

ABSTRACT
Periodontitis is one of the chronic diseases that have the greatest impact on human health, and it is associated with several other chronic diseases. Tissue damage associated with periodontitis is often connected with immune response. Immune cells are a crucial component of the human immune system and are directly involved in periodontitis during the inflammatory phase of the disease. Macrophages, as a key component of the immune system, are responsible for defence, antigen presentation and phagocytosis in healthy tissue. They are also closely linked to the development and resolution of periodontitis, through mechanisms such as macrophage polarization, pattern recognition receptors recognition, efferocytosis, and Specialized Pro-resolving Mediators (SPMs) production. Additionally, apoptosis and autophagy are also known to play a role in the recovery of periodontitis. This review aims to investigate the aforementioned mechanisms in more detail and identify novel therapeutic approaches for periodontitis.
KEYWORDS: Macrophages; cell death; pattern recognition receptors; efferocytosis, periodontitis
Introduction
Periodontitis is a multi-factor chronic inflammatory disease characterized by gingival inflammation, periodontal pocket formation, groove absorption, and tooth loosening and movement [1], which significantly affects oral health, aesthetics, and function [2]. Periodontitis is the most common cause of tooth loss, and it raises the risk of systemic disorders by triggering inflammatory responses in other tissues and organs [3]. The number of periodontal disease cases in 2019 is almost twice that in 1990, with an apparent rise in incidence and prevalence, which emphasizes the importance and urgency of the prevention and treatment of periodontitis [4]. The milestone research of Page and Schroeder announced that although plaque is the initial factor of periodontitis. It is the immune response of hosts to infection that determines the development of periodontitis [5].
Tissue destruction in periodontitis is mainly caused by the host’s immune response to infection [6,7]. Polymorphonuclear neutrophils (PMN) and macrophages play an important role in periodontitis by controlling the pathogenicity of gingival biofilm as well as activating adaptive immunity and play an important role in various physiological and pathological processes [8]. Macrophages originate from circulating monocytes, which develop from bone marrow precursor cells before entering the bloodstream. These monocytes can mature into macrophages in response to injury or inflammation, or migrate into tissues to become resident macrophages or dendritic cells. However, not all tissue-resistant macrophages are derived from monocytes. Before the formation of definitive hematopoietic stem cells, some macrophages, such as Langerhans cells, are secreted during the prenatal period from progenitor cells in the yolk sac and fetal liver of the fetus. [9,10]As an indispensable part of the human innate immune system, macrophages react phenotypically and functionally for recognizing, phagocytosing, and removing bacteria. However, they are also closely related to tissue damage and repair during diseases [11,12]. An increase in inflammatory substances produced by macrophages in the gingival tissue is associated with the advancement of periodontitis lesions, whereas inhibition of the release of these proinflammatory factors is associated with improved bone resorption and inflammatory cell infiltration status [13].
In this review, we delve into the functions and localization of macrophages. By examining the processes involved in the onset and progression of periodontitis, we attempt to discuss the possible contribution of macrophage activation to the development of periodontitis and its significance for upcoming periodontal treatment options.
Healthy periodontal condition
Polarization and plasticity of macrophages
Macrophages exhibit extremely heterogeneous characteristics in physiological and pathological conditions because they are sufficiently flexible to integrate a range of signals from bacteria, wounded tissues, and the normal tissue environment [14]. The plasticity and flexibility of macrophages characterized their activated state. Depending on various surface markers and biological activity, macrophages can be categorized into two phenotypes, including pro-inflammatory macrophages (M1) and anti-inflammatory macrophages (M2) [15,16]. Under certain conditions, M1 and M2 can be converted to each other, this dynamic process allows macrophages to maintain homeostasis in the microenvironmental changes. However, the imbalance of macrophage polarization is often connected with periodontitis progression and regulation [17]. Therefore, the ratio of M1 to M2 has been shown to indicate the progression of periodontitis [18].
Macrophage polarization is primarily stimulated by the cytokines in the micro-environment. M1 macrophages generate pro-inflammatory cytokines and chemokine to prevent the growth of neighboring cells when they are triggered by Th1 cytokines like interferon-γ(IFN-γ) or interleukin-6 (IL-6). M2 macrophages generate anti-inflammatory cytokines and Treg response in reacting to Th2 cytokines, glucocorticoids, and immune complexes for tissue repair, inflammation suppression, and immune regulation, promoting the proliferation of surrounding cells [16,19–22].
M1 macrophages have physiological functions that can be summarized as defence, pro-inflammatory responses, and tissue damage. M1 macrophages can produce cytokines and chemokines. The former include tumor necrosis factor-alpha (TNF-α), IL-6, and type I interferons. In contrast, the latter includes C-X-C motif chemokine ligand 1 (CXCL1), C-C motif chemokine ligand 2 (CCL2), and C-X3-C motif chemokine ligand 1 (CX3CL1). Additionally, M1 macrophages have strong antigen-presenting abilities, allowing them to recruit other immune cells, such as T cells, to the site of inflammation to work in concert [23]. Given their pro-inflammatory nature, an excessive inflammatory response can damage tissue or exacerbate inflammatory diseases, worsening the situation. M2 macrophages, also called anti-inflammatory macrophages, play an important role in maintaining homeostasis. The functions of M2 macrophages can be summarized as anti-inflammatory effects, promotion of tissue repair, angiogenesis, and suppression of excessive immune responses. M2 macrophages can produce various cytokines, including IL-6, IL-13, and IL-10. These cytokines have anti-inflammatory properties. M2 macrophages can also promote cell proliferation and angiogenesis, which contribute to tissue regeneration. They achieve these effects by promoting the production of interferon-gamma (IFN-γ), transforming growth factor-beta (TGF-β), and IL-10 [23,24].
The M1 or M2 classification, however, is an oversimplification, with tissue-resident macrophages being much more complex cells. With further investigation of the characteristics and functions of M2 macrophages, it is found to be developed into four other subtypes: M2a, M2b, M2c, and M2d [25].
M2a macrophages are stimulated by IL-4 or IL-13 and express markers like CD206 and IL-10. These macrophages can shift fibroblasts from a pro-inflammatory state to a reparative state, promoting tissue repair [26,27]. M2b macrophages can have both harmful and protective roles in disease. They exhibit a degree of regulatory function and can secrete both pro-inflammatory and anti-inflammatory factors. They are activated by immune complexes and Toll-like receptors. Typically, they have high expression levels of IL-10, IL-1β, and TNF-α [16,27]. M2c macrophages are activated in the presence of IL-10 or glucocorticoids, releasing transforming growth factor-beta 1 (TGF-β1) and IL-10, effectively engulfing apoptotic cells and reducing inflammation [28]. M2d macrophages are stimulated by adenosine A2 receptor (A2R) agonists and Toll-like receptor (TLR) ligands, along with IL-6. M2d macrophages can express IL-10 and vascular endothelial growth factor (VEGF), playing a role in angiogenesis and cancer metastasis [27,29,30].
The location and function of macrophages in normal conditions
The periodontium consists of gingiva, periodontal ligament, cementum, and alveolar bone. The dentogingival junction is a structure where the gingiva connects to the tooth surface and closes off the junction of the hard and soft tissues. The dentogingival junction consists of gingival epithelium, sulcular epithelium, and junctional epithelium, which prevents the invasion of bacteria in normal conditions. The slender gap between the gingiva and the tooth surface is known as the gingival sulcus. This sulcus contains a significant amount of gingival fluid, sourced from blood vessels within the sulcular and junctional epithelium. Immune cells such as polymorphonuclear leukocytes and monocytes can migrate into the gingival sulcus through the junctional epithelium. These immune cells, along with the cytokines they secreted, form the defence line of periodontium [24]. As previously mentioned, monocytes transform into macrophages in response to inflammation, thereby playing a role in the advancement of periodontitis. This implies that even in normal periodontal conditions, the connective tissue linked to the gingival epithelium is penetrated by inflammatory cells. In healthy periodontal tissues, macrophages and leukocytes are mainly located within the gingival connective tissue and are found in low concentrations. Garaicoa-Pazmino and his team employed CD68 and inducible nitric oxide synthase (iNOS) as markers to determine the number of M1 macrophages in tissue, while CD206 was used to identify M2 macrophages. The study aimed to compare the quantity and polarization status of macrophages in healthy gingival tissue. The results indicated that M2 macrophages were significantly more abundant than M1 macrophages in healthy tissue [31]. However, in cases of periodontal inflammation, there is an increased number of macrophages, especially M1 macrophages that are distributed more widely, extending deep into the connective tissue [24,32].
Macrophages are extensively dispersed throughout the body. Their roles in maintaining health can be outlined as follows. Firstly, their primary function is phagocytosis, allowing them to ingest bacteria and invading viruses, as well as to remove apoptotic cells through processes like efferocytosis. Secondly, macrophages can synthesize and release various cytokines, including anti-inflammatory and pro-inflammatory cytokines. Conversely, these cytokines can modulate the functions of macrophages. Thirdly, macrophages are a kind of professional antigen presentation cell (APC), contributing to the activation and regulation of immune responses [33,34]. Antigen presentation involves the uptake and processing of antigens by antigen-presenting cells, which then display these antigens on their surfaces as immunogenic peptides. These peptides are subsequently recognized by immune effector cells, triggering an immune response [35].
Macrophages play a significant role in periodontal health, as their presence allows for timely recognition of pathogens and recruitment to the site of infection. In different microenvironments, macrophages can differentiate into M1 or M2 phenotypes. Through interactions with pathogenic microorganisms, they secrete pro-inflammatory cytokines and chemokines, not only participating in microbial phagocytosis but also promoting inflammation.
Macrophage pattern recognition receptors(PRRs)
Pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) stimulate multiple intracellular signaling cascades by binding to the pattern recognition receptor (PRRs), which causes cell death and effector molecule synthesis to mediate inflammation and ultimately encourage the progression and regeneration of periodontitis [36]. Throughout the development of periodontitis, the innate immune response depends on identifying infectious factors and PRRs [37]. There are eight major sub-families of PRRs: stimulator of interferon genes (STING), nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), macrophages scavenger receptors (MSRs), C-type lectin receptors (CLRs), absent in melanoma 2 (AMI2)-like receptors (ALRs), retinoic acid-inducible gene (RIG)-like receptors (RLRs), and Toll-like receptors (TLRs) [38–41]. Among all pattern recognition receptors, ALRs, NLRs, and TLRs are considered the three receptors most closely associated with periodontitis [42].
According to cellular location, ten functional TLRs have been found in human beings [43]. TLR1/2/4/5/6 are expressed on plasma membranes or extracellular cell surfaces of macrophages and neutrophils. They are able to detect membrane components produced by microbes. TLR3/7/8/9 are only expressed in endolysosomal compartments, in which their primary role is to recognize nucleic acids from microorganisms [43,44]. All TLRs and members of the IL-1 receptor family, with the exception of TLR3, signal by attracting the intracellular signaling adaptor protein Myeloid differentiation factor 88 (MyD88), which encourages pro-inflammatory cascades. TLR3, on the other hand, signals through a TIR-domain-containing adapter protein (TIRAP)-inducing interferon-β pathway, inducing the expression of Type I interferons, such as IFNα/β [45–47].
Both NLR family members and ALR family members can activate the inflammasome, and they have been closely linked to the initiation and progression of periodontitis. Certain NLR family members, like the NOD-like receptor family pyrin domain-containing 3 (NLRP3), play a role in inflammasome formation. Upon recognizing bacterial lipopolysaccharides (LPS) or other metabolic byproducts, AIM2 and NLRP3 activate the inflammasome, promoting the production of activated Caspase-1, which in turn mediates the release of IL-1β and IL-18 [42,48]. Researchers have found that the expression levels of molecules such as NLRP3, Caspase-1, and IL-1β can be used as one of the indicators for detecting periodontitis. Furthermore, modulating the NLRP3 inflammasome is being considered as a new approach for treating inflammation [49].
The role of macrophages in the progression of periodontitis
Macrophage PRRs and periodontitis pathogen
Porphyromonas gingivalis has been identified as the main pathogen responsible for the development of chronic periodontitis, aggressive periodontitis, and periodontal abscess [50]. Alterations in the microbial composition of the periodontium, resulting in an imbalance in the native oral microbiota, have been closely associated with periodontal tissue destruction and the development of dental caries. The intrinsic components and metabolic byproducts of Porphyromonas gingivalis can serve as virulence factors, causing direct harm to periodontal tissues or inducing damage indirectly by provoking immune responses. The most significant pathogenic components of Porphyromonas gingivalis are capsules, proteases, LPS, and fimbriae [51]. These virulence factors act on pattern recognition receptors on the surface of macrophages, inducing cytokine production, which leads to the development of periodontitis.
Porphyromonas gingivalis has two distinct types of fimbriae on its surface: long fimbriae and short fimbriae. Although their antigenicity differs, both play a crucial role in mediating bacterial adhesion and invasion. In a rat model of oral infection, short fimbriae were observed to trigger the expression of TNF-α, along with cytokines IL-1α and IL-6, in murine macrophages. Additionally, they were found to promote the maturation of precursor cells into osteoclasts [52]. In addition to being associated with cytokine secretion, receptors on the macrophage surface are also involved in the entry of Porphyromonas gingivalis. Porphyromonas gingivalis triggers signal transduction by engaging TLR2 and CD14 on macrophage surfaces, resulting in the activation of complement receptor 3 (CR3). This interaction enables Porphyromonas gingivalis to enter macrophages through CR3, subsequently inhibiting the expression of IL-12p70 and allowing the bacterium to persist within the macrophages [53]. Porphyromonas gingivalis can exert both pro-inflammatory and pro-adhesive effects through TLRs. The former leads to the production of inflammatory cytokines, while the latter facilitates bacterial adhesion to cells. The signaling pathways mediated by TLR2 differ for these two effects. In the case of the pro-inflammatory response, the pathway hinges on the adapter protein MyD88, whereas the pro-adhesive pathway rests on phosphoinositide 3-kinase (PI3K) [54]. TLR2 forms heterodimers by combining with TLR1 and TLR6. It recognizes Porphyromonas gingivalis by recruiting TIR-domain-containing adapter proteins (TIRAP) and Myeloid differentiation primary response 88 (MyD88) to the cytoplasmic domain of TLR2, constituting the conventional TLR2-MyD88 and TIRAP-TLR inflammatory pathway. Downstream signaling events activate the NF-κB pathway, inducing host defence activities associated with periodontitis, leading to the onset of the disease [54–56]. TLRs also interact with co-receptors to induce inflammatory responses. For example, C-X-C chemokine receptor type 4 (CXCR4) serves as a co-receptor for TLR2, aiding TLR2 in recognizing specific signaling molecules and participating in the signal transduction process. Through TLR2-CXCR4 crosstalk, when TLR2 detects fimbriae, protein kinase A (PKA) suppresses the transduction of the MyD88 inflammatory signaling pathway, reducing the nitric oxide produced by macrophages and diminishing their ability to damage pathogens [37]. Furthermore, it was found that Porphyromonas gingivalis long fimbrial proteins were able to activate human gingival epithelial cells through TLR2. The long fimbriae can substantially enhance NF-κB activation and, in turn, stimulate IL-8 production [57].
Porphyromonas gingivalis is a Gram-negative bacterium, and lipid A (LPS) is a key component of the cell wall in Gram-negative bacteria, as well as one of the major virulence factors of Porphyromonas gingivalis. After the breakdown of bacteria, Porphyromonas gingivalis is capable of releasing numerous microvesicles laden with LPS, which can infiltrate periodontium and thereby contribute to immune reactions that lead to tissue damage. Earlier assumptions indicated that Porphyromonas gingivalis LPS activates macrophages solely via TLR2. However, recent findings have revealed that this activation actually requires both TLR2 and TLR4. Researchers hypothesize that this dual activation may be linked to the lipid diversity in Porphyromonas gingivalis [58]. TLR4 must form a complex with myeloid differentiation protein-2 (MD-2), known as the MD-2/TLR4 complex, which is crucial due to the multiple acyl chains of lipopolysaccharides (LPS), to bind its ligand – LPS. In this process, the lipopolysaccharide-binding protein (LBP) and CD14 complex transports LPS to the surface of the MD-2/TLR4 complex, enhancing the efficiency of LPS transport [59,60]. LPS delivered to the membrane of MD2/TLR4 in the presence of LBP-LPS-CD14 complex allows stimulation of TLR4 to initialize the immune response [37]. LPS not only acts on macrophages but can also activate neutrophils. The findings from Tomohisa and his colleagues indicate that Porphyromonas gingivalis LPS induces neutrophil extracellular traps (NETs) through TLR2 and TLR4. The expression of NETs was suppressed when cells were exposed to Porphyromonas gingivalis LPS after treatment with antibodies targeting TLR2 and TLR4, and similarly when stimulated with Escherichia coli LPS following TLR4 blockade with specific antibodies [61].
Porphyromonas gingivalis produces gingipains that include arginine-targeting enzymes as well as lysine-targeting enzymes, that is RgpA/RgpB and Kgp. As mentioned previously, CD14, serving as a co-receptor for TLR2, is significant in recognizing and transmitting signals related to pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs). Due to the purified gingipains RgpA and Kgp’s downregulation of membrane CD14 expression, macrophage response to Porphyromonas gingivalis infection is reduced. However, CD14 is crucial to the recognition and combination of fimbriae and LPS of Porphyromonas gingivalis, which means the reduction of CD14 expression benefits the reproduction of bacteria and deteriorates inflammation [37,42]. In addition, RgpA, RgpB, and Kgp can directly hydrolyze complement proteins such as C3, impacting the activation of the complement system. By degrading complement components, gingipains diminish the complement system’s capacity to attract and activate immune cells like macrophages, thus impairing the immune response and facilitating the pathogen’s evasion from immune detection [62]. From an intercellular mechanism perspective, Porphyromonas gingivalis utilizes Arginine-specific gingipain A (HRgpA) to inhibit the C4b protein on the bacterial plasma membrane, thus attenuating the classical complement activation pathway by altering the C3 convertase. Concurrently, HRgpA and HRgpB produce bioactive C5a, leading to the suppression of macrophage function through crosstalk between the complement component 5a receptor (C5aR) and TLR2. Porphyromonas gingivalis mitigates TLR2-mediated signal transduction and its associated biological processes via this C5a-C5aR-TLR2 crosstalk. It suppresses TLR2-driven IL-12p70 secretion while simultaneously enhancing the production of cytokines linked to bone degradation, like IL-6, consequently fostering alveolar bone loss [63]. Except for cytokine expression, Gingipains can also enhance macrophage cyclic adenosine monophosphate (cAMP) production through the crosstalk mechanism of C5a-C5aR-TLR2 with the aid of certain receptors (such as C-X-C chemokine receptor type 4, CXCR4). To capture the limited binding site of cAMP response element-binding protein (CREB) binding protein (CBP), cAMP promotes the activation of cAMP-dependent protein kinase A (PKA) and cAMP response element-binding protein (CREB), while inhibiting the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway. As a result, this process reduces macrophage nitric oxide (NO) production and weakens its ability to kill bacteria [37,64]. In addition to interacting with the TLRs, Porphyromonas gingivalis can also interact with the NLRs. Researchers have discovered that Porphyromonas gingivalis can stimulate the NLRP3 inflammasome, triggering the release of IL-1β and TNF-α, thereby inducing inflammation. This activation process, however, does not depend on gingipains. Despite this, gingipains can break down IL-1β and TNF-α in tissues, serving as a bacterial self-protection strategy. With this mechanism, Porphyromonas gingivalis can initiate the host’s inflammatory response while ensuring that the inflammation does not escalate excessively, thereby promoting bacterial proliferation [65].
Macrophages polarization and the progression of periodontitis
Periodontal pathogens have a significant impact on macrophage polarization. Porphyromonas gingivalis drives the differentiation of macrophages toward the M1 phenotype through four key mechanisms. As previously discussed, M1 differentiation is regulated by cytokines such as IFN-γ. When pathogens invade the periodontium, they disturb the equilibrium of the local microbiome, which in turn disrupts the cytokine balance within the periodontal environment. This imbalance leads to elevated levels of certain cytokines like IL-6, which promote M1 polarization. Moreover, the interaction between Porphyromonas gingivalis and TLRs also promotes the shift toward the M1 phenotype [21].
Different signaling molecules and pathways can influence the polarization of macrophages. When the signaling pathways are activated by interferons or TLRs, macrophages tend to polarize towards the M1 phenotype, which is characterized by pro-inflammatory and antimicrobial properties. However, when the pathways are activated by IL-4 or IL-13, macrophages lean towards the M2 phenotype. Both pathways rely on the interferon regulatory factor (IRF)/signal transducer and activator of transcription (STAT) pathway, but the former is mediated by STAT1 protein while the latter operates through STAT6 protein [21].
Researchers have found that when polymorphonuclear neutrophils (PMNs) from periodontitis patients are stimulated with Porphyromonas gingivalis lipopolysaccharide, these cells produce significant amounts of superoxide, a type of reactive oxygen species (ROS). During the development of periodontitis, large numbers of PMNs, part of the innate immune response, accumulate at the site of inflammation, and this infiltration results in substantial ROS production. ROS serves a dual function. On the one hand, they can eliminate bacteria, aiding in the defence against infections; on the other hand, they can harm tissues. Elevated levels of ROS can activate osteoclasts, leading to the degradation of periodontal tissue and bone. Moreover, ROS can trigger the NF-κB signaling pathway [66]. It activates the NF-κB signaling pathway upstream, leading to increased transcription of downstream genes responsible for pro-inflammatory cytokine secretion, which drives macrophage polarization toward the M1 phenotype [67].
Previously, we examined the influence of pathogens on macrophage polarization. Next, we will explore the specific mechanisms through which cytokines produced by polarized M1 macrophages affect periodontal tissue and their role in periodontal damage. M1 macrophages can secrete various pro-inflammatory cytokines, such as IL-6, TNF-α, and IL-1β, which can damage periodontal tissue. IL-1β is primarily produced by macrophages and can stimulate the formation of osteoclasts and the production of proteases, resulting in the breakdown of alveolar bone [68]. Research has found that significantly elevated levels of IL-1β can be detected in both gingival crevicular fluid and saliva from patients with periodontitis [68]. As mentioned above, Porphyromonas gingivalis is able to activate both TLRs and NLRs to start an immune response. The NLRP3 inflammasome can be activated through two steps (Figure 1). The first step involves pattern recognition receptors on macrophages identifying pathogen-associated molecular patterns (PAMPs) on the surface of Porphyromonas gingivalis, leading to the activation of the NF-κB signaling pathway. This activation not only triggers the NLRP3 inflammasome but also stimulates the production of pro-interleukin-1 beta (pro-IL-1β) and pro-interleukin-18 (pro-IL-18) [69]. It also reduces the activation threshold for NLRP3 [70]. The second step can be activated by a variety of signals, including the activation of the P2X7 receptor, pore-forming toxins, damaged mitochondria, and ruptured lysosomes. These activation signals lead to increased K+ efflux and increased Ca2+ influx. The increased K+ efflux activates NEK7, an important regulator in inflammasome formation. The elevated Ca2+ influx can lead to mitochondrial damage, resulting in the release of excessive reactive oxygen species (ROS) and mitochondrial DNA (mtDNA). All of these factors can trigger NLRP3, leading to the formation of the NLRP3 inflammasome. The rupture of lysosomes not only causes an increase in K+ efflux but also releases cathepsins, which further activate NLRP3 [71]. Once the inflammasome is activated, it generates caspase-1, which in turn leads to the secretion of IL-1β and IL-18 [69]. The interaction between Th17 cells and macrophages also plays a crucial role in the progression of periodontitis. Once activated, Th17 cells can recruit neutrophils and macrophages to the site of inflammation. They can also secrete IL-17, which amplifies the inflammatory response, while IL-17 can further induce macrophages to secrete TNF-α, IL-1β, and intercellular adhesion molecule 1 (ICAM-1). In addition, IL-23 and IL-6 secreted by macrophages can stimulate the activation of Th17 cells, creating a cycle of immune activation that perpetuates the inflammatory response [72].
Figure 1.
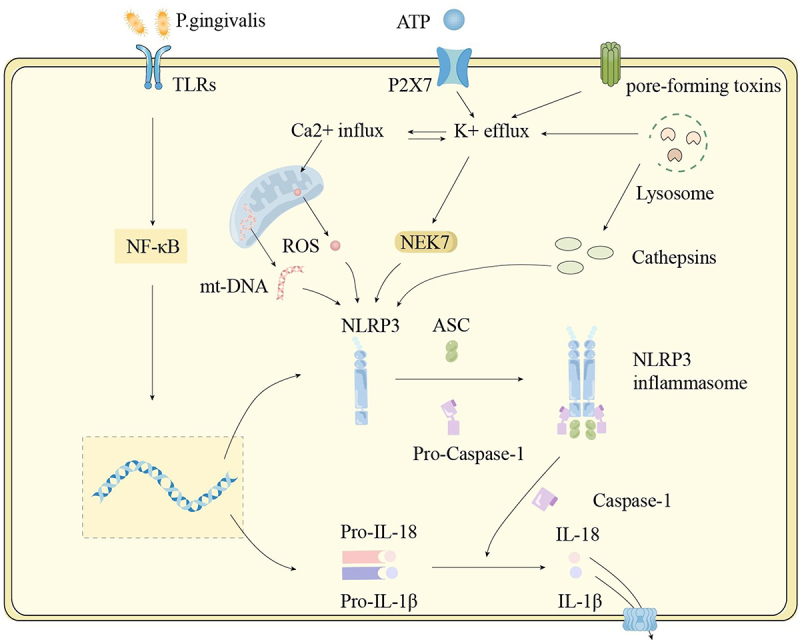
Activation of the NLRP3 inflammasome. The first step in inflammasome activation is initiation. Pattern recognition receptors (PRRs) recognize pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) from periodontal pathogens, triggering the NF-κB signaling pathway. This leads to an increase in the gene expression of NLRP3 and the precursors of interleukin-1 beta (IL-1β) and interleukin-18 (IL-18). The second step is activation. A number of activation signals have been identified, including ATP activating the P2X7 receptor, mitochondrial damage, lysosomal rupture, and pore-forming toxins. These signals cause NLRP3 proteins to aggregate and assemble with the adaptor protein ASC (apoptosis-associated speck-like protein containing a CARD) and caspase-1, which contains a cysteine protease activation domain, forming the inflammasome. Once the inflammasome is activated, caspase-1 is also activated, cleaving the precursors of IL-1β and IL-18 into their active forms.
Researchers identified the presence of soluble IL-6 receptor (sIL-6 R) in the gum tissue of patients with periodontitis. IL-6 operates by attaching to the IL-6 receptor on the membrane of human gingival fibroblasts (HGF). The existence of sIL-6 R ensures that even when the number of IL-6 receptors on the membrane of HGF cells is low, IL-6 can still efficiently activate these cells. This interaction could lead to the degradation of key proteins generated by HGF, affecting their capacity to mend damaged periodontal tissue. At the same time, TNF-α and IL-1β also target HGF, and these cytokines collectively impact the ability of fibroblasts to repair damaged periodontal tissue [73]. Additionally, IL-6 can inhibit protein synthesis in osteoblasts, impacting bone formation. When destruction outweighs repair, periodontal tissues exhibit signs of alveolar bone resorption, with periodontal pockets receding toward the root and significant subcellular infiltration [73,74].
Neutrophil changes in periodontitis
Neutrophils are the most prevalent immune cells in normal periodontal tissue. As the first line of the innate immune system, neutrophils are quickly drawn to the infection site in response to several inflammatory triggers to fight against the bacteria or fungi that try to invade our body [75].
The migration of neutrophils toward and across the vascular wall to the site of inflammation is closely related to cell adhesion molecules, such as triggering receptors expressed on myeloid cells 1 (TREM-1) [76]. The strong chemotaxis of neutrophils depends on interactions between chemokines and their receptors as well as cell adhesion molecules. However, the ability of neutrophil chemotaxis drops in chronic periodontitis, even with successful treatment [77]. Leukocyte recruitment to infected tissues is significantly mediated by TLRs, while E-selectin and intercellular adhesion molecule 1 (ICAM-1), which are necessary for leukocyte rolling and adherence, may be expressed on the surface in the guidance of TLRs on endothelium [78]. TLRs not only mediate neutrophil chemotaxis and adhesion, but they also play a significant role in modulating neutrophil function. Neutrophils recognize Porphyromonas gingivalis PAMPs through TLR2, triggering C5a-C5aR-TLR2 crosstalk. Through the crosstalk, Porphyromonas gingivalis induces ubiquitination and proteasomal breakdown of MyD88. It affects the signaling transduction of the TLR2-MyD88 inflammatory pathway, thereby reducing the anti-inflammatory potency of neutrophils. Therefore, neutrophils are controlled by Porphyromonas gingivalis in order to maintain microbial populations and prolong inflammation [79].
Neutrophils are among the primary cells that express the NLRP3 gene, and Porphyromonas gingivalis has been shown to promote NLRP3 expression. When the NLRP3 inflammasome is activated, neutrophils are induced to secrete IL-1β, which mediates the activation of osteoclasts, leading to alveolar bone resorption and destruction. Neutrophils primarily eliminate pathogens by phagocytosing them, releasing neutrophil extracellular traps (NETs), degranulating, and generating reactive oxygen species (ROS) [80]. Neutrophils can also release IL-17, promoting the recruitment of neutrophils. IL-23 stimulates Th17 cells to secrete IL-17, indirectly affecting IL-17 levels. As a result, when IL-17 and IL-23 are overactive, large numbers of neutrophils are recruited to the site of inflammation. This leads to significant production of reactive oxygen species (ROS) by the neutrophils. In the periodontal tissue of patients with periodontitis, reactive oxygen species (ROS) levels are elevated, but the levels of antioxidants such as superoxide dismutase (SOD) remain unchanged. This imbalance between oxidation and antioxidation leads to excessive ROS, causing cells in periodontal tissue to be damaged or to rupture and die [66,72].
Periodontitis not only leads to an increase in the number of neutrophils but also causes them to exhibit hyper-reactivity [81]. Trained immunity refers to the phenomenon wherein activation of the innate immune system fosters responsiveness to subsequent challenges [82]. Investigation has revealed that it was able to be triggered in hematopoietic stem and progenitor cells (HSPCs) located in the bone marrow through the interactions between PAMPs and PPRs [81]. The increase in myeloid-biased differentiation and generation of hyper-reactive myeloid cells (neutrophils) can be initiated by inflammation-induced metabolic, epigenetic, and transcriptomic adaptations in HSPCs [83]. Innate immune training of HSPCs can be triggered in the direct interaction with periodontal pathogens or the indirect activation of hematopoietic system through specialized cells located in the bone marrow or peripheral tissue, which was activated by the release of cytokines [81]. In patients with periodontitis, peripheral neutrophils exhibit hyper-reactivity, resulting in excessive production of reactive oxygen species (ROS). The release of ROS is partly due to their inherent properties and partly due to the stimulation of Fc receptors by immune complexes or antibodies. The inherent property of neutrophils is that, even without Fc receptor stimulation, those from periodontitis patients tend to release more ROS compared to neutrophils from individuals with healthy periodontal tissue. This tendency persists even after periodontal treatment has been completed [84].
The function of macrophages in the regeneration of periodontitis
Efferocytosis and periodontitis
Efferocytosis is the term used to explain the process by which both professional and non-professional phagocytes eliminate apoptotic cells. It maintains the dynamic balance of our body rapidly and efficiently [85]. Many human autoimmune or auto-inflammatory illnesses, including periodontal disease, atherosclerosis, etc. are characterized by defective elimination of dead cells [86]. The process of efferocytosis involves “find me,” “keep out,” “eat me,” and “do not eat me” signal molecules and can be mainly divided into four steps (recruitment, recognition, engulfment, and clearance) [85].
In the recruitment phase, apoptotic cells release “find-me” signals, such as fragmented chemokines and adenosine triphosphate (ATP), which attract macrophages to the site of cell death. Receptors on macrophages can detect these signals, triggering macrophage migration. Additionally, to specifically recruit phagocytes and prevent the accumulation of inflammatory cells, apoptotic cells also release “keep away” signals, such as lactoferrin. Once macrophages arrive at the site of the apoptotic cells, the recognition phase begins. Apoptotic cells interact with specific receptors on macrophages by releasing “eat-me” signals, such as phosphatidylserine, triggering the engulfment response. Conversely, “don’t eat me” markers are expressed on living cells to distinguish between live and dying targets [87,88]. In the engulfment phase, the phagocytic signals activate actin polymerization and cytoskeletal rearrangement via the CRK-like protein II (CRKII)-dedicator of cytokinesis 1 (DOCK180)-engulfment and cell motility 1 (ELMO) complex, or the ATP-binding cassette transporter A1 (ABCA1)/engulfment adapter protein GULP complex, promoting the phagocytosis process [89]. In the final phase, macrophages digest apoptotic cells through endocytosis; phagosomes fuse with lysosomes to break down the apoptotic cells into nucleotides, lipids, sterols, and peptides. During this process, anti-inflammatory mediators, such as TGF-β, are released [90].
The balance in the number of polymorphonuclear neutrophils (PMNs) in tissues depends on the equilibrium between PMN production and PMN elimination. This balance is maintained by a negative feedback mechanism (Figure 2) [91]. To maintain neutrophil stability, efferocytosis regulates neutrophil generation and elimination in healthy or inflammatory conditions [85]. Tissue macrophages engulf extravasated apoptotic PMNs for phagocytosis. This activity inhibits IL-23 expression, which in turn inhibits the production of IL-17 and Granulocyte-Macrophage colony-stimulating factor. IL-23 stimulates Th17 cells to secrete IL-17, indirectly affecting IL-17 levels. IL-17 promotes the recruitment of neutrophils. IL-17 can also increase the expression of receptor activator of nuclear factor-kappa B ligand (RANKL) while inhibiting the expression of osteoprotegerin (OPG), thereby promoting osteoclast activity (Figure 2) [72]. Efferocytosis prevents excessive activation of neutrophils caused by the overproduction of IL-17 and IL-23. As a result, it decreases neutrophils, eosinophils, and basophils in the bone marrow [92]. By inhibiting neutrophil release, this process creates an ongoing equilibrium between the formation and elimination of neutrophils.
Figure 2.
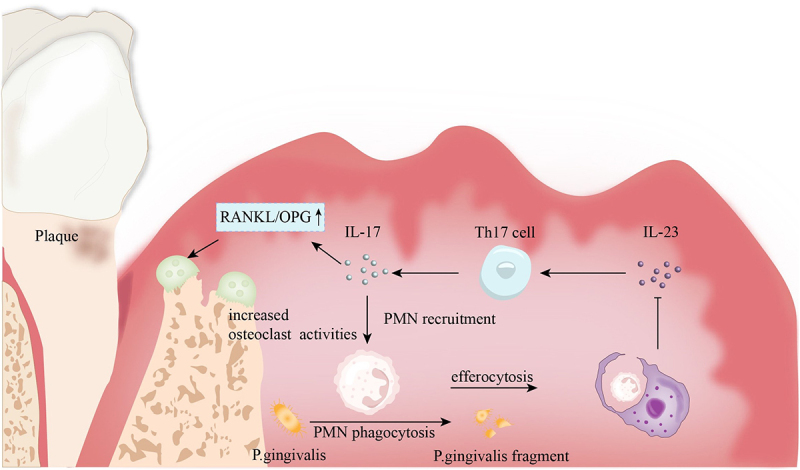
Efferocytosis and the negative feedback of PMNs. In periodontitis, apoptotic polymorphonuclear neutrophils (PMNs) are cleared through efferocytosis, a process that promotes the release of interleukin-23 (IL-23). IL-23 can activate Th17 cells, stimulating the secretion of interleukin-17 (IL-17). IL-17, on one hand, promotes the recruitment of PMNs, while on the other hand, it can increase the RANKL/OPG ratio, activating osteoclasts and leading to alveolar bone resorption.
Polymorphonuclear neutrophils (PMNs) undergo apoptosis after engulfing periodontal pathogens, and efferocytosis is the process by which macrophages clear apoptotic PMNs. On one hand, if PMNs that have ingested pathogens are not promptly cleared, they may release the harmful substances they have engulfed, causing secondary damage to the body. Additionally, the process of macrophages recognizing and phagocytizing apoptotic PMNs triggers the synthesis and release of specialized pro-resolving mediators (SPMs). SPMs may be involved in inflammatory responses through various pathways, playing a significant role in resolving inflammation by enhancing the phagocytosis of apoptotic cells by macrophages, thereby promoting the resolution of inflammation [93].
Production of specialized pro-resolving mediators
Specialized pro-resolving mediators (SPMs), enzymatically derived from polyunsaturated fatty acids, play a crucial role in anti-inflammatory processes, promoting inflammation resolution, and enhancing phagocytic capacity. The phagocytosis of apoptotic cells triggers the production of SPMs, which act as signals to facilitate the restoration of vascular integrity and the regeneration and repair of tissues, including Maresin (MaR), Resolvin E (RvE), Resolvin D (RvD), and Lipoxin (LX). SPMs, along with other molecular factors, work to maintain homeostasis in the body. Efferocytosis enhances the generation of SPMs by metabolizing apoptotic cell components, elevating SPM precursors, and facilitating their conversion. SPMs such as LXA4and RvD1 can enhance efferocytosis, creating a feedback loop between SPM production and efferocytosis. This loop enhances the continuous clearance of apoptotic cells within tissues [94]. When the balance between SPMs and pro-inflammatory lipid mediators is disrupted, it can lead to persistent leukocyte infiltration, inflammatory macrophage polarization, and reduced efferocytosis. As shown in Table 1, each type of Specialized Pro-resolving Mediator plays a distinct role in the recovery process from periodontitis.
Table 1.
Functions of different SPMs.
| SPM type | Subtype | Function | References |
|---|---|---|---|
| LX | LXA4 and LXB4 | Limit neutrophil recruitment and neutrophil-mediated tissue injury (Increased production and accumulation of prostaglandin E2) | [95,96] |
| LXA4 and LXB4 | Enhance macrophage recognition of apoptotic polymorphonuclear neutrophils (PMNs) by increasing the activity of the αvβ3-CD36 complex. | [97] | |
| LXA4 and LXB4 | Promote the polarization of macrophages from the M1 phenotype to the M2 phenotype. | [97] | |
| LXA4 | Promote the phagocytosis of apoptotic PMN in macrophages | [98,99] | |
| LXA4 | The LXA4-ALX/FPR2 pathway controls the regeneration ability of hPDLSCs | [100,101] | |
| LXA4 | Enhance macrophage phagocytic capacity, and reduced production of pro-inflammatory cytokines (IL-8) during phagocytosis, while increasing anti-inflammatory factors. | [102] | |
| Rv | RvE1 | Effects on bone: Prevents bone loss and can even reverse bone loss in already diagnosed periodontitis cases. | [103] |
| RvE1 | Effects on inflammatory cells: Reduces infiltration of inflammatory cells and lowers the expression of inflammation-associated genes. | ||
| RvE1 | Effects on microbial composition: Regulates local inflammation to shape the subgingival microbiota in patients with periodontitis. | ||
| RvE1 | Reduce the expression of L-selectin and integrin CD18, thus inhibiting binding to peripheral blood PMNs and monocytes | [104] | |
| RvE1 | Restore the regenerative property of human periodontal ligament stem cells (hPDLSCs) | [100] | |
| RvE1 | Boost cell viability, speed up wound healing, and accelerate cell migration. | [100] | |
| RvD | RvD1 | Overcame IL-1β’s negative effects on PDLF proliferation and wound healing | [105] |
| RvD1 | Promote the production of anti-inflammatory proteins in Mφs; block the secretion of Th1 cytokines |
[106] | |
| RvD2 | Reduce the RANKL/OPG ratio to decrease alveolar bone resorption. | [107,108] | |
| MaR | MaR1 | Induce the regeneration of Mφ PPAR-γ(Reduce inflammation to encourage tissue repair and regeneration. Regulate lipid synthesis and degradation to maintain cellular metabolic balance. Promote the polarization of macrophages toward the M2 phenotype.) |
[109] |
| MaR1 | Limit PMN migration and decrease ROS generation. | [102] | |
| MaR1 | Enhance the phagocytic capacity of macrophages for both pathogens and apoptotic cells. | [109] | |
| MaR1 | Promote the formation of cementum and bone. | [100,102] | |
| MaR1 | Boost cell viability, speed up wound healing, and accelerate cell migration. | [100] | |
| MaR1 | Restore the regenerative property of human periodontal ligament stem cells (hPDLSCs). | [100] | |
| MaR1 and MaR2 | Promote the formation of cementum and bone. Encourages macrophage polarization toward the M2 phenotype. |
[102] |
Chronic inflammation resulting from periodontitis is closely linked to cardiovascular diseases, such as atherosclerosis. Specialized pro-resolving mediators can regulate inflammation and play a role in maintaining periodontal health. However, SPMs also have a protective role in the cardiovascular system, potentially helping to prevent cardiovascular diseases by reducing inflammation and promoting its resolution. Thus, the function of SPMs extends beyond periodontitis and could have implications for cardiovascular health. ADP has a dual role in platelet aggregation. Firstly, it can trigger the translocation of P-selectin to the surface of platelets, thereby initiating platelet adhesion and aggregation. P-selectin is a cell adhesion molecule that plays a pivotal role in platelet activation, facilitating interactions among platelets and with other cells. Secondly, ADP can promote actin polymerization. Actin is a component of the cytoskeleton, and its polymerization aids in the shape changes and aggregation processes of platelets. Actin polymerization is a critical step in the formation of platelet clots during platelet activation. Resolvin E1 (RvE1) inhibits both of these ADP-induced processes, thereby suppressing platelet activation and aggregation. This helps prevent unwanted platelet aggregation and reduces the risk of thrombosis [110]. Experiments with mice showed that the use of Resolvin D4 (RvD4) leads to a decrease in the number of neutrophils within thrombi and an increase in cells that promote inflammation resolution. Additionally, mouse neutrophils become less sensitive to releasing neutrophil extracellular traps (NETs), which are closely linked to the formation of deep vein thrombosis. The application of RvD4 can help to resolve thrombi, suggesting a novel approach for treating cardiovascular diseases [111].
Modes of cell death and periodontitis recovery
When PAMPs or DAMPs stimulate macrophages, PRR-induced signal pathways cause cell death, which involves the development of periodontitis. Apoptosis and autophagy are the two periodontitis-related subtypes of programmed cell death, which is a form of active cellular death driven by an accumulation of gene expression events [112].
Apoptosis is a programmed and autonomous cell death process controlled by genes in the progress of normal survival [113]. There are two main apoptotic processes that are related to and interact with one another: death receptor-mediated apoptosis and mitochondria-mediated apoptosis, also known as the mitochondrial pathway and the death receptor pathway, respectively [114]. The intrinsic pathway is typically triggered by internal stress, oxidative stress, and similar factors. Upon cellular stimulation, the mitochondrial membrane becomes more permeable, leading to the leakage of substances such as cytochrome C. Cytochrome C combines with apoptosis-inducing factor (AIF) to form the apoptosome, which activates the caspase cascade. Once caspase-9 is activated, it further activates caspase-3, resulting in DNA fragmentation, cytoskeletal changes, and the formation of apoptotic bodies. Exogenous death ligands interacting with cell surface death receptors initiate the death receptor apoptosis. The most typical ligands and their receptors include FasL/Fas and TNF-α/TNFRl [115]. When cytokines or other external signals bind to death receptors on the cell surface, it triggers an internal cascade of apoptosis, activating caspase-8, which then activates caspase-3. The activation of caspase-3 leads to apoptosis [116]. After apoptosis, “find-me” signals will be released to promote macrophage migration [117]. Apoptotic bodies formed during apoptosis consist of membrane-encased cellular components. Once formed, they are eliminated through efferocytosis. This process helps prevent the leakage of harmful cellular contents that could lead to the spread of inflammation.
Apoptotic bodies function like trash bags, packaging and removing cellular debris from apoptosis. Yet, recent research indicates that the contents within these apoptotic bodies could offer therapeutic benefits [118]. Marin-Gallen et al. demonstrated that when anti-apoptotic dendritic cells engulf apoptotic bodies derived from in vitro apoptotic cells, these dendritic cells produce fewer pro-inflammatory cytokines, lowering their inherent immune response and enhancing immune tolerance. The self-antigens contained in apoptotic bodies can modulate the immune system, helping to control inflammatory responses and prevent tissue damage [118,119]. Apoptotic bodies can also mediate the transfer of intracellular contents. Research has shown that apoptotic bodies containing miRNA-126 can induce the expression of CXCL12 and recruit vascular endothelial cells, providing a potential treatment for atherosclerosis [120]. This implies that in the treatment of periodontitis, apoptotic bodies could be considered as a carrier system to deliver anti-inflammatory factors or specific functional molecules, such as miRNA, to promote the regeneration of periodontal tissues.
Kato et al. proved that macrophage apoptosis was induced by A. actinomycetemcomitans Y4 [121]. Numerous bacteria found in the oral cavity have the ability to stimulate macrophage apoptosis [42]. Macrophage apoptosis helps the body kill and clear pathogens, thereby promoting tissue recovery. Mycobacterium tuberculosis has been shown to prolong its survival within the body by inhibiting host macrophage apoptosis. In this case, the key to the survival of Mycobacterium tuberculosis is the suppression of the macrophage apoptosis mechanism, allowing the pathogen to proliferate and spread. If pathogens can inhibit macrophage apoptosis and multiply within them, it could ultimately lead to a more destructive form of cell death, namely necrosis, a type of non-programmed cell death, resulting in a more severe inflammatory response and tissue damage. However, it is uncertain if a comparable mechanism is found among periodontal pathogens. If such a mechanism does exist among periodontal pathogens, then researching how to prevent pathogens from inhibiting macrophage apoptosis could be a novel approach to treating periodontitis [42].
The process of autophagy consists of four stages: formation, membrane nucleation, fusion, and degradation (Figure 3). When the autophagy mechanism is activated, an isolation membrane is formed in the cytoplasm, driven by autophagy-related gene (AGT) products such as LC3 and Beclin-1. The isolation membrane gradually expands and envelops pathogens or damaged necrotic cells, forming a double-membrane-bound vesicle known as an autophagosome. Sequestration separates the target material from the rest of the cell, making it one of the critical steps in autophagosome formation [122]. Many genes are involved in this process, autophagy-related gene ATG5-ATG12 conjugation and the ATG8 (LC3) conjugation system is important for the production of autophagosome [123]. The autophagosome then fuses with a lysosome, which releases hydrolases to break down the enclosed material. Finally, the contents of the autophagosome are degraded into smaller molecules, such as amino acids and fatty acids, which the cell can reuse, completing the autophagy process [122].
Figure 3.
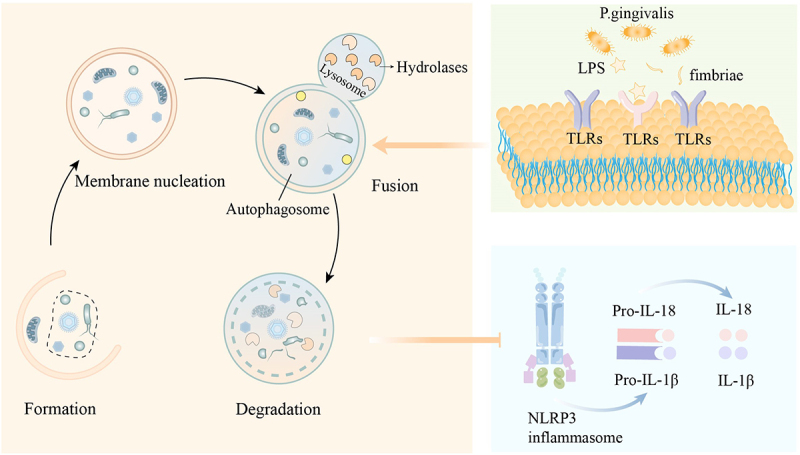
Autophagy and its related mechanism. The autophagy process includes formation, membrane nucleation, fusion, and degradation. When toll-like receptors (TLRs) are activated, intracellular autophagy increases. This increase in autophagy can inhibit the release of interleukin-1 beta (IL-1β) and interleukin-18 (IL-18) caused by the activation of the NLRP3 inflammasome.
Autophagy limits pathogen invasion and serves as an important component of innate immunity. Pattern recognition receptors play a critical role in recognizing pathogens in periodontitis (Figure 3). It has been proven to be crucial in promoting macrophage autophagy as well. Studies have shown that when macrophages are exposed to TLR ligands, the level of autophagy increases. Agonists for TLR1 and TLR2 can effectively inhibit the survival of Porphyromonas gingivalis in dendritic cells while enhancing the occurrence of autophagy in these cells [124]. As it was discovered, single-stranded DNA and bacterial lipopolysaccharide (LPS) activating TLR4 and TLR7 respectively lead to autophagy in RAW264.7 cells. Adaptors MyD88 and Trif are drawn to activated TLR4, whereupon they can bind to Atg6/Beclin-1 to initiate autophagy [42,125]. This indicates that when macrophage TLRs are stimulated, increased autophagy in macrophages facilitates the elimination of pathogens, controlling the number of intracellular pathogens. After Porphyromonas gingivalis invasion, THP-1-derived macrophages expressed more autophagy-related proteins such Beclin-1 and LC3-II. In this way, with increased ATG5-ATG12 conjugation, Porphyromonas gingivalis killing was decreased when autophagy was inhibited [126].
In addition to TLRs, autophagy can also influence cellular activities through NLRs, particularly in the activation of the NLRP3 inflammasome (Figure 3). The initiation of the NLRP3 inflammasome involves two steps: receiving the initial signal and the triggering signal. The cell detects activation signals (such as damaged mitochondria or ruptured lysosomes), leading to the assembly and construction of the inflammasome. When mitochondria are damaged, the production of reactive oxygen species (ROS) increases, activating NLRP3-dependent caspase-1 and promoting the release of IL-1β from macrophages. When ROS is cleared, this activation of caspase-1 and the release of IL-1β are significantly reduced [127]. As a pro-inflammatory mediator, IL-1β drives damage to periodontal tissue and affects osteoclast activity [68]. Autophagy can also inhibit the activation of the inflammasome by removing key components required for its assembly, such as apoptosis-associated speck-like protein containing a CARD (ASC) [127]. Controlling NLRP3-mediated inflammasome activation and reducing the production of pro-inflammatory mediators is clearly beneficial for the repair of periodontal tissue. This suggests that autophagy plays a crucial role in controlling inflammation and preventing excessive inflammatory activation. The communication between autophagy and the inflammasome can help to limit the progression of inflammation [127]. Thus, promoting autophagy in macrophages not only helps clear invading pathogens but also facilitates inflammation resolution by suppressing inflammasome activity and reducing the production of pro-inflammatory mediators.
Possible therapeutic strategy for periodontitis
The primary goals of periodontitis treatment are to control inflammation and promote tissue regeneration. Clinical treatment methods are categorized into non-surgical approaches (home care and scaling and root planing for plaque control), surgical approaches (resective, regenerative, and plastic surgery), and maintenance therapy. Several adjunctive therapies are also employed in periodontal treatment, including the use of antibiotics, host modulation through nutritional support (supplementation with antioxidants and vitamins), and laser therapy [128]. The development of immunomodulatory periodontal therapies is considered a highly promising approach in the treatment of periodontitis. These therapies can be broadly categorized into four types: stem cell therapy, gene therapy, multi-targeted therapy, and microbiome therapy [129]. Microbiome therapy aims to control dental plaque by either balancing the oral microbiome through the administration of probiotics or inhibiting quorum sensing via quorum quenching [130]. Stem cell therapy promotes tissue repair by harnessing stem cells to induce the secretion of anti-inflammatory cytokines, facilitate the polarization of macrophages toward the M2 phenotype, and enhance the proliferation of Treg cells. Gene therapy regulates genes associated with periodontitis through targeted gene editing techniques. Multi-targeted therapy aims to specifically address particular biomarkers, cell types, or molecular mechanisms involved in the immune response and cell death processes of immune cells such as macrophages and neutrophils [129]. Building on the preceding discussion of the role of macrophages in periodontitis restoration and development, this review will subsequently examine several promising multi-targeted therapeutic strategies for periodontitis.
Modulating macrophage polarization
The transition between different phenotypes of macrophages is closely related to tissue destruction in periodontitis, and the regulation of tissue destruction and repair is influenced by the different cytokines and inflammatory mediators that they secrete. Yang et al. have found that an elevated M1/M2 phenotypic proportion is associated with periodontal inflammation. Meanwhile, elevated IL-1β, an M1-related chemokine that is typically linked to inflammatory cell movement and attachment loss in human periodontitis is also found to be related to periodontal inflammation [18,131]. The polarization of pro-inflammatory (M1) macrophages can be developed in the way of unactivated macrophage differentiation as well as in the way of conversion from anti-inflammatory (M2) macrophages [16]. One potential therapy option for periodontitis is to promote the M2 anti-inflammatory impact.
When developing treatment agents to promote M2 macrophage polarization, the anti-inflammatory effects are achieved through a multifaceted and multi-mechanistic approach, rather than simply through immune suppression. The delivery of ROS-sensitive drugs using polydopamine-functionalized mesoporous silica has been shown to effectively modulate inflammatory responses by reducing the expression levels of M1 phenotype markers IL-6 and TNF-α, while simultaneously increasing the expression of M2 phenotype markers IL-10. These effects are achieved through multiple mechanisms, including ROS scavenging, antibacterial action, and the promotion of M2 polarization. Histopathological analysis and immunohistochemical detection have confirmed that this drug delivery system does not induce noticeable toxicity or tissue damage when administered in vivo [132]. The Quercetin-loaded ceria nanocomposite markedly elevates the expression of the anti-inflammatory cytokine anti-inflammatory factor arginase-1 (Arg-1) while suppressing the pro-inflammatory cytokine IL-1β, thereby enhancing the M2/M1 ratio and facilitating the resolution of inflammation. This innovative material achieves dual-directional immunoregulation, ensuring that macrophage polarization remains balanced and does not compromise the overall immune functionality of the organism [133]. Stem cell therapy represents a promising approach for treating periodontitis. Research has demonstrated that PDLSCs are capable of inducing M2 macrophage polarization without upregulating the expression of M1 macrophage markers such as CD80 and HLA-DR. Moreover, they do not elevate the expression or secretion of pro-inflammatory cytokines, including TNF-α and IL-1β, associated with M1 macrophages. This indicates that PDLSCs can promote M2 polarization while preserving the essential M1 immune responses [134]. All-Trans Retinoic Acid (ATRA) exerts anti-inflammatory effects by modulating macrophage polarization through multiple mechanisms, including the activation of the p38MAPK/STAT6 signaling pathway, inhibition of the NF-κB pathway, and regulation of RAR/RXR receptors [135]. This shows that ATRA’s therapeutic impact on periodontitis is not just due to immunosuppression.
According to these results, M2 macrophages might be vital in encouraging tissue regeneration or minimizing inflammatory damage. Therefore, sustaining the M1/M2 balance could be an innovative treatment option for controlling periodontal inflammation and preserving a harmonious microenvironment [17]. This tactic involves the regulation of signaling pathways, modulation of cytokine production, anti-inflammatory mechanisms, and ROS scavenging. Additionally, various stem cells have been found to induce the generation of anti-inflammatory cytokines, promote M2 macrophage polarization, and increase Treg cells, thereby enhancing tissue repair [129]. Modulating macrophage polarization as a therapeutic approach for periodontitis remains a challenge at present. While current efforts primarily focus on the M1/M2 transition, the reality is that macrophage subtypes are more complex, necessitating a shift toward more precise regulation of polarization in future strategies. Additionally, the complexity of the periodontal microenvironment and individual variability place higher demands on biomaterials designed to deliver therapeutics. This underscores the need for innovative materials that can cater to the distinct immunological requirements at each stage of periodontitis healing [136].
Specialized pro-resolving mediators
The synthesis of Specialized Pro-Resolving Mediators is considered crucial for promoting an anti-inflammatory and pro-resolution condition [137]. Studies on how to utilize SPMs for periodontal treatments have been taken up. Currently, two primary approaches are being explored to alleviate inflammation using SPMs: the first involves increasing the levels of SPMs in the body by supplementing with SPM precursors; the second involves using nanomedicines carrying SPMs or SPM analogs.
SPMs are enzymatically produced from polyunsaturated fatty acids (PUFA), thus researchers have looked into the advantages of PUFAs in diet for the management of inflammatory diseases. Both omega-3 and omega-6 PUFAs are present in acutely inflamed areas, where they are transformed into bioactive SPMs [137]. The necessary nutrient polyunsaturated fatty acids can only be obtained through food because they cannot be synthesized by humans [138]. A study supplemented healthy individuals with PUFAs using either a high-dose short-term regimen or a low-dose long-term regimen. In the subsequent endotoxin challenge, those who received supplementation produced SPMs more rapidly and in greater quantities compared to the placebo group [139]. Deore et al. have discovered that providing omega-3 fatty acid supplements to the diet may assist with avoiding or alleviating chronic periodontitis, which can reduce pocket depth and gingival index [140]. Therefore, dietary modification is meant to be a supplement to periodontal treatment. However, the optimal dosage of omega-3 and other precursor substances required to achieve the most effective anti-inflammatory response is still under investigation. Further research is needed to determine the appropriate supplementation level that maximizes anti-inflammatory effects [141]. The effects of SPMs are influenced by factors such as age, gender, and race, making the development of personalized treatment strategies for different individuals one of the challenges for the future [142].
SPMs play crucial roles in anti-inflammatory processes, promoting inflammation resolution, and enhancing phagocytic capacity. In contrast to the corticosteroid anti-inflammatory drugs we are familiar with, SPMs focus on the resolution of inflammation, which involves promoting the restoration of inflammation rather than completely suppressing the immune response. SPMs alleviate inflammation not only by modulating the biological functions of immune cells such as macrophages and neutrophils, but also by directly combating microorganisms. This indicates that SPMs can control inflammation while preserving the immune system’s ability to defend against pathogens [143]. RvE1, MaR1 and LX can all limit macrophage or neutrophil recruitment, and promote the phagocytosis of apoptotic PMN by macrophages, contributing to the resolution of inflammation [95,96,102–104,144]. MaR1 and LX can promote macrophage polarization from the M1 phenotype to the M2 phenotype [97,102]. In animal models and experiments, the application of SPMs (RvE1, MaR1) is proven to effectively promote the restoration of inflammatory conditions [145,146]. Recently, researchers have developed an LXA4 analog (BLXA4) mouthwash and confirmed its effectiveness in reducing gingivitis and decreasing periodontal pocket depth. Higher concentrations and abundance of SPMs were found in the serum of subjects who orally ingested BLXA4 [147]. Additionally, studies have revealed that individuals with periodontitis had higher levels of SPMs biosynthesis pathway indicators, suggesting that the occurrence of periodontitis can stimulate the release of SPMs [148]. Given these findings, SPMs can serve as a biomarker of periodontitis, and targeting enhancement of SPMs may be a promising future research direction for the treatment of periodontitis. However, the effects of SPMs are spatially and temporally constrained, highlighting the need for the development of SPM derivatives that target specific receptors or tissues [141]. Additionally, certain pathogens, such as Candida albicans, have evolved immune evasion mechanisms to enhance their survival, which complicates the balance between pathogen control and host immune regulation in the context of SPM production [93]. It has been found that long-term use of supra-physiological levels of SPMs may impair the host’s pathogen-mediated immune response. Therefore, it is critical to identify the optimal timing, dosage, and duration of SPM use to avoid excessive immune suppression [93].
Transplantation of PDLSCs has been proven to improve periodontal regeneration in animal models and human clinical trials for its ability to restore periodontal tissues [149,150] Human periodontal ligament stem cells (hPDLSCs) produce SPMs (resolvin D1, D2, D5, etc.). Meanwhile, the regeneration ability of hPDLSCs is controlled through the LXA4-ALX/FPR2 pathway while Mar1 and RvE1 are able to promote the regenerative ability of PDLCs under the inflammatory condition [100,101]. Periodontal ligament fibroblasts (PDLF) exhibit attachment properties, which help in developing periodontal treatments [151]. RvD1 overcame IL-1β’s negative effects on PDLF proliferation and wound healing [105].
In summary, regulating SPMs holds significant potential for periodontitis recovery. This can be achieved by increasing the levels of SPM precursors, administering SPM-carrying nanotherapeutics, or providing treatment agents containing SPM analogs to modulate SPM levels within the patient. Additionally, SPMs exert positive effects on various stem cells, thereby promoting the regeneration and repair of periodontal tissues.
Mediating PRRs
Pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) stimulate multiple intracellular signaling cascades by binding to the pattern recognition receptor (PRRs), which causes cell death and effector molecule synthesis [36]. The expression levels of TLR2, TLR4, and TLR9 are significantly elevated in patients with chronic periodontitis, prompting researchers to recommend the assessment of TLR expression in gingival crevicular fluid as a biomarker for periodontitis [152]. The scavenger receptor A (SR-A) and TLRs on phagocytes often work in concert to recognize and engulf pathogens. When macrophages are stimulated by Porphyromonas gingivalis, the expression of SR-A protein on their surface increases, thereby assisting in the production of TNF-α by the macrophages [153]. When different cells are stimulated by Porphyromonas gingivalis lipopolysaccharide, they exhibit distinct receptor expression changes. Macrophages exhibit elevated levels of CD14, TLR4, and SR-A on their cell surface, whereas, in human gingival fibroblasts, the expression of TLR4, MMP-9, COX-2, and MMP-2 is upregulated [154,155]. In the subepithelial connective tissue of patients with periodontitis, NOD1 receptors are markedly increased, [48] and the activation levels of the NLRP3 inflammasome are significantly elevated [156].
The use of inhibitors or modulators targeting these receptors can curtail the progression of inflammation. Experimental findings indicate that resveratrol is capable of downregulating TLR4 expression and inhibiting the activity of key protein kinases, such as p38 MAPK and AKT, thereby suppressing the production of inflammatory mediators by human gingival fibroblasts [155]. Curcumin can inhibit inflammation by suppressing the activation of the TLR4 downstream NF-κB signaling pathway [157]. Chen and colleagues have designed a nanomedicine, MCC950, that targets M1 macrophages to reduce the expression of NLRP3, thereby inhibiting the release of various pro-inflammatory factors, including IL-1β and caspase-1. Additionally, this nanomedicine can increase the expression of the Arg-1 in M2 macrophages [158]. Interactions between co-receptors are closely linked to the pathogenesis of periodontitis. In preclinical research models, the use of C5aR antagonists to block C5aR-TLR2 crosstalk has proven effective in countering the immune evasion mechanisms of periodontal pathogens, thereby reducing the production of pro-inflammatory cytokines [159]. CXCR4-specific antagonists, such as AMD300, intervene in immune evasion mechanisms and reduce bacterial adhesion by blocking interactions between bacteria and TLR2, as well as between CXCR4 and CR3. This intervention effectively restores antimicrobial activity and diminishes the infectivity of periodontal pathogens [160]. Besides regulating the receptors themselves, inflammation can also be influenced by controlling the downstream signaling pathways activated after receptor stimulation. In terms of signaling pathways, the ability of TGF-β to block TLR-NF-κB signaling is due to Smad6 methylation caused by protein arginine methyltransferase 1 (PRMT1). The modification of PRMT1-Smad6 signaling is a brand-new, highly effective method to control the host immune response in periodontitis [161].
Modulating cell death related to periodontitis
Apoptosis can clear invading pathogens and limit their proliferation within the host. If the invading pathogens suppress macrophage apoptosis, they can proliferate and spread more extensively. In chronic periodontitis, the extended lifespan of inflammatory cells may be associated with the suppression of apoptosis. This inhibition is primarily achieved through two mechanisms: first, the tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptotic signaling is blocked by TRAIL decoy receptors; second, the elevated expression of apoptosis inhibitors, such as cleaved caspase-3 inhibitors, hinders the execution of apoptosis [162]. However, increased apoptosis can also exacerbate the progression of periodontitis. Caspases are responsible for cleaving various intracellular proteins during apoptosis, thereby initiating and executing cell death. Elevated expression and activity of Caspase-3 and Caspase-8 have been observed in periodontitis, indicating that increased apoptosis contributes to the worsening of the disease [163]. In periodontitis, both excessive and inhibited apoptosis hinder disease recovery. The transition from apoptosis initiation to execution is closely related to a complex network of proteins, including cell ligands, receptors, and enzymes. Targeting and selectively blocking specific nodes or receptors in this network to maintain the balance of apoptosis holds promise for achieving targeted apoptosis and addressing periodontitis. With this in mind, a future challenge lies in further investigating specific molecular targets in periodontitis that lead to the biological effects of apoptosis, as well as the upstream and downstream signaling pathways involved. Developing a biological mechanism network that illustrates the interaction between apoptosis and periodontitis could help identify precise therapeutic strategies for regulating apoptosis. Moreover, the contents encapsulated in apoptotic bodies can act as signals. When immune cells ingest apoptotic bodies, they can lower the intensity of the immune response and reduce the production of pro-inflammatory cytokines. Apoptotic bodies can carry specific functional molecules, such as miRNAs and anti-inflammatory cytokines, and they also act as self-antigens. Transferring these contents to other cells, such as macrophages, can trigger specific immune responses or promote tissue regeneration. This implies that apoptotic bodies could serve as regulatory systems that interact with cells to mitigate pro-inflammatory reactions.
Autophagy is another form of cell death that has a complex role in the progression of periodontitis. It helps sustain the survival of periodontal ligament stem cells and osteocytes, protecting them from apoptosis [124,164]. It can also inhibit the activation of the NLRP3 inflammasome, reducing the production of pro-inflammatory cytokines such as IL-1β, which are typically released due to inflammasome activation. However, researchers have also found that autophagy supports the formation of new blood vessels in periodontitis and influences the activity of osteoclasts involved in bone tissue degradation [124,165]. Hence, investigating the mechanisms by which autophagy modulates the host’s immune response to pathogens presents two critical challenges: the dual functionality of autophagy in periodontitis and the intricate nature of its regulatory pathways. Autophagy can alleviate inflammation in periodontitis, but in certain contexts, it may also exacerbate the progression of inflammation. Additionally, autophagy regulation involves numerous signaling pathways and proteins, and the cross-talk among these pathways adds to the complexity of precisely controlling autophagy. Therefore, developing regulators that can specifically modulate autophagy to control its activity without disrupting other cellular functions is one of the significant challenges for the future.
Methodology for human macrophage research
By gaining a deeper understanding of the mechanisms underlying the response of macrophages to periodontitis, we can explore new therapeutic approaches and provide fresh perspectives for future periodontitis management. These studies all rely on extensive experimentation, where the methodology plays a particularly crucial role. The structural parallels between the immune systems of human and mouse models have positioned the latter as the predominant organisms for human disease research, largely due to their clinical research utility and economic viability [166]. When studying macrophage behavior in periodontitis using mouse models, the inherent immunological and physiological discrepancies between species pose significant challenges to the clinical translation of research outcomes. These differences can be summarized as variations in macrophage generation and polarization between humans and mice, the distinct mechanisms of immune regulation at both the molecular mediator and genetic levels, and inconsistencies in anatomical and physiological structures. Human macrophages originate from a broader temporal and spatial context and require polarization under specific agonists, such as TLR agonists, to secrete either pro-inflammatory or anti-inflammatory factors [167]. There are notable differences between human and murine macrophages in the expression of surface receptors (like TLR9 and Fc receptors) and in their capacity to produce specific cytokines, such as IFN-γ [166,168]. Furthermore, although the expression profiles of genes are comparable, the regulatory frameworks governing immune and metabolic functions exhibit distinct variations between human and mouse models. Anatomical differences between humans and mice, including variations in body size and gingival microbiota, necessitate careful consideration of how metabolic factors influence disease outcomes when translating model research findings to clinical applications [166].
For a more precise study of human macrophage dynamics in periodontitis, many innovative cross-disciplinary techniques have emerged and been employed, such as organoid-macrophage co-culture systems [169], single-cell RNA sequencing, spatial transcriptomics [170], humanized mouse models [171], and machine learning [170]. Organoid-macrophage co-culture systems simulate the periodontal environment, revealing how macrophages influence inflammation and tissue repair [169]. Spatial transcriptomics, integrated with spatial information, investigates the localization and functional changes of macrophages within periodontal tissues. Single-cell RNA sequencing provides insights into the biological behavior and genetic changes of macrophages at different stages of periodontitis, while machine learning and statistical methods are employed to analyze the extensive data generated from single-cell RNA sequencing [170]. A single-cell lineage tracing system, which detects mitochondrial DNA mutations and provides real-time readings of transcriptional states and chromatin accessibility, has been applied in hematopoietic stem cell research. If this technology is extended to study the clonal structure of human macrophages, it holds promise for a detailed understanding of the crosstalk between macrophages and periodontitis [172]. Humanized mouse models are pivotal for examining intricate immune responses in periodontitis, offering a system that closely mimics human physiology. The three primary types of humanized mouse models include the human peripheral blood lymphocyte model, the bone marrow/liver/thymus model, and the human stem cell model. Notably, the human stem cell model has been utilized to explore macrophage roles and dynamics in periodontitis [171]. As more accurate assessments of human macrophages are conducted, we will acquire a more nuanced comprehension of the host immune system, essential for advancing the treatment and prevention of periodontitis.
Conclusion
In this review, we discuss the relationship between macrophages and the development of periodontitis. As a crucial component of the body, the immune response is responsible for controlling pathogen invasion, but excessive activation of the immune system can also cause tissue damage. In response to different stimuli, macrophages exhibit significant plasticity and flexibility, allowing them to adapt in phenotype and function to recognize, phagocytize, and clear bacteria. Pattern recognition receptors on the surface of macrophages identify pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), mediating a series of intracellular activities to address periodontitis. To maintain homeostasis, macrophages remove apoptotic polymorphonuclear neutrophils (PMNs) through efferocytosis and produce specialized pro-resolving mediators (SPMs). These SPMs mainly enhance the phagocytic capacity of phagocytes, suppress excessive recruitment of immune cells, and promote the polarization of macrophages toward the M2 phenotype. These changes brought about by SPMs are beneficial for the recovery from periodontitis. Furthermore, autophagy and apoptosis have been demonstrated to be intricately linked to the recovery process in periodontitis.
By gaining a deeper understanding of the mechanisms underlying the response of macrophages to periodontitis, we can explore new therapeutic approaches and provide fresh perspectives for future periodontitis management. Through the continuous deepening of research methodologies, our understanding of the differences between animal models and humans in preclinical studies has become increasingly comprehensive. A thorough understanding of these differences is crucial for the successful translation from preclinical to clinical practice. Currently, numerous interdisciplinary approaches are being applied to medical research, aiding in a comprehensive understanding of the mechanisms by which human macrophages contribute to the onset and progression of periodontitis. An accurate assessment of human macrophages enables us to leverage the host immune system for the treatment and prevention of periodontitis.
Funding Statement
This work was supported by Guangdong Basic and Applied Basic Research Foundation [Grant No. 2022A1515220047].
Disclosure statement
The authors report there are no competing interests to declare
Authorship
Conceptualization: Zejian Li
Visualization: Keyin Mo
Writing – original draft: Keyin Mo, Yijue Wang
Writing – review & editing: Keyin Mo, Chunting Lu, Zejian Li
All authors contributed to the article and approved the submitted version.
Data availability statement
Data availability does not apply to this article as no new data were created or analyzed in this study.
References
- [1].Joudi A, Sargeran K, Pouraskari Z, et al. Illness perception amongst individuals with periodontal diseases. Community Dent Health. 2023;40(1):37–23. doi: 10.1922/CDH_00147Joudi05 [DOI] [PubMed] [Google Scholar]
- [2].O’Dowd LK, Durham J, McCracken GI, et al. Patients’ experiences of the impact of periodontal disease. J Clin Periodontol. 2010;37(4):334–339. doi: 10.1111/j.1600-051X.2010.01545.x [DOI] [PubMed] [Google Scholar]
- [3].Pihlstrom BL, Michalowicz BS, Johnson NW.. Periodontal diseases. Lancet. 2005;366(9499):1809–1820. doi: 10.1016/S0140-6736(05)67728-8 [DOI] [PubMed] [Google Scholar]
- [4].Zhang X, Wang X, Wu J, et al. The global burden of periodontal diseases in 204 countries and territories from 1990 to 2019. Oral Dis. 2022;30(2):754–768. doi: 10.1111/odi.14436 [DOI] [PubMed] [Google Scholar]
- [5].Page RC, Schroeder HE. Pathogenesis of inflammatory periodontal disease. A summary of current work. Lab Invest. 1976;34:235–249. [PubMed] [Google Scholar]
- [6].Shaddox LM, Spencer WP, Velsko IM, et al. Localized aggressive periodontitis immune response to healthy and diseased subgingival plaque. J Clin Periodontol. 2016;43(9):746–753. doi: 10.1111/jcpe.12560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hienz SA, Paliwal S, Ivanovski S. Mechanisms of bone resorption in periodontitis. J Immunol Res. 2015;2015:1–10. doi: 10.1155/2015/615486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sima C, Viniegra A, Glogauer M. Macrophage immunomodulation in chronic osteolytic diseases—the case of periodontitis. J Leukoc Biol. 2019;105(3):473–487. doi: 10.1002/JLB.1RU0818-310R [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Schulz C, Perdiguero EG, Chorro L, et al. A lineage of myeloid cells independent of myb and hematopoietic stem cells. Science. 2012;336(6077):86–90. doi: 10.1126/science.1219179 [DOI] [PubMed] [Google Scholar]
- [10].Yona S, Kim K-W, Wolf Y, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38(1):79–91. doi: 10.1016/j.immuni.2012.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8(12):958–969. doi: 10.1038/nri2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mantovani A, Biswas SK, Galdiero MR, et al. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol. 2013;229(2):176–185. doi: 10.1002/path.4133 [DOI] [PubMed] [Google Scholar]
- [13].Sima C, Glogauer M. Macrophage subsets and osteoimmunology: tuning of the immunological recognition and effector systems that maintain alveolar bone. Periodontol 2000. 2013;63(1):80–101. doi: 10.1111/prd.12032 [DOI] [PubMed] [Google Scholar]
- [14].Wang Y, Smith W, Hao D, et al. M1 and M2 macrophage polarization and potentially therapeutic naturally occurring compounds. Int Immunopharmacol. 2019;70:459–466. doi: 10.1016/j.intimp.2019.02.050 [DOI] [PubMed] [Google Scholar]
- [15].Murray PJ. Macrophage polarization. Annu Rev Physiol. 2017;79(1):541–566. doi: 10.1146/annurev-physiol-022516-034339 [DOI] [PubMed] [Google Scholar]
- [16].Shapouri‐Moghaddam A, Mohammadian S, Vazini H, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. 2018;233(9):6425–6440. doi: 10.1002/jcp.26429 [DOI] [PubMed] [Google Scholar]
- [17].Zhang W, Guan N, Zhang X, et al. Study on the imbalance of M1/M2 macrophage polarization in severe chronic periodontitis. THC. 2023;31(1):1–8. doi: 10.3233/THC-220092 [DOI] [PubMed] [Google Scholar]
- [18].Yang J, Zhu Y, Duan D, et al. Enhanced activity of macrophage M1/M2 phenotypes in periodontitis. Arch Oral Biol. 2018;96:234–242. doi: 10.1016/j.archoralbio.2017.03.006 [DOI] [PubMed] [Google Scholar]
- [19].Wang W, Liu H, Liu T, et al. Insights into the role of macrophage polarization in the pathogenesis of osteoporosis. Oxid Med Cell Longev. 2022;2022:1–11. doi: 10.1155/2022/2485959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496(7446):445–455. doi: 10.1038/nature12034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wang N, Liang H, Zen K. Molecular mechanisms that influence the macrophage M1–M2 polarization balance. Front Immunol. 2014;5:614. doi: 10.3389/fimmu.2014.00614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3(1):23–35. doi: 10.1038/nri978 [DOI] [PubMed] [Google Scholar]
- [23].Atri C, Guerfali FZ, Laouini D. Role of human macrophage polarization in inflammation during infectious diseases. Int J Mol Sci. 2018;19(6):1801. doi: 10.3390/ijms19061801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Nanci A, Bosshardt DD. Structure of periodontal tissues in health and disease. Periodontol 2000. 2006;40(1):11. doi: 10.1111/j.1600-0757.2005.00141.x [DOI] [PubMed] [Google Scholar]
- [25].Chistiakov DA, Bobryshev YV, Nikiforov NG, et al. Retracted: macrophage phenotypic plasticity in atherosclerosis: the associated features and the peculiarities of the expression of inflammatory genes. Int J Cardiol. 2015;184:436–445. doi: 10.1016/j.ijcard.2015.03.055 [DOI] [PubMed] [Google Scholar]
- [26].Liang C, Wu S, Xia G, et al. Engineered M2a macrophages for the treatment of osteoarthritis. Front Immunol. 2022;13:1054938. doi: 10.3389/fimmu.2022.1054938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wang L-X, Zhang S-X, Wu H-J, et al. M2b macrophage polarization and its roles in diseases. Journal Of Leukocyte Biology. 2019;106(2):345–358. doi: 10.1002/JLB.3RU1018-378RR [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zizzo G, Hilliard BA, Monestier M, et al. Efficient clearance of early apoptotic cells by human macrophages requires M2c polarization and MerTK induction. J Immunol. 2012;189(7):3508–3520. doi: 10.4049/jimmunol.1200662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Duluc D, Delneste Y, Tan F, et al. Tumor-associated leukemia inhibitory factor and IL-6 skew monocyte differentiation into tumor-associated macrophage-like cells. Blood, J Am Soc of Hematol. 2007;110(13):4319–4330. doi: 10.1182/blood-2007-02-072587 [DOI] [PubMed] [Google Scholar]
- [30].Wang Q, Ni H, Lan L, et al. Fra-1 protooncogene regulates IL-6 expression in macrophages and promotes the generation of M2d macrophages. Cell Res. 2010;20(6):701–712. doi: 10.1038/cr.2010.52 [DOI] [PubMed] [Google Scholar]
- [31].Garaicoa‐Pazmino C, Fretwurst T, Squarize CH, et al. Characterization of macrophage polarization in periodontal disease. J Clin Periodontol. 2019;46(8):830–839. doi: 10.1111/jcpe.13156 [DOI] [PubMed] [Google Scholar]
- [32].Kornman KS, Page RC, Tonetti MS. The host response to the microbial challenge in periodontitis: assembling the players. Periodontol 2000. 1997;14(1):33–53. doi: 10.1111/j.1600-0757.1997.tb00191.x [DOI] [PubMed] [Google Scholar]
- [33].Cavaillon J. Cytokines and macrophages. Biomed & Pharmacother. 1994;48(10):445–453. doi: 10.1016/0753-3322(94)90005-1 [DOI] [PubMed] [Google Scholar]
- [34].Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity. 2014;41(1):21–35. doi: 10.1016/j.immuni.2014.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Unanue ER. Antigen-presenting function of the macrophage. Annu Rev Immunol. 1984;2(1):395–428. doi: 10.1146/annurev.iy.02.040184.002143 [DOI] [PubMed] [Google Scholar]
- [36].Amarante-Mendes GP, Adjemian S, Branco LM, et al. Pattern recognition receptors and the host cell death molecular machinery. Front Immunol. 2018;9:2379. doi: 10.3389/fimmu.2018.02379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Jia L, Han N, Du J, et al. Pathogenesis of important virulence factors of Porphyromonas gingivalis via toll-like receptors. Front Cell Infect Microbiol. 2019;9:262. doi: 10.3389/fcimb.2019.00262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Fu YL, Harrison RE. Microbial phagocytic receptors and their potential involvement in cytokine induction in macrophages. Front Immunol. 2021;12:662063. doi: 10.3389/fimmu.2021.662063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Li D, Wu M. Pattern recognition receptors in health and diseases. Signal transduction and targeted therapy. Signal Transduct Target Ther. 2021;6(1):291. doi: 10.1038/s41392-021-00687-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140(6):805–820. doi: 10.1016/j.cell.2010.01.022 [DOI] [PubMed] [Google Scholar]
- [41].Hu W, Spaink HP. The role of TLR2 in infectious diseases caused by mycobacteria: from cell biology to therapeutic target. Biology (Basel). 2022;11(2):246. doi: 10.3390/biology11020246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Yin L, Li X, Hou J. Macrophages in periodontitis: a dynamic shift between tissue destruction and repair. Jpn Dent Sci Rev. 2022;58:336–347. doi: 10.1016/j.jdsr.2022.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol. 2010;11(5):373–384. doi: 10.1038/ni.1863 [DOI] [PubMed] [Google Scholar]
- [44].Rakoff-Nahoum S, Medzhitov R. Toll-like receptors and cancer. Nat Rev Cancer. 2009;9(1):57–63. doi: 10.1038/nrc2541 [DOI] [PubMed] [Google Scholar]
- [45].Deguine J, Barton GM. MyD88: a central player in innate immune signaling. F1000prime reports. 2014; 6. [DOI] [PMC free article] [PubMed]
- [46].Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126 [DOI] [PubMed] [Google Scholar]
- [47].Saikh KU. MyD88 and beyond: a perspective on MyD88-targeted therapeutic approach for modulation of host immunity. Immunol Res. 2021;69(2):117–128. doi: 10.1007/s12026-021-09188-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Park E, Na HS, Song Y-R, et al. Activation of NLRP3 and AIM2 inflammasomes by porphyromonas gingivalis infection. Infect Immun. 2014;82(1):112–123. doi: 10.1128/IAI.00862-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Mangan MS, Olhava EJ, Roush WR, et al. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov. 2018;17(8):588–606. doi: 10.1038/nrd.2018.97 [DOI] [PubMed] [Google Scholar]
- [50].Mysak J, Podzimek S, Sommerova P, et al. Porphyromonas gingivalis: major periodontopathic pathogen overview. J Immunol Res. 2014;2014:1–8. doi: 10.1155/2014/476068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].How KY, Song KP, Chan KG. Porphyromonas gingivalis: an overview of periodontopathic pathogen below the gum line. Front Microbiol. 2016;7:53. doi: 10.3389/fmicb.2016.00053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Amano A, Nakagawa I, Okahashi N, et al. Variations of Porphyromonas gingivalis fimbriae in relation to microbial pathogenesis. J Periodontal Res. 2004;39(2):136–142. doi: 10.1111/j.1600-0765.2004.00719.x [DOI] [PubMed] [Google Scholar]
- [53].Wang M, Shakhatreh M-A, James D, et al. Fimbrial proteins of Porphyromonas gingivalis mediate in vivo virulence and exploit TLR2 and complement receptor 3 to persist in macrophages. J Immunol. 2007;179(4):2349–2358. doi: 10.4049/jimmunol.179.4.2349 [DOI] [PubMed] [Google Scholar]
- [54].Hajishengallis G, Wang M, Liang S. Induction of distinct TLR2-mediated proinflammatory and proadhesive signaling pathways in response to Porphyromonas gingivalis fimbriae. J Immunol. 2009;182(11):6690–6696. doi: 10.4049/jimmunol.0900524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Carpenter S, O’Neill LA. How important are Toll‐like receptors for antimicrobial responses? Cell Microbiol. 2007;9(8):1891–1901. doi: 10.1111/j.1462-5822.2007.00965.x [DOI] [PubMed] [Google Scholar]
- [56].O’Neill LA, Bowie AG. The family of five: tir-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7(5):353–364. doi: 10.1038/nri2079 [DOI] [PubMed] [Google Scholar]
- [57].Kusumoto Y, Hirano H, Saitoh K, et al. Human gingival epithelial cells produce chemotactic factors interleukin‐8 and monocyte chemoattractant protein‐1 after stimulation with porphyromonas gingivalis via toll‐like receptor 2. J Periodontol. 2004;75(3):370–379. doi: 10.1902/jop.2004.75.3.370 [DOI] [PubMed] [Google Scholar]
- [58].Darveau RP, Pham T-T, Lemley K, et al. Porphyromonas gingivalis lipopolysaccharide contains multiple lipid a species that functionally interact with both toll-like receptors 2 and 4. Infect Immun. 2004;72(9):5041–5051. doi: 10.1128/IAI.72.9.5041-5051.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Yang J, Yang C, Yang J, et al. RP105 alleviates myocardial ischemia reperfusion injury via inhibiting TLR4/TRIF signaling pathways. Int J Mol Med. 2018;41:3287–3295. doi: 10.3892/ijmm.2018.3538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Nativel B, Couret D, Giraud P, et al. Porphyromonas gingivalis lipopolysaccharides act exclusively through TLR4 with a resilience between mouse and human. Sci Rep. 2017;7(1):15789. doi: 10.1038/s41598-017-16190-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Sakayori T, Ito H, Numabe Y. Porphyromonas gingivalis lipopolysaccharide expresses neutrophil extracellular traps via Toll-like receptors 2 and 4. Nihon Shishubyo Gakkai Kaishi (J Jpn Soc Of Periodontology). 2024;66:29–42. [Google Scholar]
- [62].Ruan Q, Guan P, Qi W, et al. Porphyromonas gingivalis regulates atherosclerosis through an immune pathway. Front Immunol. 2023;14:1103592. doi: 10.3389/fimmu.2023.1103592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Hajishengallis G, Lambris JD. Complement and dysbiosis in periodontal disease. Immunobiology. 2012;217(11):1111–1116. doi: 10.1016/j.imbio.2012.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Dyson HJ, Wright PE. Role of intrinsic protein disorder in the function and interactions of the transcriptional coactivators creb-binding protein (CBP) and p300. J Biol Chem. 2016;291(13):6714–6722. doi: 10.1074/jbc.R115.692020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Jung YJ, Jun HK, Choi BK. Contradictory roles of P orphyromonas gingivalis gingipains in caspase‐1 activation. Cell Microbiol. 2015;17(9):1304–1319. doi: 10.1111/cmi.12435 [DOI] [PubMed] [Google Scholar]
- [66].Waddington RJ, Moseley R, Embery G. Periodontal disease mechanisms: reactive oxygen species: a potential role in the pathogenesis of periodontal diseases. Oral Dis. 2000;6(3):138–151. doi: 10.1111/j.1601-0825.2000.tb00325.x [DOI] [PubMed] [Google Scholar]
- [67].Chen M, Zhang Y, Zhou P, et al. Substrate stiffness modulates bone marrow-derived macrophage polarization through nf-κB signaling pathway. Bioact Mater. 2020;5(4):880–890. doi: 10.1016/j.bioactmat.2020.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Cheng R, Wu Z, Li M, et al. Interleukin-1β is a potential therapeutic target for periodontitis: a narrative review. Int J Oral Sci. 2020;12(1):2. doi: 10.1038/s41368-019-0068-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Netea MG, Nold-Petry CA, Nold MF, et al. Differential requirement for the activation of the inflammasome for processing and release of IL-1β in monocytes and macrophages. Blood, The J Am Soc of Hematol. 2009;113(10):2324–2335. doi: 10.1182/blood-2008-03-146720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Bauernfeind FG, Horvath G, Stutz A, et al. Cutting edge: nf-κB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009;183(2):787–791. doi: 10.4049/jimmunol.0901363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].He Y, Hara H, Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. 2016;41(12):1012–1021. doi: 10.1016/j.tibs.2016.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Iman M, Rezaei R, Azimzadeh Jamalkandi S, et al. Th17/Treg immunoregulation and implications in treatment of sulfur mustard gas-induced lung diseases. Expert Rev Clin Immunol. 2017;13(12):1173–1188. doi: 10.1080/1744666X.2017.1389646 [DOI] [PubMed] [Google Scholar]
- [73].Takashiba S, Naruishi K, Murayama Y. Perspective of cytokine regulation for periodontal treatment: fibroblast biology. J Periodontol. 2003;74(1):103–110. doi: 10.1902/jop.2003.74.1.103 [DOI] [PubMed] [Google Scholar]
- [74].Kishimoto T. The biology of interleukin-6. Blood. 1989;74(1):1–10. doi: 10.1182/blood.V74.1.1.1 [DOI] [PubMed] [Google Scholar]
- [75].Silva LM, Brenchley L, Moutsopoulos NM. Primary immunodeficiencies reveal the essential role of tissue neutrophils in periodontitis. Immunol Rev. 2019;287(1):226–235. doi: 10.1111/imr.12724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Hajishengallis G, Chavakis T. Endogenous modulators of inflammatory cell recruitment. Trends Immunol. 2013;34(1):1–6. doi: 10.1016/j.it.2012.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Roberts HM, Ling MR, Insall R, et al. Impaired neutrophil directional chemotactic accuracy in chronic periodontitis patients. J Clin Periodontol. 2015;42(1):1–11. doi: 10.1111/jcpe.12326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Picker LJ, Butcher EC. Physiological and molecular mechanisms of lymphocyte homing. Annu Rev Immunol. 1992;10(1):561–591. doi: 10.1146/annurev.iy.10.040192.003021 [DOI] [PubMed] [Google Scholar]
- [79].Usui M, Onizuka S, Sato T, et al. Mechanism of alveolar bone destruction in periodontitis—periodontal bacteria and inflammation. Jpn Dent Sci Rev. 2021;57:201–208. doi: 10.1016/j.jdsr.2021.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Rosales C. Neutrophils at the crossroads of innate and adaptive immunity. J Leucocyte Biol. 2020;108(1):377–396. doi: 10.1002/JLB.4MIR0220-574RR [DOI] [PubMed] [Google Scholar]
- [81].Irwandi RA, Chiesa ST, Hajishengallis G, et al. The roles of neutrophils linking periodontitis and atherosclerotic cardiovascular diseases. Front Immunol. 2022;13:915081. doi: 10.3389/fimmu.2022.915081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Netea MG, Domínguez-Andrés J, Barreiro LB, et al. Defining trained immunity and its role in health and disease. Nat Rev Immunol. 2020;20(6):375–388. doi: 10.1038/s41577-020-0285-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Hajishengallis G, Chavakis T. Local and systemic mechanisms linking periodontal disease and inflammatory comorbidities. Nat Rev Immunol. 2021;21(7):426–440. doi: 10.1038/s41577-020-00488-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Matthews J, Wright H, Roberts A, et al. Neutrophil hyper-responsiveness in periodontitis. J Dent Res. 2007;86(8):718–722. doi: 10.1177/154405910708600806 [DOI] [PubMed] [Google Scholar]
- [85].Doran AC, Yurdagul JA, Tabas I. Efferocytosis in health and disease. Nat Rev Immunol. 2020;20(4):254–267. doi: 10.1038/s41577-019-0240-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Rymut N, Heinz J, Sadhu S, et al. Resolvin D1 promotes efferocytosis in aging by limiting senescent cell-induced MerTK cleavage. FASEB J: off Publ Of The Fed of Am Soc For Exp Biol. 2020;34(1):597. doi: 10.1096/fj.201902126R [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Bournazou I, Mackenzie KJ, Duffin R, et al. Inhibition of eosinophil migration by lactoferrin. Immunol Cell Biol. 2010;88(2):220–223. doi: 10.1038/icb.2009.86 [DOI] [PubMed] [Google Scholar]
- [88].Tajbakhsh A, Yousefi F, Abedi SM, et al. The cross‐talk between soluble “find me” and “keep out” signals as an initial step in regulating efferocytosis. J Cell Physiol. 2022;237(8):3113–3126. doi: 10.1002/jcp.30770 [DOI] [PubMed] [Google Scholar]
- [89].Gheibi Hayat SM, Bianconi V, Pirro M, et al. Efferocytosis: molecular mechanisms and pathophysiological perspectives. Immunol Cell Biol. 2019;97(2):124–133. doi: 10.1111/imcb.12206 [DOI] [PubMed] [Google Scholar]
- [90].Hochreiter-Hufford A, Ravichandran KS. Clearing the dead: apoptotic cell sensing, recognition, engulfment, and digestion. Cold Spring Harb Perspect Biol. 2013;5(1):a008748. doi: 10.1101/cshperspect.a008748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Hong C, Kidani Y, Noelia A, et al. Coordinate regulation of neutrophil homeostasis by liver X receptors in mice. J Clin Invest. 2012;122(1):337–347. doi: 10.1172/JCI58393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Moutsopoulos NM, Konkel J, Sarmadi M, et al. Defective neutrophil recruitment in leukocyte adhesion deficiency type I disease causes local IL-17–driven inflammatory bone loss. Sci Transl Med. 2014;6(229):ra22940–ra40. doi: 10.1126/scitranslmed.3007696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Basil MC, Levy BD. Specialized pro-resolving mediators: endogenous regulators of infection and inflammation. Nat Rev Immunol. 2016;16(1):51–67. doi: 10.1038/nri.2015.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Schilperoort M, Ngai D, Sukka SR, et al. The role of efferocytosis-fueled macrophage metabolism in the resolution of inflammation. Immunol Rev. 2023;319(1):65–80. doi: 10.1111/imr.13214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Kantarci A, Van Dyke TE. Lipoxin signaling in neutrophils and their role in periodontal disease. Prostaglandins, Leukotrienes And Essent Fat Acids. 2005;73(3–4):289–299. doi: 10.1016/j.plefa.2005.05.019 [DOI] [PubMed] [Google Scholar]
- [96].Chávez-Castillo M, Ortega Á, Cudris-Torres L, et al. Specialized pro-resolving lipid mediators: The future of chronic pain therapy? Int J Mol Sci. 2021;22(19):10370. doi: 10.3390/ijms221910370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Mitchell S, Thomas G, Harvey K, et al. Lipoxins, Aspirin-Triggered Epi-lipoxins, Lipoxin stable analogues, and the resolution of inflammation: stimulation of macrophage phagocytosis of apoptotic neutrophils: in vivo. J Am Soc of Nephrol. 2002;13(10):2497–2507. doi: 10.1097/01.ASN.0000032417.73640.72 [DOI] [PubMed] [Google Scholar]
- [98].Takano T, Fiore S, Maddox JF, et al. Aspirin-triggered 15-epi-lipoxin A4 (LXA4) and LXA4 stable analogues are potent inhibitors of acute inflammation: evidence for anti-inflammatory receptors. J Exp Med. 1997;185(9):1693–1704. doi: 10.1084/jem.185.9.1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Godson C, Mitchell S, Harvey K, et al. Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. The J Immunol. 2000;164(4):1663–1667. doi: 10.4049/jimmunol.164.4.1663 [DOI] [PubMed] [Google Scholar]
- [100].Albuquerque-Souza E, Schulte F, Chen T, et al. Maresin-1 and resolvin E1 promote regenerative properties of periodontal ligament stem cells under inflammatory conditions. Front Immunol. 2020;11:585530. doi: 10.3389/fimmu.2020.585530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Cianci E, Recchiuti A, Trubiani O, et al. Human periodontal stem cells release specialized proresolving mediators and carry immunomodulatory and prohealing properties regulated by lipoxins. Stem Cells Transl Med. 2016;5(1):20–32. doi: 10.5966/sctm.2015-0163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Sousa AB, Barbosa JN. The use of specialized pro-resolving mediators in biomaterial-based immunomodulation. J Funct Biomater. 2023;14(4):223. doi: 10.3390/jfb14040223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Lee C-T, Teles R, Kantarci A, et al. Resolvin E1 reverses experimental periodontitis and dysbiosis. The J Immunol. 2016;197(7):2796–2806. doi: 10.4049/jimmunol.1600859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Salas-Hernández A, Espinoza-Pérez C, Vivar R, et al. Resolvin D1 and E1 promote resolution of inflammation in rat cardiac fibroblast in vitro. Mol Biol Rep. 2021;48(1):57–66. doi: 10.1007/s11033-020-06133-8 [DOI] [PubMed] [Google Scholar]
- [105].Zarrough AE, Hasturk H, Stephens DN, et al. Resolvin D1 modulates periodontal ligament fibroblast function. J Periodontol. 2023;94(5):683–693. doi: 10.1002/JPER.22-0462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Titos E, Rius B, González-Périz A, et al. Resolvin D1 and its precursor docosahexaenoic acid promote resolution of adipose tissue inflammation by eliciting macrophage polarization toward an M2-like phenotype. The J Immunol. 2011;187(10):5408–5418. doi: 10.4049/jimmunol.1100225 [DOI] [PubMed] [Google Scholar]
- [107].Alshibani N. Resolvins as a treatment modality in experimental periodontitis: a systematic review of preclinical studies. Cureus. 2022;14. doi: 10.7759/cureus.21095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Daniel B, Nagy G, Czimmerer Z, et al. The nuclear receptor PPARγ controls progressive macrophage polarization as a ligand-insensitive epigenomic ratchet of transcriptional memory. Immunity. 2018;49(4):615–26. e6. doi: 10.1016/j.immuni.2018.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Yao D, Zou Y, Lv Y. Maresin 1 enhances osteogenic potential of mesenchymal stem cells by modulating macrophage peroxisome proliferator-activated receptor-γ-mediated inflammation resolution. Biomater Adv. 2022;141:213116. doi: 10.1016/j.bioadv.2022.213116 [DOI] [PubMed] [Google Scholar]
- [110].Fredman G, Van Dyke TE, Serhan CN. Resolvin E1 regulates adenosine diphosphate activation of human platelets. Arterioscler Thromb Vasc Biol. 2010;30(10):2005–2013. doi: 10.1161/ATVBAHA.110.209908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Cherpokova D, Jouvene CC, Libreros S, et al. Resolvin D4 attenuates the severity of pathological thrombosis in mice. Blood, The J Am Soc of Hematol. 2019;134(17):1458–1468. doi: 10.1182/blood.2018886317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Song B, Zhou T, Yang W, et al. Programmed cell death in periodontitis: recent advances and future perspectives. Oral Dis. 2017;23(5):609–619. doi: 10.1111/odi.12574 [DOI] [PubMed] [Google Scholar]
- [113].Wu C, Zhang Y, Sun Z, et al. Molecular evolution of cide family proteins: novel domain formation in early vertebrates and the subsequent divergence. BMC Evol Biol. 2008;8(1):1–16. doi: 10.1186/1471-2148-8-159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Fan D, Fan T-J. Clonidine induces apoptosis of human corneal epithelial cells through death receptors-mediated, mitochondria-dependent signaling pathway. Toxicological Sci. 2017;156:252–260. doi: 10.1093/toxsci/kfw249 [DOI] [PubMed] [Google Scholar]
- [115].Wajant H, Cotter T. Death receptors. Essays Biochem. 2003;39:53–71. doi: 10.1042/bse0390053 [DOI] [PubMed] [Google Scholar]
- [116].Hotchkiss RS, Nicholson DW. Apoptosis and caspases regulate death and inflammation in sepsis. Nat Rev Immunol. 2006;6(11):813–822. doi: 10.1038/nri1943 [DOI] [PubMed] [Google Scholar]
- [117].Boatright KM, Salvesen GS. Mechanisms of caspase activation. Curr Opin Cell Biol. 2003;15(6):725–731. doi: 10.1016/j.ceb.2003.10.009 [DOI] [PubMed] [Google Scholar]
- [118].Xu X, Lai Y, Hua Z-C. Apoptosis and apoptotic body: disease message and therapeutic target potentials. Biosci Rep. 2019;39:BSR20180992. 1). doi: 10.1042/BSR20180992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Marin-Gallen S, Clemente-Casares X, Planas R, et al. Dendritic cells pulsed with antigen-specific apoptotic bodies prevent experimental type 1 diabetes. Clin & Exp Immunol. 2010;160(2):207–214. doi: 10.1111/j.1365-2249.2009.04082.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Zernecke A, Bidzhekov K, Noels H, et al. Delivery of MicroRNA-126 by apoptotic bodies induces CXCL12-dependent vascular protection. Sci Signal. 2009;2(100):ra81–ra. doi: 10.1126/scisignal.2000610 [DOI] [PubMed] [Google Scholar]
- [121].Kato S, Muro M, Akifusa S, et al. Evidence for apoptosis of murine macrophages by actinobacillus actinomycetemcomitans infection. Infect Immun. 1995;63(10):3914–3919. doi: 10.1128/iai.63.10.3914-3919.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Tsuda H, Ning Z, Yamaguchi Y, et al. Programmed cell death and its possible relationship with periodontal disease. J Oral Sci. 2012;54(2):137–149. doi: 10.2334/josnusd.54.137 [DOI] [PubMed] [Google Scholar]
- [123].Tschan M, Simon HU. The role of autophagy in anticancer therapy: promises and uncertainties. J Intern Med. 2010;268(5):410–418. doi: 10.1111/j.1365-2796.2010.02266.x [DOI] [PubMed] [Google Scholar]
- [124].Yang Y, Huang Y, Li W. Autophagy and its significance in periodontal disease. J Periodontal Res. 2021;56(1):18–26. doi: 10.1111/jre.12810 [DOI] [PubMed] [Google Scholar]
- [125].Mihalache CC, Simon H-U. Autophagy regulation in macrophages and neutrophils. Exp Cell Res. 2012;318(11):1187–1192. doi: 10.1016/j.yexcr.2011.12.021 [DOI] [PubMed] [Google Scholar]
- [126].Park M, Jeong S, Na H, et al. Porphyromonas gingivalis induces autophagy in THP -1-derived macrophages. Mol Oral Microbiol. 2017;32(1):48–59. doi: 10.1111/omi.12153 [DOI] [PubMed] [Google Scholar]
- [127].Biasizzo M, Kopitar-Jerala N. Interplay between NLRP3 inflammasome and autophagy. Front Immunol. 2020;11:591803. doi: 10.3389/fimmu.2020.591803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Graziani F, Karapetsa D, Alonso B, et al. Nonsurgical and surgical treatment of periodontitis: how many options for one disease? Periodontol 2000. 2017;75(1):152–188. doi: 10.1111/prd.12201 [DOI] [PubMed] [Google Scholar]
- [129].Yang B, Pang X, Li Z, et al. Immunomodulation in the treatment of periodontitis: progress and perspectives. Front Immunol. 2021;12:781378. doi: 10.3389/fimmu.2021.781378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Kim WJ, Soh Y, Heo S-M. Recent advances of therapeutic targets for the treatment of periodontal disease. Biomol Ther (Seoul). 2021;29(3):263. doi: 10.4062/biomolther.2021.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Stashenko P, Fujiyoshi P, Obernesser M, et al. Levels of interleukin 1β in tissue from sites of active periodontal disease. J Clin Periodontol. 1991;18(7):548–554. doi: 10.1111/j.1600-051X.1991.tb00088.x [DOI] [PubMed] [Google Scholar]
- [132].Bai B, Gu C, Lu X, et al. Polydopamine functionalized mesoporous silica as ros-sensitive drug delivery vehicles for periodontitis treatment by modulating macrophage polarization. Nano Res. 2021;14(12):1–7. doi: 10.1007/s12274-021-3376-1 [DOI] [Google Scholar]
- [133].Wang Y, Li C, Wan Y, et al. Quercetin‐loaded ceria nanocomposite potentiate dual‐directional immunoregulation via macrophage polarization against periodontal inflammation. Small. 2021;17(41):2101505. doi: 10.1002/smll.202101505 [DOI] [PubMed] [Google Scholar]
- [134].Liu J, Wang H, Zhang L, et al. Periodontal ligament stem cells promote polarization of M2 macrophages. J Leukoc Biol. 2022;111(6):1185–1197. doi: 10.1002/JLB.1MA1220-853RR [DOI] [PubMed] [Google Scholar]
- [135].Zhu Y-N, Gu X-I, Wang L-Y, et al. All-trans Retinoic acid promotes M2 macrophage polarization in vitro by activating the p38MAPK/STAT6 signaling pathway. Immunological Investigations. 2023;52(3):298–318. doi: 10.1080/08820139.2023.2173077 [DOI] [PubMed] [Google Scholar]
- [136].Peng S, Fu H, Li R, et al. A new direction in periodontitis treatment: biomaterial-mediated macrophage immunotherapy. J Nanobiotechnology. 2024;22(1):1–29. doi: 10.1186/s12951-024-02592-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Molfino A, Amabile MI, Monti M, et al. Omega-3 polyunsaturated fatty acids in critical illness: anti-inflammatory, proresolving, or both? Oxid Med Cell Longev. 2017;2017(1). doi: 10.1155/2017/5987082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Nguyen QV, Malau-Aduli BS, Cavalieri J, et al. Enhancing omega-3 long-chain polyunsaturated fatty acid content of dairy-derived foods for human consumption. Nutrients. 2019;11(4):743. doi: 10.3390/nu11040743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Norris PC, Skulas-Ray AC, Riley I, et al. Identification of specialized pro-resolving mediator clusters from healthy adults after intravenous low-dose endotoxin and omega-3 supplementation: a methodological validation. Sci Rep. 2018;8(1):18050. doi: 10.1038/s41598-018-36679-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Deore GD, Gurav AN, Patil R, et al. Omega 3 fatty acids as a host modulator in chronic periodontitis patients: a randomised, double-blind, palcebo-controlled, clinical trial. J Periodontal Implant Sci. 2014;44(1):25–32. doi: 10.5051/jpis.2014.44.1.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Serhan CN, Chiang N, Dalli J. The resolution code of acute inflammation: Novel pro-resolving lipid mediators in resolution. Seminars in immunology. 2015;27(3):200–215. doi: 10.1016/j.smim.2015.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Chiang N, Serhan CN, Harwood J, et al. Specialized pro-resolving mediator network: an update on production and actions. Essays Biochem. 2020;64(3):443–462. doi: 10.1042/EBC20200018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Julliard WA, Myo YPA, Perelas A, et al. Specialized pro-resolving mediators as modulators of immune responses. Seminars in immunology. 2022;59:101605. doi: 10.1016/j.smim.2022.101605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Norling LV, Dalli J, Flower RJ, et al. Resolvin D1 limits polymorphonuclear leukocyte recruitment to inflammatory loci: receptor-dependent actions. Arterioscler Thromb Vasc Biol. 2012;32(8):1970–1978. doi: 10.1161/ATVBAHA.112.249508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Hasturk H, Kantarci A, Goguet-Surmenian E, et al. Resolvin E1 regulates inflammation at the cellular and tissue level and restores tissue homeostasis in vivo. The J Immunol. 2007;179(10):7021–7029. doi: 10.4049/jimmunol.179.10.7021 [DOI] [PubMed] [Google Scholar]
- [146].Liu D, Xu J, Figliomeni L, et al. Expression of RANKL and OPG mRNA in periodontal disease: possible involvement in bone destruction. Int J Mol Med. 2003;11:17–21. doi: 10.3892/ijmm.11.1.17 [DOI] [PubMed] [Google Scholar]
- [147].Hasturk H, Schulte F, Martins M, et al. Safety and preliminary efficacy of a novel host-modulatory therapy for reducing gingival inflammation. Front Immunol. 2021;12:12. doi: 10.3389/fimmu.2021.704163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Ferguson B, Bokka NR, Maddipati KR, et al. Distinct profiles of specialized pro-resolving lipid mediators and corresponding receptor gene expression in periodontal inflammation. Front Immunol. 2020;11:1307. doi: 10.3389/fimmu.2020.01307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Bright R, Hynes K, Gronthos S, et al. Periodontal ligament‐derived cells for periodontal regeneration in animal models: a systematic review. J Periodontal Res. 2015;50(2):160–172. doi: 10.1111/jre.12205 [DOI] [PubMed] [Google Scholar]
- [150].Liu Y, Zheng Y, Ding G, et al. Periodontal ligament stem cell-mediated treatment for periodontitis in miniature swine. Stem Cells. 2008;26(4):1065–1073. doi: 10.1634/stemcells.2007-0734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Giannopoulou C, Cimasoni G. Functional characteristics of gingival and periodontal ligament fibroblasts. J Dent Res. 1996;75(3):895–902. doi: 10.1177/00220345960750030601 [DOI] [PubMed] [Google Scholar]
- [152].Song B, Zhang Y, Chen L, et al. The role of toll-like receptors in periodontitis. Oral Dis. 2017;23(2):168–180. doi: 10.1111/odi.12468 [DOI] [PubMed] [Google Scholar]
- [153].Baer MT, Huang N, Gibson FC III. Scavenger receptor a is expressed by macrophages in response to Porphyromonas gingivalis, and participates in tnf-α expression. Oral Microbiol And Immunol. 2009;24(6):456–463. doi: 10.1111/j.1399-302X.2009.00538.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Wu C, Liu C, Luo K, et al. Changes in expression of the membrane receptors CD14, MHC-II, SR-A, and TLR4 in tissue-specific Monocytes/Macrophages following porphyromonas gingivalis–lps stimulation. Inflammation. 2018;41(2):418–431. doi: 10.1007/s10753-017-0698-y [DOI] [PubMed] [Google Scholar]
- [155].Bhattarai G, Poudel SB, Kook S-H, et al. Resveratrol prevents alveolar bone loss in an experimental rat model of periodontitis. Acta Biomater. 2016;29:398–408. doi: 10.1016/j.actbio.2015.10.031 [DOI] [PubMed] [Google Scholar]
- [156].He Y, Wu Z, Chen S, et al. Activation of the pattern recognition receptor NOD1 in periodontitis impairs the osteogenic capacity of human periodontal ligament stem cells via p38/MAPK signalling. Cell Prolif. 2022;55(12):e13330. doi: 10.1111/cpr.13330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Zambrano LMG, Brandao DA, Rocha FRG, et al. Local administration of curcumin-loaded nanoparticles effectively inhibits inflammation and bone resorption associated with experimental periodontal disease. Sci Rep. 2018;8(1):6652. doi: 10.1038/s41598-018-24866-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Chen Q, Zhao Y, Xie C, et al. Therapeutic effect of a novel M1 macrophage-targeted nanodrug in chronic periodontitis mice. Mol Pharm. 2024;21(4):1677–1690. doi: 10.1021/acs.molpharmaceut.3c00954 [DOI] [PubMed] [Google Scholar]
- [159].Abe T, Hosur KB, Hajishengallis E, et al. Local complement-targeted intervention in periodontitis: proof-of-concept using a C5a receptor (CD88) antagonist. The J Immunol. 2012;189(11):5442–5448. doi: 10.4049/jimmunol.1202339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Hajishengallis G, McIntosh ML, Nishiyama SI, et al. Mechanism and implications of CXCR 4‐mediated integrin activation by porphyromonas gingivalis. Molecular Oral Microbiology. 2013;28(4):239–249. doi: 10.1111/omi.12021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Zhang T, Wu J, Ungvijanpunya N, et al. Smad6 methylation represses NFκB activation and periodontal inflammation. J Dent Res. 2018;97(7):810–819. doi: 10.1177/0022034518755688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Lucas H, Bartold PM, Dharmapatni AASSK, et al. Inhibition of apoptosis in periodontitis. J Dent Res. 2010;89(1):29–33. doi: 10.1177/0022034509350708 [DOI] [PubMed] [Google Scholar]
- [163].Xu X, Zhang T, Xia X, et al. Pyroptosis in periodontitis: from the intricate interaction with apoptosis, NETosis, and necroptosis to the therapeutic prospects. Front Cell Infect Microbiol. 2022;12:12. doi: 10.3389/fcimb.2022.953277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Wei W, An Y, An Y, et al. Activation of autophagy in periodontal ligament mesenchymal stem cells promotes angiogenesis in periodontitis. J Periodontol. 2018;89(6):718–727. doi: 10.1002/JPER.17-0341 [DOI] [PubMed] [Google Scholar]
- [165].DeSelm CJ, Miller BC, Zou W, et al. Autophagy proteins regulate the secretory component of osteoclastic bone resorption. Dev Cell. 2011;21(5):966–974. doi: 10.1016/j.devcel.2011.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Rydell-Törmänen K, Johnson JR. The Applicability of Mouse Models to the Study of Human Disease. In: Bertoncello, I ed. Mouse Cell Culture: Methods and Protocols. New York, (NY): Springer New York. 2019. p. 3–22. doi: 10.1007/978-1-4939-9086-3_1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Monnier M, Paolini L, Vinatier E, et al. Antitumor strategies targeting macrophages: the importance of considering the differences in differentiation/polarization processes between human and mouse macrophages. J Immunother Cancer. 2022;10(10):10. doi: 10.1136/jitc-2022-005560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. The J Immunol. 2004;172(5):2731–2738. doi: 10.4049/jimmunol.172.5.2731 [DOI] [PubMed] [Google Scholar]
- [169].Gao X, Wu Y, Liao L, et al. Oral organoids: progress and challenges. J Dent Res. 2021;100(5):454–463. doi: 10.1177/0022034520983808 [DOI] [PubMed] [Google Scholar]
- [170].Bao Y, Wang G, Li H. Approaches for studying human macrophages. Trends Immunol. 2024;45(4):237–247. doi: 10.1016/j.it.2024.02.007 [DOI] [PubMed] [Google Scholar]
- [171].Rojas C, García MP, Polanco AF, et al. Humanized mouse models for the study of periodontitis: an opportunity to elucidate unresolved aspects of its immunopathogenesis and analyze new immunotherapeutic strategies. Front Immunol. 2021;12:663328. doi: 10.3389/fimmu.2021.663328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Weng C, Yu F, Yang D, et al. Deciphering cell states and genealogies of human haematopoiesis. Nature. 2024;627(8003):389–398. doi: 10.1038/s41586-024-07066-z [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data availability does not apply to this article as no new data were created or analyzed in this study.