

Abstract
Recent studies demonstrate that oxidative inactivation of tetrahydrobiopterin (H4B) may cause uncoupling of endothelial nitric oxide synthase (eNOS) to produce superoxide (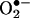 ). H4B was found recyclable from its oxidized form by dihydrofolate reductase (DHFR) in several cell types. Functionality of the endothelial DHFR, however, remains completely unknown. Here we present findings that specific inhibition of endothelial DHFR by RNA interference markedly reduced endothelial H4B and nitric oxide (NO·) bioavailability. Furthermore, angiotensin II (100 nmol/liter for 24 h) caused a H4B deficiency that was mediated by H2O2-dependent down-regulation of DHFR. This response was associated with a significant increase in endothelial
). H4B was found recyclable from its oxidized form by dihydrofolate reductase (DHFR) in several cell types. Functionality of the endothelial DHFR, however, remains completely unknown. Here we present findings that specific inhibition of endothelial DHFR by RNA interference markedly reduced endothelial H4B and nitric oxide (NO·) bioavailability. Furthermore, angiotensin II (100 nmol/liter for 24 h) caused a H4B deficiency that was mediated by H2O2-dependent down-regulation of DHFR. This response was associated with a significant increase in endothelial 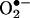 production, which was abolished by eNOS inhibitor N-nitro-l-arginine-methyl ester or H2O2 scavenger polyethylene glycol-conjugated catalase, strongly suggesting H2O2-dependent eNOS uncoupling. Rapid and transient activation of endothelial NAD(P)H oxidases was responsible for the initial burst production of
production, which was abolished by eNOS inhibitor N-nitro-l-arginine-methyl ester or H2O2 scavenger polyethylene glycol-conjugated catalase, strongly suggesting H2O2-dependent eNOS uncoupling. Rapid and transient activation of endothelial NAD(P)H oxidases was responsible for the initial burst production of 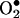 (Rac1 inhibitor NSC 23766 but not an N-nitro-l-arginine-methyl ester-attenuated ESR
(Rac1 inhibitor NSC 23766 but not an N-nitro-l-arginine-methyl ester-attenuated ESR 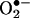 signal at 30 min) in response to angiotensin II, preceding a second peak in
signal at 30 min) in response to angiotensin II, preceding a second peak in 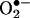 production at 24 h that predominantly depended on uncoupled eNOS. Overexpression of DHFR restored NO· production and diminished eNOS production of
production at 24 h that predominantly depended on uncoupled eNOS. Overexpression of DHFR restored NO· production and diminished eNOS production of 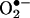 in angiotensin II-stimulated cells. In conclusion, these data represent evidence that DHFR is critical for H4B and NO· bioavailability in the endothelium. Endothelial NAD(P)H oxidase-derived H2O2 down-regulates DHFR expression in response to angiotensin II, resulting in H4B deficiency and uncoupling of eNOS. This signaling cascade may represent a universal mechanism underlying eNOS dysfunction under pathophysiological conditions associated with oxidant stress.
in angiotensin II-stimulated cells. In conclusion, these data represent evidence that DHFR is critical for H4B and NO· bioavailability in the endothelium. Endothelial NAD(P)H oxidase-derived H2O2 down-regulates DHFR expression in response to angiotensin II, resulting in H4B deficiency and uncoupling of eNOS. This signaling cascade may represent a universal mechanism underlying eNOS dysfunction under pathophysiological conditions associated with oxidant stress.
Keywords: hydrogen peroxide, tetrahydrobiopterin, superoxide
It has become apparent over the past decade that excessive production of reactive oxygen species (ROS) contributes to cardiovascular pathogenesis (1–3). Indeed, scavenging ROS restored endothelial function in animal models of hypertension and atherosclerosis (3). Antioxidant trials however produced mixed results in patients regarding disease prevention or regression, possibly because of incomplete understanding of antioxidant chemistry in vivo, inappropriate criteria for patient recruitment (4, 5), or nonspecific nature of most antioxidants. The latter is an important issue to consider because it has remained unclear whether one particular ROS is dominantly important in pathological signaling (1). Additionally, mixed results may suggest that some downstream pathological events are not readily reversible by removal of ROS. Recent studies demonstrate that endothelial nitric oxide synthase (eNOS) (and other isoforms of NOS) is capable of transforming into a prooxidant, 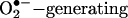 enzyme in vitro and in vivo (1, 3, 6–15). This phenomenon is now referred to as “eNOS uncoupling.” It is interesting to speculate that unsatisfactory outcomes of some antioxidant therapies are partially due to their ineffectiveness in restoring eNOS function in the vasculature or, in other words, “recoupling” of eNOS.
enzyme in vitro and in vivo (1, 3, 6–15). This phenomenon is now referred to as “eNOS uncoupling.” It is interesting to speculate that unsatisfactory outcomes of some antioxidant therapies are partially due to their ineffectiveness in restoring eNOS function in the vasculature or, in other words, “recoupling” of eNOS.
Although molecular mechanisms underlying eNOS uncoupling remain to be fully elucidated, a deficiency in eNOS cofactor tetrahydrobiopterin (H4B) appears to be a major cause (6, 8, 10–14). The precise mechanisms as to how H4B became persistently deficient under pathological conditions, however, remain obscure. Dihydrofolate reductase (DHFR) catalyzes regeneration of H4B from its oxidized form, dihydropterin, in several cell types (16, 17). Genes encoding DHFR have also been cloned. Functionality of the endothelial DHFR, however, remains completely unknown. Here we found that specific inhibition of endothelial DHFR through RNA interference (RNAi) led to a marked reduction in endothelial H4B and NO· bioavailability. In hypertension- or diabetes-related atherosclerosis, angiotensin II is a major pathological player (5). Many of the untoward effects of angiotensin II have been attributed to its ability to stimulate ROS production from vascular NAD(P)H oxidases (1, 2). Here we found that angiotensin II rapidly and transiently activated endothelial NAD(P)H oxidases to produce 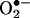 within 30 min, leading to H2O2-dependent down-regulation of DHFR, H4B deficiency, and a second phase
within 30 min, leading to H2O2-dependent down-regulation of DHFR, H4B deficiency, and a second phase 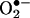 production from uncoupled eNOS after 24 h. Overexpression of DHFR restored NO· production and abolished eNOS production of
production from uncoupled eNOS after 24 h. Overexpression of DHFR restored NO· production and abolished eNOS production of 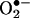 in angiotensin II-stimulated cells. These data characterize an essential role of endothelial DHFR in maintaining H4B and NO· bioavailability in the endothelium. The observation that NAD(P)H oxidase-derived H2O2 mediates angiotensin II uncoupling of eNOS via down-regulation of DHFR may have general applicability to other pathological conditions in which eNOS dysfunction occurs.
in angiotensin II-stimulated cells. These data characterize an essential role of endothelial DHFR in maintaining H4B and NO· bioavailability in the endothelium. The observation that NAD(P)H oxidase-derived H2O2 mediates angiotensin II uncoupling of eNOS via down-regulation of DHFR may have general applicability to other pathological conditions in which eNOS dysfunction occurs.
Experimental Procedures
Materials. Monoclonal antibodies for DHFR and eNOS were purchased from Research Diagnostics (Flanders, NJ) and Transduction Laboratories (San Jose, CA), respectively. Fluorescent, double-stranded small interfering RNA (siRNA) targeting human DHFR (nucleotides 35–54, 5′-cccagaacatgggcatcggc-3′; GenBank accession no. NM_000791) and scrambled control siRNA recognizing none of the known human or bovine sequences were customized at Dharmacon Research (Lafayette, CO). H4B was obtained from Sigma–Aldrich and Schircks Laboratories (Jona, Switzerland). Rac1 inhibitor NSC 23766 was obtained from EMD Biosciences (San Diego). Other chemicals were purchased from Sigma at the highest purity available. Human DHFR cDNA in pcDNA3.1 vector was a kind gift from Edward Chu and Ningwen Tai (Yale University, New Haven, CT). Aorta from mice deficient in angiotensin converting enzyme (ACE–/–) were kindly provided by Kenneth E. Bernstein and Hong Xiao (Emory University, Atlanta).
Cell Culture and Western Analysis. Bovine aortic endothelial cells (Cell Systems, Kirkland, WA) were cultured to confluence as described in refs. 18–20 and starved in 5% media overnight before experiments. For Western analysis, 20–40 μg of protein was separated by 10% SDS/PAGE, transferred to nitrocellulose membranes, and probed with DHFR (1:250) or eNOS (1:1,000) antibodies after standard Western procedure (21).
Transfection of Endothelial Cells with DHFR siRNAs or pcDNA3.1-DHFR. Proliferating endothelial cells at 85% confluency were incubated with siRNA-oligofectamine mixtures for 4 h before addition of growth media. Cells were harvested 48 h later for analysis of DHFR expression, H4B content, and NO· production. Additionally, proliferating cells were transfected with 8 μg of pcDNA3.1-DHFR plasmid by using Lipofectamine for 48 h before angiotensin II stimulation and analysis of endothelial 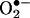 and NO· productions.
and NO· productions.
ESR Detection of NO Radical. Bioavailable NO· radical was directly measured by using ESR (18, 20). In brief, endothelial cells cultured on Petri dishes were rinsed with modified Kreb's/Hepes buffer and incubated with freshly prepared, NO·-specific spin trap Fe2+(diethyldithiocarbamate)2 colloid (0.5 mmol/liter) for 60 min. Gently collected suspensions of cells were snap-frozen in liquid nitrogen and loaded into a finger Dewar for analysis with a Miniscope 200 ESR spectrophotometer (Magnettech, Berlin) at the following settings: biofield, 3267; field sweep, 100 G (1 G = 0.1 mT); microwave frequency, 9.78 GHz; microwave power, 40 mW; modulation amplitude, 10 G; 4,096 points of resolution; and receiver gain, 900.
ESR Detection of Superoxide Radical. Gently collected endothelial cells were suspended in modified Kreb's/HEPEs buffer containing deferoximine (25 μmol/liter, metal chelator). Approximately 1 × 106 cells were mixed with 1 mmol/liter 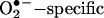 spin trap 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine in the presence or absence of 100 units/ml polyethylene glycol (PEG)-conjugated
spin trap 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine in the presence or absence of 100 units/ml polyethylene glycol (PEG)-conjugated 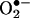 dismutase (22). The cell mixture loaded in glass capillaries was immediately analyzed for
dismutase (22). The cell mixture loaded in glass capillaries was immediately analyzed for 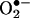 production kinetically for 10 min. The ESR settings were as follows: biofield, 3350; field sweep, 60 G; microwave frequency, 9.78 GHz; microwave power, 20 mW; modulation amplitude, 3 G; 4,096 points of resolution; receiver gain, 500; and kinetic time, 10 min. The
production kinetically for 10 min. The ESR settings were as follows: biofield, 3350; field sweep, 60 G; microwave frequency, 9.78 GHz; microwave power, 20 mW; modulation amplitude, 3 G; 4,096 points of resolution; receiver gain, 500; and kinetic time, 10 min. The 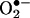 dismutase-inhibitable
dismutase-inhibitable 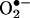 signals at a 10 min time point and normalized by protein concentrations were compared among different experimental groups.
signals at a 10 min time point and normalized by protein concentrations were compared among different experimental groups.
HPLC Determination of Endothelial H4B Content. Endothelial cells were lysed by using trichloroacetic acid containing 10 mmol/liter DTT. Lysates were subjected to differential oxidation in acidic (0.2 M trichloroacetic acid) or alkalytic (0.1 M NaOH) solutions containing 2.5% I2/10% KI or 0.9% I2/1.8% KI. After centrifugation, 20 μl of supernatant was injected into a HPLC system equipped with a 250- × 4.6-mm C18 column (Alltech, Deerfield, IL) and a highly sensitive fluorescent detector (Schimadzu model RF-10Axl, Fisher). Excitation and emission wavelengths of 350 nm and 450 nm were used to detect fluorescent H4B and its oxidized species (15). Because H4B is protected under acidic conditions and converted to pterin under alkalytic conditions, the difference in peak intensity reflects H4B content. The absolute H4B contents were calculated against a standard curve prepared by using purified H4B that went through identical extraction procedures and presented as pmol/mg protein.
Statistical Analysis. Data are presented in mean ± SEM from six to 10 independent experiments. Differences in H4B and NO· levels between control or DHFR siRNA-transfected cells were compared by using a paired Student t test. Likewise, protein levels of DHFR and DHFR/eNOS ratio in cells treated with angiotensin II or H2O2 in aortae of ACE–/– were compared with the controls by using Student's t test. H4B or 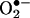 levels among control, angiotensin II- or H2O2-stimulated cells, and cells pretreated with PEG-conjugated catalase (PEG-CAT) or N-nitro-l-arginine-methyl ester (l-NAME) was compared by using ANOVA, followed by Dunnett's post hoc test. Similarly,
levels among control, angiotensin II- or H2O2-stimulated cells, and cells pretreated with PEG-conjugated catalase (PEG-CAT) or N-nitro-l-arginine-methyl ester (l-NAME) was compared by using ANOVA, followed by Dunnett's post hoc test. Similarly, 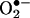 and NO· productions in pcDNA3.1-DHFR transfected cells with or without angiotensin II stimulation were compared with the controls by using ANOVA. Differences in
and NO· productions in pcDNA3.1-DHFR transfected cells with or without angiotensin II stimulation were compared with the controls by using ANOVA. Differences in 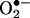 production in presence or absence the of l-NAME or NSC 23766 at different time points after angiotensin II stimulation was also analyzed with ANOVA. Statistical significance was set for P < 0.05.
production in presence or absence the of l-NAME or NSC 23766 at different time points after angiotensin II stimulation was also analyzed with ANOVA. Statistical significance was set for P < 0.05.
Results
RNAi Silencing of DHFR Reduces Endothelial H4B and NO· Bioavailability. DHFR recycles H4B in several cell types (16, 17). Its functionality within endothelium particularly regarding H4B and NO· bioavailability, however, remains unclear. We therefore first examined the consequences of specific disruption of endothelial DHFR by using RNAi. As demonstrated by the fluorescent images in Fig. 1A, siRNAs were successfully transfected into ≈95% of the cells. DHFR-targeting siRNA markedly decreased DHFR protein expression by >90% (Fig. 1B), consistent with the findings by Tai et al. (23). Endothelial H4B content was 1.89 ± 0.11 pmol/mg protein in control siRNA-transfected cells and was significantly decreased to 1.26 ± 0.11 pmol/mg protein in cells transfected with DHFR siRNA (Fig. 1C). Importantly, RNAi-induced specific disruption of DHFR was associated with a substantial decline in endothelial cell production of NO· (Fig. 2).
Fig. 1.

Effects of DHFR siRNA on endothelial H4B bioavailability. Proliferating endothelial cells were transfected with 25 nmol/liter control or DHFR siRNA for 48 h before analysis of DHFR expression and H4B content. (A) Fluorescent images of transfected cells. (B) Western blot of DHFR protein expression. (C) Endothelial H4B content determined by HPLC (n = 6). Data are presented as mean ± SEM. *, P < 0.05.
Fig. 2.

Effects of DHFR siRNA on endothelial NO· bioavailability. Proliferating endothelial cells were transfected with 25 nmol/liter control or DHFR siRNA for 48 h before analysis of NO· production by using ESR. (A) Representative ESR spectra. (B) Grouped data presented as mean ± SEM (n = 6). *, P < 0.05.
H2O2 Down-Regulation of DHFR Expression. Because DHFR recycles H4B from its oxidized form, regulation of DHFR under oxidant stress is particularly relevant to vascular pathophysiology. H2O2 directly produced or derived from 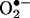 has emerged as a signaling intermediate in vascular cells, mediating a variety of gene regulatory responses (1, 2). Interestingly, exogenously applied H2O2 (at 50, 100, or 200 μmol/liter for 24 h) dose-dependently down-regulated endothelial DHFR protein expression (Fig. 3 A and B). H2O2 decreased the DHFR/eNOS ratio from 1.6 ± 0.28 to 0.6 ± 0.10 in the controls (Fig. 3C), implying that substantially less DHFR per se is available to supply recycled H4B to eNOS.
has emerged as a signaling intermediate in vascular cells, mediating a variety of gene regulatory responses (1, 2). Interestingly, exogenously applied H2O2 (at 50, 100, or 200 μmol/liter for 24 h) dose-dependently down-regulated endothelial DHFR protein expression (Fig. 3 A and B). H2O2 decreased the DHFR/eNOS ratio from 1.6 ± 0.28 to 0.6 ± 0.10 in the controls (Fig. 3C), implying that substantially less DHFR per se is available to supply recycled H4B to eNOS.
Fig. 3.

H2O2 down-regulation of DHFR expression. Confluent endothelial cells were exposed to H2O2 (50–200 μmol/liter) for 24 h before Western analysis of DHFR expression. (A) Representative Western blot of DHFR protein expression. (B) Grouped densitometric data of DHFR expression in response to 100 μmol/liter H2O2 (n = 8). (C) Grouped data on DHFR/eNOS ratio (n = 8). Data are presented at mean ± SEM. *, P < 0.05.
Angiotensin II Down-Regulation of DHFR Expression. Elevated angiotensin II levels have been observed in patients with atherosclerotic risk factors, including hypertension, diabetes, and renal dysfunction (2, 5). Interestingly, angiotensin II stimulation (100 nmol/liter for 24 h) of endothelial cells decreased DHFR expression by >50%, as reflected by representative Western blot and grouped data (Fig. 4 A and B). Similar to H2O2, angiotensin II shifted DHFR/eNOS ratio from 1.5 ± 0.17 to 0.6 ± 0.16 in the controls (Fig. 4C). In ACE–/– mice, for which angiotensin II production was diminished, aortic expression of DHFR was more than doubled compared with wild-type controls (Fig. 4D). Taken together, these data strongly suggest that angiotensin II down-regulates DHFR expression under physiological circumstances and that a pathological level of angiotensin II further reduces DHFR abundance.
Fig. 4.

Angiotensin II (Ang II) down-regulation of DHFR expression. Confluent endothelial cells were exposed to angiotensin II (100 nmol/liter) for 24 h before analysis of DHFR and eNOS protein expression. (A) Representative Western blot of DHFR protein expression. (B) Grouped densitometric data of DHFR expression (n = 6). (C) Grouped data on DHFR/eNOS ratio (n = 6). (D) Representative Western blot and grouped data on aortic DHFR expression from wild-type and ACE–/– mice (n = 6). Data are presented at mean ± SEM. *, P < 0.05.
Angiotensin II-Induced H4B Deficiency Is Mediated by H2O2 Down-Regulation of DHFR. Loss of DHFR is anticipated to blunt H4B recycling. Thus, total biopterin and H4B contents in angiotensin II-stimulated (100 nmol/liter for 24 h) or H2O2-stimulated (100 μmol/liter for 24 h) cells were determined by HPLC. Angiotensin II had little effect on endothelial total biopterin content (Fig. 5A). H2O2 however increased biopterin content modestly by 30% (Fig. 5A). These data seem consistent with the previous report that H2O2 up-regulated H4B synthetic enzyme GTP cyclohydrolase I (GTP-CH I) (24). Interestingly, endothelial H4B levels were significantly decreased by angiotensin II or H2O2 (1.38 ± 0.22 in angiotensin II- vs. 1.24 ± 0.27 in H2O2-treated cells, compared with 2.42 ± 0.34 pmol/mg protein in confluent control cells) (Fig. 5B). Importantly, scavenging of intracellular H2O2 with a pretreatment of 100 units/ml PEG-CAT for 1 h preserved H4B content (1.93 ± 0.15 pmol/mg protein) while having no effect on total biopterin, clearly implicating an intermediate role of H2O2 in angiotensin II induction of H4B deficiency (Fig. 5B). In addition, PEG-CAT prevented angiotensin II down-regulation of DHFR (Fig. 5C).
Fig. 5.

Role of H2O2 in angiotensin II (Ang II) induction of H4B deficiency. Confluent endothelial cells were exposed to angiotensin II (100 nmol/liter) or H2O2 (100 μmol/liter) for 24 h before analysis of endothelial H4B. Some cells were pretreated with 100 units/ml PEG-CAT for 1 h before angiotensin II stimulation. (A) Total biopterin content (n = 6). (B) H4B content. (C) DHFR expression in angiotensin II- or H2O2-stimulated cells with or without PEG-CAT preincubation (n = 6). Data are presented as mean ± SEM. *, P < 0.05.
Angiotensin II Uncoupling of eNOS. Role of H2O2 and DHFR. Because H2O2 mediates angiotensin II induction of H4B deficiency, we hypothesized that H2O2 may contribute to eNOS uncoupling by angiotensin II. As illustrated in Fig. 6, angiotensin II (100 nmol/liter for 24 h) induced a significant increase in endothelial 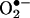 production. This response was completely attenuated by the eNOS inhibitor l-NAME (100 μmol/liter) or PEG-CAT (100 units/ml), strongly implying H2O2-dependent uncoupling of eNOS.
production. This response was completely attenuated by the eNOS inhibitor l-NAME (100 μmol/liter) or PEG-CAT (100 units/ml), strongly implying H2O2-dependent uncoupling of eNOS.
Fig. 6.

Role of H2O2 in angiotensin II (Ang II) uncoupling of eNOS. Confluent endothelial cells were exposed to angiotensin II (100 nmol/liter) for 24 h in the presence or absence of preincubation with NOS inhibitor l-NAME (100 μmol/liter for 1 h) or PEG-CAT (100 units/ml for 1 h) and subsequently analyzed for 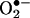 production by using ESR. (A) Representative ESR spectra. (B) Grouped data presented as mean ± SEM (n = 6). *, P < 0.05.
production by using ESR. (A) Representative ESR spectra. (B) Grouped data presented as mean ± SEM (n = 6). *, P < 0.05.
To directly examine the role of DHFR in angiotensin II uncoupling of eNOS, we transfected endothelial cells with mammalian expression vector containing human DHFR, pcDNA3.1-DHFR. Whereas angiotensin II (100 nmol/liter for 24 h) consistently stimulated 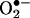 production in reagent-only-cells, it failed to evoke endothelial production of
production in reagent-only-cells, it failed to evoke endothelial production of 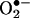 transfected in DHFR-transfected cells (Fig. 7A). DHFR overexpression also restored NO· production in angiotensin II-stimulated cells (Fig. 7B). These data strongly suggest that DHFR availability is critical in maintaining the coupling state of eNOS.
transfected in DHFR-transfected cells (Fig. 7A). DHFR overexpression also restored NO· production in angiotensin II-stimulated cells (Fig. 7B). These data strongly suggest that DHFR availability is critical in maintaining the coupling state of eNOS.
Fig. 7.

Role of DHFR in angiotensin II-induced H4B deficiency and eNOS uncoupling. Proliferating endothelial cells were transfected with pcDNA3.1-DHFR for 48 h before exposure to 100 nmol/liter angiotensin II (Ang II) for 24 h. (A) Endothelial 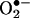 production determined by ESR (n = 4). (B) Endothelial NO· production determined by ESR (n = 6). Data are presented as mean ± SEM. *, P < 0.05.
production determined by ESR (n = 4). (B) Endothelial NO· production determined by ESR (n = 6). Data are presented as mean ± SEM. *, P < 0.05.
Role of Endothelial NAD(P)H Oxidases. To examine whether a transient or continuous production of 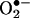 is required for H2O2-dependent eNOS uncoupling and to determine the enzymatic source responsible for the initial
is required for H2O2-dependent eNOS uncoupling and to determine the enzymatic source responsible for the initial 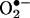 production, were exposed to cells angiotensin II for 30 min, 6 h, or 24 h. As is evident in Fig. 8A, angiotensin II stimulated a rapid production of
production, were exposed to cells angiotensin II for 30 min, 6 h, or 24 h. As is evident in Fig. 8A, angiotensin II stimulated a rapid production of 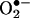 at 30 min, which was unaffected by 100 μmol/liter l-NAME but abolished by 50 μmol/liter Rac1 inhibitor NSC 23766, indicating a role of endothelial NAD(P)H oxidases. This initial production of
at 30 min, which was unaffected by 100 μmol/liter l-NAME but abolished by 50 μmol/liter Rac1 inhibitor NSC 23766, indicating a role of endothelial NAD(P)H oxidases. This initial production of 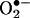 was transient because it diminished at 6 h (Fig. 8A). Twenty-four hours after angiotensin II, there was a potent second peak in
was transient because it diminished at 6 h (Fig. 8A). Twenty-four hours after angiotensin II, there was a potent second peak in 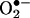 production, which was completely and consistently attenuated by l-NAME and unaffected by NSC 23766, implicating that uncoupled eNOS was predominantly responsible for
production, which was completely and consistently attenuated by l-NAME and unaffected by NSC 23766, implicating that uncoupled eNOS was predominantly responsible for 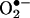 production after prolonged exposure to angiotensin II (Fig. 8A). Of note, DHFR expression did not change at 30 min, showed a slight downward trend at 6 h, but was markedly diminished at 24 h (Fig. 8B). Take together, these data demonstrate that an initial and transient activation of endothelial NAD(P)H oxidases in response to angiotensin II lies upstream of H2O2-dependent down-regulation of DHFR and eNOS uncoupling. The overall findings of the study are summarized thematically in Fig. 9.
production after prolonged exposure to angiotensin II (Fig. 8A). Of note, DHFR expression did not change at 30 min, showed a slight downward trend at 6 h, but was markedly diminished at 24 h (Fig. 8B). Take together, these data demonstrate that an initial and transient activation of endothelial NAD(P)H oxidases in response to angiotensin II lies upstream of H2O2-dependent down-regulation of DHFR and eNOS uncoupling. The overall findings of the study are summarized thematically in Fig. 9.
Fig. 8.

Rapid and transient activation of endothelial NAD(P)H oxidases precedes angiotensin II (Ang II) uncoupling of eNOS. Confluent endothelial cells were exposed to angiotensin II (100 nmol/liter) for 30 min, 6 h, and 24 h before ESR analysis of 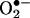 production in the presence or absence of Rac1 antagonist NSC 23766 (50 μmol/liter) or l-NAME (100 μmol/liter). (A) Grouped data on
production in the presence or absence of Rac1 antagonist NSC 23766 (50 μmol/liter) or l-NAME (100 μmol/liter). (A) Grouped data on 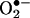 production at each time point (n = 8–10). (B) Representative Western blot and grouped data of DHFR protein expression at each time point (n = 10). Data are presented as mean ± SEM. *, P < 0.05.
production at each time point (n = 8–10). (B) Representative Western blot and grouped data of DHFR protein expression at each time point (n = 10). Data are presented as mean ± SEM. *, P < 0.05.
Fig. 9.

Schematic mechanisms underlying angiotensin II uncoupling of eNOS. Angiotensin II induces a rapid and transient activation of endothelial NAD(P)H oxidases (attenuatable by Rac1 inhibitor NSC 23766), leading to an initial burst production of 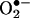 and formation of H2O2. H2O2 in turn down-regulates DHFR to induce H4B deficiency and eNOS uncoupling. These findings may represent a universal mechanism whereby eNOS dysfunction occurs under conditions associated with oxidant stress.
and formation of H2O2. H2O2 in turn down-regulates DHFR to induce H4B deficiency and eNOS uncoupling. These findings may represent a universal mechanism whereby eNOS dysfunction occurs under conditions associated with oxidant stress.
Discussion
The present study elucidated that DHFR is critical for H4B and NO· bioavailability in the endothelium. Angiotensin II down-regulates DHFR in a H2O2-dependent manner, and this response is involved in angiotensin II-induced H4B deficiency and eNOS uncoupling. The initiating source of H2O2 appears to be the endothelial NAD(P)H oxidases, which lie upstream of uncoupled eNOS in response to angiotensin II. This cascade of signaling events, as illustrated in Fig. 9, may represent a common mechanism underlying eNOS dysfunction under situations associated with oxidant stress.
H4B belongs to a large pterin family. Pterins are 2-amino-4-oxo derivatives of pteridines, which, in turn, represent a large and structurally varied group of natural compounds involved in the biosynthetic pathways of cofactors and vitamins (25). Although both oxidized and reduced pterins are detectable in physiological fluid and tissues, only H4B is biologically active (25). The de novo synthesis of H4B involves the rate-limiting GTP-CH I, 6-pyruvoyl-H4B synthase and sepiapterin reductase (16, 17). When H4B is oxidized to dihydropterin, it is recycled by DHFR in several cell types (16, 17). Previous studies demonstrated that inhibition of GTP-CH I impaired endothelium-dependent relaxation (26). Overexpression of GTP-CH I was found partially effective in improving endothelial function in diabetic (27) and hypertensive rodents (28). However, functional regulation of the salvage enzyme DHFR, which could be more crucial in determining H4B and NO· bioavailability under pathophysiological conditions associated with oxidant stress, remains obscure. Here we found that RNAi silencing of DHFR substantially reduced H4B and NO· levels in cultured endothelial cells, implying an important role of DHFR in maintaining NO· bioavailability under physiological conditions.
Furthermore, endothelial DHFR expression was significantly down-regulated by pathological concentrations of angiotensin II. The ACE–/– mice had a doubling in aortic expression of DHFR compared with the wide-type controls. These data indicate that angiotensin II modulates DHFR under pathophysiological circumstances. Of caution, these observations do not necessarily rule out potential compensatory regulations of DHFR in angiotensin II-associated hypertension, where mechanical forces or inflammatory substances may regulate the enzyme differentially. Angiotensin II may also regulate DHFR differently in other vascular cells, such as vascular smooth muscle, whose H4B bioavailability, however, is unrelated to endothelial NO· production. Of note, angiotensin II down-regulation of endothelial DHFR was H2O2-dependent. This observation shares similarity with previous findings that angiotensin II modulates expression of endothelial adhesion molecules by means of H2O2-dependent mechanisms (29).
In addition, H2O2-dependent down-regulation of DHFR mediated angiotensin II induction of H4B deficiency and eNOS uncoupling. Angiotensin II stimulation led to a significant reduction in endothelial H4B content, which was preventable by removal of H2O2. It was previously reported that exogenous H2O2 up-regulates GTP-CH I expression (23), which likely explains a modest increase in total biopterin content observed in the current study. Endogenously produced H2O2 by angiotensin II, however, did not seem to modulate GTP-CH I because angiotensin II had no effect on total biopterin content. These data may suggest that DHFR is much more sensitive to lower endogenous H2O2 compared to GTP-CH I, which was found regulated only in response to exogenous H2O2. Furthermore, we found that endothelial 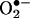 production after prolonged angiotensin II (24 h) was completely dependent on uncoupled eNOS and was correctable by scavenging H2O2 or overexpressing DHFR. It is interesting to speculate that this H2O2 down-regulation of DHFR may partially underlie eNOS uncoupling in atherosclerotic or hypertension blood vessels.
production after prolonged angiotensin II (24 h) was completely dependent on uncoupled eNOS and was correctable by scavenging H2O2 or overexpressing DHFR. It is interesting to speculate that this H2O2 down-regulation of DHFR may partially underlie eNOS uncoupling in atherosclerotic or hypertension blood vessels.
These data also suggest that, although angiotensin II potently activates vascular NAD(P)H oxidases in endothelial cells and vascular smooth muscle cells within minutes, uncoupled eNOS is the major enzymatic source of ROS within endothelium after prolonged angiotensin II. Indeed, we found that angiotensin II induced an initial burst production of 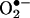 at 30 min which was abolished by Rac1 inhibitor NSC 23766 but unaffected by l-NAME. This increase in
at 30 min which was abolished by Rac1 inhibitor NSC 23766 but unaffected by l-NAME. This increase in 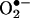 diminished at 6 h. At 24 h, however, endothelial cells had a second peak in producing
diminished at 6 h. At 24 h, however, endothelial cells had a second peak in producing 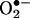 , which was completely attenuated by l-NAME but unaffected by NSC 23766. These data clearly suggest that a rapid and transient activation of endothelial NAD(P)H oxidases precedes H2O2-dependent eNOS uncoupling. These observations seems consistent with the earlier observation by Mollnau et al. (30) that ROS production in angiotensin II infused rat aorta was markedly reduced by inhibiting eNOS. However, these data are not contradictory to the findings that vascular NAD(P)H oxidases remain the predominant sources for vascular smooth muscle-derived ROS in response to prolonged angiotensin II (31). The latter finding seems to also involve up-regulation of NAD(P)H oxidases subunits (32).
, which was completely attenuated by l-NAME but unaffected by NSC 23766. These data clearly suggest that a rapid and transient activation of endothelial NAD(P)H oxidases precedes H2O2-dependent eNOS uncoupling. These observations seems consistent with the earlier observation by Mollnau et al. (30) that ROS production in angiotensin II infused rat aorta was markedly reduced by inhibiting eNOS. However, these data are not contradictory to the findings that vascular NAD(P)H oxidases remain the predominant sources for vascular smooth muscle-derived ROS in response to prolonged angiotensin II (31). The latter finding seems to also involve up-regulation of NAD(P)H oxidases subunits (32).
In summary, the present study provided evidence that endothelial DHFR critically regulates endothelial H4B and NO· bioavailability under physiological and pathological conditions. By using angiotensin II as a model system, we found that a rapid and transient activation of endothelial NAD(P)H oxidases precedes H2O2-dependent down-regulation of DHFR and eNOS uncoupling in the endothelium. This signaling cascade, as illustrated in Fig. 9, may represent a universal mechanism whereby eNOS uncouples under conditions associated with oxidant stress, such as atherosclerosis, hypertension, and diabetes, for which activation of vascular NAD(P)H oxidases has been documented.
Acknowledgments
This work was supported by American Heart Association Scientist Development Grant 0435189N, an American Diabetes Association Research Award, a Schweppe Foundation Career Development Award, and a start-up fund from the University of Chicago (all to H.C.).
Author contributions: L.C. designed research; K.C. and L.C. performed research; K.C. and L.C. analyzed data; and L.C. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: ROS, reactive oxygen species; H4B, tetrahydrobiopterin; NOS, nitric oxide synthase; eNOS, endothelial NOS; DHFR, dihydrofolate reductase; RNAi, RNA interference; siRNA, small interfering RNA; ACE, angiotensin converting enzyme; GTP-CH I, GTP cyclohydrolase I; PEG, polyethylene glycol; PEG-CAT, PEG-conjugated catalase; l-NAME, N-nitro-l-arginine-methyl ester.
References
- 1.Cai, H. (2005) Circ. Res. 96, 818–822. [DOI] [PubMed] [Google Scholar]
- 2.Cai, H., Griendling, K. K. & Harrison, D. G. (2003) Trends Pharmacol. Sci. 24, 471–478. [DOI] [PubMed] [Google Scholar]
- 3.Cai, H. & Harrison, D. G. (2000) Circ. Res. 87, 840–844. [DOI] [PubMed] [Google Scholar]
- 4.Lassegue, B. & Griendling, K. K. (2004) Am. J. Hypertens. 17, 852–860. [DOI] [PubMed] [Google Scholar]
- 5.Harrison, D. G., Cai, H., Landmesser, U. & Griendling, K. K. (2003) J. Renin Angiotensin Aldosterone Syst. 4, 51–61. [DOI] [PubMed] [Google Scholar]
- 6.Heinzel, B., John, M., Klatt, P., Bohme, E. & Mayer, B. (1992) Biochem. J. 281, 627–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pou, S., Pou, W. S., Bredt, D. S., Snyder, S. H. & Rosen, G. M. (1992) J. Biol. Chem. 267, 24173–24176. [PubMed] [Google Scholar]
- 8.Xia, Y., Dawson, V. L., Dawson, T. M., Snyder, S. H. & Zweier, J. L. (1996) Proc. Natl. Acad. Sci. USA 93, 6770–6774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xia, Y. & Zweier, J. L. (1997) Proc. Natl. Acad. Sci. USA 94, 6954–6958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xia, Y., Tsai, A. L., Berka, V. & Zweier, J. L. (1998) J. Biol. Chem. 273, 25804–25808. [DOI] [PubMed] [Google Scholar]
- 11.Wever, R. M., van Dam, T., van Rijn, H. J., de Groot, F. & Rabelink, T. J. (1997) Biochem. Biophys. Res. Commun. 237, 340–344. [DOI] [PubMed] [Google Scholar]
- 12.Vasquez-Vivar, J., Kalyanaraman, B., Martasek, P., Hogg, N., Masters, B. S., Karoui, H., Tordo, P. & Pritchard, K. A., Jr. (1998) Proc. Natl. Acad. Sci. USA 95, 9220–9225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Milstien, S. & Katusic, Z. (1999) Biochem. Biophys. Res. Commun. 263, 681–684. [DOI] [PubMed] [Google Scholar]
- 14.Laursen, J. B., Somers, M., Kurz, S., McCann, L., Warnholtz, A., Freeman, B. A., Tarpey, M., Fukai, T. & Harrison, D. G. (2001) Circulation 103, 1282–1288. [DOI] [PubMed] [Google Scholar]
- 15.Landmesser, U., Dikalov, S., Price, S. R., McCann, L., Fukai, T., Holland, S. M., Mitch, W. E. & Harrison, D. G. (2003) J. Clin. Invest. 111, 1201–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thony, B., Auerbach, G. & Blau, N. (2000) Biochem. J. 347, 1–16. [PMC free article] [PubMed] [Google Scholar]
- 17.Werner-Felmayer, G., Golderer, G. & Werner, E. R. (2002) Curr. Drug Metab. 3, 159–173. [DOI] [PubMed] [Google Scholar]
- 18.Cai, H., Li, Z., Dikalov, S., Holland, S. M., Hwang, J., Jo, H., Dudley, S. C., Jr., & Harrison, D. G. (2002) J. Biol. Chem. 277, 48311–48317. [DOI] [PubMed] [Google Scholar]
- 19.Cai, H., Li, Z., Davis, M. E., Kanner, W., Harrison, D. G. & Dudley, S. C., Jr. (2003) Mol. Pharmacol. 63, 325–331. [DOI] [PubMed] [Google Scholar]
- 20.Cai, H., McNally, J. S., Weber, M. & Harrison, D. G. (2004) J. Mol. Cell. Cardiol. 37, 121–125. [DOI] [PubMed] [Google Scholar]
- 21.Cai, H., Li, Z., Goette, A., Mera, F., Honeycutt, C., Feterik, K., Wilcox, J. N., Dudley, S. C., Jr., Harrison, D. G. & Langberg, J. J. (2002) Circulation 106, 2854–2858. [DOI] [PubMed] [Google Scholar]
- 22.Landmesser, U., Cai, H., Dikalov, S., McCann, L., Hwang, J., Jo, H., Holland, S. M. & Harrison, D. G. (2002) Hypertension 40, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tai, N., Schmitz, J. C., Chen, T. M. & Chu, E. (2004) Biochem. J. 378, 999–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shimizu, S., Shiota, K., Yamamoto, S., Miyasaka, Y., Ishii, M., Watabe, T., Nishida, M., Mori, Y., Yamamoto, T. & Kiuchi, Y. (2003) Free Radical Biol. Med. 34, 1343–1352. [DOI] [PubMed] [Google Scholar]
- 25.Blau, N., Thony, B., Cotton, R. G. H. & Hyland, K. (2003) in The Metabolic and Molecular Bases of Inherited Disease, eds. Scriver, C. R., Beaudet, A. L., Sly, W. S., Valle, D., Childs, B. & Vogelstein, B. (McGraw–Hill, New York), pp. 1725–1776.
- 26.Kinoshita, H., Milstien, S., Wambi, C. & Katusic, Z. S. (1997) Am. J. Physiol. 273, H718–H724. [DOI] [PubMed] [Google Scholar]
- 27.Alp, N. J., Mussa, S., Khoo, J., Cai, S., Guzik, T., Jefferson, A., Goh, N., Rockett, K. A. & Channon, K. M. (2003) J. Clin. Invest. 112, 725–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zheng, J. S., Yang, X. Q., Lookingland, K. J., Fink, G. D., Hesslinger, C., Kapatos, G., Kovesdi, I. & Chen, A. F. (2003) Circulation 108, 1238–1245. [DOI] [PubMed] [Google Scholar]
- 29.Pueyo, M. E., Gonzalez, W., Nicoletti, A., Savoie, F., Arnal, J. F. & Michel, J. B. (2000) Arterioscler. Thromb. Vasc. Biol. 20, 645–651. [DOI] [PubMed] [Google Scholar]
- 30.Mollnau, H., Wendt, M., Szocs, K., Lassegue, B., Schulz, E., Oelze, M., Li, H., Bodenschatz, M., August, M., Kleschyov, A. L., et al. (2002) Circ. Res. 90, E58–E65. [DOI] [PubMed] [Google Scholar]
- 31.Griendling, K. K. & Ushio-Fukai, M. (2000) Regul. Pept. 91, 21–27. [DOI] [PubMed] [Google Scholar]
- 32.Fukui, T., Ishizaka, N., Rajagopalan, S., Laursen, J. B., Capers, Q., IV, Taylor, W. R., Harrison, D. G., de Leon, H., Wilcox, J. N. & Griendling, K. K. (1997) Circ. Res. 80, 45–51. [DOI] [PubMed] [Google Scholar]