

Abstract
Background
The efficacy of immune checkpoint blockade therapy in patients with hepatocellular carcinoma (HCC) remains poor. Although serine‐ and arginine‐rich splicing factor (SRSF) family members play crucial roles in tumors, their impact on tumor immunology remains unclear. This study aimed to elucidate the role of SRSF10 in HCC immunotherapy.
Methods
To identify the key genes associated with immunotherapy resistance, we conducted single‐nuclear RNA sequencing, multiplex immunofluorescence, and The Cancer Genome Atlas and Gene Expression Omnibus database analyses. We investigated the biological functions of SRSF10 in immune evasion using in vitro co‐culture systems, flow cytometry, various tumor‐bearing mouse models, and patient‐derived organotypic tumor spheroids.
Results
SRSF10 was upregulated in various tumors and associated with poor prognosis. Moreover, SRSF10 positively regulated lactate production, and SRSF10/glycolysis/ histone H3 lysine 18 lactylation (H3K18la) formed a positive feedback loop in tumor cells. Increased lactate levels promoted M2 macrophage polarization, thereby inhibiting CD8+ T cell activity. Mechanistically, SRSF10 interacted with the 3′‐untranslated region of MYB, enhancing MYB RNA stability, and subsequently upregulating key glycolysis‐related enzymes including glucose transporter 1 (GLUT1), hexokinase 1 (HK1), lactate dehydrogenase A (LDHA), resulting in elevated intracellular and extracellular lactate levels. Lactate accumulation induced histone lactylation, which further upregulated SRSF10 expression. Additionally, lactate produced by tumors induced lactylation of the histone H3K18la site upon transport into macrophages, thereby activating transcription and enhancing pro‐tumor macrophage activity. M2 macrophages, in turn, inhibited the enrichment of CD8+ T cells and the proportion of interferon‐γ+CD8+ T cells in the tumor microenvironment (TME), thus creating an immunosuppressive TME. Clinically, SRSF10 could serve as a biomarker for assessing immunotherapy resistance in various solid tumors. Pharmacological targeting of SRSF10 with a selective inhibitor 1C8 enhanced the efficacy of programmed cell death 1 (PD‐1) monoclonal antibodies (mAbs) in both murine and human preclinical models.
Conclusions
The SRSF10/MYB/glycolysis/lactate axis is critical for triggering immune evasion and anti‐PD‐1 resistance. Inhibiting SRSF10 by 1C8 may overcome anti‐PD‐1 tolerance in HCC.
Keywords: Glycolysis, Histone lactylation, Immune checkpoint blockade, Serine and arginine rich splicing factor 10, Tumor‐Associated Macrophage
Abbreviations
- 2‐DG
2‐Deoxy‐D‐glucose
- 3’UTR
3' Untranslated Regions
- 3’UTR
3′‐untranslated region
- Adgre1
adhesion G protein‐coupled receptor E1
- ALT
alanine aminotransferase
- ARG1
arginase 1
- ARG1
arginase 1
- AST
aspartate aminotransferase
- BMDMs
Bone marrow derived macrophages
- CD206
mannose receptor C‐type 1
- CFSE
Carboxyfluorescein Succinimidyl Ester Ester
- ChIP
Chromatin immunoprecipitation
- CHOL
Cholangio carcinoma
- Clec4f
C‐type lectin domain family 4 member F
- CLIP
cross‐linking and immune‐precipitation
- CXCL10
C‐X‐C motif chemokine ligand 10
- DEGs
Differentially expressed genes
- DFS
disease‐free survival
- ECAR
extracellular acidification rate
- ELISA
enzyme‐linked immunosorbent assay
- FDR
false discovery rate
- FPKM
Fragments Per Kilobase of exon model per Million mapped fragments
- GEO
Gene Expression Omnibus
- GLUT1
Glucose transporter 1
- GPCR
G‐protein‐coupled receptor
- GSEA
Gene Set Enrichment Analysis
- HCC
hepatocellular carcinoma
- HDTVi
hydrodynamic tail‐vein injection
- HK1
hexokinase 1
- ICB
immune checkpoint blockade
- iCPD
immune confirmed progressive disease
- iCR
immune complete response
- IFN‐γ
interferon gamma
- IHC
Immunohistochemistry
- iPR
immune partial response
- iRECIST
Response criteria for use in trials testing immunotherapeutics
- iSD
immune stable disease
- Itgam
integrin subunit alpha M
- iUPD
immune unconfirmed progressive disease
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- LA
lactic acid
- LDHA
Lactate dehydrogenase A
- LPS
lipopolysaccharide
- Ly6c1
lymphocyte antigen 6 family member C1
- mAbs
monoclonal antibodies
- M‐CSF
Macrophage colony stimulating factor
- MCT1
solute carrier family 16 member 1
- MCT4
solute carrier family 16 member 3
- mIF
Multicolor immunofluorescence
- MRI
Magnetic Resonance Imaging
- MSigDB
Molecular Signatures Database
- MYB
MYB proto‐oncogene
- NES
Normalized enrichment score
- OCR
Oxygen Consumption Rate
- OS
overall survival
- PD‐1
Programmed cell death 1
- PD‐L1
Programmed cell death 1 ligand 1
- PDO
patient‐derived organoids
- PDOTS
Patient‐derived organotypic tumor spheroids
- PFS
progression‐free survival
- PFS
progression‐free survival
- PMA
phorbol 12‐myristate 13‐acetate
- PRF1
Perforin 1
- qRT‐PCR
quantitative real‐time polymerase chain reaction
- RIP
RNA Immunoprecipitation
- RIP‐seq
RIP sequencing
- RNA‐seq
RNA‐sequencing
- RT‐PCR
reverse transcription PCR
- scRNA‐seq
single‐cellar RNA‐sequencing
- snRNA‐seq
single‐nuclear RNA sequencing
- SRSF10
serine and arginine rich splicing factor 10
- ssGSEA
single‐sample gene set enrichment analysis
- TAMs
Tumor‐associated macrophages
- TCGA
The Cancer Genome Atlas
- TCM
tumor‐conditioned medium
- TGFB1
transforming growth factor beta 1
- TMAs
tissue microarrays
- TME
tumor microenvironment
- tSNE
t‐distributed stochastic neighbor embedding
1. BACKGROUND
Hepatocellular carcinoma (HCC) ranks sixth in incidence worldwide, with tumor‐related death ranking third [1, 2]. Hepatectomy, the primary treatment for HCC, significantly enhances overall survival (OS). However, a high postoperative recurrence rate markedly undermines patient prognosis [3]. Consequently, there is an urgent need for more effective treatment for HCC. Immunotherapy, which has proven effective in various cancer types such as melanoma and lung cancer, is gaining prominence in HCC management [4].
Recently, immune checkpoint blockade (ICB) therapy, especially the use of antibodies against programmed cell death (/ligand) 1 (PD‐1/PD‐L1) signaling, has achieved remarkable advancements in cancer treatment [5]. However, in the context of immunotherapy for HCC, the response rate to PD‐1/PD‐L1 blockade is less than 30% [6, 7, 8] with no significant improvement in OS. This limitation stems primarily from the heterogeneity of the tumor microenvironment (TME) and its immunosuppressive nature. The liver, traditionally considered an immune‐exempt organ, hosts a substantial population of macrophages, including resident (Kupffer cells) and recruited macrophages [9]. Macrophages exhibit considerable heterogeneity and plasticity. M1 macrophages are primarily activated by lipopolysaccharide, and interferon‐γ (IFN‐γ), among other stimuli, and contribute to pro‐inflammatory processes [10]. Within tumor environments, M1 macrophages stimulate Th1‐type immune responses, activate effector T cells through the release of interleukin 1 (IL1), IL6, IL12, tumor necrosis factor (TNF), and C‐X‐C motif chemokine ligand 9 (CXCL9), and promote anti‐tumor immune reactions [11]. In contrast, M2 macrophages are induced by IL4, IL10, IL13, and glucocorticoids, and possess anti‐inflammatory properties [11]. M2 macrophages secrete immunosuppressive factors such as IL10 and transforming growth factor beta 1 (TGFB1), and express CD163, arginase 1 (ARG1), and CD206, leading to immune suppression and promotion of angiogenesis [12]. Tumor‐associated macrophages (TAMs) share functional and phenotypic similarities with M2 macrophages and contribute significantly to tumor progression, immune evasion, and resistance to immunotherapy [13]. Consequently, strategies aimed at shifting M2 macrophages toward the M1 phenotype to enhance immune inflammatory responses or inhibit TAM recruitment and infiltration hold promise as novel approaches for tumor therapy.
Metabolic adaptation to aerobic glycolysis is a common characteristic of cancer cells [14]. The glycolytic pathway facilitates immune evasion via various mechanisms. First, there is competition for nutrients in the TME between tumor and immune cells. For example, within the TME, cancer cells can compete with T cells for glucose, leading to a detrimental effect on the activation of T cells, which is dependent on glucose metabolism [15, 16]. Second, byproducts of aerobic glycolysis, such as lactate, can impede the activity of CD8+ T cells by promoting the creation of an immunosuppressive TME [17]. Thirdly, the metabolic pathway of tumors can influence the levels of various genes involved in immune regulation, such as PD‐L1 and CXCL10 [18, 19]. Although the involvement of the glycolytic pathways in immune evasion has been suggested, the mechanism by which these pathways directly confer resistance to immune‐mediated killing remains unclear.
The serine‐ and arginine‐rich splicing factor (SRSF) family is a crucial splicing factor and RNA‐binding protein. Classical SR proteins comprise 12 evolutionarily conserved proteins, characterized by at least one RNA recognition motif and a serine/arginine‐rich domain [20]. The abnormal expression of several SRSF family members has been observed in various tumor types. For example, SRSF3 has shown high expression levels in colon cancer [21] and oral cancer [22], whereas SRSF5 exhibits elevated expression in prostate cancer [23], lung cancer [24], and pancreatic cancer [25], and has implications for cancer promotion [26]. SRSF10 influences tumorigenesis via alternative splicing regulation [27, 28, 29, 30]. However, the precise role of SRSF10 in modulating tumor metabolic imbalance and its impact on the TME remainunderstood unclear. To determine the association between SRSF10 and glycolytic reprogramming, we conducted a comprehensive analysis using data from multiple transcriptomic and metabolomic levels. Additionally, we performed in vitro and in vivo assays to investigate the involvement of SRSF10 in the TME. This study aimed to elucidate the clinical relevance of SRSF10 in HCC and the molecular mechanisms underlying SRSF10‐mediated glycolysis and its effects on the TME.
2. MATERIALS AND METHODS
2.1. Cell lines
Human HCC cell lines Huh7, PLC/PRF/5 and mouse HCC cell line Hepa1‐6 were procured from the Liver Cancer Institute, Fudan University (Shanghai, China). Human embryonic kidney cell line 293T (HEK‐293T) and myeloid line THP1 were purchased from the Chinese Academy of Sciences Shanghai Branch Cell Bank (Shanghai, China). All cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM, KFMI12800N, KeyGEN BioTECH, Jiangsu, China) or RPMI‐1640 (C11875500CP, Gibco, FL, USA) medium supplemented with 10% fetal bovine serum (FBS, F8318, Sigma, MO, USA), 100 U/mL penicillin, and 100 U/mL streptomycin (C0222, Beyotime, Shanghai, China) at 37°C in a humidified incubator with 5% CO2. All cell lines used in this study were tested to confirm that they were free of mycoplasma and authenticated by short‐tandem repeat analysis.
2.2. Mice and in vivo experiments
Male C57BL/6 mice aged 6‐7 weeks and male BALB/c nude mice aged 8 weeks (Modelorg, Shanghai, China) were utilized for this study. BALB/c nude mice were randomly assigned to two groups (n = 6), with each receiving a 150 μL injection of a single‐cell suspension comprising 1×106 Huh7 cells in 1× PBS into the right flank. In the case of C57BL/6 mice, for the subcutaneous HCC model, each mouse received a 150 μL injection of a single‐cell suspension containing 5×105 Hepa1‐6 cells in 1× PBS into the right flank. Tumor measurements were performed every two days using the formula: length × width2 × 0.5 (mm3). In the orthotopic HCC model, mice were initially anesthetized, and their abdominal hair was removed. A 9 mm transverse incision was made in the upper abdomen, following which a 20 μL single‐cell suspension of Hepa1‐6 cells (5 × 105 per mouse) was introduced into the left lobe of the liver in C57BL/6 mice. A suspension was prepared by combining Matrigel (356234, Corning, NY, USA) with 1× PBS at a 1:1 ratio. For the spontaneous HCC model, a sterile 0.9% NaCl solution/plasmid mix was prepared, containing 10 μg DNA of pT3‐EF1a‐Myc, 10 μg of px330‐p53, 10 μg of Ctnnb1‐N90, and 10 μg of PT2/C‐Luc//PGK‐SB13 (General Biol, Anhui, China). C57BL/6 mice were administered the 2 mL mixed solution via the lateral vein within 7 s. The plasmid was generously provided by Dr. Guiqi Zhu of the Jia Fan Research Group at Zhongshan Hospital Affiliated with Fudan University. All constructs were validated through nucleotide sequencing. After the experiment, mice were euthanized by rapid cervical dislocation under anesthesia to obtain HCC tumors for weighing, in accordance with the experimental objectives.
For macrophage depletion, mice received intraperitoneal injections of clodronate liposomes (40337ES08, Yeasen, Shanghai, China) or control liposomes (40338ES08, Yeasen) at a dose of 10 mg/kg on days 1 and 7 following tumor inoculation. To inhibit SRSF10 in vivo, mice were administered intraperitoneal injections of 1C8 at a dose of 15 mg/kg (HY‐134851, MedChemExpress, NJ, USA) or dimethyl sulfoxide (DMSO, HY‐Y0320, MedChemExpress) every two days starting from day 3 after tumor inoculation or day 7 after lateral vein injection. For anti‐PD‐1 therapy, mice received intraperitoneal injections of anti‐mouse PD‐1 antibody (200 μg per injection, BE0146, Bio X cell, CA, USA) or IgG isotype control (200 μg per injection, BE0089, Bio X cell) every 3rd day following tumor inoculation in the subcutaneous HCC tumor and every 3 days beginning on the 10th day in the spontaneous HCC model. All animals were housed in the Laboratory Animal Center of Zhongshan Hospital, Fudan University, which met specific pathogen‐free standards. The handling of all animals strictly adhered to the Principles for the Utilization and Care of Vertebrate Animals and the Guide for the Care and Use of Laboratory Animals. The Institutional Animal Care and Use Committee of Zhongshan Hospital, Fudan University, approved all animal experiments (Approval No. 2020‐133).
2.3. Clinical tissue samples
For this study, we utilized two separate tissue microarrays (TMAs) for HCC treatment analysis. TMA1 comprised a total of 239 pairs of tumor and adjacent normal tissues, obtained through curative resection from patients at Zhongshan Hospital (Shanghai, China). These patients underwent surgery between March 2010 and December 2010, with no prior anticancer treatment, and were followed up until December 2015. For this study, tissue specimens were collected from patients with HCC who underwent liver resection. Specimens from patients who had undergone any HCC‐related treatment prior to surgery, such as chemotherapy, radiotherapy, or targeted therapy, were excluded to eliminate potential confounding effects on the biological markers and outcomes being studied. Ethics approval for this study was obtained from the Research Ethics Committee of Zhongshan Hospital, Fudan University, and written informed consent was acquired from all participants. TMA2 consisted of 75 biopsy specimens from patients with HCC scheduled for anti‐PD‐1 treatment at Zhongshan Hospital, Fudan University, between September 2017 and December 2018. Written informed consent was obtained, and sample usage was authorized by the Institutional Review Board of Zhongshan Hospital, Fudan University. Prior to anti‐PD‐1 treatment, biopsy specimens were collected guided by computed tomography. Patients underwent bimonthly magnetic resonance imaging (MRI) examinations to assess the therapeutic effects of anti‐PD‐1 treatment according to iRECIST guidelines [31]. Specifically, immune complete response (iCR) and immune partial response (iPR) were classified as responders, while immune stable disease (iSD), immune unconfirmed progressive disease (iUPD), and immune confirmed progressive disease (iCPD) were classified as non‐responders. For single‐nuclear RNA sequencing (snRNA‐seq), surgical specimens were obtained from 6 patients with HCC who had not undergone any interventions. Following hepatectomy, these patients received the anti‐PD‐1 antibody sintilimab (Innovent Biologics, Jiangsu, China) at a dose of 200 mg each time, administered via slow intravenous infusion every two weeks for a duration of 8 weeks. Regular MRI follow‐ups were conducted to monitor disease progression and assess treatment efficacy. Among them, 2 patients exhibited progression‐free survival (PFS) exceeding 26 months, categorized as immunotherapy responders, while 4 experienced disease progression within 8 months post‐treatment and were classified as immunotherapy non‐responders. Ethical approval for this study was obtained from the Research Ethics Committee of Zhongshan Hospital (Approval No. B2021‐248), and written informed consent was obtained from all participants. Detailed clinical characteristics of the patients are listed in Supplementary Table S1.
2.4. Download and analysis of publicly available datasets
To identify target genes for further analysis, we employed a three‐step screening process: First, we identified tumor‐specific highly expressed genes by analyzing four publicly available datasets from the Gene Expression Omnibus (GEO): GSE10143, GSE29721, GSE46408, and GSE74656. Genes that were significantly overexpressed in tumor tissues were selected using the criteria of fold change (FC) > 1 and P value < 0.05. Next, to identify genes highly correlated with glycolysis, we used the glycolysis Hallmark gene set from the Molecular Signatures Database (MSigDB, http://www.gsea‐msigdb.org/gsea/msigdb/). Single‐sample Gene Set Enrichment Analysis (ssGSEA) was performed to calculate glycolysis scores for each sample in The Cancer Genome Atlas (TCGA)‐liver hepatocellular carcinoma (LIHC) dataset. Pearson correlation analysis was then conducted to identify genes with a correlation coefficient greater than 0.4, indicating a strong association with glycolysis. Finally, we assessed the prognostic significance of these genes through univariate Cox regression analysis on the TCGA‐LIHC dataset, with a significance threshold set at P < 0.05. The intersection of genes identified through these three methods was considered the final set of target genes for subsequent analyses.
RNA expression data for SRSF10 and MYB across 33 different tumor types were obtained from GEPIA 2 (http://gepia2.cancer‐pku.cn/#index). Protein expression data for these genes in various tumor types were acquired from the Human Protein Atlas (HPA, https://www.proteinatlas.org). GEPIA 2 was utilized for the analysis and visualization of RNA expression levels, while the HPA provided protein expression profiles based on immunohistochemistry data across multiple cancer types.
For immunotherapy datasets: In this study, three bulk RNA‐seq datasets and two scRNA‐seq datasets treated with immunotherapy were collected for analysis. The bulk RNA‐seq and corresponding clinical data were obtained from Riaz‐GSE91061, PRJNA482620, and Gide‐PRJEB23709, all downloaded from the TIDE (http://tide.dfci.harvard.edu) database. For scRNA‐seq, the dataset GSE123813 was downloaded from GEO database. To define the responders and non‐responders, we aimed to faithfully use the criteria defined in the original clinical trials as much as possible. For the cohorts where Response Evaluation Criteria in Solid Tumors (RECIST1.1) [32] information is available, we used complete response (CR), partial response (PR) as responders and stable disease (SD), progressive disease (PD) as non‐responders (Riaz‐GSE91061, Gide‐PRJEB23709 and GSE123813 cohorts. For PRJNA482620 dataset, Patients were classified as responders if they met at least one of the following two criteria: 1): Tissue sampled during surgery after PD‐1 inhibitor therapy grossly showed only an inflammatory response and very few to no tumor cells. 2): Tumor volumes as seen from magnetic resonance imaging (MRI) were either stable or shrinking continually over at least six months.
2.5. 5 snRNA‐seq
HCC tissues were promptly frozen in liquid nitrogen for 30 min and subsequently stored at −80°C. Tissue lysates, consisting of 40 units/mL RNase inhibitor, 0.1% NP40 (P0013F, Beyotime), 1 mmol/L CaCl2 (ST365, Beyotime), and 1 mmol/L NaCl (ST1641, Beyotime), were utilized to lyse the HCC tissues. The lysates were then centrifuged at 4°C and 500 ×g for 5 min to collect the nuclear precipitate. The Chromium Next GEM Single Cell 3ʹ Reagent Kits v3 (1000268, 10× Genomics, CA, USA) were utilized for constructing the snRNA‐seq libraries. Following various experimental procedures, including cell counting and quality control, gel bead‐in‐emulsion (GEMs) generation and barcoding, post‐GEM‐RT cleanup, cDNA amplification, gene expression library construction, high‐quality snRNA‐seq data were obtained. Sequencing was conducted using the NovaSeq platform (Illumina, CA, USA).
2.6. Plasmids, lentiviral construction, and cell transfection
The shRNA sequences were synthesized by Obio (Shanghai, China). Human and mouse plasmids targeting SRSF10 (shSRSF10, shSrsf10) and MYB (shMYB, shMyb) were constructed using the pSLenti‐U6‐CMV‐EGFP‐F2A‐Puro vector. Recombinant plasmids were synthesized by Obio. For MYB/Myb overexpression, the pcDNA3.1‐C‐Myc‐DDK‐IRES‐Puro vector was utilized, while SRSF10 overexpression plasmids were generated using the pcDNA3.1‐C‐Myc‐FLAG‐IRES‐Puro vector. Detailed sequences of the inserts are provided in Supplementary Table S2. Subsequently, the aforementioned target plasmids and empty vectors were separately co‐transfected into 293T cells with lentiviral packaging plasmids pMD2.G (TSPLA10280, Testobio‐tech, Ningbo, Zhengjiang, China) and pxPAX2 (TSPLA10280, Testobio‐tech) to produce lentiviruses. To enhance infection efficiency, lentiviruses were combined with 0.1% polybrene (5 μL/mL, C0351, Beyotime) and incubated with the cells for 16 h. Stable strains were established by treating the cells with a medium containing 6 μg/mL of puromycin (ST551, Beyotime).
2.7. Single‐cellar RNA sequencing (scRNA‐seq)
For in‐house datasets, control and Srsf10‐Knockdown Hepa1‐6 cells (5 × 105) were injected into the livers of C57BL/6 mice. Following a 14‐day period, HCC tumors were dissected and digested in DMEM medium containing 0.1% type 4 Collagenase (1:1,000, C5138, Sigma), 0.05% Hyaluronidase (1:2,000, H3506, Sigma), and 0.01% Deoxyribonuclease (1:10,000, DN25, Sigma) for 30 min at 37°C. The resulting cells were counted, and 2 × 104 primary cells from each tumor were loaded onto a 10× Genomics Chromium Platform. Subsequently, the samples were processed following the manufacturer's protocol, utilizing the Single Cell 3’v2 reagent, and then sequenced on an Illumina NextSeq sequencer. Raw data were aligned to the mouse genome mm10‐3.0.0 using cellranger3.1.0. Downstream analyses were conducted using R (v.4.1.2) with Seurat (v.3.2.2) and Scran (v.1.14.6), following the reported methodologies [33]. Publicly available scRNA‐seq datas of tumor patients were obtained from Lu et al. [34] and Ma et al. [35]. Standard Seurat settings were employed for normalization, principal component analysis (PCA), t‐distributed stochastic neighbor embedding (tSNE) analysis, and clustering. Differential expression analysis was performed utilizing the Wilcoxon rank‐sum test, with an average fold change threshold of 2 and an adjusted P value cutoff of 0.05.
2.8. Analysis of scRNA‐seq and snRNA‐seq
We applied the DoubletFinder (v2.0.4) for Double cell removal and the Harmony (v1.0) package for batch correction, followed by utilizing the FindNeighbors and FindClusters functions within the Seurat package (v4) for cluster analysis. Subsequently, visualization was conducted using the RunTSNE function. To infer cell‐cell communication patterns among different cell types in the scRNA‐seq data, we employed the CellChat package (https://www.cellchat.org/). Receptors and ligands expressed in more than 20 cells within specific clusters were selected for subsequent analyses. For the macrophage signature score, the average gene expression values for each cell were calculated using the M1 and M2 macrophage gene sets constructed by Bi et al. [36]. Finally, we utilized the ggplot2 package to visualize the putative ligand‐receptor pairs between macrophages and CD8+ T cell subpopulations directly.
2.9. Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis
We utilized the “clusterProfiler” R package to annotate the genes with KEGG. Statistical threshold was set as P value < 0.05. The hallmark gene sets were obtained from the MSigDB database (https://www.gsea‐msigdb.org/gsea/index.jsp).
2.10. Multiplex immunofluorescence (mIF) assay
To analyze the mIF samples, the slides were deparaffinized in xylene, rehydrated in ethanol, and incubated with 0.3% hydrogen peroxide. Antigen retrieval was performed using citrate buffer, and the slides were then blocked with 5% bovine serum albumin (BSA, ST025, Beyotime). Primary antibodies (Supplementary Table S3) were applied and incubated for 60 min in a humidified chamber at 37°C, followed by incubation with the corresponding secondary antibody. To remove excess antibodies, the slides were immersed in citrate buffer before use. Finally, DAPI solution was added to the slides and incubated away from light for 10 min at 37°C. The CaseViewer (Biossci, Hubei, China) software was used to detect and capture images.
2.11. Flow cytometry
To generate a single‐cell suspension, harvested tumors were initially digested using 0.1% type 4 collagenase, 0.05% hyaluronidase, and 0.01% deoxyribonuclease. The digestion process occurred on a horizontal platform at 37°C for 30 min, followed by filtration using a 40 mm cell strainer. The cells were washed twice with PBS. Next, single‐cell suspensions were incubated with Fixable Viability Dye eFluor (65‐0866‐14, Thermo Fisher Scientific, MA, USA). Before surface labeling with antibodies, the Fc gamma receptor was blocked with CD16/CD32 monoclonal antibody (mAb, 1:50, MFCR00‐4, Thermo Fisher Scientific) at 4°C for 15 min. To analyze specific membrane molecules, the indicated antibodies were added to the cells at appropriate dilutions. Staining was conducted for 30 min on ice in the dark. For intracellular staining, a Fixation/Permeabilization Kit (554714, BD Biosciences, NJ, USA) was utilized. The cell suspension was then incubated with intracellular antibodies at 4°C for 30 min, followed by two washes and suspension in PBS. Immune cells within the CD45+ live cell population were focused on, and diverse cell populations were identified. Flow cytometric analysis was performed using a FACSAria™ III flow cytometer (BD Biosciences), and compensation settings were adjusted using single‐staining samples. In flow cytometry analysis of various macrophage subsets, there was no difference in antibody autofluorescence among different groups. Data were collected and subsequent analysis was conducted using FlowJo software (Tree Star, OR, USA). All fluorophore‐conjugated antibodies are listed in Supplementary Table S3.
2.12. Bone marrow‐derived macrophages (BMDMs)
The femurs and tibias of 6–8‐week‐old mice were dissected to extract bone marrow cells. These cells were cultured in DMEM supplemented with 10% FBS, 100 U/mL penicillin, 100 U/mL streptomycin, and 40 ng/mL macrophage colony‐stimulating factor (M‐CSF) (abs04383, Absin, Shanghai, China) for 7 days to induce macrophage differentiation.
2.13. RNA extraction and quantitative real‐time polymerase chain reaction (qRT‐PCR)
Total RNA was extracted using TRIzol reagent (15596026CN, Thermo Fisher Scientific) and reverse‐transcribed into cDNA using a cDNA synthesis kit (11120ES60, Yeasen). The quantity and concentration of RNA were measured using a spectrophotometer. qRT‐PCR was performed using the SYBR Green Master Mix Kit (11202ES08, Yeasen) following the manufacturer's guidelines. Each 10 μL reaction mixture contained 1 μL of DNA extract, 1 μL of forward and reverse primers for each gene (gene‐specific primers), 5 μL SYBR, and 3 μL ddH2O. Thermal cycling conditions consisted of an initial denaturation step at 95°C for 5 min, followed by 38 cycles of denaturation at 95°C for 10 s, annealing at 57°C for 25 s, and extension at 72°C for 20 s. The specific primers used for qRT‐PCR are listed in Supplementary Table S4. Each assay was repeated at least three times. β‐actin was used as an internal reference, and the data were analyzed using the 2−ΔΔCT method.
2.14. Chromatin immunoprecipitation (ChIP)
ChIP was performed using the SimpleChIP® Plus Enzymatic Chromatin IP Kit (9005, Cell Signaling Technology, MA, USA) following the manufacturer's instructions. Initially, cells were crosslinked with 1% formaldehyde and sonicated to extract and fragment chromatin. Subsequently, the samples underwent immunoprecipitation using specific antibodies. SYBR Green PCR Master Mix was utilized for quantitative PCR analysis. To standardize the value of immunoprecipitated DNA, either an input signal or a control group was employed. The specific primer sequences utilized for ChIP‐qPCR are detailed in Supplementary Table S4.
2.15. Transwell assay
To assess cell migratory capability, Transwell assays were conducted. Initially, 320 nmol/L phorbol 12‐myristate 13‐acetate (PMA, S1819, Beyotime)‐treated THP1 cells (5 × 104) or BMDMs (2 × 105) were seeded in the upper chamber (3422, Corning), while Huh7 cells or Hepa1‐6 cells were seeded in the lower chamber. After 48 h of incubation, non‐migrating cells were removed using cotton swabs. Subsequently, the cells at the bottom of the chamber were fixed with paraformaldehyde, stained with crystal violet, and observed under an inverted microscope (Olympus, Tokyo, Japan). Each experiment was performed in triplicate to ensure the reliability of the results.
2.16. Coculture assay
To conduct the coculture experiment, we utilized 6‐well Transwell chambers with 0.4 μm pores (PTHT06H48, Corning). THP1 cells (1 × 106) were treated with PMA at a concentration of 320 nmol/L for 24 h. Similarly, BMDMs were introduced into the lower Transwell chambers. In the upper chamber of the coculture system, we seeded 5 × 105 Huh7 or Hepa1‐6 cells for 48 h. Subsequently, we separately collected macrophages for qRT‐PCR and flow cytometry analyses.
2.17. T cell suppression assay
The assay utilized carboxyfluorescein succinimidyl ester (CFSE) dilution analysis. Human T cells were isolated from peripheral venous blood of healthy human volunteers using an immunomagnetic negative selection with the EasySep Direct Human T Cell Isolation Kit (19661; STEMCELL Technologies, British Columbia, Canada). Then, T cell were cultured in CTS™ AIM V™ SFM (A3021002, Gibco) supplemented with ImmunoCult Human CD3/CD28 T cell activator (10971, STEMCELL Technologies) and recombinant human IL‐2 (78220, STEMCELL Technologies) for 48 h. Mouse CD8 T cells were isolated from the spleens of wild‐type C57BL/6 mice using CD8 T Cell Isolation Kit (480035, Biolegend, CA, USA) according to the manufacture's protocols. To initiate preactivation, CD8 T cells were enumerated and plated onto a 6‐well culture plate precoated with 2 μg/mL of anti‐CD3 antibody (100340, BioLegend) and 2 μg/mL of anti‐CD28 antibody (102116, BioLegend). The cells were cultured in a complete medium consisting of RPMI 1640 supplemented with 10% FBS, 100 U/mL interleukin‐2 (212‐12, Peprotech, NJ, USA), 2 mmol/L L‐glutamine (07100, STEMCELL Technologies), 50 μmol/L β‐mercaptoethanol (GNM21985‐1, GENOM Biotech, CA, USA), 1 mmol/L sodium pyruvate (C0331, Beyotime), 100 μmol/L MEM non‐essential amino acids (07600, STEMCELL Technologies), and 10 mmol/L HEPES (07200, STEMCELL Technologies) for 48 h. Then human T cells and mouse CD8 T cells were labeled with CellTrace CFSE Cell Proliferation Kit (C34570, Thermo Fisher Scientific) following the manufacturer's instructions. CFSE levels in T cells were assessed by flow cytometry after 96 h of culture.
2.18. Immunohistochemistry (IHC) assay
Deparaffinization and rehydration of slides for IHC were performed using xylene and ethanol, respectively. After a 30‐min incubation with 0.3% hydrogen peroxide, antigen retrieval was carried out by subjecting the slides to citrate buffer at a sub‐boiling temperature for 15 min. Subsequently, the membranes were blocked with 5% BSA for 60 min. The next step involved overnight incubation of slides with primary antibodies (Supplementary Table S3) at 4°C, followed by incubation with HRP‐conjugated secondary antibodies at 37°C for 1 h. To develop color, we utilized a 3,3’‐diaminobenzidine tetrahydrochloride kit (Gene Tech, Shanghai, China), and for nuclear counterstaining, hematoxylin was used. Images were acquired using either a standard microscope (Olympus) or CaseViewer software (3DHISTECH, Budapest, Hungary). Staining intensity was measured by analyzing digital photographs of antigen expression using Image‐Pro Plus software (version 6.0; Media Cybernetics, Inc., Rockville, MD, USA). Quantification involved evaluating the total area of brown staining in pixels, and the output provided integrated optical density (IOD) values for each section. Two pathologists, blinded to the patients' clinical information, independently double‐checked the IOD values. Any discrepancies were resolved through consensus.
2.19. RNA‐sequencing (RNA‐seq)
Total RNA from shCtrl and shSRSF10 Huh7 cells (5×106 cells, n = 3 per group) was extracted using TRIzol reagent according to the manufacturer's protocol. The collected RNA samples were subjected to quality control prior to analysis. After quality control, magnetic beads with oligo (dT) were used to enrich mRNA and prepare the library. Paired‐end sequencing was performed using an Illumina sequencing platform. All sequenced reads were quality‐checked using FastQC, followed by mapping to the human reference genomes GRCh38 and HISAT2 (https://ccb.jhu.edu/software/hisat2/index.shtml). Fragments per kilobase of exon model per million mapped fragments (FPKM) were used to normalize gene expression. Differentially expressed genes (DEGs) were defined as genes with a P value < 0.05 and an absolute log2 (fold change) > 1.
2.20. Western blotting
For Western blotting, cells were lysed using cell lysis buffer (P0013B, Beyotime) supplemented with phenylmethylsulfonyl fluoride (PMSF, ST506, Beyotime) and protease and phosphatase inhibitor cocktails (P1048, Beyotime). The lysates were then boiled in a 5× SDS loading buffer (P0286, Beyotime) for 10 min. Ten microliters of protein were loaded into each well of the Bis‐Tris SDS/PAGE gel for electrophoresis, and the gel was transferred to PVDF membranes (IPVH00010, Millipore, MA, USA). After blocking with a quick blocking solution (Beyotime, P0252), the membranes were incubated overnight at 4°C with primary antibodies, as described in Supplementary Table S3. Subsequently, species‐matched secondary antibodies (RS0001 or RS0002, ImmunoWay, CA, USA) were applied for 60 min at room temperature approximately 25°C. The bands were visualized using a chemiluminescent HRP substrate (P10100, NCM Biotech, Suzhou, China) and an electrogenerated chemiluminescence (ECL) imaging system (Tanon, Shanghai, China). For assays involving protein dephosphorylation, lysates were incubated with calf intestinal phosphatase (CIP, D7027, Beyotime) at 37°C for 1 h before boiling. Following dephosphorylation, the lysates were processed for Western blotting as described above.
2.21. RNA immunoprecipitation (RIP) and RIP sequencing (RIP‐seq)
The Millipore MagnaRIP Kit (17‐700, Millipore) was utilized for RIP following the manufacturer's protocol. Briefly, Huh7 cells, PLC/PRF/5 cells and Hepa1‐6 cells were cultured in 10‐cm dishes until they reached 90% confluence. Subsequently, the cells were washed with ice‐cold PBS and scraped off the dishes. After centrifugation at 250 ×g (4°C) for 5 min, the resulting pellet was suspended in 115 μL of the provided Lysis Buffer and incubated on ice for 5 min to complete the lysis. For each immunoprecipitation reaction, 6 μg of Antibody and 50 μL of magnetic beads were combined and cross‐linked at room temperature (approximately 25°C) for 1 h. A total of 100 μL of thawed lysate was used for each IP reaction, with 10 μL of lysate preserved and stored at –80°C for subsequent RNA elution. The IP procedure was conducted overnight with rotation at 4°C, followed by five washes of the beads using RIP Wash Buffer. The washed beads and the input lysate were then resuspended in 150 μL of RIP Wash Buffer supplemented with 0.1% SDS and 180 μg of Proteinase K. After a 30‐min incubation at 55°C following proteinase digestion, RNA extraction was performed using Trizol, followed by RT‐qPCR analysis.
For RIP‐seq analysis, RNA extracted from Huh7 cells using Trizol was utilized for repairing DNA fragment ends, adding A bases to the 3' end, and connecting sequencing adapters. PCR amplification and selection of DNA product size typically ranged from 300‐400 bp, including adapter sequences. Subsequently, library quality inspection was performed on the constructed libraries. Upon successful quality inspection, sequencing was conducted using the Illumina HiSeq/NextSeq platform. The selection criteria for analysis included a false discovery rate (FDR) of less than 0.05 and an absolute log2 fold change greater than 2.
2.22. Gene Set Enrichment Analysis (GSEA) pathway enrichment
Gene sets were obtained from the MSigDB. Data were acquired from bulk RNA‐seq analysis of shCtrl and shSRSF10 Huh7 cells. GSEA was conducted using the GSEA software (https://www.broadinstitute.org/gsea/).
2.23. RNA stability assay
Huh7, PLC/PRF/5 and Hepa1‐6 cells were seeded in 6‐well plates to achieve 50% confluence after 24 h. The cells were treated with 10 μg/mL of actinomycin D (SBR00013, Sigma) for the specified durations. Total RNA was extracted every 4 h and analyzed using qRT‐PCR. Actin served as the internal reference, and the data were analyzed using the 2−ΔΔCT method.
2.24. Untargeted energy metabolism sequencing
shCtrl or shSrsf10 Hepa1‐6 cells (1 × 106) were seeded in 100 mm2 culture dishes and cultured until reaching approximately 80%–90% confluence. Subsequently, they were washed twice with PBS and harvested. A mixture of 200 μL pre‐cooled water and 800 μL pre‐cooled pure methanol was added to the cells, followed by vortexing, sonication (600N2218, Sxsonic, Shanghai, China) in an ice bath for 20 min, and incubation at ‐20°C for 1 h to precipitate proteins. After centrifugation at 16,000 ×g for 20 min at 4°C, the supernatant was collected and standardized based on protein concentration. The supernatant was then dried using a high‐speed vacuum concentration centrifuge. Prior to mass spectrometry detection, 50 μL of methanol‐water solution (1:1, v/v) was added to reconstitute the sample. The mixture was centrifuged at 20,000 ×g at 4°C for 15 min, and the supernatant was used for sample analysis. Throughout the analysis, samples were maintained in an autosampler at 4°C. The SHIMADZU‐LC30 ultra‐high performance liquid chromatography system (UHPLC) was employed, with the ACQUITY UPLC® HSS T3 (2.1×100 mm, 1.8 μm) (Waters, Milford, MA, USA) chromatographic column. Specific operational procedures were followed as per the manufacturer's instructions. Differentially metabolites were defined as genes with a P value < 0.05 and an absolute log2 (fold change) > 1.
2.25. Lactate production
Huh7, PLC/PRF/5 and Hepa1‐6 cells were plated onto 100 mm plates and cultured for 24 h until reaching 80% confluence. Upon reaching the desired confluence, cells were lysed. For intracellular lactate measurement, cells were resuspended in 1 mL of assay buffer and sonicated for 3 min (3 s on, 7 s off, at 300 W). The supernatant was collected by centrifugation at 4°C and 12,000 ×g for 10 min. For extracellular lactate measurement in the culture medium, 100 μL of culture medium was taken and mixed with 1 mL of assay buffer, followed by centrifugation at 4°C and 12,000 ×g for 10 min to collect the supernatant. To inhibit lactate secretion, Monocarboxylate Transporter 1 (MCT1) inhibitor BAY‐8002 (50 μmol/L, T5208, Targetmol, Boston, USA) and Monocarboxylate Transporter 4 (MCT4) inhibitor AZD0095 (20 μmol/L, T64378, Targetmol) were added to DMEM cultured Huh7, PLC/PRF/5 and Hepa1‐6 cells. After incubation for 24 h, cell supernatants were collected to extract extracellular lactate. Lactate detection was performed using a lactic acid content assay kit (D799099, Sangon Biotech, Shanghai, China) following the manufacturer's instructions. Finally, the obtained precipitate was dissolved in 200 ml absolute ethanol, and the absorbance was detected at 570 nm absorption peak by enzyme‐labeled instrument. The lactic acid concentration was calculated according to the standard curve and the results were normalized to total protein content.
2.26. Measurement of extracellular acidification rate (ECAR)
ECAR was measured using a 96‐well Extracellular Flux Analyzer (Seahorse, Agilent Technologies, CA, USA) following the manufacturer's instructions. Briefly, 2 × 104 Huh7 and 1 × 104 Hepa1‐6 cells per well were cultured overnight in Seahorse XFe96 cell culture microplates. The following day, the cells were pre‐incubated at 37°C for a minimum of 45 min in the absence of CO2 in DMEM (103680, Agilent) supplemented with 200 μmol/L glutamine (103579, Agilent), 1 mmol/L pyruvate solution (103578, Agilent), and 1 mmol/L glucose solution (103577, Agilent). ECAR was measured using 10 mmol/L glucose (103020, Agilent), 1 mmol/L oligomycin (103020, Agilent), and 50 mmol/L 2‐Deoxy‐D‐glucose (2‐DG, 103020, Agilent). After equilibration of the temperature and pH, measurements were recorded with the analyzer according to the manufacturer’ protocol.
2.27. Oxygen Consumption Rate (OCR)
OCR was conducted using an OCR Plate Assay Kit (E297, Dojindo, Kanagawa Prefecture, Japan) following the manufacturer's protocol. Briefly, 4 × 104 Huh7 cells and Hepa1‐6 cells were seeded in a 96‐well plate and cultured overnight. Then, 100 μL of medium containing the Oxygen Probe was added, and measurements were taken every 10 min at 37°C using a fluorescence plate reader (Thermo Fisher Scientific). The OCR values were calculated based on the obtained curves.
2.28. Ultraviolet cross‐linking and immune‐precipitation (CLIP)
CLIP was conducted using a CLIP Kit (Bes3014, Bersinbio, Guangzhou, China) following the manufacturer's protocol. Briefly, Prior to ultraviolet cross‐linking, cells were pretreated with 4‐thiouridine (HY‐W011793, MedChemExpress) for 16 h. Subsequently, RNA was fragmented into 300 nt fragments. Following overnight antibody precipitation, the protein‐RNA complex was isolated using magnetic beads. RNA within the complex was extracted using the TRIzol reagent. MYB mRNA expression was detected using RT‐qPCR. The primers used for CLIP‐qPCR are listed in Supplementary Table S4.
2.29. Analysis of JASPAR for TF binding to targeted gene promoter regions
The binding sites within the promoters of the glucose transporter 1 (GLUT1), hexokinase 1 (HK1), and lactate dehydrogenase A (LDHA) genes with MYB were predicted utilizing the online prediction tool JASPAR (http://jaspar.genereg.net). Initially, the base sequence located 2000 bp upstream of each gene was identified as the promoter region. Subsequently, this promoter region sequence was aligned with the assumed binding sequence of MYB based on the principle of complementarity. Pairing sites were then ranked according to their scores, with the site exhibiting the highest score deemed as the putative binding site for further verification. The binding sites within the promoters of Glut1, Hk1, and Ldha with Myb were determined by employing the same protocol.
2.30. Patient‐derived organotypic tumor spheroids (PDOTS)
PDOTS were prepared and cultured following the methodology outlined by Jenkins et al. [37]. In brief, tumor specimens were submerged in DMEM on ice and fragmented within a standard 6 cm dish using sterile forceps and a scalpel. The minced tumors were subsequently suspended in high‐glucose DMEM supplemented with 10% FBS, 1% penicillin‐streptomycin, 0.1% type 4 collagenase, 0.05% hyaluronidase, and 0.01% deoxyribonuclease. Following a 20‐min incubation period, the minced tumor specimens were mixed with an equal volume of media. The resulting cell suspension was then centrifuged at 400 ×g for 5 min at 4°C to pellet the cells, after which it was resuspended in fresh DMEM and sequentially filtered through 100 mm and 40 mm filters to isolate spheroid fractions ranging from 40‐100 μm. The spheroid fraction was pelleted once more and resuspended in type I rat tail collagen (08‐115, Corning) at a concentration of 3 mg/mL. Subsequently, the spheroid‐collagen mixture was injected into the central gel region of a 24‐well plate. After incubating in sterile humidity chambers for 30 min at 37°C, collagen hydrogels containing PDOTS were cultured with fresh DMEM supplemented with 10% FBS and corresponding treatments (anti‐PD‐1 at 50 μg/mL, sintilimab and/or 1C8 at 50 μmol/L) for 48 h. The supernatant was then collected, and the secretion levels of Perforin 1 (PRF1) and Interferon gamma (IFN‐γ) were assessed via enzyme‐linked immunosorbent assay (ELISA). Subsequently, the PDOTS were digested into single cells and analyzed using flow cytometry.
2.31. ELISA
The ELISA was conducted following the manufacturer's guidelines. For PDOTS, supernatants were harvested after 48 h and any suspended cells were eliminated using a 0.45 μm filter. The concentrations of secreted IFN‐γ and PRF1 proteins in the culture media supernatant were quantified using the IFN‐γ human ELISA Kit (KHC4021, Thermo Fisher Scientific) and the PRF1 human ELISA Kit (BMS2306, Thermo Fisher Scientific), respectively. For murine blood, allow clot formation at room temperature (approximately 25°C), followed by centrifugation at 4°C and 10,000 × g for 10 min to collect the supernatant. Serum levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were measured using a mouse Alanine aminotransferase assay kit (R01502, Rayto, Shenzhen, China) and an aspartate aminotransferase assay kit (R01702, Rayto), respectively. Data were analyzed using an automatic biochemical analyzer (Chemray 240, Rayto).
2.32. OS, disease‐free survival (DFS), and PFS
To compare survival differences between high and low expressions of SRSF10 or MYB, Kaplan–Meier curves were employed. The study's endpoint was OS, defined as the period between surgery and patient death or last follow‐up. DFS was defined as the period between surgery and disease recurrence, death for any reason, or last follow‐up (whichever occurred first). PFS was defined as the period between surgery and disease progression, death for any reason, or last follow‐up (whichever occurred first).
2.33. Statistical analysis
All statistical analyses were performed using the SPSS software (version 23, IBM, Armonk, NY, USA) or GraphPad Prism (version 8, San Diego, CA, USA). Bar graphs were presented as the mean ± standard deviation. The D'Agostino‐Pearson omnibus normality test and Brown‐Forsythe test were performed before parametric or nonparametric tests. The relationship between SRSF10 expression and clinicopathological parameters was analyzed by the chi‐square test. Cox's proportional hazard regression model (α = 0.05) was used to analyze the independent prognostic factors. When data distribution normality and equal variances were met, mean differences were analyzed by Student's t test or one‐way ANOVA test. Otherwise, a nonparametric test (Wilcoxon) was applied. P < 0.05 was considered significant.
3. RESULTS
3.1. SRSF10 was a crucial gene associated with anti‐PD‐1 resistance and immunosuppressive microenvironment with HCC
We conducted snRNA‐seq on 6 patients undergoing anti‐PD‐1 treatment, among whom 2 exhibited a positive response to immunotherapy and 4 did not (Figure 1A). Cluster analysis was employed to distinguish the cell populations within the TME. Clusters exhibiting similar expression patterns were characterized using classical marker genes (Supplementary Figure S1A). The primary cellular constituents were identified as malignant liver cells (Figure 1B), acknowledging the inherent technical limitations of our approach. Cluster analysis specifically targeting tumor cells revealed five discrete groups (Figure 1C). KEGG enrichment analysis unveiled that cells within clusters 2 and 3, which were more prevalent in non‐responsive than in responsive patients (Figure 1C), were enriched in pathways associated with glucose metabolism (Figure 1D and Supplementary Figure S1B). Furthermore, enrichment analysis of a bulk RNA‐sequencing (RNA‐seq) HCC cohort treated with anti‐PD‐1 comfirmed the enrichment of the glucose metabolism pathway in the non‐responsive group (Supplementary Figure S1C). Based on these findings, we inferred a close relationship between glucose metabolism and immunotherapy resistance in HCC. Moving forward, our focus shifted toward identifying the key genes governing glycolysis, the primary pathway of glucose metabolism. Integration of bulk RNA‐seq data from TCGA and GEO databases with prognostic information led to the identification of SRSF10 as a gene significantly associated with glycolysis (Figure 1E).
FIGURE 1.
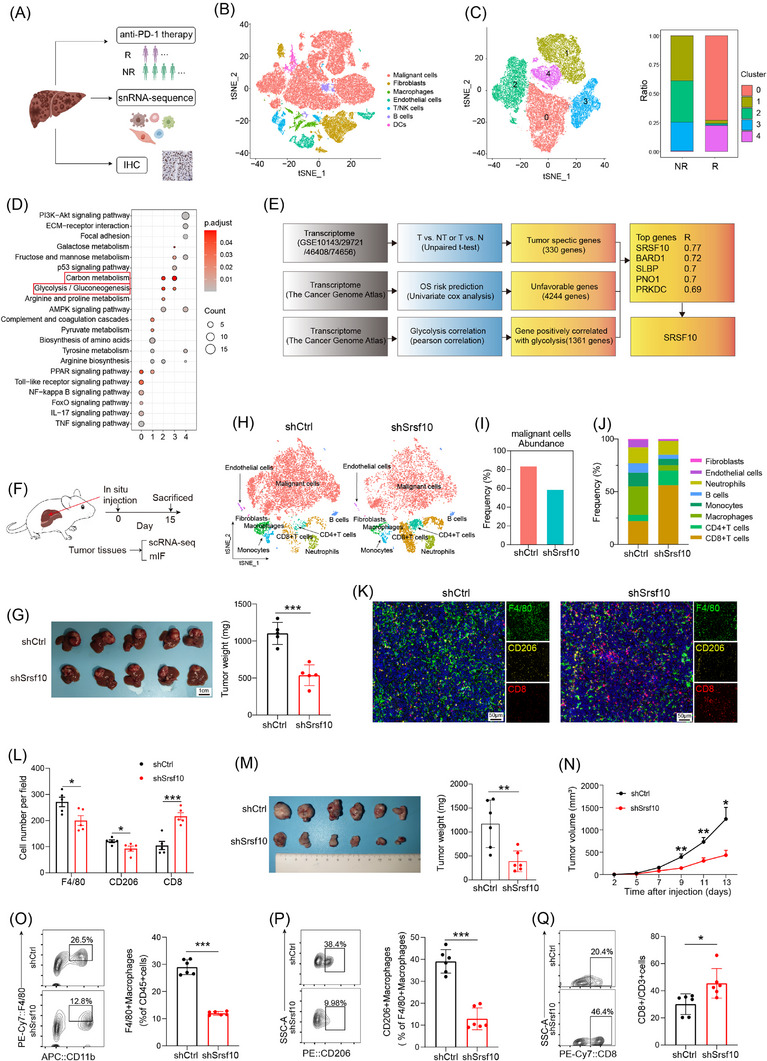
SRSF10 was a crucial gene associated with anti‐PD‐1 resistance and immunosuppressive microenvironment with HCC. (A) Schematic diagram of the recruitment of patients and specimens in the training cohort. (B‐C) t‐SNE analysis of snRNA‐seq data from HCC tumor tissues. (D) KEGG analysis of genes associated with tumor cells from snRNA‐seq. (E) Model diagram for screening of glycolysis‐related genes. (F) Workflow of the HCC model in situ. (G) Tumor picture and statistical analysis of orthotopic HCC model (n = 6). (H) t‐SNE analysis of scRNA‐seq data from mouse HCC tumor tissues. (I) Comparative abundance of the cancer cells in control versus Srsf10‐knockdown HCC tumors, extracted from the scRNA‐seq data. (J) Comparative abundance of the tumor microenvironment in control versus Srsf10‐knockdown HCC tumors, extracted from the scRNA‐seq data. (K) Representative pictures of mIF analysis for F4/80, CD206 and CD8 markers (n = 5). (L) Quantification of corresponding immune cells by mIF analysis (n = 5). (M) Representative image and statistical tests of a Hepa1‐6 cell‐derived tumor harvested from a C57BL/6 mouse on day 14 (n = 6). (N) Tumor growth curves of subcutaneous HCC tumor (n = 6). (O‐Q) Representative plots and percentages of tumor‐infiltrating F4/80+ macrophages, CD206+ macrophages and CD8+ T cells from control and shSrsf10 HCC tumor (n = 6). Two‐tailed unpaired Student's t test (G, L ‐Q); ns, not significant; * P < 0.05, ** P < 0.01, *** P < 0.001. Abbreviations: IHC, Immunohistochemistry; DC, Dendritic cell; NR, non‐response; SRSF10, serine and arginine rich splicing factor 10; BARD1, BRCA1 associated RING domain 1; SLBP, stem‐loop histone mRNA binding protein; PNO1, partner of NOB1 homolog; PRKDC, protein kinase, DNA‐activated, catalytic subunit; OS, overall survival; scRNA‐seq, single‐cellar RNA sequencing; mIF, multicolor immunofluorescence; tSNE, t‐distributed Stochastic Neighbor Embedding; HCC, hepatocellular carcinoma; snRNA‐seq, snRNA‐seq: single‐nuclear RNA sequencing.
Initially, we examined the expression of SRSF10 across various cell types in snRNA‐seq cohort and found that it was predomintantly expressed in malignant cells (Supplementary Figure S1D). Subsequently, leveraging a publicly available scRNA‐seq dataset GSE149614 (Supplementary Figure S1E‐F), we discovered that although SRSF10 was expressed in cells other than malignant cells (Supplementary Figure S1G), its significant upregulation was observed exclusively in tumor cells upon scrutinizing the expression data from tumor and adjacent normal tissues (Supplementary Figure S1H). This underscored the phenomenon wherein SRSF10 primarily exerted its effects on tumor cells. Subsequently, our findings underscored the prevalent overexpression of SRSF10 across diverse tumor types, evident at both the RNA and protein levels (Supplementary Figure S1I‐J). Subsequent analysis of another publicly available scRNA‐seq cohort, GSE125449, revealed a consistent association between increased SRSF10 expression and the immunosuppressive TME (Supplementary Figure S1K‐L). To investigate its functional implications, we generated Srsf10‐knockdown Hepa1‐6 cells and orthotopically transplanted them to induce orthotopic HCC tumors (Figure 1F). Our investigations unveiled a significant reduction in orthotopic tumor size in the shSrsf10 group compared with that in the control group (Figure 1G). To delve deeper into the role of SRSF10 within the TME, we conducted scRNA‐seq on both control and shSrsf10 HCC tumors. Subsequent dimensionality reduction clustering and cell annotation yielded a comprehensive dataset comprising 15,490 cells (Figure 1H and Supplementary Figure S1M). The results unequivocally demonstrated a marked decrease in tumor cell abundance within the TME following shSrsf10 (84% in control tumors vs. 59% in shSrsf10 tumors, Figure 1I). Additionally, our analysis revealed pronounced enrichment of CD4+ and CD8+ T cells in the knockdown tumors, in contrast to the predominant macrophage and monocyte population in control tumors (Figure 1J). Additionally, we observed that integrin alpha M (Itgam), lymphocyte antigen 6 family member C1 (Ly6c1), and lymphocyte antigen 6 family member G (Ly6g) exhibited low expression in the macrophages of control tumors, whereas C‐type lectin domain family 4 member F (Clec4f) and adhesion G protein‐coupled receptor E1 (Adgre1) demonstrated elevated expression (Supplementary Figure S1N). These findings suggest that alterations in macrophages influenced by SRSF10 predominantly involve tissue‐resident macrophages. The scRNA‐seq findings were validated using mIF. These corroborative analyses showed a heightened presence of CD8+ T cells in Srsf10‐knockdown HCC tumors, whereas control tumors exhibited a predominance of F4/80+ and CD206+ macrophages (Figure 1K‐L). Additionally, no significant changes were observed in Ly6C+CD11b+ monocytes (Supplementary Figure S1O).
To further validate these conclusions, Hepa1‐6 cells were used to establish subcutaneous HCC tumors. Initially, we observed that Srsf10‐knockdown HCC tumors exhibited a decreased tumor growth rate and weight (Figure 1M‐N). Subsequently, the tumor tissue was enzymatically digested into single cells for flow cytometric analysis, which further corroborated the findings of the scRNA‐seq analysis conducted on the control and shSrsf10 HCC tumors. Specifically, CD8+ T cells were significantly enriched in shSrsf10 HCC tumors, whereas the control group demonstrated a higher proportion of F4/80+ and CD206+ macrophages (Figure 1O‐Q, Supplementary Figure S2A). Consequently, these collective results indicate a pivotal role for SRSF10 in shaping the suppressive TME within HCC tumors and suggest a potential association with resistance to anti‐PD‐1 immunotherapy.
3.2. SRSF10 induced an immunosuppressive TME via M2 macrophage polarization
Based on these results, we hypothesized that SRSF10 mediated the cytotoxic killing effect of CD8+ T cells on tumor cells by regulating M2 macrophage polarization. Initially, we analyzed the expression of common macrophage‐related genes in macrophages using the scRNA‐seq results of control and shSrsf10 HCC tumors. We found that in tumors from Srsf10‐knockdown mice, the expression of pro‐tumoral macrophage markers was lower than that in tumors from control mice, whereas the expression of anti‐tumoral macrophage markers was higher in tumors from shSrsf10 mice than in control tumors (Figure 2A), indicating a switch from a pro‐tumoral to an anti‐tumoral macrophage phenotype mediated by shSrsf10. Additionally, shSrsf10 tumors exhibited an elevated M1 macrophage scores, whereas control tumors were skewed toward higher M2 macrophage scores (Figure 2B). Moreover, analysis of TCGA database revealed positive correlations between SRSF10 and biomarkers of M2 macrophages (Supplementary Figure S2B). Subsequently, THP1 cells were treated with PMA to induce differentiation into macrophages, whereas bone marrow cells were isolated from mouse bone marrow and treated with M‐CSF for 7 days to obtain BMDMs. After co‐culturing with SRSF10‐knockdown HCC cells, both THP1 cells and BMDMs were collected, and RNA was extracted. qPCR analysis validated the mRNA expression of pro‐tumor macrophage markers, including CD206, ARG1, TGFB1, IL10, vascular endothelial growth factor A (VEGFA) and adrenomedullin (ADM), which were significantly downregulated in human and mouse macrophages co‐cultured with shSRSF10 tumor cells compared with control tumor cells (Figure 2C‐D). Flow cytometry demonstrated a significant downregulation of CD206 in SRSF10‐knockdown cells compared to control cells, both in THP1 cells and BMDMs (Figure 2E). Besides, PCR analysis revealed a significantly upregulated expression of M2 macrophage biomarkers, including CD206, ARG1, TGFB1, IL10, VEGFA and ADM, along with a notable increase in CD206 protein levels, as detected using flow cytometry after co‐culturing THP1 cells with PLC/PRF/5 cells overexpressing SRSF10, compared with control tumor cells (Supplementary Figure S2C‐D). Furthermore, using a chemotactic migration assay, we observed that shSRSF10 HCC cells inhibited the chemotactic recruitment of macrophages (Figure 2F). To further demonstrate the indispensability of macrophages in SRSF10‐mediated carcinogenesis, we depleted macrophages by intraperitoneal injection of clodronate liposomes and observed that upon macrophage depletion, tumor volume and weight were no longer attenuated in shSrsf10 tumors, eventually becoming comparable to that in control tumors (Figure 2G‐H and Supplementary Figure S2E‐F). These findings indicate an essential role of macrophages in the adoption of a pro‐tumor phenotype mediated by SRSF10, and their depletion abolished the inhibitory effect of SRSF10 knockdown on HCC tumors.
FIGURE 2.
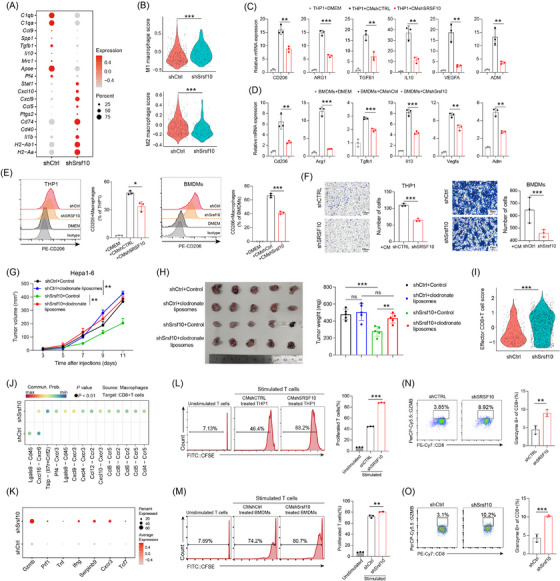
SRSF10 induced an immunosuppressive TME via M2 macrophage polarization. (A) Dot plots showing the expression of representative marker genes of antitumoral and protumoral macrophages, as in Figure 1H. Dot size indicates the percentage of cells expressing the indicated gene, and color intensity represents the relative gene expression level. (B) Violin plot of enrichment score for indicated gene sets. (C) The expression of CD206, ARG1, TGFB1, IL10, VEGFA, and ADM mRNA in THP1 cells co‐cultured with shCTRL or shSRSF10 Huh7 cells, detected by qRT‐PCR analyses (n = 3). (D) The expression of Cd206, Arg1, Tgfb1, Il10, Vegfa, and Adm mRNA in BMDMs co‐cultured with shCtrl or shSrsf10 Hepa1‐6 cells, detected by qRT‐PCR analyses (n = 3). (E) CD206 expression in macrophages detected by flow cytometry stimulated with the supernatant of control or SRSF10‐knockdown Huh7 or Hepa1‐6 cells (n = 3). (F) Chemotactic migration assays and statistical analysis of macrophages stimulated with the supernatant of control or SRSF10‐knockdown Huh7 or Hepa1‐6 cells (n = 3). (G) Tumor growth curves of HCC tumor treated with control or clodronate liposome (n = 6). (H) Tumor picture and statistical analysis of subcutaneous HCC model treated with control or clodronate liposome (n = 6). (I) Violin plot of enrichment score for indicated gene sets using scRNA‐seq data acquired from control and shSrsf10 HCC tumors. (J) Comparison of the significant ligand‐receptor involved in chemokines sending from macrophages to CD8+ T cells. (K) Dot plots showing the expression of representative marker genes of CD8+ T cells, as in Figure 1H. Dot size indicates the percentage of cells expressing the indicated gene, and color intensity represents the relative gene expression level. (L) CFSE histograms detecting the inhibition of T‐cell proliferation by THP1 after treatment with the supernatant of Huh7‐shCTRL or ‐shSRSF10 cells (n = 3). (M) CFSE histograms detecting the inhibition of T cell proliferation by BMDMs after treatment with the supernatant of Hepa1‐6‐shCtrl or ‐shSrsf10 cells (n = 3). (N) Percentages of Gzmb+ CD8+ T cells in the assay in Figure 2L, measured by flow cytometry (n = 3). (O) Percentages of Gzmb+ CD8+ T cells in the assay in Figure 2M, measured by flow cytometry (n = 3). Two‐tailed unpaired Student's t test (B‐I, L‐O); ns, not significant; * P < 0.05, ** P < 0.01, *** P < 0.001. Abbreviations: C1qa, complement C1q A chain; C1qb, complement C1q B chain; Ccl9, C‐C motif chemokine ligand 9; Spp1, secreted phosphoprotein 1; Tgfb1, transforming growth factor beta 1; Il10, interleukin 10; Mrc1, mannose receptor C‐type 1; Apoe, apolipoprotein E; Pf4, platelet factor 4; Stat1, signal transducer and activator of transcription 1; Cxcl10, C‐X‐C motif chemokine ligand 10; Ccl5, C‐C motif chemokine ligand 5; Ptgs2, prostaglandin‐endoperoxide synthase 2; Cd74, CD74 molecule; Cd40, CD40 molecule; Il1b, interleukin 1 beta; H2‐Ab1, histocompatibility 2, class II antigen A, beta 1; H2‐Aa, histocompatibility 2, class II antigen A, alpha; CD206, CD206 molecule; ARG1, arginase 1; ADM, adrenomedullin; DMEM, Dulbecco's Modified Eagle Medium; BMDM, Bone marrow‐derived macrophages; Lgals9, lectin, galactose binding, soluble 9; Cd45, CD45 molecule; Cxcl16, C‐X‐C motif chemokine ligand 16; Cxcr6, C‐X‐C motif chemokine receptor 6; Tslp, thymic stromal lymphopoietin; Il7, interleukin 7; Crlf2, cytokine receptor‐like factor 2; Cxcr3, C‐X‐C motif chemokine receptor 3;Cxcl9, C‐X‐C motif chemokine ligand 9; Cxcl4, platelet factor 4; Ccl12, C‐C motif chemokine ligand 12; Ccr2, C‐C motif chemokine receptor 2; Ccl8, C‐C motif chemokine ligand 8; Ccr5, C‐C motif chemokine receptor 5; Ccl6, C‐C motif chemokine ligand 6; Ccl5, C‐C motif chemokine ligand 5; Ccl4, C‐C motif chemokine ligand 4; Gzmb, granzyme B; Prf1, perforin 1; Tnf, tumor necrosis factor; Ifng, interferon gamma; Serpinb9, serine (or cysteine) peptidase inhibitor, clade B, member 9; Tcf7, transcription factor 7; CFSE, Carboxyfluorescein Succinimidyl Ester; qRT‐PCR, Quantitative Real‐Time Reverse Transcription Polymerase Chain Reaction; Srsf10, serine and arginine‐rich splicing factor 10.
Subsequently, our focus shifted towards on how CD8+ T cells function in Srsf10‐knockdown HCC tumors. First, effector CD8+ T cells in shSrsf10 tumors demonstrated a higher enrichment score than those in control tumors (Figure 2I). We conducted a cell‐cell communication analysis and identified stronger immune‐promoting chemokine intercellular connections between macrophages and CD8+ T cells in shSrsf10 tumors (Figure 2J). Additionally, CD8+ T cells from shSrsf10 tumors exhibited elevated expression of granzyme B (Gzmb), Prf1, Tnf, and Ifng (Figure 2K). Moreover, we observed an increase in the expression of Tcf7 in CD8+ T cells within shSrsf10 tumors (Figure 2K). TCF7 is a hallmark of CD8+ T‐cell stemness [38]. Multiple data points indicated an inverse relationship between SRSF10 and cytotoxic T lymphocytes (Supplementary Figure S2G‐H). To investigate the influence of CD8+ T cells on the immune microenvironment mediated by SRSF10, we conducted a co‐culture experiment involving macrophages stimulated by tumor cell supernatants and CD8+ T cells. These findings demonstrate that macrophages co‐cultured with control HCC cells significantly suppressed the proliferation (Figure 2L‐M) and activation (Figure 2N‐O) of CD8+ T cells, in contrast to those co‐cultured with shSRSF10 HCC cells. Collectively, these results suggest that SRSF10 expression in tumor cells impedes the cytotoxic function of CD8+ T cells by promoting the transformation of macrophages into cells with a pro‐tumor phenotype.
3.3. SRSF10 promoted M2 polarization of macrophages through lactate
Next, we investigated the mechanism by which SRSF10 influences macrophage polarization. Given the close association between SRSF10 and glycolysis, which plays a crucial role in regulating TME through lactate production, we hypothesized that SRSF10 regulates macrophage polarization via lactate. We then conducted co‐culture experiments by introducing lactate into shSRSF10 HCC cells. PCR and flow cytometry assays revealed a significant decrease in the expression of M2 macrophage markers in macrophages co‐cultured with shSRSF10 tumor cells (Figure 3A‐D). However, this decrease was reversed by the addition of lactate (Figure 3A‐D and Supplementary Figure S3A‐B). Transwell assays demonstrated a significant reduction in the migration ability of macrophages after co‐culturing with shSRSF10 tumor cells (Figure 3E). Nevertheless, the addition of lactate effectively restored the migration ability (Figure 3E). Further experiments involved culturing the tumor cells in low‐glucose DMEM or with the addition of 2‐DG. shSRSF10 did not affect macrophage polarization under these conditions (Supplementary Figure S3C‐D).
FIGURE 3.
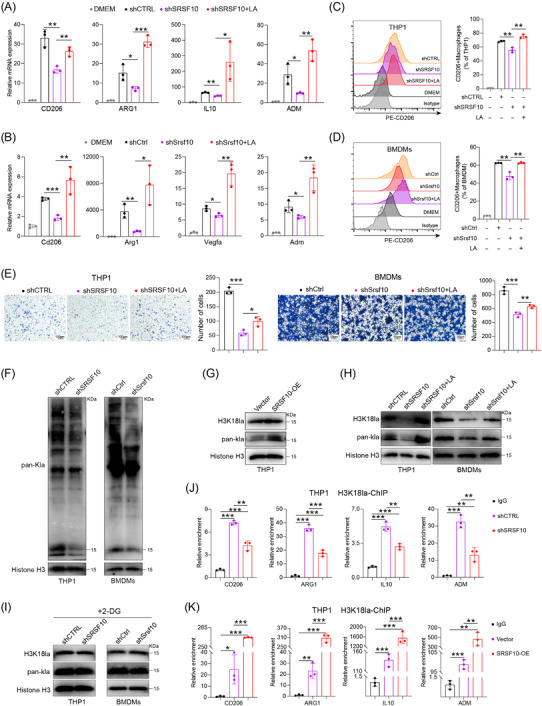
SRSF10 promoted M2 macrophage polarization through LA. (A) qRT‐PCR detected the expression of CD206, ARG1, IL10, and ADM mRNA in THP1 cells co‐cultured with Huh7 cells from shCTRL, shSRSF10, and shSRSF10 with LA groups (n = 3). (B) qRT‐PCR detected the expression of Cd206, Arg1, Vegfa, and Adm mRNA in BMDMs co‐cultured with Hepa1‐6 cells from shCtrl, shSrsf10, and shSrsf10 with LA groups (n = 3). (C) Expression of CD206 in THP1 cells, detected by flow cytometry analyses (n = 3). (D) Expression of Cd206 in BMDMs, detected by flow cytometry analyses (n = 3). (E) Chemotactic migration assays and statistical analysis of macrophages stimulated with the supernatant of Huh7 or Hepa1‐6 cells (n = 3). (F) Western blotting analysis shows the levels of Pan Kla in THP1 cells and BMDMs cultured with control or shSRSF10 HCC cells. (G‐I) Western blotting analysis shows the levels of Pan Kla and H3K18la in THP1 cells and BMDMs. (J‐K) qChIP analysis of the indicated promoters was performed using antibodies against H3K18la (n = 3). Two‐tailed unpaired Student's t test (A‐E, J‐K); ns, not significant; * P < 0.05, ** P < 0.01, *** P < 0.001. 2‐DG, 2‐Deoxy‐D‐glucose; ADM, adrenomedullin; ARG1, arginase 1; BMDMs, Bone‐marrow‐derived macrophage; CD206, CD206 molecule; ChIP, Chromatin immunoprecipitation; DMEM, Dulbecco's Modified Eagle Medium; IL10, interleukin 10; LA, lactic acid; OE, over expression; qRT‐PCR, Quantitative Real‐Time Reverse Transcription Polymerase Chain Reaction; SRSF10, serine and arginine rich splicing factor 10; Vegfa, vascular endothelial growth factor A.
Recent studies have revealed that lactate functions via various mechanisms. In addition to its previously discovered role as a primary carbon fuel source [39, 40], lactate also serves as an agonist in G‐protein‐coupled receptor (GPCR) signaling [41, 42] and an inhibitor of histone deacetylase [43]. Recent research has revealed that lactate can induce epigenetic modifications through lactylation [44], including increased M2 macrophage polarization, as supported by Western blotting (Figure 3F). The changes observed at approximately 15 kDa were more pronounced and primarily associated with histone expression. Based on this observation, we hypothesized that lactate promoted M2 macrophage polarization through histone lactylation. Western blotting analysis revealed a significant increase in histone acetylation in macrophages co‐cultured with PLC/PRF/5 cells overexpressing SRSF10 (Figure 3G) and a significant reduction in histone lactylation in macrophages co‐cultured with shSRSF10 HCC cells (Figure 3H). However, supplementation with lactic acid (LA) restored their expression levels (Figure 3H). Previous studies have indicated that histone H3 lysine 18 lactylation (H3K18la) site is the primary site for histone lactylation [44]. Our results further confirmed the involvement of H3K18la in SRSF10‐mediated M2 macrophage polarization (Figure 3H). Besides, co‐culturing macrophages with tumor cells treated with 2‐DG (an inhibitor of glycolysis) did not lead to significant changes in histone lactylation (Figure 3I). Histones play a crucial role as transcriptional cofactors in gene expression. ChIP assays indicated that increasing H3K18la modification activated the transcriptional expression of M2 macrophage genes, including CD206, ARG1, IL10 and ADM (Figure 3J‐K). In conclusion, we provide evidence that SRSF10 in tumor cells can enhance histone lactylation modification of macrophages via lactate, leading to the activation of genes such as CD206 and the promotion of an immunosuppressive TME.
3.4. SRSF10/glycolysis/H3K18la formed a positive feedback loop in HCC tumor cells
To investigate the role of SRSF10 in glycolysis, we conducted bulk RNA‐seq on control and shSRSF10 Huh7 cells, and observed 793 DEGs, with 577 genes upregulated and 216 genes downregulated in the shSRSF10 cells compared with the control groups (Supplementary Figure S4A‐B). GSEA was conducted with the DEGs, demonstrating significant enrichment of the glycolysis pathway (Figure 4A). Additionally, our GSEA results indicated that macrophage activation was enriched in the control group, whereas the shSRSF10 group exhibited enrichment of the adaptive immune response pathway, cytokine production involved in the immune response pathway, and the interferon gamma pathway (Supplementary Figure S4C). The TCGA‐LIHC cohort was divided into SRSF10‐high and SRSF10‐low groups, using the median value of SRSF10 as the threshold. We found that genes associated with glycolysis were upregulated in SRSF10‐high patients (Figure 4B and Supplementary Figure S4D). To investigate the role of SRSF10 in tumor cell metabolism, we conducted untargeted energy metabolism sequencing using control and shSrsf10 Hepa1‐6 cells. We identified 329 differentially expressed metabolites, with 189 showing increased abundance and 140 exhibiting decreased abundance in control Hepa1‐6 cells compared with shSrsf10 Hepa1‐6 cells. Among these, metabolites associated with glycolysis showed significantly decreased abundance in the shSrsf10 group (Supplementary Figure S4E). Additionally, KEGG enrichment analysis demonstrated significant enrichment of differential metabolites in carbon metabolism pathways (Figure 4C).
FIGURE 4.
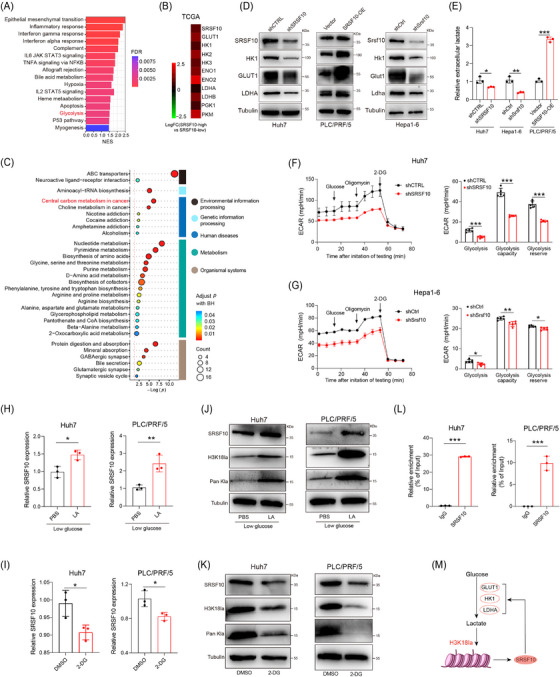
SRSF10 positively regulated glycolysis in HCC cells. (A) GSEA analysis of differentially expressed gene from bulk RNA‐seq conducted on control and SRSF10‐knockdown Huh7 cells. (B) Fold change of glycolysis‐related genes in patients with high and low expression of SRSF10 in the TCGA‐LIHC dataset. (C) KEGG analysis of differentially expressed metabolite from untargeted energy metabolism sequencing conducted on control and shSrsf10 Hepa1‐6 cells. (D) Western blotting was performed in Huh7, PLC/PRF/5, and Hepa1‐6 cells. (E) Detection of lactate content in Huh7, PLC/PRF/5, and Hepa1‐6 cells (n = 3). (F‐G) ECAR of Huh7 cells and Hepa1‐6 cells (n = 5). (H) The expression SRSF10 mRNA in Huh7 and PLC/PRF/5 cells treated with LA, detected by qRT‐PCR analyses (n = 3). (I) SRSF10 mRNA levels in Huh7 and PLC/PRF/5 cells treated with 2‐DG, detected by qRT‐PCR analyses (n = 3). (J) Western blotting was performed in Huh7 and PLC/PRF/5 cells treated with LA. (K) Western blotting was performed in Huh7 and PLC/PRF/5 cells treated with 2‐DG. (L) H3K18la binding to the SRSF10 promoter was determined by chromatin immunoprecipitation‐RT‐PCR in Huh7 and PLC/PRF/5 cells (n = 3). (M) Schematic proposing a positive feedback loop between SRSF10, glycolysis and histone lactylation in HCC cells. Two‐tailed unpaired Student's t test (E‐I, L); ns, not significant; * P < 0.05, ** P < 0.01, *** P < 0.001. 2‐DG, 2‐Deoxy‐D‐glucose; DMSO, Dimethyl Sulfoxide; ECAR, Extra Cellular Acidification Rate; ENO1, enolase 1; ENO2, enolase 2; FDR, False Discovery Rate; GLUT1, Glucose transporter 1; GSEA, Gene Set Enrichment Analysis; HK1, hexokinase 1; HK2, hexokinase 2; HK3, hexokinase 3; IL2, interleukin 2; IL6, interleukin 6; LA, lactic acid; LDHA, lactate dehydrogenase A; LDHB, lactate dehydrogenase B; LIHC, Liver hepatocellular carcinoma; NES, Normalized enrichment score; PBS, Phosphate buffered saline; PGK1, phosphoglycerate kinase 1; PKM, pyruvate kinase M1/2; qRT‐PCR, Quantitative Real‐Time Reverse Transcription Polymerase Chain Reaction; RNA‐seq, RNA Sequencing; SRSF10, serine and arginine rich splicing factor 10; STAT3, signal transducer and activator of transcription 3; STAT5, signal transducer and activator of transcription 5; TCGA, The Cancer Genome Atlas.
We further examined the expression of glycolysis‐related genes (GLUT1, HK1, and LDHA) at RNA and protein levels. In shSRSF10 Huh7 and Hepa1‐6 cells, we observed low expression of these genes. In contrast, PLC/PRF/5 cells overexpressing SRSF10 showed heightened expression of these glycolysis‐related genes (Figure 4D and Supplementary Figure S4F). Notably, ECAR and intracellular and extracellular lactate production were significantly reduced in shSRSF10 Huh7 and Hepa1‐6 cells, and the lactate content was increased in PLC/PRF/5 cells overexpressing SRSF10 (Figure 4E‐G and Supplementary Figure S4G). To better prove the regulatory effect of SRSF10 on lactate levels, we used low‐glucose DMEM or 2‐DG to inhibit glycolysis. We found that shSRSF10 did not affect the regulation of lactate content in Huh7 cells compared with the control (Supplementary Figure S4H). Lactate transporter proteins in the cell membrane mediate lactate transport [45]. To confirm which lactate transporter protein specifically mediates the upregulation of SRSF10‐induced lactate secretion, we used MCT1 and MCT4 inhibitors. The results showed that the inhibition of MCT1 with BAY‐8002 led to a significant reduction in extracellular lactate levelsin shSRSF10 HCC cells, whereas the overexpression of SRSF10 resulted in a significant increase in extracellular lactate levels. Conversely, the inhibition of MCT4 with AZD0095 did not significantly alter extracellular lactate levels upon either shSRSF10 or overexpression, demonstrating that MCT4 mediates the upregulation of lactate secretion induced by SRSF10 (Supplementary Figure S4I). These in vivo experiments further supported our findings. Control and shSRSF10 HCC cells were injected under the ribs of the mice to generate subcutaneous tumors. We observed a significant reduction in lactate production in shSRSF10 tumors compared with control tumors (Supplementary Figure S4J). In conclusion, we revealed that SRSF10 positively regulates tumor glycolysis and lactate production in vitro and in vivo.
Pandkar et al. [46] confirmed that lactate positively regulates the expression of SRSF10 in breast cancer. Therefore, we investigated whether a similar mechanism exists in HCC cells. qPCR confirmed that LA supplement upregulated SRSF10 expression, while 2‐DG downregulated SRSF10 expression (Figure 4H‐I). Western blotting revealed that LA increased SRSF10 expression in parallel with pan‐Kla and H3K18la levels, whereas 2‐DG exerted the opposite effect (Figure 4J‐K). To further understand the specific mechanism by which lactate regulates SRSF10 expression, we examined its role in histone lactylation. We hypothesized that lactate increases histone lactylation modification in HCC cells, leading to transcriptional activation of SRSF10, as demonstrated using ChIP assays (Figure 4L), providing evidence for a positive feedback loop involving the SRSF10/glycolysis/H3K18la axis (Figure 4M).
3.5. SRSF10 regulated glycolysis in tumor cells through transcription factor MYB
SRSF10 primarily functions through RNA‐binding activity. To elucidate this, RIP‐seq was conducted using Huh7 cells. KEGG enrichment analysis revealed the enrichment of DEGs in pathways related to glycolysis, such as central carbon metabolism in cancer (Supplementary Figure S5A). Subsequently, the intersection of DEGs identified from RIP‐seq (3,765 genes) and bulk RNA‐seq conducted on control and shSRSF10 Huh7 cells was obtained, followed by further intersection with genes highly correlated with SRSF10 identified through TCGA‐LIHC dataset (Figure 5A). We selected MYB, which exhibited the strongest correlation with SRSF10, as a candidate gene (Figure 5B). Moreover, our GSEA results indicated that the MYB regulatory pathway was significantly enriched in the control group compared with the SRSF10‐knockdown group (Figure 5C). Besides, MYB expression was strongly and positively correlated with glycolysis (Supplementary Figure S5B). Subsequently, we discovered the high expression of MYB in various tumor types at the RNA and protein levels acquired from GEPIA 2 for the expression of RNA and HPA for the expression of protein (Supplementary Figure S5C‐D). PCR and Western blotting analysis revealed that SRSF10 positively regulated MYB expression (Figure 5D‐E). We then elucidated the specific mechanism by which SRSF10 regulates MYB expression. As SRSF10 is a well‐known splicing factor, we initially conducted RIP assays to determine its binding affinity for MYB RNA, which confirmed that SRSF10 bound to MYB RNA (Figure 5F). Reverse transcription PCR (RT‐PCR) analyses were conducted to investigate whether SRSF10 influences MYB through alternative splicing. Nevertheless, the results indicated that shSRSF10 had a minimal impact on the expression of different MYB transcripts compared with the control group (Supplementary Figure S5E). Consequently, we shifted our focus to exploring the mechanisms by which SRSF10 regulates RNA stability in addition to splicing. To inhibit total RNA expression, we treated tumor cells with actinomycin D. The results demonstrated that after shSRSF10, MYB RNA exhibited a faster exhaustion rate (Figure 5G), suggesting that SRSF10 plays a role in regulating MYB RNA stability. CLIP assays further revealed that SRSF10 predominantly bound to the 3' untranslated regions (3’UTR) region of MYB (Figure 5H). Additionally, RIP‐seq results indicated that the primary site of downstream gene binding by SRSF10 was AAAAAAA (Supplementary Figure S5F), a base sequence primarily found in the 3’UTR. Previously, Shkreta et al. [47] demonstrated that 1C8 selectively promoted the dephosphorylation of SRSF10, thereby inhibiting SRSF10's facilitation of human immunodeficiency virus (HIV) replication. Additionally, Chang et al. [48] confirmed that 1C8 inhibited the expression of HCC tumor in vivo. Herein, we sought to ascertain whether a selective inhibitor of SRSF10, 1C8, could modulate MYB. Given the differential protein sizes of phosphorylated and dephosphorylated SRSF10, treatment with CIP facilitated total protein dephosphorylation, resulting in distinct bands on the gel. Western blotting analysis revealed a migration rate similar to that of CIP upon the addition of 1C8 to Huh7 and Hepa1‐6 cells (Supplementary Figure S5G), indicating that 1C8 inhibited the activity of SRSF10 in HCC cells. Furthermore, PCR analysis confirmed that 1C8 downregulated MYB expression in wild‐type HCC cells, whereas the application of 1C8 to shSRSF10 cells had no significant effect on MYB expression (Supplementary Figure S5H), confirming that the regulation of MYB by 1C8 was mediated through SRSF10. In summary, we demonstrated that SRSF10 can bind to the MYB 3’UTR region to stabilize its RNA, thereby increasing its protein expression.
FIGURE 5.
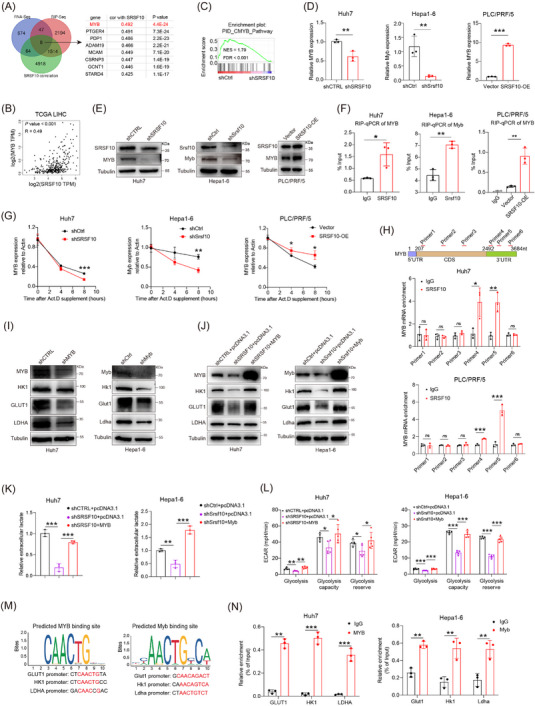
SRSF10 positively regulated glycolysis in HCC cells through transcription factor MYB. (A) Venn diagram was used to identify the overlapping genes in indicated data. (B) Correlation of SRSF10 and MYB in TCGA‐LIHC dataset. (C) GSEA analysis of CMYB pathway enriched in differential gene from RNA‐seq. (D) MYB mRNA levels in Huh7, PLC/PRF/5, and Hepa1‐6 cells, detected by qRT‐PCR analyses (n = 3). (E) Western blotting was performed in Huh7, PLC/PRF/5, and Hepa1‐6 cells. (F) RIP assays to verify the binding of SRSF10 and MYB in Huh7, PLC/PRF/5, and Hepa1‐6 cells (n = 3). (G) Huh7, PLC/PRF/5, and Hepa1‐6 cells were treated with actinomycin D as indicated times. The mRNA expression levels of MYB were examined using qRT‐PCR. Error bars are mean ± SD from three biologically independent samples (n = 3). (H) CLIP assays to verify the binding region of SRSF10 and MYB in Huh7 and PLC/PRF/5 cells (n = 3). (I) Western blotting was performed in Huh7 and Hepa1‐6 cells. (J) Western blotting was performed in control, shSRSF10, and shSRSF10 followed by MYB overexpression in Huh7 and Hepa1‐6 cells (n = 3). (K) Detection of lactate content in control, shSRSF10, and shSRSF10 followed by MYB overexpression in Huh7 and Hepa1‐6 cells. (L) ECAR of huh7 cells and hepa1‐6 cells with control, shSRSF10, and shSRSF10 followed by MYB overexpression groups (n = 5). (M) Predicted binding sites of the transcription factor MYB within the promoter sequences of GLUT1, HK1, and LDHA. (N) MYB binding to the GLUT1, HK1, LDHA promoter was determined by chromatin immunoprecipitation‐RT‐PCR in Huh7 and Hepa1‐6 cells (n = 3). Two‐tailed unpaired Student's t test (D, F‐H, K‐N); ns, not significant; * P < 0.05, ** P < 0.01, *** P < 0.001. Abbreviations: MYB, MYB proto‐oncogene, transcription factor; PTGER4, prostaglandin E receptor 4; PDP1, pyruvate dehydrogenase phosphatase catalytic subunit 1; ADAM19, ADAM metallopeptidase domain 19; MCAM, melanoma cell adhesion molecule; CSRNP3, cysteine and serine rich nuclear protein 3; GCNT1, glucosaminyl (N‐acetyl) transferase 1; STARD4, StAR related lipid transfer domain containing 4; RNA‐seq, RNA Sequencing; RIP‐seq, RNA Immunoprecipitation Sequencing; NES, Normalized enrichment score; FDR, False Discovery Rate; SRSF10, serine and arginine rich splicing factor 10; OE, over expression; TCGA, The Cancer Genome Atlas; LIHC, Liver hepatocellular carcinoma; Act D, actinomycin D; 5’UTR, 5' Untranslated Regions; 3’UTR, 3' Untranslated Regions; CDS, Coding sequence; HK1, hexokinase 1; GLUT1, Glucose transporter 1; LDHA, lactate dehydrogenase A; GSEA, Gene Set Enrichment Analysis; qRT‐PCR, Quantitative Real‐Time Reverse Transcription Polymerase Chain Reaction; CLIP, cross‐linking and immune‐precipitation; ECAR, extracellular acidification rate.
Next, we demonstrated the significance of MYB in regulating glycolysis by SRSF10. Our study showed a notable reduction in ECAR, lactate levels, and genes related to glycolysis (such as GLUT1, HK1, and LDHA) in HCC cells following the inhibition of MYB compared with control cells (Figure 5I and Supplementary Figure S6A‐D). Additionally, our study revealed a significant decrease in ECAR, lactate levels and genes associated with glycolysis in shSRSF10 HCC cells, which could be reversed by MYB (Figure 5J‐L and Supplementary Figure S6E‐G). Furthermore, upon treatment with BAY‐8002 and AZD0095, differential effects on extracellular lactate levels were observed. Specifically, in cells treated with BAY‐8002, MYB interference significantly reduced extracellular lactate levels. Conversely, in cells treated with AZD0095, MYB interference did not significantly change extracellular lactate levels compared with the control (Supplementary Figure S6H). This finding demonstrated that the increase in extracellular lactate levels induced by MYB upregulation was mediated by MCT4. Subsequently, we found that upon interference with MYB expression, the OCR of Huh7 and Hepa1‐6 cells significantly increased, whereas ATP production decreased markedly (Supplementary Figure S6I‐J), indicating that MYB‐driven glycolysis is the primary energy metabolism pathway.
In this study, we investigated the mechanism by which MYB regulates the genes involved in glycolysis. Given that MYB is a well‐known transcription factor, we used the JASPAR database to predict its transcription‐binding sequence with GLUT1, HK1, and LDHA (Figure 5M‐N). ChIP assays confirmed that MYB transcriptionally upregulated the expression of GLUT1, HK1, and LDHA (Figure 5M‐N). In summary, our experiments demonstrate that SRSF10 binds to and increases the stability of MYB RNA, thereby activating the transcriptional expression of key enzymes associated with glycolysis, such as GLUT1, and increasing intracellular and extracellular lactate content.
3.6. 6 shSRSF10 or 1C8 inhibition impeded tumor progression and enhanced immunotherapy response in HCC tumor
To investigate whether SRSF10 deficiency enhances the effect of PD‐1 mAb treatment, we administered PD‐1 mAbs or an IgG isotype control to immunocompetent mice inoculated with shSrsf10 or control Hepa1‐6 cells (Figure 6A). We noted a significant reduction in tumor volume and weight in the shSrsf10 tumor or anti‐PD‐1 treatment groups compared with the control group. Furthermore, the combination of shSrsf10 and anti‐PD‐1 led to the most pronounced suppression of tumor volume and weight (Figure 6B‐D and Supplementary Figure S7A). On the 15th day after tumor cell injection, the tumors were harvested for further analysis. Flow cytometry analysis revealed that shSrsf10 combined with PD‐1 mAbs significantly increased the tumor‐infiltrating cytotoxic CD8+ T cell population in tumors compared with shSrsf10 or PD‐1 mAbs treatment alone while decreasing F4/80+ and CD206+ macrophages (Figure 6E and Supplementary Figure S7B).
FIGURE 6.
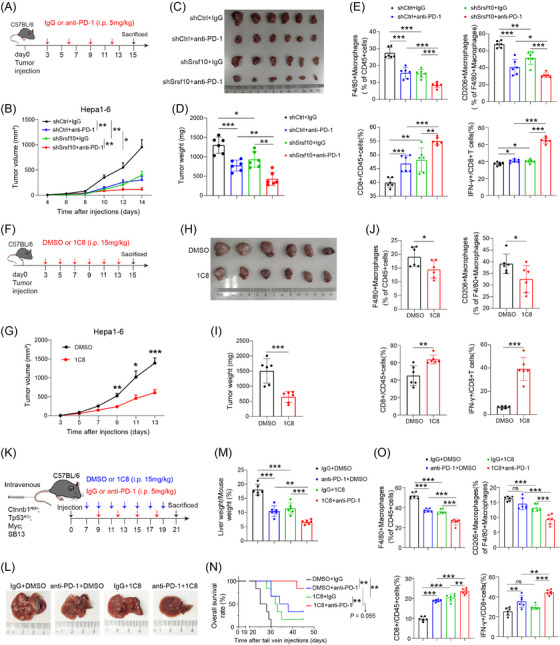
shSRSF10 or 1C8 inhibition impeded tumor progression and enhances immunotherapy response in HCC. (A) Workflow of the subcutaneous HCC tumor treated with control or PD‐1 mAbs. (B) Tumor growth curves of subcutaneous HCC tumor treated with control or PD‐1 mAbs (n = 6). (C) Representative image of a Hepa1‐6 cell‐derived tumor harvested from a C57BL/6 mouse on day 14 (n = 6). (D) Summary of weight data from Hepa1‐6 tumors collected after the mice were euthanized (n = 6). (E) Percentages of tumor‐infiltrating F4/80+ macrophages, CD206+ macrophages, CD8+ T cells IFN‐γ+ CD8+ T cells from control and shSrsf10 HCC tumor (n = 6). (F) Workflow of the subcutaneous HCC tumor treated with DMSO or 1C8. (G) Tumor growth curves of subcutaneous HCC tumor treated with DMSO or 1C8 (n = 6). (H) Representative image of a Hepa1‐6 cell‐derived tumor harvested from a C57BL/6 mouse on day 15 (n = 6). (I) Summary of weight data from Hepa1‐6 tumors collected after the mice were euthanized (n = 6). (J Percentages of tumor‐infiltrating F4/80+ macrophages, CD206+ macrophages, CD8+ T cells, IFN‐γ+ CD8+ T cells from DMSO and 1C8 treated HCC tumor (n = 6). (K) Workflow of the spontaneous HCC tumor treated with PD‐1 mAbs and/or 1C8. (L) Representative image of spontaneous HCC liver harvested from a C57BL/6 mouse on day 21 (n = 6). (M) Summary of liver weight data from spontaneous tumor collected after the mice were euthanized (n = 6). (N) Kaplan–Meier survival curves for spontaneous tumor mice (n = 6). (O) Percentages of tumor‐infiltrating F4/80+ macrophages, CD206+ macrophages, CD8+ T cells IFN‐γ+ CD8+ T cells from spontaneous HCC tumor (n = 6). Two‐tailed unpaired Student's t test (B, D‐E, G, I‐J, M, O); Log Rank test(N); ns, not significant; * P < 0.05, ** P < 0.01, *** P < 0.001. Abbreviations: PD‐1, programmed cell death 1; DMSO, Dimethyl Sulfoxide; Ctnnb1, catenin beta 1; Tp53, tumor protein p53; SRSF10, serine and arginine rich splicing factor 10; HCC, Hepatocellular carcinoma; mAbs, monoclonal antibodies; IFN‐γ, Interferon gamma.
To translate our findings into potential therapeutic strategies for inhibiting HCC growth, we subcutaneously inoculated control and shSrsf10 Hepa1‐6 cells into wild‐type C57B/6 mice and intraperitoneally administered DMSO or 1C8 (Figure 6F). We observed that tumors in the shSRSF10 and shSRSF10 plus 1C8 groups exhibited a significant reduction compared with the control group. However, there was no significant difference in tumor weight between the shSRSF10 plus 1C8 group and the shSRSF10 group (Supplementary Figure S7C), indicating that the functionality of 1C8 in vivo was mediated by SRSF10. Besides, we observed that 1C8 significantly attenuated tumor growth and weight without affecting mouse body weight (Figure 6G‐I and Supplementary Figure S7D). Moreover, the proportions of F4/80+ and CD206+ macrophages decreased significantly, while CD8+ T and IFN‐γ+ CD8+ T cells increased in 1C8‐treated tumor compared with the control tumor (Figure 6J and Supplementary Figure S7E).
To better mimic the TME of HCC, we induced spontaneous HCC tumor formation by injecting the sterile 0.9% NaCl solution/plasmid mix containing 10 μg of pX330‐p53, pT3‐N90‐beta‐catenin, and pT3‐EF1A‐MYC and 10 μg of CMV‐SB13 transposase through the tail vein (Figure 6K). From the seventh day onward, we administered 1C8 or PD‐1 mAbs alone or in combination (Figure 6K). The results demonstrated a significant reduction in tumor burden in mice treated with 1C8 or anti‐PD‐1 alone, compared with that in the control group (Figure 6L‐M). Moreover, the tumor burden was significantly lower in mice treated with the combination of 1C8 and anti‐PD‐1, compared to either treatment alone (Figure 6L‐M), with no apparent side effects (Supplementary Figure S7F‐G). Survival analysis indicated that mice receiving combination treatment exhibited prolonged OS (Figure 6N). On day 21, the mice were sacrificed, and the tumors were harvested for further analysis. Flow cytometry experiments confirmed the previous findings that either 1C8 or anti‐PD‐1 alone effectively reduced the proportion of tumor‐promoting macrophages and increased the proportion of CD8+ T cells in the TME (Figure 6O and Supplementary Figure S7H). Combined administration of 1C8 and anti‐PD‐1 exhibited an even stronger effect, reinforcing this observation (Figure 6O and Supplementary Figure S7H). Therefore, in a mouse model, we used RNA interference and pharmacological inhibition to demonstrate that SRSF10 could potentially serve as a strategy to modulate the macrophage population and restore their anti‐tumor capabilities.
3.7. SRSF10 was associated with immunotherapy resistance and poor prognosis
Based on our preclinical murine data, we conducted further investigations to determine whether 1C8 could reshape the TME in humans. Since traditional patient‐derived organoids (PDOs) have limited immune cell preservation [49, 50], we performed ex vivo profiling of PDOTS [37, 51] obtained from human HCC tumors. The PDOTS established from patients with HCC were cultured ex vivo in DMEM. One group was treated with PD‐1 mAb, and the other was treated with PD‐1 mAb and 1C8 for 48 h (Figure 7A). Flow cytometry analysis showed that combination treatment with 1C8 and anti‐PD‐1 significantly reduced the proportion of pro‐tumor CD206+ macrophages and increased the proportion of CD8+ T cells (Figure 7B and Supplementary Figure S8A‐B). Additionally, ELISA indicated that the secretion of IFN‐γ and PRF1 significantly increased when treated with 1C8 and anti‐PD‐1 compared with anti‐PD‐1 alone (Figure 7C). Therefore, our findings in human preclinical models suggest that targeting SRSF10 with 1C8 is a potential strategy to modulate macrophage polarization and restore the immune‐killing TME in patients with HCC.
FIGURE 7.
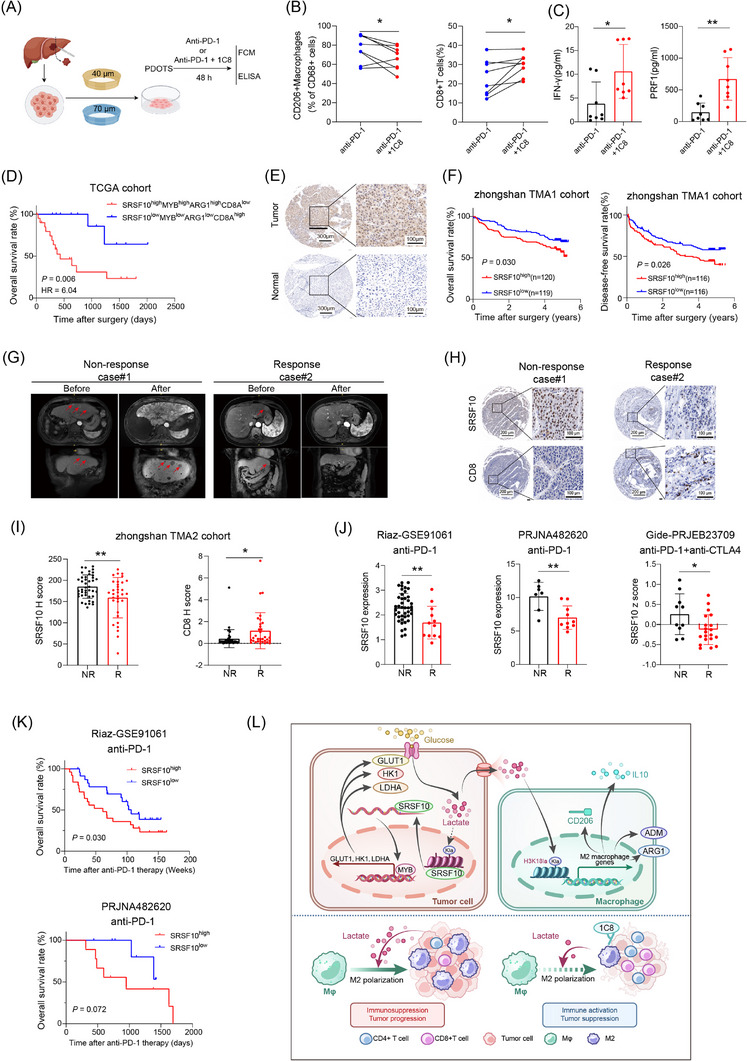
SRSF10 was associated with immunotherapy resistance and poor prognosis. (A) Schematic of PDOTS experiment. (B) Percentages of tumor‐infiltrating CD206+ macrophages and CD8+ T cells from PDOTS. (C) Secreted IFN‐γ and PRF1 in the supernatants of PDOTS detected by ELISA. (D) Kaplan–Meier curves for OS of patients with HCC from TCGA‐LIHC cohort. (E) Representative images of IHC staining for SRSF10 of HCC tissue and adjacent normal tissue. (F) Kaplan–Meier curves for OS and Disease‐free survival (DFS) of patients with HCC. (G) Magnetic resonance imaging scans of a responder (patient 1) and non‐responder (patient 2) to PD‐1 mAbs before and after treatment. (H) Representative images of IHC staining for SRSF10 and CD8 of two patients with HCC. (I) The expression of SRSF10 and CD8 in response patients and non‐response patients of Zhongshan TMA2 cohort. (J) Bulk RNA‐seq data revealed that responders had high expression of SRSF10 in publicly available datasets. (K) The prognostic prediction performances of SRSF10 for OS were assessed by Kaplan‐Meier curve analysis using a melanoma cohort and GBM cancer cohort. (L) Illustration of the proposed working model. Two‐tailed Paried Student's t test (B); two‐tailed unpaired Student's t test (C, I‐J); Log Rank test (D, F); Gehan‐Breslow‐Wilcoxon test (L); ns, not significant; * P < 0.05, ** P < 0.01, *** P < 0.001. Abbreviations: PDOTS, Patient‐derived organotypic tumor spheroids; FCM, flow cytometry; ELISA, enzyme‐linked immunosorbent assay; PD‐1, programmed cell death 1; SRSF10, serine and arginine rich splicing factor 10; OS, overall survival; DFS, Disease‐free survival; IFN‐γ, Interferon gamma; PRF1, perforin 1; TCGA, The Cancer Genome Atlas; LIHC, Liver hepatocellular carcinoma; IHC, immunohistochemistry; TMA, tissue microarray; GBM, Glioblastoma multiforme.
Our study found that low expression of SRSF10 or MYB was associated with a more favorable prognosis in patients with HCC, which was supported by data from TCGA database. Survival analysis revealed longer OS and progression‐free survival (PFS) in patients with low SRSF10 or MYB expression than in those with high expression (Supplementary Figure S8C). Additionally, patients with high SRSF10‐MYB‐ARG1 expression combined with low CD8 axis expression had significantly worse survival outcomes (Figure 7D). Moreover, our research showed that elevated SRSF10 associated with malignant phenotype in patients with HCC in the TCGA LIHC cohort (Supplementary Table S5). The TMA1 cohort of Zhongshan Hospital was used for IHC staining to examine the expression of SRSF10 in tumor tissues compared with that in adjacent normal tissues. The findings revealed that SRSF10 expression was higher in tumor tissues (Figure 7E and Supplementary Figure S8D). Additionally, we observed that low SRSF10 expression indicated a significantly better prognosis in patients with HCC than in those with high expression, as evidenced by longer OS and disease‐free survival (DFS) in the TMA1 cohort (Figure 7F). Besides, univariate and multivariate Cox regression analyses showed that higher expression of SRSF10 was an independent predictor for postoperative OS (Supplementary Table S6). Next, we collected specimens from 75 patients with HCC who received anti‐PD‐1 immunotherapy (33 responders and 42 non‐responders) at Zhongshan Hospital (Figure 7G). IHC assays demonstrated that patients with HCC who responded to anti‐PD‐1 immunotherapy had lower expression of SRSF10 and higher expression of CD8 (Figure 7H‐I).
To further support the association between SRSF10 and the response to immunotherapy, we utilized three publicly available immunotherapy cohorts for verification. The findings revealed that SRSF10 expression was elevated in non‐responders than in responders (Figure 7J). Patients exhibiting lower SRSF10 expression also demonstrated extended OS (Figure 7K). Furthermore, we performed a publicly available scRNA‐seq cohort study of patients treated with PD‐1 mAbs (GSE123813) to corroborate the association between SRSF10 and immunotherapy. Our analysis revealed heightened SRSF10 expression in tumor cells from non‐responsive patients, with GLUT1, HK1, and LDHA displaying similar patterns to SRSF10 (Supplementary Figure S8E).
These data collectively confirmed that SRSF10 expression associated with the immunotherapy response and patient survival in tumors treated with anti‐PD‐1. Consequently, the aforementioned findings established that SRSF10, a gene overexpressed in HCC, is a key player in promoting glycolysis via MYB. Furthermore, this illustrates that the SRSF10/glycolysis/H3K18la axis forms a positive feedback loop, resulting in lactate accumulation, a glycolysis byproduct, in the TME. Subsequently, lactate is transported into the macrophages, where it induces histone modification through lactylation. This modification further enhances pro‐tumor macrophage polarization, thus fostering an immunosuppressive TME and contributing to PD‐1 immunotherapy resistance (Figure 7L).
4. DISCUSSION
The efficacy of anti‐PD‐1 therapies in the treatment of various solid tumors is limited to a small subset of patients with HCC. Our study revealed a robust correlation between elevated SRSF10 levels and resistance to anti‐PD‐1 treatment, alongside a negative prognosis among patients with HCC. Specifically, we found that SRSF10 binds to MYB RNA, thereby stabilizing and amplifying its expression. Consequently, MYB, activates key enzymes that are pivotal in glycolysis and fosters lactate accumulation in the TME. Subsequently, lactate is transported into macrophages, inducing histone lactylation and culminating in the transcriptional upregulation of ARG1 and other genes. This polarization of M2 macrophages impedes the function of CD8+ T cells, thereby facilitating immune evasion and diminishing the response to anti‐PD‐1 therapy. Clinical observations indicate that patients with HCC exhibiting high SRSF10 expression frequently encounter resistance to anti‐PD‐1 treatment and exhibit a poor prognosis.
Resistance to T‐cell‐mediated killing constitutes a crucial mechanism of immune evasion and resistance to ICB therapy [52]. This resistance primarily stems from the immunosuppressive microenvironment, fostered by prolonged T‐cell activation and tumor adaptation. Macrophages, as the principal immune cells within the liver TME, play intricate roles in tumor progression and immune microenvironmental remodeling. Mainstream studies suggest that macrophages can be broadly classified into two major subsets: classically activated M1 macrophages and alternatively activated M2 macrophages [53]. M1 macrophages secrete pro‐inflammatory cytokines such as IL6, IL12, and TNF‐α, promoting CD8+ T cell activation. Additionally, M1 macrophages may directly interact with CD8+ T cells through surface receptors, thereby influencing their activity. These interactions may involve the expression and signaling of costimulatory genes such as CD40, CD80, and CD86. In contrast, M2 macrophages secrete immunosuppressive factors like IL10 and TGFB1, directly inhibiting CD8+ T cell activity. Thus, shifting M2 macrophages toward the M1 phenotype to activate CD8+ T cells and overcome resistance to immune therapyis a promising strategy. Our study indicates that interfering with SRSF10 expression in tumor cells reduces the proportion of M2 macrophages in the tumor microenvironment, increases CD8+ T cell proportion, and elevates the proportion of IFN‐γ+ CD8+ T cells, suggesting enhanced immune activity. Single‐cell sequencing results demonstrated decreased expression of M2 polarization‐related genes, such as ADM, and upregulation of M1 polarization‐related genes, such as CXCL9, in macrophages following SRSF10 interference. Furthermore, immunoreactive substances such as Gzmb were upregulated in CD8+ T cells. These findings were partially validated through in vitro and in vivo experiments. Thus, SRSF10 has emerged as a key gene mediating T cell exhaustion and immune evasion, and interfering with its expression may alleviate immune evasion and enhance the efficacy of immune therapy.
Lactate exerts dual effects on CD8+ T cells through various mechanisms. Most studies have reported that increasing the LA concentration impairs the viability of T and NK cells [54]. Alternatively, reducing lactate levels in the TME significantly enhances the activation and proliferation of CD8+ T cells [55]. However, a recent study has suggested that sodium lactate increases the expression of TCF7 in CD8+ T cells, thereby enhancing the immune‐activating function of the effector T cell population [56]. Consequently, the effects of lactate on CD8+ T cells may be biphasic, and further investigation is required to fully understand its comprehensive role. Regarding the regulation of macrophages, previous studies have primarily focused on lactate serving as a carbon source or activating pathways such as hypoxia‐inducible factor 1 [57] and extracellular signal‐regulated kinase/signal transducer and activator of transcription 3 [58] through GPCR receptors like G protein‐coupled receptor 81, promoting macrophage polarization toward the M2 phenotype. Recent studies have indicated that lactate increases the expression of ARG1 through histone lactylation in M1 macrophages [44]. We found that elevated lactate levels mediated by SRSF10 enhanced histone lactylation modification in macrophages, directly transcriptionally activating the expression of various M2 macrophage markers, such as CD206, ARG1, IL10, and ADM. Moreover, inhibition of tumor cell glycolysis using low‐glucose culture media or 2‐DG, interfering with SRSF10 expression, did not significantly alter the expression of M2 macrophage markers, indicating that this effect is indeed mediated by lactate.
Shkreta et al. [47] discovered that inhibitor 1C8 selectively blocked the phosphorylation of SRSF10 at site 132, thereby inhibiting its activity. This inhibition decreases the regulatory effects of SRSF10 on HIV proliferation. Our findings provide evidence of the involvement of SRSF10 in regulating the immune microenvironment. Therefore, we propose that 1C8 functions as a specific SRSF10 inhibitor, potentially modulating the immune microenvironment and improving the efficacy of PD‐1 immunotherapy. Our experiments demonstrated that 1C8 suppressed the pro‐tumor polarization of macrophages and activated CD8+ T cells, leading to tumor elimination. Besides, we observed that tumors in the shSRSF10 and shSRSF10 plus 1C8 groups exhibited a significant reduction compared with the control group. However, there was no significant difference in tumor weight or growth rate between the shSRSF10 plus 1C8 and shSRSF10 groups, indicating that the functionality of 1C8 in vivo is mediated through SRSF10. When combining SRSF10 inhibition with 1C8 and anti‐PD‐1 treatment, we observe a significant reduction in the infiltration of CD206+ macrophages and a notable increase in the infiltration of CD8+ T cells, including IFN‐γ+ CD8+ T cells. To better simulate the TME within HCC, we used hydrodynamic tail vein injection of plasmids to establish spontaneous HCC tumors. These results further supported our previous conclusions. Moreover, 1C8 exhibited no obvious side effects in the heart or spleen of the mice.
Due to disparities between mouse and human genomes, in vivo mouse experiments cannot fully replicate humanized tumors. In response, researchers have developed PDOs, a method for directly culturing primary tumor cells from tumor samples. However, traditional PDOs exhibit limited similarities to the TME [50]. Subsequently, researchers introduced PDOTS [37], which effectively simulated changes in the human TME. We validated the effect of the SRSF10 inhibitor, 1C8, on the constructed model. Our results revealed that compared with anti‐PD‐1 alone, the combined application of 1C8 and PD‐1 mAbs reduced the proportion of tumor‐promoting CD206+ macrophages while enhancing the enrichment of CD8+ T cells. Additionally, there was an increased secretion of IFN‐γ and PRF1 within the TME.
To investigate the effect of SRSF10 expression in tumor cells on TAMs, we established an in vitro culture system using tumor‐conditioned medium (TCM) and conducted a series of functional assays. However, existing experimental methods do not provide precise measurements of the response and functional state of TAMs within the tumor upon stimulation by tumor cells. Consequently, the direct effects of high SRSF10 levels within tumors in vivo could not be demonstrated. Additionally, since SRSF10 is expressed in other cells in the TME, limited knowledge exists regarding how these cells would respond to the effects of the SRSF10 inhibitor 1C8. Since SRSF10 is naturally expressed in the cardiovascular system, modifying a selective inhibitor before systemic administration may be necessary to enhance its therapeutic efficacy.
5. CONCLUSION
Our study revealed that the SRSF10/MYB/glycolysis axis inhibited TME by promoting M2 macrophage polarization. We also discovered that RNA interference or pharmacological inhibition of SRSF10 reshaped the TME and enhanced the efficacy of anti‐PD‐1 therapy. Furthermore, our research revealed that the SRSF10 inhibitor 1C8 exhibited promising antitumor effects in humanized tumor preclinical models. Therefore, these findings unveil a promising anti‐PD‐1 combination treatment regimen.
AUTHOR CONTRIBUTIONS
Jialiang Cai, Lina Song, Feng Zhang, Suiyi Wu and Zhi Dai designed the study. Jialiang Cai, Lina Song, Feng Zhang and Suiyi Wu conducted the majority of the experments. Guiqi Zhu and Shiping Chen assisted in molecular biology and functions analysis performed in vivo. Peiling Zhang analyzed the bioinformatic data. Junxian Du, Biao Wang and Yufan Cai collected tissue samples and data. Feng Zhang and Suiyi Wu did the statistical analyses. Jialiang Cai, Guiqi Zhu, Peiling Zhang and Shiping Chen prepared figures, reviewed the results, interpreted data, and wrote the manuscript. Yi Yang and Jinglei Wan revised the paper. Zhi Dai, Jia Fan and Jian Zhou contributed to study supervision. Zhi Dai provided financial support. All authors have made an intellectual contribution to the manuscript and approved the submission.
CONFLICT OF INTEREST STATEMENT
All authors: no conflicts.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Ethical approval for this study was obtained from the Research Ethics Committee of Zhongshan Hospital (Approval No. B2021‐248), and written informed consent was obtained from all participants. The handling of all animals strictly adhered to the Principles for the Utilization and Care of Vertebrate Animals and the Guide for the Care and Use of Laboratory Animals. The Institutional Animal Care and Use Committee of Zhongshan Hospital, Fudan University, approved all animal experiments (Approval No. 2020‐133).
Supporting information
Supporting Information
ACKNOWLEDGEMENTS
This work was supported by the National Natural Science Foundation of China (No. 82372946 and No. 82072670), the Leading Project of the Science and Technology Committee of Shanghai Municipality (No. 21Y21900100), and the Project of Shanghai Municipal Health Commission (No. 202140269).
Cai J, Song L, Zhang F, Wu S, Zhu G, Zhang P, et al. Targeting SRSF10 might inhibit M2 macrophage polarization and potentiate anti‐PD‐1 therapy in hepatocellular carcinoma. Cancer Commun. 2024;44:1231–1260. 10.1002/cac2.12607
Jialiang Cai, Lina Song, Feng Zhang and Suiyi Wu contributed equally to this work.
DATA AVAILABILITY STATEMENT
Data were collected from independent databases, including The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov) for all 33 tumor types and the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) for data under the accession codes GSE10143, GSE29721, GSE46408, GSE74656, GSE202069, GSE125449, GSE149614, and GSE123813. The Riaz‐GSE91061, PRJNA482620, and Gide‐PRJEB23709 datasets were downloaded from TIDE (http://tide.dfci.harvard.edu). The raw data of RNA‐seq conduceted on control and shSRSF10 Huh7 cells were uploaded on GEO with accession number GSE272166. The raw data of scRNA‐seq conducted on this study were uploaded on GEO with accession number GSE273080. All other data supporting the findings of this study are available from the corresponding author upon request.
REFERENCES
- 1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71(3):209‐49. [DOI] [PubMed] [Google Scholar]
- 2. Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet (London, England). 2018;391(10127):1301‐14. [DOI] [PubMed] [Google Scholar]
- 3. Allemani C, Matsuda T, Di Carlo V, Harewood R, Matz M, Nikšić M, et al. Global surveillance of trends in cancer survival 2000‐14 (CONCORD‐3): analysis of individual records for 37 513 025 patients diagnosed with one of 18 cancers from 322 population‐based registries in 71 countries. Lancet (London, England). 2018;391(10125):1023‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pinter M, Scheiner B, Pinato DJ. Immune checkpoint inhibitors in hepatocellular carcinoma: emerging challenges in clinical practice. Lancet Gastroenterol Hepatol. 2023;8(8):760‐70. [DOI] [PubMed] [Google Scholar]
- 5. Eggermont AMM, Blank CU, Mandala M, Long GV, Atkinson V, Dalle S, et al. Adjuvant Pembrolizumab versus Placebo in Resected Stage III Melanoma. N Engl J Med. 2018;378(19):1789‐801. [DOI] [PubMed] [Google Scholar]
- 6. Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N Engl J Med. 2020;382(20):1894‐905. [DOI] [PubMed] [Google Scholar]
- 7. Yau T, Park JW, Finn RS, Cheng AL, Mathurin P, Edeline J, et al. Nivolumab versus sorafenib in advanced hepatocellular carcinoma (CheckMate 459): a randomised, multicentre, open‐label, phase 3 trial. Lancet Oncol. 2022;23(1):77‐90. [DOI] [PubMed] [Google Scholar]
- 8. Qin S, Chan SL, Gu S, Bai Y, Ren Z, Lin X, et al. Camrelizumab plus rivoceranib versus sorafenib as first‐line therapy for unresectable hepatocellular carcinoma (CARES‐310): a randomised, open‐label, international phase 3 study. Lancet. 2023;402(10408):1133‐46. [DOI] [PubMed] [Google Scholar]
- 9. Tian Z, Hou X, Liu W, Han Z, Wei L. Macrophages and hepatocellular carcinoma. Cell & Bioscience. 2019;9:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yunna C, Mengru H, Lei W, Weidong C. Macrophage M1/M2 polarization. Eur J Pharmacol. 2020;877:173090. [DOI] [PubMed] [Google Scholar]
- 11. Tarique AA, Logan J, Thomas E, Holt PG, Sly PD, Fantino E. Phenotypic, functional, and plasticity features of classical and alternatively activated human macrophages. Am J Respir Cell Mol Biol. 2015;53(5):676‐88. [DOI] [PubMed] [Google Scholar]
- 12. Shapouri‐Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili S‐A, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. 2018;233(9):6425‐40. [DOI] [PubMed] [Google Scholar]
- 13. Xiao H, Guo Y, Li B, Li X, Wang Y, Han S, et al. M2‐Like Tumor‐Associated Macrophage‐Targeted Codelivery of STAT6 Inhibitor and IKKβ siRNA Induces M2‐to‐M1 Repolarization for Cancer Immunotherapy with Low Immune Side Effects. ACS Central Science. 2020;6(7):1208‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hanahan D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022;12(1):31‐46. [DOI] [PubMed] [Google Scholar]
- 15. Chang CH, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell. 2015;162(6):1229‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Buck MD, Sowell RT, Kaech SM, Pearce EL. Metabolic Instruction of Immunity. Cell. 2017;169(4):570‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Qian Y, Galan‐Cobo A, Guijarro I, Dang M, Molkentine D, Poteete A, et al. MCT4‐dependent lactate secretion suppresses antitumor immunity in LKB1‐deficient lung adenocarcinoma. Cancer Cell. 2023;41(7):1363‐80 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guo D, Tong Y, Jiang X, Meng Y, Jiang H, Du L, et al. Aerobic glycolysis promotes tumor immune evasion by hexokinase2‐mediated phosphorylation of IkappaBalpha. Cell Metab. 2022;34(9):1312‐24 e6. [DOI] [PubMed] [Google Scholar]
- 19. Cascone T, McKenzie JA, Mbofung RM, Punt S, Wang Z, Xu C, et al. Increased Tumor Glycolysis Characterizes Immune Resistance to Adoptive T Cell Therapy. Cell Metab. 2018;27(5):977‐87 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Du JX, Zhu GQ, Cai JL, Wang B, Luo YH, Chen C, et al. Splicing factors: Insights into their regulatory network in alternative splicing in cancer. Cancer Lett. 2021;501:83‐104. [DOI] [PubMed] [Google Scholar]
- 21. Zhang C, Chen Y, Li F, Yang M, Meng F, Zhang Y, et al. B7‐H3 is spliced by SRSF3 in colorectal cancer. Cancer Immunol Immunother. 2021;70(2):311‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Peiqi L, Zhaozhong G, Yaotian Y, Jun J, Jihua G, Rong J. Expression of SRSF3 is Correlated with Carcinogenesis and Progression of Oral Squamous Cell Carcinoma. Int J Med Sci. 2016;13(7):533‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gao X, Dai C, Huang S, Tang J, Chen G, Li J, et al. Functional Silencing of HSD17B2 in Prostate Cancer Promotes Disease Progression. Clin Cancer Res. 2019;25(4):1291‐301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim HR, Lee GO, Choi KH, Kim DK, Ryu JS, Hwang KE, et al. SRSF5: a novel marker for small‐cell lung cancer and pleural metastatic cancer. Lung Cancer. 2016;99:57‐65. [DOI] [PubMed] [Google Scholar]
- 25. Chen S, Yang C, Wang ZW, Hu JF, Pan JJ, Liao CY, et al. CLK1/SRSF5 pathway induces aberrant exon skipping of METTL14 and Cyclin L2 and promotes growth and metastasis of pancreatic cancer. J Hematol Oncol. 2021;14(1):60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen Y, Huang Q, Liu W, Zhu Q, Cui CP, Xu L, et al. Mutually exclusive acetylation and ubiquitylation of the splicing factor SRSF5 control tumor growth. Nat Commun. 2018;9(1):2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu F, Dai M, Xu Q, Zhu X, Zhou Y, Jiang S, et al. SRSF10‐mediated IL1RAP alternative splicing regulates cervical cancer oncogenesis via mIL1RAP‐NF‐kappaB‐CD47 axis. Oncogene. 2018;37(18):2394‐409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhou X, Li X, Cheng Y, Wu W, Xie Z, Xi Q, et al. BCLAF1 and its splicing regulator SRSF10 regulate the tumorigenic potential of colon cancer cells. Nat Commun. 2014;5:4581. [DOI] [PubMed] [Google Scholar]
- 29. Chang C, Rajasekaran M, Qiao Y, Dong H, Wang Y, Xia H, et al. The aberrant upregulation of exon 10‐inclusive SREK1 through SRSF10 acts as an oncogenic driver in human hepatocellular carcinoma. Nat Commun. 2022;13(1):1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chabrolles H, Auclair H, Vegna S, Lahlali T, Pons C, Michelet M, et al. Hepatitis B virus Core protein nuclear interactome identifies SRSF10 as a host RNA‐binding protein restricting HBV RNA production. PLoS Pathog. 2020;16(11):e1008593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Seymour L, Bogaerts J, Perrone A, Ford R, Schwartz LH, Mandrekar S, et al. iRECIST: guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol. 2017;18(3):e143‐e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228‐47. [DOI] [PubMed] [Google Scholar]
- 33. Amezquita RA, Lun ATL, Becht E, Carey VJ, Carpp LN, Geistlinger L, et al. Orchestrating single‐cell analysis with Bioconductor. Nat Methods. 2020;17(2):137‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lu Y, Yang A, Quan C, Pan Y, Zhang H, Li Y, et al. A single‐cell atlas of the multicellular ecosystem of primary and metastatic hepatocellular carcinoma. Nat Commun. 2022;13(1):4594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ma L, Hernandez MO, Zhao Y, Mehta M, Tran B, Kelly M, et al. Tumor Cell Biodiversity Drives Microenvironmental Reprogramming in Liver Cancer. Cancer Cell. 2019;36(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bi K, He MX, Bakouny Z, Kanodia A, Napolitano S, Wu J, et al. Tumor and immune reprogramming during immunotherapy in advanced renal cell carcinoma. Cancer Cell. 2021;39(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jenkins RW, Aref AR, Lizotte PH, Ivanova E, Stinson S, Zhou CW, et al. Ex Vivo Profiling of PD‐1 Blockade Using Organotypic Tumor Spheroids. Cancer Discov. 2018;8(2):196‐215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pais Ferreira D, Silva JG, Wyss T, Fuertes Marraco SA, Scarpellino L, Charmoy M, et al. Central memory CD8+ T cells derive from stem‐like Tcf7hi effector cells in the absence of cytotoxic differentiation. Immunity. 2020;53(5). [DOI] [PubMed] [Google Scholar]
- 39. Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG, et al. Lactate Metabolism in Human Lung Tumors. Cell. 2017;171(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hui S, Ghergurovich JM, Morscher RJ, Jang C, Teng X, Lu W, et al. Glucose feeds the TCA cycle via circulating lactate. Nature. 2017;551(7678):115‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ahmed K, Tunaru S, Tang C, Müller M, Gille A, Sassmann A, et al. An autocrine lactate loop mediates insulin‐dependent inhibition of lipolysis through GPR81. Cell Metab. 2010;11(4):311‐9. [DOI] [PubMed] [Google Scholar]
- 42. Vardjan N, Chowdhury HH, Horvat A, Velebit J, Malnar M, Muhič M, et al. Enhancement of Astroglial Aerobic Glycolysis by Extracellular Lactate‐Mediated Increase in cAMP. Front Mol Neurosci. 2018;11:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Latham T, Mackay L, Sproul D, Karim M, Culley J, Harrison DJ, et al. Lactate, a product of glycolytic metabolism, inhibits histone deacetylase activity and promotes changes in gene expression. Nucleic Acids Res. 2012;40(11):4794‐803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574(7779):575‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fang Y, Liu W, Tang Z, Ji X, Zhou Y, Song S, et al. Monocarboxylate transporter 4 inhibition potentiates hepatocellular carcinoma immunotherapy through enhancing T cell infiltration and immune attack. Hepatology (Baltimore, Md). 2023;77(1):109‐23. [DOI] [PubMed] [Google Scholar]
- 46. Pandkar MR, Sinha S, Samaiya A, Shukla S. Oncometabolite lactate enhances breast cancer progression by orchestrating histone lactylation‐dependent c‐Myc expression. Transl Oncol. 2023;37:101758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shkreta L, Blanchette M, Toutant J, Wilhelm E, Bell B, Story BA, et al. Modulation of the splicing regulatory function of SRSF10 by a novel compound that impairs HIV‐1 replication. Nucleic Acids Res. 2017;45(7):4051‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chang C, Rajasekaran M, Qiao Y, Dong H, Wang Y, Xia H, et al. The aberrant upregulation of exon 10‐inclusive SREK1 through SRSF10 acts as an oncogenic driver in human hepatocellular carcinoma. Nat Commun. 2022;13(1):1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. van de Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A, et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell. 2015;161(4):933‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Broutier L, Mastrogiovanni G, Verstegen MM, Francies HE, Gavarro LM, Bradshaw CR, et al. Human primary liver cancer‐derived organoid cultures for disease modeling and drug screening. Nat Med. 2017;23(12):1424‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Aref AR, Campisi M, Ivanova E, Portell A, Larios D, Piel BP, et al. 3D microfluidic ex vivo culture of organotypic tumor spheroids to model immune checkpoint blockade. Lab Chip. 2018;18(20):3129‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kalbasi A, Ribas A. Tumour‐intrinsic resistance to immune checkpoint blockade. Nat Rev Immunol. 2020;20(1):25‐39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li M, Yang Y, Xiong L, Jiang P, Wang J, Li C. Metabolism, metabolites, and macrophages in cancer. J Hematol Oncol. 2023;16(1):80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, et al. LDHA‐Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016;24(5):657‐71. [DOI] [PubMed] [Google Scholar]
- 55. Ma J, Tang L, Tan Y, Xiao J, Wei K, Zhang X, et al. Lithium carbonate revitalizes tumor‐reactive CD8+ T cells by shunting lactic acid into mitochondria. Nat Immunol. 2024;25(3):552‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Feng Q, Liu Z, Yu X, Huang T, Chen J, Wang J, et al. Lactate increases stemness of CD8 + T cells to augment anti‐tumor immunity. Nat Commun. 2022;13(1):4981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhang L, Li S. Lactic acid promotes macrophage polarization through MCT‐HIF1α signaling in gastric cancer. Exp Cell Res. 2020;388(2):111846. [DOI] [PubMed] [Google Scholar]
- 58. Gou Y, Wang H, Wang T, Wang H, Wang B, Jiao N, et al. Ectopic endometriotic stromal cells‐derived lactate induces M2 macrophage polarization via Mettl3/Trib1/ERK/STAT3 signalling pathway in endometriosis. Immunology. 2023;168(3):389‐402. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
Data were collected from independent databases, including The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov) for all 33 tumor types and the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) for data under the accession codes GSE10143, GSE29721, GSE46408, GSE74656, GSE202069, GSE125449, GSE149614, and GSE123813. The Riaz‐GSE91061, PRJNA482620, and Gide‐PRJEB23709 datasets were downloaded from TIDE (http://tide.dfci.harvard.edu). The raw data of RNA‐seq conduceted on control and shSRSF10 Huh7 cells were uploaded on GEO with accession number GSE272166. The raw data of scRNA‐seq conducted on this study were uploaded on GEO with accession number GSE273080. All other data supporting the findings of this study are available from the corresponding author upon request.