

Abstract
We report the discovery of an imidazopyridine series of IRAK1/4/pan-FLT3 kinase inhibitors. Optimization of this series has produced compound 31 which displays potent and selective inhibition of IRAK1, IRAK4, FLT3, and all mutant forms of FLT3, as well as good in vitro ADME and pharmacokinetic properties. In a mouse xenograft model of AML, 31 produces survival prolongation equal to that of Gilteritinib, the leading marketed FLT3 inhibitor currently used to treat AML.
Keywords: IRAK1, IRAK4, FLT3, kinase, AML, leukemia
Acute myeloid leukemia (AML) is a blood cell cancer that is estimated to cause ∼100,000 deaths worldwide each year.1 Current standard of care includes treatment with chemotherapeutic agents, newer targeted therapies such as FLT3 kinase inhibitors, and in some cases, hematopoietic stem cell transplantation.2 Despite the availability of these treatment options, a diagnosis of AML is, for most patients, a death sentence, with only 32% surviving at least 5 years postdiagnosis.3
FLT3 kinase can initiate signaling through a variety of pathways associated with cell growth and proliferation.4 In healthy individuals, FLT3 signaling is tightly controlled through restricted expression and autoinhibition. In AML, a variety of FLT3 mutations can disrupt the autoinhibition process, resulting in constitutive activation of the kinase and its associated downstream signaling pathways.5 That aberrant, unchecked signaling promotes the rapid growth and proliferation of AML tumor cells, and is a key driver of disease progression.6
Given the prominent role FLT3 signaling plays in AML, significant efforts have been aimed at discovering FLT3 inhibitors as targeted AML therapies.7 Several FLT3 inhibitors have recently been approved by the FDA as first- or second-line treatments for AML. These include Midostaurin, a first-generation inhibitor of FLT3 and numerous other kinases, as well as Gilteritinib and Quizartinib, more selective second-generation inhibitors. Treatment of AML patients with these and other FLT3 inhibitors, either alone or in combination with other agents, can produce good initial response rates. However, after relatively brief periods of remission, patients often relapse with treatment-resistant AML and eventually succumb to the disease.
Prior work has revealed the importance of IRAK-mediated signaling as a key adaptive resistance pathway that emerges in patients treated with FLT3 inhibitors.8 In these patients, signaling through the FLT3-mediated pathway is blocked, but compensatory signaling through IRAK1 and IRAK4 is upregulated, and this signaling promotes the continued growth and proliferation of the cancer. While selective inhibition of IRAK4 can attenuate this signaling, potent inhibition of both IRAK1 and IRAK4 is required to maximize block of signaling through this key adaptive resistance pathway, and to maximize antileukemic efficacy in cell and mouse models of AML.9
Our goal is to discover agents that potently and selectively inhibit IRAK1, IRAK4, FLT3, and key mutant forms of FLT3. These targeted inhibitors should effectively block signaling through both the IRAK- and FLT3-mediated pathways, and should thus offer improved AML efficacy and more durable remission than the FLT3 inhibitors that have been advanced into the clinic to date.
We report the optimization of a series of imidazopyridine IRAK-FLT3 kinase inhibitors. Prior work in this series led to the identification of early tool compounds such as NCGC-1481 (Figure 1, compound 1) that were used to establish the importance of IRAK-mediated signaling in adaptive resistance.8,10 Further optimization, described herein, has produced more advanced compounds that display potent and selective inhibition of IRAK1, IRAK4, and FLT3, good in vitro ADME and pharmacokinetic (PK) properties, and efficacy equal to that of Gilteritinib in a mouse xenograft model of AML.11
Figure 1.

IRAK-FLT3 inhibitor NCGC-1481 (1).
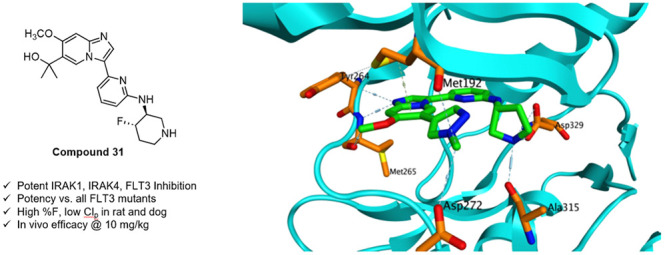
We began our SAR campaign by elucidating the minimum pharmacophore necessary for potent inhibition of IRAK1, IRAK4, and FLT3. At this early juncture we were fortunate to possess an X-ray cocrystal structure of 1 bound to IRAK4 (Figure 2, PDB code 6MOM). The structure indicated that 1 bound as a Type I inhibitor, forming a single H-bond from the imidazopyridine N1 to the amide NH of Met265 in the IRAK4 hinge region. Additional hydrogen bonds are formed between the linker NH and Asp329, and between the pyrrolidine NH and Ala315 of IRAK4. Finally, π-CH interactions are observed between aromatic portions of the molecule and the Met192 residue in the G loop, and with Leu318 in the catalytic loop.
Figure 2.
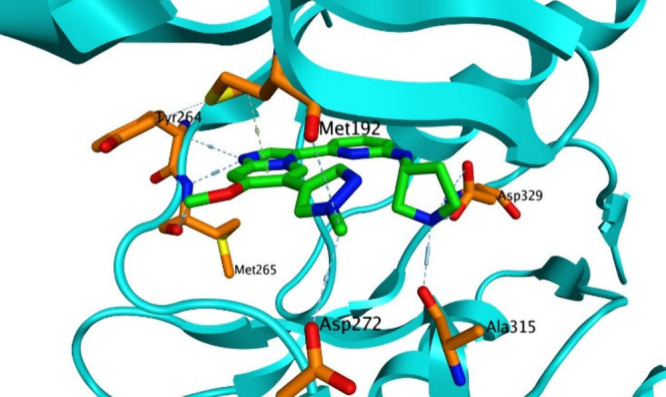
X-ray cocrystal structure of compound 1 bound to IRAK4.
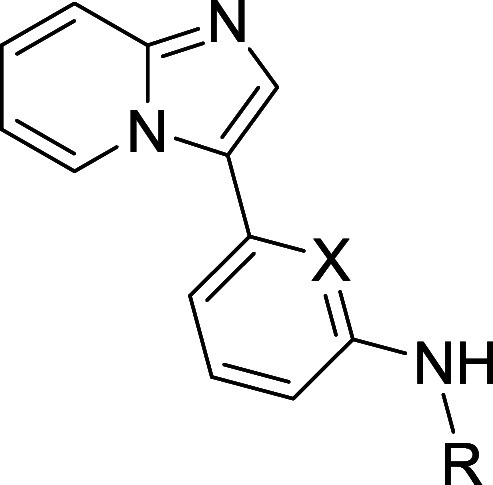
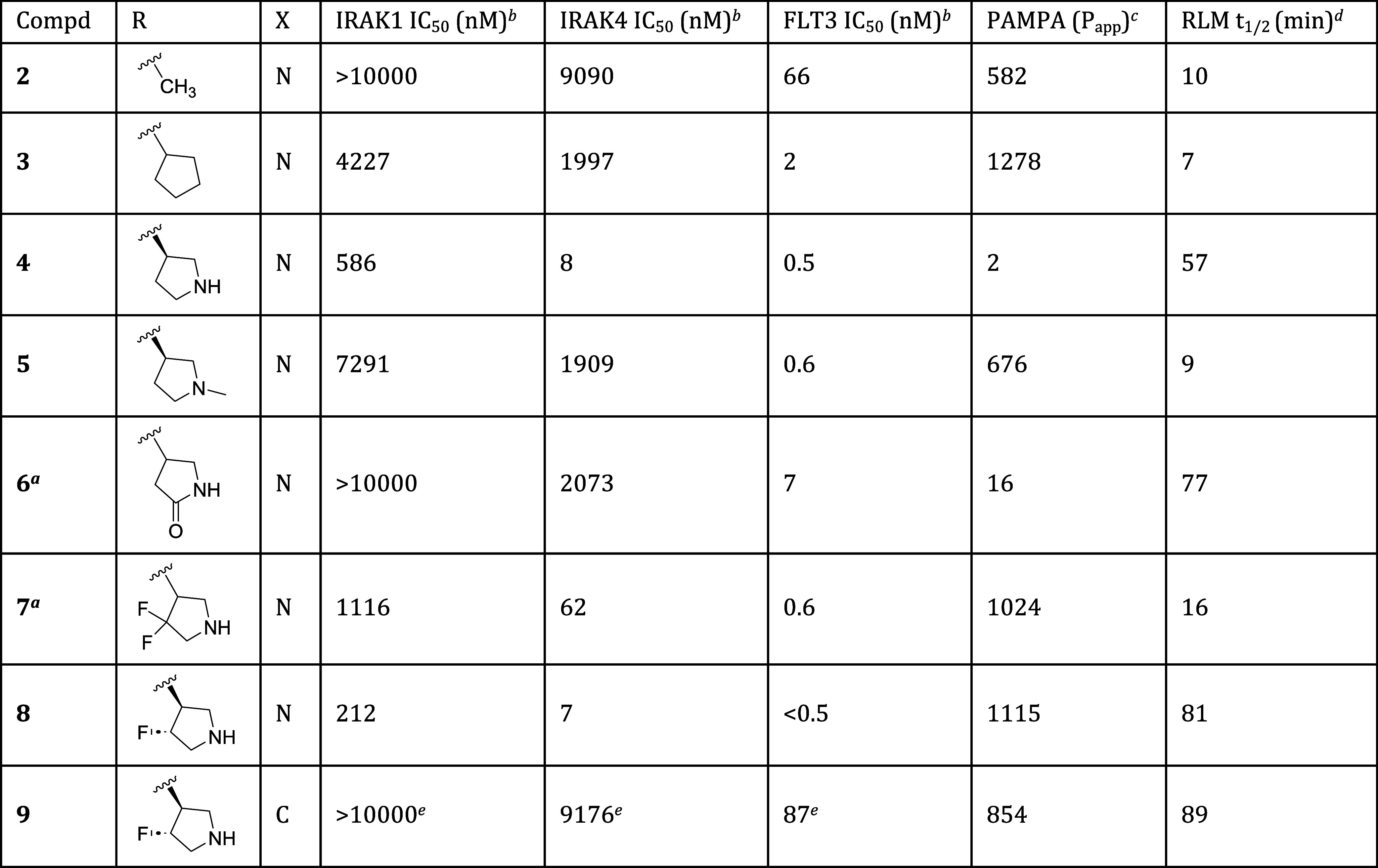
To quantify the contributions of these interactions to on-target potency, we synthesized a set of truncated analogs as shown in Table 1. The parent compound in this series, compound 2, displayed moderately potent functional inhibition of FLT3, but no significant inhibition of IRAK1 or IRAK4. Enlargement of the amine R group from methyl to cyclopentyl (compound 3) afforded an increase in FLT3 potency, but did little to improve IRAK1 or IRAK4 potency. Incorporation of a secondary amine as in pyrrolidine 4, on the other hand, yielded a 250-fold increase in IRAK4 potency, as well as a more modest increase in IRAK1 potency. This empirical result is consistent with the aforementioned cocrystal structure, which shows the pyrrolidine NH of 1 forming a hydrogen bond to the Ala315 residue of IRAK4. Finally, methylation of the pyrrolidine amine led to significant reductions in IRAK1 and IRAK4 potency (compound 5), a result again consistent with the importance of that NH to binding.
Table 1. Elucidation of Minimum Pharmacophore.
Compounds 6 and 7 are racemates.
Data are the geometric mean of three independent experiments (n = 3) unless otherwise specified.
Permeability (Papp × 10–6 cm/s) as measured in PAMPA assay.
Half-life of compound incubated with rat liver microsomes (RLM).
Data are the geometric mean of two independent experiments (n = 2).
While the pyrrolidine NH imparted improved IRAK4 potency, it also proved detrimental to membrane permeability, as evidenced by PAMPA data indicating high permeability for cyclopentyl analog 3 and low permeability for pyrrolidine 4.12 The basic pyrrolidine amine (pKa = 11.3) is protonated at physiological pH (7.4), and that charge can inhibit passive permeation through the lipophilic cell membrane. To temper the amine’s basicity, we prepared analogs that contained electron-withdrawing groups at positions alpha- or beta- to the amine. As shown in Table 1, conversion of the amine to the nonbasic lactam effected only a modest improvement in permeability, and even that came at the cost of attenuated IRAK1 and IRAK4 inhibition (compound 6). Difluorination at the pyrrolidine 4-position was better tolerated. As exemplified by compound 7, this change improved permeability while imparting a potency profile better than that of lactam 6, but still not as good as unsubstituted pyrrolidine 4. All four monofluoro diastereomers were prepared, and of those, the best overall profile was provided by compound 8, which displayed equally high permeability as well as improved potencies and metabolic stability when compared to compound 7.
Finally, to test whether the pyridine nitrogen of the central ring was required, we prepared phenyl analog 9. Analysis of the cocrystal structure indicated that the pyridine nitrogen did not interact with IRAK4. It was thus plausible that the nitrogen might be removed without incident. Testing of compound 9, however, revealed a catastrophic loss of potency at all 3 targets, indicating an essential role for the nitrogen. While it does not appear to engage in bonding interactions with IRAK4, the pyridine nitrogen may, for instance, modulate the pKa of the adjacent linker NH in a way that is critical for maintaining the H-bonding interaction to Asp329 in IRAK4, and to analogous residues in IRAK1 and FLT3.
With the minimum pharmacophore thus established and embodied in compound 8, we moved next to improve its properties. We began by optimizing substitution at the imidazopyridine C6 position. As shown in Table 2, the parent compound in this series (compound 8, R = H) displayed moderate inhibition of IRAK1, and more potent inhibition of IRAK4 and FLT3. Substitution of C6 with small electron-donating groups such as methoxy or difluoromethoxy (compounds 10 and 11) afforded improved IRAK1 and IRAK4 potency, but at the cost of reduced metabolic stability in rat liver microsome (RLM) incubations. Hypothesizing that the electron-donating ethers of 10 and 11 might promote oxidative metabolism, we excised the ether oxygen from 11 to afford difluoromethyl analog 12. That change proved beneficial, as 12 displayed improved metabolic stability relative to 11 while maintaining equivalent potencies at all key targets. The slightly larger cyclopropyl analog 13 exhibited a potency and in vitro ADME profile that was broadly similar to that of 12. Methyl- and trifluoromethyl-substituted cyclopropyl analogs 14 and 15 displayed potencies similar to those of 13, but suffered from reduced metabolic stability as measured in the RLM assay. Finally, to investigate the effects of polarity at the periphery of the molecule, we prepared tertiary alcohol 16. This analog displayed a potency profile comparable to that of cyclopropyl compound 13. Unfortunately, it also displayed greatly reduced membrane permeability as measured in the PAMPA assay.
Table 2. Effect of C6 Substitution on Potency and In Vitro ADME Properties.
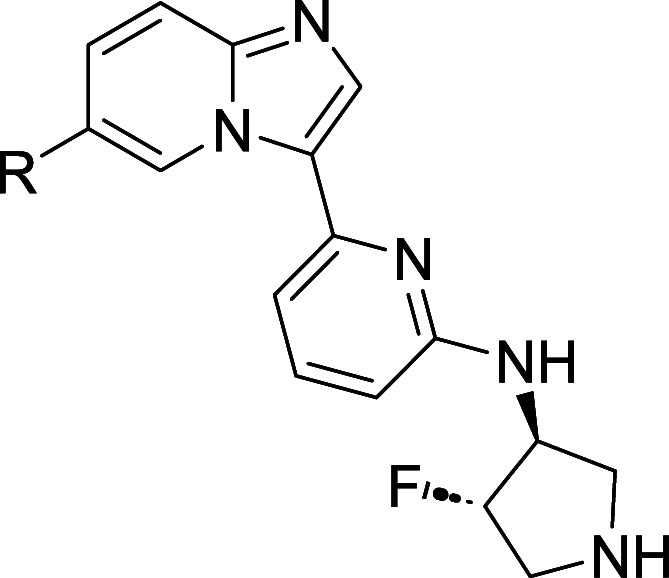
Data are the geometric mean of three independent experiments (n = 3) unless otherwise specified.
Permeability (Papp × 10–6 cm/s) as measured in PAMPA assay.
Half-life of compound incubated with rat liver microsomes (RLM).
With a set of improved C6 substituents in hand, we next optimized the cyclic amine portion of the molecule. As shown in Table 3, monocyclic azetidine, pyrrolidine, and piperidine derivatives were prepared (compounds 17–19), and from this set, pyrrolidine 18 displayed optimal potency. Though less potent than 17 or 18, piperidine 19 offered a useful permeability advantage as indicated by PAMPA data. Several spirobicyclic amines were also prepared (compounds 20–22). These offered the potential advantages of increased three-dimensionality and structural novelty, an important IP consideration. Unfortunately, they displayed significantly reduced IRAK1 and IRAK4 potency as well as low permeability.
Table 3. Effect of Cyclic Amine on Potency and In Vitro ADME Properties.
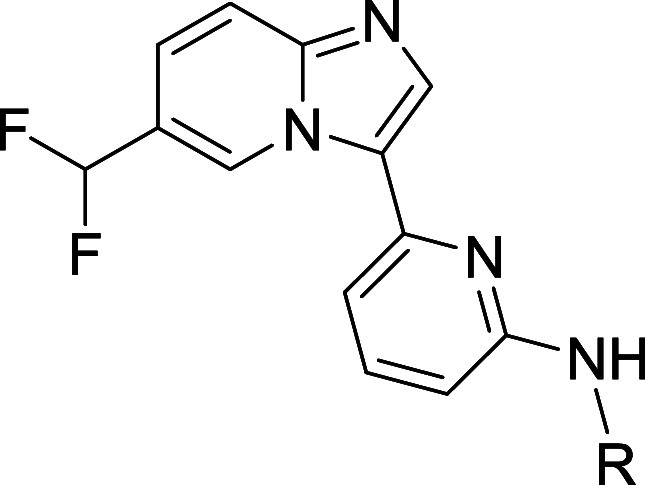
Compounds 18, 21, 22, and 25 are racemates.
Data are the geometric mean of three independent experiments (n = 3) unless otherwise specified.
Permeability (Papp × 10–6 cm/s) as measured in PAMPA assay.
Half-life of compound incubated with rat (R), mouse (M), or human (H) liver microsomes (LM).
Data are from a single experiment (n = 1). N.D. = not determined.
We next explored substitution at various positions on the pyrrolidine and piperidine rings. Substitution at the pyrrolidine 4-position was tolerated, with the less basic analogs 12 (Table 2) and 23 both showing comparable potencies and improved permeability relative to 18. Of the two, the less lipophilic 4-fluoro derivative 12 was preferred due to its higher metabolic stability. Substitution at the pyrrolidine 2- and 5-positions was less well tolerated. As illustrated by compounds 24 and 25, incorporation of even small groups like methyl at these positions typically resulted in diminished potency, possibly due to steric hindrance around the site of critical H-bond formation. Transferring these learnings to the piperidine series, we prepared the 4-fluoro analog 26, and were gratified to find that it maintained the high permeability of parent piperidine 19 while also showing improved IRAK1 and IRAK4 inhibition.
With optimization at several positions complete, we conducted a final round of SAR studies to probe the effect of substitution at C7. As shown in Table 4, installation of a methoxy group at C7 consistently imparted improved IRAK1 inhibition. Addition of a C7 methoxy group to parent compounds 13, 16 and 30 afforded methoxy derivatives 27, 28, and 31, respectively. In each case, the C7-methoxy derivative displayed improved IRAK1 potency. In the C6-tertiary alcohol series, pyrrolidine-containing compounds 16 and 28 displayed good potency, but low to moderate membrane permeability (PAMPA values <100). We had previously observed that piperidine derivatives afforded improved permeability relative to their pyrrolidine counterparts. We thus prepared piperidine-containing analogs 29–31, and were pleased to find that they each displayed high passive membrane permeability as measured in the PAMPA assay, as well as moderate to high permeability as measured in the bidirectional Caco-2 assay. From this set, compounds 27, 29 and 31 displayed optimal potency and in vitro ADME properties, and were thus selected for advanced profiling.
Table 4. Potency and In Vitro ADME Properties of Optimized Analogs.

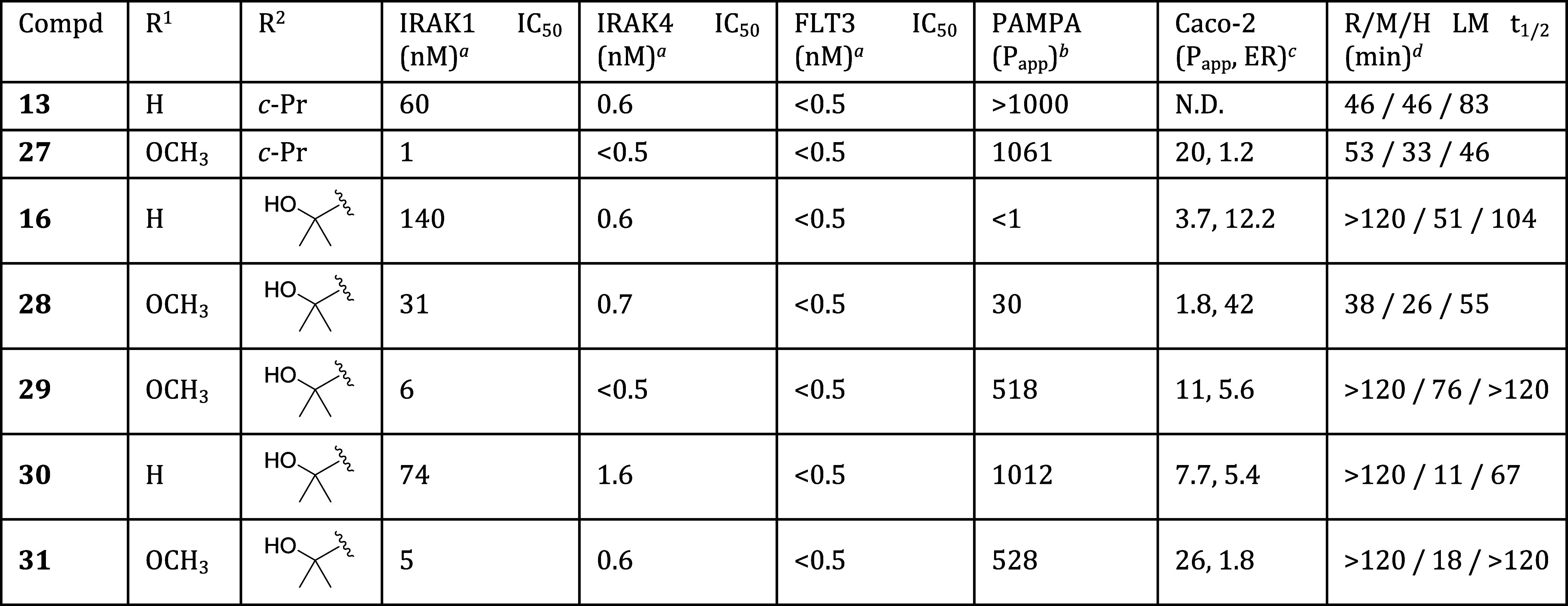
Data are the geometric mean of three independent experiments (n = 3) unless otherwise specified.
Permeability (Papp × 10–6 cm/s) as measured in PAMPA assay.
Permeability (Papp × 10–6 cm/s) and efflux ratio (ER) as measured in bidirectional Caco-2 assay.
Half-life of compound incubated with rat (R), mouse (M), or human (H) liver microsomes (LM).
Key compounds 27, 29 and 31 exhibited potent binding to IRAK1, IRAK4, FLT3, and a broad panel of FLT3 mutant isoforms. As shown in Table 5, compounds 27, 29 and 31 displayed highly potent binding to IRAK1, IRAK4 and FLT3, with low-nM or sub-nM Kds observed in all cases. These compounds also displayed highly potent binding to clinically important FLT3 (D835X) and FLT3 (ITD) mutant isoforms, including the (ITD, D835V) and (ITD, F691L) double mutants.
Table 5. Binding Potencies of Key Compounds.
| Compd | 27 | 29 | 31 |
|---|---|---|---|
| IRAK1 Kd (nM)a | 3.8 | 2.6 | 0.53 |
| IRAK4 Kd (nM)a | 0.44 | 0.25 | 0.096 |
| FLT3 Kd (nM)a | <0.17 | 0.22 | 0.074 |
| FLT3 (D835H) Kd (nM)a | 0.95 | 0.19 | 0.16 |
| FLT3 (D835V) Kd (nM)a | <0.17 | <0.17 | 0.034 |
| FLT3 (D835Y) Kd (nM)a | 1.1 | 0.20 | 0.10 |
| FLT3 (ITD) Kd (nM)a | 2.3 | <0.17 | 0.18 |
| FLT3 (ITD, D835V) Kd (nM)a | <0.17 | <0.17 | 0.017 |
| FLT3 (ITD, F691L) Kd (nM)a | <0.17 | <0.17 | 0.056 |
| FLT3 (K663Q) Kd (nM)a | 0.72 | 1.6 | 1.5 |
| FLT3 (N841I) Kd (nM)a | 0.81 | 0.58 | 0.24 |
| FLT3 (R834Q) Kd (nM)a | 0.71 | 0.74 | 0.33 |
Data are the geometric mean of two independent experiments (n = 2).
Key compounds from this series displayed potent activity in a variety of cell-based assays. As shown in Table 6, compounds 27, 29, and 31 were tested in NanoBRET assays that measure IRAK4, FLT3, and FLT3 (D835Y) potencies in a cellular context. These compounds all displayed high potency in the IRAK4, FLT3, and FLT3 (D835Y) NanoBRET assays, thus building confidence that the potencies previously observed in binding and functional biochemical assays would translate to in vivo settings.
Table 6. Cellular Potencies of Key Compounds.
| Compd | 27 | 29 | 31 |
|---|---|---|---|
| NanoBRET IRAK4 IC50 (nM)a | 0.096 | 2.5 | 0.90 |
| NanoBRET FLT3 IC50 (nM)a | 2.2 | 51 | 1.7 |
| NanoBRET FLT3 (D835Y) IC50 (nM)a | 0.009 | 0.41 | 0.22 |
| NF-κB (Pam3SCK4) IC50 (nM)b | 3c | 27 | 18 |
| NF-κB (IL1B) IC50 (nM)b | 6c | 38 | 13 |
| MOLM14 (D835Y) IC50 (nM)d | 3 | 7e | 7 |
| MOLM14 (F691L) IC50 (nM)f | 42 | 59 | 44 |
Data are the geometric mean of two independent experiments (n = 2).
Data are the geometric mean of three independent experiments (N = 3) unless otherwise specified.
Data are the geometric mean of four independent experiments (N = 4).
Data are the geometric mean of four independent experiments (N = 4) unless otherwise specified.
Data are the geometric mean of three independent experiments (N = 3).
Data are the geometric mean of two independent experiments (N = 2).
Key compounds also displayed potent and complete block of IRAK1/4-mediated signaling in a cellular NF-κB assay (Table 6). In this assay, either Pam3SCK4 or IL1B were used to initiate signaling at the TLR2 or IL1R receptors, respectively, and through the pathway mediated by IRAK1 and IRAK4. Production of NF-κB, which is downstream of IRAK1 and IRAK4, was then measured. Compounds 27, 29, and 31 all showed potent and complete inhibition of NF-κB production, and thus potent and complete block of signaling through the IRAK1/4 mediated pathway, when signaling was initiated with either Pam3SCK4 or IL1B.
Finally, key compounds were tested for their ability to inhibit the growth of MOLM14 AML tumor cells that contained FLT3 (D835Y) or (F691L) mutations. As shown in Table 6, compounds 27, 29, and 31 all potently inhibited the growth of these tumor cell lines, suggesting that they might prove efficacious in animal models and ultimately human patients whose cancers contain these mutations.
In Western blot experiments employing MOLM14 (D835Y) or THP1 cells, compounds 27, 29 and 31 inhibited phosphorylation of FLT3 or IRAK substrates at concentrations similar to their IC50s in the MOLM14 (D835Y) or NF-kB assays (see Supporting Information Figure S1 for details).
Compounds 28 and 31 displayed high selectivity for inhibition of key targets relative to other kinases. These compounds were tested for inhibition of 369 kinases (see Supporting Information for full details). In the same panel, the starting point for these efforts, compound 1, displayed only moderate selectivity. As shown in Figure 3, relative to its inhibition of IRAK4, 1 displayed >100-fold selectivity vs 55% of the 369 kinase panel, and <10-fold selectivity vs 21% of the panel. Optimized compound 31 proved more selective, exhibiting >100-fold selectivity vs 76% of the panel. Related compound 28 displayed >100-fold selectivity vs 85% of the panel, thus demonstrating the potential for even higher selectivity in this series.
Figure 3.

Kinase selectivity profiles of key compounds. For each off-target kinase, selectivity was calculated vs IRAK4 and represented as fold selectivity (IC50 of off-target kinase/IC50 IRAK4). Based on fold selectivity, each off-target kinase was placed in one of four bins (<10-fold, 10–100 fold, >100–1000 fold, >1000 fold). Percentages refer to percent of 369 kinase panel.
Select compounds were profiled in rat pharmacokinetic (PK) experiments. As shown in Table 7, compounds 27 and 28 displayed moderate stability when incubated in the presence of rat liver microsomes (RLM) or hepatocytes. These compounds also displayed suboptimal rat PK profiles that suffered from high plasma clearance rates and correspondingly low oral bioavailabilities and exposures. Compound 29 is more stable in RLM and hepatocyte incubations. It also exhibits an improved rat PK profile with reduced plasma clearance and increased oral exposure and bioavailability. Finally, compound 31 displays a markedly improved rat PK profile that boasts greatly reduced clearance and greatly improved oral exposure. Notably, the intrinsic clearance rates in RLM and hepatocytes are similar for compounds 29 and 31. The higher rate of plasma clearance observed for 29in vivo thus appears to result from a nonhepatic clearance mechanism that is attenuated in compound 31.
Table 7. In Vitro ADME and In Vivo Pharmacokinetic Properties of Key Compounds.
| |
In Vitro
ADME |
In
Vivo PKd |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Compd | Species | LM Clint (μL/min)a | Hep Clint (μL/min)b | PPB (% free)c | Dose (iv/po) (mg/kg) | F (%) | AUC (po) (μM*h) | Clp (mL/min/kg) | t1/2 (h) |
| 27 | Rat | 26 | 59 | 23.2 | 1/3 | 10 | 0.29 | 46 | 2.0 |
| 28 | Rat | 36 | 42 | 17.8 | 1/3 | 9 | 0.25 | 47 | 2.1 |
| 29 | Rat | <12 | 23 | 11.0 | 1/3 | 30 | 1.30 | 30 | 4.2 |
| 31 | Rat | <12 | 17 | 7.4 | 1/3 | 60 | 31.8 | 3 | 4.0 |
| 31 | Dog | N.D. | N.D. | 0.8 | 1/3 | 100 | 237.5 | 0.3 | 12.2 |
| 31 | Mouse | 77 | 65 | 15.0 | 1/3 | 25 | 1.39 | 120 | 2.4 |
| Gilteritinib | Mouse | 39 | N.D. | 9.9 | 1/10 | 57 | 3.89 | 44 | 4.1 |
Intrinsic clearance rate in liver microsomes (μL/min/mg protein).
Intrinsic clearance in hepatocytes (μL/min/106 cells).
Plasma protein binding, expressed as percent free (unbound) compound.
See Supporting Information for PK experiment details.
The pharmacokinetic profile of key compound 31 was also examined in dog and mouse. In dog, 31 exhibits low clearance, high oral exposure, and high oral bioavailability. In mouse, compound 31 displays a higher rate of clearance, with concomitantly lower oral exposure and bioavailability. Among the preclinical species examined, the weaker PK profile in mouse is thus the outlier.
In a mouse xenograft model of AML, compound 31 exhibited efficacy comparable to that of Gilteritinib, the leading marketed FLT3 inhibitor. In this model, NSG-SGM3 mice were intravenously xenografted with MOLM14 AML tumor cells containing FLT3 (ITD) and (D835Y) mutations. Compound dosing began 21 days post cell injection. Mice were dosed once daily Monday - Friday with either vehicle (PBS), 10 mg/kg IP compound 31, or 10 mg/kg IP Gilteritinib (n = 10 mice per dosing arm).13 As shown in Figure 4, the median survival of mice dosed with vehicle was 11 days from the start of dosing. In comparison, mice dosed with compound 31 survived a median of 33 days, and those dosed with Gilteritinib survived a median of 22 days. Compound 31 thus produces significant prolongation of survival relative to vehicle. Although the median survival time for mice dosed with 31 is longer than that of mice dosed with Gilteritinib, the difference is not statistically significant.
Figure 4.

Kaplan–Meier survival analysis of NSGS mice engrafted with MOLM14 (ITD, D835Y) cells and dosed with 10 mg/kg IP compound 31 or Gilteritinib.
In a separate PK arm of the study, NSG-SGM3 mice were dosed once daily Monday - Friday with 10 mg/kg IP compound 31 or Gilteritinib. Blood samples were collected 1 h postdose on dosing days 1, 5, 22 and 26, and 4 h postdose on day 5. The unbound plasma concentrations of compound 31 1 h postdose ranged from 112–127 nM. The unbound plasma concentrations of Gilteritinib were similar, albeit more variable, and ranged from 41–146 nM. Thus, at equivalent doses, and at similar concentrations, compound 31 produced prolongation of survival comparable to that of Gilteritinib.
Compound 31’s unbound plasma concentrations 1 h post -dose were 9–10x above the compound’s MOLM14 (D835Y) IC50 (13 nM) in the cell viability assay. On day 5, the unbound plasma concentration of 31 was 26 nM at 4 h postdose, or 2x above the target IC50. Based on those data, we estimate that unbound plasma concentrations of 31 were maintained at or above the target IC50 for the first 6–8 h of every 24 h period postdose. Factoring in the lack of dosing on Saturdays and Sundays, free drug plasma concentrations were likely maintained at or above target for at most 40 h per week. In that context, the significant prolongation of survival produced by 31 relative to vehicle is notable.
In summary, we have reported the discovery and optimization of a series of imidazopyridine IRAK-FLT3 inhibitors. Key compound 31 from this series displays potent and selective inhibition of IRAK1, IRAK4, and FLT3, good pharmacokinetic properties, and efficacy equal to that of Gilteritinib in a mouse xenograft model of AML. Further optimization in this series is ongoing, and will be reported in due course.
Acknowledgments
The authors thank Nishita Rao, Shane Biesecker, and Chris LeClair for assistance with compound purification.
Glossary
Abbreviations
- ADME
absorption, distribution, metabolism and elimination
- AML
acute myeloid leukemia
- BRET
bioluminescence resonance energy transfer
- FLT
FMS-like tyrosine kinase
- IRAK
interleukin 1 receptor associated kinase
- NF-κB
nuclear factor kappa B
- PAMPA
parallel artificial membrane permeability assay
- PK
pharmacokinetic
- SAR
structure–activity relationship.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.4c00269.
Synthesis schemes; representative procedures and characterization data for compounds 8, 10, 12–16, 27 and 31; kinase selectivity data for compounds 1, 28 and 31; procedures for biological assays, pharmacokinetic assays, and mouse xenograft assay (PDF)
Author Contributions
The manuscript was written by S.B.H. with contributions from all authors.
This work was supported in part by NIH (U54DK126108, R35HL135787, R01CA275007), Cincinnati Children’s Hospital Research Foundation, and Cancer Free Kids grants to D.T.S., and by NCATS grant 1ZIATR000044. This project has been funded in part with Federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261201500003I. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This contract number represents work performed within the scope of work of the nonseverable IDIQ contract.
The authors declare the following competing financial interest(s): D.T.S. serves on the scientific advisory board at Kurome Therapeutics; is a consultant for and/or received funding from Kurome Therapeutics, Captor Therapeutics, Treeline Biosciences, and Tolero Therapeutics; and has equity in Kurome Therapeutics. L.C.B. consulted for Kurome Therapeutics. J.R. is employed by, and holds equity in, Kurome Therapeutics; holds equity in Airway Therapeutics; and is a consultant for Radius Health and MoglingBio. G.G.-M. was employed by, and holds equity in, Kurome Therapeutics. A.K. is employed by, and holds equity in, Kurome Therapeutics. S.B.H. and C.J.T. are inventors on patent WO 2022026935. Their rights have been assigned to the U.S. government, but they may receive royalties on the patent. The remaining authors declare no competing financial interests.
Supplementary Material
References
- Fitzmaurice C.; et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived without disability, and disability-adjusted life-years for 29 cancer groups, 1990–2017. A systematic analysis for the global burden of disease study. JAMA Oncol. 2019, 5, 1749–1768. 10.1001/jamaoncol.2019.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levis M.; Perl A. E. Gilteritinib: potent targeting of FLT3 mutations in AML. Blood Adv. 2020, 4, 1178–1191. 10.1182/bloodadvances.2019000174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Cancer Institute (NIH-NCI) Website. https://seer.cancer.gov/statfacts/html/amyl.html (accessed 2023-10-06).
- Zhong Y.; Qiu R.-Z.; Sun S.-L.; Zhao C.; Fan T.-Y.; Chen M.; Li N.-G.; Shi Z.-H. Small-molecule Fms-like tyrosine kinase 3 inhibitors: an attractive and efficient method for the treatment of acute myeloid leukemia. J. Med. Chem. 2020, 63, 12403–12428. 10.1021/acs.jmedchem.0c00696. [DOI] [PubMed] [Google Scholar]
- Kennedy V. E.; Smith C. C. FLT3 mutations in acute myeloid leukemia: key concepts and emerging controversies. Front. Oncol. 2020, 10, 612880. 10.3389/fonc.2020.612880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight T. E.; Edwards H.; Meshinchi S.; Taub J. W.; Ge Y. FLipping” the story: FLT3-mutated acute myeloid leukemia and the evolving role of FLT3 inhibitors. Cancers 2022, 14, 3398–3423. 10.3390/cancers14143398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acharya B.; Saha D.; Armstrong D.; Lakkaniga N. R.; Frett B. FLT3 inhibitors for acute myeloid leukemia: successes, defeats, and emerging paradigms. RSC Med. Chem. 2022, 13, 798–816. 10.1039/D2MD00067A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melgar K.; Walker M. M.; Jones L. M.; Bolanos L. C.; Hueneman K.; Wunderlich M.; Jiang J.-K.; Wilson K. M.; Zhang X.; Sutter P.; Wang A.; Xu X.; Choi K.; Tawa G.; Lorimer D.; Abendroth J.; O’Brien E.; Hoyt S. B.; Berman E.; Famulare C. A.; Mulloy J. C.; Levine R. L.; Perentesis J. P.; Thomas C. J.; Starczynowski D. T. Overcoming adaptive therapy resistance in AML by targeting immune response pathways. Sci. Transl. Med. 2019, 11, eaaw8828. 10.1126/scitranslmed.aaw8828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett J.; Ishikawa C.; Agarwal P.; Yeung J.; Sampson A.; Uible E.; Vick E.; Bolanos L. C.; Hueneman K.; Wunderlich M.; Kolt A.; Choi K.; Volk A.; Greis K. D.; Rosenbaum J.; Hoyt S. B.; Thomas C. J.; Starczynowski D. T. Paralog-specific signaling by IRAK1/4 maintains MyD88-independent functions in MDS/AML. Blood 2023, 142, 989–1007. 10.1182/blood.2022018718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones L. M.; Melgar K.; Bolanos L.; Hueneman K.; Walker M. M.; Jiang J.-K.; Wilson K. M.; Zhang X.; Shen J.; Jiang F.; Sutter P.; Wang A.; Xu X.; Tawa G. J.; Hoyt S. B.; Wunderlich M.; O’Brien E.; Perentesis J. P.; Starczynowski D. T.; Thomas C. J. Targeting AML-associated FLT3Mutations with a Type I Kinase Inhibitor. J. Clin. Invest. 2020, 130, 2017–2023. 10.1172/JCI127907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compound syntheses and select bioassay data have been reported previously: Hoyt S. B.; Thomas C. J.; Finocchio C. J.; Starczynowski D. T.; Tawa G. J.; Rosenbaum J. S.; Gracia Maldonado G.. Multi-cyclic IRAK and FLT3 Inhibiting Compounds and Uses Thereof. WO 2022026935, Feb 3, 2022.
- As described in the Supporting Information, our PAMPA assay incorporates stirring, resulting in higher Papp values than nonstirred models. In this assay, Papp < 10 = low permeability, Papp 10–100 = moderate permeability, and Papp > 100 = high permeability.
- Mice were not dosed on weekends for logistical/staffing reasons.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.