

Abstract
Ligand binding to a chemokine receptor triggers signaling events through heterotrimeric G-proteins. The mechanisms underlying receptor-mediated G-protein activation in the heterogeneous microenvironments of the plasma membrane are unclear. Here, using live-cell fluorescence resonance energy transfer imaging to detect the proximity between CXCR1-cyan fluorescent protein (CFP) and fluorescence probes that label lipid raft or non-lipid raft microdomains and using fluorescence recovery after photobleaching analysis to measure the lateral diffusion of CXCR1-CFP, we found that interleukin-8 induces association between the receptors and lipid raft microenvironments. Disruption of lipid rafts impaired G-protein-dependent signaling, such as Ca2+ responses and phosphatidylinositol 3-kinase activation, but had no effect on ligand-binding function and did not completely abolish ligand-induced receptor phosphorylation. Our results suggest a novel mechanism by which ligand binding to CXCR1 promotes lipid raft partitioning of receptors and facilitates activation of heterotrimeric G-proteins.
Chemokine receptors belong to a family of seven-transmembrane G-protein-coupled receptors (GPCRs) that are differentially expressed by a number of immune and nonimmune cell populations and mediate cell responses to a family of soluble chemoattractant molecules called chemokines (22, 29, 45). Chemokines and their receptors control the trafficking of leukocytes and T and B lymphocytes. Cell movement and positioning, two fundamental properties of immune cells, play critical roles in many processes, including inflammation, allergy, and T- and B-cell and dendritic cell interactions that are necessary for self-tolerance and immune responses to various pathogens (10, 21, 27, 42). Extracellular chemokines binding to cell surface receptors initiate dissociation of heterotrimeric G-proteins into Gαi and Gβγ subunits. These subunits, in turn, induce downstream intracellular signaling components to generate biochemical responses (29, 42). CXCR1 is one of the human chemokine receptors that is selectively expressed in neutrophils and directs leukocytes to sites of inflammation (2, 47). Several chemokines, such as interleukin-8 (IL-8), and acP-2, bind to CXCR1 and activate G-protein-dependent intracellular signaling pathways, for example, Ca2+ influx and phosphatidylinositol 3-kinase (PI3K) activation, and also trigger G-protein-independent CXCR1 phosphorylation (2, 47).
The plasma membranes of eukaryotic cells consist of a complex assembly of various lipids and proteins that are distributed in regions of distinct lipid microenvironments, known as lipid raft or non-lipid raft microdomains (8, 15, 24, 32, 40, 43). Lipids in rafts possess long and saturated fatty acyl chains and are organized in a tightly packed, liquid-ordered manner, whereas non-lipid raft microdomains contain shorter, unsaturated fatty acyl chains and are in a loosely packed, disordered manner (8, 15, 24, 32, 40, 43). Lipid rafts are defined as microdomains that are enriched in cholesterol, glycosphingolipids, and sphingomyelin and are often isolated in detergent-resistant membrane (DRM) fractions. Both lipid and non-lipid raft microdomains contain multiple proteins that play critical roles in signal transduction via complex protein-protein interactions between ligands, receptors, and signaling components (32, 40). G-protein signaling components, such as Gα subunits, have been shown to be more concentrated in the lipid rafts (15). In order to effectively transmit signals, a chemokine receptor could interact with lipid rafts in the plasma membrane in different ways. For example, (i) receptors could localize in lipid raft microdomains and interact with G-proteins upon ligand binding (23, 30, 31). (ii) Receptors could localize in non-lipid raft microdomains and associate with lipid raft microdomains upon ligand binding. (iii) Activated receptors could result in the formation of clustered lipid rafts so that more signaling components become aggregated (40). Using detergent insolubility or immunostaining with fluorescence microscopy, studies of chemokine receptors (CCR5 and CXCR4) suggest that the receptors localize in lipid rafts and this localization is important for ligand binding and receptor signaling (23, 30, 31). However, recent studies suggest that membrane microdomains isolated from detergent do not reliably reflect the organization of the lipids in the cell membrane. It has been very difficult to demonstrate the existence of lipid rafts in cells because their size is too small to be resolved by light microscopy and their stability and motion in live cells are unclear (8, 17, 24, 32). Due to the dynamic and submicroscopic nature of lipid microdomains in living cells, the physiological function of lipid microdomains in regulating chemokine receptor signaling remains unclear.
Ligands binding to chemokine G-protein-coupled receptors dissociate Gαi and Gβγ subunits of G-proteins, leading to signaling. Activated receptors then induce interaction between the receptors and G-protein-coupled-receptor kinases, leading to phosphorylation at the receptor's C-terminal tails (18). Following phosphorylation, the receptors recruit β-arrestins to desensitize the receptors by blocking their interaction with heterotrimeric G-proteins. Binding of β-arrestins to the receptors initiates receptor internalization via clathrin-mediated endocytosis (18). Previous studies have shown that IL-8-triggered CXCR1 internalization (endocytosis) requires G-protein-coupled receptor kinase 2, β-arrestins, and dynamin (4). In HEK293 cells transiently expressing CXCR1-green fluorescent protein (GFP), the fusion receptors do not internalize when the cells are stimulated with IL-8 unless both β-arrestins and G-protein-coupled receptor kinases (GRKs) are coexpressed (4). To investigate the function of lipid microdomains in regulating the early events in CXCR1 receptor signaling in living cells without the complication of IL-8-induced endocytic trafficking of the receptors, we chose HEK293 cells for our study.
Here, by monitoring live-cell fluorescence resonance energy transfer (FRET) between CXCR1-cyan fluorescent protein (CFP) and fluorescence lipid analog DiIC16 or FastDiI to label lipid raft or non-lipid raft microdomains, respectively, we show that ligand binding to CXCR1-CFP induces a partitioning of activated receptors into lipid raft microenvironments. By analyzing fluorescence recovery after photobleaching (FRAP), we observed that activated CXCR1-CFP displayed a reduced mobility on the plasma membrane, further supporting the notion that the activated receptors associate with lipid raft microenvironments. Disruption of lipid raft microdomains by methyl-β-cyclodextrin (MβCD) treatment did not significantly affect ligand binding to the receptor and did not completely abolish ligand-induced receptor phosphorylation, which is G-protein independent. However, this treatment impaired ligand-induced G-protein-dependent Ca2+ response and PI3K activation. Taken together, our results suggest that ligand-induced partitioning of chemokine-occupied receptors into lipid raft microenvironments is critical for the interaction of activated receptors with G-proteins leading to G-protein-dependent signaling.
MATERIALS AND METHODS
Chemicals and reagents.
pEF6/v5-his-topo vector and Lipofectamine 2000 were purchased from Invitrogen (Grand Island, NY). IL-8 was purchased from Biosource International (Camarillo, CA). Anti-human CXCR1 monoclonal antibody (5A12) was from BD Biosciences Pharmingen (San Diego, CA). Anti-GFP monoclonal antibody (A.v. monoclonal antibody JL-8) was from BD Biosciences Clontech (Palo Alto, CA). Anti-Gαi3 and anti-Gβ rabbit polyclonal antibody and anti-GFP monoclonal antibody protein G beads were from Santa Cruz Biotechnology (Santa Cruz, CA). Akt and phospho-Akt (Ser 473) antibody were from Cell Signaling Technology (Beverly, MA). Fluo-4-AM, DiIC16, and FastDiI were from Molecular Probes (Eugene, OR). 125I-labeled IL-8 was from Perkin-Elmer Life and Analytical Sciences (Torrance, CA). MβCD, ionomycin, and EGTA were from Sigma-Aldrich (St. Louis, MO). Horseradish peroxidase-conjugated goat anti-mouse immunoglobulin G with γ light chain [IgG(γ)], goat anti-rabbit IgG (heavy and light chains), milk diluent/blocking solution concentrate, and LumiGLO chemiluminescent substrate were from Kirkegaard & Perry Laboratories (Gaithersburg, MD). All of the other reagents were reagent grade and were obtained from standard suppliers.
Plasmid, cell line, cellular labeling, and cholesterol extraction.
The plasmid encoding CXCR1-CFP was constructed by inserting the CXCR1-CFP gene into the pEF6/v5-his-topo vector. HEK293T cells were cultured in Dulbecco modified Eagle medium supplemented with fetal calf serum (10%), penicillin (5 μg/ml), and streptomycin (5 μg/ml) and were grown in 5% CO2 at 37°C. Cells were transfected with the plasmid encoding CXCR1-CFP mediated by Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions and cloned by limiting dilutions and blastcidin selection (10 μg/ml). The positive clones were screened on the basis of CFP fluorescence with a fluorescence microscope. For DiIC16 or FastDiI labeling experiments, cells were prelabeled with DiIC16 or FastDiI at 4°C for 5 min and then washed, which is a modification of the previously described method to label plasma membranes of neutrophils (11, 37). For cholesterol extraction, cells in serum-free medium were treated with 10 mM of MβCD for 30 min at 37°C (8, 24, 32). They were then rinsed and incubated in the growth medium with 1% lipid-free bovine serum albumin. The efficiency of cholesterol extraction was reported in previous studies (7, 38, 44).
Imaging.
Live cells cultured in a four-well chamber were imaged using a Zeiss laser-scanning microscope, LSM510META, with a 40× oil immersion Doc Plan-Neofluar lens objective with a numerical aperture of 1.3. To monitor CXCR1-CFP (see Fig. 1a, 4a, and 6b), the specimens were excited with a 458-nm laser line, and images were recorded in two channels: channel one, fluorescence emission from 475 to 525 nm for CFP; and channel two, differential interference contrast. To monitor both CFP and DiIC16 or FastDiI (see Fig. 2a), the specimens were excited with two laser lines, 458 nm for CFP and 543 nm for DiI. Images were simultaneously recorded in three channels: channel one, emission filter of 475 to 525 nm for CFP; channel two, emission filter of 560 to 615 nm for DiIC16 or FastDiI; and channel three, differential interference contrast. To detect CFP and Fluo-4 in Ca2+ assay (see Fig. 1e and 5a), cells were excited with a 458-nm laser line, and the images were recorded in 16 channels (lambda stack) from 464 to 621 nm. To detect FRET between CFP and DiIC16 or FastDiI (see Fig. 3), we used time-lapse and lambda stack acquisition linked with the photobleaching command. In the time series, the cells were first excited with the 454-nm laser at about 7.5% power in order to limit photobleaching, and images were recorded in 16 channels from 464 to 621 nm. Spectral images were recorded three times before photobleaching, the selected regions were illuminated with 100% 514-nm laser power 20 to 30 times to photobleach DiI, and then images were recorded three more times with excitation by the 454-nm laser line. To determine the contribution of each fluorophore, the spectrally resolved images of the lambda stack were processed using the linear unmixing function of LSM510META. To separate multifluorescence signals, each fluorescence image was collected using lambda stack acquisition. The spectral emissions of fluorescence images were simultaneously recorded in a CHS-1 from 464 to 621 nm. The spectra of CFP, Fluo-4, and DiI were obtained and used as references for the linear unmixing function. The digitally separated images of CFP, Fluo-4, and DiIC were obtained. The intensity of each fluorophore in the regions of interest in the time-lapse experiments were measured and expressed as a function of time using the software of LSM510META.
FIG. 1.

A stable HEK293 cell line expressing a functional CXCR1-CFP receptor. (a) Confocal images of living cells expressing membrane-localized CXCR1-CFP (cyan). (b) Fusion proteins of CXCR1-CFP were detected by Western blotting using anti-GFP (αGFP) monoclonal antibodies that cross-react with CFP and anti-CXCR1 monoclonal antibodies in whole CX1-HEK cell lysates. The positions of molecular mass markers (in kilodaltons) are shown to the left of the gel. (c) Binding curve of IL-8 with CXCR1-CFP receptors expressed in CX1-HEK cells. (d) Time course of CXCR1-CFP on the plasma membranes of living cells upon the stimulation of IL-8 (50 nM) using time-lapse live-cell confocal microscopy. CX1-HEK cells were illuminated with a 458-nm laser line for monitoring fluorescence signals from CFP. Frames were captured at 12-s intervals for more than 10 min. IL-8 was added at time zero. The entire plasma membrane was selected as the ROI, and the intensity of CFP fluorescence reflects the amount of CXCR1-CFP on the plasma membrane. The graph shows means and standard errors of the relative fluorescence (It/I0) as a function of time, where It is the intensity at any time point on the plasma membrane and I0 is the intensity at time zero. (e) IL-8-induced Ca2+ response in living CX1-HEK cells. IL-8 was added at time zero, and intracellular Ca2+ changes were detected as intensity changes of Fluo-4. Means ± standard errors (error bars) are shown (n = 12). (f) IL-8-induced Ca2+ response in two living CX1-HEK cells. IL-8 was added at time zero, and fluorescence images of transient intracellular Ca2+ elevation, detected as intensity changes of Fluo-4, are shown as rainbow pseudocolor. A color bar shows the relative intensity of Fluo-4. The time following IL-8 stimulation is shown in the upper left corner of each image. A video showing a complete sequence of this time-lapse experiment is available upon request.
FIG. 4.

FRAP measurement shows that CXCR1-CFP's mobility becomes limited on the plasma membranes of live cells upon IL-8 stimulation. (a) FRAP experiment on the CX1-HEK cells in the absence (control) or presence of IL-8. Prebleach images (−10 s) and postbleach images (0 s and 100 s) are shown. Arrowheads indicate selected bleaching areas. (b) Quantitation and kinetics of CXCR1-CFP recovering on the plasma membrane after photobleaching. The mean fluorescence intensity of CXCR1-CFP in each selected area was determined at the times indicated and expressed as a ratio, (Ft − F0/Fi − F0). Ft is the mean intensity at any time point. F0 is the mean intensity immediately after bleaching. Fi is the mean intensity before bleaching. The black dots and solid line represent data from cells in the absence of IL-8, while the gray dots and dashed line are from cells in the presence of IL-8. (c) Kinetic parameters of CXCR1-CFP in FRAP experiments. T1/2 is the time for 50% recovery. A kinetic parameter of a protein can be discerned from quantitative studies using FRAP. The mobile fraction (R) is the fraction of fluorescent proteins that can diffuse into the bleached region during the time course of the experiment (20). We determined the R value as the percentage of fluorescence recovery at the last time point of the experiments. Data were obtained from 10 (control) and 16 (IL-8) independent experiments, respectively. Means ± standard errors (error bars) are shown. In the presence of IL-8, T1/2 is longer and R is smaller.
FIG. 6.

Depletion of cholesterol from the plasma membrane does not impair the ligand-binding ability of CXCR1-CFP and did not completely abolish IL-8-triggered receptor phosphorylation. (a) (Left) Binding curve of IL-8 with CXCR1-CFP receptors expressed in CX1-HEK cells that were treated with MβCD. (Right) Dissociation constant (Kd) and maximal binding sites (Bmax) from nontreated (control) and treated (MβCD) cells. Note that the treatment affects Bmax, but not Kd. (b) MβCD treatment results in a loss of about 20% of CXCR1-CFP receptors from the cell membrane, measured by CFP intensity on the cell surface. DIC, differential interference contrast. (c) IL-8 stimulation induced CXCR1-CFP phosphorylation in both nontreated and treated CX1-HEK cells.
FIG. 2.

CXCR1-CFP and a lipid raft fluorescent dye, DiIC16, are colocalized on the plasma membrane regardless of IL-8 stimulation. (a) CX1-HEK cells were labeled with DiIC16 for 5 min. Labeled cells in the absence of IL-8 (control) or in the presence of IL-8 were visualized for CFP and DiIC16 with a confocal laser-scanning microscope. CX1-HEK cells labeled with Fluo-4 (green) are shown as a negative control of colocalization analysis. (b) Quantitative analysis of colocalization of two images expressed as a scatter diagram (LSM 510META software; Carl Zeiss). Identical images produce a clean diagonal line running from the bottom left to the top right. Differences between the images cause an irregular distribution in the scatter diagram (for example, CXCR1-CFP and Fluo-4 images of CX1-HEK cells labeled with Fluo-4 serve as a negative control). CXCR1-CFP and DiIC16 images, in the absence of IL-8 (control) and presence of IL-8, display similarly high degrees of colocalization. (c) Correlation coefficients of CXCR1-CFP and DiIC16 images with or without IL-8 show high degrees of colocalization, whereas CXCR1-CFP and Fluo-4 images show little colocalization. Correlation coefficients provide information on the intensity distribution within the colocalizing region. Values range from 0 to 1, with 1 indicating that all pixels are found on a straight line in the scattergram while 0 indicates that the pixels in the scattergram are distributed in a cloud.
FIG. 5.

MβCD treatment impairs IL-8-triggered Ca2+ response and Akt phosphorylation and chemotaxis in response to IL-8 gradient. (a) IL-8 failed to induce CXCR1-CFP-mediated Ca2+ response in the MβCD-treated cells. CX1-HEK cells labeled with Fluo-4 (green) were stimulated with 50 nM of IL-8. Non-MβCD-treated cells (control) and MβCD-treated cells are shown. Upon IL-8 stimulation, changes in the intracellular Ca2+ concentration are shown in the graph on the right. (b) IL-8 stimulation induced CXCR1-CFP-mediated Akt phosphorylation in control cells but not in MβCD-treated cells. CX1-HEK cells were stimulated with IL-8 and subjected to Western blotting with antibodies for Akt and the phosphorylated form of Akt (pAkt). (c) Quantification of IL-8-induced Akt phosphorylation is shown. (d) Effect of MβCD on chemotaxis. CX1-HEK cell migration across 10-mm-pore-size filter membranes was measured in response to gradient of IL-8 of three different concentrations. In the absence of IL-8, the chemotaxis index of CX1-HEK cells is 1.
FIG. 3.

FRET measurement between CXCR1-CFP and DiIC16 or FastDiI on the plasma membranes of live cells with or without IL-8 stimulation. (a) Images of CXCR1-CFP (FRET donor) and DiIC16 before and after photobleaching DiIC16 (FRET acceptor) in the CX1-HEK cells exposed to IL-8. CX1-HEK cells labeled with DiIC16 were under IL-8 stimulation. The entire area of cells was illuminated to photobleach DiIC16 (with a 514-nm laser line), and FRET was monitored as increased CFP emission. The cells were excited with a 454-nm laser line, and spectral images were acquired before and after photobleaching DiIC16. Spectrally resolved images were processed into digitally separated CXCR1-CFP and DiIC16 channels. The plasma membrane was selected as the ROI for quantitative analyses shown in panels c and d. Before the photobleaching experiment, the level of DiIC16 labeling was monitored by acquiring an image with a 514-nm laser line, which does not excite CFP. This image is shown in the lower right corner. (b) Images of CFP intensity increase on the cell surface after photobleaching DiIC16. (c) Emission spectra of the membrane region (ROI) before (gray line) and after (black line) photobleaching DiIC16. After photobleaching DiIC16, a significant decrease near 565 nm and a clear increase in the CFP emission signal near 475 nm was observed. (d) Fluorescence intensities of CXCR1-CFP (black) and DiIC16 (gray) in the ROI before and after photobleaching DiIC16. Fluorescence intensity of CXCR1-CFP and DiIC16 were recorded before (two time points) and immediately after (three time points) photobleaching. (e) FRET efficiency of CXCR1-CFP alone (a negative control) and between CXCR1-CFP and either DiIC16 or FastDiI in cells unstimulated (control) (−) or stimulated with IL-8 (+). FRET efficiency is calculated on the basis of the intensity increase in CFP emission after photobleaching DiIC16. After IL-8 stimulation, FRET efficiency between CXCR1-CFP and DiIC16 increases (n = 12; P < 0.001), while that between CXCR1-CFP and FastDiI decreases (n = 15; P < 0.05). (f) Fluorescence intensity of DiIC16 or FastDiI on the plasma membranes of living cells in the FRET measurements shown in panel d. Means and standard errors (error bars) are shown.
Image data processing.
Images were processed and analyzed by LSM510META software and converted to TIFF files by Photoshop 7.0. All frames of any given series were processed identically. Selected frames from the series were assembled as montages using Photoshop 7.0. Quantification of the fluorescence intensities of each fluorophore was performed using LSM510META software.
Calcium assay.
CX1-HEK cells were seeded in four-well chambers at 104/ml, 2 days before the experiments. After 3 hours of starvation, the cells were labeled by incubation with Fluo-4-AM in Hanks balanced salt solution for half an hour, washed twice, and incubated for half an hour before imaging under the microscope. Upon the addition of IL-8 (50 nM) to the cell chamber, time-lapse images were collected using a lambda mode, and CFP and Fluo-4 images were digitally separated as described above. Ca2+ concentration, determined by the intensity of Fluo-4, was calibrated in situ using ionomycin (5 μg/ml) combined with EGTA (20 mM) and MnCl2 (20 mM) and calculated by the following equation according to the manufacturer's instructions: [Ca2+] = Kd (F − Fmin)/(Fmax − F). Kd is the dissociation constant of Ca2+ with Fluo-4. Fmin is the fluorescence intensity of Fluo-4 in the absence of Ca2+, which was achieved by adding ionomycin and EGTA. Fmax is the Ca2+-saturated Fluo-4 fluorescence intensity that equaled 5 times Mn2+-saturated Fluo-4's fluorescence intensity. F is the fluorescence of the sample.
FRET and FRAP assays.
FRET between CFP and DiIC16 or FastDiI was measured by intensity increase of the donor (CFP) after photobleaching the acceptor (DiI). CX1-HEK cells were labeled with DiIC16 or FastDiI. Levels of labeling were determined on the basis of the intensity of DiI using excitation with the 514-nm laser line. The cells displaying similar levels of labeling throughout the plasma membrane were selected for the FRET experiments. Spectrally resolved images before and after photobleaching DiI were acquired, and the digitally separated images of CFP and DiI were obtained as described above. Fluorescence intensity of CFP on the entire plasma membrane (region of interest [ROI]) was obtained. The FRET efficiency (EFRET) was calculated according to the following equation: EFRET (as a percentage) = (FB − F0)/FB × 100. FB is the fluorescence intensity of CFP after photobleaching, and F0 is the fluorescence intensity of CFP before photobleaching. FRAP was used to monitor the mobility of CXCR1-CFP on the membranes of live CX1-HEK cells. Selective photobleaching was carried out using consecutive scans for 20 seconds with the full power of the 458-nm laser line. Intensity of CFP in the selected regions was measured in a time-lapse experiment and quantitatively analyzed. To determine the time for 50% recovery (T1/2) and the percentage of the mobile fraction, the mean fluorescence intensity in each selected area was expressed as the ratio of (Ft − F0)/(Fi − F0) as a function of time. Ft is the mean intensity at any given time. F0 is the mean intensity immediately after bleaching. Fi is the mean intensity before bleaching. T1/2 and R, the mobile fraction, are determined from the kinetic curve.
IL-8 binding assay.
The ligand binding assays were carried out by the method of Grimm et al. (12) with modifications. The cells were preincubated with IL-8 for 30 min at 37°C, washed extensively, and resuspended in binding medium at 107 cells/ml. The assay was carried out on ice using 0.5 nM 125I-labeled IL-8 in the presence of increasing concentrations of competing unlabeled IL-8. The cells were then incubated at 4°C for 30 min, and unbound ligands were separated from the cells by a 10% sucrose gradient. The level of binding was determined by the 125I count using a γ-counter. Nonlinear regression analysis of the data was performed by a PRISM3.0 program by fitting the following equation: total binding = Bmax × [hot]/([hot] + [cold] + Kd) + nonspecific binding, where Bmax is the number of maximal binding sites.
Isolation of DRM.
Cells were lysed on ice in lysis buffer (0.5% Triton X-100, 10 mM Tris-HCl, pH 7.5, 150 mM NaCl, 5 mM EDTA, CLAP [2.5 mg/ml each of chymostatin, leupeptin, antipain, and pepstatin A in dimethyl sulfoxide], and 1 mM sodium orthovanadate). The cell lysates were further homogenized by pipetting. Nuclei and cellular debris were removed from the supernatant after centrifugation at 900 × g for 11 min. To prepare a discontinuous sucrose gradient, 1 ml of cleared supernatant was mixed with 1 ml of 85% sucrose in TNEV and transferred to the bottom of a Beckman centrifuge tube (14 by 89 mm). The diluted cell lysate was overlaid with 6 ml 35% sucrose in TNEV and finally 3.5 ml 5% sucrose in lysis buffer without Triton X-100 (TNEV). The samples were centrifuged in an SW41 rotor at 200,000 × g for 16 to 20 h at 4°C. One-milliliter fractions were collected from the top of the gradient.
Western blot.
The expression of CXCR1-CFP in stable CXCR1-CFP transfecting cell lines, the distribution of G-proteins in lipid rafts, and the phosphorylation of Akt under IL-8 stimulation were all detected by Western blotting. The blots were visualized with horseradish peroxidase-conjugated secondary antibodies and enhanced chemiluminescence. All films were scanned with a scanner and quantitatively analyzed by Image J software.
Phosphorylation of CXCR1-CFP.
Phosphorylation of CXCR1 was detected as described previously (12). Cells were washed, incubated in phosphate-free medium for 3 h, and then incubated with 150 μCi/7.5 × 106 cells of [32P]orthophosphate for 90 min at 37°C before stimulation with IL-8 for 5 min. Equivalent numbers of cells for each treatment were lysed in radioimmunoprecipitation assay buffer containing protease and phosphatase inhibitors. CXCR1-CFP was immunoprecipitated with anti-GFP monoclonal antibody protein G beads. Immunoprecipitated protein was separated electrophoretically by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The gels were dried and exposed to radiographic film.
Statistical analysis.
Statistical significance was determined with Student's t test.
RESULTS
CXCR1 tagged with CFP is functional.
To visualize CXCR1 in living cells, we fused CFP to the C terminus of CXCR1 and established a stable HEK293 cell line expressing a CXCR1-CFP fusion protein designated CX1-HEK. CXCR1-CFP exhibited plasma membrane localization with no gross heterogeneity in distribution. In some cells, CXCR1-CFP accumulated in the Golgi apparatus under nonstimulated conditions (Fig. 1a), as previously described (4). CXCR1-CFP fusion proteins were detected with the expected molecular mass by Western blotting (Fig. 1b). Two bands were detected by anti-GFP. The upper one is CXCR1-CFP, and the lower one, about 30 kDa, is likely degraded products of CXCR1-CFP. Since they were not detected by the anti-CXCR1 antibody, the degraded products do not contain a portion of CXCR1; therefore, they have no transmembrane domain. They should not affect our analyses of the plasma membrane. To test functionality of the CFP-tagged receptor, we first examined the ability of CXCR1-CFP to bind its ligand, IL-8. CX1-HEK cell line bound to IL-8 with an apparent Kd of 3.9 nM (Fig. 1c), which is similar to the Kd for wild-type CXCR1 receptors expressed in RBL-2H3 cells (2.3 nM) or in neutrophils (1 to 2 nM) (34). Activated GPCRs undergo clathrin-dependent endocytotic internalization process. The process is initiated by receptor phosphorylation by GRKs, followed by binding of proteins called arrestins, which bind the phosphorylated receptor and inhibit further G-protein activation. Desensitized receptor-arrestin complexes undergo arrestin-dependent targeting for sequestered receptor through clathrin-coated pits (18). IL-8-induced internalization of CXCR1-GFP has been previously visualized by confocal microscopy (4). In RBL-1H3 cells, agonist-occupied CXCR1-GFP conjugates and forms specific membrane-associated vesicles that gradually translocate from the plasma membrane to the cytosol following IL-8 stimulation. However, in HEK 293 cells, CXCR1-GFP does not internalize when stimulated with IL-8 in the absence of overexpressed GRK2 and β-arrestins (4). Our results here were consistent with the previous study. We found that there was no significant decrease in membrane CFP fluorescent signal in response to IL-8 stimulation (Fig. 1d). We then measured IL-8-induced Ca2+ responses in single CX1-HEK living cells. We imaged IL-8-induced fluorescence intensity change of Fluo-4, a fluorescent calcium indicator, and simultaneously monitored localization of CXCR1-CFP using a spectral-confocal microscope (Fig. 1d and e). CX1-HEK cells were first labeled with Fluo-4, and fluorescence images of Fluo-4 (green) and CXCR1-CFP (red) of two living cells were simultaneously recorded in a time-lapse experiment (Fig. 1f). Fluo-4 was distributed throughout the entire cytosol, and CXCR1-CFPs were uniformly distributed on the cell surface. Upon addition of IL-8 to the cell chamber, the green fluorescence signal transiently increased in the cytosol, indicating that IL-8 triggered changes in the intracellular Ca2+ concentration (Fig. 1e and f). Temporal changes of green fluorescence in the cell showed similar kinetics to the IL-8-stimulated CXCR1-mediated Ca2+ response (Fig. 1e) (2, 47). The distribution of CFP signal in the two living cells was unchanged upon stimulation (unpublished data), indicating that ligand binding immediately triggered downstream signaling events without altering the membrane localization of the receptor. To ensure that IL-8-elicited Ca2+ responses are specifically mediated by the expressed CXCR1-CFP, we did a control experiment with the parental HEK293 cells. No increase in green fluorescence signal was observed after IL-8 stimulation (data not shown). These results demonstrated that the CXCR1 receptor fused with CFP at its C terminus retained its signaling functions. Therefore, CX1-HEK cells provide a system for probing ligand-induced changes in the dynamic distribution and signaling of CXCR1 receptors on the plasma membrane without interference with the formation of desensitized receptor-arrestin complexes that undergo arrestin-dependent targeting for internalization through clathrin-coated pits.
CXCR1-CFP and DiIC16 are uniformly distributed on the plasma membrane.
We investigated membrane distribution of CXCR1 and its relationship to fluorescence probes DiIC16 and FastDiI. DiIC16 is incorporated primarily in relatively ordered membrane regions, lipid raft microdomains, whereas FastDiI partitions into the more fluid regions, non-lipid raft microdomains (28, 37). Using a detergent insolubility assay, we demonstrated that DiIC16 and FastDiI specifically labeled raft-like and non-raft microdomains in CX1-HEK living cells, respectively (unpublished data). Using live-cell confocal microscopy, we examined colocalization of CXCR1-CFP and DiIC16 in living cells that were incubated with or without IL-8 (Fig. 2). CX1-HEK cells were labeled with DiIC16 to visualize lipid raft microdomains (37) (Materials and Methods). We found that both CXCR1-CFP (cyan) and DiIC16 (red) colocalized and were evenly distributed on the living cell surface (Fig. 2a). IL-8, when added into the cell chamber, did not change the pattern of membrane distribution of either CXCR1-CFP or DiIC16 (Fig. 2a). There was some variation in the intensities of either CXCR1-CFP or DiIC16, which is consistent with cell surface projections. Using a quantitative colocalization analysis, CXCR1-CFP and DiIC16 channels of an image were computed and expressed as a scatter diagram (Fig. 2b), indicating that CXCR1-CFP and DiIC16 were colocalized on the plasma membrane of the cells regardless of IL-8 stimulation, with colocalization correlations of 0.72 and 0.82, respectively (Fig. 2b and c). As a negative control, CXCR1-CFP and Fluo-4 displayed little colocalization, with a correlation of 0.01. Our colocalization studies found that FastDiI also uniformly labeled the plasma membrane, and CXCR1-CFP and FastDiI were colocalized on the membrane regardless of IL-8 (data not shown). Thus, CXCR1-CFP colocalized with both lipid raft and non-lipid raft fluorescence probes. These results suggest that the microdomains on the plasma membranes of living cells are very small and the spatial separation between different microdomains was beyond the limit of the spatial resolution of fluorescence microscopy, which is 200 nm at best. Therefore, the colocalization studies cannot determine the microenvironment surrounding CXCR1 on the plasma membrane and whether ligand binding alters the receptor's microenvironment on the plasma membranes of living cells.
Ligand binding changes the receptor's microenvironments on the plasma membrane.
To probe the microenvironment of CXCR1 receptors on the plasma membrane with greater sensitivity, we applied FRET to detect the distance between CXCR1-CFP (FRET donor) and DiIC16 or FastDiI (FRET acceptor) on the membranes of living cells. FRET between a donor and acceptor typically occurs over distances of less than 10 nm and thus offers a way of detecting very small clusters of FRET donors and acceptors (20, 36, 46). We reasoned that if CXCR1-CFP receptors localize in a lipid raft-like microenvironment, CFPs would be surrounded by more DiIC16 probes than FastDiI probes so that FRET efficiency between CXCR1-CFP and DiIC16 would be greater than that between CXCR1-CFP and FastDiI. In our experiments, FRET efficiency is measured on the basis of the increase in the intensity of the donor (CFP) upon photobleaching the acceptor with either DiIC16 or Fast DiI. Increased donor (CXCR1-CFP) fluorescence after destruction of the acceptor (DiIC16 or FastDiI) indicates that donor fluorescence had been quenched by the surrounding acceptors because of energy transfer. Using this FRET measurement, concentrations of the donor (CXCR1-CFP) and acceptor (DiIC16 or FastDiI) throughout the entire plasma membrane, the ROI, need to be comparable. We used the stable cell line CX1-HEK, which provides a similar expression level of CXCR1-CFP among all cells, standardized the labeling conditions of DiIC16 and FastDiI, and analyzed the image data only from cells that displayed similar levels of fluorescence labeling throughout the entire plasma membrane (ROI).
Data from a typical FRET experiment are shown in Fig. 3a, b, c and d. CX1-HEK cells were starved in serum-free buffer for 3 hours before the addition of IL-8. The cells were then labeled with DiIC16 for 5 min. Using a time-lapse live-cell image experiment coupled with a photobleaching function, the cell was illuminated with a 458-nm laser line for monitoring fluorescence signals from CFP and DiIC16 and with a 514-nm laser line specifically for photobleaching DiIC16. Spectral images in 16 channels from 464 to 624 nm were simultaneously recorded. Each pixel of the image contains data corresponding to an emission spectrum resulting from both CFP and DiIC16. The emission spectra across the entire plasma membrane showed a clear increase in the CFP signal at 475 nm after destruction of DiIC16, which has an emission peak around 575 nm (Fig. 3c). The digitally separated CFP and DiIC16 channels showed that CFP emission increases were observed on the entire plasma membrane after photobleaching DiIC16 (Fig. 3b and c). In our quantitative analysis, FRET efficiency is defined as (FB − F0)/FB × 100%, where FB is the intensity of the donor (CFP) after photobleaching and F0 is the intensity of the donor before photobleaching. We selected the entire plasma membrane as the ROI. As a negative control, when CX1-HEK cells without DiIC16 or FastDiI labeling were photobleached, a low level of CFP fluorescence increase on the entire membrane (5.4% ± 1.4%; n = 10) was detected, suggesting the existence of a small amount of false quenching of CFP on the membranes of CX1-HEK cells under our experimental conditions (Fig. 3d). Using the same experimental design, we found that the measured FRET efficiency between CXCR1-CFP and DiIC16 was 23.6 ± 3.3 (n = 12) on the plasma membrane of the cell incubated with IL-8, and the calculated FRET efficiency, which is defined as the measured FRET minus the false FRET (5.4%), was 18.2% ± 3.3% (Fig. 3c). We also measured FRET between CXCR1-CFP and DiIC16 on the plasma membranes of cells in the absence of IL-8, and the calculated FRET efficiency was 3% ± 1.1% (n = 27), whereas the calculated FRET between CXCR1-CFP and FastDiI on the plasma membranes of cells with or without IL-8 is 3.9% ± 1.1% (n = 15) and 8.9% ± 1.7% (n = 24), respectively (Fig. 3e). FRET efficiency between CXCR1-CFP and DiIC16 on the plasma membranes was significantly higher in the presence of IL-8 than in the absence (P < 0.001); however, FRET efficiency between CXCR1-CFP and FastDiI was lower when IL-8 was present (P < 0.05) (Fig. 3d). FRET efficiency as measured in our experiments depends on the distance between CXCR1-CFP and DiIC16 or FastDiI as well as the concentration of DiIC16 or FastDiI in the plasma membrane (ROI). To ensure that differences in FRET efficiency were compared with the similar concentrations of FRET acceptors on the membrane, we determined the fluorescence intensity of DiIC16 or FastDiI on the cell membrane in our FRET measurements. Cells were illuminated with a 514-nm laser line for monitoring fluorescence of DiIC16 or FastDiI before the photobleaching experiments, and fluorescence intensity is shown in Fig. 3f. Our results indicate that empty CXCR1 receptors are surrounded by the non-lipid raft environment, whereas IL-8 binding to the CXCR1 receptors induces a microenvironmental change in which activated receptors are surrounded by lipid rafts.
Differences in the dynamic interaction of unbound and ligand-bound CXCR1-CFP with raft microenvironments.
Lipid raft microdomains, which are enriched with cholesterol and sphingomyelin, are more rigid and less flexible. When a protein is associated with a lipid raft microenvironment, its mobility becomes slower and limited (39). To further explore the microenvironments of inactive or activated CXCR1 receptors, lateral diffusion of CXCR1-CFP on the plasma membrane was measured by FRAP (17, 20). We used selective photobleaching and time-lapse imaging to analyze diffusion of CXCR1-CFP on the plasma membranes of live cells in the absence or presence of IL-8. A series of images were rapidly collected after bleaching CXCR1-CFP in selected regions of the plasma membrane (Fig. 4a). We found complete recovery of the fluorescence of CXCR1-CFP (R = 101.2% ± 5.2%; n = 10) with a T1/2 of 20.6 ± 1.6 seconds (n = 10) on the plasma membranes of the cells without the addition of IL-8 (Fig. 4b and c). In the presence of IL-8, the recovery of fluorescence decreased (R = 77.3% ± 6.0% [n = 15]; P < 0.01) and the rate of recovery for a region of the same size was much slower (T1/2 = 39.4 ± 4.2 seconds [n = 15]; P < 0.001) (Fig. 4b and c). These FRAP results show that active CXCR1 receptors diffuse more slowly than inactive receptors as their mobility becomes restricted on the plasma membrane. It is possible that proteins interacting with the cytoskeletons could affect their diffusion on the plasma membrane. It is reported that activation of chemokine receptors could lead to the plasma membrane reorganization of DRM in neutrophils. Reorganization of the plasma membrane may direct signaling events that control cytoskeletal rearrangements (37). However, to our knowledge, there is no report that a chemokine GPCR directly interacts with the cytoskeleton. It is known that activated chemokine receptors dissociate Gα and Gβγ subunits, which induce F-actin polymerization via two possible pathways, one involving free Gβγ subunits signaling to small G-proteins, Cdc42, Rac, or Rho, and the other involving free Gβγ activating PI3Kγ (6). Since activated receptors are no longer coupled with free Gβγ, it is unlikely that the receptors directly interact with actin cytoskeletons. Taken together, it is most likely that IL-8 induced a decrease in the mobility of CXCR1-CFP on the plasma membrane as a result of the association between the receptors and raft-like microenvironments.
Depletion of cholesterol impairs CXCR1-triggered Ca2+ responses and activation of PI3K.
Activation of chemokine receptors dissociates Gαi and Gβγ subunits of G-proteins and initiates multiple signal transduction pathways including activation of phospholipase C (PLC) leading to intracellular Ca2+ response and activation of PI3K resulting in phosphorylation of Akt/protein kinase B (PKB) (2, 14, 19, 35, 47). To examine whether lipid raft microenvironments are essential for CXCR1-mediated signaling events, we disrupted these microenvironments on the plasma membrane by depleting cholesterol with MβCD and measured IL-8-triggered Ca2+ responses and activation of PI3K (Fig. 5). As previously shown, IL-8 triggered a clear Ca2+ response in CX1-HEK cells (Fig. 1). Upon the addition of IL-8 to the cell chamber, intracellular Ca2+ concentrations transiently increased from 25 nM to 190 nM (Fig. 5a). After CX1-HEK cells were treated with 10 mM MβCD for 10 min, the same stimulation failed to trigger the Ca2+ response, although CXCR1-CFP receptors and Fluo-4 were appropriately localized (Fig. 5a). The effects of MβCD treatment on IL-8-induced Ca2+ response could be reversed. After the treated cells were incubated in complete medium for 3 h to replenish cholesterol in the plasma membrane, IL-8-induced Ca2+ response was restored (unpublished data). These results suggest that this acute MβCD treatment is generally nontoxic but can temporarily deplete cholesterol from the plasma membrane, which impairs certain steps in the signal transduction pathway mediated by CXCR1. We then tested whether acute MβCD treatment affects CXCR1-triggered activation of PI3K (Fig. 5b). Activation of PI3K produces phosphatidylinositol-3,4,5-triphosphate (PIP3) through phosphorylation of phosphatidylinositol-4,5-bisphosphate (PIP2). Akt/PKB, a serine/threonine kinase that binds to PIP3 through its pleckstrin homology (PH) domain, is phosphorylated by phosphoinositol-dependent kinase I (PDK1) (3, 5). IL-8 was added to CX1-HEK cells that were untreated or treated with 10 mM MβCD for 30 min, and the amount of phosphorylated Akt/PKB in cell lysates was detected by Western blotting using an antibody to the phosphorylated form of Akt/PKB (Fig. 5b). IL-8 stimulation induced Akt/PKB phosphorylation in control cells, and this response was significantly impaired in the MβCD-treated cells. Since activation of either PLC or PI3K is triggered by and diverged from dissociation of Gαi and Gβγ subunits (2, 3, 5, 14, 19, 35, 47) and the MβCD treatment impaired both pathways, we suggest that depletion of cholesterol from the plasma membrane likely impairs a signaling step(s) involved in ligand binding to the receptor-mediated G-protein dissociation. We also examined the effect of MβCD treatment on the chemotaxis of CX1-HEK cells (Fig. 5d). CX1-HEK cells migrated across the filter membrane in response to the gradient of IL-8, while MβCD treatment impaired the CXCR1 receptor-mediated chemotaxis toward the IL-8 gradient.
MβCD treatment does not impair binding of CXCR1 to IL-8 and does not completely abolish IL-8-induced CXCR1 phosphorylation.
It is possible that disruption of lipid raft microenvironments on the plasma membrane causes conformational changes of CXCR1 receptors so that they can no longer bind IL-8, therefore impairing all receptor-mediated signaling functions. We measured the IL-8 binding curve of MβCD-treated CX1-HEK cells and compared the dissociation constant (Kd) and maximal binding sites (Bmax) to Kd and Bmax for nontreated cells (Fig. 6a). MβCD treatment reduced the Bmax on the cell surface but did not affect the dissociation constant (Fig. 6a). The difference in Bmax between nontreated and MβCD-treated CX1-HEK cells was due to the loss of receptors from the plasma membrane. Using live-cell imaging, we found that CFP fluorescence intensity of the plasma membrane gradually decreased, with a loss of about 20% after 30 min of treatment (Fig. 6b). In nontreated cells, such a difference in the receptor number on the plasma membrane did not affect the Ca2+ response upon IL-8 stimulation (data not shown).
Ligands binding to chemokine receptors induce interaction between the C-terminal tails of the receptors and G-protein-coupled-receptor kinases leading to receptor phosphorylation, which is important for receptor desensitization and internalization (25, 33). We found that IL-8-triggered CXCR1-CFP phosphorylation could still be detected in the MβCD-treated cells (Fig. 6c). These results indicate that disruption of lipid raft microenvironments on the plasma membrane does not affect IL-8 binding to CXCR1-CFP and does not completely abolish ligand-induced and G-protein-independent receptor phosphorylation.
G-protein subunits associate with DRM fractions.
Interaction between chemokine receptors and heterotrimeric G-proteins is an essential step in triggering multiple pathways upon ligands binding to the receptors. To examine the distribution of G-proteins on the plasma membrane, cells were treated with 0.5% cold Triton X-100 and whole-cell lysates were subjected to a flotation gradient. We found that over 75% of the Gβ subunits were associated with DRMs of the cells, indicating that the Gβγ subunits are present in the lipid raft microenvironments on the plasma membrane (Fig. 7). Gα subunits were also associated with DRMs, as previously reported (data not shown). In addition, we also found that IL-8-induced enrichment of CXCR1-CFP in the DRMs by flotation assay (Fig. 7b). Addition of IL-8 did not change the distribution pattern of G-protein subunits but induced enrichment of CXCR1-CFP in the DRM. Therefore, the redistribution of the ligand-bound CXCR1 to the lipid rafts rapidly brings the receptor to the site enriched in G-proteins, thus enabling it to signal.
FIG. 7.

Gβ subunits of heterotrimeric G-proteins are associated with DRM fractions, and IL-8-induced enrichment of CXCR1-CFP are associated with DRM fractions. CX1-HEK cells stimulated with IL-8 or unstimulated (control) were treated with 0.5 percent cold Triton X-100, and whole-cell lysates were subjected to a flotation gradient. (a) Fractions of each sample were subjected to Western blotting with antibodies for Gβ subunits. Quantification of the percentage of Gβ subunits in each fraction is shown. (b) Fractions of each sample were subjected to Western blotting with GFP antibodies to detect CXCR1-CFP. A faint band of CXCR1-CFP was detected in fractions 4 and 5 in IL-8-stimulated samples (a longer exposure). A shorter exposure showed that there was a nonspecific band, which is bigger than CXCR1-CFP, appearing in fractions 10, 11, and 12. The positions of molecular mass (MM) markers (in kilodaltons) are shown to the left of the gels.
DISCUSSION
One of the fundamental questions regarding GPCR signaling is how ligand binding induces an interaction between one kind of GPCRs with a set of heterotrimeric G-proteins and triggers activation of the G-proteins in the plasma membrane. Here, we applied live-cell imaging techniques to probe ligand-induced changes in the dynamic distribution and signaling of CXCR1 receptors on the plasma membrane. Using live-cell FRET to monitor the proximity between CXCR1-CFP and either DiIC16 or FastDiI, we found that upon IL-8 stimulation, FRET efficiency between CXCR1-CFP and DiIC16 increased, while that between CXCR1-CFP and FastDiI decreased, indicating that IL-8 binding triggers a microenvironmental change surrounding CXCR1-CFP with lipid raft microdomains. FRAP measurement of the lateral diffusion of a protein in live cells can detect weak, dynamic interactions between the protein and the lipid raft microdomains, which is not detectable by biochemical methods (39). We showed that IL-8-bound CXCR1-CFPs diffuse more slowly than nonbound receptors.
FRET and FRAP measurements are in good agreement, indicating that ligand binding induces CXCR1-CFP partitioning into lipid raft microdomains in the plasma membranes of live cells. We also demonstrated that association between ligand-bound receptors and lipid raft microdomains is essential for heterotrimeric G-protein-dependent signaling pathways. We found that acute MβCD treatment, which disrupts lipid raft microdomains by depleting cholesterol from membranes of live cells, impaired two G-protein-dependent pathways: Ca2+ response and PI3K activation. Both pathways are activated by the CXCR1 receptor through Gβγ subunits that dissociate from Gαi subunits and activate different downstream signaling components (2, 14, 19, 35, 47). However, this MβCD treatment did not affect the ligand-binding ability of CXCR1 and did not completely abolish ligand-induced receptor phosphorylation, which is a G-protein-independent signaling event (3, 5). Furthermore, we found that Gβγ and Gαi subunits are enriched in DRM fractions isolated from live cells with or without exposure to IL-8, suggesting that subunits of G-proteins persistently associate with lipid raft microdomains. We also found that IL-8 induced enrichment of CXCR1-CFP in the DRM fraction. Taken together, our results suggest that ligand-induced changes in the microenvironment surrounding the CXCR1-CFP receptor may facilitate the critical interaction between the chemokine-bound receptors and the heterotrimeric G-proteins in the plasma membrane. This provides a mechanism to spatially separate receptors and G-proteins in the resting state and selectively assemble ligand-bound receptors with G-proteins when the cell is exposed to a chemokine (Fig. 8).
FIG. 8.
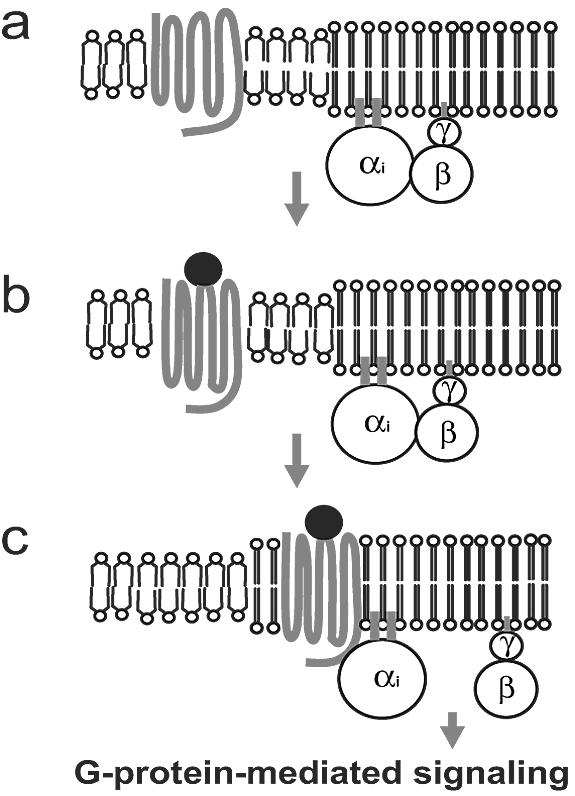
Model of ligand-induced redistribution of chemokine receptors into lipid raft microenvironments triggering G-protein activation. (a) Unbound chemokine receptors localize in the non-lipid raft microenvironment. Subunits of G-proteins are concentrated in the lipid raft microenvironment. (b) When a ligand binds to a chemokine receptor, the receptor undergoes a conformational change that enables it to attract raft lipids. (c) Ligand-bound chemokine receptors associate with lipid raft microenvironments and activate G-proteins, leading to G-protein-mediated signaling.
GPCRs undergo a conformational change when extracellular ligands bind. The molecular mechanism of agonist-induced conformational transition from an inactive state to an active state is shown best by rhodopsin (25). This mechanism can be generalized to other GPCRs, since the structure-function relationship in the basic seven-transmembrane structural motifs is conserved (16). A receptor in the active state has a higher affinity for G-proteins than a receptor in the inactive state. It has been shown in vitro that an agonist-bound human δ-opioid receptor has the highest affinity toward G-proteins (1). Previous studies suggest that the lipid raft microdomains provide a microenvironment for the assembly of multiple signaling components downstream of GPCRs. The chemokine receptors CXCR4 and CCR5 have been identified in lipid rafts both by colocalization with the raft lipid GM1 and by copurification in the DRM fraction. It has been reported that cholesterol extraction by hydroxypropyl-β-cyclodextrin inhibits ligand binding to CXCR4 and CCR5, suggesting that both unbound and ligand-bound receptors are surrounded by lipid raft microdomains (30, 31). However, our results showed that both the ligand-binding ability of CXCR1-CFP and ligand-triggered receptor phosphorylation were not impaired by cholesterol extraction. In addition, unbound CXCR1-CFPs are localized in non-lipid raft microenvironments, whereas ligand-bound receptors are surrounded by lipid raft microenvironments.
It is important to underscore the technical significance of live-cell FRET and FRAP approaches in probing the dynamic distribution of a protein in the plasma membrane. It has been well documented that in the case of tyrosine kinase signaling (for example, T-cell and B-cell receptors for antigen), adaptors, scaffolds, and enzymes are assembled in lipid raft microdomains as a result of receptor activation (8, 15, 24, 32, 40, 43). Although the involvement of lipid rafts in receptor-mediated signaling is compelling, it has become increasingly clear that operational definitions for lipid rafts, such as detergent insolubility to define components of rafts, cholesterol depletion to define raft functions, and immunostaining to define colocalization with rafts on the cell surface, are not adequate to investigate lipid rafts in native cell membrane (8, 24). Our FRET and FRAP analyses revealed that unbound and chemokine-bound receptors are surrounded by different microenvironments in live cells, although both unbound and chemokine-bound CXCR1 receptors were colocalized with DiIC16 and FastDiI. Our results are consistent with the view that different microdomains are dynamic and submicroscopic in the plasma membrane of living cells (8, 17, 24). Our data suggest that ligand binding triggers the conformational changes that alter the receptor's affinity not only to signaling components, such as G-proteins and GRKs, but also to different lipids in the plasma membrane, leading to a partitioning of the receptor with lipid raft microenvironments in which G-proteins are activated.
Our proposed mechanism potentially provides cells with the capability to respond to different ligands through various GPCRs and a common set of G-protein subunits. Leukocytes have the ability to respond to multiple chemokines through different chemokine receptors and Gi proteins (9). How cells direct their path when encountering multiple chemoattractant signals is still unclear. One concept is that GPCRs, including chemokine receptors, exist and potentially function as dimers or oligomers (13, 26, 41). Such a mechanism of interaction between receptors may contribute to the ability of cells to integrate various directional signals from different attractants and thus migrate to the correct destination. This study cannot address the issue of receptor dimerization. Our studies here indicate that the interaction between chemokine receptors and G-proteins is spatially regulated by ligand binding in the plasma membranes of living cells. This mechanism may selectively bring ligand-bound receptors and G-proteins together to effectively transduce and integrate signals through multiple chemokine receptors and a limited set of G-proteins, which could play a crucial role for leukocyte navigation in a complex chemoattractant field.
Acknowledgments
We thank Susan Pierce, Lauren Nelson, and Susan Pierce's lab members for stimulating discussion and reading the manuscript.
REFERENCES
- 1.Alves, I. D., Z. Salamon, E. Varga, H. I. Yamamura, G. Tollin, and V. J. Hruby. 2003. Direct observation of G-protein binding to the human delta-opioid receptor using plasmon-waveguide resonance spectroscopy. J. Biol. Chem. 278:48890-48897. [DOI] [PubMed] [Google Scholar]
- 2.Baggiolini, M., P. Loetscher, and B. Moser. 1995. Interleukin-8 and the chemokine family. Int. J. Immunopharmacol. 17:103-108. [DOI] [PubMed] [Google Scholar]
- 3.Barlic, J., J. D. Andrews, A. A. Kelvin, S. E. Bosinger, M. E. DeVries, L. Xu, T. Dobransky, R. D. Feldman, S. S. Ferguson, and D. Kelvin. 2000. Regulation of tyrosine kinase activation and granule release through beta-arrestin by CXCRI. Nat. Immunol. 1:227-233. [DOI] [PubMed] [Google Scholar]
- 4.Barlic, J., M. H. Khandaker, E. Mahon, J. Andrews, M. E. DeVries, G. B. Mitchell, R. Rahimpour, C. M. Tan, S. S. Ferguson, and D. J. Kelvin. 1999. β-Arrestins regulate interleukin-8-induced CXCR1 internalization. J. Biol. Chem. 274:16287-16294. [DOI] [PubMed] [Google Scholar]
- 5.Cantley, L. C. 2002. The phosphoinositide 3-kinase pathway. Science 296:1655-1657. [DOI] [PubMed] [Google Scholar]
- 6.Chodniewicz, D., and D. V. Zhelev. 2003. Novel pathways of F-actin polymerization in the human neutrophil. Blood 102:2251-2258. [DOI] [PubMed] [Google Scholar]
- 7.Christian, A. E., M. P. Haynes, M. C. Phillips, and G. H. Rothblat. 1997. Use of cyclodextrins for manipulating cellular cholesterol content. J. Lipid Res. 38:2264-2272. [PubMed] [Google Scholar]
- 8.Edidin, M. 2003. Lipids on the frontier: a century of cell-membrane bilayers. Nat. Rev. Mol. Cell Biol. 4:414-418. [DOI] [PubMed] [Google Scholar]
- 9.Foxman, E. F., J. J. Campbell, and E. C. Butcher. 1997. Multistep navigation and the combinatorial control of leukocyte chemotaxis. J. Cell Biol. 139:1349-1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gerard, C., and B. J. Rollins. 2001. Chemokines and disease. Nat. Immunol. 2:108-115. [DOI] [PubMed] [Google Scholar]
- 11.Gomez-Mouton, C., R. A. Lacalle, E. Mira, S. Jimenez-Baranda, D. F. Barber, A. C. Carrera, A. C. Martinez, and S. Manes. 2004. Dynamic redistribution of raft domains as an organizing platform for signaling during cell chemotaxis. J. Cell Biol. 164:759-768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grimm, M. C., A. Ben-Baruch, D. D. Taub, O. M. Howard, J. H. Resau, J. M. Wang, H. Ali, R. Richardson, R. Snyderman, and J. J. Oppenheim. 1998. Opiates transdeactivate chemokine receptors: delta and mu opiate receptor-mediated heterologous desensitization. J. Exp. Med. 188:317-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hernanz-Falcon, P., J. M. Rodriguez-Frade, A. Serrano, D. Juan, A. del Sol, S. F. Soriano, F. Roncal, L. Gomez, A. Valencia, A. C. Martinez, and M. Mellado. 2004. Identification of amino acid residues crucial for chemokine receptor dimerization. Nat. Immunol. 5:216-223. [DOI] [PubMed] [Google Scholar]
- 14.Hirsch, E., V. L. Katanaev, C. Garlanda, O. Azzolino, L. Pirola, L. Silengo, S. Sozzani, A. Mantovani, F. Altruda, and M. P. Wymann. 2000. Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science 287:1049-1053. [DOI] [PubMed] [Google Scholar]
- 15.Jacobson, K., and C. Dietrich. 1999. Looking at lipid rafts? Trends Cell Biol. 9:87-91. [DOI] [PubMed] [Google Scholar]
- 16.Karnik, S. S., C. Gogonea, S. Patil, Y. Saad, and T. Takezako. 2003. Activation of G-protein-coupled receptors: a common molecular mechanism. Trends Endocrinol. Metab. 14:431-437. [DOI] [PubMed] [Google Scholar]
- 17.Kenworthy, A. K., B. J. Nichols, C. L. Remmert, G. M. Hendrix, M. Kumar, J. Zimmerberg, and J. Lippincott-Schwartz. 2004. Dynamics of putative raft-associated proteins at the cell surface. J. Cell Biol. 165:735-746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lefkowitz, R. J., and E. J. Whalen. 2004. β-Arrestins: traffic cops of cell signaling. Curr. Opin. Cell Biol. 16:162-168. [DOI] [PubMed] [Google Scholar]
- 19.Li, Z., H. Jiang, W. Xie, Z. Zhang, A. V. Smrcka, and D. Wu. 2000. Roles of PLC-β2 and -β3 and PI3Kγ in chemoattractant-mediated signal transduction. Science 287:1046-1049. [DOI] [PubMed] [Google Scholar]
- 20.Lippincott-Schwartz, J., E. Snapp, and A. Kenworthy. 2001. Studying protein dynamics in living cells. Nat. Rev. Mol. Cell Biol. 2:444-456. [DOI] [PubMed] [Google Scholar]
- 21.Luther, S. A., and J. G. Cyster. 2001. Chemokines as regulators of T cell differentiation. Nat. Immunol. 2:102-107. [DOI] [PubMed] [Google Scholar]
- 22.Mackay, C. R. 2001. Chemokines: immunology's high impact factors. Nat. Immunol. 2:95-101. [DOI] [PubMed] [Google Scholar]
- 23.Manes, S., R. A. Lacalle, C. Gomez-Mouton, G. del Real, E. Mira, and A. C. Martinez. 2001. Membrane raft microdomains in chemokine receptor function. Semin. Immunol. 13:147-157. [DOI] [PubMed] [Google Scholar]
- 24.Maxfield, F. R. 2002. Plasma membrane microdomains. Curr. Opin. Cell Biol. 14:483-487. [DOI] [PubMed] [Google Scholar]
- 25.Meng, E. C., and H. R. Bourne. 2001. Receptor activation: what does the rhodopsin structure tell us? Trends Pharmacol. Sci. 22:587-593. [DOI] [PubMed] [Google Scholar]
- 26.Milligan, G., D. Ramsay, G. Pascal, and J. J. Carrillo. 2003. GPCR dimerisation. Life Sci. 74:181-188. [DOI] [PubMed] [Google Scholar]
- 27.Moser, B., and P. Loetscher. 2001. Lymphocyte traffic control by chemokines. Nat. Immunol. 2:123-128. [DOI] [PubMed] [Google Scholar]
- 28.Mukherjee, S., T. T. Soe, and F. R. Maxfield. 1999. Endocytic sorting of lipid analogues differing solely in the chemistry of their hydrophobic tails. J. Cell Biol. 144:1271-1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murphy, P. M. 1994. The molecular biology of leukocyte chemoattractant receptors. Annu. Rev. Immunol. 12:593-633. [DOI] [PubMed] [Google Scholar]
- 30.Nguyen, D. H., and. D. Taub. 2002. Cholesterol is essential for macrophage inflammatory protein 1 beta binding and conformational integrity of CC chemokine receptor 5. Blood 99:4298-4306. [DOI] [PubMed] [Google Scholar]
- 31.Nguyen, D. H., and D. Taub. 2002. CXCR4 function requires membrane cholesterol: implications for HIV infection. J. Immunol. 168:4121-4126. [DOI] [PubMed] [Google Scholar]
- 32.Pierce, S. K. 2002. Lipid rafts and B-cell activation. Nat. Rev. Immunol. 2:96-105. [DOI] [PubMed] [Google Scholar]
- 33.Richardson, R. M., H. Ali, B. C. Pridgen, B. Haribabu, and R. Snyderman. 1998. Multiple signaling pathways of human interleukin-8 receptor A. Independent regulation by phosphorylation. J. Biol. Chem. 273:10690-10695. [DOI] [PubMed] [Google Scholar]
- 34.Richardson, R. M., R. A. DuBose, H. Ali, E. D. Tomhave, B. Haribabu, and R. Snyderman. 1995. Regulation of human interleukin-8 receptor A: identification of a phosphorylation site involved in modulating receptor functions. Biochemistry 34:14193-14201. [DOI] [PubMed] [Google Scholar]
- 35.Sasaki, T., J. Irie-Sasaki, R. G. Jones, A. J. Oliveira-dos-Santos, W. L. Stanford, B. Bolon, A. Wakeham, A. Itie, D. Bouchard, I. Kozieradzki, N. Joza, T. W. Mak, P. S. Ohashi, A. Suzuki, and J. M. Penninger. 2000. Function of PI3Kγ in thymocyte development, T cell activation, and neutrophil migration. Science 287:1040-1046. [DOI] [PubMed] [Google Scholar]
- 36.Sekar, R. B., and A. Periasamy. 2003. Fluorescence resonance energy transfer (FRET) microscopy imaging of live cell protein localizations. J. Cell Biol. 160:629-633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seveau, S., R. J. Eddy, F. R. Maxfield, and L. M. Pierini. 2001. Cytoskeleton-dependent membrane domain segregation during neutrophil polarization. Mol. Biol. Cell 12:3550-3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sheets, E. D., D. Holowka, and B. Baird. 1999. Critical role for cholesterol in Lyn-mediated tyrosine phosphorylation of FɛRI and their association with detergent-resistant membranes. J. Cell Biol. 145:877-887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shvartsman, D. E., M. Kotler, R. D. Tall, M. G. Roth, and Y. I. Henis. 2003. Differently anchored influenza hemagglutinin mutants display distinct interaction dynamics with mutual rafts. J. Cell Biol. 163:879-888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Simons, K., and E. Ikonen. 1997. Functional rafts in cell membranes. Nature 387:569-572. [DOI] [PubMed] [Google Scholar]
- 41.Terrillon, S., and M. Bouvier. 2004. Roles of G-protein-coupled receptor dimerization. EMBO Rep. 5:30-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thelen, M. 2001. Dancing to the tune of chemokines. Nat. Immunol. 2:129-134. [DOI] [PubMed] [Google Scholar]
- 43.van Meer, G. 2002. Cell biology. The different hues of lipid rafts. Science 296:855-857. [DOI] [PubMed] [Google Scholar]
- 44.Venkatesan, S., J. J. Rose, R. Lodge, P. M. Murphy, and J. F. Foley. 2003. Distinct mechanisms of agonist-induced endocytosis for human chemokine receptors CCR5 and CXCR4. Mol. Biol. Cell 14:3305-3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wells, T. N., C. A. Power, and A. E. Proudfoot. 1998. Definition, function and pathophysiological significance of chemokine receptors. Trends Pharmacol. Sci. 19:376-380. [DOI] [PubMed] [Google Scholar]
- 46.Zacharias, D. A., J. D. Violin, A. C. Newton, and R. Y. Tsien. 2002. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science 296:913-916. [DOI] [PubMed] [Google Scholar]
- 47.Zeilhofer, H. U., and W. Schorr. 2000. Role of interleukin-8 in neutrophil signaling. Curr. Opin. Hematol. 7:178-182. [DOI] [PubMed] [Google Scholar]