

Abstract
One of the major obstacles to membrane protein (MP) structural studies is the destabilizing effect of detergents. Amphipols (APols) are short amphipathic polymers that can substitute for detergents to keep MPs water-soluble under mild conditions. In the present work, we have explored the feasibility of studying the structure of APol-complexed MPs by NMR. As a test MP, we chose the 171-residue transmembrane domain of outer MP A from Escherichia coli (tOmpA), whose x-ray and NMR structures in detergent are known. 2H,15N-labeled tOmpA was produced as inclusion bodies, refolded in detergent solution, trapped with APol A8-35, and the detergent removed by adsorption onto polystyrene beads. The resolution of transverse relaxation-optimized spectroscopy–heteronuclear single-quantum correlation spectra of tOmpA/A8-35 complexes was found to be close to that of the best spectra obtained in detergent solutions. The dispersion of chemical shifts indicated that the protein had regained its native fold and retained it during the exchange of surfactants. MP–APol interactions were mapped by substituting hydrogenated for deuterated A8-35. The resulting dipolar broadening of amide proton linewidths was found to be limited to the β-barrel region of tOmpA, indicating that A8-35 binds specifically to the hydrophobic transmembrane surface of the protein. The potential of this approach to MP studies by solution NMR is discussed.
Keywords: membrane proteins, amphipols, OmpA, surfactant
Integral membrane proteins (MPs) are involved in such essential cell functions as energy transduction, import and export of nutrients and drugs, signal detection, cell-to-cell communication, etc. They comprise 20–30% of the proteins encoded in the genome of cells and a majority of the targets of currently marketed drugs (1). A detailed knowledge of their structure is essential to understanding their function and dysfunction, as well as to a wide range of biomedical and biotechnological applications. The scarcity of high-resolution MP structures (which represent <0.3% of currently available structures) can be traced to three main factors: low levels of natural abundance, difficult overexpression, and a poor stability in the presence of detergent. Detergents are generally used to handle MPs in aqueous solutions, because the highly hydrophobic character of their transmembrane surface renders MPs water-insoluble. By adsorbing onto this surface, detergents make it hydrophilic (2). However, the dissociating character of detergents, combined with the need to maintain an excess of them, frequently results in more or less rapid inactivation of solubilized MPs (3).
Inactivation by detergents is a particularly serious problem in the field of solution-state NMR for the following reasons: (i) to keep highly concentrated MPs (in the mM range) from aggregating, high concentrations of detergents must generally be used (usually in the 200- to 600-mM range; see, for instance, ref. 4); (ii) high temperatures are usually resorted to, to improve the resolution of the spectra; and (iii) those detergents that tend to be less destabilizing, such as digitonin or surfactants of the Tween series, are unsuitable for solution NMR, where a primary requirement is that the MP/detergent complex be as small as possible. As a result, the only MPs whose native structure has been studied by solution NMR to date are exceptionally robust ones, such as the transmembrane domains of glycophorin A (5) and OmpA (6), OmpX (7), and PagP (8), all of which are sturdy enough to resist denaturation by SDS at room temperature (9–12). It appears likely that a more general extension of solution NMR to MP studies will depend primarily on three major types of technical progress: (i) higher magnetic fields and improved pulse sequences (13–15), (ii) more efficient approaches to producing micromolar amounts of isotopically labeled MPs (16–19), and (iii) novel surfactants, milder than classical detergents but nevertheless allowing the acquisition of NMR spectra of a quality sufficient for structure determination. The latter issue is the focus of the present work.
The frequent instability of MPs in detergent solutions has prompted the development of alternative media based on the use of nondetergent surfactants and/or nonmicellar phases (for reviews, see, e.g., refs. 20 and 21). Over the past few years, we have endeavored to develop a novel family of surfactants dubbed “amphipols” (APols) (22). APols are amphiphilic polymers designed to bind to the transmembrane surface of MPs in a noncovalent but quasi-irreversible manner. APols are not (or are extremely weak) detergents and, as a rule, they are unable to extract MPs from biological membranes (reviewed in ref. 23). Nevertheless, they can maintain in solution MPs extracted by classical detergents after being substituted to the latter (22). MPs complexed by APols are in their native state, generally much more stable than in detergent solution, and they remain water-soluble in the absence of detergent or free APols (23). Although several families of APols have now been described (21, 23), the best-characterized ones feature a polyacrylate backbone derived with fatty amines (22). The synthesis and physicochemical properties of one of them, A8-35 (Fig. 1a) have been described in detail (24, 25).
Fig. 1.

Trapping tOmpA with amphipol A8-35. (a) Chemical structure of A8-35, a polyacrylate-based APol. The average degree of polymerization (n) is ≈80, and the average molecular weight, ≈9 kDa. The molar percentage of each type of unit (x, y, and z), randomly distributed along the chain, is indicated. It corresponds, respectively, to ≈20, ≈32, and ≈28 units per A8-35 molecule. HAPol is the hydrogenated form; in DAPol, the octyl and isopropyl side chains are perdeuterated (ellipses). (b) Trapping procedure; see text.
As a model MP, we chose the transmembrane domain of outer MP A from Escherichia coli, tOmpA (≈19 kDa), a protein that has been well studied by x-ray crystallography (26–28) and NMR spectroscopy (6, 29–32).
Materials and Methods
APol Synthesis. Protonated and deuterated forms of APol A8-35 (hereafter HAPol and DAPol, respectively; Fig. 1a) were synthesized in our laboratory by grafting either hydrogenated or deuterated (≥99%) octylamine and isopropylamine groups onto a hydrogenated poly(acrylic acid) precursor (22, 25).
Expression, Refolding, and Purification of [2H,15N]tOmpA. The plasmid containing the coding sequence of tOmpA was kindly provided by G. E. Schulz (Freiburg University, Freiburg-im-Brisgau, Germany; ref. 27). The protein contains three mutations (F23L, Q34K, and K107Y). An N-terminal His8 tag was added to it in our laboratory. It was overexpressed as inclusion bodies in E. coli. Freshly transformed cells were grown in 1 liter of 99.9% D2O M9 media containing 0.1% 15NH4Cl (≥98%) and 0.7% nondeuterated glucose (33). D2O and 15NH4Cl were purchased from Spectra Stable Isotope, Columbia, MD. Purification and refolding procedures (34) were similar to those described in ref. 27. Yields were ≈25 mg of pure tOmpA per liter of culture.
Preparation of tOmpA/Detergent NMR Samples. tOmpA/detergent NMR samples were obtained as follows. At the end of the purification procedure, the protein solution, containing 0.6% (wt/vol) n-octylpolyoxyethylene (C8POE, Bachem) in 20 mM Tris·HCl (pH 8) was loaded onto a 1-ml ion exchange column (Source-30Q, Amersham Pharmacia) equilibrated with the same buffer. The column was washed with 1 ml of a solution containing 50 mM dihexanoyl phosphatidylcholine (DHPC, Avanti Polar Lipids) in 20 mM Tris·HCl buffer (pH 8), followed by 40 ml of 28 mM DHPC, and eluted with 300 mM NaCl in the same buffer. The NaCl concentration was adjusted to 100 mM on a 5-ml desalting column (Hi-Trap, Amersham Pharmacia) equilibrated with 20 mM phosphate buffer (pH 6.5 or 7.9) and 28 mM DHPC. The solutions were then transferred to Centricon ultra-filtration devices (Millipore; 10-kDa cutoff) and concentrated down to a volume of 300 μl. The tOmpA/DHPC NMR sample buffer solution contained 20 mM phosphate, pH either 6.5 or 7.9, 100 mM NaCl, 0.1 mM NaN3, and 5% D2O. The final DHPC concentration was ≈300 mM. The protein concentration (1 mM at either pH) was determined by using ε280 = 46,470 M–1·cm–1, established by amino acid analysis.
Preparation of tOmpA/APol NMR Samples. tOmpA/HAPol and tOmpA/DAPol NMR samples were prepared as schematized in Fig. 1b. The initial protein/detergent solution contained 20 mM phosphate buffer (pH 7.9), 0.6% (wt/vol) C8POE (19.6 mM), 100 mM NaCl, and 0.1 mM NaN3. A slightly basic solution is required to prevent MP/A8-35 from aggregating (24). tOmpA was trapped by using a 1:4 wt/wt protein/A8-35 ratio (34). After 15-min incubation, polystyrene beads were added at a 10:1 bead/detergent mass ratio. They were removed by centrifugation after 2-h incubation at room temperature. Absorption measurements at 205 nm for a protein-free control sample of 19.6 mM C8POE supplemented with the same amount of polystyrene beads indicated that, after 2 h, the detergent concentration had fallen to ≈4.5 mM, i.e., half its critical micellar concentration. The samples were then concentrated by using Centricon devices with 10-kDa molecular mass cutoff. To extensively remove any remaining detergent, five dilution/concentration cycles were performed (final theoretical concentration of C8POE ≤0.1 mM, i.e., ≤0.1 mol per mol tOmpA). The characteristic signal of the polyethyleneglycol polar moiety of C8POE was undetectable in 1H NMR spectra. The NMR buffer contained 20 mM phosphate, pH 7.9, 100 mM NaCl, 0.1 mM NaN3 and 5% D2O. Protein recovery was close to 100%. In the final samples (300 μl), protein concentration was ≈1 mM and total APol concentration ≈70 g/liter (≈8 mM). Unbound APol was not separated from tOmpA/A8-35 complexes, because its removal induces particle aggregation (34). The final pH was checked by using a microelectrode dedicated to concentrated protein solutions (Spintrode P, Hamilton).
NMR Spectroscopy. NMR experiments were carried out at a probe temperature of 30°C on Bruker (Wissembourg, France) DRX600 and DRX800 spectrometers equipped with a 5-mm triple-resonance gradient probe. [1H,15N]-transverse relaxation-optimized spectroscopy (TROSY)–heteronuclear single-quantum correlation (HSQC) experiments (35, 36) were performed with 32 transients per increment and a time domain data size of 128 × 2,048 complex points [t1max(15N) = 27 ms, t2max(1H) = 122 ms at 600 MHz, and t1max (15N) = 20 ms, t2max (1H) = 92 ms at 800 MHz]. Linewidth measurements were extracted from [1H,15N]-TROSY-HSQC experiments performed at 800 MHz with 128 transients per increment. Data were processed with nmrpipe (37) and analyzed with nmrview (38) softwares.
The viscosity of the NMR samples was estimated from the water translational diffusion coefficient, evaluated by a pulsed-field-gradient NMR approach (39). Measurements were carried out on a Bruker AMX 400 MHz WB spectrometer at 20°C, to avoid or attenuate convective phenomena in the NMR Shigemi tube. Gradient field strengths were calibrated by using a standard sample of H2O (40).
Numerical Simulations. The extent of 1HN line broadening to be expected from tOmpA/HAPol interactions was predicted by lineshape simulations for the narrow 1HN line of the 15N-1H moiety at 800 MHz 1H frequency. A rigid spherical rotor was assumed, with a rotational correlation time (τc) of 50 ns. Other parameters, such as scalar coupling constant, components of 1H and 15N axially symmetric tensors, etc., were identical to those used in ref. 35. Relaxation due to dipole–dipole coupling with other protons was taken into account in two ways: first, by adjusting the calculated lineshapes to those experimentally observed for β-barrel residues in tOmpA/DAPol complexes; second, by simulating the broadening induced by the substitution of DAPol by HAPol. In the first case, a simple relaxation rate constant was added to the relaxation matrix; in the second, a relaxation term was added to describe dipole–dipole interactions with k protons at a distance rk (equation 6 of ref. 35).
Results
2D [1H,15N]-TROSY-HSQC spectra of [2H,15N]tOmpA were recorded in the following four environments: (i) in DHPC solution at either pH 7.9 or 6.5 and (ii) at pH 7.9 after trapping the protein with either a fully hydrogenated (HAPol) or a partially deuterated (DAPol) form of APol A8-35 (Fig. 1a). Their analysis yielded information about the conformation of tOmpA, factors affecting the quality of the spectra of tOmpA/A8-35 complexes, and the distribution of A8-35 around the protein.
[1H,15N]-TROSY-HSQC Spectra of tOmpA in A8-35 vs. Detergent. Because there is no functional test for tOmpA in solution, ascertaining whether it lies in its native state must rely on structural investigations. NMR studies have established that tOmpA solubilized either in dodecylphosphocholine (DPC) (6) or DHPC (30) adopts the same eight-strand β-barrel structure as in 3D crystals of tOmpA/C8E4 complexes (26). Using conditions (see Materials and Methods) optimized by K. Wüthrich and coworkers (29, 30), the DHPC-solubilized preparations yielded well resolved [1H,15N]-TROSY-HSQC spectra (34). These conditions differ somewhat from those (50°C, 600 mM DPC) used by L. Tamm and colleagues (6) to establish the structure of tOmpA by NMR.
Because preservation of a good monodispersity imposes that NMR experiments with MP/A8-35 complexes be carried out in a slightly basic solution (24), measurements in DHPC were repeated at pH 7.9 to provide a benchmark against which to gauge the effect of substituting APol for detergent under otherwise identical conditions. As expected, the TROSY-HSQC spectra of tOmpA/DHPC obtained at pH 7.9 were less intense than those recorded at pH 6.5 (34), due to faster exchange of amide protons with bulk water (41, 42). Using the 1HN and 15N chemical shifts for tOmpA kindly communicated by the groups of K. Wüthrich and L. Tamm (personal communication), we observed that, again as expected, the pH change affected much more strongly those protons belonging to residues located in the loops and turns than those belonging to the barrel (Fig. 2 c and d). Linewidths measured at 600 MHz tended to be narrower than at 800 MHz, whatever the residues concerned (for instance, see residue Ala-130; Fig. 2 c and d). This anomalous decrease could arise from the presence of rapid chemical exchange, whose broadening effect increases with the strength of the static magnetic field (43, 44).
Fig. 2.

NMR 2D spectrum of A8-35-trapped tOmpA. (a) [1H,15N]-TROSY-HSQC spectrum of [2H,15N]tOmpA/HAPol recorded at 30°C, pH 7.9, and 800 MHz 1H frequency. (b) Zooms on two 1H rows, one showing Ala-130, a residue belonging to the β-barrel (Left), the other Gly-22, a residue from an extracellular loop (Right), extracted from the spectrum shown in a.(c and d) Signals of the same residues obtained with a tOmpA/DHPC sample at pH 7.9 and 600 MHz (c) and pH 6.5 and 800 MHz (d). TROSY spectra were aligned relative to each other by taking the 1HN-15N line of Gly-22 as an internal reference.
[1H,15N]-TROSY-HSQC spectra of tOmpA/HAPol and tOmpA/DAPol complexes featured a high resolution and a wide spectral dispersion in both dimensions (Fig. 2a). In addition to signals due to indole and imino protons, they showed ≈110 correlation peaks, a number equivalent to that obtained with tOmpA/DHPC complexes at pH 7.9 (to be compared with ≈150 peaks observed in DHPC at pH 6.5; ref. 34). For tOmpA solubilized in detergent, intermediate levels of chemical exchange on an NMR chemical-shift timescale are invoked to explain the lack of assignment for ≈60 residues, mostly located at the barrel/loop boundary region (6, 30, 31). In both tOmpA/APol and tOmpA/DHPC spectra recorded at pH 7.9, the broader lines are mostly located at the center of the amide 1H dimension (Fig. 2a); they originate from amino acids located in the extracellular loops, whose amide protons experience faster chemical exchange with bulk water 1H at high pH.
Amide 1H and 15N chemical shifts for tOmpA/A8-35 and tOmpA/DHPC were very similar (34). The spectra in DHPC were identical to those recorded by Wüthrich and colleagues under similar conditions on wild-type tOmpA (personal communication), indicating that our his-tagged construct (which also contains three mutations as compared with wild type; see Materials and Methods) had properly refolded. By reference to tOmpA/detergent NMR data, we identified 73 correlation peaks in the tOmpA/APol spectra (colored residues in Fig. 4). Unassigned peaks mostly concern broad signals lying toward the center of the spectrum. The 1H- and 15N-weighted-average chemical-shift differences (45–47) between tOmpA/HAPol (pH 7.9) and tOmpA/DHPC (pH 6.5) lie between 0.003 and 0.13 ppm, with an average of 0.04 ppm. The 3D structure of tOmpA, therefore, is not affected by trapping with APols. That no additional signals are observed as compared with tOmpA/DHPC spectra suggests that, in tOmpA/APol complexes, residues located at the top of the barrel experience intermediate chemical exchange, as they are thought to do in detergent solution. As for tOmpA/DHPC, linewidths increased between 600 and 800 MHz, particularly for loop residues.
Fig. 4.

Mapping of contacts with APol alkyl chains onto the structure of tOmpA. Residues are color-coded depending on whether dipolar line broadening upon substituting HAPol for DAPol (Fig. 3) is strong (red), weak (yellow), or undetectable (blue). Residues giving rise to no assigned line are white. The data are plotted on a topology sketch (adapted from ref. 26; the side chains of residues shown in italics point toward the exterior of the barrel) and on a ribbon representation of the 3D structure (Protein Data Bank code 1G90; ref. 6). The 3D diagram was realized with the open-source software pymol (DeLano Scientific, San Carlos, CA).
On average, tOmpA/HAPol spectra feature slightly larger linewidths (12% broader at midheight in the 1H dimension) than those of tOmpA/DHPC complexes recorded at the same pH. A closer look reveals that the residues most affected by the overall correlation time of the particles, i.e., those belonging to the β-barrel, exhibit lines 30–40% broader in the 1H dimension as compared with tOmpA/DHPC spectra recorded at the same pH (Fig. 2 b and c). In the case of tOmpA/APol complexes, 15N relaxation measurements indicate an overall correlation time (τc) ≈60% longer than that of tOmpA/DHPC complexes. This slowing down of particle tumbling is likely due to tOmpA/A8-35 particles being larger than tOmpA/DHPC ones, because the viscosity of the two samples, estimated by NMR at 20°C, is similar: ≈1.14 × 10–3 N·s·m–2 for tOmpA/DHPC vs. ≈1.20 × 10–3 N·s·m–2 for tOmpA/A8-35. This conclusion is consistent with other estimates, obtained by pulsed-field-gradient NMR, analytical ultracentrifugation, and size-exclusion chromatography (34). Overall, however, the broadening effect of moving from DHPC, pH 6.5, to A8-35, pH 7.9, is more pronounced for loop residues, due to the unfavorable chemical exchange (Fig. 2 b and d).
Mapping the Distribution of A8-35 at the Surface of tOmpA. Dipole–dipole coupling between amide protons (1HN) and nearby 1H plays a major role in 1HN spin relaxation phenomena. Among those, transverse relaxation can be substantially slowed down by deuterium labeling (33, 48). We therefore compared [1H,15N]-TROSY-HSQC spectra obtained by using two forms of A8-35: HAPol, which is fully hydrogenated, and DAPol, whose isopropylamine and octylamine chains are perdeuterated (Fig. 1a). Linewidth differences between the two spectra contain information about the proximity of the alkyl chains of the polymer to the surface of the protein. This information is patchy, because less than half of the amide protons are assigned (colored residues in Fig. 4), but it nevertheless provides a first atomic-level description of the way an APol interacts with the MP it keeps soluble.
Two sets of [1H,15N]-TROSY-HSQC spectra were recorded at high magnetic field (νH = 800 MHz), one with HAPol- and the other with DAPol-trapped tOmpA. Half-height linewidths were compared in the 1H dimension, because the coupling with remote protons is more pronounced with 1H than with 15N nuclei (35). The extent of relative broadening induced by substituting HAPol for DAPol is plotted in Fig. 3. Color-coded representations are shown in Fig. 4, superimposed onto models of the secondary and tertiary structures. Strikingly, only residues belonging to β-strands displayed any dipolar broadening (on average, by 22 ± 8%), whereas no statistically significant broadening was observed for amide protons located in the periplasmic turns (1 ± 6%) or the extracellular loops (–1 ± 6%) (Fig. 3). Even though this experiment does not report directly on Van der Waals contacts between amino acid side chains and the alkyl chains of the APol, the latter are obviously close enough to the amide protons of the protein for the broadening effect of substituting HAPol for DAPol to be detected. Using the (simplifying) assumption that broadening is mainly due to interactions with the terminal methyl group of a single alkyl chain of HAPol, numerical simulations (see Materials and Methods) indicate that broadening by ≥20% should correspond to distances ≤3.5 Å.
Fig. 3.

Dipolar broadening due to tOmpA/APol interactions. Changes in 1HN linewidth variations, measured at midheight (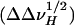 ), for tOmpA trapped with a fully protonated APol (HAPol) with respect to signals obtained after complexation with an APol with perdeuterated alkyl chains (DAPol) are plotted against residue number. 2D TROSY experiments were repeated at least three times. Average variations are shown ± SD. Squares, diamonds, and circles refer to amino acids belonging to β-strands, periplasmic turns, and external loops, respectively. Averages for each secondary structure element are shown at the top, under a schematic representation of the secondary structure.
), for tOmpA trapped with a fully protonated APol (HAPol) with respect to signals obtained after complexation with an APol with perdeuterated alkyl chains (DAPol) are plotted against residue number. 2D TROSY experiments were repeated at least three times. Average variations are shown ± SD. Squares, diamonds, and circles refer to amino acids belonging to β-strands, periplasmic turns, and external loops, respectively. Averages for each secondary structure element are shown at the top, under a schematic representation of the secondary structure.
Interestingly, some amide protons belonging to the β-barrel do not show much linewidth variation in the presence of HAPol (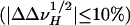 ). Such is the case both for residues that lie very close to the periplasmic side, like Tyr-6, Leu-91, or Ala-130, and for residues located more deeply within β-strands, like Ala-39 or Tyr-137 (Fig. 4). A lesser broadening effect may reflect either the dearth of APol alkyl chains in the vicinity of the amide protons, as could be the case for residues located at the border of the barrel or that are partially screened due to steric hindrance or electrostatic repulsion, or faster local dynamics. These observations will deserve to be extended to MPs whose assignments are more complete than is the case for tOmpA, because they can lead to improving our understanding of MP/APol interactions and, thereby, can help in optimizing the methodology.
). Such is the case both for residues that lie very close to the periplasmic side, like Tyr-6, Leu-91, or Ala-130, and for residues located more deeply within β-strands, like Ala-39 or Tyr-137 (Fig. 4). A lesser broadening effect may reflect either the dearth of APol alkyl chains in the vicinity of the amide protons, as could be the case for residues located at the border of the barrel or that are partially screened due to steric hindrance or electrostatic repulsion, or faster local dynamics. These observations will deserve to be extended to MPs whose assignments are more complete than is the case for tOmpA, because they can lead to improving our understanding of MP/APol interactions and, thereby, can help in optimizing the methodology.
Discussion
The primary goal of the present work was to examine the feasibility of using APols as an environment for MP solution-state studies by NMR. For these particular experiments, we resorted to a polyacrylate-based polymer, A8-35, because it is by far the best characterized APol with respect to its synthesis, purification, and physicochemical properties, as well as to the preparation and properties of MP/APol complexes (23–25, 34). It was known from the onset, however, that using A8-35 instead of detergents like DPC or DHPC entailed two disadvantages: a larger particle size and the impossibility of working in neutral or acidic solution. For that vast majority of MPs that do not stand detergents well, however, these two drawbacks might conceivably be offset by the stabilizing effect of APols. As a test protein, we chose tOmpA because of its small size, β-barrel fold, easy synthesis, and the wealth of x-ray and NMR data already available. It is also a MP whose NMR studies in detergent solution have met with some problems, including difficulties in assigning all backbone nuclei. It therefore provided a good opportunity to compare the limits of the use of APols with that of detergent for an NMR structural study.
The lower quality of the signal observed in APol for residues participating to the loops (and turns) and, to a lesser extent for β-strands, as illustrated in Fig. 2, is partly due to increasing proton exchange with the solvent. Indeed, changing the pH from 6.5 to 7.9 gives rise, in the case of tOmpA/DHPC samples, to an average decrease of intensity by ≈60% for residues belonging to the loops and turns. Despite the hydrogen bond network and the presence of the detergent layer, the average decrease for β-barrel residues reaches ≈40% (34). This problem might be alleviated by using APols that remain monodisperse in acidic solution. The increase in particle size, and therefore linewidth, upon substituting DHPC with HAPol at constant pH (Fig. 2) may be more difficult to avoid. This inconvenience, however, may become more and more tolerable as spectrometers operating at higher magnetic fields and improved pulse sequences become available, as testified to by recent emblematic studies (49, 50). Overall, these data, which will need to be extended to other proteins, seem to bode well for the use of APols for MP structural studies by solution NMR.
The analysis of [1H,15N]-TROSY-HSQC spectra of tOmpA/A8-35 complexes led to a number of interesting conclusions. First, the spectral dispersions observed on the TROSY spectrum of Fig. 2a are equivalent to those observed with tOmpA/DHPC samples (34), as well as in other NMR studies of β-barrel MPs (6–8). The same number of peaks were observed as with the tOmpA/DHPC (pH 7.9) sample, with very limited chemical-shift differences. This indicates that tOmpA adopts essentially the same 3D structure in the two environments. Whether the dynamics of MPs is affected by complexation with APols remains an open question. Functional data suggest that large-scale MP movements, which are unlikely in the case of the β-barrel region of tOmpA, may be slowed down by association with APols (23, 51). The anomalous increase of NMR linewidths observed for tOmpA β-barrel residues between 600 and 800 MHz is slightly more pronounced in a DHPC than in an A8-35 environment (data not shown). This could be due either to these residues experiencing a higher rate of chemical exchange in DHPC or to a shift in relative populations of exchanging conformational states. If the lack of observable amide proton signals for residues located at the border between the β-barrel and the loops indeed results from the particular dynamics of tOmpA in this region (30), it does not seem, however, that their dynamics are slowed down upon trapping with APols. This interesting but complex question will probably better be addressed by using other model proteins.
APols were designed to associate with MP transmembrane surfaces (22). Until now, MP/APol complexes have been studied by such methods as size-exclusion chromatography, ultracentrifugation, and neutron scattering, using either radioactive, fluorescent, or deuterated APols (see refs. 23, 34, 52, and refs. therein). These approaches have made it possible to estimate MP/APol mass ratios in the complexes, as well as to study some kinetic aspects of APol binding, but they have yielded little information about the distribution of the polymer around MPs. The present work shows that, as they were designed to do, the alkyl chains of A8-35 interact exclusively with the strongly hydrophobic transmembrane surface of tOmpA. The approach used in the present work aimed at obtaining an overall view of the distribution of the APol at the protein surface, using limited spectrometer time and a low-cost biochemical labeling strategy. More sophisticated NMR experiments, such as transverse relaxation measurements, polarization transfer in HSQC-edited filtered NOESY experiment, and/or saturation transfer methods (53), would probably lead to a more accurate description of the organization of the complexes. Knowledge of the 13C–1H chemical-shift assignments of the methyl group of amino acid side chains, in particular, would make it possible to obtain a more quantitative description of methyl–methyl contacts between the surfactant and the protein.
Conclusion
The present work establishes that replacing detergents by APols is a viable approach to the study of MPs by solution-state NMR. In the current state of the methodology, its interest lies probably mostly in structural studies of those MPs whose stability in detergent solution is limited, or of ligands bound to them. The approach offers ample room for methodological improvements. First, it is possible to develop APols that do not present the undesirable sensitivity to pH of first-generation molecules, the use of which would lead to an increased sensitivity (unpublished work). Second, APols and/or the conditions of their use can likely still be optimized to reduce the size of the particles, thereby improving resolution. APols present, in addition to their stabilizing effects on MPs, the useful characteristic of great chemical versatility. For NMR purposes, this means, for instance, that deuterating the groups in contact with the protein is straight-forward and much less costly than acquiring equivalent amounts of a deuterated detergent. In the present work, we have used this opportunity to map the distribution of the polymer at the surface of the protein. It would be straightforward to carry out similar experiments using, for instance, a spin-labeled APol.
Acknowledgments
We are indebted to G. E. Schulz (Freiburg University, Freiburg-im-Brisgau, Germany) for his kind gift of a tOmpA plasmid and to F. Zito (Unité Mixte de Recherche 7099, Institut de Biologie Physico-Chimique, Paris) for constructing its histidine-tagged variant. We are extremely grateful to K. Wüthrich (Eidgenössische Technische Hochschule, Zurich, and Scripps Research Institute, La Jolla, CA) and L. Tamm (University of Virginia, Charlottesville) for communication of 1HN and 15N chemical shifts. We thank C. Lebreton (Unité Mixte de Recherche 7099) for technical assistance; E. Guittet, C. Van Heijenoort, and N. Birlirakis (Institut de Chimie de Substances Naturelles, Gif-sur-Yvette, France) for access to a DRX800 Bruker spectrometer, for helpful discussions, and for technical support; and G. Bodenhausen and P. Pelupessy for access to a DRX600 Bruker spectrometer (Unité Mixte de Recherche 8642, Ecole Normale Supérieure, Paris). Special thanks are due to S. Hiller (Scripps Research Institute), F. Damberger (Eidgenössische Technische Hochschule, Zürich), and D. Picot (Unité Mixte de Recherche 7099) for information and discussions. This work was supported by the Centre National de la Recherche Scientifique (CNRS), Paris-7 University, and by a fellowship from the Ministère de la Recherche et de la Technologie (to M.Z.). The support of the Human Frontier Science Program Organization (Grant RG00223/2000-M) and the Fondation Rothschild (to J.-L.P.) is gratefully acknowledged.
Abbreviations: APol, amphipol; DAPol, a partially deuterated form of A8-35; DHPC, dihexanoyl phosphatidylcholine; HAPol, fully hydrogenated form of A8-35; HSQC, heteronuclear single-quantum correlation; MP, membrane protein; tOmpA, transmembrane domain of outer membrane protein A (residues 1–171); TROSY, transverse relaxation-optimized spectroscopy.
References
- 1.Liu, J. & Rost, B. (2001) Protein Sci. 10, 1970–1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.le Maire, M., Champeil, P. & Møller, J. V. (2000) Biochim. Biophys. Acta 1508, 86–111. [DOI] [PubMed] [Google Scholar]
- 3.Bowie, J. U. (2001) Curr. Opin. Struct. Biol. 11, 397–402. [DOI] [PubMed] [Google Scholar]
- 4.McDonnell, P. A. & Opella, S. J. (1993) J. Magn. Reson. B102, 120–125. [Google Scholar]
- 5.MacKenzie, K. R., Prestegard, J. H. & Engelman, D. M. (1997) Science 276, 131–133. [DOI] [PubMed] [Google Scholar]
- 6.Arora, A., Abildgaard, F., Bushweller, J. H. & Tamm, L. K. (2001) Nat. Struct. Biol. 8, 334–338. [DOI] [PubMed] [Google Scholar]
- 7.Fernández, C., Adeishvili, K. & Wüthrich, K. (2001) Proc. Natl. Acad. Sci. USA 98, 2358–2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hwang, P. M., Choy, W. Y., Lo, E. I., Chen, L., Forman-Kay, J. D., Raetz, C. R., Privé, G. G., Bishop, R. E. & Kay, L. E. (2002) Proc. Natl. Acad. Sci. USA 99, 13560–13565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schnaitman, C. A. (1973) Arch. Anal. Biochem. 152, 541–552. [DOI] [PubMed] [Google Scholar]
- 10.Schweizer, M., Hindennach, I., Garten, W. & Henning, U. (1978) Eur. J. Biochem. 82, 211–217. [DOI] [PubMed] [Google Scholar]
- 11.Schulte, T. H. & Marchesi, V. T. (1978) Biochim. Biophys. Acta 508, 425–430. [DOI] [PubMed] [Google Scholar]
- 12.Bishop, R. E., Gibbons, H. S., Guina, T., Trent, M. S., Miller, S. I. & Raetz, C. R. H. (2000) EMBO J. 19, 5071–5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Riek, R., Pervushin, K. & Wüthrich, K. (2000) Trends Biochem. Sci. 25, 462–468. [DOI] [PubMed] [Google Scholar]
- 14.Fernández, C. & Wider, G. (2003) Curr. Opin. Struct. Biol. 13, 570–580. [DOI] [PubMed] [Google Scholar]
- 15.Tugarinov, V., Hwang, P. M. & Kay, L. E. (2004) Annu. Rev. Biochem. 73, 107–146. [DOI] [PubMed] [Google Scholar]
- 16.Goto, N. K. & Kay, L. E. (2000) Curr. Opin. Struct. Biol. 10, 585–592. [DOI] [PubMed] [Google Scholar]
- 17.Hewitt, L. & McDonnell, J. M. (2004) Methods Mol. Biol. 278, 1–16. [DOI] [PubMed] [Google Scholar]
- 18.Pickford, A. R. & O'Leary, J. M. (2004) Methods Mol. Biol. 278, 17–34. [DOI] [PubMed] [Google Scholar]
- 19.Matthews, S. (2004) Methods Mol. Biol. 278, 35–46. [DOI] [PubMed] [Google Scholar]
- 20.Gohon, Y. & Popot, J.-L. (2003) Curr. Opin. Colloid Interface Sci. 8, 15–22. [Google Scholar]
- 21.Sanders, C. R., Hoffmann, A. K., Gray, D. N., Keyes, M. H. & Ellis, C. D. (2004) ChemBioChem 5, 423–426. [DOI] [PubMed] [Google Scholar]
- 22.Tribet, C., Audebert, R. & Popot, J.-L. (1996) Proc. Natl. Acad. Sci. USA 93, 15047–15050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Popot, J.-L., Berry, E. A., Charvolin, D., Creuzenet, C., Ebel, C., Engelman, D. M., Flötenmeyer, M., Giusti, F., Gohon, Y., Hervé, P., et al. (2003) Cell Mol. Life Sci. 60, 1559–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gohon, Y. (2002) Ph.D. thesis (Université Pierre et Marie Curie, Paris).
- 25.Gohon, Y., Pavlov, P., Timmins, P., Tribet, C., Popot, J.-L. & Ebel, C. (2004) Anal. Biochem. 334, 318–334. [DOI] [PubMed] [Google Scholar]
- 26.Pautsch, A. & Schulz, G. E. (1998) Nat. Struct. Biol. 5, 1013–1017. [DOI] [PubMed] [Google Scholar]
- 27.Pautsch, A., Vogt, J., Model, K., Siebold, C. & Schulz, G. E. (1999) Proteins 34, 167–172. [PubMed] [Google Scholar]
- 28.Pautsch, A. & Schulz, G. E. (2000) J. Mol. Biol. 298, 273–282. [DOI] [PubMed] [Google Scholar]
- 29.Pervushin K., Braun, D., Fernández, C. & Wüthrich, K. (2000) J. Biomol. NMR 17, 195–202. [DOI] [PubMed] [Google Scholar]
- 30.Fernández, C., Hilty, C., Bonjour, S., Adeishvili, K., Pervushin, K. & Wüthrich, K. (2001) FEBS Lett. 504, 173–178. [DOI] [PubMed] [Google Scholar]
- 31.Tamm, L. K., Abildgaard, F., Arora, A., Blad, H. & Bushweller, J. H. (2003) FEBS Lett. 555, 139–143. [DOI] [PubMed] [Google Scholar]
- 32.Cierpicki, T. & Bushweller, J. H. (2004) J. Am. Chem. Soc. 126, 16259–16266. [DOI] [PubMed] [Google Scholar]
- 33.Gardner, K. H. & Kay, L. E. (1998) Annu. Rev. Biophys. Biomol. Struct. 27, 357–406. [DOI] [PubMed] [Google Scholar]
- 34.Zoonens, M. (2004) Ph.D. thesis (Université Pierre et Marie Curie, Paris).
- 35.Pervushin, K., Riek, R., Wider, G. & Wüthrich, K. (1997) Proc. Natl. Acad. Sci. USA 94, 12366–12371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pervushin, K., Wider, G. & Wüthrich, K. (1998) J. Biomol. NMR 12, 345–348. [DOI] [PubMed] [Google Scholar]
- 37.Delaglio, F., Grzesiek, S., Vuister, G. W., Zhu, G., Pfeifer, J. & Bax, A. (1995) J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- 38.Johnson, B. A. & Blevins, R. A. (1994) J. Biomol. NMR 4, 603–614. [DOI] [PubMed] [Google Scholar]
- 39.Altieri, A. S., Hinton, D. P. & Byrd, R. A. (1995) J. Am. Chem. Soc. 117, 7566–7567. [Google Scholar]
- 40.Price, W. S. (1997) Concepts Magn. Reson. 10, 197–237. [Google Scholar]
- 41.Wüthrich, K. (1986) NMR of Proteins and Nucleic Acids (Wiley, New York).
- 42.Dempsey, C. E. (2001) Prog. Nucl. Mag. Res. Sp. 39, 135–170. [Google Scholar]
- 43.Phan, I. Q. H., Boyd, J. & Campbell, I. D. (1996) J. Biomol. NMR 8, 369–378. [DOI] [PubMed] [Google Scholar]
- 44.Millet, O., Loria, J. P., Kroenke, C. D., Pons, M. & Palmer A. G. (2000) J. Am. Chem. Soc. 122, 2867–2877. [Google Scholar]
- 45.Grzesiek, S., Bax, A., Clore, G. M., Gronenborn, A. M., Hu, J. S., Kaufman, J., Palmer, I., Stahl, S. J. & Wingfield, P. T. (1996) Nat. Struct. Biol. 3, 340–345. [DOI] [PubMed] [Google Scholar]
- 46.Garrett, D. S., Seok, Y. J., Peterkofsky, A., Clore, G. M. & Gronenborn, A. M. (1997) Biochemistry 36, 4393–4398. [DOI] [PubMed] [Google Scholar]
- 47.Foster, M. P., Wuttke, D. S., Clemens, K. R., Jahnke, W., Radhakrishnan, I., Tennant, L., Reymond, M., Chung, J. & Wright, P. E. (1998) J. Biomol. NMR 12, 51–71. [DOI] [PubMed] [Google Scholar]
- 48.Grzesiek, S., Anglister, J., Ren, H. & Bax, A. (1993) J. Am. Chem. Soc. 115, 4369–4370. [Google Scholar]
- 49.Fiaux, J., Bertelsen, E. B., Horwich, A. L. & Wüthrich, K. (2002) Nature 418, 207–211. [DOI] [PubMed] [Google Scholar]
- 50.Tugarinov, V., Muhandiram, R., Ayed, A. & Kay L. E. (2002) J. Am. Chem. Soc. 124, 10025–10035. [DOI] [PubMed] [Google Scholar]
- 51.Champeil, P., Menguy, T., Tribet, C., Popot, J.-L. & le Maire, M. (2000) J. Biol. Chem. 275, 18623–18637. [DOI] [PubMed] [Google Scholar]
- 52.Tribet, C., Audebert, R. & Popot, J.-L. (1997) Langmuir 13, 5570–5576. [Google Scholar]
- 53.Walters, K. J., Ferentz, A. E., Hare, B. J., Hidalgo, P., Jasanoff, A., Matsuo, H. & Wagner, G. (2001) Methods Enzymol. 339, 238–258. [DOI] [PubMed] [Google Scholar]