

Abstract
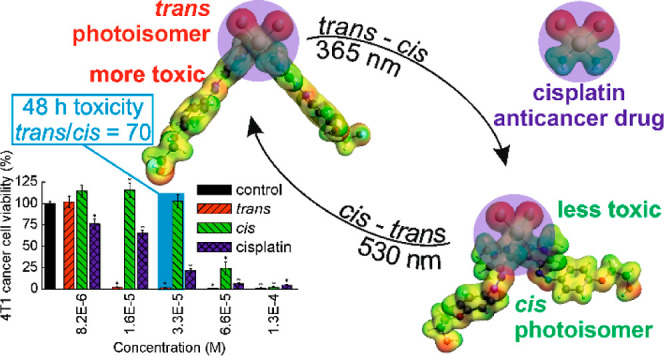
A photoactive analogue of cisplatin was synthesized with two arylazopyrazole ligands, able to undergo trans–cis/cis–trans photoisomerizations. The cis photoisomer showed a dark half-life of 9 days. The cytotoxicities of both photoisomers of the complex were determined in several cancer and normal cell lines and compared to that of cisplatin. The trans photoisomer of the complex was much more cytotoxic than both the cis photoisomer and cisplatin, and was more toxic for cancer (4T1) than for normal (NMuMG) murine breast cells. 4T1 cell death occurred through necrosis. Photoisomerization of the trans and cis photoisomers internalized by the 4T1 cells increased and decreased their viability, respectively. The cellular uptake of the trans photoisomer was stronger than that of both the cis photoisomer and cisplatin. Both photoisomers interacted with DNA faster than cisplatin. The trans photoisomer was bound stronger by bovine serum albumin and induced a greater decrease in cellular glutathione levels than the cis photoisomer.
Introduction
Cisplatin (cis-diamminedichloroplatinum(II), CDDP), a planar complex composed of a Pt(II) cation coordinating two chloride ions and two ammonia molecules, was first described by Michele Peyrone almost 200 years ago.1 In 1965, Rosenberg et al. found its strong bacteriostatic properties,2 but soon they also realized its anticancer potential. Its application in Swiss white mice with sarcoma resulted in the tumor remission and survival of the animals.3 After 6 months of the treatment, the mice did not show any signs of cancer. In 1978, cisplatin was approved by the FDA and a year later in Europe. Tests of over 4000 platinum compounds resulted in the introduction of two other Pt(II) complexes, carboplatin and oxaliplatin, into the clinic. Now cisplatin and other platinum-based chemotherapeutics are mainstay first-line anticancer drugs used to treat almost 50% of all cancers.4
The mechanism of the anticancer activity of cisplatin is well recognized. Cisplatin enters cells via passive diffusion through the cell membrane5 or is actively transferred by the copper transporter protein (CTR1).6 Inside the cell, Cl– ligands of cisplatin are replaced with water molecules forming a hydrated, positively charged complex. It reacts with two nucleobase moieties in the DNA, primarily in the same strand, forming a cyclic adduct7 which leads to cell apoptosis or necrosis.
Unfortunately, the therapeutic effects of cisplatin are hindered by its adverse effects, which can be both serious sequelae such as cardiotoxicity as well as milder symptoms such as nausea and vomiting.8 Also, the therapeutic efficiency of cisplatin is limited due to resistance to this drug, which results in the need to escalate the doses, aggravating adverse effects.
The possibility of improving the therapeutic effects and limiting the adverse effects of cisplatin is offered by the photopharmacological approach.9 It involves the application of a photoactive molecule, which itself is a drug (as opposed to the photosensitizers used in photodynamic therapy, PDT). It may be also attached to a drug molecule or may replace a structurally similar part of a drug molecule (the procedures known as “azo-extension” and “azologization”, respectively, in the case of azobenzene-based photoactive compounds10,11). The pharmacological activity of such photoactive systems can be modified by absorption of light followed by irreversible cleavage (termed as “uncaging”, “photoactivation”, or “photocleavage”) of a bond and release of an activated (or inactivated) species.12,13 Light may also trigger a reversible change in the shape of the molecule, a photoswitch (PS), and its physicochemical properties, which in turn may change (increase or decrease) its pharmacological activity.10,11 The opposite change of activity (decrease or increase, respectively) can be achieved by irradiation with light of another wavelength.
The discovery that the biological activity of the platinum compounds may be controlled with light has been made concurrently with the discovery of bacteriostatic activity of cisplatin by Rosenberg et al. who found that irradiation of a solution of PtCl62– and NH4+ ions with UV light leads to the formation of a mixture of different Pt(IV) complexes, of which cis-[PtCl4(NH3)2], i.e., cisplatin, shows bacteriostatic activity.14 Later, photoresponsible platinum compounds were synthesized based both as photocages and photoswitches.15 The first photoactive Pt(IV) compounds employing the former principle were the Pt(IV) iodides which upon irradiation for 6 h with >375 nm light released iodide ligands with the formation of a product that bound to DNA and was more cytotoxic to bladder cancer cells than the nonirradiated compound, bringing about a 22% enhancement in the antiproliferative activity.16 Pt(IV) photoactive complexes were extensively studied by Sadler et al. These compounds contained a diazido ligand which was released upon irradiation with a 420 nm light, thereby activating the molecule.17−20 Puddephatt et al. have thoroughly studied Pt(II) and Pt(IV) complexes, including those containing one photoisomerizable azobenzene-based moiety, however, not in the context of the photoswitchability of their biological properties.21−24 To the best of our knowledge, there are only a few papers on photoswitchable platinum complexes whose toxicity in cancer cells was studied. One of them describes a complex containing a photoswitchable 1,2-dithienylethene motif whose open and closed forms showed different cytotoxicity.25 Samper et al. obtained a Pt complex containing a photoswitchable azobenzene group; however, they did not find statistically significant differences between the cytotoxicities of both photoisomers.26
In this study, we have designed and synthesized a Pt complex that may be considered as an analogue of cisplatin in which both ammonia ligands were replaced with an arylazopyrazole (AAP)-based photoswitchable ligand (1) able to undergo trans–cis and cis–trans photoisomerizations, both with high yield. AAPs are a relatively new class of photoswitches offering several advantages over azobenzenes widely used so far.27,28 We have hypothesized that, on the one hand, the complex will be cytotoxic due to the presence of the cisplatin motif in its molecule (i.e., two hydrolyzable chloride ligands cis-coordinated to a Pt atom expected to give the complex the ability to cross-link DNA). On the other hand, the photoisomerization of two AAP ligands in the complex is accompanied by a remarkable change in the complex geometry, as shown by preliminary theoretical calculations. We expected that photoisomerization may result in a significant and reversible change in the complex toxicity which could be thus spatiotemporally controlled with light. We have positively verified these hypotheses in a series of in vitro experiments, including the determination of the cytotoxicity of the complex photoisomerized both before cellular uptake and upon internalization by the cells.
Results and Discussion
PS Design and Synthesis
A crucial step in the development of a photoswitchable cisplatin analogue was to design a PS ligand that could be easily coordinated by the Pt(II) ion to form a complex. It was assumed that the complex should significantly change its molecular shape and size and consequently physicochemical properties upon photoswitching, which would in turn result in the change of its biological properties. Finally, the photoswitch should have favorable photochemical properties, i.e., undergo fast, quantitative, and reversible photoswitching under irradiation with light of possibly long wavelength. Moreover, both photoisomers should be thermally stable in physiological conditions for a time long enough so that the difference in their biological properties, cytotoxicity in particular, could be detected, if any. Moreover, the PS should be nontoxic if released from the complex.
To fulfill as many as possible of these requirements, we considered arylazopyrazole (AAP)-based PSs as ligands. One of the two hetero nitrogen atoms in the pyrazole ring of AAPs can coordinate with the Pt(II) atom, forming a planar complex. The complexes formed between PtCl42– ions, used in our synthesis, and pyrazole and imidazole have preferentially cis geometry29−31 since the trans effect of pyrazole is weaker than that of the chloride anion.32 Thus, compounds containing pyrazole rings in their structure can form complexes which are analogues of cisplatin. On the other hand, it was recently found that pyrazolylazophenyl alkyl ethers, which are AAPs with phenyl ring substituted with alkoxy groups, have both photostationary states (PSS) composed of almost pure trans and cis photoisomers when irradiated with 365 and 530 nm light, respectively.33 Their thermally unstable cis photoisomers show exceptionally long half-lives (up to 3 months in organic media at room temperature).33 In fact, their trans–cis photoisomerization could be also achieved using a visible 400 nm light, although photoswitching was then not complete (PSS contained about 80% of the cis photoisomer33). Of potential importance were also a variety of beneficial pharmacological activities of pyrazole derivatives34 including antiviral,35 anti-inflammatory,35 antimicrobial,36 and, the most relevant from the point of view of this research, anticancer ones.37 Importantly, the N=N bond in AAPs, unlike that in the azobenzene-based PSs, shows excellent resistance to reduction with cellular glutathione (GSH).27
Photoswitch Structure and Properties
Considering the above facts, we have designed an AAP-based photoswitch (1, Figure 1a) with a –OCH2CH2OH substituent at the para position of the phenyl ring to achieve a long half-life of the cis photoisomer, characteristic of pyrazolylazophenyl alkyl ethers. The alkyl chain was terminated with a hydroxyl group and was possibly short to maximize the solubility of the complex in aqueous media. The pyrazolyl ring with a N atom substituted with a methyl group was used to additionally increase the half-life of the photoswitch (the presence of a N–H bond in unsubstituted pyrazole leads to fast cis–trans thermal isomerization through an azo-hydrazone tautomerization mechanism38). Moreover, the methyl group provided a steric hindrance close to the coordination center which had been found to protect the anticancer planar complexes of Pt(II) from the axial nucleophilic attack of the cellular thiols, GSH and metallothionein in particular, which could result in the complex inactivation and the development of drug resistance.39
Figure 1.

Structures of the (a) designed photoswitch (trans-1) with important structural elements responsible for its favorable photopharmacological properties, (b) (trans-2), and (c) cisplatin.
The synthesis of trans-1 was described in our previous work.40 Its structure was confirmed with EA, 1H NMR, 13C NMR,40 LC–MS,40 and UV–vis spectroscopy (see Table S1, Figure S3, and Figure S4 in the Supporting Information). trans-1 was soluble in water well enough to give measurable UV–vis spectra (solubility in water 0.58 mg/mL, see Figure S20). The yield of the 1 photoisomerizations was estimated based on 1H NMR spectra. The spectrum of trans-1 (nonirradiated) in DMF is shown in Figure S5a. The trans-1 solution was then irradiated with a 365 nm light to switch it to the cis configuration. The progress of the photoisomerization could also be followed using UV–vis spectra (Figure S4). The singlets of the pyrazole protons shifted significantly from δ = 8.40 and 7.91 ppm in trans-1 to 7.89 and 6.48 ppm in cis-1 (Figure S5b). The doublets of the phenylene protons shifted from 7.76, 7.74, 7.11, and 7.09 ppm in trans-1 to 7.10, 7.08, 6.78, and 6.76 ppm in cis-1, respectively. Since the pyrazole and phenylene proton signals of trans-1 are absent in the spectrum of cis-1, the trans–cis photoisomerization of trans-1 under 365 nm light can be considered as quantitative (100%). Irradiation of cis-1 with 530 nm light to reverse its configuration back to the trans one resulted in opposite shifts of the signals (Figure S5c); however, the presence of the residual signals of pyrazole and phenylene protons of cis-1 indicated that the cis–trans photoisomerization was not quantitative, but its yield, calculated based on the signal integration, was still very high (89%). Thus, the 1H NMR measurements confirmed the possibility to photoswitch 1 with high yield in both directions, which was a prerequisite for its application in the synthesis of the complex with photocontrollable biological activity.
Photoswitchable Complex of Pt(II) with 1 and Cl– as Ligands
1 was used as a ligand to obtain a Pt(II) complex by substituting two Cl– ligands in PtCl42– with two 1 molecules (Figure 1b). Dichloroplatinum Pt(II) complexes in either cis or trans configuration complexes can be obtained selectively, depending on the experimental conditions applied.29−31,41 The synthesis was based on the literature procedure,41 allowing formation of the dichloroplatinum Pt(II) complex with two highly substituted pyrazole derivative ligands in cis configuration which was proven by the authors with the Kurnakov test.42 The complex may be thus considered as an analogue of cisplatin (Figure 1c) with both ammonia molecules replaced with 1. The complex is hereafter referred to as 2, if the cis/trans configuration of 1 is irrelevant, while the complex, in which 1 is known to have cis or trans configuration, is denoted as cis-2 and trans-2, respectively. The complex structure was confirmed/characterized by 1H NMR, ATR-FT-IR, and LC–MS techniques (see Figures S5d,e,f and S6–S11). The EA results confirmed the assumed elemental compositions of 1 and 2 (Tables S1 and S2), which are significantly different.
In the spectrum of the nonirradiated complex (i.e., trans-2) in DMF-d7, the most intense signals correspond to those of trans-1 (compare Figure S5a,d). In the spectrum of the complex, both aromatic pyrazole singlets (at 8.81 and 8.79 ppm) and the methyl proton singlet (4.55 ppm) are significantly shifted downfield compared to their signals in trans-1. This is due to the proximity of the coordination center in the complex. On the other hand, the doublets of distant phenylene protons (7.76, 7.74, 7.12, and 7.10 ppm) and triplets of ethylene protons (4.15 and 3.85) are not shifted. The spectrum of the complex irradiated with a 365 nm light (i.e., cis-2) became more complicated (Figure S5e). Compared to the trans photoisomer of the complex, the pyrazole singlets in the cis photoisomer of the complex shifted upfield to 8.36 and 6.73 ppm, and the methyl singlet shifted upfield to 4.18 ppm. The position of these signals was shifted downfield compared to that of the respective signals of cis-1 due to the proximity of the coordination center. The phenylene doublets appear at 7.03, 7.01, 6.79, and 6.76 and are accompanied by smaller signals (see below). Their position and that of the ethylene protons is similar to that in the free cis-1 Based on the integration of the pyrazole proton signal of the cis and trans photoisomer (at 8.36 ppm and a residual signal at 8.79 ppm, respectively) in Figure S5f, the yield of the complex trans–cis photoisomerization could be estimated as at least 98%. After irradiation of the cis photoisomer of the complex with a 530 nm light, resulting in cis–trans photoisomerization, the positions of the main signals were the same as those of the nonirradiated complex (compare Figures S5f,d), except for the disappearance of the hydroxyl proton signal at 5.0 ppm. This confirms the possibility of achieving back-and-forth switching between trans and cis forms of the complex. The pyrazole proton signals of the cis photoisomer are completely absent in Figure S5f; thus cis–trans photoisomerization of the complex can be achieved with 100%, even higher than for 1 itself.
The presence of a singlet at 3.97 ppm in the spectrum of the nonirradiated complex (Figure S5d), characteristic of methyl protons of trans-1 (Figure S5a), indicates that the complex contained about 3.8 mol % (about 1.2 wt %) of trans-1 based on signal integration. The content of trans-1 in the complex solution irradiated first with 365 nm and then with 530 nm light increased up to 9 mol % (about 3 wt %) (Figure S5f). This indicates that the complex underwent photodissociation to a small extent with the formation of free trans-1 and the complex monosubstituted with trans-1 and DMF molecules as ligands, i.e., cis-PtCl2(trans-1) (DMF).43 The formation of cis-PtCl2(trans-1) (DMF) may also explain the presence of a small singlet accompanying the pyrazole singlets (8.84 ppm) and two small doublets accompanying phenylene doublets (7.84 and 7.82; 7.16 and 7.13 ppm) of the trans complex photoisomer (Figures S5d,f). The small doublets (7.10 and 7.08; 6.87 and 6.84 ppm) are also present close to the phenylene doublets of the cis complex (Figure S5e). The fact that the position of the small doublets shifted after both irradiations and that their relative intensity increased after irradiation confirms the assumption that they originate from a monosubstituted complex. However, this increase is difficult to quantify due to the signal overlap and its low intensity.
The ATR-FT-IR spectrum of the complex is dominated by bands of the trans-1 ligand (Figure S7). The trans-1 bands do not shift due to complexation, or shifts are very small and occur in both directions. The relative intensities of some of the bands of the complex changed, as observed in analogous Pt(II) complexes with N-heteroaromatic compounds.44 The bands characteristic of Pt–Cl stretching vibrations occur in the far-infrared region (450–100 cm–1),44 outside of the accessible wavenumber range of the apparatus. The LC chromatogram (Figure S8a) reveals a single dominant signal at a retention time of 5.73 min. In the mass spectrum of the compound showing this LC signal (Figure S8b), the peaks with the highest m/z values are grouped around m/z of 757–762 which agrees with the assumed molecular weight of the complex of 758.82 Da. The identity of the complex is additionally supported by the presence of groups of signals around m/z of 722–725 and 686–689, corresponding to the complex with one and two Cl atoms detached, respectively (Figure S8b), and by a signal at m/z of 247.13 corresponding to the detached protonated 1 ligand (molecular weight 246.27 Da). The presence of weak signals close to the complex signal and a weak signal at 4.67 min in the chromatogram, whose mass spectrum features a single signal at m/z of 247.26 (Figure S8c) corresponding to 1, may be due to the fact that the complexes of Pt(II) with pyrazoles may partially degrade on interaction with the stationary phase of the chromatographic columns, as observed by others.30,41 We also observed that the complex is susceptible to degradation in different analytical conditions (e.g., when analyzed using other columns, data not shown). The region of the HR-MS spectrum corresponding to the molecular ion of the complex showed an isotope pattern (Figure S9a) characteristic of a compound with one Pt and two Cl atoms (Figure S9b), confirming the predicted composition of the complex. In spite of intensive efforts, so far we have not succeeded in obtaining a single crystal of a quality sufficient for an XRD structural measurement (Figure S12).
Since the complex is poorly soluble in water (<3.7 μg/mL), it had to be introduced into the cell culture medium in DMSO (see the Cytotoxicity Tests section below). It is, however, well known that DMSO displaces ligands in Pt complexes, including cisplatin.45 Therefore, we decided to verify its stability in this solvent and 1% v/v DMSO in PBS. A 1.0 × 10–4 M solution of trans-2 in 100% DMSO was stored in dark Eppendorf probes, to protect it from daylight, at ambient temperature for 0, 3, 4, and 24 h from sample preparation and its LC chromatograms were recorded (Figure 2).
Figure 2.

HPLC chromatograms of trans-2 in 100% DMSO. The stability was measured after 0, 3, 4, and 24 h from sample preparation at ambient temperature. Peak identifications: 17.60 min—trans-2; 13.65 min—trans-1; 10.13 min—cis-1 (see Figure S10 in the Supporting Information for mass spectra).
Figure 2 suggests that trans-2 is unfortunately unstable in a 100% DMSO solution. Indeed, over time, the complex underwent degradation with the major formation of trans-1. Additionally, a small amount of the cis-1 ligand was observed (see Figure 2, the signal at 10.13 min). After 24 h from solution preparation, trans-2 decayed almost completely, despite protection from daylight. It was thus verified that in pure DMSO, the trans-1 ligands in trans-2 were substituted by the DMSO molecules. We therefore checked whether the complex was stable in PBS solution with 1% v/v DMSO. We found that the complex is completely stable under these conditions (Figure 3) which enabled the cytotoxicity tests to be performed.
Figure 3.

LC chromatograms of trans-2 in 1% v/v DMSO in PBS. The stability was measured after 0, 3, 4, and 24 h from sample preparation at ambient temperature. Peak identification: 17.60 min—trans-2; 13.65 min—trans-1.
Photoswitching of 2 Studied with UV–Vis Spectroscopy
UV–vis spectra of the trans-2 and cis-2 solutions in 1% v/v DMSO in PBS irradiated with 365 and 530 nm light are shown in Figure 4a,b, respectively.
Figure 4.

(a) UV–vis spectra of trans-2 and (b) cis-2 solution in 1% v/v DMSO in PBS (6.5 μg/mL, 8.6 μM) irradiated with 365 and 530 nm light, respectively. A spectrum of trans-1 in PBS normalized to the same intensity at the 346 nm band maximum is given for comparison; (c) relative absorbance at 350 nm of 2 undergoing cyclic irradiation with 365 and 530 nm for up to 5 cycles (0.0125 mg/mL, 16.5 μM, 1% v/v DMSO). The irradiation times were 30 and 300 s, respectively, long enough to reach complete conversion; (d) first-order kinetic analysis of cis-2 → trans-2 thermal isomerization in pH 7.4 PBS containing 1% v/v DMSO.
They confirm that irradiation with 365 and 530 nm light leads to effective and reversible trans–cis and cis–trans photoisomerization, respectively, of the 1 ligands. The comparison of the spectrum of trans-2 and that of trans-1 normalized at the maximum of the π → π* absorption band (Figure 4a) reveals that at longer wavelengths, the absorption of the complex is relatively stronger than that of the respective photoisomer of 1, resulting in a slight difference in color (yellow and orange, respectively) and, advantageously, much faster cis–trans photoisomerization of the complex compared to that of free trans-1 at the same irradiation conditions. It should be pointed out that photoisomerizations of both 1 ligands in the complex are independent, so both photoisomerizations proceed through an intermediate complex structure in which both ligands have different configurations, i.e., cis-PtCl2(trans-1)(cis-1). However, since isolating this intermediate unstable product would be very challenging while its photochemical and biological properties are of secondary importance at the current stage of this study, we disregarded this point in further studies.
From the point of view of photoreversibility of the complex structure and consequently its biological properties, an important question is the photochemical stability of its 1 ligands and resistance to side photoreactions such as degradation, cyclization, dimerization, etc. during possible multiple back-and-forth trans–cis photoisomerizations. To address this problem, the complex was subjected to cyclic photoisomerizations, and the changes in the relative absorbance at the maximum of the trans-2 absorption band (Figure 4c) were measured. The absorbance of trans-2 after 5 cycles decreased by 2.3% which means that the linearly extrapolated cyclability,10Z50, of the complex 1 ligand is about 116 cycles. Resistance to fatigue of 1 as a ligand is comparable to that of other AAPs described in the literature46 and thus much higher than that required in any conceivable photopharmaceutical application.
Another important prerequisite for the photopharmacological application is that both photoisomers of the complex should also be thermally stable. To determine the kinetic parameters of thermal (dark) cis-2 → trans-2 isomerization, the UV–vis spectra of cis-2 solutions were measured at 25 and 37 °C at pH 7.4. The absorption data fitted well to the assumed first-order kinetics (Figures 4d and S11). The values of the corresponding rate constants and half-lives obtained therefrom are given in Table 1.
Table 1. First-Order Rate Constants and Half-Lives for cis-2 → trans-2 Thermal Isomerization Obtained from the Data Shown in Figure 4D.
| 25 °C | 37 °C | |
|---|---|---|
| k [1/h] | 9.01 × 10–4 | 3.18 × 10–3 |
| τ1/2 [h/days] | 770/32 | 218/9 |
The thermal half-life of the cis photoisomer of the complex under physiological conditions is 218 h (9 days). This half-life is similar to that of the free cis-1 found in our previous work40 which means that the complexation of cis-1 does not significantly influence its thermal stability. Thus, the thermal stability of both photoisomers of the complex is high enough to enable detection of possible differences in their biological properties (half-lives of the order of a few hours were considered as already long enough to determine biological properties of much less thermally stable photoisomers36).
Results of Theoretical (Density Functional Theory) Calculations
To get some insight into the geometry of the ligand and the complex, electronic structure, photochemical properties, and stability of the complex, we have performed theoretical calculations with the density functional theory (DFT) method. In Figure 5, the optimized geometries of the models used in this study are shown. According to these calculations, the trans and cis isomers adopt significantly different geometries. Like in the case of trans-azobenzene, the aromatic scaffold of trans-1 is planar, which is the effect of conjugation of π electrons from the diazo bond and both aromatic rings. In contrast to the similar geometries of trans-azobenzene and trans-1, those of cis-1 and cis-azobenzene are quite different. Figure 5a shows that the pyrazole ring in cis-1 is situated in the same plane as the diazo bond, almost perpendicular to the benzene moiety. This result is in agreement with the X-ray diffraction data obtained by Weston et al.46 It implies low oscillatory strengths of the π → π* transitions in cis-1, resulting from a decrease in the conjugation and the symmetry of the orbitals with π character. On the other hand, in cis-azobenzene, the two phenyl groups attached to the azo bonds are too hindered sterically to assume the relative perpendicular conformation. As a result, their mutual conformation is twisted. Selected structural parameters of 1 and azobenzene as a reference photoswitch are given in Section ST2 in the Supporting Information.
Figure 5.

(a) Optimized geometries of trans-1 and cis-1 photoisomers, (b) molecular electrostatic potential (MEP) for trans-1 and cis-1 photoisomers (Δρ = 0.05), (c) optimized geometries of trans-2 and cis-2, and (d) MEP for trans-2 and cis-2.
The above-described structural changes occurring upon photoisomerization cause the differences in the charge distribution between the 1 photoisomers. The MEP for both 1 photoisomers has been depicted in Figure 5b. The color contours show that the main nucleophilic areas within both 1 photoisomers are located on the nitrogen atoms of the azo-bond, the unmethylated nitrogen atom of pyrazole, and two oxygen atoms in the ether moiety. The differences in the spatial arrangement of these areas between trans and cis photoisomers together with the heterogenicity of the charge distribution in both photoisomers additionally result in the significant difference in their dipole moments which are equal to 8.01 and 5.03 D for trans-1 and cis-1, respectively. The nucleophilic character of the pyrazole nitrogen atom is also an important chemical feature of 1 visible on the MEP contour map and is correlated with the ability of the moiety to coordinate the transition metals. The analysis of the photochemical properties of 1 and the comparison with those of azobenzene are given in the Supporting Information (Section ST2).
The optimization procedure of the complex geometry was performed for two systems in which the configuration of the 1 ligand was the same; therefore, the hypothetical situation when only one of the two 1 ligands undergoes photoisomerization was not taken into consideration (as decided above). Figure 5c presents optimized geometries of trans-2 and cis-2. There are remarkable structural differences between both photoisomers of the complex, which are the cause of the differences in their biological activity described further. The planar aromatic scaffold appearing in the optimized unbound trans-1 system is distorted in the complex and both ligands are no longer planar. However, it should be pointed out that ab initio MD results show that both conformers are stable at room temperature. The detailed analysis of structural parameters along with simulations has been discussed in the Supporting Information (Section ST3). MEP (shown in Figure 5d) qualitatively depicts the difference in the charge distributions of both photoisomers, resulting in the difference in their dipole moments. Similarly to the results obtained for unbound 1, trans-2 exhibits a notably larger dipole moment than cis-2 (Table 2). The values of relative energies, Erel, and solvation energies, Esolv, of trans and cis isomers are presented in Table 2. The trans-2 system has a lower electronic energy, which is mostly due to the higher stability of the diazo bond configuration in the ligand. However, the Esolv of the trans photoisomer is less negative, indicating weaker solvation in comparison to that of the cis photoisomer. A higher absolute value of Esolv, observed for cis-2 indicates a greater stabilizing effect of solvation resulting from stronger spatial exposition of the 1 nucleophilic groups. This was also demonstrated in the MEP visualization (Figure 5d).
Table 2. Comparison of the Selected Properties (Dipole Moment, |μD|, Relative Stability, Erel, and Solvation Energy, Esolv) Obtained from the DFT Calculations for Both Complex Photoisomers.
| trans-2 | cis-2 | ||
|---|---|---|---|
| |μD| [D] | 18.13 | 6.88 | |
| 0 | 20.92 | ||
| –46.09 | –55.24 |
The quantitative decomposition of the Pt–Cl and Pt–N bonds in the complex has been done via a two-ETS system scheme. ETS-1 provided the characteristics of the bonds between 1 ligands and the rest of the molecule, while ETS-2 corresponded to the interaction between chloride anions and platinum cation bonded to two 1 moieties (right panels in Figure 6).
Figure 6.

Differential density functions, Δρ, for the partitioning scheme depicting interaction between (a) 1 ligands and PtCl2 group and (b) chloride anions and Pt coordinated with 1 ligands (Δρ = 0.003).
The values of the energy decomposition terms, proposed by the Ziegler–Rauk energy decomposition analysis, are presented in Table 3. The interaction strengths, ΔEbond, between 1 ligands and the PtCl2 group for trans and cis complex photoisomers differ only slightly. The balance between steric and orbital interaction contributions elucidates the interaction character. In this partitioning, the electrostatic contribution is counterbalanced by the Pauli repulsion term, which is reflected in the small steric terms, similar for both photoisomers. The main stabilization comes from the orbital interaction contribution, ΔEorb. Qualitatively, the same results have been obtained for cisplatin. The donor character of this interaction and charge flow from the 1 ligand to the metal center is shown in Figure 6a. The second partitioning reflects the interactions between the Pt center and leaving group (Cl–) (Figure 6b) and expresses the tendency of the complex to undergo hydrolysis (substitution of chloride anions with water molecules), which is an important process activating Pt(II) compounds within cells. In trans-2, the interaction with the chloride leaving group is weaker than that in the cis-2 photoisomer (−225.78 and −228.29 kcal/mol, respectively). This difference is, however, too small to explain the observed differences in the toxicity between the complex photoisomers (see the following sections). The difference between cis and trans photoisomers of the complex is again the largest in the electrostatic contribution, which is equal to −217 and −222 kcal/mol, for trans-2 and cis-2, respectively. The energy terms obtained from the chosen energy decomposition approach were compared to the values obtained for cisplatin, indicating that in the complex, the 1 ligands are more strongly bound than the corresponding NH3 ligands in cisplatin. On the other hand, chloride ligands are less strongly bound in the complex than in cisplatin, suggesting that the complex has a stronger tendency for hydrolysis in the cells than cisplatin, which should contribute to its higher toxicity. The obtained values of the chloride ligand binding energy do not, however, explain the experimental observation that generally the trans and cis photoisomers are, respectively, more and less toxic than cisplatin (see the following sections).
Table 3. Calculated Values of ETS-Energy Decomposition Analysis Performed on the Considered Structures (All Values in kcal/mol per Ligand).
| ΔEorb | ΔEelstat | ΔEPauli | ΔEdisp | ΔEbond | |
|---|---|---|---|---|---|
| ΔEster | |||||
| ETS-1 | |||||
| Cisplatin | –47.97 | –107.22 | +117.65 | –1.97 | –39.51 |
| +10.43 | |||||
| trans-2 | –53.11 | –102.43 | +117.96 | –6.74 | –44.31 |
| +15.53 | |||||
| cis-2 | –54.48 | –108.89 | +124.92 | –6.88 | –45.33 |
| +16.03 | |||||
| ETS-2 | |||||
| Cisplatin | –79.22 | –269.88 | +85.72 | –1.31 | –264.68 |
| –184.16 | |||||
| trans-2 | –112.10 | –217.29 | +106.00 | –2.39 | –225.78 |
| –111.29 | |||||
| cis-2 | –113.03 | –221.82 | +109.00 | –2.44 | –228.29 |
| –112.82 | |||||
The energy decomposition calculations performed explain the character of the considered metal–ligand interactions. First, the bonding between the PtCl2 moiety and the 1 ligands was shown to be the charge-transfer one, as supported by the values of the ΔEorb term obtained and differential density visualizations. On the other hand, the interaction between the platinum cation attached to ligands with the chloride anions has an ionic character with strong polarization in the metal fragment. The Pt–Cl bonding in 2 is much weaker than that in cisplatin, which indicates a higher tendency for the Cl– dissociation and a stronger electrophilic character of 2. Additionally, the trans/cis conformation of the N-donating ligands does not impact the platinum center character; therefore, the photoisomerization should not significantly influence the reactivity of the metal complex center, as indeed found in the studies of the complex interaction with DNA (see below).
In Vitro Cytotoxicity of the Complex
The ultimate goal of the studies was to test whether the desirable significant difference exists between the toxicities of both photoisomers of the complex and how they compare to that of cisplatin. For this purpose, the cytotoxicity of both photoisomers of the complex was extensively tested in a few cell lines, murine and human, both cancer and normal ones, in a culture medium with and without the addition of 10% FBS (referred to later as serum- and serum-free conditions, respectively). The selected cell lines originated from organs whose cancers may be treated with cisplatin, i.e., breast cancer, melanoma, and ovary cancer. Only the NMuMG murine mammary normal cells could not be cultured in a completely serum-free medium, and to grow these cells, the addition of 1% FBS was necessary (referred to later as near-serum-free conditions). Before the cytotoxicity tests were started, it was not possible to predict which of the photoisomers would be more toxic. Moreover, literature data indicate that it is difficult to anticipate which photoisomer will be more active, even having some prior theoretical modeling data favoring one of the photoisomers.36 The metabolism of the complex upon uptake by the cells is unknown, and one should consider some release of the 1 ligand from the complex, as found from the 1H NMR study of the complex photoisomerization (Figure S5). Therefore, to rule out the possibility that the cytotoxicity of the complex may be due to the cytotoxicity of the released 1 ligand, the latter was determined in the B16-F10 murine melanoma, 4T1 murine mammary cancer, and NMuMG murine mammary cell lines, both in serum and serum-free conditions, after both 24 and 48 h of exposure (Figures S13–S15). Both photoisomers of 1 ligand were found nontoxic in all tested cell lines in serum conditions for up to 48 h. In serum-free conditions, cis-1 showed cytotoxicity that grew with time and was significantly different from those of the control and trans-1. However, in none of the cell lines, the cytotoxicity of the cis-1 photoisomer exceeded 50%.
The expected difference between the toxicities of complex photoisomers was indeed found in all studied cell lines, human and murine, normal and cancer ones, after both tested culture times (24 and 48 h) and irrespective of the presence of serum in the culture medium. Namely, it was the trans photoisomer of the complex that was always more toxic than the cis photoisomer and, importantly, also more toxic than cisplatin. In the B16-F10 murine melanoma cell line after 24 h of exposition (Figure 7A), the cytotoxicity of the trans photoisomer was significantly higher than that of the cis photoisomer at all concentrations and the ratio of the cell viabilities for the trans and cis photoisomers (hereafter referred to as the cis/trans viability ratio) grew with increasing concentration, reaching 2.7 at 1.3 × 10–4 M.
Figure 7.
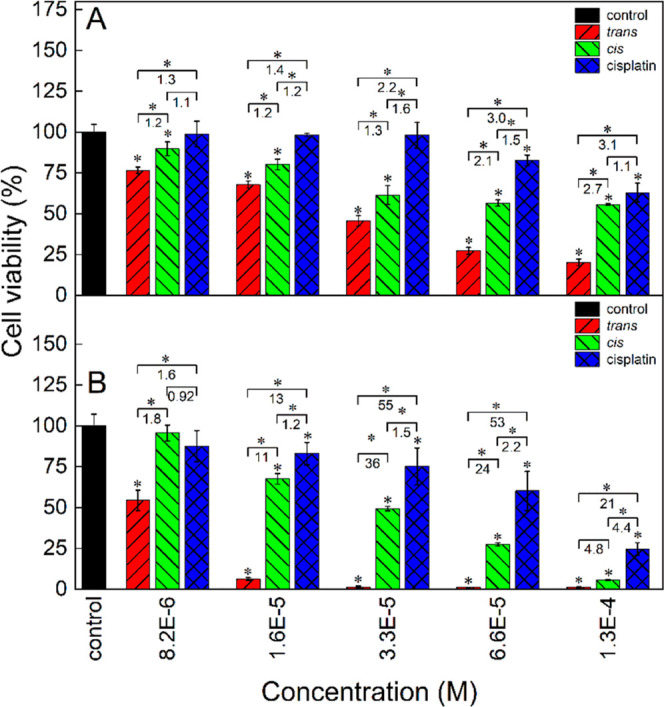
Cytotoxicity of trans-2, cis-2, and cisplatin in the B16-F10 murine melanoma cell line after 24 (A) and 48 h (B) of cell culture determined with the MTT test. The culture medium (DMEM–high glucose) was supplemented with 10% FBS. The numbers denote the cis/trans, cisplatin/cis, and cisplatin/trans viability ratios at respective concentrations. Statistically significant differences are indicated with asterisks above the bars (difference from the control) and above the brackets (difference between the photoisomers and between cisplatin and either of the photoisomers), *p < 0.05.
Both photoisomers were more toxic than cisplatin; the trans photoisomer was up to 3.1 times more toxic, while the cis photoisomer was up to 1.6 times. After 48 h of cell culture (Figure 7B), the differences among the cytotoxicities of the three species were much more pronounced than those after 24 h. At 3.3 × 10–5 M, the cis/trans and cisplatin/trans viability ratios reached maximum values as high as 36 and 55, respectively. The cisplatin/cis cytotoxicity ratio grew steadily with the concentration reaching 4.4 at 1.3 × 10–4 M.
In serum-free conditions (Figure S16), the relationships among the cytotoxicities of the three species were qualitatively similar; however, after 48 h, the cytotoxicity of the trans complex photoisomer was very high already at the lowest concentration of 8.2 × 10–6 M, much higher (13% viability) than in serum conditions (54% viability).
The opposite relationship between the cytotoxicities of both forms of 2 (trans photoisomer being always more toxic than the cis one) and 1 (the cis photoisomer more toxic than the trans one and significantly toxic only at longer culture time, at serum-free conditions, and at highest concentrations, Figures S13–S15) indicates that the cytotoxicity observed in the cells supplemented with the complex should be ascribed to the presence of the complex itself and not to the 1 ligand released due to irradiation.
The analogous cytotoxicity tests were performed in the 4T1 murine mammary cancer cells after 24 and 48 h of cell culture, and the cytotoxicities of both photoisomers were again compared to that of cisplatin as a reference drug (Figure 8A,B).
Figure 8.

Cytotoxicity of trans-2, cis-2, and cisplatin in the 4T1 murine mammary cancer (A,B) and normal NMuMG (C,D) cell line after 24 (A,C) and 48 h (B,D) of cell culture determined with the MTT test. The culture medium (DMEM—high glucose) was supplemented with 10% FBS. The numbers denote the cis/trans, cisplatin/cis, and cisplatin/trans viability ratios at respective concentrations. Statistically significant differences are indicated with asterisks above the bars (difference from the control) and above the brackets (difference between the photoisomers and between cisplatin and either of the photoisomers), *p < 0.05.
The cis/trans viability ratios for 24 h culture were even higher than in the B16-F10 cell line and ranged from 1.6 to 11 (Figure 8A). The cytotoxicity of the cis photoisomer was very similar to that of cisplatin with a cisplatin/cis viability ratio ranging from 0.7 to 1.1. The trans photoisomer was generally much more toxic than cisplatin, and the cisplatin/trans viability ratio ranged from 1.4 to almost 10. The differences between the toxicities of both photoisomers and between cisplatin and the trans photoisomer dramatically increased upon prolonging the exposition time to 48 h (Figure 8B). The cis/trans and cisplatin/trans viability ratios reached very high values of 70 at 3.3 × 10–5 and 28 at 1.6 × 10–5 M, respectively. After 48 h, the trans photoisomer killed almost all cancer cells, except for its lowest concentration (8.2 × 10–6 M), while the cis photoisomer was nontoxic at concentrations up to 3.3 × 10–5 M. Thus, there was a concentration window (between 8.2 × 10–6 and 6.6 × 10–5 M) where the trans photoisomer was toxic to almost 100% cells, while the cis photoisomer was completely nontoxic to these cancer cells. Beneficially, after 48 h of exposition, the cytotoxicity of cisplatin was found to be intermediate between the cytotoxicities of both complex photoisomers at most of the concentrations (except for the concentration of 8.2 × 10–6 M at which it was higher than the cytotoxicity of the trans photoisomer).
In serum-free conditions after 24 h, the results for 4T1 cells were qualitatively comparable to those obtained for 24 h culture time in serum conditions (compare Figures S17A and 8A). After 24 h, the cisplatin/cis cytotoxicity ratios ranged from 1.0 to 1.3, indicating that the cytotoxicity of the cis photoisomer was equal to or only slightly higher than that of cisplatin. The respective cisplatin/trans ratios ranged from 1.4 to 16, denoting much higher toxicity of the trans photoisomer than that of cisplatin. The ranges of cis/trans viability ratios in serum and serum-free conditions were similar, indicating much higher cytotoxicity of the trans photoisomer in both conditions.
For 48 h, the cytotoxicity of cisplatin in serum-free conditions was similar to that of the cis photoisomer (Figure S17B), in contrast to serum conditions (Figure 8B) at which it was higher. Like in the serum conditions, there was a concentration window at which the trans photoisomer killed almost all the cells and the cis photoisomer was nontoxic, although it was narrower than in serum conditions (between 8.2 × 10–6 and 3.3 × 10–5 M).
Cytotoxicity of cisplatin and both forms of the complex was also tested in the normal murine mammary cell line (NMuMG) both in serum (Figure 8C,D) and in near-serum-free conditions (with the addition of 1% FBS, Figure S17C,D). In the serum conditions, the toxicity of the trans photoisomer after 24 h was, like in previously described cell lines, higher than that of the cis photoisomer and the cis/trans viability ratios ranged from 1.3 to 2.2 (Figure 8C), so the cytotoxicities of both photoisomers did not differ as much as in the case of corresponding 4T1 cancer cells at the same culture conditions (Figure 8A). After 48 h of the cell culture, the cis/trans viability ratios for NMuMG cells increased (Figure 8D) compared to those after 24 h and ranged from 3.4 to 7.4, reflecting a greater difference in the toxicities of both complex photoisomers, but these ratios were still much lower than the respective ratios for the 48 h culture of the corresponding cancer cells (4T1, Figure 8B).
In near-serum-free conditions after 24 h of NMuMG cell culture, the toxicity of cisplatin was comparable to that of the cis photoisomer of the complex with cisplatin/cis viability ratios ranging from 0.68 to 1.0 (Figure S17C). The trans photoisomer was moderately toxic at the whole range of concentrations and was only up to 2.0 times more toxic than the cis photoisomer. After 48 h of NMuMG cell culture (Figure S17D), the trans photoisomer was very toxic at the whole range of concentrations, while the cis photoisomer was nontoxic up to 3.3 × 10–5 M, and at higher concentrations, it was very toxic. The toxicity of cisplatin did not change significantly with culture time except for the highest concentrations.
By comparing the respective data for cancer (4T1) and normal (NMuMG) murine breast cells, an important conclusion may be reached that the trans photoisomer was always more toxic for cancer than for normal cells, for both 24 and 48 h incubation times and for both serum- and serum-free conditions (compare red bars in Figure 8A,C, in Figure 8B,D, in Figures S17A,C, and in Figures S17B,D).
Preliminary measurements of the cytotoxicity of both photoisomers were also done in the A2780 human ovary carcinoma cell line (Figure S18) after 24 h of culture under serum-free conditions. Also in this case, the trans photoisomer was more toxic than the cis one and the cis/trans viability ratios were quite high reaching about 9.
The fact that the trans photoisomer was found to be always more toxic has another important implication. Since the cis–trans thermal isomerization of AAPs is strongly accelerated by acids (by up to 5 orders of magnitude47), it may be also expected that the cis–trans dark isomerization of the complex in vivo will be faster in the extracellular matrix of cancerous than of normal tissue because of the Warburg effect.48 Thus, the half-life of the cis photoisomer in cancer tissue may be shorter than 9 days. The potential consequences of this fact are yet to be determined in in vivo studies.
Cellular Accumulation of the Complex
The results of the above experiments could not answer the question of whether the observed significant difference in the cytotoxicity of both complex photoisomers could be due to a higher uptake of the trans photoisomer (assuming their similar toxicity), higher intrinsic toxicity of the trans photoisomer, or both factors. To resolve this question, we have measured the uptake of both complex photoisomers and cisplatin after exposing the 4T1 cells to these compounds for various times. The results are shown in Figure 9.
Figure 9.
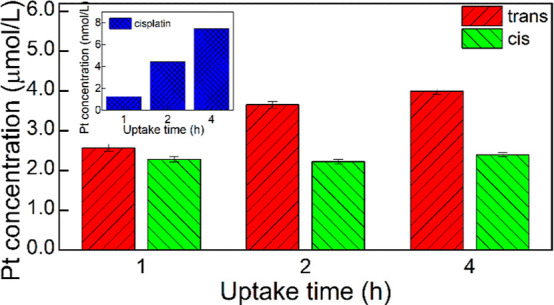
Pt concentration in the lysates of 4T1 cells incubated for various times with trans-2, cis-2, and cisplatin. Since the Pt concentration in the lysates of cells exposed to cisplatin was 3 orders of magnitude lower than in cells exposed to either of the complex photoisomers, the cisplatin bars would be not visible on the common Y-scale; therefore, the data for cisplatin are shown in the inset.
It was found that the uptake of both photoisomers is much stronger than that of cisplatin. This difference is particularly high for 1 h uptake time (Pt concentrations are 2.0 × 103 and 1.8 × 103 higher for trans-2 and cis-2, respectively, than for cisplatin) and decreases with uptake time (down to 5.3 ×102 and 3.2 ×102, respectively, for a 4 h uptake time). Also, the uptake of the trans photoisomer grew with incubation time while that of the cis one was completed within 1 h and did not change within 4 h. Importantly, the uptake of the trans photoisomer was higher after 1 h of incubation (by about 12%) and this difference increased with the incubation time, reaching 67% after 4 h. Thus, a significant and time-dependent difference in the uptake of both photoisomers was found, which probably contributed to the observed higher toxicity of the trans photoisomer.
Photoswitching of the Cytotoxicity of the Complex upon Uptake by the 4T1 Cells
Our next goal was to investigate if there are also differences in the intrinsic toxicity between the photoisomers, not related to the differences in their uptake. To answer this question, the 4T1 cells were incubated for different times with trans and cis photoisomers. Next, the medium was replaced with a clean one, the cells were irradiated in the incubator with 365 and 530 nm light, respectively, to induce trans–cis and cis–trans photoisomerization, respectively, and the cells were cultured for 48 h. Thus, the light-induced changes in the toxicity of only the complex that was internalized by the cells could be studied.
The trans–cis photoisomerization of the trans photoisomer resulted in a significant decrease in toxicity (18 and 8.5 times for the uptake time of both 0.5 and 1 h, respectively, Figure 10A). Conversely, the irradiation of the cells incubated with the cis photoisomer with 530 nm followed by its cis–trans photoisomerization resulted in a decrease in the toxicity (by 33 and 38% for the uptake time of 0.5 and 1 h, respectively, Figure 10B). It was also verified that doubling the irradiation time of the trans and cis photoisomers (to 10 and 400 s, respectively) did not statistically change the results (data not shown), which additionally excluded the influence of the toxicity of light on the results and confirmed complete photoisomerization of both complex forms within the cells. The data obtained indicate unambiguously that the trans photoisomer is intrinsically more toxic than the cis photoisomer and that it is possible to both activate and deactivate the complex with light. Also, we found that the effect of decreasing the complex toxicity by its trans–cis photoisomerization is much stronger than the effect of increasing the complex toxicity through its cis–trans photoisomerization in the cells. The smaller toxicity change upon irradiation for the cis photoisomer can be at least in part due to its lower uptake (Figure 9) and in part due to the photodissociation of the complex (Figure S5).
Figure 10.
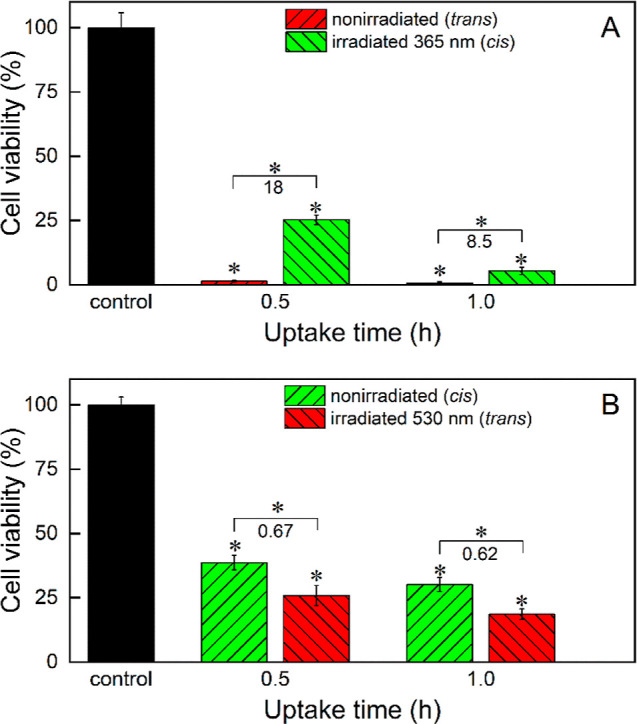
Cytotoxicity of (a) trans-2 and (b) cis-2 nonirradiated and irradiated upon internalization by the 4T1 cells after 48 h of culture in DMEM–high glucose determined with the MTT test. The cells were irradiated directly after complex uptake for 5 and 200 s to convert the trans photoisomer into the cis one and vice versa, respectively. The numbers denote the irradiated/nonirradiated viability ratios at a respective uptake time. Statistically significant differences are indicated with asterisks above the bars (difference from the control) and above the brackets (difference between nonirradiated and irradiated photoisomers), *p < 0.05.
Mechanism of Cellular Death Induced by the Complex
One of the most important questions that should be raised upon finding high toxicity of the complex concerns the mode of cellular death it induced. Cisplatin was reported to trigger mostly apoptosis,49−51 but it can also induce necrosis,52 or both.53 On the other hand, the pyrazole complexes of Pt(II), which are structurally related to 2, were found to cause apoptosis.54 The annexin V binding test carried out using 4T1 cells indicated that both 2 photoisomers caused necrosis of the 4T1 cells (Figure S19), with the number of necrotic cells being higher for the trans photoisomer. Since the type of cell death induced by Pt complexes may vary for different cancer cells and even within the heterogeneous collection of the cells within a tumor, further studies on the cell death mechanism in different cancer cells are required.
Interaction of the Complex and Cisplatin with DNA and Albumin Studied Using Circular Dichroism Spectroscopy
The impact of trans-2 and cis-2 on naked DNA from the calf thymus was analyzed using circular dichroism (CD) spectroscopy and compared to that of cisplatin. Adding the studied compounds to DNA causes significant changes in its CD spectra, as shown in Figure 11. The initial DNA spectrum is characteristic of the B form of the double helix with a minimum in the 250 nm band and a maximum in the 270 nm band. In the presence of both complex photoisomers and cisplatin, the minimum disappeared while the maximum was reduced in amplitude and red-shifted. These changes indicate that both the complex and cisplatin induce changes in the DNA structure,55 however, for the complex, they are more pronounced. On the other hand, in the range above 320 nm, in which 1 ligands absorb, no induced CD signal was observed; thus, no chirality was induced in the structure of the ligand. This may indicate that the part of the complex molecule involved in the interaction with DNA is that opposite to the 1 ligands, i.e., chloride (or more probably water) ligands. It may therefore be speculated that the nature of the interaction with DNA is the same for both the complex and cisplatin. However, there were significant differences in the kinetics of the interaction between the complex and cisplatin, as shown in the inset in Figure 11. For trans-2 and cis-2, the characteristic spectral changes were obtained already after 2 h, while for cisplatin after 20 h of incubation of the sample, the changes in the CD spectrum of the DNA were still less pronounced than for both complex photoisomers. On the other hand, the rate of the changes in the DNA structure caused by both complex photoisomers is very similar. Thus, the complex favorably shows much stronger and faster interaction with naked DNA than cisplatin. The higher rate of this interaction for the complex can be contrasted with another cisplatin analogue, AMD473, containing a 2-methylpyridine ligand, which was bound by DNA at a much slower rate than cisplatin.39 This difference is probably related to the difference in the electronic effects, thermal stability, and release of the 1N-methylpyrazole and 2-methylpyridine ligands under biological conditions.
Figure 11.
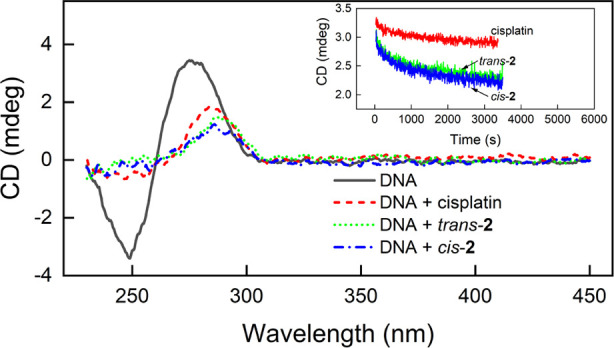
CD spectra of free calf thymus DNA (230 μM base pair) before and after its incubation with both complex photoisomers (2 h of incubation) and with cisplatin (20 h of incubation) as a positive reference. The concentration of all platinum compounds was 250 μM. Inset: the interaction kinetics tracked at 275 nm. All measurements were performed in 10 mM sodium phosphate buffer pH 7.4 at 25 °C.
Interaction of the Complex with Bovine Serum Albumin
Binding to the plasma proteins, mainly albumin, is an important aspect of cisplatin activity. It was thus important to verify whether 2 is also bound by this protein, as expected, and whether there are some differences in the binding of both photoisomers. The binding of trans-2 and cis-2 to BSA was monitored by measuring the CD spectra. The results are shown in Figure 12. Upon the addition of trans-2 to the BSA solution, the minimum centered at 370 nm on the spectrum appeared. In the case of cis-2, two minima centered at 330 and 400 nm appeared. The amplitudes of these extremes depended on the concentration of the added compounds.
Figure 12.
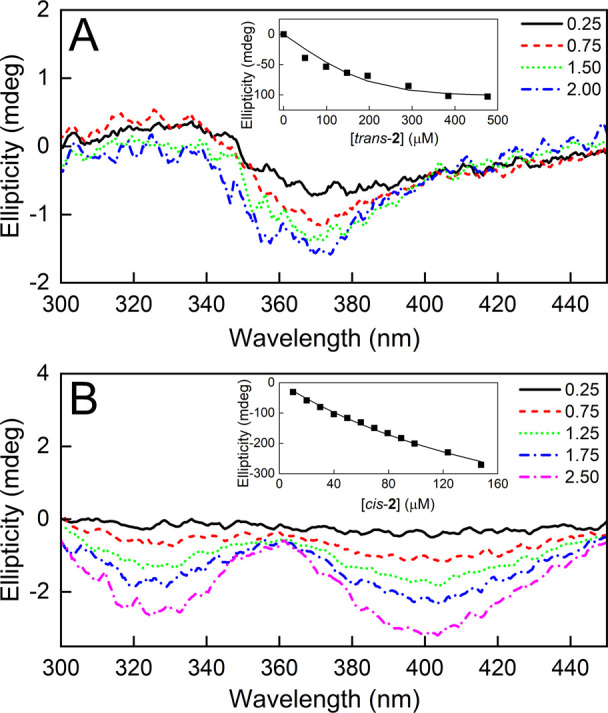
CD spectra of BSA complexes with trans-2 (A) and cis-2 (B). Measurements were done in PBS pH 7.4 at 25 °C. Numbers shown in the legends indicate the molar excess of 2 relative to BSA. Insets: exemplary dependence of the CD signal intensity relative to 2 concentration. Solid lines represent the best fit of the measurement data to the single-site binding model.
In the selected spectral range, BSA does not absorb light and the complexes do not have CD spectra (data not shown). Thus, the appearance of a CD signal indicates the interaction of both complex photoisomers with BSA. The CD signal obtained is of low intensity but sufficiently strong to allow affinity analysis. Global analysis of three measurements performed for different concentrations of reactants using a single-binding-site model yields satisfactory fits (examples of single titration with the fit are shown in the insets in Figure 12). According to this model, BSA binds trans-2 and cis-2 with KD values equal to 30 ± 15 and 184 ± 25 μM, respectively. It is generally assumed that the binding of cisplatin to serum proteins is irreversible; therefore, the relevance of the KD value obtained for the complex may seem questionable at first glance considering the structural analogy of the coordination center of both compounds. However, one should take into account that the irreversible binding of cisplatin to proteins occurs within a time scale of several hours, while for the first few hours, both irreversible and reversible binding mechanisms are expected.56 CD measurements indicate that the binding of the complex occurs within the time scale of seconds, and importantly, the complex binds to BSA through the ligand. Thus, the binding of the complex to BSA in the second time scale can be considered reversible. The fact that the trans photoisomer is more strongly bound indicates that, first, trans–cis photoisomerization of the trans photoisomer bound to BSA should result in its partial release from the complex with BSA (and probably also from the complex with HSA) and, second, the difference in binding of both photoisomers to BSA present in FBS added to the culture medium cannot be the cause of the difference in their cytotoxicity since the more strongly bound trans photoisomer is also more toxic for both cancer and normal cells cultured in the presence of FBS (in serum conditions) than the cis photoisomer. Also, there is no clear difference in the toxicity of the trans photoisomer between serum-free and serum conditions.
Influence of the Complex on the Glutathione and Cysteine Level
The level of cellular thiols, of which GSH is the most abundant, is an important factor influencing the anticancer activity of platinum-based drugs since they are deactivated by these compounds. The change in the level of cysteine (CYS) and both reduced (GSH) and oxidized (GSSG) forms of glutathione induced by the complex was studied in 4T1 cells. The data obtained are shown in Table 4.
Table 4. Levels of Reduced and Oxidized Glutathione and Cysteine (nmol/mg Protein) in the 4T1 Cells Treated with the Complex.
| CYS | GSH | GSSG | |||
|---|---|---|---|---|---|
| control | 3.0 ± 0.6 | 65.0 ± 5.3 | 0.94 ± 0.2 | 70.8 ± 11.9 | |
| cis-2 | 3.7 ± 0.5 | 38.3 ± 2.3 | 0.32 ± 0.1 | 120.1 ± 16.5 | |
| trans-2 | 7.3 ± 0.8 | 29.0 ± 3.0 | 0.59 ± 0.02 | 52.9 ± 6.6 |
A decrease in GSH (greater for the trans complex) and GSSG (greater for the cis complex) levels was significant for both photoisomers of the complex. However, the level of GSH was reduced more by the more toxic trans photoisomer of the complex (down to about half of its control level). It is an important observation since, on the one hand, the reaction of cisplatin with GSH is one of the mechanisms of its deactivation, and, second, the concentration of GSH in cisplatin-resistant cancer cells is often increased compared to nonresistant ones.57 It is known that GSH actively participates in the metabolism of xenobiotics, mainly by forming conjugates with electrophilic metabolites and scavenging free radicals formed in the cells.58 The cause of this decrease in GSH is unknown, but it may likely be the result of, for example, a direct reaction of GSH with the complex. It may also be the result of the detoxification of reactive metabolites formed in cells during the metabolism of the tested compounds or the sum of the above-mentioned factors. As the cysteine residue of GSH can be readily oxidized nonenzymatically to disulfide (GSSG), the level of the latter compound was also determined, and it was found that the level of GSSG was decreased more profoundly by the cis photoisomer of the complex (down to about one-third of its control level). The level of reduced GSH is higher after the addition of the cis photoisomer, and the level of GSSG is lower compared to the test group treated with the trans photoisomer. Consequently, the ratio of GSH/GSSG, the major redox couple that determines the antioxidative capacity of the cells59 which is high when the redox state in cells is maintained, is more favorable for the cis complex photoisomer than for the trans one. The GSH/GSSG ratio of the trans photoisomer is the lowest among those analyzed, which may be partially responsible for its higher toxicity. The lowest GSH/GSSG ratio reflects poorer cellular conditions for maintaining the redox state; the equilibrium is shifted toward the formation of GSSG, which may contribute to the development of oxidative stress in the cells. On the other hand, in cells treated with the trans photoisomer, the CYS level significantly increased compared to the control, while for the cis photoisomer, its increase was not considerable. The bioavailability of CYS is a limiting factor for the synthesis of new GSH molecules. However, the 2-fold increase in CYS level in the trans photoisomer group compared to the control and cis photoisomer groups does not imply that the excess CYS is used for GSH synthesis (and probably is not used as the GSH level in the trans photoisomer group is the lowest). In this case, CYS is probably diverted to other metabolic pathways or is a substrate for the formation of compounds such as metallothioneins, thioredoxin, or coenzyme A.
Conclusions
A photoactive cisplatin analogue was synthesized which is a square planar Pt(II) complex with two chloride ligands and two photoswitchable arylazopyrazole (AAP) ligands in the cis configuration in the coordination center. The complex efficiently undergoes trans–cis and cis–trans photoisomerizations when irradiated with 365 and 530 nm light, respectively. Photoisomerization is accompanied by significant changes in the molecule geometry and, consequently, in complex toxicity versus several cell lines, both normal and cancer ones (B16-F10, 4T1, NMuMG, and A2780). The trans photoisomer was found to be always more toxic than the cis one and also always more toxic than cisplatin. It was also more toxic to cancer 4T1 cells than to normal cells of the murine mammary gland. The uptake of the trans photoisomer by 4T1 cells was stronger than that of the cis photoisomer, while both photoisomers were taken up much more strongly than cisplatin. The irradiation with 365 nm light of the trans photoisomer internalized by the 4T1 cells resulted in a decrease in the toxicity of the complex, while the irradiation of the internalized cis photoisomer with 530 nm light resulted in an increased toxicity. The complex caused cellular death in the necrotic mechanism.
Experimental Section
Reagents and Materials
K2PtCl4 (98%, Angene), cisplatin (99%, AmBeed), acetonitrile (POCH), formic acid (Sigma-Aldrich), DMSO (Sigma-Aldrich), pH 7.4 PBS tablets (Sigma-Aldrich), BSA (Acros), MTT reagent (3[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide, AmBeed), Dulbecco’s modified Eagle’s medium—high glucose (DMEM, Sigma-Aldrich), RPMI-1640 cell culture medium (Sigma-Aldrich), fetal bovine serum (FBS, Sigma-Aldrich), l-glutamine (99.0–101.0%, Sigma-Aldrich), insulin (human, Sigma-Aldrich), and trypsin–EDTA solution (Sigma-Aldrich) were the reagents used.
Synthesis of trans-2
Synthesis of (E)-2-(4-((1-methyl-1H-pyrazol-4-yl)diazenyl)phenoxy)ethan-1-ol (trans-1) was performed according to the procedure described in our previous paper.40 The synthesis of trans-2 (Figure S1) was performed based on modified literature procedures.29−31,41 Briefly, 2.31 g (9.35 mmol) of trans-1 was suspended in 300 mL of methanol, added to the solution of 1.96 g (4.72 mmol) K2PtCl4 in 150 mL of water, and mixed at room temperature for 48 h. Then, half of the volume of methanol was removed under reduced pressure. The precipitated product was centrifuged (5 min, 10 000 rpm), and the supernatant was decanted. 5 mL of water was added, the suspension was mixed, and the supernatant was removed. The washing procedure was repeated thrice. Next, the product was washed seven times with 5 mL of diethyl ether and dried at 22 °C for 24 h in a vacuum oven. Yield: 1.92 g (55%). Elemental analysis (%): theoretical: C, 38.00; H, 3.72; N, 14.77; experimental: C, 38.25; H, 3.98; N, 14.21. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.16 (s, 2H), 8.07 (s, 2H), 7.77 (d, J = 9.0 Hz, 4H), 7.00 (d, J = 9.0 Hz, 4H), 4.49 (s, 6H), 4.16 (m, 4H), 4.06–3.93 (m, 4H) (see Figure S6 in the Supporting Information). FT-IR bands ν (cm–1), trans-1/trans-2 (Figure S7): 3415/3292 (O–H stretching), 3113/3124 (C–H stretching, aromatic), 2937/2940 (C–H stretching), 1596/1600, 1543/1536 (C–C aromatic), 1499/1497 (N=N stretching), 1446/1444 (C–H bending), 1246/1246 (C–O stretching), 1024/1019 (C–O – C stretching). HR-MS (ESI+) m/z calcd for C24H28Cl2N8O4Pt [M + Na]+, 781.51; found, 781.11.
All compounds are >95% pure by elemental and NMR analyses (since the Pt complexes are susceptible to decomposition on a chromatographic column, HPLC is not the optimal method for the determination of their purity).
Apparatus
NMR spectra of both photoisomers of 1 and 2 were measured in CDCl3 or DMF-d7 using either a 400 or 600 MHz spectrometer (JEOL). FT-IR spectra of trans-1 and trans-2 were collected by using a Nicolet iS10 spectrophotometer (Thermo Scientific). UV–vis spectra were recorded using a Varian Cary 50 UV–vis spectrophotometer (Agilent Technologies). HPLC analyses were carried out with an HPLC Dionex UltiMate 3000 equipped with a diode array (Sunnyvale, CA, USA). The analyses were carried out using an analytical Wakopak Handy ODS column (150 × 4.6 mm) and a gradient elution. Mobile phase A was 0.1% HCOOH in water, and mobile phase B was 80% ACN in water (v/v). A linear gradient from 0 to 70% of mobile phase B in 20 min, at a flow rate of 1 mL/min, was applied. LC–MS measurements were performed using a UPLC-MS/MS system composed of ACQUITY UPLC and TQD mass spectrometers, and a TripleTOF 5600+ (SCIEX, Framingham, MA, USA) high-resolution mass spectrometer coupled to a UPLC system (Nexera X2, Shimadzu, Canby, OR, USA). The HR-MS measurement was performed using a MicroTOF II mass spectrometer (Bruker, Bremen, Germany) equipped with a time-of-flight (TOF) analyzer. The MS detection was performed using the positive ion mode (ESI+), and the profile spectra were acquired within the mass range of 50–3000 m/z. The ESI conditions were as follows: nebulizer pressure 0.4 bar, dry gas 4.0 L/min heated up to 180 °C, and capillary voltage −4500 V. Mass calibration was carried out using sodium formate clusters according to the procedure given by a manufacturer. Data were collected by Compass DataAnalysis 3.2 software (Bruker). Expected ions attributed to analytes were predicted by the IsotopicPattern software (Bruker, Germany). Before the analysis, samples were dissolved in acetonitrile (LC–MS grade, Honeywell). The solutions were irradiated with 365 and 530 nm LED plates (Bio Research Center, Japan). Cytotoxicity was measured using a microplate reader (Synergy HTX, BioTek, Winooski, VT, USA).
Circular Dichroism Spectroscopy
All CD measurements were taken using a J-710 spectropolarimeter (JASCO Co., Japan) equipped with a circulating water bath (JULABO Labortechnik GmbH, Germany). Spectra were recorded at 25 °C with a data pitch of 0.5 nm and a bandwidth of 2 nm. Each spectrum was the result of three averaged scans. The spectrum of the appropriate solvent was subtracted.
Interaction of 2 with BSA
BSA solutions were titrated with both photoisomers of the complex in PBS using an automatic pipette. A 10 mM solution of the complex in DMSO was used for titration. The initial protein concentration ranged between 40 and 200 μM. The concentration of the complex increased by 10–50 μM per each titration step. After each addition of the titrant, the CD spectrum was measured in the range of 300–450 nm with an optical path length of 0.1 or 1 cm. The scanning speed was 100 nm/min. The binding constant was determined from changes in the summed signal in the range of 350–450 nm. The analysis was performed by using the one-binding-site model assuming a 1:1 interaction stoichiometry. Global analysis of three independent titrations was performed assuming a shared value of the equilibrium constant (KD) using OriginPro software (Version 2021b, OriginLab Corporation, Northampton, MA, USA).
Interaction of 2 and Cisplatin with DNA
To a 230 μM bp solution of calf-thymus DNA in 10 mM sodium phosphate, pH 7.4, 10 mM solutions of both photoisomers of the complex and cisplatin in DMSO were added to a concentration of 250 μM. CD spectra were measured over time until a stable signal was obtained. For the analysis of interaction kinetics, the measurement was carried out in duplicate by recording the signal at 275 nm for 1 h with a resolution of 5 s at 25 °C. The process was initiated manually by adding a portion of the compound using an automatic pipette.
Cytotoxicity Tests
Cytotoxicity studies of the resulting photocomplex and cisplatin were performed on the normal and cancer cell lines: murine mammary normal (NMuMG, ATCC CRL-1636), murine melanoma (B16-F10, ATCC CRL-6323), murine mammary cancer (4T1, ATCC CRL-2539), and human ovary cancer (A2780, 93112519, Merck). Cells were cultured on Petri dishes in a culture medium (for A2780: RPMI-1640 + 2 mM glutamine, for the rest: DMEM (high glucose, for NMuMG extra 10 μg/mL insulin was added) supplemented with 10% v/v fetal bovine serum (FBS), at 37 °C in a humidified atmosphere containing 5% v/v CO2. The cells were then seeded in 96-well plates and cultured for 24 h. After this time, the cells were treated with both photoisomers of 1, 2, or cisplatin in 1% v/v solution of DMSO in the medium (obtained by diluting the solution in DMSO, with suitable serum content) at different concentrations and incubated for 24 or 48 h. The solution of the complex in DMSO was always diluted with PBS immediately after preparation and promptly supplemented to the cell cultures. Following removal of the media, a 100 μL solution of MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) in the medium to each well was added and kept in the dark for 4 h at 37 °C. Then, the MTT solution was gently aspirated and 100 μL of DMSO/isopropanol 1:1 v/v was added to each well. After dissolving the crystals of purple formazan, the absorbance at 570 nm was measured using a plate reader. Measurements were made in triplicate, *p < 0.05.
Annexin V Binding Assessment
Annexin V-Cy3 Apoptosis Detection Kit (Merck) was used following the manufacturer’s protocol to determine the mechanism of cellular death caused by both photoisomers. Briefly, the 4T1 cells were seeded on coverslips in 6-well plates and incubated with both photoisomers at the concentration of 33 μM for 1 h at 37 °C. Next, cells were washed twice with 1 mL of PBS and three times with 50 μL of binding buffer (1 mM HEPES pH 7.5, 14 mM NaCl, and 0.25 mM CaCl2). Then, 50 μL of the double-label staining solution (1 μg/mL of AnnCy3 and 500 μM of 6-CFDA in binding buffer) was added, and cells were incubated for 10 min at RT. After staining, cells were washed five times with 50 μL of binding buffer, and coverslips were mounted on glass slides and sealed for confocal imaging. Fluorescent images were acquired using an A1-Si Nikon (Tokyo, Japan) confocal laser scanning system coupled with a Nikon inverted microscope Ti-E and processed using NIS-Elements AR 3.2 software (Nikon Europe BV, Amsterdam, The Netherlands).
Determination of Low-Molecular-Weight Cellular Thiols
The 4T1 cells were seeded in Petri dishes and cultured for 72 h to 90% confluency. After this time, the cells were treated with both photoisomers of 2 in 1% v/v solution of DMSO in the medium (without serum) at a concentration of 11.8 μM and incubated for 2.5 h. The control experiment was performed with cells treated with a 1% v/v solution of DMSO in the medium. For RP-HPLC analysis, pellets of cells (3.5–5 × 106 cells) were suspended in 0.25 mL of 0.9% NaCl/70% perchloric acid/1 mM bathophenanthroline–disulfonic acid disodium salt and sonicated three times for 5 s at 4 °C. The sediment was separated by centrifugation at 1600 g at 4 °C for 10 min, and the supernatant was stored at −80 °C until analysis. The levels of low-molecular-weight thiols, i.e., reduced (GSH) and oxidized (GSSG) glutathione, and cysteine, in the incubation mixtures were determined using the RP-HPLC method of Dominic and others60 with modifications described by Bronowicka-Adamska and others.61 The samples were separated on a 4.6 mm × 250 mm Luna C18 (5 μm) column (Phenomenex, Warsaw, Poland) with a Phenomenex Security Guard column filled with the same packing material. The chromatographic system consisted of LC-10 Atvp Shimadzu Corp. (Kyoto, Japan) pumps, four channel degassers, a column oven, a Shimadzu SIL-10 Advp autosampler, and a Shimadzu Corp. SIL-10 SPD-M10Avp-diode array detector. LabSolutions LC software (Shimadzu, Kyoto, Japan) was used for system operation and data collection.
Protein Content Determination
The pellets of cells (3.5–5 × 106 cells) were suspended in 0.1 M phosphate buffer, pH 7.5, in proportion to 1 million cells/0.04 mL of the buffer and sonicated three times for 5 s at 4 °C. After centrifugation at 1600 g at 4 °C for 10 min, the supernatant was used for the determination of protein content. Total protein content was determined by the method of Lowry and others.62 The crystalline BSA was used as a standard.
Uptake of 2 and Cisplatin by 4T1 Cells
The 4T1 cells were seeded in 6-well plates and cultured for 24 h. After this time, the cells were treated with both complex photoisomers and cisplatin as a reference drug at the concentration of 66 μM and incubated for 1–4 h in the dark at 37 °C. Cells incubated in the medium with 1% v/v DMSO in PBS served as a negative control. After that time, the medium containing the respective Pt compound was removed, the cells were washed three times with PBS, harvested by trypsinization, and centrifuged, and the supernatant was removed. The samples of cellular pellets were mineralized using concentrated nitric acid and a high-pressure mineralization technique supported by microwave radiation (Anton Paar, Ultrawave 3000). Obtained digests were diluted with deionized water up to 10 mL, and platinum was determined using an ICP–MS spectrometer (PerkinElmer, Nexion 5000). All experiments were conducted in triplicate. *p = 0.05.
Theoretical Calculations
DFT calculations were carried out for both photoisomers of 1 ligand and 2. All structures have been optimized with the Amsterdam density functional (ADF) program, version 2014.07.63 For each structure, B3LYP as an exchange–correlation functional64,65 was applied. The evaluation of the functional has been previously performed (see Section ST1 in the Supporting Information). The Grimme dispersion correction was used.66 Solvation effects were modeled with the conductor-like screening model (COSMO).67 The zero-order regular approximation (ZORA) to the Dirac equation was applied.68 For all of the atom types in the system, the triple-ζ basis set (TZP) was used. To visualize the results of geometry optimization, such as electronic features of considered systems like molecular electrostatic potential (MEP)69 or molecular orbitals (MOs), the ADF View package was used. Bonding between ligands and metal centers was analyzed with the Ziegler–Rauk bond energy decomposition scheme (ETS method).70,71 In this approach, the overall bonding energy, ΔEbonding, was calculated as ΔEbonding = ΔEorb + ΔEsteric + ΔEdisp = ΔEorb + (ΔEelstat + ΔEPauli) + ΔEdisp, where ΔEbonding is the total interaction energy of fragments (in the geometry of the complete interacting system). The orbital interaction term, ΔEorb, describes the energetic effect related to the charge transfer between the fragments and their internal polarization (thus, leading to the formation of the optimized orbitals of the whole system), and the ΔEdisp is the dispersion correction based on the Grimme approximation. ΔEsteric can be decomposed into the electrostatic term, ΔEelstat, describing the electrostatic interaction between the unperturbed fragments and the Pauli repulsion term, ΔEPauli (mutual orthogonalization of the fragment orbitals, without any charge transfer between the fragments).
The CP2K software was used for MD simulations.72 The DFT methodology was chosen as an approach to electronic structure evaluation using the Quickstep procedure scheme73 with PBE as an exchange–correlation functional74 and the Grimme3 dispersion correction. The DZVP-MOLOPT-GTH basis set75 was applied for all atoms except Pt. Platinum was described by a short-range version basis set, DZVP-MOLOPT-SR-GTH.75 The cutoff for plane waves was setup as 350 Ry.
The simulation was carried out in a canonical ensemble (NVT). The time step of the simulation was 1 fs, and the temperature was set to 300 K. The full length of the simulation for both considered compounds and their photoisomers was 25,000 fs, and to obtain a relatively constant temperature, the Nose–Hoover thermostat was applied.76 All simulations obtained from the applied methodology were visualized and analyzed using the Visual Molecular Dynamics (VMD) software.77
Acknowledgments
The authors gratefully acknowledge the financial support from the Polish National Science Centre, grant no. UMO-2021/41/B/NZ7/02484. The Strategic Programme Excellence Initiative at Jagiellonian University has supported the maintenance and service costs of the scientific equipment.
Glossary
Abbreviations used
- 4T1
cancer murine breast cells
- 6-CFDA
6-carboxyfluorescein diacetate
- AAP
arylazopyrazole
- ADF
Amsterdam density functional
- ATCC
American Type Culture Collection
- ACN
acetonitrile
- AMD473
cis-amminedichloro(2-methylpyridine) platinum(II), picoplatin
- AnnCy3
Annexin V-Cy3 conjugate
- ATR
attenuated total reflectance
- B16-F10
murine melanoma cells
- B3LYP
Becke, 3-parameter, Lee–Yang–Parr
- COSMO
conductor-like screening model
- CD
circular dichroism
- CDDP
cis-diamminedichloroplatinum(II)
- CRL
cell repository line
- CTR1
copper transporter 1
- DMEM
Dulbecco’s modified Eagle’s medium
- DZVP
double-zeta valence with polarization
- EA
elemental analysis
- ESI
electrospray ionization
- ETS
extended transition state
- FBS
fetal bovine serum
- FT
Fourier transform
- GSH
reduced glutathione
- GSSG
oxidized glutathione
- GTH
Goedecker–Teter–Hutter pseudopotentials
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HPLC
high-performance liquid chromatography
- HR-MS
high-resolution mass spectrometry
- ICP–MS
inductively coupled plasma mass spectrometry
- IR
infrared
- LC–MS
liquid chromatography–mass spectroscopy
- LED
light-emitting diode
- MEP
molecular electrostatic potential
- MOLOPT
molecularly optimized
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NMuMG
normal murine breast cells
- NVT
number of particles, volume, and temperature
- NMR
nuclear magnetic resonance
- PBE
Perdew–Burke–Ernzerhof
- PDT
photodynamic therapy
- PS
photoswitch
- PSS
photostationary state
- RP
reverse phase
- RPMI
Roswell Park Memorial Institute
- RT
room temperature
- SR
short-range
- TOF
time of flight
- TZP
triple-zeta with polarization
- UPLC
ultraperformance liquid chromatography
- VMD
Visual Molecular Dynamics
- XRD
X-ray diffraction
- ZORA
zero-order regular approximation
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.4c01575.
Results of EA of 1 and 2; HPLC chromatogram of trans-2;1H NMR spectra of 1 and trans-2 in CDCl3, UV–vis spectra of trans-1 and cis-1 in 1% DMSO in PBS; 1H NMR spectra of nonirradiated and irradiated 1 and the complex; FT-IR spectra of trans-1 and trans-2; LC–MS of trans-2; HR-MS spectrum of trans-2; the mass spectra of cis-1, trans-1, trans-2, and the complex isotopic profile obtained in LC–MS analysis of the trans-2 solutions in DMSO; UV–vis spectra of trans-2 kept in the dark at 25 and 37 °C in 1% v/v DMSO in PBS; description of the crystallization attempts; cytotoxicity of 1 in B16–F10, 4T1, and NMuMG cells after 24 and 48 h in serum- and serum-free conditions; cytotoxicity of trans-2, cis-2, and cisplatin in B16–F10, 4T1, and NMuMG cells after 24 and 48 h in serum-free conditions; cytotoxicity of trans-2 and cis-2 in the A2780 ovary cancer cell line after 24 h in serum-free conditions; determination of 1’s solubility in water; evaluation of the exchange–correlation functional of 1; comparison of selected structural properties of 1 and azobenzene; and dynamic stability of the Pt(1)2 group (PDF)
Molecular formula strings (CSV)
Ab initio MD of the cis complex photoisomer (MPG)
Ab initio MD of the trans complex photoisomer (MPG)
The authors declare the following competing financial interest(s): The title compound is protected by Polish and European patents granted to the coauthors of this paper.
Supplementary Material
References
- Peyrone M. Ueber Die Einwirkung Des Ammoniaks Auf Platinchlorür. Adv. Cycloaddit. 1844, 51 (1), 1–29. 10.1002/jlac.18440510102. [DOI] [Google Scholar]
- Rosenberg B.; Van Camp L.; Krigas T. Inhibition of Cell Division in Escherichia coli by Electrolysis Products from a Platinum Electrode. Nature 1965, 205 (4972), 698–699. 10.1038/205698a0. [DOI] [PubMed] [Google Scholar]
- Rosenberg B.; Vancamp L.; Trosko J. E.; Mansour V. H. Platinum Compounds: A New Class of Potent Antitumour Agents. Nature 1969, 222 (5191), 385–386. 10.1038/222385a0. [DOI] [PubMed] [Google Scholar]
- Barry N. P. E.; Sadler P. J. 100 years of metal coordination chemistry: from Alfred Werner to anticancer metallodrugs. Anticancer Metallodrugs 2014, 86 (12), 1897–1910. 10.1515/pac-2014-0504. [DOI] [Google Scholar]
- Gately D. P.; Howell S. B. Cellular Accumulation of the Anticancer Agent Cisplatin: A Review. Br. J. Cancer 1993, 67 (6), 1171–1176. 10.1038/bjc.1993.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida S.; Lee J.; Thiele D. J.; Herskowitz I. Uptake of the Anticancer Drug Cisplatin Mediated by the Copper Transporter Ctr1 in Yeast and Mammals. Proc. Natl. Acad. Sci. U.S.A. 2002, 99 (22), 14298–14302. 10.1073/pnas.162491399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambley T. W. Platinum Binding to DNA: Structural Controls and Consequences. J. Chem. Soc., Dalton Trans. 2001, (19), 2711–2718. 10.1039/b105406f. [DOI] [Google Scholar]
- Dasari S.; Bernard Tchounwou P. Cisplatin in Cancer Therapy: Molecular Mechanisms of Action. Eur. J. Pharmacol. 2014, 740, 364–378. 10.1016/j.ejphar.2014.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brieke C.; Rohrbach F.; Gottschalk A.; Mayer G.; Heckel A. Light-Controlled Tools. Angew. Chem., Int. Ed. 2012, 51 (34), 8446–8476. 10.1002/anie.201202134. [DOI] [PubMed] [Google Scholar]
- Volarić J.; Szymanski W.; Simeth N. A.; Feringa B. L. Molecular Photoswitches in Aqueous Environments. Chem. Soc. Rev. 2021, 50 (22), 12377–12449. 10.1039/D0CS00547A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchter M. J. On the Promise of Photopharmacology Using Photoswitches: A Medicinal Chemist’s Perspective. J. Med. Chem. 2020, 63 (20), 11436–11447. 10.1021/acs.jmedchem.0c00629. [DOI] [PubMed] [Google Scholar]
- Sharma M.; Friedman S. H. The Issue of Tissue: Approaches and Challenges to the Light Control of Drug Activity. ChemPhotoChem 2021, 5 (7), 611–618. 10.1002/cptc.202100001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klán P.; Šolomek T.; Bochet C. G.; Blanc A.; Givens R.; Rubina M.; Popik V.; Kostikov A.; Wirz J. Photoremovable Protecting Groups in Chemistry and Biology: Reaction Mechanisms and Efficacy. Chem. Rev. 2013, 113 (1), 119–191. 10.1021/cr300177k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg B.; Van Camp L.; Grimley E. B.; Thomson A. J. The Inhibition of Growth or Cell Division in Escherichia Coli by Different Ionic Species of Platinum(IV) Complexes. J. Biol. Chem. 1967, 242 (6), 1347–1352. 10.1016/S0021-9258(18)96186-7. [DOI] [PubMed] [Google Scholar]
- Gurruchaga-Pereda J.; Martínez A. ´.; Terenzi A.; Salassa L. Anticancer Platinum Agents and Light. Inorg. Chim. Acta 2019, 495, 118981. 10.1016/j.ica.2019.118981. [DOI] [Google Scholar]
- Kratochwil N. A.; Zabel M.; Range K.-J.; Bednarski J.; Synthesis P. Synthesis and X-ray Crystal Structure of trans,cis-[Pt(OAc)2I2(en)]: A Novel Type of Cisplatin Analog That Can Be Photolyzed by Visible Light to DNA-Binding and Cytotoxic Species in Vitro. J. Med. Chem. 1996, 39 (13), 2499–2507. 10.1021/jm9509105. [DOI] [PubMed] [Google Scholar]
- Shi H.; Wang Q.; Venkatesh V.; Feng G.; Young L. S.; Romero-Canelón I.; Zeng M.; Sadler P. J. Photoactive Platinum(IV) Complex Conjugated to a Cancer-Cell-Targeting Cyclic Peptide. Dalt. Trans. 2019, 48 (24), 8560–8564. 10.1039/C9DT00909D. [DOI] [PubMed] [Google Scholar]
- Shi H.; Imberti C.; Huang H.; Hands-Portman I.; Sadler P. J. Biotinylated Photoactive Pt(IV) Anticancer Complexes. Chem. Commun. 2020, 56 (15), 2320–2323. 10.1039/C9CC07845B. [DOI] [PubMed] [Google Scholar]
- Shi H.; Imberti C.; Clarkson G. J.; Sadler P. J. Axial Functionalisation of Photoactive Diazido Platinum(Iv) Anticancer Complexes. Inorg. Chem. Front. 2020, 7 (19), 3533–3540. 10.1039/D0QI00685H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H.; Clarkson G. J.; Sadler P. J. Dual Action Photosensitive Platinum(II) Anticancer Prodrugs with Photoreleasable Azide Ligands. Inorg. Chim. Acta 2019, 489, 230–235. 10.1016/j.ica.2019.02.016. [DOI] [Google Scholar]
- Moustafa M. E. M. E.; McCready M. S. M. S.; Boyle P. D. P. D.; Puddephatt R. J. R. J. Photoswitchable and PH Responsive Organoplatinum(Ii) Complexes with Azopyridine Ligands. Dalt. Trans. 2017, 46 (26), 8405–8414. 10.1039/C7DT01290J. [DOI] [PubMed] [Google Scholar]
- Moustafa M. E. M. E.; Boyle P. D. P. D.; Puddephatt R. J. R. J. Photoswitchable Organoplatinum Complexes with an Azobenzene Derivative of Di-2-Pyridylamine. New J. Chem. 2020, 44 (7), 2882–2889. 10.1039/C9NJ05313A. [DOI] [Google Scholar]
- Moustafa M. E.; Boyle P. D.; Puddephatt R. J. Photoswitchable Organoplatinum Complexes Containing an Azobenzene Derivative of 3,6-Di(2-Pyridyl)Pyridazine. Can. J. Chem. 2014, 92 (8), 706–715. 10.1139/cjc-2013-0588. [DOI] [Google Scholar]
- Moustafa M. E.; McCready M. S.; Puddephatt R. J. Switching by Photochemical Trans-Cis Isomerization of Azobenzene Substituents in Organoplatinum Complexes. Organometallics 2012, 31 (17), 6262–6269. 10.1021/om3005405. [DOI] [Google Scholar]
- Presa A.; Vázquez G.; Barrios L. A.; Roubeau O.; Korrodi-Gregório L.; Pérez-Tomás R.; Gamez P. Photoactivation of the Cytotoxic Properties of Platinum(II) Complexes through Ligand Photoswitching. Inorg. Chem. 2018, 57 (7), 4009–4022. 10.1021/acs.inorgchem.8b00146. [DOI] [PubMed] [Google Scholar]
- Samper K. G.; Lorenzo J.; Capdevila M.; Palacios O. `.; Bayón P. Functionalized Azobenzene Platinum(II) Complexes as Putative Anticancer Compounds. J. Biol. Inorg. Chem. 2021, 26 (4), 435–453. 10.1007/s00775-021-01865-9. [DOI] [PubMed] [Google Scholar]
- Stricker L.; Böckmann M.; Kirse T.; Doltsinis N.; Ravoo B. Arylazopyrazole Photoswitches in Aqueous Solution: Substituent Effects, Photophysical Properties, and Host–Guest Chemistry. Chem.—A Eur. J. 2018, 24 (34), 8639–8647. 10.1002/chem.201800587. [DOI] [PubMed] [Google Scholar]
- Boelke J.; Hecht S. Designing Molecular Photoswitches for Soft Materials Applications. Adv. Opt. Mater. 2019, 7 (16), 1900404. 10.1002/adom.201900404. [DOI] [Google Scholar]
- van Kralingen C. G.; de Ridder J. K.; Reedijk J. Coordination Compounds of Platinum(II) and Palladium(II) with Pyrazole as a Ligand. New Synthetic Procedures and Characterisation. Transition Met. Chem. 1980, 5 (1), 73–77. 10.1007/BF01396873. [DOI] [Google Scholar]
- Boixassa A.; Pons J.; Solans X.; Font-Bardia M.; Ros J. Reaction of platinum(II) derivatives with 1-hydroxyalkyl-3,5-dimethylpyrazole ligands. Cleavage of the N(pz)—C(sp3) bond. X-ray crystal structure of cis-[PtCl2(HL2)2] (HL2=1-(2-hydroxyethyl)-3,5-dimethylpyrazole) and trans-[PtCl2(dmpz)2] (dmpz=3,5-dimethylpyrazole). Inorg. Chim. Acta 2003, 355, 254–263. 10.1016/S0020-1693(03)00346-3. [DOI] [Google Scholar]
- Sakai K.; Tomita Y.; Ue T.; Goshima K.; Ohminato M.; Tsubomura T.; Matsumoto K.; Ohmura K.; Kawakami K. Syntheses, antitumor activity, and molecular mechanics studies of cis-PtCl2(pzH)2 (pzH=pyrazole) and related complexes. Crystal structure of a novel Magnus-type double-salt [Pt(pzH)4] [PtCl4] [cis-PtCl2(pzH)2]2 involving two perpendicularly aligned 1D chains. Inorg. Chim. Acta 2000, 297 (1–2), 64–71. 10.1016/S0020-1693(99)00287-X. [DOI] [Google Scholar]
- Adams C. J.; Haddow M. F.; Hughes R. J. I.; Kurawa M. A.; Orpen A. G. Coordination Chemistry of Platinum and Palladium in the Solid-State: Synthesis of Imidazole and Pyrazole Complexes. Dalt. Trans. 2010, 39 (15), 3714–3724. 10.1039/b919665j. [DOI] [PubMed] [Google Scholar]
- Zhang Z.-Y.; He Y.; Zhou Y.; Yu C.; Han L.; Li T. Pyrazolylazophenyl Ether-Based Photoswitches: Facile Synthesis, (Near-)Quantitative Photoconversion, Long Thermal Half-Life, Easy Functionalization, and Versatile Applications in Light-Responsive Systems. Chem.—A Eur. J. 2019, 25 (58), 13402–13410. 10.1002/chem.201902897. [DOI] [PubMed] [Google Scholar]
- Ansari A.; Ali A.; Asif M.; Shamsuzzaman S. Review: biologically active pyrazole derivatives. New J. Chem. 2017, 41 (1), 16–41. 10.1039/c6nj03181a. [DOI] [Google Scholar]
- Karati D.; Mahadik K. R.; Kumar D. Pyrazole Scaffolds: Centrality in Anti-Inflammatory and Antiviral Drug Design. Med. Chem. 2022, 18 (10), 1060–1072. 10.2174/1573406418666220410181827. [DOI] [PubMed] [Google Scholar]
- Weston C. E.; Krämer A.; Colin F.; Yildiz O. ¨.; Baud M. G. J.; Meyer-Almes F.-J.; Fuchter M. J. Toward Photopharmacological Antimicrobial Chemotherapy Using Photoswitchable Amidohydrolase Inhibitors. ACS Infect. Dis. 2017, 3 (2), 152–161. 10.1021/acsinfecdis.6b00148. [DOI] [PubMed] [Google Scholar]
- Kumar H.; Saini D.; Jain S.; Jain N. Pyrazole Scaffold: A Remarkable Tool in the Development of Anticancer Agents. Eur. J. Med. Chem. 2013, 70, 248–258. 10.1016/j.ejmech.2013.10.004. [DOI] [PubMed] [Google Scholar]
- Chen J.; Yin Z. Cooperative Intramolecular Hydrogen Bonding Induced Azo-Hydrazone Tautomerism of Azopyrrole: Crystallographic and Spectroscopic Studies. Dyes Pigm. 2014, 102, 94–99. 10.1016/j.dyepig.2013.10.036. [DOI] [Google Scholar]
- Holford J.; Sharp S. Y.; Murrer B. A.; Abrams M.; Kelland L. R. In Vitro Circumvention of Cisplatin Resistance by the Novel Sterically Hindered Platinum Complex AMD473. Br. J. Cancer 1998, 77 (3), 366–373. 10.1038/bjc.1998.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolarek M.; Pycior A.; Bonarek P.; Opydo M.; Kolaczkowska E.; Kamiński K.; Mogielnicki A.; Szczubiałka K. Biological Properties of Heparins Modified with an Arylazopyrazole-Based Photoswitch. J. Med. Chem. 2023, 66 (3), 1778–1789. 10.1021/acs.jmedchem.2c01616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budzisz E.; Malecka M.; Nawrot B. Synthesis and Structure of Highly Substituted Pyrazole Ligands and Their Complexes with Platinum(II) and Palladium(II) Metal Ions. Tetrahedron 2004, 60 (8), 1749–1759. 10.1016/j.tet.2003.12.044. [DOI] [Google Scholar]
- Arpalahti J.; Lippert B. An Alternative HPLC Method for Analysing Mixtures of Isomeric Platinum(II) Diamine Compounds. Inorg. Chim. Acta 1987, 138 (3), 171–173. 10.1016/S0020-1693(00)81218-9. [DOI] [Google Scholar]
- Rochon F. D.; Melanson R.; Doyon M.; Butler I. S. Synthesis of Pt(amine)(DMF)Cl2 and [Pt(amine)X2]n (X = Cl, I) and Crystal Structure of cyclo-Tris(.mu.-chloro)tris[chloro(dimethylamine)platinum(II)]. Inorg. Chem. 1994, 33 (20), 4485–4493. 10.1021/ic00098a014. [DOI] [Google Scholar]
- Jawad S. H.; Al-Adilee K. J. Synthesis and Characterization of a New 1-Methyl Imidazole Derived Ligand with Its Ionic Complexes Pd(II) and Pt(IV) and Study of Biological Activity as Anticancer and Antioxidant. Results Chem. 2022, 4, 100573. 10.1016/j.rechem.2022.100573. [DOI] [Google Scholar]
- Hall M. D.; Telma K. A.; Chang K.-E.; Lee T. D.; Madigan J. P.; Lloyd J. R.; Goldlust I. S.; Hoeschele J. D.; Gottesman M. M. Say No to DMSO: Dimethylsulfoxide Inactivates Cisplatin, Carboplatin, and Other Platinum Complexes. Cancer Res. 2014, 74 (14), 3913–3922. 10.1158/0008-5472.CAN-14-0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston C.; Richardson R.; Haycock P.; White A.; Fuchter M. Arylazopyrazoles: Azoheteroarene Photoswitches Offering Quantitative Isomerization and Long Thermal Half-Lives. J. Am. Chem. Soc. 2014, 136 (34), 11878–11881. 10.1021/ja505444d. [DOI] [PubMed] [Google Scholar]
- Ludwanowski S.; Ari M.; Parison K.; Kalthoum S.; Straub P.; Pompe N.; Weber S.; Walter M.; Walther A. PH Tuning of Water-Soluble Arylazopyrazole Photoswitches. Chem.—A Eur. J. 2020, 26 (58), 13203–13212. 10.1002/chem.202000659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz L.; Supuran C. T.; Alfarouk K. O. The Warburg Effect and the Hallmarks of Cancer. Anticancer Agents Med. Chem. 2017, 17 (2), 164–170. 10.2174/1871520616666161031143301. [DOI] [PubMed] [Google Scholar]
- Jordan P.; Carmo-Fonseca M. Molecular Mechanisms Involved in Cisplatin Cytotoxicity. Cell. Mol. Life Sci. 2000, 57 (8), 1229–1235. 10.1007/PL00000762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddik Z. H. Cisplatin: Mode of Cytotoxic Action and Molecular Basis of Resistance. Oncogene 2003, 22 (47), 7265–7279. 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- Ghosh S.Cisplatin: The First Metal Based Anticancer Drug; Academic Press Inc., 2019; Vol. 88, p 102925. 10.1016/j.bioorg.2019.102925. [DOI] [PubMed] [Google Scholar]
- Ozkok A.; Edelstein C. L. Pathophysiology of Cisplatin-Induced Acute Kidney Injury. Anticancer Agents Med. Chem. 2014, 2014, 1–17. 10.1155/2014/967826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda V.; Fuertes M. A.; Castilla J.; Alonso C.; Quevedo C.; Perez J. M. Biochemical Mechanisms of Cisplatin Cytotoxicity. Anticancer Agents Med. Chem. 2007, 7 (1), 3–18. 10.2174/187152007779314044. [DOI] [PubMed] [Google Scholar]
- Querino A. L. D. A.; Enes K. B.; Chaves O. A.; Dittz D.; Couri M. R. C.; Diniz R.; Silva H. Modified Pyrazole Platinum(II) Complex Can Circumvent Albumin and Glutathione: Synthesis, Structure and Cytotoxic Activity. Bioorg. Chem. 2020, 100, 103936. 10.1016/j.bioorg.2020.103936. [DOI] [PubMed] [Google Scholar]
- Brabec V.; Kleinwächter V.; Butour J.-L.; Johnson N. P. Biophysical Studies of the Modification of DNA by Antitumour Platinum Coordination Complexes. Biophys. Chem. 1990, 35 (2–3), 129–141. 10.1016/0301-4622(90)80003-P. [DOI] [PubMed] [Google Scholar]
- Takada K.; Kawamura T.; Inai M.; Yoshikawa Y.; Ike O.; Wada H.; Hitomi S. Irreversible Binding of Cisplatin in Rat Serum. Pharm. Pharmacol. Commun. 1999, 5 (7), 449–453. 10.1211/146080899128735153. [DOI] [Google Scholar]
- Godwin A. K.; Meister A.; O’Dwyer P. J.; Huang C. S.; Hamilton T. C.; Anderson M. E. High Resistance to Cisplatin in Human Ovarian Cancer Cell Lines Is Associated with Marked Increase of Glutathione Synthesis. Proc. Natl. Acad. Sci. U.S.A. 1992, 89 (7), 3070–3074. 10.1073/pnas.89.7.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinkai Y.; Kumagai Y. Sulfane Sulfur in Toxicology: A Novel Defense System Against Electrophilic Stress. Toxicol. Sci. 2019, 170 (1), 3–9. 10.1093/toxsci/kfz091. [DOI] [PubMed] [Google Scholar]
- Wu G.; Lupton J. R.; Turner N. D.; Fang Y. Z.; Yang S. Glutathione Metabolism and Its Implications for Health. J. Nutr. 2004, 134 (3), 489–492. 10.1093/jn/134.3.489. [DOI] [PubMed] [Google Scholar]
- Dominick P. K.; Cassidy P. B.; Roberts J. C. A New and Versatile Method for Determination of Thiolamines of Biological Importance. J. Chromatogr. B: Biomed. Sci. Appl. 2001, 761 (1), 1–12. 10.1016/S0378-4347(01)00298-5. [DOI] [PubMed] [Google Scholar]
- Bronowicka-Adamska P.; Zagajewski J.; Czubak J.; Wróbel M. RP-HPLC Method for Quantitative Determination of Cystathionine, Cysteine and Glutathione: An Application for the Study of the Metabolism of Cysteine in Human Brain. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2011, 879 (21), 2005–2009. 10.1016/j.jchromb.2011.05.026. [DOI] [PubMed] [Google Scholar]
- Lowry O. H.; Rosenbrough N. J.; Farr A. L.; Randall R. J. Protein Measurement with the Folin Phenol Reagent. J. Biol. Chem. 1951, 193 (1), 265–275. 10.1016/s0021-9258(19)52451-6. [DOI] [PubMed] [Google Scholar]
- Fonseca Guerra C.; Snijders J. G.; Te Velde G.; Baerends E. J. Towards an Order-N DFT Method. Theor. Chem. Acc. 1998, 99 (6), 391–403. 10.1007/s002140050353. [DOI] [Google Scholar]
- Becke A. D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98 (7), 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37 (2), 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Hansen A.; Brandenburg J. G.; Bannwarth C. Dispersion-Corrected Mean-Field Electronic Structure Methods. Chem. Rev. 2016, 116 (9), 5105–5154. 10.1021/acs.chemrev.5b00533. [DOI] [PubMed] [Google Scholar]
- Klamt A.; Schüürmann G. COSMO: A New Approach to Dielectric Screening in Solvents with Explicit Expressions for the Screening Energy and Its Gradient. J. Chem. Soc., Perkin Trans. 2 1993, (5), 799–805. 10.1039/P29930000799. [DOI] [Google Scholar]
- Van Lenthe E.; Ehlers A.; Baerends E. J. Geometry Optimizations in the Zero Order Regular Approximation for Relativistic Effects. J. Chem. Phys. 1999, 110 (18), 8943–8953. 10.1063/1.478813. [DOI] [Google Scholar]
- Politzer P.; Laurence P. R.; Jayasuriya K. Molecular Electrostatic Potentials: An Effective Tool for the Elucidation of Biochemical Phenomena. Environ. Health Perspect. 1985, 61, 191–202. 10.1289/ehp.8561191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler T.; Rauk A. On the Calculation of Bonding Energies by the Hartree Fock Slater Method: I. The Transition State Method. Theor. Chim. Acta 1977, 46 (1), 1–10. 10.1007/BF02401406. [DOI] [Google Scholar]
- Ziegler T.; Rauk A. Carbon Monoxide, Carbon Monosulfide, Molecular Nitrogen, Phosphorus Trifluoride, and Methyl Isocyanide as.Sigma. Donors and.Pi. Acceptors. A Theoretical Study by the Hartree-Fock-Slater Transition-State Method. Inorg. Chem. 1979, 18 (7), 1755–1759. 10.1021/ic50197a006. [DOI] [Google Scholar]
- Kühne T. D.; Iannuzzi M.; Del Ben M.; Rybkin V. V.; Seewald P.; Stein F.; Laino T.; Khaliullin R. Z.; Schütt O.; Schiffmann F.; Golze D.; Wilhelm J.; Chulkov S.; Bani-Hashemian M. H.; Weber V.; Borštnik U.; Taillefumier M.; Jakobovits A. S.; Lazzaro A.; Pabst H.; Müller T.; Schade R.; Guidon M.; Andermatt S.; Holmberg N.; Schenter G. K.; Hehn A.; Bussy A.; Belleflamme F.; Tabacchi G.; Glöß A.; Lass M.; Bethune I.; Mundy C. J.; Plessl C.; Watkins M.; VandeVondele J.; Krack M.; Hutter J. CP2K: An Electronic Structure and Molecular Dynamics Software Package -Quickstep: Efficient and Accurate Electronic Structure Calculations. J. Chem. Phys. 2020, 152 (19), 194103. 10.1063/5.0007045. [DOI] [PubMed] [Google Scholar]
- Vandevondele J.; Krack M.; Mohamed F.; Parrinello M.; Chassaing T.; Hutter J. Quickstep: Fast and Accurate Density Functional Calculations Using a Mixed Gaussian and Plane Waves Approach. Comput. Phys. Commun. 2005, 167 (2), 103–128. 10.1016/j.cpc.2004.12.014. [DOI] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77 (18), 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Krack M. Pseudopotentials for H to Kr Optimized for Gradient-Corrected Exchange-Correlation Functionals. Theor. Chem. Acc. 2005, 114 (1–3), 145–152. 10.1007/s00214-005-0655-y. [DOI] [Google Scholar]
- Evans D. J.; Holian B. L. The Nose-Hoover Thermostat. J. Chem. Phys. 1985, 83 (8), 4069–4074. 10.1063/1.449071. [DOI] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual Molecular Dynamics. J. Mol. Graphics 1996, 14 (1), 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.