

Abstract
MicroRNAs (miRNAs) are regulators of gene expression, and their dysregulation is linked to cancer and other diseases, making them important therapeutic targets. Several strategies for targeting and modulating miRNA activity are being explored. For example, steric-blocking antisense oligonucleotides (ASOs) can reduce miRNA activity by either blocking binding sites on specific mRNAs or base-pairing to the miRNA itself to prevent its interaction with the target mRNAs. ASOs have been less explored as a tool to elevate miRNA levels, which could also be beneficial for treating disease. In this study, using the PKD1/miR-1225 gene locus as an example, where miR-1225 is located within a PKD1 intron, we demonstrate an ASO-based strategy that increases miRNA abundance by enhancing biogenesis from the primary miRNA transcript. Disruptions in PKD1 and miR-1225 are associated with autosomal dominant polycystic kidney disease (ADPKD) and various cancers, respectively, making them important therapeutic targets. We investigated PKD1 sequence variants reported in ADPKD that are located within the sequence shared by miR-1225 and PKD1, and identified one that causes a reduction in miR-1225 without affecting PKD1. We show that this reduction in miR-1225 can be recovered by treatment with a steric-blocking ASO. The ASO-induced increase in miR-1225 correlates with a decrease in the abundance of predicted miR-1225 cellular mRNA targets. This study demonstrates that miRNA abundance can be elevated using ASOs targeted to the primary transcript. This steric-blocking ASO-based approach has broad potential application as a therapeutic strategy for diseases that could be treated by modulating miRNA biogenesis.
Keywords: PKD1, antisense oligonucleotides, autosomal dominant polycystic kidney disease, miR-1225, microRNA
INTRODUCTION
MicroRNAs (miRNAs) are short, noncoding RNAs that regulate gene expression posttranscriptionally by base-pairing to RNAs and thereby targeting them for degradation (Shang et al. 2023). Destabilization of RNA by miRNA targeting is an important mechanism for controlling gene expression, and artificially modulating miRNA activity is a means to alter expression of genes that are regulated by a particular miRNA. Targeting miRNA is also a therapeutic strategy in cases where the deregulation of miRNAs is linked to cancer and other conditions. Considering these potential applications, miRNA modulation has become an active area of therapeutic development (Sempere et al. 2021).
Steric-blocking antisense oligonucleotides (ASOs), which have had success in the clinic as splice-switching tools to treat disease, can also be used to target miRNA biogenesis and activity. This type of ASO can be designed to base-pair to and sequester miRNAs to reduce their targeting capacity and stabilize their target RNAs (Winkle et al. 2021). ASOs can also be designed to base-pair with miRNA-binding sites on mRNA, thereby blocking miRNA binding and protecting the RNA from miRNA-induced degradation. These strategies all aim to limit miRNA activity. However, in some cases, increasing miRNA abundance and targeting may be desirable. Available methods to increase miRNA concentration and targeting include both vector-based and vector-independent strategies to deliver miRNAs exogenously. However, regulating the miRNA biogenesis to alter endogenous levels is a relatively unexplored potential therapeutic avenue.
One example of a miRNA that is implicated in disease is miR-1225, which is housed within another disease-associated gene, PKD1. PKD1 mutations are associated with autosomal dominant polycystic kidney disease (ADPKD) and dysregulation of miR-1225 has been implicated in numerous cancers, in most cases acting as a tumor suppressor by targeting genes associated with cell growth and proliferation (Zheng et al. 2016; Sun et al. 2019; Wang et al. 2019, 2020; Zhong et al. 2019; Li et al. 2020; Zhang et al. 2020, 2022; Gab et al. 2021; Gong et al. 2021; Yang et al. 2021; Xia et al. 2023). Thus, miR-1225 may be a therapeutic target for diseases associated with abnormal cellular proliferation. In addition, ADPKD-associated mutations that fall within both miR-1225 and PKD1 sequences have been reported (Badenas et al. 1999; Aguiari et al. 2000; Burtey et al. 2002), but their effects on miRNA processing, function, and link to disease pathology are unknown. PKD1 is not a predicted target of miR-1225, thus any role for the miRNA in ADPKD would likely involve the targeting of other genes.
PKD1/miR-1225 is a particularly interesting target for steric-blocking ASOs, not only because of the disease association of both genes, but also because of the unusual co-regulation of their RNA transcripts. MiR-1225, a type of miRNA called a simtron, spans the entire length of intron 45 in its host gene, PKD1 (Havens et al. 2012). Two pathways exist for its maturation: the mirtron pathway, in which splicing liberates the pre-miRNA (Westholm and Lai 2011), and the simtron pathway (Havens et al. 2012), in which Drosha cleavage directly releases the pre-miRNA. The latter method precludes splicing because it removes the consensus sequences necessary for splicing. Depending on which pathway predominates, there can be implications for disease progression and severity due to competition between miRNA biogenesis and mRNA production in the simtron pathway.
To explore the potential of steric-blocking ASOs in regulating miRNA biogenesis, we focused on miR-1225. We evaluated the ability of ASOs to modulate miR-1225 levels, particularly when its sequence is disrupted by PKD1 variants associated with ADPKD. Our study examined the interaction between PKD1 and miR-1225, potential ADPKD-related co-dysregulation of the two transcripts, and screened for ASOs that could have possible therapeutic effects on disrupted miR-1225 production. We identified an ADPKD-linked PKD1 variant that selectively disrupts miR-1225 without affecting PKD1 and discovered ASOs that effectively restored miR-1225 levels disrupted by the ADPKD-associated variant. This ASO-mediated recovery of miR-1225 correlated with a reduction in the RNA of predicted miR-1225 targets, demonstrating that specific ASO-based targeting of the pri-miRNA transcript can modulate downstream miRNA activity. These findings suggest a new targeting strategy for steric-blocking ASOs, which could be applied to diseases that may benefit from modulating miRNA biogenesis or targeting.
RESULTS
PKD1 and miR-1225 are differentially expressed in human and mouse tissue
As a first step toward devising an ASO-based approach for modulating miRNA biogenesis from the primary RNA transcript, we considered target miRNAs with potential disease associations and selected miR-1225, which has been implicated in a number of cancers and is located within the intron of the ADPKD-related gene, PKD1. Assessing the abundance of PKD1 and miR-1225 is a first step toward understanding a potential regulatory interplay between these two RNAs and establishing a rationale for selectively modulating the miRNA.
Previous studies have found that the expression of PKD1 varies in human tissues and during development of the kidney (Ward et al. 1996; Chauvet et al. 2002). We measured PKD1 and miR-1225-5p expression in 20 adult human tissues (non-ADPKD) using semiquantitative RT-PCR and stem–loop RT-PCR, respectively. The abundance of PKD1 and miR-1225 was variable across tissues (Fig. 1A; Supplemental Fig. S1). Some tissues, such as thymus and brain, had high miR-1225 levels compared to PKD1, and others had detectable PKD1, without miR-1225 expression (Fig. 1A,B). These data reveal that PKD1 and miR-1225 are expressed systemically in adult tissue and at levels that may suggest differential tissue-specific regulation.
FIGURE 1.
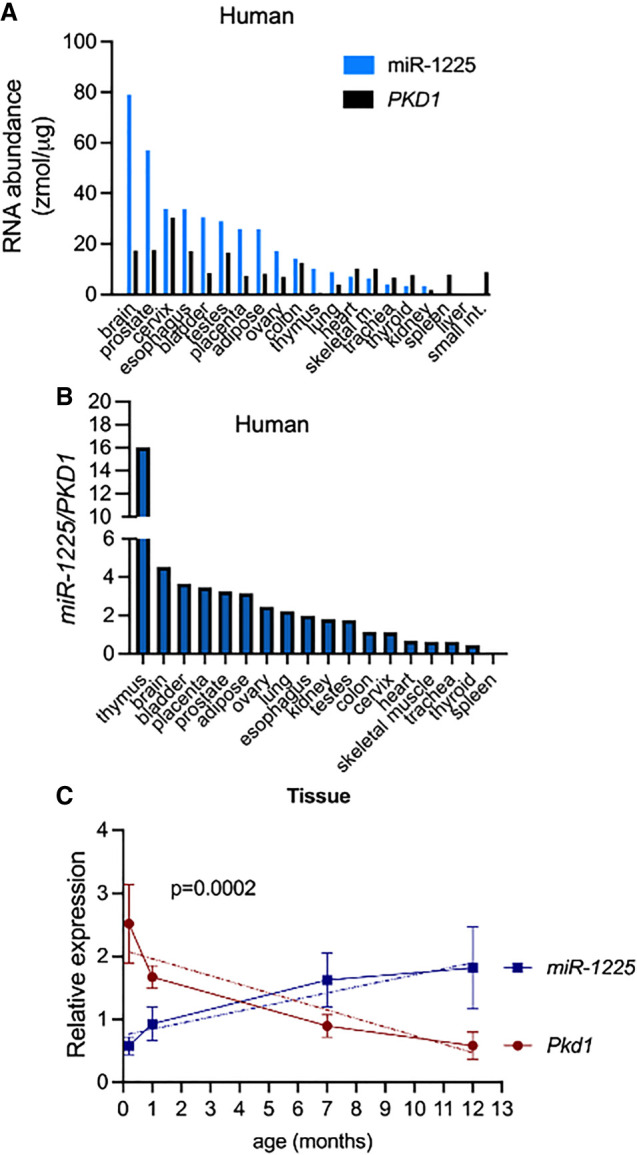
Interactions between miR-1225 and PKD1 RNA expression. (A) miR-1225 and PKD1 are expressed in human tissues. Quantification of RT-PCR and stem–loop RT-PCR analysis of total human mRNA isolated from various tissues. The absolute amount of PKD1 mRNA and mature miR-1225 are calculated based on comparison to a standard curve generated by amplification of different concentrations of a synthetic sequence of known concentration and graphed as zmol/μg of total RNA. (B) The relative expression of miR-1225 to PKD1 is graphed as (zmol of miR-1225/μg of total RNA)/(zmol of PKD1/μg of total RNA). (C) Mouse miR-1225 and Pkd1 are dynamically expressed across the mouse life-span. Quantitation of radiolabeled stem–loop RT-PCR of mouse miR-1225 and RT-PCR of Pkd1 relative to sno65 and Gapdh, respectively (see Supplemental Fig. S1). Error bars represent standard error of the mean SEM. Postnatal day 2 (P2), n = 4; P23, n = 5; 7 months, n = 5; and 11–13 months, n = 6. Simple linear regression analysis indicates that the slopes are significantly different, P = 0.0002.
Mir-1225 has been predicted but not experimentally identified in mice. Pre-miR-1225 (intron 45), is 94% identical between mice and humans (Supplemental Fig. S2A), more highly conserved than the flanking exons (Rodova et al. 2003), suggesting a functional importance of the sequence that may restrict genetic drift. The hairpin structure of intron 45 is also conserved (Supplemental Fig. S2B). We detected the mature miRNA species with the same sequence and location in mouse as in humans (Supplemental Fig. S2C,D).
To evaluate the dynamic temporal expression of miR-1225 and PKD1, we quantified their abundance in the kidney of mice at different ages to assess their developmental expression pattern. It has been reported that PKD1 expression decreases with age in humans (Geng et al. 1996; Chauvet et al. 2002) and mice (Geng et al. 1997), but the dynamics of miR-1225 expression are not known. We analyzed PKD1 and miR-1225 expression in mouse kidneys at postnatal day 2 (P2), day 23 (P23), 6–7 months, and 11–13 months of age. As mice age, the expression of Pkd1 decreased, as expected (Fig. 1C; Supplemental Fig. S2D). Conversely, miR-1225 expression, which was low in the kidney after birth, increased with age (Fig. 1C; Supplemental Fig. 2D). In general, Pkd1 levels were low when miR-1225 levels were high, and vice versa (Fig. 1C) in the kidney, demonstrating an inverse expression correlation between the two RNA species from P2 to 13 months (Fig. 1C).
ADPKD variants disrupt miR-1225 but not PKD1 abundance in vitro
Given that miR-1225 is located within PKD1, and PKD1 expression is dysregulated in ADPKD, we sought to determine whether any disease-associated variants within intron 45 (pre-miR-1225) might affect miR-1225 expression. In order to study intron 45 variants that might impact miR-1225 but not PKD1 splicing, we did not consider those that removed the miRNA or disrupted the splice sites. We selected four variants for further analysis: IVS45-14T > C (Burtey et al. 2002), IVS45 + 34_+47del14 (Athena Diagnostics, unpubl., http://pkd.mayo.edu), IVS45-36_12del25 (Aguiari et al. 2000), and IVS45-3C < G (Athena Diagnostics, unpubl., http://pkd.mayo.edu) (Fig. 2A). We first examined predicted structural changes that occur with the introduction of these variants using an RNA folding algorithm (Zuker 2003). The hairpin structure remains relatively intact for all the variants, though branching of the hairpin is predicted in IVS45-14T > C and IVS45 + 34_+47del14 (Supplemental Fig. S3).
FIGURE 2.

The human PKD1 variant IVS45-14T > C decreases miR-1225 abundance and biogenesis without altering host gene (PKD1) splicing. (A) Location of the ADPKD variants in intron 45 of PKD1. Capitalized nucleotides are exonic, lower case nucleotides are intron 45. The red line represents the mature 5p miRNA sequence. (B) Radiolabeled RT-PCR (left) and stem–loop RT-PCR (right) of RNA isolated from HEK-293T cells transiently transfected with WT and mutant minigenes to detect PKD1 splicing and miR-1225 abundance, respectively. Samples were quantitated and normalized to GAPDH, miR-16, and sno65. The quantity of PKD1 is shown relative to expression from the WT plasmid using the equation: (PKD1/GAPDH)mutant/(PKD1/GAPDH)wild-type. The quantity of miR-1225 is shown relative to expression from WT using the equation: (miR-1225/miR-16)mutant/(miR-1225/miR-16)wild-type (top right) or (miR-1225/sno65)mutant/(miR-1225/sno65)wild-type (bottom right). Panel (C) refers to a mock transfection with vehicle. Error bars represent SEM, n = 3 (shown as individual points). (*) P < 0.05, one sample T and Wilcoxon test, (***) P < 0.001, one sample T and Wilcoxon test; all other groups were not significantly different than WT.
To determine whether miR-1225 expression is affected by these ADPKD patient variants, we constructed a series of PKD1 minigene expression vectors comprised of exons 43–46 of PKD1 and the intervening introns. We created a WT PKD1 minigene (Havens et al. 2012), and minigenes containing the selected mutations (Fig. 2A; Supplemental Fig. S3). The minigenes were transfected into HEK-293T cells and PKD1 pre-mRNA splicing and miR-1225 abundance were analyzed. IVS45-14T > C and +34_47del14 had no apparent effect on PKD1 mRNA splicing, and −36_−12del25 and −3C > G caused intron 45 retention (Fig. 2B). Despite a lack of effect on PKD1 mRNA splicing, the IVS45-14T > C variant resulted in a significant reduction in the abundance of miR-1225 using either miR-16 (decreased by 50%) or snoRNA65 (sno65) (decreased by 40%) as controls (Fig. 2B).
Steric-blocking antisense oligonucleotides can modulate miR-1225 abundance
Because miR-1225 is spatially linked to PKD1 and may also be differentially regulated relative to splicing of its host intron in PKD1, we tested whether we could modulate miR-1225 biogenesis without affecting PKD1 expression or splicing, a goal that may be important for any therapeutic targeting strategy. For this, we designed a series of tiled 18 nt, chemically modified steric-blocking ASOs, each off-set by 4 nt and complementary to intron 45 of PKD1. The first ASO base-pairs at the first position of the intron, and the last ASO in the series base-pairs to 17 nt upstream of the 3′ splice site (Fig. 3A). The WT human PKD1 minigene, described above, was transfected into HeLa cells with the individual human-specific ASOs. After 2 days, RNA was collected and PKD1 splicing and miR-1225 abundance were analyzed. Treatment with several ASOs resulted in a decrease in miR-1225 abundance and others increased miR-1225 (Fig. 3B,C). Some ASOs increased intron 45 retention and others do not alter PKD1 splicing (Fig. 3B,C). Treatment with ASO-16 resulted in the greatest increase in miR-1225 abundance, with no apparent change in PKD1 splicing, and thus was selected for further testing.
FIGURE 3.

Modulation of human miR-1225 abundance using steric-blocking ASOs. (A) ASO sequences and target site on PKD1 RNA are shown. (B) Radiolabeled stem–loop RT-PCR (top, miR-1225/miR-16) and RT-PCR (bottom, PKD1/GAPDH) of RNA isolated from HeLa cells that were transiently transfected with a WT human PKD1 minigene and 50 nM ASO, or minigene alone (mock) as a control. GAPDH and miR-16 are controls for normalization of total RNA quantity among samples. Intron 45 retained indicates amplicon from RNA with unspliced intron 45. (C) Quantitation of spliced PKD1 mRNA and miR-1225 normalized to GAPDH and miR-16 values, respectively, and graphed relative to mock-treated sample.
ASO-16 increases miR-1225 produced from ADPKD-associated PKD1 variant
As a first step in demonstrating the utility of an ASO for modulating miRNA biogenesis, we tested whether ASO-16 could increase miR-1225 derived from PKD1 variants in intron 45 that have been reported in ADPKD patients and might impact miR-1225 (Fig. 2). ASO-16 was cotransfected with the WT and variant PKD1 minigenes including PKD1 IVS45-14T > C, which produces less miR-1225 than WT. ASO-16 treatment resulted in a significant increase in miR-1225 produced from PKD1 IVS45-14T > C without significantly changing PKD1 abundance or splicing (Fig. 4). This result suggests that ASO-16 may be a viable approach for restoring miRNA expression that is disrupted by mutations.
FIGURE 4.

ASO-16 restores miR-1225 derived from the ADPKD variant IVS45-14T > C. (A) Stem–loop RT-PCR and RT-PCR analysis of miR-1225 and PKD1 mRNA, respectively, of RNA isolated from HEK-293T cells transiently transfected with plasmids expressing WT, IVS45-14T > C, IVS45 + 34_27del14, IVS45-36_-12del25, or IVS45-3C > G PKD1 minigenes with (+) or without (−) ASO-16 cotransfection. (B) Quantitation of miR-1225 and PKD1 normalized to miR-16 and GAPDH, respectively. Error bars represent SEM, n = 4, (*) P<0.05 multiple paired t-test comparing control (C) and ASO-16 for each minigene.
Mouse ASOs modulate miR-1225 expression in vitro
Next, we tested whether ASOs could modulate miR-1225 biogenesis in mouse cells. For this, we assessed the activity of a series of 16 ASOs in two different mouse cell lines using mouse-specific ASOs (Fig. 5A; Supplemental Fig. S4; Lentz et al. 2013). Though the mouse and human intron are highly conserved, the mouse intron is 4 nt longer than the human intron, thus ASO-12 and 13 in mouse base-pair with sequences that are represented by a single ASO (12/13) in the human sequence (Fig. 3). ASOs were transfected into cells, and RNA was collected 48 h later. RT-PCR analysis revealed that, similar to results with ASOs targeting the human minigene (Figs. 3 and 4), ASO-16 increased endogenous miR-1225 in mouse cells (Fig. 5B,C). ASO-5 decreased miR-1225 expression in human cells (Fig. 3) but had little effect on the endogenous miR-1225 in mouse cells (Fig. 5B,C). Neither ASO-5 nor ASO-16 affected intron 45 splicing in mouse cells (Fig. 5B,C). ASO-15 and ASO-16, the two ASOs with the highest activity, increased endogenous miR-1225 in a dose-responsive manner with EC50 values of 99 and 44 nM, respectively (Fig. 6A,B).
FIGURE 5.

Modulating mouse miR-1225 abundance with ASOs. (A) ASO sequences and target sequences are shown. (B) Mouse 89N cells were transiently transfected with 50 nM of ASO or lipofectamine alone (mock) as a control. miR-1225 abundance and Pkd1 splicing were assessed by radiolabeled stem–loop RT-PCR and RT-PCR, respectively. Gapdh, snoRNA65, and miR-877 are controls for loading. C indicates a mock transfection where no ASO was added, and – indicates untransfected cells. (C) Graph represents quantitation of amplicons in (B). miR-1225 abundance was normalized miR-877 and graphed relative to mock-treated cells. Pkd1 abundance is normalized Gapdh and graphed relative to mock-treated sample.
FIGURE 6.

ASO-mediated increase in mir-1225 corresponds with a decrease in the abundance of predicted target RNAs. (A) Gel of endogenous miR-1225 and Pkd1 stem–loop RT-PCR and RT-PCR products, respectively, amplified from RNA isolated from cultured cells treated with 12.5, 25, 50, 100, 200 nM of ASO-15 or -16. miR-877 is a loading control. (B) Quantitation of results in (A). Half-maximal effective concentration (EC50) was determined after fitting the data using nonlinear regression with a variable slope. (C) RT-PCR analysis of Jak1, Keap1, and Clcn5 cDNA from cultured cells treated with 12.5, 50, or 200 nM of ASO-16. C refers to control, vehicle treatment. (D) Quantitation of amplicons in (C) with each sample normalized to Gapdh and graphed relative to mock-treated sample. Error bars are SEM. n = 3–6 as indicated by individual points. (*) P < 0.05, (**) P < 0.01, (***) P < 0.0001. One-sample t-test relative to control-treated cells.
To determine whether ASO-16 has a biologically relevant effect on miR-1225 abundance, we analyzed the expression of putative miR-1225 target mRNAs in a mouse cell line following treatment with increasing amounts of ASO-16 (Fig. 6C,D). JAK1 and Keap1 are reported targets of miR-1225 in humans and mice, respectively (Supplemental Fig. S5; Reziwan et al. 2019; Zhong et al. 2019). Though the reported target site in JAK1 is not conserved in mice, we identified a nearby seed match (Supplemental Fig. S5A). CLCN5 is a predicted target for human miR-1225 based on both miRDB and Targetscan (Agarwal et al. 2015; Chen and Wang 2020). The predicted miR-1225 target site in Clcn5 is conserved in mice (Supplemental Fig. S5B). We found that treatment of a mouse cell line with ASO-16 resulted in a dose-responsive reduction in Jak1, Clcn5, and Keap1 RNA (Fig. 6C,D). These findings indicate that the ASO-16-induced increase in miR-1225 causes a decrease in the abundance of RNAs with predicted miR-1225 binding sites and show that ASO-16 can not only increase miR-1225 abundance, but also its activity.
Mouse ASOs modulate miR-1225 expression in vivo
To test the activity of ASO-5 and ASO-16 in vivo, we treated neonatal mice with the ASOs by intraperitoneal injection at postnatal day 2 (P2) and an additional dose at P14. We collected the mouse kidneys at P21 and quantitated miR-1225 abundance (Fig. 7). ASO-5 did not significantly reduce miR-1225 abundance in mouse kidney as expected (Fig. 7A,B). However, ASO-16 treatment caused a significant increase in miR-1225 (Fig. 7A,B). Pkd1 mRNA abundance and splicing did not change with ASO treatment (Fig. 7A,C; Supplemental Fig. S6). There were no obvious gross morphological defects in the mouse kidneys following treatment or changes in kidney size relative to body weight (Supplemental Fig. S7). Taken together, these results demonstrate that ASO-16 can be used to increase miR-1225 abundance in vivo.
FIGURE 7.
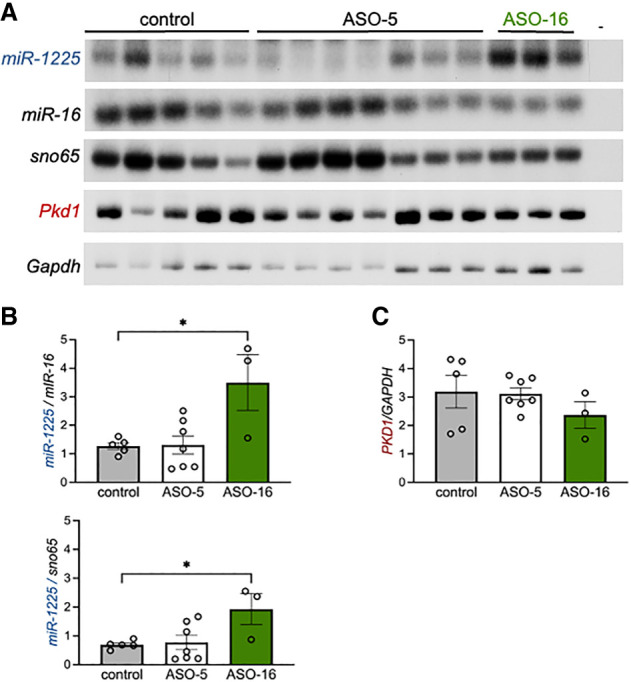
ASO-16 increases miR-1225 abundance in vivo. (A) Analysis of RNA isolated from kidneys of P21 mice treated with indicated ASO or vehicle control (C). miR-1225, miR-16 and sno65 were analyzed by radiolabeled stem–loop RT-PCR. Pkd1 and Gapdh were analyzed by radiolabeled RT-PCR. (–) refers to a control PCR reaction with no cDNA. Amplicons were separated by PAGE. (B) Quantitation of miR-1225 (left) normalized to miR-16 (top) or sno65 (bottom). (C) Quantitation of Pkd1 normalized to Gapdh. Error bars represent SEM, n = 3–7 mice. (*) P ≤ 0.05, one-way ANOVA with Dunnet's post hoc analysis samples compared to control.
DISCUSSION
Here, we report on steric-blocking ASOs that can modulate miR-1225 abundance in vitro and in vivo. Our findings demonstrate that ASOs can be used strategically to augment miRNA biogenesis in a disease-relevant gene locus. More broadly, this approach positions steric-blocking ASOs as a tool to control miRNA biogenesis and activity, offering a potential therapeutic recourse for treating disease.
Steric-blocking ASOs are becoming a valuable therapeutic platform, recognized for their specificity, favorable safety profile, and long-lasting effects (Havens and Hastings 2016; Bennett et al. 2017). Mechanistically, they can be designed to alter almost every step in the gene expression process, from transcription and splicing to translation and stability, to achieve a desired outcome. Though successful in the clinic as modulators of splicing, steric-blocking ASOs have been less explored for their potential to alter miRNA abundance for treating disease. Our demonstration that ASOs can be used to either increase or decrease miRNA levels, reveals an alternative way to modulate gene expression by modulating miRNA targeting. This approach could have therapeutic potential in cases where increasing miRNA to downregulate target gene expression would be beneficial.
The exact mechanistic principles behind the enhancement or inhibition of miRNA processing via ASOs remain under investigation. One possibility is that ASO-RNA base-pairing alters the structure or accessibility to RNA-binding proteins subsequently affecting microprocessor activity (Michlewski and Caceres 2019). Genetic variants affecting miRNA structure, protein binding and biogenesis, not unlike the case with IVS45-14T > C, have been described (Fernandez et al. 2017). In addition, G-rich tracts of RNA can form G-quadruplexes (G4), which can interfere with miRNA maturation (Pandey et al. 2015; Rouleau et al. 2018; Liu et al. 2020a,b). ASOs targeted to the G-repeats can relieve this inhibition by presumably preventing G4 folding (Rouleau et al. 2018). We note that miR-1225 has a predicted G4 repeat that could be forming such structures. However, ASO-16, which increases miR-1225 abundance, does not base-pair in the G4 region. Nonetheless, base-pairing of this ASO in the strand complementary to the G4 in the hairpin structure, may block folding into a quadruplex or other inhibitory structures.
Changes in PKD1 expression are a pivotal component of ADPKD, with both overexpression (Lanoix et al. 1996; Ward et al. 1996; Ong et al. 1999; Thivierge et al. 2006; Kurbegovic et al. 2010) and loss of expression or activity (Latinga-van Leeuwen et al. 2004; Parnell et al. 2018) linked to the pathological formation of cysts (Latinga-van Leeuwen et al. 2004; Parnell et al. 2018). Given the observed inverse relationship between PKD1 and miR-1225 expression in correlation with mouse age, that we report here, it is conceivable that miR-1225 is also dysregulated in ADPKD, acting as a potential disease modifier, though further evidence is needed to support this hypothesis conclusively. The PKD1 variant IVS45-14T > C, identified in a patient with ADPKD and no other known mutations, highlights this possibility (Burtey et al. 2002). The initial discovery and analysis of this variant indicated that it caused PKD1 intron 45 retention in an ADPKD patient-derived sample (Burtey et al. 2002). However, the introduction of this variant in our minigene expression system led to a 40%–50% reduction in miR-1225 levels without altering PKD1 mRNA splicing, possibly implicating miR-1225 biogenesis as a cause of disease pathogenesis in this case of ADPKD. While the broader implications of miR-1225 dysregulation in ADPKD remain to be determined, our evidence supports the need to consider miR-1225 in the genetic analysis of ADPKD patients.
The discovery that ASOs can restore WT miR-1225 levels from PKD1 IVS45-14T > C opens the door to new treatment strategies for pathologies associated with miR-1225 depletion, including the various cancers linked to the miRNA (Zheng et al. 2016; Sun et al. 2019; Wang et al. 2019, 2020; Li et al. 2020; Zhang et al. 2020; Gao et al. 2021; Yang et al. 2021; Xia et al. 2023). Overall, our study demonstrates an ASO-based approach to increase the abundance of a miRNA species by targeting biogenesis. This strategy, which manipulates pri- or pre-miRNAs, provides a promising avenue for treatment. Our findings also highlight the need for a comprehensive analysis of genetic co-regulation in the development of therapeutics strategies.
MATERIALS AND METHODS
Human tissue RNA
FirstChoice Human Total RNA Survey Panel (Applied Biosystems) was used for RNA expression analysis in human tissues. The RNA was reverse transcribed with the GoScript Reverse Transcription kit (Promega) according to manufacturer instructions. Synthetic oligonucleotides of known concentrations were used for the standard curves of miR-1225 (Thermo Fisher Scientific). The standard curve for PKD1 was created using PKD1 cDNA of known concentration.
Mice
Mice were bred and treated at Rosalind Franklin University of Medicine and Science; all procedures met the NIH guidelines for the care and use of laboratory animals and were approved by the Institutional Animal Care and Use Committee. Male and female mice (C57BL/6/CBA; heterozygous Ush1C.216A knock-in) were used in approximately equal proportions (Lentz et al. 2013). Mice used to examine Pkd1 and miR-1225 expression between p2 and 13 months were untreated. Neonatal mice were given intraperitoneal injections of 500 mg/kg of ASO at P2 and again at P14, and tissue was collected at P21. Control mice were saline injected. RNA from mouse kidneys was isolated using TRIzol (Thermo Fisher Scientific), as per manufacturer instructions.
RT-PCR
RT-PCR and stem–loop RT-PCR were performed as previously described (Havens et al. 2012). Primer sequences are provided in Supplemental Table S1. PCR products were verified by sequencing. Exogenous human PKD1 was detected with a forward primer in exon 43 and a vector-specific (pTT3) reverse primer to detect only PKD1 expressed from the minigenes. Endogenous mouse mRNA was detected with a forward primer in exon 43 and the reverse in exon 46 and endogenous human mRNA with primers in exon 45 and 46. All miRNAs and snoRNAs were detected using stem–loop RT-PCR (Chen et al. 2005; Havens et al. 2012).
Expression plasmids
The human wild-type (WT) PKD1 minigene was generated previously (Havens et al. 2012). In brief, the human genomic PKD1 from exons 44 to 46, including the intervening introns, were amplified by RT-PCR with Phire PCR Kit (New England Biolabs) and ligated into a pTT3 plasmid. ADPKD-associated variants were generated using the Quik Change Lightening kit (Agilent Technologies) per manufacturer instructions. Plasmid constructs were verified by sequencing.
Antisense oligonucleotides
Synthesis and purification of all ASOs were performed as previously described (Baker et al. 1997; Swayze et al. 2007; Rigo et al. 2014). ASOs were uniformly modified with 2′-O-methoxyethyl sugars (2′MOE) with phosphorothioate (PS) backbone and dissolved in sterile phosphate-buffered saline and sterilized through a 0.2 μm filter. ASO sequences are provided in Supplemental Table S2. A BLAST search with the ASO-16 target sequence against the mouse and human genomes revealed no other perfect sequences matches. The ASOs were diluted to the desired concentration required for dosing mice in sterile 0.9% saline.
Cell culture and transfection
HEK-293T, HeLa, NSC-34, and 89N cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. The 89N cell line was derived from cultured brain tissue of a Ush1c 216G > A mouse (Lentz et al. 2013). Lipofectamine 2000 (Thermo Fisher Scientific) was used for transfection of cells with plasmids and ASOs. Where indicated, cells were transfected with 3 μg of plasmids according to manufacturer instructions. For ASO walks, HeLa cells were cotransfected with a plasmid expressing a human PKD1 minigene and ASO (50 nM). Mouse 89N cells were transfected with ASO alone using Lipofectamine 2000 (Thermo Fisher Scientific). For analysis of the ADPKD1 variants in PKD1 intron 45 and the effect of ASO treatment, the PKD1 minigene plasmid was transfected with or without 100 nM of ASO in HEK-293T cells. In all cases, RNA was collected 48 h after the last transfection using TRIzol (Thermo Fisher Scientific).
Statistics
All statistics were conducted using Prism 10. Specific statistical tests are indicated in the figure legends. Values were considered significant when P < 0.05.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
COMPETING INTEREST STATEMENT
F.R. is an employee of Ionis Pharmaceuticals. A.J.H. is an employee of Abbott Molecular. M.L.H. and F.R. are inventors on patent applications on ASOs filed by RFUMS and Ionis Pharmaceuticals.
ACKNOWLEDGMENTS
We thank James Hogan for editing the manuscript and members of the Hastings and Havens labs for valuable discussions. This work was supported by the National Institutes of Health (NS069759 to M.L.H.; 1F31NS076237 to M.A.H.)
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080021.124.
Freely available online through the RNA Open Access option.
REFERENCES
- Agarwal V, Bell GW, Nam JW, Bartel DP. 2015. Predicting effective microRNA target sites in mammalian mRNAs. Elife 4: e05005. 10.7554/eLife.05005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguiari G, Savelli S, Garbo M, Bozza A, Augello G, Penolazzi L, De Paoli Vitali E, La Torre C, Cappelli G, Piva R, et al. 2000. Novel splicing and missense mutations in autosomal dominant polycystic kidney disease 1 (PKD1) gene: expression of mutated genes. Hum Mutat 16: 444–445. [DOI] [PubMed] [Google Scholar]
- Badenas C, Torra R, San Millan JL, Lucero L, Mila M, Estivill X, Darnell A. 1999. Mutational analysis within the 3′ region of the PKD1 gene. Kidney Int 55: 1225–1233. 10.1046/j.1523-1755.1999.00368.x [DOI] [PubMed] [Google Scholar]
- Baker BF, Lot SS, Condon TP, Cheng-Flournoy S, Lesnik EA, Sasmor HM, Bennett CF. 1997. 2'-O-(2-methoxy)ethyl-modified anti-intercellular adhesion molecule 1 (ICAM-1) oligonucleotides selectively increase the ICAM-1 mRNA level and inhibit formation of the ICAM-1 translation initiation complex in human umbilical vein endothelial cells. J Biol Chem 272: 11994–2000. 10.1074/jbc.272.18.11994 [DOI] [PubMed] [Google Scholar]
- Bennett CF, Baker BF, Pham N, Swayze E, Geary RS. 2017. Pharmacology of antisense drugs. Annu Rev Pharmacol Toxicol 57: 81–105. 10.1146/annurev-pharmtox-010716-104846 [DOI] [PubMed] [Google Scholar]
- Burtey S, Lossi AM, Bayle J, Berland Y, Fontes M. 2002. Mutation screening of the PKD1 transcript by RT-PCR. J Med Genet 39: 422–429. 10.1136/jmg.39.6.422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauvet V, Qian F, Boute N, Cai Y, Phakdeekitacharoen B, Onuchic LF, Attie-Bitach T, Guicharnaud L, Devuyst O, Germino GG, et al. 2002. Expression of PKD1 and PKD2 transcripts and proteins in human embryo and during normal kidney development. Am J Pathol 160: 973–983. 10.1016/S0002-9440(10)64919-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Wang X. 2020. miRDB: an online database for prediction of functional microRNA targets. Nucleic Acids Res 48: D127–D131. 10.1093/nar/gkz757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, et al. 2005. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res 33: e179. 10.1093/nar/gni178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez N, Cordiner RA, Young RS, Hug N, Macias S, Caceres JF. 2017. Genetic variation and RNA structure regulate microRNA biogenesis. Nat Commun 8: 15114. 10.1038/ncomms15114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S, Shi P, Tian Z, Yang X, Liu N. 2021. Overexpression of miR-1225 promotes the progression of breast cancer, resulting in poor prognosis. Clin Exp Med 21: 287–296. 10.1007/s10238-020-00676-7 [DOI] [PubMed] [Google Scholar]
- Geng L, Segal Y, Peissel B, Deng N, Pei Y, Carone F, Rennke HG, Glucksmann-Kuis AM, Schneider MC, Ericsson M, et al. 1996. Identification and localization of polycystin, the PKD1 gene product. J Clin Invest 98: 2674–2682. 10.1172/JCI119090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng L, Segal Y, Pavlova A, Barros EJ, Lohning C, Lu W, Nigam SK, Frischauf AM, Reeders ST, Zhou J. 1997. Distribution and developmentally regulated expression of murine polycystin. Am J Physiol 272: F451–F459. 10.1152/ajprenal.1997.272.4.F451 [DOI] [PubMed] [Google Scholar]
- Gong Y, Wei Z, Liu J. 2021. MiRNA-1225 inhibits osteosarcoma tumor growth and progression by targeting YWHAZ. Onco Targets Ther 14: 15–27. 10.2147/OTT.S282485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havens MA, Hastings ML. 2016. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res 44: 6549–6563. 10.1093/nar/gkw533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havens MA, Reich AA, Duelli DM, Hastings ML. 2012. Biogenesis of mammalian microRNAs by a non-canonical processing pathway. Nucleic Acids Res 40: 4626–4240. 10.1093/nar/gks026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurbegovic A, Côté O, Couillard M, Ward CJ, Harris PC, Trudel M. 2010. Pkd1 transgenic mice: adult model of polycystic kidney disease with extrarenal and renal phenotypes. Hum Mol Genet 19: 1174–1189. 10.1093/hmg/ddp588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanoix J, D'Agati V, Szabolcs M, Trudel M. 1996. Dysregulation of cellular proliferation and apoptosis mediates human autosomal dominant polycystic kidney disease (ADPKD). Oncogene 13: 1153–1160. [PubMed] [Google Scholar]
- Latinga-van Leeuwen IS, Dauwerse G, Baelde HJ, Leonhard WN, van de Wal A, Ward CJ, Verbeek S, Deruiter MC, Breuning MH, et al. 2004. Lowering of Pkd1 expression is sufficient to cause polycystic kidney disease. Hum Mol Genet 13: 3069–3077. 10.1093/hmg/ddh336 [DOI] [PubMed] [Google Scholar]
- Lentz JJ, Jodelka FM, Hinrich AJ, McCaffrey KE, Farris HE, Spalitta MJ, Bazan NG, Duelli DM, Rigo F, Hastings ML. 2013. Rescue of hearing and vestibular function by antisense oligonucleotides in a mouse model of human deafness. Nat Med 19: 345–350. 10.1038/nm.3106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Zhang F, Li H. 2020. miR-1225-5p inhibits non-small cell lung cancer cell proliferation, migration and invasion, and may be a prognostic biomarker. Exp Ther Med 20: 172. 10.3892/etm.2020.9302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Du W, Xu H, Sun Q, Tang D, Zou S, Zhang Y, Ma M, Zhang G, Du X, et al. 2020a. RNA G-quadruplex regulates microRNA-26a biogenesis and function. J Hepatol 73: 371–382. 10.1016/j.jhep.2020.02.032 [DOI] [PubMed] [Google Scholar]
- Liu L, Zhang W, Hu Y, Ma L, Xu X. 2020b. Downregulation of miR-1225-5p is pivotal for proliferation, invasion, and migration of HCC cells through NFκB regulation. J Clin Lab Anal 34: e23474. 10.1002/jcla.23474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michlewski G, Cáceres JF. 2019. Post-transcriptional control of miRNA biogenesis. RNA 25: 1–16. 10.1261/rna.068692.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong AC, Ward CJ, Butler RJ, Biddolph S, Bowker C, Torra R, Pei Y, Harris PC. 1999. Coordinate expression of the autosomal dominant polycystic kidney disease proteins, polycystin-2 and polycystin-1, in normal and cystic tissue. Am J Pathol 154: 1721–1729. 10.1016/S0002-9440(10)65428-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey S, Agarwala P, Jayaraj GG, Gargallo R, Maiti S. 2015. The RNA stem-loop to G-quadruplex equilibrium controls mature microRNA production inside the cell. Biochemistry 54: 7067–7078. 10.1021/acs.biochem.5b00574 [DOI] [PubMed] [Google Scholar]
- Parnell SC, Magenheimer BS, Maser RL, Pavlov TS, Havens MA, Hastings ML, Jackson SF, Ward CJ, Peterson KR, Staruschenko A, et al. 2018. A mutation affecting polycystin-1 mediated heterotrimeric G-protein signaling causes PKD. Hum Mole Genet 27: 3313–3324. 10.1093/hmg/ddy223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reziwan K, Sun D, Zhang B, Zhao Z. 2019. MicroRNA-1225 activates Keap1-Nrf2-HO-1 signalling to inhibit TNFα-induced osteoclastogenesis by mediating ROS generation. Cell Biochem Funct 37: 256–265. 10.1002/cbf.3394 [DOI] [PubMed] [Google Scholar]
- Rigo F, Chun SJ, Norris DA, Hung G, Lee S, Matson J, Fey RA, Gaus H, Hua Y, Grundy JS, et al. 2014. Pharmacology of a central nervous system delivered 2'-O-methoxyethyl-modified survival of motor neuron splicing oligonucleotide in mice and nonhuman primates. J Pharmacol Exp Ther 350: 46–55. 10.1124/jpet.113.212407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodova M, Islam MR, Peterson KR, Calvet JP. 2003. Remarkable sequence conservation of the last intron in the PKD1 gene. Mol Biol Evol 20: 1669–1674. 10.1093/molbev/msg191 [DOI] [PubMed] [Google Scholar]
- Rouleau SG, Garant JM, Bolduc F, Bisaillon M, Perreault JP. 2018. G-Quadruplexes influence pri-microRNA processing. RNA Biol 15: 198–206. 10.1080/15476286.2017.1405211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sempere LF, Azmi AS, Moore A. 2021. microRNA-based diagnostic and therapeutic applications in cancer medicine. Wiley Interdiscip Rev RNA 12: e1662. 10.1002/wrna.1662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang R, Lee S, Senavirathne G, Lai EC. 2023. microRNAs in action: biogenesis, function and regulation. Nat Rev Genet 24: 816–833. 10.1038/s41576-023-00611-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun P, Zhang D, Huang H, Yu Y, Yang Z, Niu Y, Liu J. 2019. MicroRNA-1225-5p acts as a tumor-suppressor in laryngeal cancer via targeting CDC14B. Biol Chem 400: 237–246. 10.1515/hsz-2018-0265 [DOI] [PubMed] [Google Scholar]
- Swayze EE, Siwkowski AM, Wancewicz EV, Migawa MT, Wyrzykiewicz TK, Hung G, Monia BP, Bennett CF. 2007. Antisense oligonucleotides containing locked nucleic acid improve potency but cause significant hepatotoxicity in animals. Nucleic Acids Res 35: 687–700. 10.1093/nar/gkl1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thivierge C, Kurbegovic A, Couillard M, Guillaume R, Coté O, Trudel M. 2006. Overexpression of PKD1 causes polycystic kidney disease. Mol Cell Biol 26: 1538–1548. 10.1128/MCB.26.4.1538-1548.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Chen X, Zhang Z, Wu Z. 2019. MicroRNA-1225-5p inhibits the development and progression of thyroid cancer via targeting sirtuin 3. Pharmazie 74: 423–427. [DOI] [PubMed] [Google Scholar]
- Wang GH, Wang LY, Zhang C, Zhang P, Wang CH, Cheng S. 2020. MiR-1225-5p acts as tumor suppressor in glioblastoma via targeting FNDC3B. Open Med (Wars) 15: 872–881. 10.1515/med-2020-0156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward CJ, Turley H, Ong AC, Comley M, Biddolph S, Chetty R, Ratcliffe PJ, Gattner K, Harris PC. 1996. Polycystin, the polycystic kidney disease 1 protein, is expressed by epithelial cells in fetal, adult, and polycystic kidney. Proc Natl Acad Sci 93: 1524–1528. 10.1073/pnas.93.4.1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westholm JO, Lai EC. 2011. Mirtrons: microRNA biogenesis via splicing. Biochimie 93: 1897–1904. 10.1016/j.biochi.2011.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkle M, El-Daly SM, Fabbri M, Calin GA. 2021. Noncoding RNA therapeutics - challenges and potential solutions. Nat Rev Drug Discov 20: 629–651. 10.1038/s41573-021-00219-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia L, Ge M, Shan G, Qian H, Xia Y. 2023. The effects of circ_000558/miR-1225-5p/ARL4C on regulating the proliferation of renal cell carcinoma cells. J Oncol 2023: 1–12. 10.1155/2023/1303748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Shen Z, Zou Y, Gao K. 2021. Rosmarinic acid inhibits migration, invasion, and p38/AP-1 signaling via miR-1225-5p in colorectal cancer cells. J Recept Signal Transduct Res 41: 284–293. 10.1080/10799893.2020.1808674 [DOI] [PubMed] [Google Scholar]
- Zhang W, Wei L, Sheng W, Kang B, Wang D, Zeng H. 2020. miR-1225-5p functions as a tumor suppressor in osteosarcoma by targeting Sox9. DNA Cell Biol 39: 78–91. 10.1089/dna.2019.5105 [DOI] [PubMed] [Google Scholar]
- Zhang D, Zhang Y, Zhang X, Zhai H, Sun X, Li Y. 2022. Circ_0091579 serves as a tumor-promoting factor in hepatocellular carcinoma through miR-1225-5p/PLCB1 axis. Dig Dis Sci 67: 585–597. 10.1007/s10620-021-06861-2 [DOI] [PubMed] [Google Scholar]
- Zheng H, Zhang F, Lin X, Huang C, Zhang Y, Li Y, Lin J, Chen W, Lin X. 2016. MicroRNA-1225-5p inhibits proliferation and metastasis of gastric carcinoma through repressing insulin receptor substrate-1 and activation of β-catenin signaling. Oncotarget 7: 4647–4663. 10.18632/oncotarget.6615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong R, Li S, Fang K, Yang L, Wang L. 2019. microRNA-1225 inhibit apoptosis of pancreatic cancer cells via targeting JAK1. Cell Cycle 18: 990–1000. 10.1080/15384101.2019.1608127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31: 3406–3415. 10.1093/nar/gkg595 [DOI] [PMC free article] [PubMed] [Google Scholar]