs 400 kb), suggesting fewer condensins are recruited per Mb in mouse cells as compared to human cells. Intriguingly, the difference may be related to the fact that mouse chromosomes are all acrocentric and thus the longest chromosome arm in the mouse genome is much longer than the longest arm in the human genome. Increasing loop size through regulating condensin recruitment genome-wide may ensure that even the longest chromosomes are short enough to facilitate their segregation during anaphase.
A similar adaptive scaling of mitotic chromosome dimensions appears to occur during Xenopus development249. During early cleavage stages of development, the cells are very large and mitotic chromosomes are relatively long. At later stages of development, when cells are much smaller, mitotic chromosomes become increasingly short. Again, analysis of loop sizes showed that the difference is due to formation of small loops in early stages, and larger loops at later stages. Differential recruitment of condensin complexes would explain this phenomenon.
Interestingly, factors on the chromatin in differentiated cells appear to reduce condensin loading. One such factor could be Histone H1.8. In vitro reconstitution experiments showed that in Xenopus egg extracts, depletion of H1.8 resulted in increased condensin recruitment, longer chromosomes, and smaller loops250.
These examples show that by simply regulating the recruitment of loop extruding factors, the same process of loop extrusion can produce mitotic chromosomes of distinct dimensions. This makes mitotic chromosome architecture adaptable to ensure condition-appropriate scaling of chromosome arm length.
Mitosis versus meiosis
Mitotic and meiotic chromosomes are both folded as arrays of loops to form rod-shaped compacted chromatids. While transcription ceases during mitosis, and compartments become undetectable, during meiotic prophase I transcription continues, and a form of compartmentalization remains present251,252. Another key difference is how sister chromatids are arranged with respect to each other: in mitosis, by prometaphase sister chromatids are connected through cohesin-mediated connections within their loops. Microscopically this can be deduced from the fact that cohesin complexes are localized in between the masses of each sister chromatid, and away from the condensin complexes that are located at the bases of the loops in the center of each chromatid24,253. In contrast, during meiotic prophase, sisters are cohesed at the bases of the loops254. We recently proposed that the mitotic arrangement could arise naturally when actively extruding condensins step over cohesin complexes that hold sister chromatids together (so-called cohesive cohesin complexes)24. This will result in cohesive cohesin being localized inside condensin-mediated loops. Modeling showed that when condensin, or any other loop extruding complex (cohesin likely in meiosis) cannot bypass cohesive cohesins, the extruding complexes and cohesive cohesins will both localize at the base of the loops, a scenario that may be present during meiotic prophase. It is therefore possible that by simply modulating the ability to bypass cohesive cohesins, one can obtain either the mitotic or meiotic arrangement of sister chromatids (Figure 4B). Consistent with this proposal is that during meiosis cohesin plays a significant role in loop array formation, and recent study showed that (mitotic) cohesin cannot bypass cohesive cohesin183. CTCF, that remains chromatin-bound during meiotic prophase and is also found at the conjoined axes of sister chromatids may also contribute to this arrangement of cohesin-mediated sister loops255.
Finally, it is noteworthy that at early prophase stages in mitosis in human cells, sister chromatids are also transiently connected at their loop bases256. Possibly condensins initially stall at cohesive cohesin complexes and only later bypass. Clearly, how SMC complexes resolve encounters between them during interphase, mitosis and meiosis can lead to very distinct chromosome conformations at the macro scale. Several of such “rules of engagement” have now been described 24, but surely additional ones remain to be discovered.
Across the tree of life: Prokaryotes vs Eukaryotes
Although bacterial nucleoids and eukaryotic chromosomes appear very different in size and conformation, similar processes fold these genomes. Such similarities were already recognized and reviewed a number of years ago257. Loop extrusion by SMC-like complexes act on bacterial chromosomes, and as in eukaryotic chromosomes, cis-elements can determine where these complexes load and where they are blocked (for review258). For instance, in Bacillus subtilis, the ParS sites at the centromere recruits SMC complexes that then start to extrude DNA bidirectionally, leading to coalignment of the arms of the chromosomes174. In E. coli, SMC-like complexes extrude DNA within each arm, thereby condensing the nucleoid. Interestingly, in B. subtilis, with engineered arrangement of ParS sites complicated patterns of folding have been observed that can be explained when SMC complexes can bypass one another184. Such bypassing can be critical to avoid traffic jams between SMCs loaded at nine native proximal ParS sites, providing another example of a “rule of engagement” whereby resolution of molecular encounters between extruding complexes can determine folding of entire genomes.
In bacteria, topological features appear to play a much more dominant role in chromosome folding than in interphase in eukaryotes. Supercoiling will compact the nucleoid and is determined by transcription and replication, but also directly by enzymes such as gyrase that introduce positive supercoiling. Interestingly, chromosomal interaction domains (CIDs) have been observed along the C. crescentus and B. subtilis chromosomes that in Hi-C resembles topologically associating domains175. CIDs, however, could be formed in an SMC-independent manner as dense arrays of plectonemes that are separated by plectoneme-free regions at highly expressed genes.
Finally, even though conventional compartmentalization is not observed, some form of affinity driven clustering of loci can occur in bacteria. For instance, nucleoid-associated proteins, such as HU and H-NS, can act as bridging factors condensing chromosomal domains259.
Across the tree of life: more variations of folding mechanisms, and possibly additional mechanisms?
In a recent study, chromosome folding for 24 eukaryotic species from across the tree of life was studied by Hi-C51. These included several vertebrate classes and animal phyla, plants and fungi. Two main types of folding architectures were described: one type is defined by a Rabl-like organization with centromeres clustered, telomeres clustered, and/or chromosome arms being aligned to each other from centromere to telomere. The second type lacks these features but has chromosome territories. Interestingly, a single factor, the presence or absence of condensin II defines whether the first type or the second type is formed. The authors proposed a model where condensin II mediates length-wide compaction during mitosis that then facilitates chromosome territory formation in the next G1, while preventing centromere and telomere clustering. This study shows how genome-wide chromosome folding patterns in eukaryotes can be altered by simply turning one extrusion complex on or off.
These results show that studying chromosome folding in a range of species can be fruitful for gaining a better understanding of the basic chromosome folding mechanisms discussed here. Through such an approach we can discover additional ways these conserved mechanisms can be regulated and implemented. Possibly new variants of the basic machinery, e.g., additional SMC complexes, can be discovered. Finally, it is possible that entirely new and yet-to-be-discovered mechanisms of folding chromosomes remain to be discovered in groups of species with highly divergent chromosome conformations.
We envision two ways to select groups of organisms for such evolutionary studies. First, one can study groups of organisms which contain distinct variants of the conserved machineries for chromosome folding. An example are the two major groups of archaea, the euryarchaea and crenarchaea. These single cell organisms differ in how they organize chromatin and are only recently being studied using 3C-based assays. Interestingly, both archaeal groups express SMC-related proteins and these proteins may play roles in chromosome folding260. In most cases in euryarchaea these complexes are clearly related to condensins. Intriguingly, the crenarchaea appear to have lost the condensin-like SMC complex and instead have acquired a poorly characterized SMC-like complex called coalescin261. Hi-C studies show that coalescin plays roles in chromosome folding and compartmentalization. The fact that an SMC-related complex may be involved in compartmentalization may point to a new role for an SMC complex and highlights how study of divergent species can provide opportunities to discover new roles, or new ways to employ these otherwise conserved machineries.
In a second approach one can select species that display chromosome conformations that appear particularly different from any other groups of species. One example are the dinoflagellates. Dinoflagellates are single cell eukaryotes with very large genomes (up to hundreds of gigabases), and that do not wrap the bulk of their genome around nucleosomes. Macroscopically, dinoflagellate chromosomes appear very distinct from any other group262,263: the chromosomes are permanently condensed through the cell cycle, and they have optical properties that suggest a liquid crystalline arrangement of chromatin fibers within them. Recent Hi-C studies show that the chromosomes are composed of structural domains, resembling TADs and CIDs, each of which contains a pair of divergently transcribed gene arrays7,8. Very little is known about the mechanism of chromosome folding in these organisms. They express condensin and cohesin-like complexes, and therefore it is possible that they represent yet another example where new ways have evolved to employ these conserved folding machines. On the other hand, given that their chromosomes appear so different from any other group, it is also possible that new mechanisms to fold chromosomes have emerged in this lineage.
Structure - function relationships
Higher order chromosome folding in eukaryotes is linked to genomic functions, including for instance chromosome compaction, segregation and regulation of transcription. However, despite extensive efforts it has proven difficult to demonstrate that higher order folding has consistent, conserved, and genome-wide consequences for gene expression. This is likely because the primary function of chromosome folding is not gene regulation, but instead is for cells to manage long DNA molecules to ensure their replication, compaction, segregation and subsequent decompaction. Consistent with this, the proteins involved in chromosome compaction, especially the factors that perform loop extrusion appear conserved in all organisms. Further, we propose that once mechanisms for folding and unfolding genomes were in place, these same mechanisms, e.g., loop extrusion and affinity-driven clustering of loci, have subsequently been co-opted for roles in additional processes including long-range regulation of genes by distal regulatory elements, DNA replication, etc. Given that these latter functions are secondary, and possibly ad hoc, chromosome folding will not necessarily have consistent, conserved, and genome-wide roles for regulating expression of all genes ,and different organisms. Such roles of chromosome-folding processes in gene regulation and, broadly, in epigenetic mechanisms, have started to emerge in disperate biological systems, with universal effects yet to be discovered.
Functional roles for mitotic chromosome compaction
The most obvious functional role of chromosome folding is related to chromosome duplication and segregation. In large vertebrate genomes, loci replicate at different times during S-phase. This phenomenon shows a clear connection with chromosome folding: DNA replication timing is strongly correlated with compartmentalization264, and when replication timing is disrupted or altered this can lead to changes in compartmentalization265. After replication, sister chromatids are to a significant level topologically intertwined. These interlinks need to be resolved to facilitate their segregation during anaphase. In addition, each sister chromatid needs to compact into mitotic chromatids. In eukaryotes this involves formation of arrays of loops mostly by the condensin complexes (above). These loops can become topologically interlinked (i.e., mitotic chromatids can become self-entangled). These two processes are likely mechanistically linked as loop extrusion in cis, in the presence of topoisomerase II enzymes, will automatically drive the sister chromatids to become unlinked while they are still held together by cohesive cohesin complexes147. The function of this elaborate process of folding, self-entangling and unlinking sister chromatids is to facilitate chromosome segregation to daughter cells. Any functional role of self-entanglement is less understood. It has been shown that through micromechanical measurements of single isolated mitotic chromosomes, that human mitotic chromosomes are self-entangled, and that these entanglements contribute to mechanical rigidity of chromosomes211. Such rigidity may be important for chromosome segregation to counteract spindle forces.
Roles of chromosome folding in controlling gene expression
In eukaryotic cells, genes can be regulated by enhancers that are located up to hundreds of kilobases from their promoter. A long-standing model has been that the spatial folding of chromosomes would allow enhancers to loop to gene promoters and through physical interactions between complexes bound to the enhancer and the promoter, and possibly through factors bridging them such as the Mediator complex, activate transcription266,267. Other mechanisms have also been put forth, such as models where factors recruited at an enhancer can somehow travel along chromatin over considerable distances and in that way reach target promoters that are then activated without needing a direct physical interaction between these elements (“tracking models”). In the latter case, chromosome folding may not be critical for long-range gene regulation. It now appears multiple mechanisms may contribute.
One of the first applications of 3C-based assays had been to determine whether looping interactions between genes and their distal regulatory elements occur. In a very early study, it was found that the Locus Control Region of the mouse beta-globin locus physically touches a target beta-globin gene only in cell types where that gene is expressed268. Further work showed that when different beta-globin genes become active during development, the LCR switches its long-range interactions accordingly269. This locus remains one of the best studied examples of long-range interactions in relation to gene expression. In a demonstration for the importance of physical interactions between enhancers and promoters, it was shown that direct tethering of the distal LCR to the target gene is sufficient for activation of the gene270.
Since then, numerous studies have detected many more long-range promoter-enhancer interactions, e.g., in single gene studies for the alpha-globin locus128, in higher throughput studies for hundreds of genes throughout numerous targeted regions of the human genome (5C271), or genome-wide analyses using targeted approaches (Capture-C272, and ChiaPET59). From these studies one would conclude that the looping model is firmly established. However, other lines of experimentation produced observations that were at first glance not consistent with the “activation by physical contact” model for long-range control of gene expression. First, imaging-based studies showed that 3D spatial distances between enhancers and promoters do not correlate well with gene activation, neither in life cells in real time nor over cell populations (reviewed here: 273). Furthermore, a relatively small (~2–3 fold) increase in the contact frequency within a TAD results in much more frequent intra-TAD enhancer-promoter interactions.This lack of correlation between contacts, or close proximity, and gene activation may appear to be inconsistent with a mechanistic role of looping contacts. Second, even when enhancers and promoters have been observed in close spatial proximity, the distance that separates them was relatively large, i.e., 300 nm274.
Several new insights, experimental approaches and models, are now attempting to unify these observations. First, the discovery that transcription complexes can form condensates that can be several hundreds of nm in diameter suggests that such condensates can potentially mediate connections between enhancer and promoters274. By imaging the enhancer and promoter can then appear not directly touching, yet 3C-based assays that employ formaldehyde cross-linking may still detect these contacts as looping interactions.
Second, several models have been proposed to explain the lack of simple correlation between the frequency of enhancer-promoter contacts (measured by Hi-C and microscopy) and transcription from the target promoter275,276. The idea of these models is that transient and rare, enhancer-promoter interactions result in the incremental but cumulative changes in the multi-state promoter, with only the final state resulting in transcription. These changes may, for example, reflect accumulation of marks at the promoter and enhancer. Importantly, such multi-state models show sigmoidal response to contact frequency with small changes in contact frequency resulting in large changes in transcription. when a critical level of marks is delivered will the promoter become active. These models of multi-state promoters await validation by experimental live-cell dynamic measurements of distance and transcription at high temporal and spatial resolutions.
What is the mechanism by which an enhancer can loop to target promoters? One attractive proposal is that cohesin-mediated loop extrusion actively brings elements together. The observation that transcriptional elements such as enhancers, promoters, insulators (CTCF sites), and 3’ ends of active genes all play roles in recruiting, pausing, blocking, and unloading cohesin respectively, already suggests links between transcriptional control and cohesin-mediated loop extrusion182,185. However, acute depletion of cohesin was found to have little immediate effect on transcription153,213. Most recent studies, however, demonstrated that the loss of cohesin activity in postmitotic cells has a profound effect on cell and organism physiology such as neuronal maturation277 and differentiation and response to activation by dendritic cells of the innate immune system278.
After years of conflicting observations, a unified view is now emerging how cohesin plays a role in long-range gene regulation (Figure 6). In this view, cohesin can be loaded at random positions, with some preference for cis elements such as enhancers. These complexes can then extrude loops and through this process reach distal target promoters. This can lead to detectable enhancer-promoter loops in 3C-based assays. However, in such assays these interactions appear very weak, suggesting these are either rare or very transient contacts. As stated above, repeated transient interactions may be sufficient to trigger changes of promoter states that will become active after several interactions.
Figure 6: The interplay of Loop extrusion, topologically associating domain formation, and guidance of promoter-enhancer interactions.
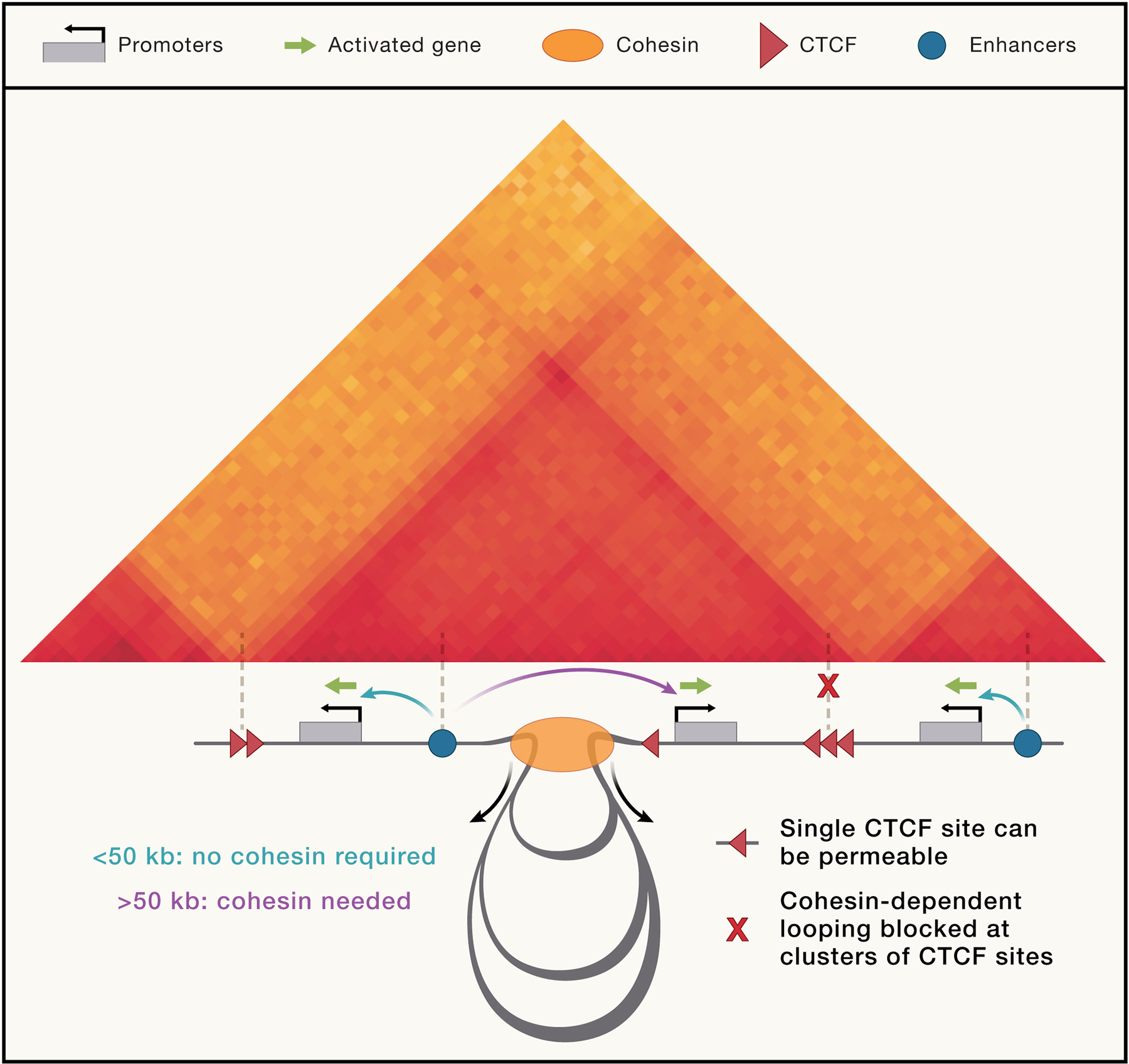
Enhancers can reach nearby promoters (separated by less than 50 kb) and interact via affinity-driven contacts, without a need for loop extrusion to facilitate their interaction. Cohesin facilitates contacts between enhancers and promoters when they are separated by more than 50 kb. Repeated enhancer-promoter contacts may be required to activate the gene (green arrows), leading to reduced correlation between contact and transcription in real time. For longer-range enhancer-promoter pairs: when clusters of multiple CTCF sites are located in between them, they will block loop extrusion, induce a TAD boundary, and inhibit enhancer-promoter contacts, preventing enhancers from activating the gene. Single CTCF sites can create weak and permeable boundaries that may not fully prevent enhancer-mediated activation leading to some activation (smaller green arrow). Orange triangles, in different shades represent Hi-C interaction data and TADs.
Another factor is that enhancer-promoter communication may require a different type of interactions than contacts captured by 3C-based assays or microscopy. For example functional communication may require close interactions between molecular complexes, i.e. ~5–10 nm separation, or a prolonged contact, while 3C-based and microscopy detect mere proximity of 100–300 nm. It remains to be studied how proximity, spatial, cohesin- or affinity-mediated, may translate into functional communications. We note that it is also possible that cohesin will travel in cis from the enhancer to the promoter carrying transcription activating complexes picked up at the enhancer that will be delivered to the promoter, and this “tracking” process would not necessarily involve physical promoter-enhancer interactions.
The observation that acute depletion of cohesin has little direct effect on transcription genome-wide is now understood to be at least in part because most promoters are regulated by enhancers that are located relatively close to the start site (< 50kb). Several studies have now shown that enhancers can interact with promoters over such short genomic distances without the assistance of cohesin 185,279,280, while cohesin-mediated extrusion becomes important only for enhancers to reach target genes located >50 kb away. Enhancer-promoter interactions are also likely assisted by affinity-based interactions, e.g., like compartmentalization, without a need for cohesin. The frequency with which affinity-based interactions occur in the population, will be inversely related to the genomic distance between them127. It appears that when they are less than 50 kb apart, the random dynamics and conformation of chromatin fibers ensures that they interact sufficiently frequently to enable gene activation, whereas enhancers located farther away only contact their targets when loci are actively brought together through extrusion. Direct visualization and models of enhancer-promoter interactions and resulting transcription (e.g.223,281) will be instrumental in elucidating this interplay of extrusion-mediated and spatial contacts in gene regulation.
If cohesin assists enhancers to reach, and activate, genes that can be located hundreds of kilobases away in the linear genome, how is specificity controlled? CTCF-bound elements, when located in between genes and enhancers, can block both enhancer action towards a promoter, and block loop extrusion in an orientation-dependent manner (see above). This has led to models that pairs of convergent CTCF sites form the boundaries of structural domains282. An enhancer located within a domain would contact promoters in the same domain, but interactions with promoters outside the domain would be less frequent (though not completely inhibited).
The Hox loci in mouse provide particularly well studied and characterized examples of how CTCF boundaries prevent or allow enhancer-promoter interactions (see 283 for a recent review). These clusters of multiple hox genes are regulated by progressive movement of CTCF boundaries in cells positioned along the posterior-anterior body axis, allowing progressive activation of the hox genes in order of their location along the genomic locus. Another example is the regulation of the protocadherin clusters where regulated stochastic CTCF binding to different protocadherins promoters, in combination of cohesin-mediated loop extrusion, defines cell-to-cell variation in expression of different variants of protocadherins (284, and see 285, and see for a review).
The model also predicts that loss of a domain boundary, e.g., in disease through deletion of CTCF-binding sites, would allow formation of new enhancer-promoter contacts that in normal cells would rarely occur. There are multiple examples where this prediction is fulfilled. For instance, in T cell acute lymphoblastic leukemia recurrent deletions around the TAL1 and LMO2 oncogenes result in loss of CTCF-bound domain boundaries282. Deletion of these boundaries exposes these genes to enhancers in adjacent domains that they normally rarely interact with. Inappropriate DNA methylation of CTCF binding sites can also prevent CTCF binding, and in that way prevent domain boundary formation. An example is DNA hypermethylation observed in gastrointestinal stromal tumors286,287. This leads to inactivation of CTCF-dependent domain boundaries and formation of new enhancer-promoter interactions, including at the FGF8 locus, which in turns drives tumorigenesis. Thus, loop extrusion can facilitate very long-range enhancer-promoter interactions in general, and CTCF-dependent blocking of extrusion can impose specificity by preventing some interactions.
Long-range gene regulation is clearly complex and driven by a multitude of processes: for some cases loop extrusion is critical, for others it is not. CTCF can prevent long-range interactions between enhancers and promoters, but sometimes affinity-driven interactions can override this. Not all CTCF-bound sites act in the same manner, possibly dependent on how many CTCF sites are near each other, their orientation, the presence of other DNA binding sites, and other factors.
Other processes that have co-opted the loop extrusion process
Loop extrusion has been co-opted by other more specialized genomic processes. We highlight one example.
Immunoglobulin locus rearrangement
B and T cells can express an enormous diversity of antigen receptor molecules: antibodies in B cells, and T-cell receptors in T-cells. This diversity is the result of rearrangements of immunoglobin and T-cell receptor loci. For instance, the IgH locus is composed of three sets of loci: V, D, and J segments. There are 13 V segments, 4 D segments and 113 J segments. In a given B-cell the locus recombines so that a single V region gets placed next to a single D segment, and then 1 J segment is selected to be recombined adjacent to the D segment. This progressive recombination occurs only on 1 allele. As a result, a given B-cell will express only IgH polypeptide that is encoded by 1 V, D, and J segment. Wood and Tonegawa proposed, already in 1983137, that loop extrusion could play a role in bringing V, D, and J segments, spread out over a large 2.5 Mb region, in close spatial proximity for their subsequence recombination. Their argument was based on the fact that all V, D, and J segments are in the same orientation. A tracking model was proposed where a complex would track along the locus and bring distal loci together for subsequent recombination.
There is now strong evidence that cohesin plays a key role in immunoglobulin locus rearrangements (reviewed more comprehensively here288,289. First, IgH recombination is dependent on cohesin. Depletion of cohesin subunits inhibits V(D)J recombination290. Second, CTCF sites are located throughout the locus, and they occur in convergent orientations: CTCF sites throughout the section of the locus encoding J segments is oriented towards the 3’ end of the locus, while CTCF sites at the 3’ end of the locus are oriented towards the 5’ end containing the J segments. This convergent orientation is strongly predictive of a role of cohesin-mediate loop extrusion. Third, inversion of large sections of the locus changes the selection of segments due to inversion of CTCF sites291. Fourth, deletion of individual CTCF sites, or ectopic insertion of CTCF sites, changes the selection probability of the adjacent segment ways that are consistent with roles of CTCF in blocking loop extrusion292. Fifth, in B-cells expression of WAPL is reduced through Pax5-mediated repression of the WAPL gene, and this increases the residence time of cohesin on chromatin genome-wide. As a result, cohesin mediated loops can become larger and this facilitates the very long-range looping that is required in the IgH locus to select the most distal J segments for recombination293.
Epigenetic memory, a functional role of compartmentalization?
A recent study has put forward a hypothesis that spatial segregation and higher heterochromatin density can be key for the maintenance of repressive heterochromatin marks, and broadly epigenetic memory294. Since marked histones are constantly lost and replaced with non-marked ones, particularly during replications, the patterns of histone marks need to be restored by the activity of reader-writer histone methyltransferases. Models show that such activity could lead to either uncontrolled spreading or to complete loss of marks. Yet creating dense and spatially segregated heterochromatin by virtue of making heterochromatic regions attract each other, can put this process under control; with another key ingredient being the limited amount of the reader-writer enzyme. Polymer models show that such a mechanism can be self-sustained in maintaining “epigenetic memory”, i.e. preserving an initially deposited pattern of marks for up to hundreds of cell divisions. Importantly, this mechanism suggests that the memory is preserved in the form of spatial folding (segregation and density) of chromosomes during interphase, and in the form of the pattern of marks when chromosomes are refolded into mitosis and back into the interphase. Perturbing chromosome folding experimentally would test this hypothesis. Broadly, this study suggests that one of the functions of compartmentalization may be to help maintain the pattern of epigenetic marks.
Future perspective
We described the biophysical and molecular mechanisms that drive chromosome folding, and allow cells to solve their chromosome folding problem. Many open questions remain to be answered on how these mechanisms are regulated, and how they influence each other. Going forward, a better understanding of the molecular mediators of compartmentalization, and its function in genomic activities is needed. How the basic process of loop extrusion works at the nanometer scale is increasingly clear, but we are only just beginning to understand how this process, with interplay with other mechanisms, leads to a variety of chromosome conformations at the micrometer scale. Regulation of loop extrusion likely involves cell cycle-, condition-, and species-dependent expression of extrusion complexes, their targeted and non-targeted recruitment and their post-translational modifications. Interactions between extruders and with other DNA-bound complexes must occur frequently, and how they are resolved determines to a large extent the conformation of whole chromosomes. Such rules of engagements are only now starting to be defined, but likely will be key to our understanding how activities at the nanometer scale lead to differential folding at the micrometer scale.
Much has been learned from the study of chromosome folding, and changes in folding throughout the cell cycle. We anticipate that additional insights will come from studies across the tree of life, where different organisms may have evolved additional solutions to the chromosome folding problem. On the one hand it is possible that the same mechanisms fold chromosomes in all species, but that these mechanisms are employed in different ways to give rise to chromosomes that can look very different. In that case studying chromosome folding in different species will provide deeper insights into such universal mechanisms. On the other hand, it is possible that entirely different mechanisms remain to be discovered in highly divergent species. The coming 50 years promise to be as exciting for the chromosome folding field as the last few decades have been.
Acknowledgements
We thank past and present members of our labs, and our many collaborators for sharing and discussing ideas and results over many years. Our current research is funded by the National Human Genome Research Institute (HG003143 to JD; HG011536 to JD and LM), the National Institute of General Medicine (GM 114190 to LM) and the National Science Foundation (Physics of Living Systems grant 1504942 to LM). JD is an investigator of the Howard Hughes Medical Institute. LM is the Simons Investigator in Theoretical Physics in Life Sciences.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
Job Dekker is a member of the scientific advisory board of Arima Genomics, San Diego, CA, USA and Omega Therapeutic, Cambridge, MA, USA.
Job Dekker is listed as co-inventor of patents describing the 5C and Hi-C technologies.
References
- 1.Ris H, and Kubai DF (1970). Chromosome structure. Annu Rev Genet 4, 263–294. 10.1146/annurev.ge.04.120170.001403. [DOI] [PubMed] [Google Scholar]
- 2.Thomas CA Jr. (1971). The genetic organization of chromosomes. Annu Rev Genet 5, 237–256. 10.1146/annurev.ge.05.120171.001321. [DOI] [PubMed] [Google Scholar]
- 3.Oudet P, Gross-Bellard M, and Chambon P (1975). Electron microscopic and biochemical evidence that chromatin structure is a repeating unit. Cell 4, 281–300. 10.1016/0092-8674(75)90149-x. [DOI] [PubMed] [Google Scholar]
- 4.Weintraub H, Palter K, and Van Lente F (1975). Histones H2a, H2b, H3, and H4 form a tetrameric complex in solutions of high salt. Cell 6, 85–110. 10.1016/0092-8674(75)90077-x. [DOI] [PubMed] [Google Scholar]
- 5.Ryder OA, and Smith DW (1975). Properties of membrane-associated folded chromosomes of E. coli related to initiation and termination of DNA replication. Cell 4, 337–345. 10.1016/0092-8674(75)90154-3. [DOI] [PubMed] [Google Scholar]
- 6.Allen JR, Roberts M, Loeblich AR 3rd, and Klotz LC (1975). Characterization of the DNA from the dinoflagellate Crypthecodinium cohnii and implications for nuclear organization. Cell 6, 161–169. 10.1016/0092-8674(75)90006-9. [DOI] [PubMed] [Google Scholar]
- 7.Nand A, Zhan Y, Salazar OR, Aranda M, Voolstra CR, and Dekker J (2021). Genetic and spatial organization of the unusual chromosomes of the dinoflagellate Symbiodinium microadriaticum. Nat Genet 53, 618–629. 10.1038/s41588-021-00841-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marinov GK, Trevino AE, Xiang T, Kundaje A, Grossman AR, and Greenleaf WJ (2021). Transcription-dependent domain-scale three-dimensional genome organization in the dinoflagellate Breviolum minutum. Nat Genet 53, 613–617. 10.1038/s41588-021-00848-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dekker J (2008). Mapping in vivo chromatin interactions in yeast suggests an extended chromatin fiber with regional variation in compaction. J. Biol. Chem 283, 34532–34540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arbona JM, Herbert S, Fabre E, and Zimmer C (2017). Inferring the physical properties of yeast chromatin through Bayesian analysis of whole nucleus simulations. Genome Biol 18, 81. 10.1186/s13059-017-1199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dekker J, and Mirny LA (2016). The 3D genome as moderator of chromosomal communication. Cell 164, 1110–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gabriele M, Brandao HB, Grosse-Holz S, Jha A, Dailey GM, Cattoglio C, Hsieh TS, Mirny L, Zechner C, and Hansen AS (2022). Dynamics of CTCF- and cohesin-mediated chromatin looping revealed by live-cell imaging. Science 376, 496–501. 10.1126/science.abn6583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delbruck M (1954). On the Replication of Desoxyribonucleic Acid (DNA). Proc Natl Acad Sci U S A 40, 783–788. 10.1073/pnas.40.9.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mirny LA (2023). Chromosome and protein folding: In search for unified principles. Curr Opin Struct Biol 81, 102610. 10.1016/j.sbi.2023.102610. [DOI] [PubMed] [Google Scholar]
- 15.Naumova N, Imakaev M, Fudenberg G, Zhan Y, Lajoie BR, Mirny LA, and Dekker J (2013). Organization of the mitotic chromosome. Science 342, 948–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gibcus JH, Samejima K, Goloborodko A, Samejima I, Naumova N, Nuebler J, Kanemaki MT, Xie L, Paulson JR, Earnshaw WC, et al. (2018). A pathway for mitotic chromosome formation. Science 359. 10.1126/science.aao6135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shintomi K, Takahashi TS, and Hirano T (2015). Reconstitution of mitotic chromatids with a minimum set of purified factors. Nat Cell Biol 17, 1014–1023. 10.1038/ncb3187. [DOI] [PubMed] [Google Scholar]
- 18.Kinoshita K, and Hirano T (2017). Dynamic organization of mitotic chromosomes. Curr Opin Cell Biol 46, 46–53. 10.1016/j.ceb.2017.01.006. [DOI] [PubMed] [Google Scholar]
- 19.Swedlow JR, and Hirano T (2003). The making of the mitotic chromosome: modern insights into classical questions. Mol Cell 11, 557–569. [DOI] [PubMed] [Google Scholar]
- 20.Dekker J (2014). Two ways to fold the genome during the cell cycle: insights obtained with chromosome conformation capture. Epigenetics Chromatin. 7, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sood V, and Misteli T (2022). The stochastic nature of genome organization and function. Curr Opin Genet Dev 72, 45–52. 10.1016/j.gde.2021.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Misteli T (2020). The Self-Organizing Genome: Principles of Genome Architecture and Function. Cell 183, 28–45. 10.1016/j.cell.2020.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gibcus JH, and Dekker J (2013). The hierarchy of the 3D genome. Mol Cell 49, 773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Samejima K, Gibcus JH, Abraham S, Cisneros-Soberanis F, Samejima I, Beckett AJ, Pucekova N, Abad MA, Medina-Pritchard B, Paulson JR, et al. (2024). Rules of engagement for condensins and cohesins guide mitotic chromosome formation. bioRxiv. 10.1101/2024.04.18.590027. [DOI] [Google Scholar]
- 25.Abramo K, Valton AL, Venev SV, Ozadam H, Fox AN, and Dekker J (2019). A chromosome folding intermediate at the condensin-to-cohesin transition during telophase. Nat Cell Biol 21, 1393–1402. 10.1038/s41556-019-0406-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang H, Emerson DJ, Gilgenast TG, Titus KR, Lan Y, Huang P, Zhang D, Wang H, Keller CA, Giardine B, et al. (2019). Chromatin structure dynamics during the mitosis-to-G1 phase transition. Nature 576, 158–162. 10.1038/s41586-019-1778-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pelham-Webb B, Polyzos A, Wojenski L, Kloetgen A, Li J, Di Giammartino DC, Sakellaropoulos T, Tsirigos A, Core L, and Apostolou E (2021). H3K27ac bookmarking promotes rapid post-mitotic activation of the pluripotent stem cell program without impacting 3D chromatin reorganization. Mol Cell 81, 1732–1748 e1738. 10.1016/j.molcel.2021.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DuPraw EJ (1970). DNA and chromosomes (Holt, Rinehart and Winston, Inc.). [Google Scholar]
- 29.Manuelidis L, and Chen TL (1990). A unified model of eukaryotic chromosomes. Cytometry 11, 8–25. 10.1002/cyto.990110104. [DOI] [PubMed] [Google Scholar]
- 30.Marsden MP, and Laemmli UK (1979). Metaphase chromosome structure: evidence for a radial loop model. Cell. 17, 849–858. [DOI] [PubMed] [Google Scholar]
- 31.Bak AL, Zeuthen J, and Crick FH (1977). Higher-order structure of human mitotic chromosomes. Proc Natl Acad Sci U S A 74, 1595–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Belmont AS, Sedat JW, and Agard DA (1987). A three-dimensional approach to mitotic chromosome structure: evidence for a complex hierarchical organization. J. Cell Biol 105, 77–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kireeva N, Lakonishok M, Kireev I, Hirano T, and Belmont AS (2004). Visualization of early chromosome condensation: a hierarchical folding, axial glue model of chromosome structure. J Cell Biol 166, 775–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Strukov YG, Wang Y, and Belmont AS (2003). Engineered chromosome regions with altered sequence composition demonstrate hierarchical large-scale folding within metaphase chromosomes. J Cell Biol 162, 23–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van den Engh G, Sachs R, and Trask BJ (1992). Estimating genomic distance from DNA sequence location in cell nuclei by a random walk model. Science 257, 1410–1412. [DOI] [PubMed] [Google Scholar]
- 36.Hahnfeldt P, Hearst JE, Brenner DJ, Sachs RK, and Hlatky LR (1993). Polymer models for interphase chromosomes. Proc Natl Acad Sci U S A 90, 7854–7858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yokota H, van den Engh G, Hearst JE, Sachs RK, and Trask BJ (1995). Evidence for the organization of chromatin in megabase pair-sized loops arranged along a random walk path in the human G0/G1 interphase nucleus. J. Cell. Biol 130, 1239–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sachs RK, van den Engh G, Trask B, Yokota H, and Hearst JE (1995). A random-walk/giant-loop model for interphase chromosomes. Proc. Natl. Acad. Sci. USA 92, 2710–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, Galibert F, Hoheisel JD, Jacq C, Johnston M, et al. (1996). Life with 6000 genes. Science 274, 546, 563–547. 10.1126/science.274.5287.546. [DOI] [PubMed] [Google Scholar]
- 40.Consortium, C.e.S. (1998). Genome sequence of the nematode C. elegans: a platform for investigating biology. Science 282, 2012–2018. 10.1126/science.282.5396.2012. [DOI] [PubMed] [Google Scholar]
- 41.Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, et al. (2000). The genome sequence of Drosophila melanogaster. Science 287, 2185–2195. 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- 42.McPherson JD, Marra M, Hillier L, Waterston RH, Chinwalla A, Wallis J, Sekhon M, Wylie K, Mardis ER, Wilson RK, et al. (2001). A physical map of the human genome. Nature 409, 934–941. [DOI] [PubMed] [Google Scholar]
- 43.Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al. (2001). The sequence of the human genome. Science 291, 1304–1351. [DOI] [PubMed] [Google Scholar]
- 44.Mouse Genome Sequencing C, Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, et al. (2002). Initial sequencing and comparative analysis of the mouse genome. Nature 420, 520–562. 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 45.Christmas MJ, Kaplow IM, Genereux DP, Dong MX, Hughes GM, Li X, Sullivan PF, Hindle AG, Andrews G, Armstrong JC, et al. (2023). Evolutionary constraint and innovation across hundreds of placental mammals. Science 380, eabn3943. 10.1126/science.abn3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stiller J, Feng S, Chowdhury AA, Rivas-Gonzalez I, Duchene DA, Fang Q, Deng Y, Kozlov A, Stamatakis A, Claramunt S, et al. (2024). Complexity of avian evolution revealed by family-level genomes. Nature 629, 851–860. 10.1038/s41586-024-07323-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang T, Antonacci-Fulton L, Howe K, Lawson HA, Lucas JK, Phillippy AM, Popejoy AB, Asri M, Carson C, Chaisson MJP, et al. (2022). The Human Pangenome Project: a global resource to map genomic diversity. Nature 604, 437–446. 10.1038/s41586-022-04601-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kaplan N, and Dekker J (2013). High-throughput genome scaffolding from in vivo DNA interaction frequency. Nat Biotechnol 31, 1143–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Burton JN, Adey A, Patwardhan RP, Qiu R, Kitzman JO, and Shendure J (2013). Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat Biotechnol 31, 1119–1125. 10.1038/nbt.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dudchenko O, Batra SS, Omer AD, Nyquist SK, Hoeger M, Durand NC, Shamim MS, Machol I, Lander ES, Aiden AP, and Aiden EL (2017). De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95. 10.1126/science.aal3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hoencamp C, Dudchenko O, Elbatsh AMO, Brahmachari S, Raaijmakers JA, van Schaik T, Sedeno Cacciatore A, Contessoto VG, van Heesbeen R, van den Broek B, et al. (2021). 3D genomics across the tree of life reveals condensin II as a determinant of architecture type. Science 372, 984–989. 10.1126/science.abe2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Obinu L, Trivedi U, and Porceddu A (2023). Benchmarking of Hi-C tools for scaffolding de novo genome assemblies. BioRxiv 10.1101/2023.05.16.540917. [DOI] [Google Scholar]
- 53.Dekker J, Rippe K, Dekker M, and Kleckner N (2002). Capturing Chromosome Conformation. Science 295, 1306–1311. [DOI] [PubMed] [Google Scholar]
- 54.Simonis M, Klous P, Splinter E, Moshkin Y, Willemsen R, de Wit E, van Steensel B, and de Laat W (2006). Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nat. Genet 38, 1348–1354. [DOI] [PubMed] [Google Scholar]
- 55.Dostie J, Richmond TA, Arnaout RA, Selzer RR, Lee WL, Honan TA, Rubio EH, Krumm A, Lamb J, Nusbaum C, et al. (2006). Chromosome Conformation Capture Carbon Copy (5C): A Massively Parallel Solution for Mapping Interactions between Genomic Elements. Genome Res 16, 1299–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hsieh TS, Weiner A, Lajoie BR, Dekker J, Friedman N, and Rando OJ (2015). Mapping nucleosome resolution chromosome folding in yeast by Micro-C. Cell 162, 108–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ma W, Ay F, Lee C, Gulsoy G, Deng X, Cook S, Hesson J, Cavanaugh C, Ware CB, Krumm A, et al. (2015). Fine-scale chromatin interaction maps reveal the cis-regulatory landscape of human lincRNA genes. Nat Methods 12, 71–78. 10.1038/nmeth.3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fullwood MJ, Liu MH, Pan YF, Liu J, Xu H, Mohamed YB, Orlov YL, Velkov S, Ho A, Mei PH, et al. (2009). An oestrogen-receptor-alpha-bound human chromatin interactome. Nature 462, 58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mumbach MR, Rubin AJ, Flynn RA, Dai C, Khavari PA, Greenleaf WJ, and Chang HY (2016). HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat Methods 13, 919–922. 10.1038/nmeth.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fang R, Yu M, Li G, Chee S, Liu T, Schmitt AD, and Ren B (2016). Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP-seq. Cell Res 26, 1345–1348. 10.1038/cr.2016.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.van Steensel B, and Henikoff S (2000). Identification of in vivo DNA targets of chromatin proteins using tethered dam methyltransferase. Nat Biotechnol 18, 424–428. [DOI] [PubMed] [Google Scholar]
- 63.Chen Y, Zhang Y, Wang Y, Zhang L, Brinkman EK, Adam SA, Goldman R, van Steensel B, Ma J, and Belmont AS (2018). Mapping 3D genome organization relative to nuclear compartments using TSA-Seq as a cytological ruler. J Cell Biol 217, 4025–4048. 10.1083/jcb.201807108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Beagrie RA, Scialdone A, Schueler M, Kraemer DC, Chotalia M, Xie SQ, Barbieri M, de Santiago I, Lavitas LM, Branco MR, et al. (2017). Complex multi-enhancer contacts captured by genome architecture mapping. Nature 543, 519–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Quinodoz SA, Ollikainen N, Tabak B, Palla A, Schmidt JM, Detmar E, Lai MM, Shishkin AA, Bhat P, Takei Y, et al. (2018). Higher-Order Inter-chromosomal Hubs Shape 3D Genome Organization in the Nucleus. Cell 174, 744–757 e724. 10.1016/j.cell.2018.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nagano T, Lubling Y, Stevens TJ, Schoenfelder S, Yaffe E, Dean W, Laue ED, Tanay A, and Fraser P (2013). Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 502, 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nagano T, Lubling Y, Varnai C, Dudley C, Leung W, Baran Y, Mendelson Cohen N, Wingett S, Fraser P, and Tanay A (2017). Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature 547, 61–67. 10.1038/nature23001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ramani V, Deng X, Qiu R, Gunderson KL, Steemers FJ, Disteche CM, Noble WS, Duan Z, and Shendure J (2017). Massively multiplex single-cell Hi-C. Nat Methods 14, 263–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li G, Liu Y, Zhang Y, Kubo N, Yu M, Fang R, Kellis M, and Ren B (2019). Joint profiling of DNA methylation and chromatin architecture in single cells. Nat Methods 16, 991–993. 10.1038/s41592-019-0502-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kind J, Pagie L, de Vries SS, Nahidiazar L, Dey SS, Bienko M, Zhan Y, Lajoie B, de Graaf CA, Amendola M, et al. (2015). Genome-wide maps of nuclear lamina interactions in single human cells. Cell 163, 134–147. 10.1016/j.cell.2015.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tan L, Xing D, Chang CH, Li H, and Xie XS (2018). Three-dimensional genome structures of single diploid human cells. Science 361, 924–928. 10.1126/science.aat5641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang S, Su JH, Beliveau BJ, Bintu B, Moffitt JR, Wu CT, and Zhuang X (2016). Spatial organization of chromatin domains and compartments in single chromosomes. Science 353, 598–602. 10.1126/science.aaf8084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bintu B, Mateo LJ, Su JH, Sinnott-Armstrong NA, Parker M, Kinrot S, Yamaya K, Boettiger AN, and Zhuang X (2018). Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science 362. 10.1126/science.aau1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Beliveau BJ, Joyce EF, Apostolopoulos N, Yilmaz F, Fonseka CY, McCole RB, Chang Y, Li JB, Senaratne TN, Williams BR, et al. (2012). Versatile design and synthesis platform for visualizing genomes with Oligopaint FISH probes. Proc Natl Acad Sci U S A 109, 21301–21306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nir G, Farabella I, Perez Estrada C, Ebeling CG, Beliveau BJ, Sasaki HM, Lee SD, Nguyen SC, McCole RB, Chattoraj S, et al. (2018). Walking along chromosomes with super-resolution imaging, contact maps, and integrative modeling. PLoS Genet 14, e1007872. 10.1371/journal.pgen.1007872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mateo LJ, Murphy SE, Hafner A, Cinquini IS, Walker CA, and Boettiger AN (2019). Visualizing DNA folding and RNA in embryos at single-cell resolution. Nature 568, 49–54. 10.1038/s41586-019-1035-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mach P, Kos PI, Zhan Y, Cramard J, Gaudin S, Tunnermann J, Marchi E, Eglinger J, Zuin J, Kryzhanovska M, et al. (2022). Cohesin and CTCF control the dynamics of chromosome folding. Nat Genet 54, 1907–1918. 10.1038/s41588-022-01232-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sikorav JL, and Jannink G (1994). Kinetics of chromosome condensation in the presence of topoisomerases: a phantom chain model. Biophys J. 66, 827–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Grosberg AY, Nechaev SK, and Shakhnovich EI (1988). The role of topological constraints in the kinetics of collapse of macromolecules. J. Phys. France 49, 2095–2100. [Google Scholar]
- 80.Vologodskii AV, Levene SD, Klenin KV, Frank-Kamenetskii M, and Cozzarelli NR (1992). Conformational and thermodynamic properties of supercoiled DNA. J Mol Biol 227, 1224–1243. 10.1016/0022-2836(92)90533-p. [DOI] [PubMed] [Google Scholar]
- 81.Goychuk A, Kannan D, Chakraborty AK, and Kardar M (2023). Polymer folding through active processes recreates features of genome organization. Proc Natl Acad Sci U S A 120, e2221726120. 10.1073/pnas.2221726120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chan B, and Rubinstein M (2023). Theory of chromatin organization maintained by active loop extrusion. Proc Natl Acad Sci U S A 120, e2222078120. 10.1073/pnas.2222078120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chan B, and Rubinstein M (2024). Activity-driven chromatin organization during interphase: Compaction, segregation, and entanglement suppression. Proc Natl Acad Sci U S A 121, e2401494121. 10.1073/pnas.2401494121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Polovnikov KE, Slavov B, Belan S, Imakaev M, Brandao HB, and Mirny LA (2023). Crumpled polymer with loops recapitulates key features of chromosome organization. Phys Rev X 13. 10.1103/physrevx.13.041029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Keizer VIP, Grosse-Holz S, Woringer M, Zambon L, Aizel K, Bongaerts M, Delille F, Kolar-Znika L, Scolari VF, Hoffmann S, et al. (2022). Live-cell micromanipulation of a genomic locus reveals interphase chromatin mechanics. Science 377, 489–495. 10.1126/science.abi9810. [DOI] [PubMed] [Google Scholar]
- 86.Denker A, and de Laat W (2016). The second decade of 3C technologies: detailed insights into nuclear organization. Genes Dev 30, 1357–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Goel VY, and Hansen AS (2021). The macro and micro of chromosome conformation capture. Wiley Interdiscip Rev Dev Biol 10, e395. 10.1002/wdev.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jerkovic I, and Cavalli G (2021). Understanding 3D genome organization by multidisciplinary methods. Nat Rev Mol Cell Biol 22, 511–528. 10.1038/s41580-021-00362-w. [DOI] [PubMed] [Google Scholar]
- 89.Bouwman BAM, Crosetto N, and Bienko M (2022). The era of 3D and spatial genomics. Trends Genet 38, 1062–1075. 10.1016/j.tig.2022.05.010. [DOI] [PubMed] [Google Scholar]
- 90.Dekker J, Alber F, Aufmkolk S, Beliveau BJ, Bruneau BG, Belmont AS, Bintu L, Boettiger A, Calandrelli R, Disteche CM, et al. (2023). Spatial and temporal organization of the genome: Current state and future aims of the 4D nucleome project. Mol Cell 83, 2624–2640. 10.1016/j.molcel.2023.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Heitz E (1928). Das Heterochromatin der Moose. I. Jahrb Wiss Bot 69, 762–818. [Google Scholar]
- 92.Croft JA, Bridger JM, Boyle S, Perry P, Teague P, and Bickmore WA (1999). Differences in the localization and morphology of chromosomes in the human nucleus. J. Cell Biol 145, 1119–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Boyle S, Gilchrist S, Bridger JM, Mahy NL, Ellis JA, and Bickmore WA (2001). The spatial organization of human chromosomes within the nuclei of normal and emerinmutant cells. Hum. Mol. Genet 10, 211–219. [DOI] [PubMed] [Google Scholar]
- 94.Cremer T, and Cremer C (2001). Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat. Rev. Genet 2, 292–301. [DOI] [PubMed] [Google Scholar]
- 95.Dimitrova DS, and Gilbert DM (1999). The spatial position and replication timing of chromosomal domains are both established in early G1 phase. Mol Cell 4, 983–993. 10.1016/s1097-2765(00)80227-0. [DOI] [PubMed] [Google Scholar]
- 96.van Steensel B, and Belmont AS (2017). Lamina-Associated Domains: Links with Chromosome Architecture, Heterochromatin, and Gene Repression. Cell 169, 780–791. 10.1016/j.cell.2017.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, de Klein A, Wessels L, de Laat W, and van Steensel B (2008). Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 453, 948–951. [DOI] [PubMed] [Google Scholar]
- 98.Harris HL, Gu H, Olshansky M, Wang A, Farabella I, Eliaz Y, Kalluchi A, Krishna A, Jacobs M, Cauer G, et al. (2023). Chromatin alternates between A and B compartments at kilobase scale for subgenic organization. Nat Commun 14, 3303. 10.1038/s41467-023-38429-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, and Lieberman Aiden E (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Spracklin G, Abdennur N, Imakaev M, Chowdhury N, Pradhan S, Mirny LA, and Dekker J (2023). Diverse silent chromatin states modulate genome compartmentalization and loop extrusion barriers. Nat Struct Mol Biol 30, 38–51. 10.1038/s41594-022-00892-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hildebrand EM, and Dekker J (2020). Mechanisms and Functions of Chromosome Compartmentalization. Trends Biochem Sci 45, 385–396. 10.1016/j.tibs.2020.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Haddad N, Jost D, and Vaillant C (2017). Perspectives: using polymer modeling to understand the formation and function of nuclear compartments. Chromosome Res 25, 35–50. 10.1007/s10577-016-9548-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Falk M, Feodorova Y, Naumova N, Imakaev M, Lajoie BR, Leonhardt H, Joffe B, Dekker J, Fudenberg G, Solovei I, and Mirny LA (2019). Heterochromatin drives organization of conventional and inverted nuclei. Nature In 570, 395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Esposito A, Bianco S, Chiariello AM, Abraham A, Fiorillo L, Conte M, Campanile R, and Nicodemi M (2022). Polymer physics reveals a combinatorial code linking 3D chromatin architecture to 1D chromatin states. Cell Rep 38, 110601. 10.1016/j.celrep.2022.110601. [DOI] [PubMed] [Google Scholar]
- 105.Barbieri M, Chotalia M, Fraser J, Lavitas LM, Dostie J, Pombo A, and Nicodemi M (2012). Complexity of chromatin folding is captured by the strings and binders switch model. Proc Natl Acad Sci U S A 109, 16173–16178. 10.1073/pnas.1204799109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Takemata N, and Bell SD (2021). Multi-scale architecture of archaeal chromosomes. Mol Cell 81, 473–487 e476. 10.1016/j.molcel.2020.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang Y, Zhang Y, Zhang R, van Schaik T, Zhang L, Sasaki T, Peric-Hupkes D, Chen Y, Gilbert DM, van Steensel B, et al. (2021). SPIN reveals genome-wide landscape of nuclear compartmentalization. Genome Biol 22, 36. 10.1186/s13059-020-02253-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gibson BA, Doolittle LK, Schneider MWG, Jensen LE, Gamarra N, Henry L, Gerlich DW, Redding S, and Rosen MK (2019). Organization of Chromatin by Intrinsic and Regulated Phase Separation. Cell 179, 470–484 e421. 10.1016/j.cell.2019.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Strom AR, Emelyanov AV, Mir M, Fyodorov DV, Darzacq X, and Karpen GH (2017). Phase separation drives heterochromatin domain formation. Nature 547, 241–245. 10.1038/nature22989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Strom AR, Biggs RJ, Banigan EJ, Wang X, Chiu K, Herman C, Collado J, Yue F, Ritland Politz JC, Tait LJ, et al. (2021). HP1alpha is a chromatin crosslinker that controls nuclear and mitotic chromosome mechanics. Elife 10. 10.7554/eLife.63972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Larson AG, Elnatan D, Keenen MM, Trnka MJ, Johnston JB, Burlingame AL, Agard DA, Redding S, and Narlikar GJ (2017). Liquid droplet formation by HP1alpha suggests a role for phase separation in heterochromatin. Nature 547, 236–240. 10.1038/nature22822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sanulli S, Trnka MJ, Dharmarajan V, Tibble RW, Pascal BD, Burlingame AL, Griffin PR, Gross JD, and Narlikar GJ (2019). HP1 reshapes nucleosome core to promote phase separation of heterochromatin. Nature 575, 390–394. 10.1038/s41586-019-1669-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Keenen MM, Brown D, Brennan LD, Renger R, Khoo H, Carlson CR, Huang B, Grill SW, Narlikar GJ, and Redding S (2021). HP1 proteins compact DNA into mechanically and positionally stable phase separated domains. Elife 10. 10.7554/eLife.64563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhao H, Lin Y, Lin E, Liu F, Shu L, Jing D, Wang B, Wang M, Shan F, Zhang L, et al. (2024). Genome folding principles uncovered in condensin-depleted mitotic chromosomes. Nat Genet 56, 1213–1224. 10.1038/s41588-024-01759-x. [DOI] [PubMed] [Google Scholar]
- 115.Akilli N, Cheutin T, and Cavalli G (2024). Phase separation and inheritance of repressive chromatin domains. Curr Opin Genet Dev 86, 102201. 10.1016/j.gde.2024.102201. [DOI] [PubMed] [Google Scholar]
- 116.Schuettengruber B, Bourbon HM, Di Croce L, and Cavalli G (2017). Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell 171, 34–57. 10.1016/j.cell.2017.08.002. [DOI] [PubMed] [Google Scholar]
- 117.Girelli G, Custodio J, Kallas T, Agostini F, Wernersson E, Spanjaard B, Mota A, Kolbeinsdottir S, Gelali E, Crosetto N, and Bienko M (2020). GPSeq reveals the radial organization of chromatin in the cell nucleus. Nat Biotechnol 38, 1184–1193. 10.1038/s41587-020-0519-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bantignies F, Roure V, Comet I, Leblanc B, Schuettengruber B, Bonnet J, Tixier V, Mas A, and Cavalli G (2011). Polycomb-dependent regulatory contacts between distant Hox loci in Drosophila. Cell 144, 214–226. [DOI] [PubMed] [Google Scholar]
- 119.Schoenfelder S, Sugar R, Dimond A, Javierre BM, Armstrong H, Mifsud B, Dimitrova E, Matheson L, Tavares-Cadete F, Furlan-Magaril M, et al. (2015). Polycomb repressive complex PRC1 spatially constrains the mouse embryonic stem cell genome. Nat Genet 47, 1179–1186. 10.1038/ng.3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bonev B, Mendelson Cohen N, Szabo Q, Fritsch L, Papadopoulos GL, Lubling Y, Xu X, Lv X, Hugnot JP, Tanay A, and Cavalli G (2017). Multiscale 3D Genome Rewiring during Mouse Neural Development. Cell 171, 557–572 e524. 10.1016/j.cell.2017.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jost D, Carrivain P, Cavalli G, and Vaillant C (2014). Modeling epigenome folding: formation and dynamics of topologically associated chromatin domains. Nucleic Acids Res 42, 9553–9561. 10.1093/nar/gku698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Solovei I, Kreysing M, Lanctôt C, Kösem S, Peichl L, Cremer T, Guck J, and Joffe B (2009). Nuclear architecture of rod photoreceptor cells adapts to vision in mammalian evolution. Cell. 137, 356–368. [DOI] [PubMed] [Google Scholar]
- 123.Belaghzal H, Borrman T, Stephens AD, Lafontaine DL, Venev SV, Weng Z, Marko JF, and Dekker J (2019). Compartment-dependent chromatin interaction dynamics revealed by liquid chromatin Hi-C. BioRxiv doi: 10.1101/704957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Krietenstein N, Abraham S, Venev SV, Abdennur N, Gibcus JH, Hsieh T-H, Parsi KM, Yang L, Maehr R, Mirny LA, et al. (2020). Ultrastructural details of mammalian chromosome architecture. Mol. Cell in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hsieh TS, Cattoglio C, Slobodyanyuk E, Hansen AS, Darzacq X, and Tjian R (2022). Enhancer-promoter interactions and transcription are largely maintained upon acute loss of CTCF, cohesin, WAPL or YY1. Nat Genet 54, 1919–1932. 10.1038/s41588-022-01223-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Friman ET, Flyamer IM, Marenduzzo D, Boyle S, and Bickmore WA (2023). Ultra-long-range interactions between active regulatory elements. Genome Res 33, 1269–1283. 10.1101/gr.277567.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Goel VY, Huseyin MK, and Hansen AS (2023). Region Capture Micro-C reveals coalescence of enhancers and promoters into nested microcompartments. Nat Genet 55, 1048–1056. 10.1038/s41588-023-01391-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hua P, Badat M, Hanssen LLP, Hentges LD, Crump N, Downes DJ, Jeziorska DM, Oudelaar AM, Schwessinger R, Taylor S, et al. (2021). Defining genome architecture at base-pair resolution. Nature 595, 125–129. 10.1038/s41586-021-03639-4. [DOI] [PubMed] [Google Scholar]
- 129.Rosencrance CD, Ammouri HN, Yu Q, Ge T, Rendleman EJ, Marshall SA, and Eagen KP (2020). Chromatin Hyperacetylation Impacts Chromosome Folding by Forming a Nuclear Subcompartment. Mol Cell 78, 112–126 e112. 10.1016/j.molcel.2020.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Schooley A, Venev SV, Aksenova V, Navarrete E, Dasso M, and Dekker J (2024). Interphase chromosome conformation is specified by distinct folding programs inherited via mitotic chromosomes or through the cytoplasm. BioRxiv doi: 10.1101/2024.09.16.613305. [DOI] [Google Scholar]
- 131.Goel VY, Aboreden NG, Jusuf JM, Zhang H, Mori LP, Mirny LA, Blobel GA, Banigan EJ, and Hansen AS (2024). Dynamics of microcompartment formation at the mitosis-to-G1 transition. BioRxiv doi: 10.1101/2024.09.16.611917. [DOI] [Google Scholar]
- 132.Paulson JR, and Laemmli UK (1977). The structure of histone-depleted metaphase chromosomes. Cell. 12, 817–828. [DOI] [PubMed] [Google Scholar]
- 133.Earnshaw WC, and Laemmli UK (1983). Architecture of metaphase chromosomes and chromosome scaffolds. J Cell Biol 96, 84–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cook PR, and Brazell IA (1975). Supercoils in human DNA. J Cell Sci 19, 261–279. 10.1242/jcs.19.2.261. [DOI] [PubMed] [Google Scholar]
- 135.Nelson WG, Pienta KJ, Barrack ER, and Coffey DS (1986). The role of the nuclear matrix in the organization and function of DNA. Annu Rev Biophys Biophys Chem 15, 457–475. 10.1146/annurev.bb.15.060186.002325. [DOI] [PubMed] [Google Scholar]
- 136.Ostashevsky J (1998). A polymer model for the structural organization of chromatin loops and minibands in interphase chromosomes. Mol. Biol. Cell. 9, 3031–3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wood C, and Tonegawa S (1983). Diversity and joining segments of mouse immunoglobulin heavy chain genes are closely linked and in the same orientation: implications for the joining mechanism. Proc Natl Acad Sci U S A 80, 3030–3034. 10.1073/pnas.80.10.3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Riggs AD (1990). DNA methylation and late replication probably aid cell memory, and type I DNA reeling could aid chromosome folding and enhancer function. Philos Trans R Soc Lond B Biol Sci 326, 285–297. [DOI] [PubMed] [Google Scholar]
- 139.Blackwood EM, and Kadonaga JT (1998). Going the Distance: A Current View of Enhancer Action. Science 281, 61–63. [DOI] [PubMed] [Google Scholar]
- 140.Kimura K, Rybenkov VV, Crisona NJ, Hirano T, and Cozzarelli NR (1999). 13S condensin actively reconfigures DNA by introducing global positive writhe: implications for chromosome condensation. Cell 98, 239–248. [DOI] [PubMed] [Google Scholar]
- 141.Nasmyth K (2001). Disseminating the genome: joining, resolving, and separating sister chromatids during mitosis and meiosis. Annu Rev Genet 35, 673–745. [DOI] [PubMed] [Google Scholar]
- 142.Kagey MH, Newman JJ, Bilodeau S, Zhan Y, Orlando DA, van Berkum NL, Ebmeier CC, Goossens J, Rahl PB, Levine SS, et al. (2010). Mediator and cohesin connect gene expression and chromatin architecture. Nature 467, 430–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Merkenschlager M (2010). Cohesin: a global player in chromosome biology with local ties to gene regulation. Curr Opin Genet Dev 20, 555–561. [DOI] [PubMed] [Google Scholar]
- 144.Hirano T (2002). The ABCs of SMC proteins: two-armed ATPases for chromosome condensation, cohesion, and repair. Genes Dev 16, 399–414. [DOI] [PubMed] [Google Scholar]
- 145.Strick TR (2004). Real-time detection of single-molecule DNA compaction by condensin I. Curr Biol 14, 874–880. [DOI] [PubMed] [Google Scholar]
- 146.Alipour E, and Marko JF (2012). Self-organization of domain structures by DNA-loop-extruding enzymes. Nucleic Acids Res 40, 11202–11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Goloborodko A, Imakaev MV, Marko JF, and Mirny L (2016). Compaction and segregation of sister chromatids via active loop extrusion. Elife 5. 10.7554/eLife.14864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Fudenberg G, Imakaev M, Lu C, Goloborodko A, Abdennur N, and Mirny LA (2016). Formation of chromosomal domains by loop extrusion. Cell Rep 15, 2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sanborn AL, Rao SS, Huang SC, Durand NC, Huntley MH, Jewett AI, Bochkov ID, Chinnappan D, Cutkosky A, Li J, et al. (2015). Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc Natl Acad Sci U S A 112, E6456–6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, et al. (2012). Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485, 381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, and Ren B (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Nora EP, Goloborodko A, Valton AL, Gibcus JH, Uebersohn A, Abdennur N, Dekker J, Mirny LA, and Bruneau BG (2017). Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell 169, 930–944 e922. 10.1016/j.cell.2017.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Rao SSP, Huang SC, Glenn St Hilaire B, Engreitz JM, Perez EM, Kieffer-Kwon KR, Sanborn AL, Johnstone SE, Bascom GD, Bochkov ID, et al. (2017). Cohesin Loss Eliminates All Loop Domains. Cell 171, 305–320 e324. 10.1016/j.cell.2017.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Fudenberg G, Abdennur N, Imakaev M, Goloborodko A, and Mirny LA (2017). Emerging Evidence of Chromosome Folding by Loop Extrusion. Cold Spring Harb Symp Quant Biol 82, 45–55. 10.1101/sqb.2017.82.034710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Davidson IF, and Peters JM (2021). Genome folding through loop extrusion by SMC complexes. Nat Rev Mol Cell Biol 22, 445–464. 10.1038/s41580-021-00349-7. [DOI] [PubMed] [Google Scholar]
- 156.Hoencamp C, and Rowland BD (2023). Genome control by SMC complexes. Nat Rev Mol Cell Biol 24, 633–650. 10.1038/s41580-023-00609-8. [DOI] [PubMed] [Google Scholar]
- 157.Goloborodko A, Marko JF, and Mirny LA (2016). Chromosome Compaction by Active Loop Extrusion. Biophys J 110, 2162–2168. 10.1016/j.bpj.2016.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hirano T, Kobayashi R, and Hirano M (1997). Condensins, chromosome condensation protein complexes containing XCAP-C, XCAP-E and a Xenopus homolog of the Drosophila Barren protein. Cell 89, 511–521. [DOI] [PubMed] [Google Scholar]
- 159.Hirano T, and Mitchison TJ (1994). A heterodimeric coiled-coil protein required for mitotic chromosome condensation in vitro. Cell 79, 449–458. [DOI] [PubMed] [Google Scholar]
- 160.Saitoh N, Goldberg IG, Wood ER, and Earnshaw WC (1994). ScII: an abundant chromosome scaffold protein is a member of a family of putative ATPases with an unusual predicted tertiary structure. J Cell Biol 127, 303–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ganji M, Shaltiel IA, Bisht S, Kim E, Kalichava A, Haering CH, and Dekker C (2018). Real-time imaging of DNA loop extrusion by condensin. Science 360, 102–105. 10.1126/science.aar7831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Davidson IF, Bauer B, Goetz D, Tang W, Wutz G, and Peters JM (2019). DNA loop extrusion by human cohesin. Science 366, 1338–1345. 10.1126/science.aaz3418. [DOI] [PubMed] [Google Scholar]
- 163.Golfier S, Quail T, Kimura H, and Brugues J (2020). Cohesin and condensin extrude DNA loops in a cell cycle-dependent manner. Elife 9. 10.7554/eLife.53885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kong M, Cutts EE, Pan D, Beuron F, Kaliyappan T, Xue C, Morris EP, Musacchio A, Vannini A, and Greene EC (2020). Human Condensin I and II Drive Extensive ATP-Dependent Compaction of Nucleosome-Bound DNA. Mol Cell 79, 99–114 e119. 10.1016/j.molcel.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Jeppsson K, Pradhan B, Sutani T, Sakata T, Umeda Igarashi M, Berta DG, Kanno T, Nakato R, Shirahige K, Kim E, and Bjorkegren C (2024). Loop-extruding Smc5/6 organizes transcription-induced positive DNA supercoils. Mol Cell 84, 867–882 e865. 10.1016/j.molcel.2024.01.005. [DOI] [PubMed] [Google Scholar]
- 166.Barth R, Davidson IF, van der Torre J, Taschner M, Gruber S, Peters J-M, and Dekker C (2023). SMC motor proteins extrude DNA asymmetrically and contain a direction switch. BioRxiv 10.1101/2023.12.21.572892. [DOI] [Google Scholar]
- 167.Banigan EJ, Tang W, van den Berg AA, Stocsits RR, Wutz G, Brandao HB, Busslinger GA, Peters JM, and Mirny LA (2023). Transcription shapes 3D chromatin organization by interacting with loop extrusion. Proc Natl Acad Sci U S A 120, e2210480120. 10.1073/pnas.2210480120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Li Y, Haarhuis JHI, Sedeno Cacciatore A, Oldenkamp R, van Ruiten MS, Willems L, Teunissen H, Muir KW, de Wit E, Rowland BD, and Panne D (2020). The structural basis for cohesin-CTCF-anchored loops. Nature 578, 472–476. 10.1038/s41586-019-1910-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Davidson IF, Barth R, Zaczek M, van der Torre J, Tang W, Nagasaka K, Janissen R, Kerssemakers J, Wutz G, Dekker C, and Peters JM (2023). CTCF is a DNA-tension-dependent barrier to cohesin-mediated loop extrusion. Nature 616, 822–827. 10.1038/s41586-023-05961-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Kim E, Kerssemakers J, Shaltiel IA, Haering CH, and Dekker C (2020). DNA-loop extruding condensin complexes can traverse one another. Nature 579, 438–442. 10.1038/s41586-020-2067-5. [DOI] [PubMed] [Google Scholar]
- 171.Pradhan B, Barth R, Kim E, Davidson IF, Bauer B, van Laar T, Yang W, Ryu JK, van der Torre J, Peters JM, and Dekker C (2022). SMC complexes can traverse physical roadblocks bigger than their ring size. Cell Rep 41, 111491. 10.1016/j.celrep.2022.111491. [DOI] [PubMed] [Google Scholar]
- 172.Dekker C, Haering CH, Peters JM, and Rowland BD (2023). How do molecular motors fold the genome? Science 382, 646–648. 10.1126/science.adi8308. [DOI] [PubMed] [Google Scholar]
- 173.Nomidis SK, Carlon E, Gruber S, and Marko JF (2022). DNA tension-modulated translocation and loop extrusion by SMC complexes revealed by molecular dynamics simulations. Nucleic Acids Res 50, 4974–4987. 10.1093/nar/gkac268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wang X, Le TBK, Lajoie BR, Dekker J, Laub MT, and Rudner DZ (2015). Condensin promotes the juxtaposition of DNA flanking its loading site in Bacillus subtilis. Genes Dev 29, 1661–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Le TB, Imakaev MV, Mirny LA, and Laub MT (2013). High-resolution mapping of the spatial organization of a bacterial chromosome. Science 342, 731–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Umbarger MA, Toro E, Wright MA, Porreca GJ, Baù D, Hong S, Fero MJ, Zhu LJ, Marti-Renom MA, McAdams HH, et al. (2011). The three-dimensional architecture of a bacterial genome and its alteration by genetic perturbation. Mol. Cell 44, 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Sullivan NL, Marquis KA, and Rudner DZ (2009). Recruitment of SMC by ParB-parS organizes the origin region and promotes efficient chromosome segregation. Cell 137, 697–707. 10.1016/j.cell.2009.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Wang X, Hughes AC, Brandao HB, Walker B, Lierz C, Cochran JC, Oakley MG, Kruse AC, and Rudner DZ (2018). In Vivo Evidence for ATPase-Dependent DNA Translocation by the Bacillus subtilis SMC Condensin Complex. Mol Cell 71, 841–847 e845. 10.1016/j.molcel.2018.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Dequeker BJH, Scherr MJ, Brandao HB, Gassler J, Powell S, Gaspar I, Flyamer IM, Lalic A, Tang W, Stocsits R, et al. (2022). MCM complexes are barriers that restrict cohesin-mediated loop extrusion. Nature 606, 197–203. 10.1038/s41586-022-04730-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Chang LH, and Noordermeer D (2024). Permeable TAD boundaries and their impact on genome-associated functions. Bioessays, e2400137. 10.1002/bies.202400137. [DOI] [PubMed] [Google Scholar]
- 181.Liu NQ, Maresca M, van den Brand T, Braccioli L, Schijns M, Teunissen H, Bruneau BG, Nora EP, and de Wit E (2021). WAPL maintains a cohesin loading cycle to preserve cell-type-specific distal gene regulation. Nat Genet 53, 100–109. 10.1038/s41588-020-00744-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Valton AL, Venev SV, Mair B, Khokhar ES, Tong AHY, Usaj M, Chan K, Pai AA, Moffat J, and Dekker J (2022). A cohesin traffic pattern genetically linked to gene regulation. Nat Struct Mol Biol 29, 1239–1251. 10.1038/s41594-022-00890-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Bastie N, Chapard C, Cournac A, Nejmi S, Mboumba H, Gadal O, Thierry A, Beckouet F, and Koszul R (2024). Sister chromatid cohesion halts DNA loop expansion. Mol Cell 84, 1139–1148 e1135. 10.1016/j.molcel.2024.02.004. [DOI] [PubMed] [Google Scholar]
- 184.Brandao HB, Ren Z, Karaboja X, Mirny LA, and Wang X (2021). DNA-loop-extruding SMC complexes can traverse one another in vivo. Nat Struct Mol Biol 28, 642–651. 10.1038/s41594-021-00626-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Rinzema NJ, Sofiadis K, Tjalsma SJD, Verstegen M, Oz Y, Valdes-Quezada C, Felder AK, Filipovska T, van der Elst S, de Andrade Dos Ramos Z, et al. (2022). Building regulatory landscapes reveals that an enhancer can recruit cohesin to create contact domains, engage CTCF sites and activate distant genes. Nat Struct Mol Biol 29, 563–574. 10.1038/s41594-022-00787-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Gruber S, and Errington J (2009). Recruitment of condensin to replication origin regions by ParB/SpoOJ promotes chromosome segregation in B. subtilis. Cell 137, 685–696. 10.1016/j.cell.2009.02.035. [DOI] [PubMed] [Google Scholar]
- 187.Galitsyna A, Ulianov SV, Bykov NS, Veil M, Gao M, Perevoschikova K, Gelfand M, Razin SV, Mirny L, and Onichtchouk D (2023). Extrusion fountains are hallmarks of chromosome organization emerging upon zygotic genome activation. BioRxiv doi: 10.1101/2023.07.15.549120. [DOI] [Google Scholar]
- 188.Gil J, Rosin LF, Navarrete E, Chowdhury N, Abraham S, Cornilleau G, Lei EP, Mozziconacci J, Mirny LA, Muller H, and Drinnenberg IA (2024). Unique territorial and compartmental organization of chromosomes in the holocentric silkmoth. bioRxiv. 10.1101/2023.09.14.557757. [DOI] [Google Scholar]
- 189.Busslinger GA, Stocsits RR, van der Lelij P, Axelsson E, Tedeschi A, Galjart N, and Peters JM (2017). Cohesin is positioned in mammalian genomes by transcription, CTCF and Wapl. Nature 544, 503–507. 10.1038/nature22063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Natale F, Rapp A, Yu W, Maiser A, Harz H, Scholl A, Grulich S, Anton T, Horl D, Chen W, et al. (2017). Identification of the elementary structural units of the DNA damage response. Nat Commun 8, 15760. 10.1038/ncomms15760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Arnould C, Rocher V, Finoux AL, Clouaire T, Li K, Zhou F, Caron P, Mangeot PE, Ricci EP, Mourad R, et al. (2021). Loop extrusion as a mechanism for formation of DNA damage repair foci. Nature 590, 660–665. 10.1038/s41586-021-03193-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Amendola M, and van Steensel B (2015). Nuclear lamins are not required for lamina-associated domain organization in mouse embryonic stem cells. EMBO Rep 16, 610–617. 10.15252/embr.201439789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Manzo SG, Dauban L, and van Steensel B (2022). Lamina-associated domains: Tethers and looseners. Curr Opin Cell Biol 74, 80–87. 10.1016/j.ceb.2022.01.004. [DOI] [PubMed] [Google Scholar]
- 194.Solovei I, Wang AS, Thanisch K, Schmidt CS, Krebs S, Zwerger M, Cohen TV, Devys D, Foisner R, Peichl L, et al. (2013). LBR and lamin A/C sequentially tether peripheral heterochromatin and inversely regulate differentiation. Cell 152, 584–598. 10.1016/j.cell.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 195.Yu R, Roseman S, Siegenfeld AP, Nguyen SC, Joyce EF, Liau BB, Krantz ID, Alexander KA, and Berger SL (2023). CTCF/cohesin organize the ground state of chromatin-nuclear speckle association. bioRxiv. 10.1101/2023.07.22.550178. [DOI] [Google Scholar]
- 196.Blobel G (1985). Gene gating: a hypothesis. Proc Natl Acad Sci U S A 82, 8527–8529. 10.1073/pnas.82.24.8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Toro E, Hong SH, McAdams HH, and Shapiro L (2008). Caulobacter requires a dedicated mechanism to initiate chromosome segregation. Proc Natl Acad Sci U S A 105, 15435–15440. 10.1073/pnas.0807448105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Trelles-Sticken E, Loidl J, and Scherthan H (1999). Bouquet formation in budding yeast: initiation of recombination is not required for meiotic telomere clustering. J Cell Sci 112, 651–658. [DOI] [PubMed] [Google Scholar]
- 199.Wasserman SA, and Cozzarelli NR (1986). Biochemical topology: applications to DNA recombination and replication. Science 232, 951–960. 10.1126/science.3010458. [DOI] [PubMed] [Google Scholar]
- 200.Wood ER, and Earnshaw WC (1990). Mitotic chromatin condensation in vitro using somatic cell extracts and nuclei with variable levels of endogenous topoisomerase II. J Cell Biol 111, 2839–2850. 10.1083/jcb.111.6.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.de Gennes P-G (1979). Scaling theory of polymer physics (Cornell University Press; ). [Google Scholar]
- 202.Grosberg AY, Rabin Y, Havlin S, and Neer A (1993). Crumpled globule model of the three-dimensional structure of DNA. Europhys. Lett 23, 373–378. [Google Scholar]
- 203.Rosa A, and Everaers R (2008). Structure and dynamics of interphase chromosomes. PLoS Comput Biol 4, e1000153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Boettiger AN, Bintu B, Moffitt JR, Wang S, Beliveau BJ, Fudenberg G, Imakaev M, Mirny LA, Wu CT, and Zhuang X (2016). Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature 529, 418–422. 10.1038/nature16496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Halverson JD, Smrek J, Kremer K, and Grosberg AY (2014). From a melt of rings to chromosome territories: the role of topological constraints in genome folding. Rep Prog Phys 77, 022601. [DOI] [PubMed] [Google Scholar]
- 206.Tavares-Cadete F, Norouzi D, Dekker B, Liu Y, and Dekker J (2020). Multi-contact 3C data reveal that the human genome is largely unentangled. BioRxiv doi: 10.1101/2020.03.03.975425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Hildebrand EM, Polovnikov K, Dekker B, Liu Y, Lafontaine DL, Fox AN, Li Y, Venev SV, Mirny LA, and Dekker J (2024). Mitotic chromosomes are self-entangled and disentangle through a topoisomerase-II-dependent two-stage exit from mitosis. Mol Cell 84, 1422–1441 e1414. 10.1016/j.molcel.2024.02.025. [DOI] [PubMed] [Google Scholar]
- 208.Orlandini E, Marenduzzo D, and Michieletto D (2019). Synergy of topoisomerase and structural-maintenance-of-chromosomes proteins creates a universal pathway to simplify genome topology. Proc Natl Acad Sci U S A 116, 8149–8154. 10.1073/pnas.1815394116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Hirano T, and Mitchison TJ (1991). Cell cycle control of higher-order chromatin assembly around naked DNA in vitro. J Cell Biol 115, 1479–1489. 10.1083/jcb.115.6.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Shintomi K, and Hirano T (2021). Guiding functions of the C-terminal domain of topoisomerase IIalpha advance mitotic chromosome assembly. Nat Commun 12, 2917. 10.1038/s41467-021-23205-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Kawamura R, Pope LH, Christensen MO, Sun M, Terekhova K, Boege F, Mielke C, Andersen AH, and Marko JF (2010). Mitotic chromosomes are constrained by topoisomerase II-sensitive DNA entanglements. J Cell Biol 188, 653–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Su JH, Zheng P, Kinrot SS, Bintu B, and Zhuang X (2020). Genome-Scale Imaging of the 3D Organization and Transcriptional Activity of Chromatin. Cell 182, 1641–1659 e1626. 10.1016/j.cell.2020.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Schwarzer W, Abdennur N, Goloborodko A, Pekowska A, Fudenberg G, Loe-Mie Y, Fonseca NA, Huber W, C HH, Mirny L, and Spitz F (2017). Two independent modes of chromatin organization revealed by cohesin removal. Nature 551, 51–56. 10.1038/nature24281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Tjong H, Gong K, Chen L, and Alber F (2012). Physical tethering and volume exclusion determine higher-order genome organization in budding yeast. Genome Res 22, 1295–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Schalbetter SA, Goloborodko A, Fudenberg G, Belton JM, Miles C, Yu M, Dekker J, Mirny L, and Baxter J (2017). SMC complexes differentially compact mitotic chromosomes according to genomic context. Nat Cell Biol 19, 1071–1080. 10.1038/ncb3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Wong H, Marie-Nelly H, Herbert S, Carrivain P, Blanc H, Koszul R, Fabre E, and Zimmer C (2012). A predictive computational model of the dynamic 3D interphase yeast nucleus. Curr Biol 22, 1881–1890. [DOI] [PubMed] [Google Scholar]
- 217.Tortora MM, Salari H, and Jost D (2020). Chromosome dynamics during interphase: a biophysical perspective. Curr Opin Genet Dev 61, 37–43. 10.1016/j.gde.2020.03.001. [DOI] [PubMed] [Google Scholar]
- 218.Grosse-Holz S (2023). The searchable chromosome. Trends Genet 39, 895–896. 10.1016/j.tig.2023.08.006. [DOI] [PubMed] [Google Scholar]
- 219.Strickfaden H, Zunhammer A, van Koningsbruggen S, Köhler D, and Cremer T (2010). 4D chromatin dynamics in cycling cells: Theodor Boveri's hypotheses revisited. Nucleus 1, 284–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Weber SC, Spakowitz AJ, and Theriot JA (2010). Bacterial chromosomal loci move subdiffusively through a viscoelastic cytoplasm. Phys Rev Lett 104, 238102. 10.1103/PhysRevLett.104.238102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Hajjoul H, Mathon J, Ranchon H, Goiffon I, Mozziconacci J, Albert B, Carrivain P, Victor JM, Gadal O, Bystricky K, and Bancaud A (2013). High-throughput chromatin motion tracking in living yeast reveals the flexibility of the fiber throughout the genome. Genome Res 23, 1829–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Khanna N, Zhang Y, Lucas JS, Dudko OK, and Murre C (2019). Chromosome dynamics near the sol-gel phase transition dictate the timing of remote genomic interactions. Nat Commun 10, 2771. 10.1038/s41467-019-10628-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Bruckner DB, Chen H, Barinov L, Zoller B, and Gregor T (2023). Stochastic motion and transcriptional dynamics of pairs of distal DNA loci on a compacted chromosome. Science 380, 1357–1362. 10.1126/science.adf5568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Mine-Hattab J, Recamier V, Izeddin I, Rothstein R, and Darzacq X (2017). Multi-scale tracking reveals scale-dependent chromatin dynamics after DNA damage. Mol Biol Cell 28, 3323–3332. 10.1091/mbc.E17-05-0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Bronshtein I, Kepten E, Kanter I, Berezin S, Lindner M, Redwood AB, Mai S, Gonzalo S, Foisner R, Shav-Tal Y, and Garini Y (2015). Loss of lamin A function increases chromatin dynamics in the nuclear interior. Nat Commun 6, 8044. 10.1038/ncomms9044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Tamm MV, and Polovnikov K (2018). Chapter 3: Dynamics of Polymers: Classic Results and Recent Developments. In Order, Disorder and Criticality - Advanced Problems of Phase Transition Theory Volume 5, Holovatch Y, ed. (National Academy of Sciences, Ukraine: ), pp. 113–172. [Google Scholar]
- 227.Lucas JS, Zhang Y, Dudko OK, and Murre C (2014). 3D trajectories adopted by coding and regulatory DNA elements: first-passage times for genomic interactions. Cell 158, 339–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Kleckner N, Zickler D, Jones GH, Dekker J, Padmore R, Henle J, and Hutchinson J (2004). A mechanical basis for chromosome function. Proc Natl Acad Sci U S A 101, 12592–15297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Marko JF, and Siggia ED (1997). Polymer models of meiotic and mitotic chromosomes. Mol Biol Cell. 8, 2217–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Marko JF (2008). Micromechanical studies of mitotic chromosomes. Chromosome Res 16, 469–497. 10.1007/s10577-008-1233-7. [DOI] [PubMed] [Google Scholar]
- 231.Stephens AD, Banigan EJ, Adam SA, Goldman RD, and Marko JF (2017). Chromatin and lamin A determine two different mechanical response regimes of the cell nucleus. Mol Biol Cell 28, 1984–1996. 10.1091/mbc.E16-09-0653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Stephens AD, Liu PZ, Banigan EJ, Almassalha LM, Backman V, Adam SA, Goldman RD, and Marko JF (2018). Chromatin histone modifications and rigidity affect nuclear morphology independent of lamins. Mol Biol Cell 29, 220–233. 10.1091/mbc.E17-06-0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Poirier MG, and Marko JF (2002). Micromechanical studies of mitotic chromosomes. J Muscle Res Cell M 23, 409–431. [PubMed] [Google Scholar]
- 234.Biggs R, Liu PZ, Stephens AD, and Marko JF (2019). Effects of altering histone posttranslational modifications on mitotic chromosome structure and mechanics. Mol Biol Cell 30, 820–827. 10.1091/mbc.E18-09-0592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Rowley MJ, Nichols MH, Lyu X, Ando-Kuri M, Rivera ISM, Hermetz K, Wang P, Ruan Y, and Corces VG (2017). Evolutionarily Conserved Principles Predict 3D Chromatin Organization. Mol Cell 67, 837–852 e837. 10.1016/j.molcel.2017.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Mirny L, and Dekker J (2022). Mechanisms of Chromosome Folding and Nuclear Organization: Their Interplay and Open Questions. Cold Spring Harb Perspect Biol 14. 10.1101/cshperspect.a040147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Nuebler J, Fudenberg G, Imakaev M, Abdennur N, and Mirny LA (2018). Chromatin organization by an interplay of loop extrusion and compartmental segregation. Proc Natl Acad Sci U S A 115, E6697–E6706. 10.1073/pnas.1717730115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.. Haarhuis JHI, van der Weide RH, Blomen VA, Yanez-Cuna JO, Amendola M, van Ruiten MS, Krijger PHL, Teunissen H, Medema RH, van Steensel B, et al. (2017). The Cohesin Release Factor WAPL Restricts Chromatin Loop Extension. Cell 169, 693–707 e614. 10.1016/j.cell.2017.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Tan L, Xing D, Daley N, and Xie XS (2019). Three-dimensional genome structures of single sensory neurons in mouse visual and olfactory systems. Nat Struct Mol Biol 26, 297–307. 10.1038/s41594-019-0205-2. [DOI] [PubMed] [Google Scholar]
- 240.Ono T, Losada A, Hirano M, Myers MP, Neuwald AF, and Hirano T (2003). Differential contributions of condensin I and condensin II to mitotic chromosome architecture in vertebrate cells. Cell 115, 109–121. [DOI] [PubMed] [Google Scholar]
- 241.Chu L, Liang Z, Mukhina M, Fisher J, Vincenten N, Zhang Z, Hutchinson J, Zickler D, and Kleckner N (2020). The 3D Topography of Mitotic Chromosomes. Mol Cell 79, 902–916 e906. 10.1016/j.molcel.2020.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Schneider MWG, Gibson BA, Otsuka S, Spicer MFD, Petrovic M, Blaukopf C, Langer CCH, Batty P, Nagaraju T, Doolittle LK, et al. (2022). A mitotic chromatin phase transition prevents perforation by microtubules. Nature 609, 183–190. 10.1038/s41586-022-05027-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Gerlich D, Koch B, Dupeux F, Peters JM, and Ellenberg J (2006). Live-cell imaging reveals a stable cohesin-chromatin interaction after but not before DNA replication. Curr Biol 16, 1571–1578. [DOI] [PubMed] [Google Scholar]
- 244.Oomen ME, Fox AN, Gonzalez I, Molliex A, Papadopoulou T, Pablo Navarro P, and Dekker J (2023). Mitotic chromosomes harbor cell type and species-specific structural features within a universal looping architecture. BioRxiv doi: 10.1101/2023.12.08.570796. [DOI] [Google Scholar]
- 245.Das M, Semple JI, Haemmerli A, Volodkina V, Scotton J, Gitchev T, Annan A, Campos J, Statzer C, Dakhovnik A, et al. (2024). Condensin I folds the Caenorhabditis elegans genome. Nat Genet 10.1038/s41588-024-01832-5. [DOI] [PubMed] [Google Scholar]
- 246.Schalbetter SA, Fudenberg G, Baxter J, Pollard KS, and Neale MJ (2019). Principles of meiotic chromosome assembly revealed in S. cerevisiae. Nat Commun 10, 4795. 10.1038/s41467-019-12629-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Fischle W, Tseng BS, Dormann HL, Ueberheide BM, Garcia BA, Shabanowitz J, Hunt DF, Funabiki H, and Allis CD (2005). Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature 438, 1116–1122. 10.1038/nature04219. [DOI] [PubMed] [Google Scholar]
- 248.Banigan EJ, and Mirny LA (2019). Limits of Chromosome Compaction by Loop-Extruding Motors. Phys. Rev. X 9, 031007. [Google Scholar]
- 249.Zhou CY, Dekker B, Liu Z, Cabrera H, Ryan J, Dekker J, and Heald R (2023). Mitotic chromosomes scale to nuclear-cytoplasmic ratio and cell size in Xenopus. Elife 12. 10.7554/eLife.84360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Choppakatla P, Dekker B, Cutts EE, Vannini A, Dekker J, and Funabiki H (2021). Linker histone H1.8 inhibits chromatin binding of condensins and DNA topoisomerase II to tune chromosome length and individualization. Elife 10. 10.7554/eLife.68918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Alavattam KG, Maezawa S, Sakashita A, Khoury H, Barski A, Kaplan N, and Namekawa SH (2019). Attenuated chromatin compartmentalization in meiosis and its maturation in sperm development. Nat Struct Mol Biol 26, 175–184. 10.1038/s41594-019-0189-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Patel L, Kang R, Rosenberg SC, Qiu Y, Raviram R, Chee S, Hu R, Ren B, Cole F, and Corbett KD (2019). Dynamic reorganization of the genome shapes the recombination landscape in meiotic prophase. Nat Struct Mol Biol 26, 164–174. 10.1038/s41594-019-0187-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Rhodes JDP, Haarhuis JHI, Grimm JB, Rowland BD, Lavis LD, and Nasmyth KA (2017). Cohesin Can Remain Associated with Chromosomes during DNA Replication. Cell Rep 20, 2749–2755. 10.1016/j.celrep.2017.08.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Zickler D, and Kleckner N (1998). The leptotene-zygotene transition of meiosis. Annu Rev Genet 32, 619–697. [DOI] [PubMed] [Google Scholar]
- 255.Cheng G, Pratto F, Brick K, Li X, Alleva B, Huang M, Lam G, and Camerini-Otero RD (2024). High resolution maps of chromatin reorganization through mouse meiosis reveal novel features of the 3D meiotic structure. bioRxiv. 10.1101/2024.03.25.586627. [DOI] [Google Scholar]
- 256.Liang Z, Zickler D, Prentiss M, Chang FS, Witz G, Maeshima K, and Kleckner N (2015). Chromosomes Progress to Metaphase in Multiple Discrete Steps via Global Compaction/Expansion Cycles. Cell 161, 1124–1137. 10.1016/j.cell.2015.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Hirano T (2016). Condensin-Based Chromosome Organization from Bacteria to Vertebrates. Cell 164, 847–857. 10.1016/j.cell.2016.01.033. [DOI] [PubMed] [Google Scholar]
- 258.Yanez-Cuna FO, and Koszul R (2023). Insights in bacterial genome folding. Curr Opin Struct Biol 82, 102679. 10.1016/j.sbi.2023.102679. [DOI] [PubMed] [Google Scholar]
- 259.Lioy VS, Cournac A, Marbouty M, Duigou S, Mozziconacci J, Espeli O, Boccard F, and Koszul R (2018). Multiscale Structuring of the E. coli Chromosome by Nucleoid-Associated and Condensin Proteins. Cell 172, 771–783 e718. 10.1016/j.cell.2017.12.027. [DOI] [PubMed] [Google Scholar]
- 260.Pilatowski-Herzing E, Samson RY, Takemata N, Badel C, Bohall PB, and Bell SD (2024). Capturing chromosome conformation in Crenarchaea. Mol Microbiol 10.1111/mmi.15245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Takemata N, Samson RY, and Bell SD (2019). Physical and Functional Compartmentalization of Archaeal Chromosomes. Cell 179, 165–179 e118. 10.1016/j.cell.2019.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Gornik SG, Hu I, Lassadi I, and Waller RF (2019). The Biochemistry and Evolution of the Dinoflagellate Nucleus. Microorganisms 7. 10.3390/microorganisms7080245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Rizzo PJ (2003). Those amazing dinoflagellate chromosomes. Cell Res 13, 215–217. 10.1038/sj.cr.7290166. [DOI] [PubMed] [Google Scholar]
- 264.Ryba T, Hiratani I, Lu J, Itoh M, Kulik M, Zhang J, Dalton S, and Gilbert DM (2010). Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Res 20, 761–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Klein KN, Zhao PA, Lyu X, Sasaki T, Bartlett DA, Singh AM, Tasan I, Zhang M, Watts LP, Hiraga SI, et al. (2021). Replication timing maintains the global epigenetic state in human cells. Science 372, 371–378. 10.1126/science.aba5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Bulger M, and Groudine M (1999). Looping versus linking: toward a model for long-distance gene activation. Genes Dev 13, 2465–2477. [DOI] [PubMed] [Google Scholar]
- 267.Grosveld F, van Staalduinen J, and Stadhouders R (2021). Transcriptional Regulation by (Super)Enhancers: From Discovery to Mechanisms. Annu Rev Genomics Hum Genet 22, 127–146. 10.1146/annurev-genom-122220-093818. [DOI] [PubMed] [Google Scholar]
- 268.Tolhuis B, Palstra RJ, Splinter E, Grosveld F, and de Laat W (2002). Looping and Interaction between Hypersensitive Sites in the Active beta-globin Locus. Mol Cell 10, 1453–1465. [DOI] [PubMed] [Google Scholar]
- 269.Palstra RJ, Tolhuis B, Splinter E, Nijmeijer R, Grosveld F, and de Laat W (2003). The beta-globin nuclear compartment in development and erythroid differentiation. Nat Genet 35, 190–194. [DOI] [PubMed] [Google Scholar]
- 270.Deng W, Lee J, Wang H, Miller J, Reik A, Gregory PD, Dean A, and Blobel GA (2012). Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell 149, 1233–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Sanyal A, Lajoie BR, Jain G, and Dekker J (2012). The long-range interaction landscape of gene promoters. Nature 489, 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Hughes JR, Roberts N, McGowan S, Hay D, Giannoulatou E, Lynch M, De Gobbi M, Taylor S, Gibbons R, and Higgs DR (2014). Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nat. Genet 46, 205–212. [DOI] [PubMed] [Google Scholar]
- 273.Chen LF, Lee J, and Boettiger A (2023). Recent progress and challenges in single-cell imaging of enhancer-promoter interaction. Curr Opin Genet Dev 79, 102023. 10.1016/j.gde.2023.102023. [DOI] [PubMed] [Google Scholar]
- 274.Du M, Stitzinger SH, Spille JH, Cho WK, Lee C, Hijaz M, Quintana A, and Cisse II (2024). Direct observation of a condensate effect on super-enhancer controlled gene bursting. Cell 187, 331–344 e317. 10.1016/j.cell.2023.12.005. [DOI] [PubMed] [Google Scholar]
- 275.Zuin J, Dixon JR, van der Reijden MI, Ye Z, Kolovos P, Brouwer RW, van de Corput MP, van de Werken HJ, Knoch TA, van IJcken WF, et al. (2014). Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells. Proc Natl Acad Sci U S A. 111, 996–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Xiao JY, Hafner A, and Boettiger AN (2021). How subtle changes in 3D structure can create large changes in transcription. Elife 10. 10.7554/eLife.64320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Calderon L, Weiss FD, Beagan JA, Oliveira MS, Georgieva R, Wang YF, Carroll TS, Dharmalingam G, Gong W, Tossell K, et al. (2022). Cohesin-dependence of neuronal gene expression relates to chromatin loop length. Elife 11. 10.7554/eLife.76539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Adams NM, Galitsyna A, Tiniakou I, Esteva E, Lau CM, Reyes J, Abdennur N, Shkolikov A, Yap GS, Khodadadi-Jamayran A, et al. (2024). Cohesin-mediated chromatin remodeling controls the differentiation and function of conventional dendritic cells. bioRxiv. 10.1101/2024.09.18.613709. [DOI] [Google Scholar]
- 279.Kane L, Williamson I, Flyamer IM, Kumar Y, Hill RE, Lettice LA, and Bickmore WA (2022). Cohesin is required for long-range enhancer action at the Shh locus. Nat Struct Mol Biol 29, 891–897. 10.1038/s41594-022-00821-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Guckelberger P, Doughty BR, Munson G, Rao SSP, Tan Y, Cai XS, Fulco CP, Nasser J, Mualim KS, Bergman DT, et al. (2024). Cohesin-mediated 3D contacts tune enhancer-promoter regulation. bioRxiv. 10.1101/2024.07.12.603288. [DOI] [Google Scholar]
- 281.Chen H, Levo M, Barinov L, Fujioka M, Jaynes JB, and Gregor T (2018). Dynamic interplay between enhancer-promoter topology and gene activity. Nat Genet 50, 1296–1303. 10.1038/s41588-018-0175-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Hnisz D, Weintraub AS, Day DS, Valton AL, Bak RO, Li CH, Goldmann J, Lajoie BR, Fan ZP, Sigova AA, et al. (2016). Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science 351, 1454–1458. 10.1126/science.aad9024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Rekaik H, and Duboule D (2024). A CTCF-dependent mechanism underlies the Hox timer: relation to a segmented body plan. Curr Opin Genet Dev 85, 102160. [DOI] [PubMed] [Google Scholar]
- 284.Kiefer L, Gaudin S, Rajkumar S, Servito GIF, Langen J, Mui MH, Nawsheen S, and Canzio D (2024). Tuning cohesin trajectories enables differential readout of the Pcdhα cluster across neurons. Science 385, eadm9802. [DOI] [PubMed] [Google Scholar]
- 285.Canzio D, and Maniatis T (2019). The generation of a protocadherin cell-surface recognition code for neural circuit assembly. Curr Opin Neurobiol, 213–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, Suva ML, and Bernstein BE (2016). Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529, 110–114. 10.1038/nature16490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Kim KL, Rahme GJ, Goel VY, El Farran CA, Hansen AS, and Bernstein BE (2024). Dissection of a CTCF topological boundary uncovers principles of enhancer-oncogene regulation. Mol Cell 84, 1365–1376 e1367. 10.1016/j.molcel.2024.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Peters JM (2021). How DNA loop extrusion mediated by cohesin enables V(D)J recombination. Curr Opin Cell Biol 70, 75–83. 10.1016/j.ceb.2020.11.007. [DOI] [PubMed] [Google Scholar]
- 289.Zhang Y, Zhang X, Dai HQ, Hu H, and Alt FW (2022). The role of chromatin loop extrusion in antibody diversification. Nat Rev Immunol 22, 550–566. 10.1038/s41577-022-00679-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Ba Z, Lou J, Ye AY, Dai HQ, Dring EW, Lin SG, Jain S, Kyritsis N, Kieffer-Kwon KR, Casellas R, and Alt FW (2020). CTCF orchestrates long-range cohesin-driven V(D)J recombinational scanning. Nature 586, 305–310. 10.1038/s41586-020-2578-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Dai HQ, Hu H, Lou J, Ye AY, Ba Z, Zhang X, Zhang Y, Zhao L, Yoon HS, Chapdelaine-Williams AM, et al. (2021). Loop extrusion mediates physiological Igh locus contraction for RAG scanning. Nature 590, 338–343. 10.1038/s41586-020-03121-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Zhang Y, Zhang X, Ba Z, Liang Z, Dring EW, Hu H, Lou J, Kyritsis N, Zurita J, Shamim MS, et al. (2019). The fundamental role of chromatin loop extrusion in physiological V(D)J recombination. Nature 573, 600–604. 10.1038/s41586-019-1547-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Hill L, Ebert A, Jaritz M, Wutz G, Nagasaka K, Tagoh H, Kostanova-Poliakova D, Schindler K, Sun Q, Bonelt P, et al. (2020). Wapl repression by Pax5 promotes V gene recombination by Igh loop extrusion. Nature 584, 142–147. 10.1038/s41586-020-2454-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Owen JA, Osmanovic D, and Mirny L (2023). Design principles of 3D epigenetic memory systems. Science 382, eadg3053. 10.1126/science.adg3053. [DOI] [PMC free article] [PubMed] [Google Scholar]