

Abstract
Background and Objectives
Cases and studies of neurologic symptoms in children caused by genetic metabolic diseases have been widely reported. Ornithine transcarbamylase deficiency (OTCD) is the most common urea cycle disorder, which is due to mutations in the OTC gene located on chromosome Xp21.1. In this study, we analyzed the clinical and genetic characteristics of 8 Chinese children diagnosed with OTCD.
Methods
A total of 8 patients (5 male and 3 female) were diagnosed with OTCD by biochemical and molecular analysis between 2015 and 2023. Clinical manifestations, biochemical features, and OTC gene sequencing analysis were reviewed retrospectively. The effect of c.664-1G>C on OTC mRNA synthesis was confirmed by a minigene splicing assay.
Results
All children were late-onset patients, with a median onset age of 3.6 years (range 1.8–17 years). Neurologic symptoms caused by hyperammonemia include vomiting, coma, dyssomnia, and seizures. The peak plasma ammonia levels ranged from 149 to 4,490 μmol/L, and alanine transaminase levels ranged from 20 to 1316 U/L. Four of them had received CRRT, and only 1 patient was admitted for liver transplantation. By December 2023, 4 patients had survived and 4 were deceased. Blood amino acids or urinary organic acids were detected in 7 cases. All patients underwent whole-exome sequencing, and 2 novel mutations were revealed (P1, c.617dupT and P2, c.664-1G>C). The alteration (c.664-1G>C) leads to the deletion of a 54-bp sequence in the exon 7 of the OTC gene (NM_000531.6: c.664_717del).
Discussion
Two novel pathogenic variants in the OTC gene were confirmed in 8 Chinese children by biochemical findings and genetic analysis. These findings will provide a better understanding of the diagnosis and treatment of pediatric patients with OTCD.
Introduction
Ornithine transcarbamylase deficiency (OTCD) is an X-linked genetic disorder and the commonest urea cycle disorder.1 The estimated incidence is approximately 1/14,000 in the population.2 According to the age at onset, the early-onset type (onset age ≤30 days) and late-onset type (onset age >30 days), or asymptomatic, are the main clinical phenotypes of OTCD. Neonatal onset typically presents tachypnea, vomiting, and lethargy, while the symptoms of the late-onset form are complicated and polymorphic.3 Because of the excretion of ammonia in OTCD, toxic levels of ammonia cause CNS symptoms such as vomiting, lethargy, headaches, and confusion.4 The severe neurologic manifestations contain convulsions, mental and behavioral disorders, encephalopathy, coma, and death.5
The OTC gene is located on chromosome Xp21.1 and encodes ornithine transcarbamylase. The mutation of OTC is the underlying cause of OTCD, and over 500 disease-causing mutations have been found: Missense and/or nonsense mutations were the most common variation, while splice-junction alterations, large deletions, and frameshift mutations have also been reported.5 Because heterozygous female patients have 1 normal X chromosome, they have lower liver OTC activity and symptom onset triggered by certain drugs and stress states, such as infection, trauma, or after surgery. Because hemizygous (not homozygous) male patients have only 1 X chromosome, male patients are more common in newborns or children and show more serious symptoms than female patients.6 The OTC gene (NM_000531) encodes 354 amino acids with a domain named PRK00779. The OTC is one of the urea cycle enzymes and participates in arginine biosynthesis, converting carbamoyl phosphate and ornithine into citrulline in liver cells. The laboratory findings of OTCD usually include elevated ammonia and liver function, decreased blood citrulline levels, and increased urinary orotic acid.7 Studies also showed that chronic presentations of OTCD on MRI were cortical atrophy and decreased myelin.8 In addition to the typical clinical signs and family history, enzyme analysis, genetic testing, or liver biopsy can also identify OTCD.
In this study, we will analyze the clinical and genetic characteristics of 8 pediatric patients from 8 unrelated families. All patients were identified by genetic analysis, and 2 novel heterozygous pathogenic mutations were found (c.617dupT and c.664-1G>C). Our study will provide more evidence for the diagnosis of OTCD.
Methods
Patients and Family
In this study, 8 pediatric patients (5 male and 3 female) with OTCD were recruited from 2015 to 2023 in this study. This study was authorized by the ethics committee of Qilu Hospital of Shandong University. Written consents were obtained from the patients or guardians of the patients. Peripheral blood of 2–3 mL was collected from the children and their parents. The clinical and biochemical characteristics of patients are presented in Table 1.
Table 1.
Clinical Characteristics of Patients With OTCD
| NO. | Type | Clinical presentation | Peak AMONa | Peak ALTa | GLNa | CITa | Urinary OA | Uracila | Therapy received | Outcome |
| P1 | L | Vomiting | 256 μmol/L (9–33 μmol/L) | 1316 U/L (5–40 U/L) | 19.22 μmol/L (1–70 μmol/L) | 36.71 μmol/L (6–45 μmol/L) | 0 | 119.19 μmol/L (0–7 μmol/L) | Drug | Alive |
| P2 | L | Dyssomnia | 373 μmol/L (9–33 μmol/L) | 903 U/L (9–50 U/L) | ND | 34.85 μmol/L (7–36 μmol/L) | 294.93 μg/mg.Cr (0–1.5 μg/mg.Cr) | 185.06 μg/mg.Cr (0–5.5 μg/mg.Cr) | Drug and CRRT | Alive |
| P3 | L | Coma | 149 μmol/L (9–33 μmol/L) | 85 U/L (9–50 U/L) | ND | ND | ND | ND | Drug | Alive |
| P4 | L | Astasia | 272 μmol/L (9–33 μmol/L) | 713 U/L (9–50 U/L) | 31.45 μmol/L (8–45 μmol/L) | 7.31 μmol/L (6–45 μmol/L) | ND | ND | Drug and liver transplantation | Death |
| P5 | L | Vomiting Seizure Coma |
1263 μmol/L (18–72 μmol/L) | 64 U/L (0–38 U/L) | ND | 3.83 μmol/L (4–30 μmol/L) | ND | ND | CRRT Mechanical ventilation |
Death |
| P6 | L | Vomiting Unconsciousness |
221 μmol/L (9–33 μmol/L) | 20 U/L (9–50 U/L) | 20.11 μmol/L (1–55 μmol/L) | 9.51 μmol/L (5.5–45 μmol/L) | 136.2 μg/mg.Cr (0–2 μg/mg.Cr) | 18.9 μg/mg.Cr (0–8 μg/mg.Cr) | Drug CRRT Mechanical ventilation |
Death |
| P7 | L | Seizure Coma |
438.5 μmol/L (9–33 μmol/L) | 54 U/L (9–50 U/L) | 68.36 μmol/L (1–55 μmol/L) | 76.68 μmol/L (5.5–45 μmol/L) | 299.4 μg/mg.Cr (0–2 μg/mg.Cr) | 79.4 μg/mg.Cr (0–8 μg/mg.Cr) | Drug | Alive |
| P8 | L | Seizure Coma |
4,490 μmol/L (9–33 μmol/L) | 297 U/L (9–50 U/L) | 20.59 (7–35 μmol/L) | 30.41 μmol/L (9–64 μmol/L) | 85.98 μg/mg.Cr (0–0.62 μg/mg.Cr) | 6.18 μg/mg.Cr (0–8.5 μg/mg.Cr) | Drug CRRT Mechanical ventilation |
Death |
Abbreviations: ALT = alanine transaminase; AMON = ammonia; CIT = citrulline; CRRT = continuous renal replacement therapy; F = female; GLN = glutamine; L = late-onset phenotype; M = male; m = month; ND = no detection; OA = orotic acid; P = patient; y = year
Different reference range according to age groups and laboratory data.
Biochemical Characteristics
The blood amino acids were detected by tandem mass spectrometry (MS/MS; AB SCIEX API 3200MD). The urinary organic acids were discovered by gas chromatography-mass spectrometry (GC-MS; Shimadzu GCMS-QP2010).
DNA Library Preparation and Sequencing of Targeted Genes
After obtaining informed consent, genomic DNA was extracted from peripheral blood according to the manufacturer's instructions by the QIAamp DNA Mini Kit (Qiagen, Shanghai, China). Genomic DNA 1–3 μg was fragmented to a size of approximately 150 bp using an ultrasonicator after the quantification of DNA (Thermal Fisher Scientific, Covaris, MA). The standard libraries were prepared using a DNA Sample Prep Reagent Set (MyGenostics, Beijing, China) and sequenced by DNBSEQ (DNBSEQ-T7) according to the manufacturer's protocol.9 The capture experiment of amplified DNA was conducted according to the manufacturer's protocol of the GenCap skeletal system capture kit (MyGenostics Inc., Beijing, China). After the purification by SPRI beads (Beckman Coulter), the PCR products were sequenced on the DNBSEQ (DNBSEQ-T7) sequencer for paired reading of 150 bp.10
Bioinformatics Analysis
After sequencing, the raw data were filtered by cutadaptor software.11 The clean reads were aligned to the human reference genome (hg19) using the parameter BWA software package. The indels and SNP were detected using Sentieon software and further annotated using ANNOVAR.12 Variants were screened by the databases, such as 1000 Genome, ESP6500, dbSNP, EXAC, Inhouse (MyGenostics), and HGMD. The functional effects of variants were predicted by SIFT, PolyPhen-2, MutationTaster, and GERP++.13,14
OTC mRNA Splicing Verification in Vitro
OTC wild-type and OTC mutant minigene plasmids were constructed and transfected into HepG2 cells, respectively. RNA was extracted and reverse transcribed into cDNA. The cDNA was amplified by PCR. OTC mRNA splicing was verified by analyzing the size of the PCR fragment and the results of PCR sequencing. The primers designed for RT-PCR amplification were MiniRT-F (5′-GGCTAACTAGAGAACCCACTGCTTA-3′) and OTC-RT-R (5′-CTTCATTGTAACCTGGTAACCTTGG-3′).
Standard Protocol Approvals, Registrations, and Patient Consents
The study was approved by the ethical committee of Qilu Hospital of Shandong University (KYLL-202204-028-1). Our study was performed according to relevant guidelines and regulations. Written informed consents were obtained from all legal guardians of the recruited subjects.
Data Availability
The data sets used during and/or analyzed during this study are available from the corresponding author on request.
Results
Clinical Characteristics of Patients With OTCD
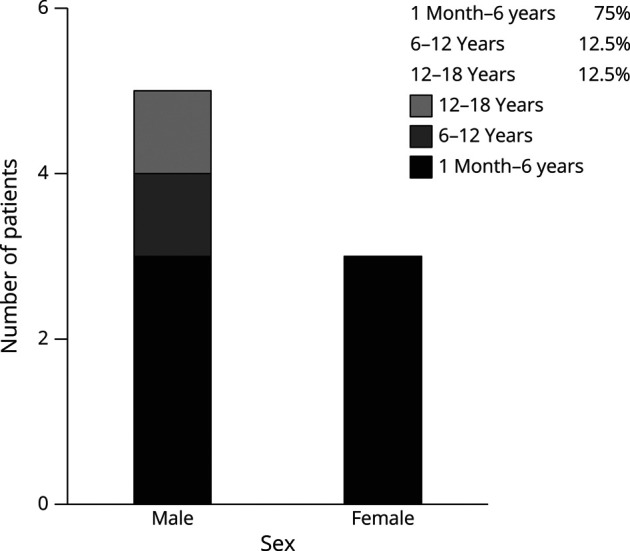
The sex and age at onset of 8 patients are shown in Figure 1. The median onset age of all the children was 3.6 years (range 1.8–17 years). Six patients (75%), 3 male and 3 female, had an onset age between 1 month and 6 years. The 2 children with the novel mutation (P1 and P2) were girls aged 1 month-6 years. The clinical presentations of 8 patients are presented in Table 1. Neurologic symptoms were the most common clinical manifestations. The acute-severe neurologic symptoms (including coma, dyssomnia, and seizures) occurred in 6 cases (75%). Vomiting was the second prominent symptom and occurred in 3 cases (37.5%). For the 2 novel variants, P1 developed severe vomiting before admission, and P2 presented hyperactivity and sleep-wake disturbances from 2 months ago before admission.
Figure 1. Sex and Age at Onset of the 8 Patients.
Biochemical Findings of Patients With OTCD
The results of the biochemical studies of patients with OTCD are presented in Table 1. The elevated levels of blood ammonia were found in all patients (range 149–4,490 μmol/L). Seven patients had elevated plasma alanine aminotransferase (ALT), and the peak ALT ranged from 20 to 1316 U/L. P1 showed an exactly high ALT level (1316 U/L) and clinical features of severe liver failure. It is suggested that the novel heterozygous frameshift mutation in the OTC gene (c.617dupT, p.M206fs*19) is closely related to the phenotype of liver damage. Blood amino acids or urinary organic acids were detected in 7 patients. Only P7 showed elevated plasma l-glutamine (68.36 µmol/L, reference range: 1–55 µmol/L) and citrulline (76.68 µmol/L, reference range: 5.5–45 µmol/L). 50% (4/8) of the patients had increased urinary orotic acid and uracil. Among them, P2 had the highest urinary orotic acid and uracil levels.
Brain MRI and Liver Biopsy of Patients With OTCD
37.5% (3/8) of patients had brain MRI examinations. Brain MRI of P2 showed swelling of the bilateral frontal, temporal, parietal, and insular gyrus, suggesting metabolic encephalopathy (Figure 2A). Brain MRI of P4 showed increased signal in the bilateral basal ganglia and brain atrophy (Figure 2B). Brain MRI of the P5 revealed increased signal intensity in the bilateral basal ganglia (Figure 2C). These findings suggest that splicing mutations and nonsense or missense mutations in the OTC gene might cause damage to different parts of the brain. Owing to increasingly severe neurologic disturbances, P4 received liver transplantation. Results of liver biopsy showed multifocal hepatic steatosis and infiltration of inflammatory cells in the hepatic portal area (Figure 3, A and B).
Figure 2. Brain MRI Results of the Patients With OTCD Detected in This Study.
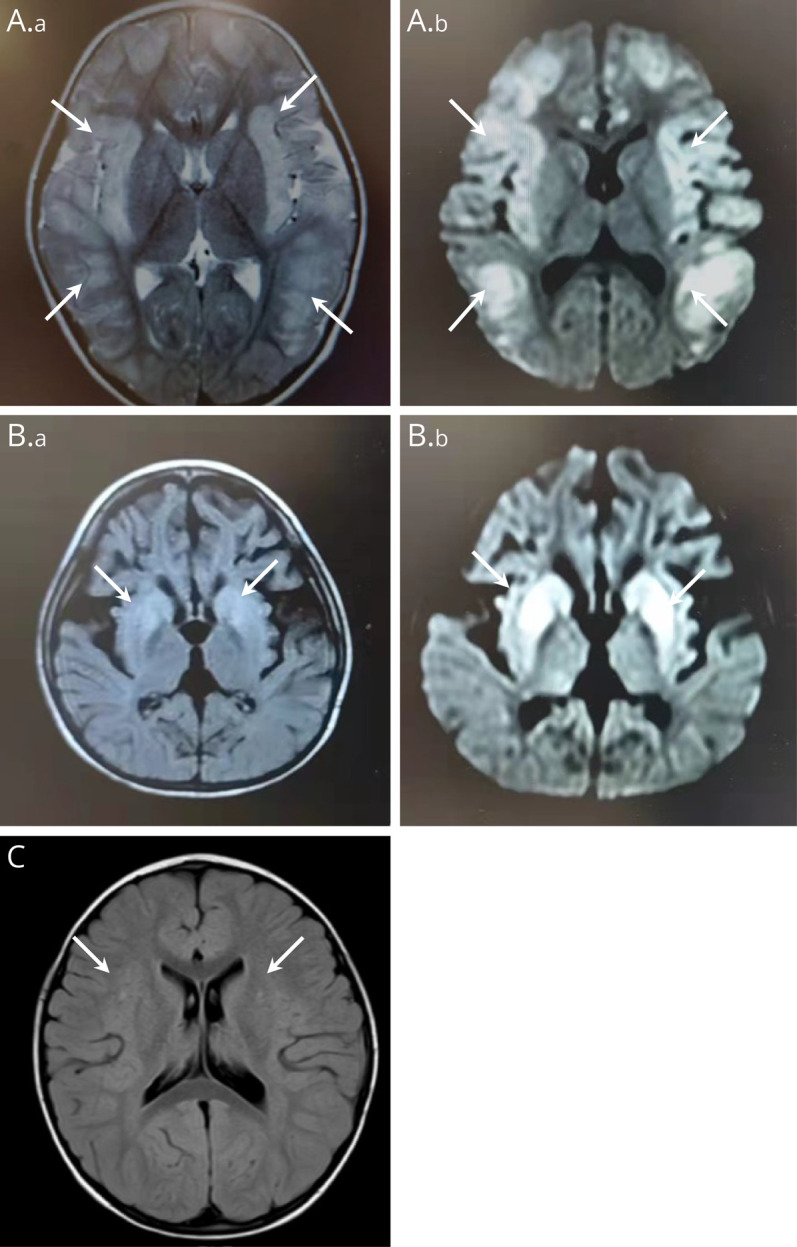
(A) T2-weighted image (a) and diffusion-weighted image (DWI, b) showed the swelling of the bilateral frontal, temporal, parietal, and insular gyrus of patient 2 (white arrows). (B) T2-FLAIR (a) and DWI (b) showed increased signal in the bilateral basal ganglia and brain atrophy of patient 4 (white arrows). (C) T2-FLAIR showed increased signal in the bilateral basal ganglia of patient 5 (white arrows).
Figure 3. Pathologic Results of Liver Biopsy of Patient 4.

The multifocal hepatic steatosis (A) and infiltration of inflammatory cells in the hepatic portal area (B) were shown in HE staining of the liver tissue of patient 4 (white arrows).
Mutation Analysis of the OTC Gene
All the patients completed whole-exome sequencing (WES) (Table 2). A novel heterozygous frameshift mutation was found in the OTC gene of P1 (c.617dupT, p.M206fs*19). PCR-Sanger sequencing confirmed the presence of this mutation in the patient, and this mutation was absent in her mother (Figure 4A).WES of P2 revealed a novel heterozygous splicing variant in the OTC gene (c.664-1G>C), and PCR-Sanger sequencing showed that this variant was not present in her mother (Figure 4B).Three missense mutations were found in P3 (c.365A>G, p.Glu122Gly), P5 (c.119G>A, p.Arg40His), and P6 (c.158T>C, p.Ile53Thr), which were previously reported.15-17 PCR-Sanger sequencing showed the variants were also present in the asymptomatic mothers of P3 and P5 (Figures 4C and 5, A and B). DNA studies of P4 revealed a single nucleotide change from C to T, and variant c.67C>T was detected in exon 1, resulting in a nonsense mutation p.Arg23*, which has previously been reported18 (Figure 4D). Two hemizygous mutations were found in the OTC genes of P7 (c.790A>G, p.Thr264Ala) and P8 (c.493G>T,p.Asp165Tyr), which had previously been reported,19,20 and these variants were also present in their asymptomatic mothers (Figures 5, C and D).
Table 2.
Molecular Characteristics of Patients With OTCD
| No. | Affected Exon/Intron | Nucleotide change | Amino acid change | Mutation type | Novelty | Maternal status |
| P1 | Exon6 | c.617dupT | p.M206fs*19 | Frameshift | Novel | Non-carrier |
| P2 | IVS6, Exon7 |
c.664-1G>C | — | Splicing | Novel | Non-carrier |
| P3 | Exon4 | c.365A>G | p.E122G | Missense | Reported | Carrier |
| P4 | Exon1 | c.67C>T | p.R23* | Nonsense | Reported | NA |
| P5 | Exon2 | c.119G>A | p.R40H | Missense | Reported | Carrier |
| P6 | Exon2 | c.158T>C | p.I53T | Missense | Reported | NA |
| P7 | Exon8 | c.790A>G | p.T264A | Missense | Reported | Carrier |
| P8 | Exon5 | c.493G>T | p.D165Y | Missense | Reported | Carrier |
Abbreviations: P, patient; NA: not available.
Figure 4. Molecular Analysis of the OTC Gene in This Study.

(A–D) The partial sequence results of P1 to P4 and their parents were detected in this study.
Figure 5. Molecular Analysis of the OTC Gene in This Study.

(A–D) The partial sequence results of P5 to P8 and their parents were detected in this study.
Corroboration of the OTC: c.664-1G>C Mutation as the Determinant of Alternative Splicing in the Splicing Assay
We used bioinformatics software dbscSNV and SpliceAI to analyze OTC: c.664-1G>C. Our results suggested that the mutation may lead to the deletion of the exon 7 in the OTC gene (Figure 6B). The abnormal splicing of the mutant mRNA was verified by the band size of PCR amplification and Sanger sequencing. The results showed that the transcribed mRNA sequence of the WT plasmid contained a complete mRNA product transcribed by exon 6, exon 7, and exon 8. The sequence of the mutant plasmid showed that the deletion of the 54-bp sequence in exon 7 and the alteration of c.664-1G>C (c.664_717del) affect mRNA splicing (Figure 6C).
Figure 6. OTC Gene Constructs for Splicing Pattern Investing of the P2.

(A) The positions of the 8 mutations in the OTC gene. (B) Splicing schematic diagram. The bioinformatics software dbscSNV and SpliceAI predicted that the mutation might lead to a splicing variation of the OTC gene. (C) OTC gene c.664-1G>C splicing pattern minigene verification experiment.
Treatment and Clinical Outcomes of Patients With OTCD
All the patients received the treatment of a protein restriction diet, liver protection, and blood hypoammonia drugs (Table 1). 50% (4/8) of patients had CRRT, and 37.5% (3/8) of patients received mechanical ventilation. Only P4 had liver transplantation. During 8 years of follow-up, 4 cases (3 male and 1 female) had died and 4 cases (2 male and 2 female) survived.
Discussion
In this study, we reported 8 unrelated and late-onset OTCD children. The clinical phenotypes of the OTCD features included neurologic disturbances and acute liver failure. Blood amino acids or urinary organic acids were detected in all patients, except P3 (Table 1). All patients had a normal karyotype and were identified as disease-associated variants (Figure 6A). Of the 8 different variants, 2 have never been previously described. Two novel alterations, c.617dupT (p.M206fs*19) and c.664-1G>C, were found in P1 and P2 (Table 2). P2, P5, P6, and P8 had received CRRT, and P4 was admitted for liver transplantation. By December 2023, P4, P5, P6, and P8 were deceased, and other patients recovered completely without any neurologic deficits. Therefore, OTCD is treatable if caught early and fatal if left undiagnosed.
To date, a total of 514 pathogenic variants in the OTC gene have been identified, including 53 splicing mutations and 2 duplications.21 Our study identified a splicing mutation (c.664-1G>C) and a duplication mutation (c.617dupT), causing the loss of OTC function. These 2 patients were both late-onset female, and the novel variants were not present in their mothers. Similar reports showed that the loss of OTC function might cause neonatal-onset disease in hemizygous male patients and symptoms of OTCD in some heterozygous female patients.1,22,23 In addition, alanine transaminase (ALT) of these 2 patients was higher than those of the other 6 children, suggesting that frameshift or splicing mutations of the OTC gene might reveal lower liver OTC activity than missense mutations. These results might contribute to finding predictors of the severity of OTCD based on the biochemical properties of mutant OTC, although numerous studies reported the correction of OTCD genotype with age at onset, liver OTC activity, incorporation of nitrogen into urea, and peak plasma ammonia levels.24,25 Otherwise, we applied an in vitro minigene splicing assay to detect the aberrant splicing caused by OTC: c.664-1G>C. This method can easily detect the aberrant exonic or intronic splicing caused by single-base substitutions.26 Our result demonstrated that the c.664-1G>C variant preserved a deletion of the 54-bp sequence in exon 7 of OTC transcripts, resulting in deletion of 18 amino acids behind the amino acid at position 222 (p.Gly 222_Glu239delGlyTyrGluProAspAlaSerValThrLysLeuAlaGluGlnTyrAlaLysGlu) (Figure 6C). Further functional studies and structural analysis will be required to understand the changes in the proteins.
Ammonia levels above 200 mmol/L are usually caused by inherited metabolic diseases.3 Because the OTC enzyme is early in the urea cycle, severe hyperammonemia is more likely to occur in OTC than most of the other UCDs. All cases had hyperammonemia, and P8 had irreversible neurologic damage with an extremely high ammonia level (4490 µmol/L). P2, P4, and P5 had abnormal brain MRI (Figure 2). A previous head CT scan of a 27-year-old patient with OTCD showed diffuse cerebral edema, diffuse sulcal effacement, and effacement of the lateral ventricles.27 Briefly, the aim of the treatment was to lower the ammonia concentrations as fast as possible. In addition to hypoammonia medication, protein-restricted diet is a key adjuvant therapy, and hemodialysis has an immediate effect on ammonia clearance. However, the above treatments do not address the underlying cause of the disease. Researchers28 reported that 2 cases of prenatal treatment of mothers who harbor severe OTCD mutations showed therapeutic levels of benzoate and phenylacetate in the newborn at delivery and without acute hyperammonemia and neurologic decompensation in conjunction with high caloric enteral nutrition. Two cases also received liver transplantation at ages 3 months and 5 months. Furthermore, liver transplantation has its own set of problems, including the requirement for lifelong immunosuppression.29 In this study, P4 died of severe pneumonia caused by long-term immunosuppression. Lately, research or trials on OTCD gene therapy have risen. For example, Prieve et al. demonstrated that hOTC mRNA/HMT administration not only reduces ammonia levels but also leads to significant survival benefit in the murine OTCD disease model.30 These experiments represent the development of human gene therapy for OTCD and other UCDs.
In conclusion, the diagnosis of OTCD should be considered when patients show unexplained behavioral, hepatic, or neurologic disturbances associated with hyperammonemia. It is best to improve gene detection in patients, especially with a similar family history of UCDs. Meanwhile, expanded carrier screening can detect asymptomatic cases without any family history. The performance of appropriate therapy is essential to prevent irreversible neurologic damage and avoid severe consequences.
Acknowledgment
The authors are grateful to the patients and their families for their participation in this study.
Glossary
- ALT
alanine transaminase
- OTCD
ornithine transcarbamylase deficiency
- WES
whole-exome sequencing
Appendix. Authors
| Name | Location | Contribution |
| Chen Zhang, PhD | Department of Pediatrics, Qilu Hospital of Shandong University, China | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data |
| Junli Shan, MS | Department of Pediatrics, Qilu Hospital of Shandong University, China | Major role in the acquisition of data |
| Jiaqi Su, MS | Department of Pediatrics, Qilu Hospital of Shandong University; Department of Respiratory Disease, Children's Hospital affiliated to Shandong University, Jinan, China | Drafting/revision of the manuscript for content, including medical writing for content; analysis or interpretation of data |
| Guan Wang, PhD | Department of Pediatrics, Qilu Hospital of Shandong University, China | Analysis or interpretation of data |
| Qin Huo, MS | Department of General Practice, Fourth People's Hospital of Jinan, China | Analysis or interpretation of data |
| Rui Xu, MS | Department of Pediatrics, Qilu Hospital of Shandong University, China | Analysis or interpretation of data |
| Meng Dong,PhD | Department of Pediatrics, Qilu Hospital of Shandong University, China | Major role in the acquisition of data; study concept or design |
Study Funding
Shandong Natural Science Foundation (grant number ZR2023QH065).
Disclosure
The authors report no relevant disclosures. Go to Neurology.org/NG for full disclosures.
References
- 1.Caldovic L, Abdikarim I, Narain S, Tuchman M, Morizono H. Genotype-phenotype correlations in ornithine transcarbamylase deficiency: a mutation update. J Genet Genomics. 2015;42(5):181-194. doi: 10.1016/j.jgg.2015.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brusilow SW, Maestri NE. Urea cycle disorders: diagnosis, pathophysiology, and therapy. Adv Pediatr. 1996;43(1):127-170. doi: 10.1016/s0065-3101(24)00072-0 [DOI] [PubMed] [Google Scholar]
- 3.Leonard JV, Morris AA. Urea cycle disorders. Semin Neonatol.2002;7(1):27-35. doi: 10.1053/siny.2001.0085 [DOI] [PubMed] [Google Scholar]
- 4.Donovan K, Guzman N. Ornithine transcarbamylase deficiency. In: StatPearls. StatPearls Publishing; 2024. [PubMed] [Google Scholar]
- 5.Chongsrisawat V, Damrongphol P, Ittiwut C, Ittiwut R, Suphapeetiporn K, Shotelersuk V. The phenotypic and mutational spectrum of Thai female patients with ornithine transcarbamylase deficiency. Gene. 2018;679:377-381. doi: 10.1016/j.gene.2018.09.026 [DOI] [PubMed] [Google Scholar]
- 6.Choi J-H, Lee BH, Kim JH, et al. . Clinical outcomes and the mutation spectrum of the OTC gene in patients with ornithine transcarbamylase deficiency. J Hum Genet. 2015;60(9):501-507. doi: 10.1038/jhg.2015.54 [DOI] [PubMed] [Google Scholar]
- 7.Gordon N. Ornithine transcarbamylase deficiency: a urea cycle defect. Eur J Paediatr Neurol. 2003;7(3):115-121. doi: 10.1016/s1090-3798(03)00040-0 [DOI] [PubMed] [Google Scholar]
- 8.Gropman A. Brain imaging in urea cycle disorders. Mol Genet Metab. 2010;100(suppl 1):S20-S30. doi: 10.1016/j.ymgme.2010.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang L, You C, Qiu S, et al. . Novel and de novo point and large microdeletion mutation in PRRT2-related epilepsy. Brain Behav. 2020;10(5):e01597. doi: 10.1002/brb3.1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang L, Yu J, Xian Y, et al. . Application of high-throughput sequencing for hereditary thrombocytopenia in southwestern China. J Clin Lab Anal. 2021;35(8):e23896. doi: 10.1002/jcla.23896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Accessed November 23, 2023. https://cutadapt.readthedocs.io/en/stable/
- 12.Annovar documentation. Accessed November 13, 2023.
- 13.Qian F-Y, Guo YD, Zu J, et al. . A novel recessive mutation affecting DNAJB6a causes myofibrillar myopathy. Acta Neuropathol Commun. 2021;9(1):23. doi: 10.1186/s40478-020-01046-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao C, Zhang Q, Zhang R, et al. . Genetic and clinical characteristics of primary hemophagocytic lymphohistiocytosis in children. Ann Hematol. 2024;103(1):17-28. doi: 10.1007/s00277-023-05499-6 [DOI] [PubMed] [Google Scholar]
- 15.Gao H, Li W, Yan ZH, Jiang MH, Rui Dr, He YS. Molecular characterization of a new mutation E122G of human ornithine transcarbamylase gene. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2003;20(1):19-22. [PubMed] [Google Scholar]
- 16.Tuchman M, Jaleel N, Morizono H, Sheehy L, Lynch MG. Mutations and polymorphisms in the human ornithine transcarbamylase gene. Hum Mutat. 2002;19(2):93-107. doi: 10.1002/humu.10035 [DOI] [PubMed] [Google Scholar]
- 17.Tuchman M, Plante RJ, McCann MT, Qureshi AA. Seven new mutations in the human ornithine transcarbamylase gene. Hum Mutat. 1994;4(1):57-60. doi: 10.1002/humu.1380040109 [DOI] [PubMed] [Google Scholar]
- 18.Grompe M, Caskey CT, Fenwick RG. Improved molecular diagnostics for ornithine transcarbamylase deficiency. Am J Hum Genet. 1991;48(2):212-222. [PMC free article] [PubMed] [Google Scholar]
- 19.Matsuura T, Hoshide R, Setoyama C, et al. . Four novel gene mutations in five Japanese male patients with neonatal or late onset OTC deficiency: application of PCR-single-strand conformation polymorphisms for all exons and adjacent introns [corrected]. Hum Genet. 1993;92(1):49-56. doi: 10.1007/BF00216144 [DOI] [PubMed] [Google Scholar]
- 20.Genet S, Cranston T, Middleton-Price HR. Mutation detection in 65 families with a possible diagnosis of ornithine carbamoyltransferase deficiency including 14 novel mutations. J Inherit Metab Dis. 2000;23(7):669-676. doi: 10.1023/a:1005614409241 [DOI] [PubMed] [Google Scholar]
- 21.HGMD. Accessed May 1, 2024. hgmd.org
- 22.Tuchman M, Morizono H, Rajagopal BS, Plante RJ, Allewell NM. Identification of ‘private’ mutations in patients with ornithine transcarbamylase deficiency. J Inherit Metab Dis. 1997;20(4):525-527. doi: 10.1023/a:1005301513465 [DOI] [PubMed] [Google Scholar]
- 23.Yamaguchi S, Brailey LL, Morizono H, Bale AE, Tuchman M. Mutations and polymorphisms in the human ornithine transcarbamylase (OTC) gene. Hum Mutat. 2006;27(7):626-632. doi: 10.1002/humu.20339 [DOI] [PubMed] [Google Scholar]
- 24.McCullough BA, Yudkoff M, Batshaw ML, Wilson JM, Raper SE, Tuchman M. Genotype spectrum of ornithine transcarbamylase deficiency: correlation with the clinical and biochemical phenotype. Am J Med Genet. 2000;93(4):313-319. doi: [DOI] [PubMed] [Google Scholar]
- 25.Gobin-Limballe S, Ottolenghi C, Reyal F, et al. . OTC deficiency in females: phenotype-genotype correlation based on a 130-family cohort. J Inherit Metab Dis. 2021;44(5):1235-1247. doi: 10.1002/jimd.12404 [DOI] [PubMed] [Google Scholar]
- 26.Wai H, Douglas AGL, Baralle D. RNA splicing analysis in genomic medicine. Int J Biochem Cell Biol. 2019;108:61-71. doi: 10.1016/j.biocel.2018.12.009 [DOI] [PubMed] [Google Scholar]
- 27.Durer S, Durer C, Hoilat GJ. Adult-onset ornithine transcarbamylase deficiency as a rare cause of fatal hyperammonaemia. Lancet. 2021;398(10302):e11. doi: 10.1016/S0140-6736(21)01606-8 [DOI] [PubMed] [Google Scholar]
- 28.Wilnai Y, Blumenfeld YJ, Cusmano K, et al. . Prenatal treatment of ornithine transcarbamylase deficiency. Mol Genet Metab. 2018;123(3):297-300. doi: 10.1016/j.ymgme.2018.01.004 [DOI] [PubMed] [Google Scholar]
- 29.Youssef D, Niazi A, Alkhouri N. Long term complications in pediatric liver transplant recipients: what every pediatrician should know. Curr Pediatr Rev. 2016;12(3):209-221. doi: 10.2174/1573396312666160627124242 [DOI] [PubMed] [Google Scholar]
- 30.Prieve MG, Harvie P, Monahan SD, et al. . Targeted mRNA therapy for ornithine transcarbamylase deficiency. Mol Ther. 2018;26(3):801-813. doi: 10.1016/j.ymthe.2017.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data sets used during and/or analyzed during this study are available from the corresponding author on request.