

Abstract
Rett syndrome (RTT) is a neurodevelopmental disorder caused by loss-of-function mutations in the methyl-CpG-binding protein 2 (MECP2) gene. Despite its severe phenotypes, studies in mouse models suggest that restoring MeCP2 levels can reverse RTT symptomology. Nevertheless, traditional gene therapy approaches are hindered by MeCP2's narrow therapeutic window, complicating the safe delivery of viral constructs without overshooting the threshold for toxicity. The 3′ untranslated region (3′ UTR) plays a key role in gene regulation, where factors like miRNAs bind to pre-mRNA and fine-tune expression. Given that each miRNA's contribution is modest, blocking miRNA binding may represent a potential therapeutic strategy for diseases with high dosage sensitivity, like RTT. Here, we present a series of site-blocking antisense oligonucleotides (sbASOs) designed to outcompete repressive miRNA binding at the MECP2 3′ UTR. This strategy aims to increase MeCP2 levels in patients with missense or late-truncating mutations, where the hypomorphic nature of the protein can be offset by enhanced abundance. Our results demonstrate that sbASOs can elevate MeCP2 levels in a dose-dependent manner in SH-SY5Y and patient fibroblast cell lines, plateauing at levels projected to be safe. Confirming in vivo functionality, sbASO administration in wild-type mice led to significant Mecp2 upregulation and the emergence of phenotypes associated with Mecp2 overexpression. In a T158M neural stem cell model of RTT, sbASO treatment significantly increased MeCP2 expression and levels of the downstream effector protein brain-derived neurotrophic factor (BDNF). These findings highlight the potential of sbASO-based therapies for MeCP2-related disorders and advocate for their continued development.
Keywords: Rett syndrome, site-blocking, antisense, oligonucleotides, MeCP2
INTRODUCTION
Rett syndrome (RTT) is a debilitating neurodevelopmental disorder that affects approximately one in 10,000 female live births. In 1999, researchers identified de novo loss-of-function mutations in the methyl-CpG-binding protein 2 (MECP2) gene as the cause of RTT (Amir et al. 1999). It is now estimated that these mutations account for ∼95% of classic RTT cases. Over two decades of intensive research has led to an improved understanding of how MeCP2 functions in health and disease; however, therapeutic advancements have been minimal, with existing treatments primarily focused on alleviating symptoms and improving quality of life.
In rodent models, the absence of Mecp2 mimics the phenotypes of RTT in humans, including acquired microcephaly, apneas, seizures, impaired sociability, cognitive deficits, and hindlimb clasping, which parallel the hand stereotypies seen in humans. (Chen et al. 2001; Guy et al. 2001). Restoring Mecp2 levels in these models has been shown to reverse many of these symptoms, even in advanced disease stages (Luikenhuis et al. 2004; Guy et al. 2007; Garg et al. 2013). These findings support the widely accepted theory that, despite its myriad of challenges, RTT is reversible. As a result, normalizing MeCP2 expression in the brain has become the primary therapeutic goal.
Several strategies to restore MeCP2 expression are currently under development. These include genome editing, base editing, RNA editing, and MECP2 delivery platforms such as gene therapy, protein replacement, and mRNA therapy (Gadalla et al. 2013; Sinnett et al. 2017, 2021; Carrette et al. 2018; Luoni et al. 2020; Grimm and Lee 2022; Bijlani et al. 2024; Flynn et al. 2024). However, many of these approaches are complicated by the precise dosage requirements of MeCP2, as both insufficient and excessive levels can cause severe neurological phenotypes. For example, even a modest 1× overexpression can lead to MeCP2 duplication syndrome (MDS), which is characterized by intellectual disability, motor dysfunction, and autistic features (Collins et al. 2004; D'Mello III 2021; Collins and Neul 2022). This narrow therapeutic window requires innovative approaches to safely and effectively elevate levels of the protein. Additionally, while genome editing therapies hold promise for treating RTT by correcting MECP2 mutations, they are difficult to implement across the entire brain and carry the risk of permanent adverse effects associated with off-target indels. These challenges highlight the need for alternative strategies that allow for more controlled modulation of MeCP2 expression.
A critical discrepancy between preclinical and clinical RTT is the routine use of the Mecp2 knockout (KO) allele in mice. While informative for basic science research, this model presents significant challenges in translating findings to clinical applications. Many of the most common patient mutations either render key functional domains hypomorphic or are late-truncating, allowing for partial activity to be retained. Eight MeCP2 mutations account for 70% of all RTT cases, with five (R106W, R133C, T158M, R294X, R306C) believed to maintain partial functionality (Neul et al. 2008; Frullanti et al. 2019). The level of function retained by each mutant form of the protein is predictive of clinical outcomes. Missense or late truncating mutations such as R133C, T158M, R294X, and R306C are associated with milder phenotypes compared to early truncating mutations like R168X and R255X (McGowan and Pang 2015; Rodrigues et al. 2020). The preserved functionality of these mutations raises the possibility that increasing levels of the mutant protein could be exploited therapeutically. This potential was demonstrated in experiments showing that a MECP2-T158M mutant transgene was able to rescue RTT-like phenotypes in Mecp2T158M/y knock-in mice (Lamonica et al. 2017).
One mechanism of increasing levels of the mutant MeCP2 protein is through post-transcriptional regulation. The 3′ untranslated region (3′ UTR) of MECP2 plays a well-defined role in regulating mRNA stability and translation (McGowan and Pang 2015; Rodrigues et al. 2020; Hong and Jeong 2023). MicroRNAs (miRNAs) and RNA-binding proteins interact with complementary sequences within the 3′ UTR, leading to mRNA degradation or translational inhibition (Shang et al. 2023). For proteins with high dosage sensitivity, these interactions are crucial for sustaining appropriate levels of expression. In RTT, this finely tuned regulatory mechanism maintains the homeostatic balance of MeCP2, which is essential for normal neuronal function (Cheng et al. 2014; Horvath et al. 2022; Jauhari et al. 2022).
There are over 35 predicted miRNA-binding sites in MECP2′s 3′ UTR, often targeted by multiple miRNAs (McGowan and Pang 2015; Rodrigues et al. 2020). Primary mutations in miRNA seed sequences have been reported in patients with RTT and MDS (Santos et al. 2008), underscoring the importance of post-transcriptional regulation. Research has shown that overexpression of miRNAs can reduce MeCP2 protein levels, while mutation in their predicted binding sites results in increased expression (Urdinguio et al. 2010; Khan et al. 2018; Horvath et al. 2022). Importantly, the regulatory effect of each miRNA is modest, creating a ceiling effect on their capacity to elevate MeCP2 levels when disrupted (Shang et al. 2023). We propose that preventing these miRNA-MECP2 interactions is a viable therapeutic approach to increase MeCP2 dosage while remaining within the narrow safety window of the protein.
To test this hypothesis, we have developed a series of site-blocking antisense oligonucleotides (sbASOs) designed to bind to miRNA seed regions in the 3′ UTR and de-repress MeCP2 expression. Our sbASOs also bind to peripheral sequences that are unique to MECP2, ensuring a highly specific interaction that allows the target miRNA to maintain its regulatory interactions at all other loci. These sbASOs are modified with phosphorothioate backbones (PS) to increase stability (Clavé et al. 2021) and locked nucleic acids (LNA) to enhance binding affinity (Koshkin et al. 1998; Vester and Wengel 2004; Obad et al. 2011). The PS modification can extend in vivo stability to over six months, allowing for a sustained effect that is also reversible as the sbASO degrades. In this paper, we explore the potential of sbASOs to regulate MeCP2 expression by targeting three distinct miRNA-binding sites within the MECP2 3′ UTR (miRNAs 22-3p, 132-3p, and 483-5p). We provide comprehensive data on the efficacy of this approach in modulating MeCP2 protein levels in vitro and in vivo, supporting the continued exploration of sbASOs as potential RTT therapeutics.
RESULTS
Differential expression of miRNAs in RTT autopsy samples
To define the miRNA expression profile in the brains of RTT patients, we obtained age-, sex-, and postmortem interval (PMI)-matched temporal cortex autopsy samples from five neurotypical controls, three individuals with the T158M mutation, and four with the R255X mutation in MeCP2 (Supplemental Table S1). Total miRNA levels were quantified using nCounter miRNA Expression Panels and compared relative to neurotypical controls. When analyzed as a combined “all RTT” group encompassing both mutations, we identified 66 significantly downregulated and 78 significantly upregulated miRNAs (Fig. 1A; Supplemental Table S2). The top 15 upregulated and downregulated miRNAs are depicted in Supplemental Figure S1. Further analysis by patient subpopulation revealed distinct miRNA expression patterns specific to each mutation. We quantified nine decreased and 19 increased miRNAs that were uniquely disrupted in patients with the T158M mutation (Fig. 1A). Conversely, there were five increased and 15 decreased miRNAs that were specific to the R255X mutation (Fig. 1A).
FIGURE 1.
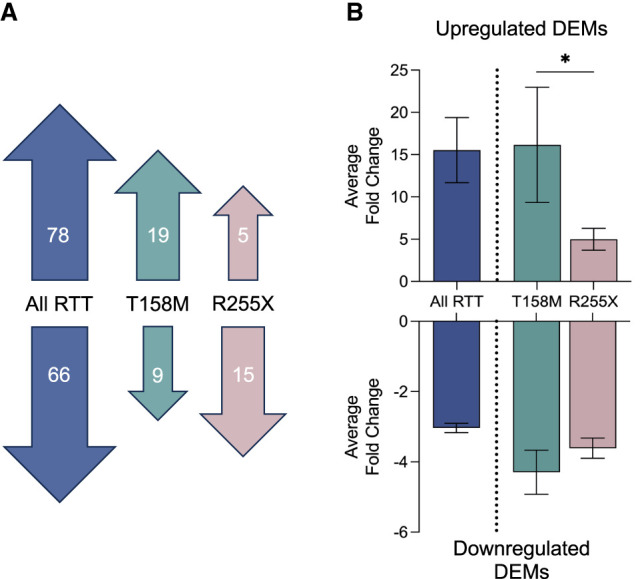
Differential miRNA expression in RTT autopsy samples. Nanostring data on total miRNA levels in temporal cortical tissue from T158M (n = 3) and R255X (n = 5) RTT patients. (A) Overview of differentially expressed microRNAs (DEMs) in the temporal cortex of RTT patients, comparing the combined “all RTT” group with neurotypical controls. The analysis identified 78 DEMs with ∼16-fold higher expression and 67 DEMs with around threefold lower expression in RTT individuals compared to controls. (B) Comparison of the average fold change of upregulated DEMs common to both T158M and R255X mutations. The data revealed a significantly greater average fold change in the T158M group compared to the R255X group ([*] P = 0.02352, Student's t-test). Data are presented as mean ± SEM. Refer to Supplemental Figure S1 and Table S2 for additional information.
When comparing the magnitude of change between the increased and decreased miRNAs, those that were upregulated exhibited a significantly greater disruption (∼16-fold) relative to those that were downregulated (approximately threefold) (Fig. 1B). This suggests that a primary function of MeCP2 may involve repressing miRNA biogenesis and that mutations in MeCP2 result in a de-repression of global miRNA levels (Cheng et al. 2014; Woo and Kim 2014). Mutation-specific effects were also evident in this analysis, with the T158M mutation causing significantly higher disruptions than the R255X mutation (Fig. 1B). The average difference between the two groups was 10-fold, with the largest difference being 275-fold. Given that the R255X mutation is among the most clinically severe (Brown et al. 2016; Frullanti et al. 2019), these results demonstrate the surprising result of a greater impact on miRNA regulation by the T158M mutation.
MECP2-targeted sbASO design
Increasing the level of mutant MeCP2 protein has been shown to rescue symptoms in RTT model mice (Lamonica et al. 2017). We hypothesized that steric blockage of miRNA repression is a viable mechanism to achieve this goal. To test this theory, we designed antisense oligonucleotides (ASOs) that were complimentary to the 3′-UTR miRNA seed sequence and the surrounding sequence specific to the MECP2 3′ UTR (Fig. 2). These site-blocking ASOs were modified with a phosphorothioate backbone to improve stability and LNA modifications to increase binding affinity (Koshkin et al. 1998; Vester and Wengel 2004; Clavé et al. 2021).
FIGURE 2.

Schematic representation of sbASO-mediated disinhibition approach on MECP2 3′ UTR. (A) RNA-induced silencing complexes (RISCs) bind to complementary microRNA response elements (MREs) on the MECP2 3′ UTR, resulting in translational repression or transcript destabilization. (B) sbASOs are designed to target these MREs, thereby preventing the binding of RISCs. This action blocks miRNA-mediated repression, allowing for increased expression of MeCP2. The schematic illustrates the potential of sbASOs to de-repress MeCP2 by competitively inhibiting miRNA binding to the MECP2 3′ UTR. (C) Sequences of sbASO target sites and percentage conservation between mice and humans are shown. (ORF) Open reading frame.
Three target miRNAs were selected for proof-of-concept studies based on two main criteria: (1) existing literature demonstrating their repressive effects on MECP2 (Hansen et al. 2010; Alvarez-Saavedra et al. 2011; Han et al. 2013; Jovicic et al. 2013; Pejhan et al. 2020; Tong et al. 2020; Rastegar-Moghaddam et al. 2022; Matson et al. 2023) and (2) Nanostring nCounter analysis showing increased expression of the target miRNA in clinical populations, suggesting an enhanced role in repressing mutant forms of MeCP2. miR-22-3p and miR-132-3p fulfilled both criteria, while miR-483-5p was not detected in the nCounter panel but had substantial preclinical evidence supporting its interaction with MECP2 (Han et al. 2013; Matson et al. 2023).
Dose-dependent efficacy of sbASOs in vitro
The efficacy of all three sbASO was initially evaluated in the human SH-SY5Y neuroblastoma cell line to establish proof of concept. This cell line was chosen for its neuron-like characteristics, high levels of MeCP2 expression, and the relative abundance of all three target miRNAs (Jung et al. 2003; Lopez-Suarez et al. 2022). sbASOs were transfected with a dose range of 1.5, 5, 50, and 125 nM, and nuclear protein was isolated 48 h later. Under identical conditions, a similarly modified fluorescent control ASO (BLOCK-iT Fluorescent Oligo) achieved ∼ 50%–75% transfection efficiency with no cell death observed at concentrations below 250 nM (Supplemental Fig. S2A).
All three sbASOs caused a significant and dose-dependent increase in MeCP2 expression, quantified via fluorescent western blot. Elevated MeCP2 protein levels reached a ceiling effect at low (40% for sbASO.miR-22), intermediate (75% for sbASO.miR-483), and high (200% for sbASO.miR-132) levels, highlighting the distinct regulatory capacity of each miRNA (Fig. 3B–D). The expression plateau observed between 50 and 125 nM suggests that the repressive effects of each miRNA can be saturated over a broad concentration range, which is a critical advantage of this approach in the context of MeCP2's narrow therapeutic index (Collins and Neul 2022).
FIGURE 3.

Dose-dependent increase of MeCP2 expression in SH-SY5Y cells. (A) Schematic of the experimental setup for evaluating the effect of sbASOs on SH-SY5Y neuroblastoma cells. (B–D) Western blot analysis showing the dose-dependent effects of sbASOs on MeCP2 protein levels. Quantification was normalized to Histone H3 (H3), with values represented as a percentage relative to vehicle treatment. (B) sbASO.miR-22 significantly increased MeCP2 levels at 125 nM ([***] P = 0.0002). (C) sbASO.miR-132 significantly increased MeCP2 levels at 1.5 nM ([*] P = 0.0468), 5 nM ([***] P = 0.0002), and 50 nM ([**] P = 0.0013). (D) sbASO.miR-483 increased MeCP2 levels at 1.5 nM ([*] P = 0.0462) and 5 nM ([*] P = 0.0375). Representative western blots showing the relative quantities of MeCP2 treated with different sbASO concentrations are located directly below each bar graph (the upper band representing MeCP2 [71 kDa] and the lower band representing H3 [17 kDa]). n = 3 per concentration per sbASO. (E) MeCP2 expression was further analyzed in SH-SY5Y cells transfected with subeffective concentrations of sbASOs (miR-22 = 5 nM, miR-483 = 1.5 nM, and miR-132 = 0.5 nM), demonstrating additive effects. V = vehicle, C = combined sbASOs. Statistical significance was determined using a two-way ANOVA with Tukey's post hoc test, (**) P < 0.01. n = 3–4 per sbASO. (F) MECP2 mRNA levels in sbASO-treated SH-SY5Y cells were measured via qRT-PCR after treatment with 50 nM sbASO.miR-22, sbASO.miR-132, and sbASO.miR-483. No significant changes in MECP2 mRNA levels were observed in response to sbASO treatment. n = 4–6 per sbASO. Outliers were identified and excluded using Grubb's test at α = 0.1. Data are presented as mean ± SEM.
To build on these findings, we explored the potential for additive effects by cotransfecting all three sbASOs simultaneously at concentrations in which no significant effect was observed individually. Cotransfection at subeffective concentrations (sbASO.miR-22 = 5 nM, sbASO.miR-483 = 1.5 nM, and sbASO.miR-132 = 0.5 nM) resulted in a significant increase in MeCP2 expression in SH-SY5Y cells, confirming the potential of sbASOs to have additive effects (Fig. 3E).
To determine whether the increase in MeCP2 protein was mediated through transcriptional or translational mechanisms, we examined MECP2 mRNA levels. Total RNA was isolated from SH-SY5Y cells treated with 50 nM sbASO, followed by cDNA synthesis and qRT-PCR of MECP2 mRNA. The 50 nM dosage was selected as it significantly increased MeCP2 protein across all sbASOs (Fig 3B–D). qRT-PCR analysis revealed that cells treated at this concentration showed no changes in MECP2 mRNA levels. These findings indicate that the sbASOs exert their effects primarily through translational repression rather than mRNA destabilization (Fig. 3F). In summary, administration of all three sbASOs in SH-SY5Y cells led to a dose-dependent increase in MeCP2 protein, which plateaued within the predicated therapeutic window for RTT.
sbASOs increase Mecp2 expression in WT mice
Having established proof of concept for the sbASO approach in vitro, we sought to establish a similar efficacy profile in vivo. The human-to-mouse homology of the sbASO.miR-22, sbASO.miR-132, sbASO.miR-483 target sequences are 100%, 100%, 55%, respectively (Fig. 2C). Consequently, at least sbASO.miR-22 and sbASO.miR-132 are predicted to function similarly in both species. Effective delivery of ASOs to the brain and spinal cord has been demonstrated via several distinct mechanisms in rodents (DeVos and Miller 2013). In our study, we used an osmotic pump implanted subcutaneously, connected via a catheter to a cannula surgically placed into the right lateral ventricle of wild-type (WT) C57BL/6J mice (Fig. 4A). This method was chosen based on its demonstrated effectiveness in seminal studies on spinal muscular atrophy (SMA) (Hua et al. 2010; Passini et al. 2011) and amyotrophic lateral sclerosis (ALS) (McCampbell et al. 2018; Miller et al. 2020). Successful catheter implantation and robust delivery potential were verified using 2.5% FastGreen dye, as coronal brain sections demonstrated the presence of the dye throughout the ventricular system (Supplemental Fig. S3). sbASO or saline solutions were infused into the cerebral spinal fluid for 5 days at a concentration of 50 µg/day. Nuclear Mecp2 protein was quantified in the frontal cortex and hippocampus using a fluorescent western blot.
FIGURE 4.
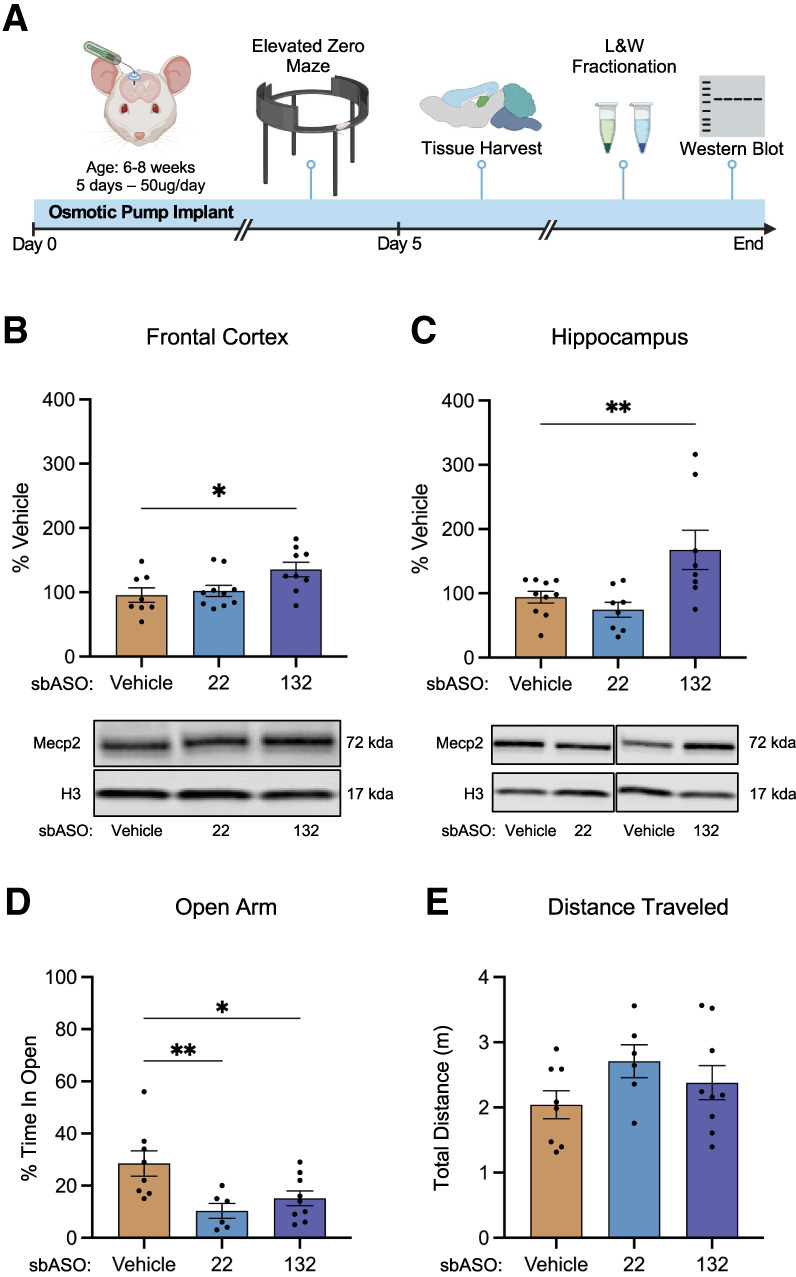
sbASOs increase Mecp2 protein expression and induce MDS-like phenotypes in WT mice. (A) Schematic illustration of the experimental workflow, detailing the implantation of osmotic pumps for continuous sbASO administration and subsequent treatment protocols. (B) Western blot analysis of Mecp2 protein expression in the nuclear fraction of the frontal cortex of WT mice treated with sbASO.miR-22 and sbASO.miR-132. Protein quantification was normalized to Histone H3 (H3), with data represented as the mean percentage relative to the vehicle-treated controls. Statistical analysis via one-way ANOVA showed a significant effect of treatment ([*] P = 0.0265), with Fisher's LSD post hoc analysis indicating no significant difference for sbASO.miR-22 (P = 0.6567), but a significant increase for sbASO.miR-132 ([*] P = 0.0137). (C) Western blot analysis of Mecp2 expression in the nuclear fraction of the hippocampus of WT mice treated with sbASO.miR-22 and sbASO.miR-132. Quantification was also normalized to H3, with values shown as mean percentages relative to vehicle controls. One-way ANOVA indicated a significant effect ([**] P = 0.0052), with Fisher's LSD revealing no significant difference for sbASO.miR-22 (P = 0.4535), but a significant increase for sbASO.miR-132 ([**] P = 0.0089). The sample size for each treatment was n = 8–10. Representative western blots showing Mecp2 levels for each treatment (vehicle, 22 = sbASO.miR-22 and 132 = sbASO.miR-132) in both the frontal cortex and hippocampus, with the upper band representing Mecp2 (71 kDa) and the lower band representing H3 (17 kDa), are located directly below each bar graph. (D) Open arm index assessment in the EZM revealed that WT animals treated with sbASO.miR-22 [Bonferroni post hoc (df,20) (**) P = 0.0078] and sbASO.miR-132 [Bonferroni post hoc (df,20) (*)P = 0.0283] exhibited a significant increase in the time spent in the open arm compared to the closed arm of the maze. (E) No significant difference in total distance traveled was detected between treatment groups for sbASO.miR-22 [Bonferroni post hoc (df,20) (P = 0.1734)] and sbASO.miR-132 [Bonferroni post hoc (df,20) (P = 0.6414)]. n = 8–10 per treatment. Outliers were removed using Grubb's tests at α = 0.1. Data are presented as mean ± SEM.
Animals treated with sbASO.miR-22 and sbASO.miR-132 recovered well postimplantation, showing no adverse effects throughout the study period. Conversely, mice administered sbASO.miR-483 exhibited severe adverse phenotypes, including ataxia and orofacial dyskinesia, resulting in euthanasia before the full 5-day postimplantation period. It is unknown whether these effects were on- or off-target mediated; however, due to their severity, sbASO.miR-483 was excluded from all remaining in vivo experiments.
Western blot analysis showed that sbASO.miR-22 did not produce a notable increase in Mecp2 protein in either brain region examined (Fig. 4B,C). In contrast, with only 5 days postimplantation, sbASO.miR-132 exhibited strong efficacy, resulting in a significant increase in Mecp2 of 42% in the frontal cortex and 80% in the hippocampus (Fig. 4B,C).
sbASOs induce anxiety-like phenotypes in WT mice
Mecp2 dosage is known to bidirectionally regulate several phenotypes in mice, including anxiety in the elevated zero maze (EZM), motor learning on the rotarod, and associative learning in contextual fear conditioning assays (Na et al. 2012; Collins and Neul 2022). In this study, anxiety-like behaviors were selected as the primary outcome measure, as the presence of an osmotic pump could confound assessments of motor and freezing behaviors. To assess whether sbASO administration led to the predicted changes in anxiety, WT mice were infused with either saline, sbASO.miR-22, or sbASO.miR-132, and tested in the EZM after 5 days. sbASO-treated mice exhibited a significant reduction in the time spent in the open sections of the maze compared to the saline group (Fig. 4D), indicative of increased anxiety-like behavior. Importantly, there were no significant differences in the total distance traveled in the EZM between any of the treatment groups (Fig. 4E), suggesting that the anxiety-like phenotype was not attributable to altered locomotor activity. Notably, the anxiety-related outcomes following sbASO.miR-22 administration were observed despite the absence of significant changes in Mecp2 protein expression in the hippocampus and frontal cortex (Fig. 4B,C). This unexpected finding suggests that sbASO.miR-22 may influence anxiety-like behaviors through mechanisms involving Mecp2 regulation in brain regions not examined or in subpopulations of cells that were too granular to detect in bulk tissue analysis.
Mutation-specific efficacy of sbASOs in RTT patient-derived fibroblasts
Our findings from female RTT autopsies suggest that the magnitude of miRNA disruption may be uniquely affected by mutation type, potentially impacting the efficacy of sbASOs in different patient subpopulations. To investigate this variable in vitro, we administered sbASOs to fibroblast lines derived from RTT patients with five of the most common MeCP2 mutations (R133C, T158M, R306C, R270X, and R294X) and compared protein levels relative to vehicle-treated controls from each line (Fig. 5A,B). Consistent with previous observations, human fibroblasts demonstrated rapid uptake of the control fluorescent ASOs, achieving ∼75% transfection efficiency after 12 h (Supplemental Fig. S2B).
FIGURE 5.

Mutation-specific sbASO efficacy in RTT patient-derived human fibroblast cells. (A) Schematic representation of the experimental design, illustrating the treatment of human fibroblast cells (hFIBs) derived from RTT patients with sbASOs for biochemical analysis of nuclear fractions. (B) List of hotspot mutations that account for 70% of all RTT mutations. The mutations used in this study are highlighted in red. Our results demonstrated that sbASOs elevated MeCP2 levels in a dose-dependent and mutation-specific manner. (C–D) The optimal concentrations of each sbASO for increasing MeCP2 levels in various RTT patient-derived cell lines. (C) sbASO.miR-22 administration increased MeCP2 levels in R133C (50 nM), R306C (125 nM), and MT R294X (1.5 nM) cell lines. No significant effect was observed in T158M, WT R270X, and WT R294X. (D) sbASO.miR-132 treatment elevated MeCP2 expression across all tested cell lines, including R133C (50 nM), T158M (27.5 nM), R306C (125 nM), WT R270X (27.5 nM), WT R294X (1.5 nM), and MT R294X (27.5 nM). (E) sbASO.miR-483 administration increased MeCP2 expression in T158M (27.5 nM), WT R270X (27.5 nM), and MT R294X (27.5 nM). No significant changes were noted in R133C, R306C, and WT R294X. n = 3–5/mutation/sbASO. Statistical significance was determined using Student's t-test, with (*) P < 0.05, (**) P < 0.01, (***) P < 0.001, (****) P < 0.0001. Outliers were identified and excluded using Grubb's test at α = 0.1. Data are presented as mean ± SEM. For representative westerns and additional details on dose-response experiments and statistical analyses, please refer to Supplemental Figures S4 and S5.
Fluorescent western blot quantification of MeCP2 levels across five different RTT fibroblast cell lines revealed several key findings. sbASO.miR-132 demonstrated a robust capacity to increase MeCP2 protein levels in all cell lines tested, although effective concentrations varied depending on the specific mutation (Fig. 5D; representative westerns: Supplemental Figs. S4 and S5). In contrast, sbASO.miR-22 and sbASO.miR-483 showed less consistent efficacy across the different cell lines (Fig. 5C,E; representative westerns: Supplemental Figs. S4 and S5). For example, in missense mutations, the minimal effective concentrations of sbASO.miR-132 were 27.5, 50, and 125 nM, in the T158M, R133C, and R306C patient cell lines, respectively (Fig. 5D; Supplemental Fig. S4B,E,H). Conversely, sbASO.miR-22 was only effective in the R133C (50 nM) and R306C (125 nM) lines, while sbASO.miR-483 showed no efficacy in cell lines with missense mutations (Fig. 5C; Supplemental Fig. S4G).
Fibroblast lines from individuals with RTT are mosaic for the WT and mutant MECP2 alleles, making it impossible to isolate sbASO effects on the mutant protein in the R133C, T158M, and R306C missense lines. However, truncating mutations produce two distinct protein sizes: the full-length (FL) WT protein and a smaller truncated protein. Analysis of the R270X cell line was limited to the FL protein, as the R270X truncated protein is unstable (Cuddapah et al. 2014), leading to rapid degradation and rendering it undetectable by western blot. Both sbASO.miR-132 and sbASO.miR-483 effectively increased FL R270X MeCP2 protein levels at 27.5 nM, while sbASO.miR-22 had no efficacy (Fig. 5E,D; Supplemental Fig. S5A–C).
In contrast, the R294X truncated protein is known to be more stable and less prone to nonsense-mediated decay (Merritt et al. 2020), making it a promising target for therapeutic interventions. In this cell line, truncated R294X MeCP2 protein levels increased in response to all three sbASOs across various concentrations: sbASO.miR-22 (1.5, 5, 27.5, 50, and 125 nM), sbASO.miR-132 (5 and 27.5 nM), and sbASO.miR-483 (27.5 nM) (Fig. 5C–E; Supplemental Fig. S5G–I). Unexpectedly, FL MeCP2 protein levels were only upregulated with sbASO.miR-132, showing efficacy at 1.5, 5, and 50 nM concentrations (Fig. 5D; Supplemental Fig. S5E). Together, these findings indicate that mutation-specific properties significantly influence sbASO responsiveness, highlighting the need to tailor therapeutic strategies based on the specific MeCP2 mutation.
To determine whether the shift in effective sbASO concentrations across mutations was linked to unique expression patterns of the target miRNAs, we quantified miR-22, miR-132, and miR-483 expression using qRT-PCR. miRNA expression in all five cell lines was compared to a WT, monoclonal cell line derived from the T158M mosaic cell line (Supplemental Fig. S6A,B). Consistent with findings in the human autopsy samples, the expression of sbASO target miRNAs was significantly increased in most cases, but the pattern varied across mutations and did not correlate with sbASO efficacy (Supplemental Fig. S6C,D). These results suggest that factors beyond the levels of the target miRNA may also contribute to the effectiveness of each sbASO, which may merit consideration with the continued development of this approach.
sbASO efficacy in MECP2-T158M neural stem cells
To confirm the efficacy of ASO treatment in a model more relevant to the neuronal pathology of RTT, we tested our sbASOs in neural stem cells (NSCs) expressing either the mutant (T158MMT) or WT isogenic control allele (T158MWT). NSC differentiation from human induced pluripotent stem cells (iPSCs) was confirmed via immunofluorescence, showing positive Nestin expression and the absence of the pluripotency marker Oct4 (Supplemental Fig. S7A–D).
LNA–ASOs have shown significant potential as central nervous system therapeutics due to their ability to enter cells without delivery reagents. This “unassisted uptake” is facilitated by clathrin-mediated, context-dependent endocytic pathways, enabling delivery to traditionally difficult-to-transfect cells, such as neurons (Juliano et al. 2013; Geary et al. 2015). Compared to transfection methods, unassisted uptake requires higher ASO concentrations and has slower kinetics of internalization. In these experiments, sbASOs or a scramble control sbASO (sbASO.Scr) were diluted directly in the culture medium at concentrations ranging from 125 to 375 nM and incubated for 5 days. After treatment, proteins were extracted for analysis (Fig. 6A).
FIGURE 6.

sbASO show efficacy in MECP2-T158M NSCs. (A) Schematic representation of NSC(s) differentiation from iPSCs and the experimental strategy to evaluate the effect of sbASOs on NSCs. (B–D, H–J) Western blot analysis of MeCP2 protein levels in the nuclear fraction of T158MMT and T158MWT NSCs. Protein quantification was normalized using Histone H3 (H3), with data represented as the mean percentage relative to vehicle treatment. The sbASOs effectively increased MeCP2 expression in both mutant (MT) and WT NSCs at various concentrations. Representative western blots show relative MeCP2 protein quantities in MT and WT NSCs treated with sbASOs at concentrations of 125 nM (E,K), 250 nM (F,L), and 375 nM (G,M). (Upper band) MeCP2 (71 kDa), (lower band) H3 (17 kDa). The analysis includes n = 3 replicates for each condition and sbASO, illustrating the relative expression levels of MeCP2 in response to the different sbASO concentrations. Outliers were removed by Grubb's tests at α = 0.1. Data presented as mean ± SEM. (B–H) Compared to scramble sbASO controls, (B) MT NSCs showed no difference in MeCP2 protein levels when treated with sbASO.miR-22 at 125 nM [t (df,4) = 1.816, P = 0.1436] and 250 nM [t (df,4) = 1.077, P = 0.3420], but there was a notable decrease at 375 nM [t (df,4) = 6.125, (**)P = 0.0036]. (H) In contrast, WT cells treated with sbASO.miR-22 showed increased MeCP2 levels at 125 nM [t (df,4) = 5.096, (**)P = 0.0070], with no notable changes at 250 nM [t (df,4) = 0.9023, P = 0.4179] or 375 nM [t (df,4) = 1.176, P = 0.3049]. (C–I) sbASO.miR-132 led to higher MeCP2 expression in both (C) MT [125 nM [t (df,4) = 5.412, (**)P = 0.0056], 250 nM [t (df,4) = 2.883, (*)P = 0.0449]] and (I) WT [250 nM [t (df,4) = 18.14, (****)P < 0.0001], 375 nM [t (df,4) = 2.778, (*)P = 0.0499]] NSCs. No significant differences were detected at 375 nM [t (df,4) = 0.3510, P = 0.7433] in MT cells, or in WT NSCs at 125 nM [t (df,4) = 2.411, P = 0.0735]. (D–J) MeCP2 expression increased following sbASO.miR-483 treatment in (D) MT at the 125 nM [t (df,4) = 5.157, (**)P = 0.0067] and (J) WT NSCs at 250 nM [t (df,4) = 14.70, (***)P = 0.0001], and 375 nM [t (df,4) = 18.78, (***)P = 0.0003]. No significant changes were observed in MT NSCs at 250 nM [t (df,4) = 1.121, P = 0.3250] and 375 nM [t (df,4) = 1.364, P = 0.2443], or in WT NSCs at 125 nM [t (df,4) = 1.979, P = 0.1189].
Using this strategy, we observed a significant increase in MeCP2 protein levels in sbASO-treated cells compared to sbASO.Scr-treated cells (Fig. 6B–J). This increase was achieved without the use of transfection agents, indicating that the sbASOs were efficiently internalized by the NSCs through unassisted uptake. Similar to what was observed in R294X fibroblasts, the sbASOs exhibited a left-shifted dose response in T158MMT NSCs relative to isogenic control lines, suggesting a more pronounced response in mutant cells at lower concentrations (Fig. 6B–D,H,J). While speculative, this finding may be attributed to the global increase in miRNA levels described in Figure 1, which likely results in overly repressed baseline levels of mutant MeCP2 protein. The heightened repressive tone in T158MMT NSCs could thus enhance the potential to respond to sbASOs, leading to a more substantial relative increase in MeCP2 at lower concentrations.
sbASOs increase total BDNF levels in T158M NSCs
We next sought to determine whether the increase in mutant T158M MeCP2 protein translates to a functional improvement in RTT pathophysiology. Although RTT is a heterogeneous disorder, one consistent finding across patient samples and animal models is decreased levels of brain-derived neurotrophic factor (BDNF) (Li and Pozzo-Miller 2014). To assess whether sbASO administration normalizes BDNF levels, we performed ELISA experiments using isolated cytoplasmic extracts from sbASO-treated WT and mutant T158M NSCs. These experiments quantified a significant elevation in BDNF levels in T158MMT NSCs at the 250 nM concentration for sbASO.miR-22 and sbASO.miR-132 (Fig. 7A–C). Consistent with their reduced efficacy on MeCP2, sbASOs also had muted effects on BDNF in isogenic control NSCs, with only sbASO.miR-22 showing an increase in expression (Fig. 7A). These data indicate that sbASOs can enhance the expression of mutant MeCP2 in a T158M NSC model of RTT, which correlates with the upregulation of a protein whose deficiency is a known contributor to the pathophysiology of the disorder in BDNF.
FIGURE 7.

sbASOs increase total BDNF levels in MECP2-T158M NSCs. (A–C) ELISA experiments using isolated cytoplasmic extracts from WT and mutant T158M NSCs revealed a significant increase in BDNF levels in mutant NSCs at the 250 nM concentration for sbASO.miR22 and sbASO.miR-132 compared to sbASO scramble controls. (A) ELISA analysis of MT and WT NSCs treated with sbASO.miR-22 demonstrated that concentration had a significant impact on BDNF levels (two-way ANOVA F(3, 24) = 12.67, [****] P < 0.0001). A Tukey's multiple comparisons test indicated an increase at the 250 nM concentration, with a notable rise in BDNF levels in WT NSCs ([*] P = 0.0331) and an even more pronounced increase in mutant NSCs ([**] P = 0.0097). Other concentrations showed no changes in WT (125 nM, P = 0.8172; 375 nM, P = 0.7082) or MT (125 nM, P = 0.9907; 375 nM, P = 0.7868) NSCs. (B) Treatment with sbASO.miR-132 showed that concentration significantly influenced BDNF levels (two-way ANOVA F(3, 29) = 10.62, [****] P < 0.0001). A Tukey's multiple comparisons test revealed no differences in WT NSCs at any concentration (125 nM, P = 0.9956; 250 nM, P = 0.1968; 375 nM, P = 0.9673), but there was a substantial increase in mutant NSCs at the 250 nM concentration (125 nM, P = 0.3801; 250 nM [***] P = 0.0002; 375 nM, P = 0.9938). (C) MT and WT NSCs treated with sbASO.miR-483 showed that concentration had a measurable effect on BDNF levels (two-way ANOVA F(3, 24) = 5.031, [**] P = 0.0076). A Tukey's multiple comparisons test showed no changes in BDNF levels in either WT (125 nM, P = 0.3424; 250 nM, P = 0.3058; 375 nM, P = 0.9949) or mutant (125 nM, P = 0.9995; 250 nM, P = 0.4183; 375 nM, P = 0.6196) NSCs at any concentration compared to sbASO scramble controls. Data are presented as mean ± SEM. Statistical significance is indicated as (*) P < 0.05, (**) P < 0.01, (***) P < 0.001, (****) P < 0.0001.
DISCUSSION
Over the past decade, numerous studies have demonstrated the remarkable potential of MeCP2 reexpression to improve disease symptoms in RTT (Luikenhuis et al. 2004; Guy et al. 2007; Garg et al. 2013). This groundbreaking discovery has fundamentally shifted the perception of RTT from a hardwired neurodevelopmental disorder to one that can potentially be reversed. As a result, there has been a surge in research focused on restoring MeCP2 expression through genome, base, and RNA editing, alongside delivery platforms such as gene therapy, protein replacement, and mRNA therapies (Palmieri et al. 2023). Despite these advancements, the dosage sensitivity of MeCP2 remains a critical challenge, as both insufficient and excessive levels result in neurological dysfunction. Moreover, these methods are difficult to implement across the entire brain and carry the risk of permanent adverse effects.
Restoring optimal MeCP2 levels is further complicated by the fact that most RTT patients carry hypomorphic mutations that result in only partial loss-of-function (Heckman et al. 2014; Brown et al. 2016). Therapeutic strategies must therefore account for the varying degrees of functionality retained by each respective mutation. For instance, the R133C mutation is associated with a mild clinical presentation. While adding a MECP2 transgene rescues null models of RTT, it evokes MDS-like motor and cognitive effects in Mecp2R133C/+ mice (Vermudez et al. 2021). The crux of the challenge lies in achieving the correct level of MeCP2 in a context where the required amount varies with factors like mutation type, developmental stage, and cell type (Ip et al. 2018). These complexities underscore the need for alternative strategies that enable precise and controlled modulation of MeCP2 expression, preferably from the endogenous locus, where the majority of regulatory elements remain intact (Shao et al. 2021).
In this study, we developed sbASOs designed to target miRNA-binding sites within the MECP2 3′ UTR. The clinical success of ASOs in SMA (Hua et al. 2010; Passini et al. 2011) and ALS (McCampbell et al. 2018; Miller et al. 2020) highlights their potential as therapeutics for genetic disorders of the nervous system. Building on these seminal advancements, our approach leverages the ability of miRNAs to fine-tune protein levels, providing a precision medicine strategy that can be tailored for each mutation to counteract diminished MeCP2 protein function by increasing its abundance.
The role of MeCP2 in miRNA biogenesis
miRNAs are small, noncoding RNAs that regulate gene expression post-transcriptionally by binding to the 3′ UTRs of target mRNAs, leading to their degradation or translational repression (Hong and Jeong 2023; Shang et al. 2023). These regulatory interactions play crucial roles in neural development, synaptic plasticity, and neuronal differentiation (Szulwach et al. 2010; Ye et al. 2016; Shang et al. 2023). Before targeting miRNA–MECP2 interactions, we first sought to define the miRNA expression profile in the brains of RTT patients. These experiments identified significant dysregulation of miRNAs in patient samples, consistent with prior in vivo and in vitro studies reporting widespread miRNA alterations in RTT (Wu et al. 2010; Mellios et al. 2018).
The genome-wide disruption of miRNAs in RTT arises from both indirect and direct consequences of MeCP2 loss-of-function. MeCP2 regulates global gene expression, triggering compensatory alterations in noncoding RNA levels, including miRNAs, to buffer against harmful changes in gene dosage (Good et al. 2021). Directly, MeCP2 represses primary miRNA (pri-miRNA) transcription by binding to methylated CpG sites within promoter regions (Szulwach et al. 2010; Ip et al. 2018). When MeCP2 is deficient, pri-miRNA transcription increases, leading to higher levels of mature miRNAs. Thus, pathogenic mutations in the methyl binding, transcriptional repression, and/or NCoR interacting domains are predicted to increase the expression of MeCP2-regulated miRNAs, as we have reported here (Ip et al. 2018). Additionally, MeCP2 can inhibit miRNA biogenesis by sequestering a key component of the microprocessor complex, DGCR8, and stalling RNA polymerase II at methylated miRNA gene boundaries (Cheng et al. 2014; Woo and Kim 2014). In patients with C-terminal mutations or truncations, the binding between MeCP2 and DGCR8 is likely disrupted, allowing Drosha to bind and activate pri-mRNA processing (Cheng et al. 2014; Tsujimura et al. 2015). While speculative, this differential regulation of miRNA biogenesis may account for the distinct miRNA expression patterns observed in T158M and R255X patient samples and could explain the variable efficacy of sbASOs depending on the specific MeCP2 mutation.
Nanostring analysis indicated that upregulated miRNAs were more significantly disrupted compared to downregulated miRNAs, including two key targets in our study: miR-22 and miR-132. This observation is consistent with earlier studies in Mecp2 KO mice (Wu et al. 2010). The inverse regulatory relationship between MeCP2 and both miRNAs has been well established. Specifically, reduced MeCP2 leads to increased expression of miR-22 and miR-132, whereas overexpression suppresses these miRNAs. For miR-22, these effects are likely mediated by MeCP2 binding to an enhancer region upstream of the miR-22 gene, resulting in increased CpG methylation and reduced miR-22 levels (Tong et al. 2020). The relationship between miR-132 and MeCP2 involves a dynamic regulatory loop where elevated MeCP2 increases BDNF levels, and BDNF, in turn, induces miR-132 expression to provide negative feedback on MECP2 (Pejhan et al. 2020). These complex feedback loops illustrate how noncoding miRNAs maintain appropriate levels of dosage-sensitive genes like MECP2.
The concerted action of many repressive miRNAs likely represents a primary mechanism for why reduced levels of mutant MeCP2 protein are commonly observed in RTT (Woo and Kim 2014; Khan et al. 2018). Loss of MeCP2-mediated repression leads to increased miRNA expression, which further suppresses MeCP2 levels, exacerbating the effects of loss-of-function mutations. sbASOs are designed to disrupt this cycle, as supported by studies showing that transgenic overexpression of the Mecp2-T158M allele rescues RTT-like phenotypes in mice (Lamonica et al. 2017). One limitation not addressed here is the relative abundance of specific miRNA and their targets at baseline, which can influence the extent of de-repression achieved by sbASOs. Future studies will focus on identifying overlaps between highly expressed miRNAs and those differentially expressed in RTT to refine our target selection criteria.
Dose-dependent increase of MeCP2 in vitro
We initially focused our studies on SH-SY5Y cells due to their high transfection efficiency and endogenous expression of MeCP2 (Hoffmann et al. 2022; Lopez-Suarez et al. 2022). In this cell line, sbASO treatment elevated MeCP2 levels in a dose-dependent manner, plateauing at subtoxic thresholds. Notably, subeffective concentrations of sbASOs exhibited additive effects, a property that could be critical in cases where the target miRNA is not uniformly expressed across all cell types or developmental stages, or where severe mutations necessitate higher therapeutic thresholds.
Interestingly, sbASO treatment in SH-SY5Y cells did not significantly alter MECP2 mRNA levels, despite the typical model wherein miRNAs induce deadenylation followed by translational repression. While unexpected, this discrepancy between transcript and protein levels is not uncommon and could result from complex regulatory mechanisms not yet fully understood (Bauernfeind and Babbitt 2017). These results suggest that in this context, sbASO may primarily impact translation without impacting mRNA stability, or that compensatory pathways modulate the response. To further validate the specificity of the sbASO designs and address the lack of mRNA effect, additional studies are required to confirm that the targeted miRNA-binding sites are functional.
To identify RTT subpopulations likely to respond to this approach, we extended our studies to patient-derived fibroblasts modeling five common RTT-causing mutations. sbASOs showed a consistent ability to increase MeCP2 levels in this model, pointing to a broad utility across mutation types. Similarly, experiments in RTT patient-derived neuronal stem cells demonstrated successful elevation of MeCP2 levels, providing further support for sbASOs in a more clinically relevant context. One notable finding was the mutation-dependent shift in dose-response curves. No overt correlation between sbASO efficacy and miRNA levels could be established in these cell lines; however, mutation-specific miRNA expression patterns were observed in human brain samples, suggesting that this relationship may merit further study. Alternatively, the fluorescent oligo used for transfection optimization may not accurately reflect each cell line's unique sbASO uptake capacity. Future studies will quantify sbASO uptake to address this important consideration.
Functional rescue
The therapeutic potential of the sbASO approach depends on whether increasing levels of mutant MeCP2 can rescue functional deficits. To address this, we quantified levels of BDNF, a downstream effector of MeCP2 that plays a critical role in RTT pathogenesis (Gonzales et al. 2012; Li and Pozzo-Miller 2014). BDNF levels are consistently reduced across various brain regions in RTT model mice (Erickson et al. 1996; Abuhatzira et al. 2007; Chahrour and Zoghbi 2007; Schmid et al. 2012), and strategies to elevate BDNF through genetic or pharmacological means have been shown to mitigate deficits in similar models (Chang et al. 2006).
In our study, treatment with sbASO.miR-22 and sbASO.miR-132 resulted in a significant increase in BDNF levels in T158MMT NSCs, suggesting that upregulating mutant MeCP2 can potentially mitigate neurotrophic deficits associated with RTT. However, this effect was not consistently mirrored across all effective sbASO concentrations, indicating a more complex regulatory mechanism at play between the two proteins. Early research suggested that MeCP2 represses BDNF by binding to its promoter, with transcription activated upon neuronal depolarization (Chen et al. 2003; Martinowich et al. 2003; Ballas et al. 2005; Zhou et al. 2006). Other studies, however, highlighted an activator role, as evidenced by reduced BDNF levels in Mecp2-null mice (Chang et al. 2006; Li et al. 2012). More recent models propose a dual function for Mecp2, capable of repressing or activating BDNF transcription depending on its phosphorylation status or other epigenetic factors (Li and Pozzo-Miller 2014).
This duality likely explains the inconsistent correlation between sbASO-induced MeCP2 increases and changes in BDNF levels. While sbASOs increased MeCP2 expression, additional factors—such as phosphorylation status, miRNA interactions, or post-transcriptional regulation—may have influenced BDNF expression. Furthermore, the use of an ELISA assay to measure total BDNF does not distinguish between its pre-, pro-, and mature forms, which may have masked more nuanced regulatory changes. These distinct BDNF isoforms may have differing relationships with MeCP2, contributing to the observed discrepancies.
sbASOs increase Mecp2 levels in vivo
Administration of sbASO.miR-132 significantly increased Mecp2 protein levels in the frontal cortex and hippocampus of WT mice. This in vivo upregulation of Mecp2 serves as a proof-of-concept for the potential use of sbASOs in a complex biological environment. Interestingly, both sbASO.miR-22 and sbASO.miR-132 administration led to the emergence of MDS-like anxiety phenotypes in WT mice. While the anxiety-related outcomes for sbASO.miR-132 were anticipated due to the robust effect on Mecp2 levels, the behavioral alterations observed with sbASO.miR-22 were unexpected given the lack of differences in protein expression compared to saline control.
One possible explanation for these findings lies in the tissue-specific and subcellular localization of miRNAs, which have distinct expression patterns in neurons, astrocytes, and other cell types. miRNAs can be segregated within specific regions of neurons, particularly at synapses, where they regulate local protein translation (Lagos-Quintana et al. 2002; Natera-Naranjo et al. 2010; He et al. 2012; Stappert et al. 2015; Alberti et al. 2018) Moreover, miRNA maturation at synapses is activity-dependent, directly linking miRNA function to synaptic plasticity (Sambandan et al. 2017). Therefore, it is plausible that sbASO.miR-22′s impact on anxiety-like behaviors results from modulation of Mecp2 levels in specific neuronal subpopulations or brain regions not assessed in this study, such as the amygdala, or within distinct subcellular compartments where Mecp2 regulation might occur independently of overall protein levels.
Future studies will prioritize evaluating the feasibility of this approach in missense or late-truncating mouse models of RTT. Notably, many of the missense models of RTT were developed using the same base Mecp2-GFP construct, which does not contain the entire 3′ UTR (Brown et al. 2016). Since the sbASO.miR-132 target site is in the distal portion of the UTR, the availability of suitable mouse models to fully test its efficacy is limited. For this reason, we propose that follow-up in vivo efficacy studies be conducted in the Mecp2-T158M-Tavi model, which does not have these limitations (Johnson et al. 2017; Lamonica et al. 2017). While RTT models will be the primary focus, we do not rule out exploring other disease models, such as Pitt Hopkins Syndrome, where increased MeCP2 levels have also been shown to be therapeutic (Dennys et al. 2024).
Risk of adverse effects
Oligonucleotide-based therapies offer a unique advantage due to their shared chemical composition, which typically results in comparable safety profiles. Nevertheless, potential risks associated with ASOs, including hepatotoxicity, renal toxicity, and hypersensitivity reaction, are well-documented (Geary et al. 2015; Collotta et al. 2023) and must be monitored during development. Although the sbASOs used in this study do not have significant homology with targets other than MECP2, assessing the potential for off-target effects is crucial, especially given the in vivo neurological phenotypes observed following sbASO.miR-483 administration. It is noteworthy, however, that sbASO.miR-483 only has 55% homology with the murine Mecp2 3′ UTR, increasing the likelihood that the observed adverse effects were not target-mediated.
Similar to viral gene therapy strategies (Gadalla et al. 2013), a primary on-target risk is that sbASOs theoretically do not distinguish between cells expressing WT and mutant MECP2 alleles. However, our in vitro experiments, which demonstrated a left-shifted dose response in mutant cells, suggest some degree of selectivity, as only one of the three sbASOs produced comparable effects in control NSCs. Additionally, the tolerance of maternal asymptomatic carriers of MDS, who exhibit a mosaic pattern with 50% of cells carrying an extra copy of MECP2, indicates that moderate overexpression of MeCP2 may not cause significant adverse effects (D'Mello III 2021). Importantly, any adverse effects, whether on- or off-target, are expected to resolve naturally as the sbASOs degrade over time, offering a potential safety benefit over more permanent gene-editing approaches.
Conclusions
Our findings highlight the promising potential of sbASOs in modulating MeCP2 expression across multiple experimental models. The observed global de-repression of miRNA quantified in patient brains, coupled with the in vitro and in vivo efficacy of sbASOs, underscores their therapeutic potential as a treatment strategy for RTT. The mutation-specific and neuronal efficacy of sbASOs further support their versatility as a personalized treatment strategy. Moving forward, efforts should prioritize optimizing sbASO delivery and assessing long-term safety to advance this approach toward clinical application. Addressing these challenges is essential to developing effective and safe therapies for RTT, offering renewed hope for affected individuals and their families.
MATERIALS AND METHODS
Animals
Animals were provided food and water ad libitum, with cages changed weekly. All mice were maintained on a 12 h light/12 h dark cycle, received routine veterinary monitoring, and were cared for in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All studies were approved by the Institutional Animal Care and Use Committee of Loyola University Chicago. Postsurgical mice were single-housed to protect sutures and tubing. Male C57BL/6J mice, obtained from Jackson Laboratories, were aged to 6 weeks for all experiments.
sbASO synthesis and design
Site-blocking ASOs were designed to cover the target miRNA seed region and a portion of the surrounding sequence that was unique to MECP2. The backbones of sbASOs were fully phosphorothioate modified to increase stability, and LNA modifications were made to all bases to increase binding affinity. The modified ASOs were synthesized by Qiagen and Integrated DNA Technologies. The sbASO sequences are: sbASO.miR-22 (5′-GAGCCAACAGCTGCCT-3′), sbASO.miR-132 (5′-AGTAACAGTCCTGGTG-3′), and sbASO.miR-483 (5′-TGTAGACGGGGCACTG-3′).
Cell culture and treatment
SH-SY5Y cells: Immortalized human neuroblastoma cells were obtained from ATCC (CRL-2266) and were cultured in IMDM (1×) (Gibco, Thermo Fisher Scientific, #12440053) supplemented with 10% heat-inactivated FBS, 1% antibiotic-antimycotic (100×), and 0.5% MEM nonessential amino acids (100×) (Gibco, Thermo Fisher Scientific, #16140071, 15240062, 11140035). These cells were maintained at 37°C and 5% CO2, and passaged using TrypLE Express (1×) (Gibco, Thermo Fisher Scientific, #12605010).
Human fibroblast cells: Primary human fibroblasts derived from RTT patients were obtained from the Rett Syndrome Research Trust (RSRT) and cultured in DMEM (1×), high glucose (Gibco, Thermo Fisher Scientific, #11965092) supplemented with 15% heat-inactivated FBS, 1% antibiotic-antimycotic (100×), and 1% MEM nonessential amino acids (100×). These fibroblasts were also maintained at 37°C and 5% CO2, and passaged using TrypLE Express (1×).
Neural stem cells: NSCs were generated from iPSCs following the Life Technologies protocol for “Induction of Neural Stem Cells from Human Pluripotent Stem Cells” (Publication number: MAN0008031). This included both the MeCP2-T158M mutation (T158MMT) and isogenic control (T158MWT) cell lines. Briefly, the differentiation process involved three stages. First, iPSCs were cultured to 90% confluency in Essential 8 Medium (Gibco, Thermo Fisher Scientific, #A1517001) on a Vitronectin substrate (Gibco, Thermo Fisher Scientific, #CTS279S3). Second, neural induction from iPSCs was initiated using PSC neural induction medium (Gibco, Thermo Fisher Scientific, #A1647801) for 7 days. Third, terminal differentiation was achieved by applying a neural differentiation medium with an 80% media change every 48 h for 30 days. The differentiation of NSCs was confirmed by immunofluorescence staining, showing positive expression of neural markers such as Nestin and the absence of pluripotent stem cell marker Oct4 in differentiated cells. Once the control and mutant lines were differentiated, ASO delivery was initiated.
ASO delivery
Assisted transfections: Cells were plated in surface-treated 6-well sterile tissue culture plates and grown to ∼80% confluency before transfection with sbASOs or BLOCK-iT Fluorescent Oligo (Invitrogen Life Technologies, #1370-070) using Lipofectamine 3000 Transfection Reagent (Invitrogen, Thermo Fisher Scientific, #L3000150) in Opti-MEM reduced serum medium (Gibco, Thermo Fisher Scientific, #31985070). Cells were treated with 3, 10, 55, 100, and 125 nM sbASO per transfection. After 12–18 h, transfected cells were washed with DPBS, maintained in growth medium for 24 h, harvested using TrypLE Express (1×), and stored at −80°C until processing.
Unassisted ASO uptake: Cells were plated in Geltrex-treated 12-well sterile tissue culture plates and grown to ∼40%–60% confluency before unassisted sbASO treatment. sbASOs or scramble control sbASOs were directly added to the iPSC-derived NSC culture medium at concentrations of 125, 250, and 375 nM for 5 days to allow for internalization. After incubation, cells were harvested using StemPro Accutase Cell Dissociation Reagent (Gibco, Thermo Fisher Scientific, #A1110501) and stored at −80°C until processing.
Osmotic pump implantation surgery
Osmotic pumps were prepared according to the manufacturer's protocol (Alzet; pump model 10007D). Mice were anesthetized with a 5% isoflurane/oxygen mixture and confirmed to be unconscious before being placed in a Kopf model stereotaxic apparatus fitted with ear bars. The osmotic pump was implanted subcutaneously and connected via a catheter to a cannula surgically inserted into the right lateral ventricle, positioned 1.1 mm lateral and 0.5 mm posterior to the bregma. sbASO or saline solutions were infused into the cerebrospinal fluid for 5 days at a rate of 50 µg/day. Five days postimplantation, brains were harvested, and total Mecp2 protein levels were quantified in the frontal cortex and hippocampus.
Elevated zero maze (EZM)
To assess anxiety, mice were placed on a continuous circular platform with two closed and two open regions for 5 min under full light conditions (>400 lux). The time spent exploring the open regions, as well as the distance traveled in both open and closed regions, was quantified by ANY-maze software.
Subcellular fractionation
Cells: Transfected cells were collected and fractionated into nuclear and cytoplasmic fractions using the Lyse and Wash protocol (Senichkin et al. 2021) to isolate cytoplasmic and nuclear protein.
Tissue: Frontal cortex and hippocampal samples were harvested from WT mice post osmotic pump implantation and behavioral assays. The tissue samples were homogenized in ice-cold hypotonic buffer and incubated on ice for 20 min. Triton X-100 was then added to a final concentration of 0.1%, followed by a 10 min incubation on ice with intermittent vortexing. The cell suspension was centrifuged at 2000 rcf at 4°C for 10 min to separate the nuclei (pellet) from the cytoplasm (supernatant). The cytoplasmic supernatant was further centrifuged at 12,000 rcf at 4°C for 10 min to remove debris, and the clean supernatant was collected as the cytoplasmic fraction. The nuclear pellets were combined and resuspended in ice-cold isotonic buffer, incubated on ice for 10 min with vortexing, and centrifuged at 12,000 rcf at 4°C for 10 min. The supernatant was discarded, and the pellet was used for DNA digestion. DNase was added to DNase buffer, the nuclear pellet was resuspended in this mixture, passed through a 27-gauge needle five times, and incubated for 1 h with vortexing to ensure thorough digestion.
Western blot
Nuclear protein concentration was measured using the Micro BCA Protein Assay Kit (Thermo Fisher Scientific, #23235). Samples were diluted to equal concentrations before gel electrophoresis in 4%–20% Criterion TGX precast polyacrylamide gels (Bio-Rad Laboratories 5671095) and subsequent immunoblotting with Histone H3 (1B1B2) mouse mAb and MeCP2 (D4F3) rabbit mAb (Cell Signaling Technologies 14269S, 3456S, respectively). Western blots were imaged using the Odyssey Imaging System (LICORbio) and quantified with Empiria Studio Software (LICORbio).
Enzyme-linked immunosorbent assay (ELISA)
The total BDNF in the cytoplasmic protein extracts from cell samples was quantified using the Human BDNF Simple Step ELISA kit (Abcam ab212166), according to the manufacturer's instructions. The absorbance was measured at 450 nM on a BioTek Cytation5 Microplate Reader (Agilent Technologies), and BDNF concentrations were calculated from a standard curve, providing precise quantification of BDNF levels in the samples.
mRNA and miRNA isolation
mRNA and miRNA were isolated from cells or tissue using the PureLink miRNA Isolation Kit (Invitrogen; Thermo Fisher Scientific, #K157001) per manufacturer instructions. Resultant mRNA and miRNA were diluted in DEPC-treated water, tested for purity and concentration using a NanoDrop Lite Spectrophotometer (Thermo Fisher Scientific), and stored at −80°C until cDNA synthesis.
mRNA reverse transcription and qRT-PCR
cDNA was synthesized using SuperScript IV VILO Master Mix (Invitrogen; Thermo Fisher Scientific, #11756050). MECP2 transcript levels were measured by qRT-PCR using the TaqMan Gene Expression Assay for MECP2 (Thermo Fisher Scientific, #4351372). qRT-PCR was performed in triplicate for each sample with the CFX96 Touch Real-Time PCR Detection System (Bio-Rad Laboratories, #1855196), using glucose-6-phosphate dehydrogenase (G6PD) levels measured by the TaqMan Gene Expression Assay (Invitrogen; Thermo Fisher Scientific, #4331182) as the internal control for normalization, with negative controls in each plate.
miRNA reverse transcription and qRT-PCR
miR-cDNA was synthesized from isolated and purified miRNA using the TaqMan MicroRNA Reverse Transcription Kit (applied Biosystems, #4366596) for target miRNAs miR-22-3p (assay 000398), miR-132-3p (assay 000457), miR-483-5p (assay 002338), and U6 snRNA (assay 001973). miRNA qRT-PCR was performed in triplicate for each sample using the CFX96 Touch Real-Time PCR Detection System (Bio-Rad Laboratories, #1855196). Quantification data were collected with CFX Maestro Software (Bio-Rad Laboratories, #12013758) and evaluated for fold changes using the ΔΔCt method, normalized to neurotypical expression levels in isogenic WT HDFa human dermal fibroblasts derived from heterozygous T158M RTT patient fibroblasts.
nCounter miRNA expression panels
Total miRNA was isolated from postmortem temporal cortices of T158M and R255X RTT patients, as well as neurotypical controls, obtained from the NIH NeuroBioBank and the University of Maryland Brain and Tissue Bank. Samples were submitted to the NUSeq Core at Northwestern University (Chicago, Illinois) for nCounter miRNA expression analysis, following manufacturer protocols. miRNA expression levels were normalized to total RNA levels and compared to neurotypical controls using nCounter Advanced Analysis Software.
Statistical analysis
Statistics were carried out using Prism 8 (GraphPad) and Excel (Microsoft). All data shown represent mean ± SEM to provide a measure of variability and accuracy. Post hoc tests were conducted for all statistical comparisons to identify specific group differences. Researchers conducting the analyses were blind to the experimental conditions to ensure unbiased results. Statistical significance was assessed using appropriate tests, and P-values were determined to evaluate the differences between experimental groups.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We acknowledge the RTT patients and families for their selfless donation of autopsy samples. We also thank the International Rett Syndrome Foundation for their continued financial and intellectual support and the Rett Syndrome Research Trust for their donation of the T158M iPSC line. This work was funded by R01 NS112171 and R21 HD111864 to R.G.G., and F32 HD113386A to A.V.P.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080220.124.
Freely available online through the RNA Open Access option.
MEET THE FIRST AUTHOR
Amanda Vanderplow.

Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Amanda Vanderplow is the first author of this paper, “Site-blocking antisense oligonucleotides as a mechanism to fine-tune MeCP2 expression.” Amanda is a postdoctoral fellow in the Gogliotti Lab at Loyola University Chicago. Amanda's research focuses on the use of site-blocking antisense oligonucleotides (sbASOs) to modulate gene expression as a therapeutic approach for neurological disorders whose therapeutic window is narrow, specifically Rett syndrome.
What are the major results described in your paper and how do they impact this branch of the field?
The major results described in our paper demonstrate that site-blocking antisense oligonucleotides (sbASOs) can effectively increase MeCP2 levels by outcompeting repressive miRNA binding at the MECP2 3′ UTR. Our findings show a dose-dependent elevation of MeCP2 in SH-SY5Y cells and patient fibroblasts, plateauing at levels projected to be safe. Similarly, in a T158M Rett syndrome neural stem cell model, sbASO treatment elevated both MeCP2 and brain-derived neurotrophic factor (BDNF) levels. Experiments in wild-type mice confirmed sbASO functionality in vivo, resulting in Mecp2 upregulation and the predicted phenotypes associated with overexpression of the protein. These results highlight the potential of sbASOs as a therapeutic strategy for MeCP2-related disorders, advancing the field by providing a method to fine-tune MeCP2 expression. We predict this approach will be useful for patients with missense or late-truncating mutations, where the hypomorphic nature of the protein can be overcome by increased abundance.
What led you to study RNA or this aspect of RNA science?
My graduate research focused on neurobiology at the level of the protein, but I wanted to broaden my expertise and move into the world of DNA and RNA. Joining Dr. Rocco Gogliotti's lab provided an ideal opportunity to expand my foundational knowledge and apply new strategies to address complex genetic disorders. This shift has allowed me to explore how RNA-based approaches, such as antisense oligonucleotides, can be leveraged to modulate gene expression and develop potential therapies for conditions like Rett syndrome.
During the course of these experiments, were there any surprising results or particular difficulties that altered your thinking and subsequent focus?
One surprising challenge we faced was the difficulty of working with mutant iPSCs differentiated into neural stem cells (NSCs), as they struggled to maintain their NSC state and tended to shift toward an astrocytic lineage. This instability altered our experimental strategies and forced us to rethink how we approach these models. Additionally, many of the missense models of Rett syndrome were developed using a Mecp2-GFP construct that lacks the full 3′ UTR. Since the sbASO.miR-132 target site is located in the distal portion of the UTR, this construct does not allow for a complete evaluation of sbASO efficacy. As a result, we have had to establish a new colony using the Mecp2-T158M-Tavi model, which includes the entire UTR. This will ultimately allow us to establish more comprehensive in vivo follow-up studies. However, establishing this new colony has been both time-consuming and costly, significantly impacting our timeline and resource allocation.
What are some of the landmark moments that provoked your interest in science or your development as a scientist?
A turning point in my scientific development was realizing the significant gender disparity in research. Many studies focused only on male subjects, assuming the results would apply to females despite key biological differences. This gap drove me to advocate for research that includes and addresses gender differences, ensuring findings apply to everyone.
Beyond my scientific goals, I am passionate about redefining what it means to succeed in research. I believe meaningful contributions come from mentally happy scientists, who have a healthy work-life balance and can take breaks without sacrificing productivity. Science often equates long hours with success, but I want to show that impactful discoveries can thrive in a lab that values well-being, respects boundaries, and embraces collaboration. Encouraging self-care, normalizing breaks, and celebrating all successes—big or small—leads to teams that are not only more productive but also more creative and resilient. My goal is to show that a people-centered, balanced approach doesn't detract from scientific rigor; it enhances it by creating a supportive environment where researchers feel valued and inspired to pursue their best ideas.
REFERENCES
- Abuhatzira L, Makedonski K, Kaufman Y, Razin A, Shemer R. 2007. MeCP2 deficiency in the brain decreases BDNF levels by REST/CoREST-mediated repression and increases TRKB production. Epigenetics 2: 214–222. 10.4161/epi.2.4.5212 [DOI] [PubMed] [Google Scholar]
- Alberti C, Manzenreither RA, Sowemimo I, Burkard TR, Wang J, Mahofsky K, Ameres SL, Cochella L. 2018. Cell-type specific sequencing of microRNAs from complex animal tissues. Nat Methods 15: 283–289. 10.1038/nmeth.4610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Saavedra M, Antoun G, Yanagiya A, Oliva-Hernandez R, Cornejo-Palma D, Perez-Iratxeta C, Sonenberg N, Cheng H-YM. 2011. miRNA-132 orchestrates chromatin remodeling and translational control of the circadian clock. Hum Mol Genet 20: 731–751. 10.1093/hmg/ddq519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. 1999. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 23: 185–188. 10.1038/13810 [DOI] [PubMed] [Google Scholar]
- Ballas N, Grunseich C, Lu DD, Speh JC, Mandel G. 2005. REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell 121: 645–657. 10.1016/j.cell.2005.03.013 [DOI] [PubMed] [Google Scholar]
- Bauernfeind AL, Babbitt CC. 2017. The predictive nature of transcript expression levels on protein expression in adult human brain. BMC Genomics 18: 322. 10.1186/s12864-017-3674-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijlani S, Pang KM, Bugga LV, Rangasamy S, Narayanan V, Chatterjee S. 2024. Nuclease-free precise genome editing corrects MECP2 mutations associated with Rett syndrome. Front Genome Ed 6: 1346781. 10.3389/fgeed.2024.1346781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown K, Selfridge J, Lagger S, Connelly J, De Sousa D, Kerr A, Webb S, Guy J, Merusi C, Koerner MV, et al. 2016. The molecular basis of variable phenotypic severity among common missense mutations causing Rett syndrome. Hum Mol Genet 25: 558–570. 10.1093/hmg/ddv496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrette LLG, Wang C-Y, Wei C, Press W, Ma W, Kelleher RJ, Lee JT. 2018. A mixed modality approach towards Xi reactivation for Rett syndrome and other X-linked disorders. Proc Natl Acad Sci 115: E668–E675. 10.1073/pnas.1716514115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahrour M, Zoghbi HY. 2007. The story of Rett syndrome: from clinic to neurobiology. Neuron 56: 422–437. 10.1016/j.neuron.2007.10.001 [DOI] [PubMed] [Google Scholar]
- Chang Q, Khare G, Dani V, Nelson S, Jaenisch R. 2006. The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron 49: 341–348. 10.1016/j.neuron.2005.12.027 [DOI] [PubMed] [Google Scholar]
- Chen RZ, Akbarian S, Tudor M, Jaenisch R. 2001. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet 27: 327–331. 10.1038/85906 [DOI] [PubMed] [Google Scholar]
- Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC, Jaenisch R, Greenberg ME. 2003. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science 302: 885–889. 10.1126/science.1086446 [DOI] [PubMed] [Google Scholar]
- Cheng T-L, Wang Z, Liao Q, Zhu Y, Zhou W-H, Xu W, Qiu Z. 2014. MeCP2 suppresses nuclear microRNA processing and dendritic growth by regulating the DGCR8/Drosha complex. Dev Cell 28: 547–560. 10.1016/j.devcel.2014.01.032 [DOI] [PubMed] [Google Scholar]
- Clavé G, Reverte M, Vasseur J-J, Smietana M. 2021. Modified internucleoside linkages for nuclease-resistant oligonucleotides. RSC Chem Biol 2: 94–150. 10.1039/D0CB00136H [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins BE, Neul JL. 2022. Rett syndrome and MECP2 duplication syndrome: disorders of MeCP2 dosage. Neuropsychiatr Dis Treat 18: 2813–2835. 10.2147/NDT.S371483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins AL, Levenson JM, Vilaythong AP, Richman R, Armstrong DL, Noebels JL, David Sweatt J, Zoghbi HY. 2004. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum Mol Genet 13: 2679–2689. 10.1093/hmg/ddh282 [DOI] [PubMed] [Google Scholar]
- Collotta D, Bertocchi I, Chiapello E, Collino M. 2023. Antisense oligonucleotides: a novel frontier in pharmacological strategy. Front Pharmacol 14: 1304342. 10.3389/fphar.2023.1304342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuddapah VA, Pillai RB, Shekar KV, Lane JB, Motil KJ, Skinner SA, Tarquinio DC, Glaze DG, McGwin G, Kaufmann WE, et al. 2014. Methyl-CpG-binding protein 2 (MECP2) mutation type is associated with disease severity in Rett syndrome. J Med Genet 51: 152–158. 10.1136/jmedgenet-2013-102113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennys CN, Vermudez SAD, Deacon RJM, Sierra-Delgado JA, Rich K, Zhang X, Buch A, Weiss K, Moxley Y, Rajpal H, et al. 2024. MeCP2 gene therapy ameliorates disease phenotype in mouse model for Pitt Hopkins syndrome. Neurotherapeutics e00376. 10.1016/j.neurot.2024.e00376 [DOI] [PubMed] [Google Scholar]
- DeVos SL, Miller TM. 2013. Direct intraventricular delivery of drugs to the rodent central nervous system. J Vis Exp 75: e50326. 10.3791/50326-v [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Mello SR III. 2021. MECP2 and the biology of MECP2 duplication syndrome. J Neurochem 159: 29–60. 10.1111/jnc.15331 [DOI] [PubMed] [Google Scholar]
- Erickson JT, Conover JC, Borday V, Champagnat J, Barbacid M, Yancopoulos G, Katz DM. 1996. Mice lacking brain-derived neurotrophic factor exhibit visceral sensory neuron losses distinct from mice lacking NT4 and display a severe developmental deficit in control of breathing. J Neurosci 16: 5361–5371. 10.1523/JNEUROSCI.16-17-05361.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn MJ, Mayfield AMH, Du R, Gradinaru V, Elowitz MB. 2024. Synthetic dosage-compensating miRNA circuits allow precision gene therapy for Rett syndrome. bioRxiv 10.1101/2024.03.13.584179 [DOI] [Google Scholar]
- Frullanti E, Papa FT, Grillo E, Clarke A, Ben-Zeev B, Pineda M, Bahi-Buisson N, Bienvenu T, Armstrong J, Roche Martinez A, et al. 2019. Analysis of the phenotypes in the Rett networked database. Int J Genomics 2019: 6956934. 10.1155/2019/6956934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadalla KKE, Bailey MES, Spike RC, Ross PD, Woodard KT, Kalburgi SN, Bachaboina L, Deng JV, West AE, Samulski RJ, et al. 2013. Improved survival and reduced phenotypic severity following AAV9/MECP2 gene transfer to neonatal and juvenile male Mecp2 knockout mice. Mol Ther 21: 18–30. 10.1038/mt.2012.200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg SK, Lioy DT, Cheval H, McGann JC, Bissonnette JM, Murtha MJ, Foust KD, Kaspar BK, Bird A, Mandel G. 2013. Systemic delivery of MeCP2 rescues behavioral and cellular deficits in female mouse models of Rett syndrome. J Neurosci 33: 13612–13620. 10.1523/JNEUROSCI.1854-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geary RS, Norris D, Yu R, Bennett CF. 2015. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv Drug Deliv Rev 87: 46–51. 10.1016/j.addr.2015.01.008 [DOI] [PubMed] [Google Scholar]
- Gonzales ML, Adams S, Dunaway KW, LaSalle JM. 2012. Phosphorylation of distinct sites in MeCP2 modifies cofactor associations and the dynamics of transcriptional regulation. Mol Cell Biol 32: 2894–2903. 10.1128/MCB.06728-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good KV, Vincent JB, Ausió J. 2021. MeCP2: the genetic driver of Rett syndrome epigenetics. Front Genet 12. 10.3389/fgene.2021.620859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm N-B, Lee JT. 2022. Selective Xi reactivation and alternative methods to restore MECP2 function in Rett syndrome. Trends Genet 38: 920–943. 10.1016/j.tig.2022.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J, Gan J, Selfridge J, Cobb S, Bird A. 2007. Reversal of neurological defects in a mouse model of Rett syndrome. Science 315: 1143–1147. 10.1126/science.1138389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J, Hendrich B, Holmes M, Martin JE, Bird A. 2001. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet 27: 322–326. 10.1038/85899 [DOI] [PubMed] [Google Scholar]
- Han K, Gennarino VA, Lee Y, Pang K, Hashimoto-Torii K, Choufani S, Raju CS, Oldham MC, Weksberg R, Rakic P, et al. 2013. Human-specific regulation of MeCP2 levels in fetal brains by microRNA miR-483-5p. Genes Dev 27: 485–490. 10.1101/gad.207456.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KF, Sakamoto K, Wayman GA, Impey S, Obrietan K. 2010. Transgenic miR132 alters neuronal spine density and impairs novel object recognition memory. PLoS ONE 5: e15497. 10.1371/journal.pone.0015497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M, Liu Y, Wang X, Zhang MQ, Hannon G, Huang ZJ. 2012. Cell-type based analysis of microRNA profiles in the mouse brain. Neuron 73: 35–48. 10.1016/j.neuron.2011.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckman LD, Chahrour MH, Zoghbi HY. 2014. Rett-causing mutations reveal two domains critical for MeCP2 function and for toxicity in MECP2 duplication syndrome mice. eLife 3: e02676. 10.7554/eLife.02676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann LF, Martins A, Majolo F, Contini V, Laufer S, Goettert MI. 2022. Neural regeneration research model to be explored: SH-SY5Y human neuroblastoma cells. Neural Regen Res 18: 1265–1266. 10.4103/1673-5374.358621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong D, Jeong S. 2023. 3′UTR diversity: expanding repertoire of RNA alterations in human mRNAs. Mol Cells 46: 48–56. 10.14348/molcells.2023.0003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath PM, Piazza MK, Kavalali ET, Monteggia LM. 2022. MeCP2 loss-of-function dysregulates microRNAs regionally and disrupts excitatory/inhibitory synaptic transmission balance. Hippocampus 32: 610–623. 10.1002/hipo.23455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Y, Sahashi K, Hung G, Rigo F, Passini MA, Bennett CF, Krainer AR. 2010. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev 24: 1634–1644. 10.1101/gad.1941310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip JPK, Mellios N, Sur M. 2018. Rett syndrome: insights into genetic, molecular and circuit mechanisms. Nat Rev Neurosci 19: 368–382. 10.1038/s41583-018-0006-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jauhari A, Singh T, Yadav S. 2022. Neurodevelopmental disorders and neurotoxicity: microRNA in focus. J Chem Neuroanatomy 120: 102072. 10.1016/j.jchemneu.2022.102072 [DOI] [PubMed] [Google Scholar]
- Johnson BS, Zhao YT, Fasolino M, Lamonica JM, Kim YJ, Georgakilas G, Wood KH, Bu D, Cui Y, Goffin D, et al. 2017. Biotin tagging of MeCP2 in mice reveals contextual insights into the Rett syndrome transcriptome. Nat Med 23: 1203–1214. 10.1038/nm.4406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovicic A, Jolissaint JFZ, Moser R, Santos M de FS, Luthi-Carter R. 2013. MicroRNA-22 (miR-22) overexpression is neuroprotective via general anti-apoptotic effects and may also target specific Huntington's disease-related mechanisms. PLoS ONE 8: e54222. 10.1371/journal.pone.0054222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juliano RL, Carver K, Cao C, Ming X. 2013. Receptors, endocytosis, and trafficking: the biological basis of targeted delivery of antisense and siRNA oligonucleotides. J Drug Targeting 21: 27–43. 10.3109/1061186X.2012.740674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung BP, Jugloff DGM, Zhang G, Logan R, Brown S, Eubanks JH. 2003. The expression of methyl CpG binding factor MeCP2 correlates with cellular differentiation in the developing rat brain and in cultured cells. J Neurobiol 55: 86–96. 10.1002/neu.10201 [DOI] [PubMed] [Google Scholar]
- Khan AW, Ziemann M, Rafehi H, Maxwell S, Ciccotosto GD, El-Osta A. 2018. MeCP2 interacts with chromosomal microRNAs in brain. Epigenetics 12: 1028–1037. 10.1080/15592294.2017.1391429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshkin AA, Singh SK, Nielsen P, Rajwanshi VK, Kumar R, Meldgaard M, Olsen CE, Wengel J. 1998. LNA (locked nucleic acids): synthesis of the adenine, cytosine, guanine, 5-methylcytosine, thymine and uracil bicyclonucleoside monomers, oligomerisation, and unprecedented nucleic acid recognition. Tetrahedron 54: 3607–3630. 10.1016/S0040-4020(98)00094-5 [DOI] [Google Scholar]
- Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. 2002. Identification of tissue-specific microRNAs from mouse. Curr Biol 12: 735–739. 10.1016/S0960-9822(02)00809-6 [DOI] [PubMed] [Google Scholar]
- Lamonica JM, Kwon DY, Goffin D, Fenik P, Johnson BS, Cui Y, Guo H, Veasey S, Zhou Z. 2017. Elevating expression of MeCP2 T158M rescues DNA binding and Rett syndrome–like phenotypes. J Clin Invest 127: 1889–1904. 10.1172/JCI90967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Pozzo-Miller L. 2014. BDNF deregulation in Rett syndrome. Neuropharmacology 76: 737–746. 10.1016/j.neuropharm.2013.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Calfa G, Larimore J, Pozzo-Miller L. 2012. Activity-dependent BDNF release and TRPC signaling is impaired in hippocampal neurons of Mecp2 mutant mice. Proc Natl Acad Sci 109: 17087–17092. 10.1073/pnas.1205271109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Suarez L, Awabdh SA, Coumoul X, Chauvet C. 2022. The SH-SY5Y human neuroblastoma cell line, a relevant in vitro cell model for investigating neurotoxicology in human: focus on organic pollutants. NeuroToxicology 92: 131–155. 10.1016/j.neuro.2022.07.008 [DOI] [PubMed] [Google Scholar]
- Luikenhuis S, Giacometti E, Beard CF, Jaenisch R. 2004. Expression of MeCP2 in postmitotic neurons rescues Rett syndrome in mice. Proc Natl Acad Sci 101: 6033–6038. 10.1073/pnas.0401626101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luoni M, Giannelli S, Indrigo MT, Niro A, Massimino L, Iannielli A, Passeri L, Russo F, Morabito G, Calamita P, et al. 2020. Whole brain delivery of an instability-prone Mecp2 transgene improves behavioral and molecular pathological defects in mouse models of Rett syndrome. eLife 9: e52629. 10.7554/eLife.52629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G, Sun YE. 2003. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 302: 890–893. 10.1126/science.1090842 [DOI] [PubMed] [Google Scholar]
- Matson K, Macleod A, Mehta N, Sempek E, Tang X. 2023. Impacts of microRNA-483 on human diseases. Non-Coding RNA 9: 37. 10.3390/ncrna9040037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCampbell A, Cole T, Wegener AJ, Tomassy GS, Setnicka A, Farley BJ, Schoch KM, Hoye ML, Shabsovich M, Sun L, et al. 2018. Antisense oligonucleotides extend survival and reverse decrement in muscle response in ALS models. J Clin Invest 128: 3558–3567. 10.1172/JCI99081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan H, Pang ZP. 2015. Regulatory functions and pathological relevance of the MECP2 3′UTR in the central nervous system. Cell Regen 4: 9. 10.1186/s13619-015-0023-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellios N, Feldman DA, Sheridan SD, Ip JPK, Kwok S, Amoah SK, Rosen B, Rodriguez BA, Crawford B, Swaminathan R, et al. 2018. MeCP2-regulated miRNAs control early human neurogenesis through differential effects on ERK and AKT signaling. Mol Psychiatry 23: 1051–1065. 10.1038/mp.2017.86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt JK, Collins BE, Erickson KR, Dong H, Neul JL. 2020. Pharmacological read-through of R294X Mecp2 in a novel mouse model of Rett syndrome. Hum Mol Genet 29: 2461–2470. 10.1093/hmg/ddaa102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller T, Cudkowicz M, Shaw PJ, Andersen PM, Atassi N, Bucelli RC, Genge A, Glass J, Ladha S, Ludolph AL, et al. 2020. Phase 1–2 trial of antisense oligonucleotide tofersen for SOD1 ALS. New Engl J Med 383: 109–119. 10.1056/NEJMoa2003715 [DOI] [PubMed] [Google Scholar]
- Na ES, Nelson ED, Adachi M, Autry AE, Mahgoub MA, Kavalali ET, Monteggia LM. 2012. A mouse model for MeCP2 duplication syndrome: MeCP2 overexpression impairs learning and memory and synaptic transmission. J Neurosci 32: 3109–3117. 10.1523/JNEUROSCI.6000-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natera-Naranjo O, Aschrafi A, Gioio AE, Kaplan BB. 2010. Identification and quantitative analyses of microRNAs located in the distal axons of sympathetic neurons. RNA 16: 1516–1529. 10.1261/rna.1833310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neul JL, Fang P, Barrish J, Lane J, Caeg E, Smith EO, Zoghbi H, Percy A, Glaze DG. 2008. Specific mutations in methyl-CpG-binding protein 2 confer different severity in Rett syndrome. Neurology 70: 1313–1321. 10.1212/01.wnl.0000291011.54508.aa [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obad S, dos Santos CO, Petri A, Heidenblad M, Broom O, Ruse C, Fu C, Lindow M, Stenvang J, Straarup EM, et al. 2011. Silencing of microRNA families by seed-targeting tiny LNAs. Nat Genet 43: 371–378. 10.1038/ng.786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri M, Pozzer D, Landsberger N. 2023. Advanced genetic therapies for the treatment of Rett syndrome: state of the art and future perspectives. Front Neurosci 17: 1172805. 10.3389/fnins.2023.1172805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passini MA, Bu J, Richards AM, Kinnecom C, Sardi SP, Stanek LM, Hua Y, Rigo F, Matson J, Hung G, et al. 2011. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci Transl Med 3: 72ra18. 10.1126/scitranslmed.3001777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pejhan S, Del Bigio MR, Rastegar M. 2020. The MeCP2E1/E2-BDNF-miR132 homeostasis regulatory network is region-dependent in the human brain and is impaired in Rett syndrome patients. Front Cell Dev Biol 8: 763. 10.3389/fcell.2020.00763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rastegar-Moghaddam SH, Ebrahimzadeh-Bideskan A, Shahba S, Malvandi AM, Mohammadipour A. 2022. MicroRNA-22: a novel and potent biological therapeutics in neurological disorders. Mol Neurobiol 59: 2694–2701. 10.1007/s12035-022-02769-8 [DOI] [PubMed] [Google Scholar]
- Rodrigues DC, Mufteev M, Ellis J. 2020. Regulation, diversity and function of MECP2 exon and 3′UTR isoforms. Hum Mol Genet 29: R89–R99. 10.1093/hmg/ddaa154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambandan S, Akbalik G, Kochen L, Rinne J, Kahlstatt J, Glock C, Tushev G, Alvarez-Castelao B, Heckel A, Schuman EM. 2017. Activity-dependent spatially localized miRNA maturation in neuronal dendrites. Science 355: 634–637. 10.1126/science.aaf8995 [DOI] [PubMed] [Google Scholar]
- Santos M, Yan J, Temudo T, Oliveira G, Vieira JP, Fen J, Sommer S, Maciel P. 2008. Analysis of highly conserved regions of the 3′UTR of MECP2 gene in patients with clinical diagnosis of Rett syndrome and other disorders associated with mental retardation. Dis Markers 24: 319–324. 10.1155/2008/738401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid DA, Yang T, Ogier M, Adams I, Mirakhur Y, Wang Q, Massa SM, Longo FM, Katz DM. 2012. A TrkB small molecule partial agonist rescues TrkB phosphorylation deficits and improves respiratory function in a mouse model of Rett syndrome. J Neurosci 32: 1803–1810. 10.1523/JNEUROSCI.0865-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senichkin VV, Prokhorova EA, Zhivotovsky B, Kopeina GS. 2021. Simple and efficient protocol for subcellular fractionation of normal and apoptotic cells. Cells 10: 852. 10.3390/cells10040852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang R, Lee S, Senavirathne G, Lai EC. 2023. microRNAs in action: biogenesis, function and regulation. Nat Rev Genet 24: 816–833. 10.1038/s41576-023-00611-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Y, Bajikar SS, Tirumala HP, Gutierrez MC, Wythe JD, Zoghbi HY. 2021. Identification and characterization of conserved noncoding cis-regulatory elements that impact Mecp2 expression and neurological functions. Genes Dev 35: 489. 10.1101/gad.345397.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnett SE, Hector RD, Gadalla KKE, Heindel C, Chen D, Zaric V, Bailey MES, Cobb SR, Gray SJ. 2017. Improved MECP2 gene therapy extends the survival of MeCP2-null mice without apparent toxicity after intracisternal delivery. Mole Ther Methods Clin Dev 5: 106–115. 10.1016/j.omtm.2017.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnett SE, Boyle E, Lyons C, Gray SJ. 2021. Engineered microRNA-based regulatory element permits safe high-dose miniMECP2 gene therapy in Rett mice. Brain 144: 3005–3019. 10.1093/brain/awab182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stappert L, Roese-Koerner B, Brüstle O. 2015. The role of microRNAs in human neural stem cells, neuronal differentiation and subtype specification. Cell Tissue Res 359: 47–64. 10.1007/s00441-014-1981-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szulwach KE, Li X, Smrt RD, Li Y, Luo Y, Lin L, Santistevan NJ, Li W, Zhao X, Jin P. 2010. Cross talk between microRNA and epigenetic regulation in adult neurogenesis. J Cell Biol 189: 127–141. 10.1083/jcb.200908151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong D, Zhang J, Wang X, Li Q, Liu L, Lu A, Guo B, Yang J, Ni L, Qin H, et al. 2020. MiR-22, regulated by MeCP2, suppresses gastric cancer cell proliferation by inducing a deficiency in endogenous S-adenosylmethionine. Oncogenesis 9: 1–16. 10.1038/s41389-019-0187-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimura K, Irie K, Nakashima H, Egashira Y, Fukao Y, Fujiwara M, Itoh M, Uesaka M, Imamura T, Nakahata Y, et al. 2015. miR-199a links MeCP2 with mTOR signaling and its dysregulation leads to Rett syndrome phenotypes. Cell Rep 12: 1887–1901. 10.1016/j.celrep.2015.08.028 [DOI] [PubMed] [Google Scholar]
- Urdinguio RG, Fernández AF, Lopez-Nieva P, Rossi S, Huertas D, Kulis M, Liu C-G, Croce CM, Calin GA, Esteller M. 2010. Disrupted microRNA expression caused by Mecp2 loss in a mouse model of Rett syndrome. Epigenetics 5: 656–663. 10.4161/epi.5.7.13055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermudez SAD, Gogliotti RG, Arthur B, Buch A, Morales C, Moxley Y, Rajpal H, Conn PJ, Niswender CM. 2021. Profiling beneficial and potential adverse effects of MeCP2 overexpression in a hypomorphic Rett syndrome mouse model. Genes Brain Behav 21: e12752. 10.1111/gbb.12752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vester B, Wengel J. 2004. LNA (locked nucleic acid): high-affinity targeting of complementary RNA and DNA. Biochemistry 43: 13233–13241. 10.1021/bi0485732 [DOI] [PubMed] [Google Scholar]
- Woo J-S, Kim VN. 2014. MeCP2 caught moonlighting as a suppressor of MicroRNA processing. Dev Cell 28: 477–478. 10.1016/j.devcel.2014.02.015 [DOI] [PubMed] [Google Scholar]
- Wu H, Tao J, Chen PJ, Shahab A, Ge W, Hart RP, Ruan X, Ruan Y, Sun YE. 2010. Genome-wide analysis reveals methyl-CpG–binding protein 2–dependent regulation of microRNAs in a mouse model of Rett syndrome. Proc Natl Acad Sci 107: 18161–18166. 10.1073/pnas.1005595107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Xu H, Su X, He X. 2016. Role of microRNA in governing synaptic plasticity. Neural Plast 2016: 4959523. 10.1155/2016/4959523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Hong EJ, Cohen S, Zhao W, Ho HH, Schmidt L, Chen WG, Lin Y, Savner E, Griffith EC, et al. 2006. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron 52: 255–269. 10.1016/j.neuron.2006.09.037 [DOI] [PMC free article] [PubMed] [Google Scholar]