

Abstract
The formation of disulfide bonds is critical to the folding of many extracytoplasmic proteins in all domains of life. With the discovery in the early 1990s that disulfide bond formation is catalyzed by enzymes, the field of oxidative folding of proteins was born. Escherichia coli played a central role as a model organism for the elucidation of the disulfide bond-forming machinery. Since then, many of the enzymatic players and their mechanisms of forming, breaking, and shuffling disulfide bonds have become understood in greater detail. This article summarizes the discoveries of the past 3 decades, focusing on disulfide bond formation in the periplasm of the model prokaryotic host E. coli.
DISULFIDE BOND FORMATION AND FOLDING
Disulfide bonds usually contribute to the three-dimensional structure of a protein and often are required for the full activity of a protein (1). Protein disulfide bonds are formed by an oxidative reaction in which the thiol (RSH) groups of two cysteines are joined by a covalent linkage. In contrast to other covalent linkages in proteins, disulfide bonds are reactive and easily reversible or isomerized by thiol:disulfide exchange reactions, both spontaneously or catalyzed by enzymes (2). Proteins that include disulfides in their mature, functional structure are usually oxidized during folding in a process known as “oxidative folding.”
In many organisms, disulfide bond formation and isomerization occur in specialized oxidizing compartments, such as the endoplasmic reticulum or mitochondrial intermembrane space (2–4). In Gram-negative bacteria, proteins that require disulfide bonds for their final folded state are translocated across the cytoplasmic membrane into the periplasm, where enzymes that catalyze the formation and isomerization of disulfide bonds reside. Although proteins with disulfide bonds are also found in the cytoplasm of both eukaryotes and prokaryotes, most of them are transient and involve cysteines located in redox-active sites of enzymes (also called oxidoreductases), with the disulfides being formed and reduced in electron transfer reactions (5). A few exceptions are certain lineages of archaea which contain disulfide-rich cytosolic proteins, presumably due to a selective pressure for thermostable proteins (6), and some viruses that encode their own oxidative folding machinery to make disulfide-containing proteins in the cytoplasm of the infected cells (7).
In vitro, disulfides are commonly formed by thiol:disulfide exchange, a reaction that has been widely studied both in vitro and in vivo and whose output is easily predictable based on the physicochemical properties of the thiols involved (8). The pathway by which disulfides are generated de novo and transferred to nascent proteins in vivo is, however, much more complicated and requires the concerted action of several oxidoreductases, electron transfer proteins, and cofactors. This article reviews the process of disulfide bond formation in the periplasm of Escherichia coli by the Dsb systems. In brief, the DsbA-DsbB pair forms disulfides in proteins that are translocated to the periplasm and passes electrons to the respiratory chain (see “Formation of Disulfides by a Thiol Oxidase” below), while the DsbC-DsbD pair uses electrons transferred through the inner membrane to isomerize mismatched disulfides (see “Isomerization of Mismatched Disulfides and Protection of Lonely Cysteines” below) (Fig. 1). The engineering of the Dsb system for improved protein expression is briefly described in “Engineering E. coli for Disulfide Bond Formation” below. During this period, several important reviews about disulfide bond formation and Dsb systems were published, and the reader is pointed toward them for details (9–15). The formation and isomerization of disulfides in eukaryotes has several similarities with the bacterial periplasmic disulfide bond formation system and was recently reviewed (2, 16) and will not be commented on in this article.
Figure 1.
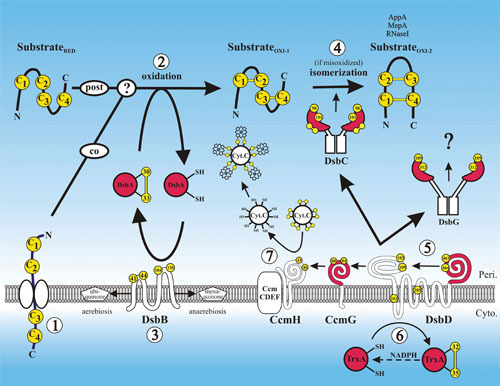
Disulfide bond formation in the periplasm. A protein requiring disulfide bonds for its stability is translocated into the periplasm via the SecYEG translocon with its cysteines (arbitrarily labeled C1 to C4) in a reduced state (substrateRED) (1). Disulfide bond formation is catalyzed by DsbA, either during translocation, after translocation, or both (2). DsbA is reoxidized back to its active oxidized state by DsbB, and DsbB is oxidized by ubiquinone in aerobic conditions or by menaquinone in anaerobic conditions (3). If the substrate is misoxidized (substrateOXI-1), its disulfide bonds are isomerized to their native oxidized states (substrateOXI-2) by DsbC (4). DsbC along with DsbG and CcmG are maintained in their active reduced states by DsbD (5). DsbD in turn is reduced by the cytoplasmic thioredoxin TrxA, which receives its reducing potential ultimately from cytoplasmic pools of NADPH (6). CcmG maintains CcmH in a reduced state. Through the interaction of CcmH with the CcmCDEF membrane complex, oxidized cytochrome-c is reduced, enabling it to form thioether covalent bonds with its heme cofactor (7). Proteins with thioredoxin folds are in red, and cysteines are in yellow. The amino acid residue numbers of the redox-active cysteines are indicated.
FORMATION OF DISULFIDES BY A THIOL OXIDASE
DsbA
For decades, disulfides were supposed to be formed in the periplasm of bacteria, due to the spontaneous reaction of reduced cysteines with oxidants, in spite of the slow kinetics and lack of specificity of this process (17–19). The discovery of a specific oxidase (DsbA) in a genetic screen looking for factors involved in membrane protein insertion (20) offered the first evidence of a disulfide-forming enzyme in bacteria, opening an entire new field that proved to be relevant for understanding both bacterial and eukaryotic oxidative folding. Shortly after the discovery of DsbA, more members of this pathway were discovered, exemplifying the power of genetic screens in E. coli (for a historical note, see reference 21).
Periplasmic proteins destined to contain structural disulfide bonds are translocated across the cytoplasmic membrane into the periplasm via the Sec machinery (for recent reviews see 22 and 23). The central component of this pathway is an evolutionarily conserved protein-conducting channel in the cytoplasmic membrane composed of three integral membrane proteins: SecY, SecE, and SecG. Substrate proteins must be in an unfolded conformation to pass through this channel during the process of translocation (22). Proteins can be translocated through SecYEG by two mechanisms: in posttranslational export, protein synthesis and translocation are uncoupled (24); in the cotranslational mechanism, protein synthesis is tightly coupled to translocation, avoiding the presence of an unfolded cytoplasmic intermediate (25). Cells have a variety of mechanisms for maintaining these proteins in an export-competent unfolded state in the cytoplasm prior to translocation (22, 26). In addition, many periplasmic proteins (e.g., alkaline phosphatase or PhoA [27]) require structural disulfide bonds, which are only formed in the periplasm. Formation of structural disulfide bonds between cysteines in proteins is catalyzed in E. coli by the periplasmic oxidoreductase DsbA. The interaction between DsbA and the cysteines of its reduced substrate has been shown to occur both co- and posttranslocationally (28). The interaction between DsbA and substrate protein may take place between a completely unfolded protein, a protein that has assumed some secondary structure, or one that has folded even more extensively in the periplasm (Haldar et al. in-press). The oxidation of a cysteine pair into a disulfide bond in the substrate protein by DsbA results in the consequent reduction of the redox-active site cysteines in DsbA. The active site cysteines in DsbA are restored to their oxidized active state by the inner membrane protein, DsbB, that channels electrons to the respiratory chain via its interaction with the quinone pool (see “DsbB” below).
DsbA is a monomeric soluble protein with 189 amino acids in its mature form that is directed into the periplasm by its 19-amino acid signal peptide. While most periplasmic proteins are translocated posttranslationally, the translocation of DsbA is cotranslational and is dependent on SRP (signal recognition particle) for its interaction with the SecYEG translocon (29). This cotranslational export of DsbA appears to be necessary to avoid it being trapped in the cytoplasm, due to its rapid folding kinetics (30). DsbA is composed of a domain with a classical thioredoxin (Trx)-fold with a helical domain inserted between its third and fourth beta strands (Fig. 2). Like most members of the Trx-fold superfamily (31), DsbA has a characteristic Cys-X-X-Cys motif (where X is any amino acid). Several structures of DsbA have been determined by both nuclear magnetic resonance and crystallography (32–34), providing structural snapshots of its mechanism (schematically presented in Fig. 3). The helical domain folds around the Trx-fold, constructing a hydrophobic patch around the active site in a fashion similar to glutathione (GSH) transferases (31). An uncharged groove running along the active site of DsbA may promote the binding of DsbA to unfolded substrates through hydrophobic interactions (35–38). This groove is believed to have no specificity supporting DsbA’s ability to oxidize a large spectrum of substrates (39).
Figure 2.
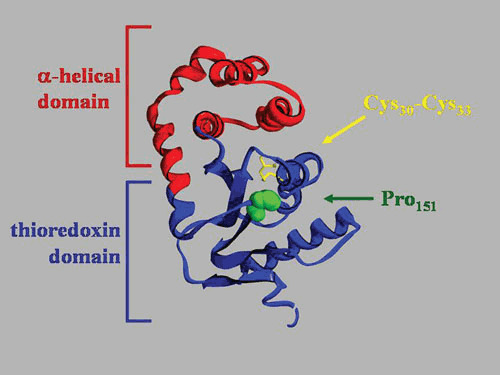
Domain organization of DsbA. Crystal structure of oxidized DsbA (PDB accession number 1FVK). The thioredoxin domain is in blue, and the α-helical domain is in red. The active site disulfide bond and the critical proline151 are indicated.
Figure 3.
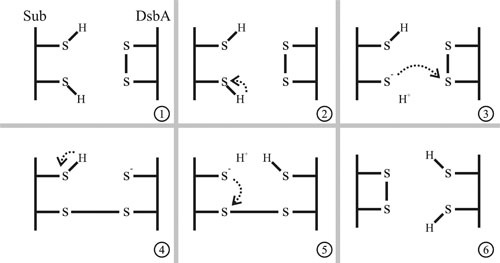
The mechanism of disulfide bond formation. DsbA catalyzes the formation of disulfide bonds in a polypeptide with reduced cysteines. The cysteines within the Cys-X-X-Cys active site of DsbA are oxidized (S-S), and the thiol side-groups of cysteine residues in the substrate are reduced (SH) (panel 1). Disulfide bond formation is initiated by deprotonation of a thiol group in the substrate (panel 2). The resulting thiolate anion can initiate a nucleophilic attack on the disulfide bond of DsbA (panel 3). The resolution of the mixed-disulfide bonded complex could occur by deprotonation of another thiol group (panel 4), which can attack the substrate-DsbA disulfide bond (panel 5). The result of this reaction is the oxidation of the substrate and the reduction of DsbA (panel 6).
Redox potential (or, more accurately, reduction potentials) are commonly used in the thiol:disulfide exchange literature, but their extrapolation to biology calls for caution. The electrochemical potential of a compound simply describes its tendency to become oxidized compared to a reference known as a “standard hydrogen electrode,” where the reversible oxidation of a hydrogen molecule to protons and electrons by a platinum electrode is given a redox potential of zero (40). Most of the Trx-fold proteins participate in thiol:disulfide exchange reactions, either reducing or catalyzing the formation of disulfide bonds. Thioredoxin 1 of E. coli (Trx1, encoded by the gene trxA) is the best-studied representative of this family, and it is usually considered a “reducing” protein because it has a redox potential of –270 mV (41); i.e., it has a high tendency to become oxidized in thiol:disulfide exchanges. Oxidized Trx1 is reduced by thioredoxin reductase, which accepts electrons from NADPH. The NADPH/NADP+ pair has a redox potential of about –370 mV, meaning that the flow of electrons from NADPH (–370 mV) to Trx1 (–270 mv) is favorable because of the higher (more positive) redox potential of Trx, resulting in a higher tendency to gain electrons. However, Trx1 is not directly reduced by NADPH; instead, the enzyme thioredoxin reductase (TrxR, encoded by the gene trxB in E. coli) relays the electrons in between.
DsbA has a redox potential of –120 mV (42), which is among the highest redox potentials reported for any Trx-fold protein (42). The redox potential differences measured in vitro are indicative of the flow of electrons in vivo. Thus, electrons flow from the higher redox potential of DsbA (–120 mV) through an enzymatic cascade that passes through the inner membrane, ultimately toward the lowest reducing potential of NADPH (–370 mV). In this way, DsbA tends to gain electrons from its partners, i.e., to be reduced by thiols, forming disulfides. However, the redox potential of reduced DsbA remains negative, and it is still a long way from highly oxidizing agents such as hydrogen peroxide (H2O2) or aqueous iron salts that have redox potentials above zero.
In conclusion, redox potentials govern what is thermodynamically feasible, but this does not unequivocally predict that the reaction will occur in vivo. The specificity of protein-protein interactions, kinetics, and thermodynamic parameters is the gatekeeper for any biologically relevant thiol:disulfide exchange reaction, which is particularly important in understanding the plethora of thiol:disulfide exchange reactions that take place in the bacterial periplasm during oxidative folding. As examples, we can point to the genetic alterations that cause cytoplasmic Trx1 to accumulate in the oxidized form in the cytoplasm, resulting in a relatively efficient enzyme that can catalyze the formation of disulfide bonds (43–46). Conversely, under appropriate in vitro conditions, DsbA can effectively reduce the disulfide bonds of insulin (20).
The N-terminal Cys30 in the active site of DsbA has a pKa of 3.5, determining that it will be a thiolate under physiological conditions when reduced (47). As in many other Trx-fold proteins, the thiolate is stabilized by hydrogen bonds and charge interactions with lateral chains from amino acids that are proximal, particularly His32 and Glu97 (48, 49). The sulfur atom of Cys30 is fully exposed in the oxidized crystal structure of DsbA, allowing it to be attacked by reduced cysteines in substrate proteins without the need for substrate-dependent interactions. The C-terminal Cys33 is buried inside the protein and is unlikely to be involved in the initial mixed-disulfide bonded complex with a substrate (47). The two amino acids flanked by the active cysteines of DsbA are key determinants of DsbA redox properties; exchanging these two amino acids with those of other thioredoxin family members results in striking effects on the redox potentials of the proteins (50). Furthermore, oxidized DsbA is more flexible and less stable than the reduced form (51–53), as addressed by equilibrium unfolding experiments. An important note on DsbA mechanisms is that, as in any thiol:disulfide exchange, the reaction is started by the nucleophilic attack of a thiol group from the substrate on the DsbA active-site disulfide. Considering that there is no evidence of activation of substrate cysteines by DsbA, this reaction is expected to have a pH-dependent kinetic like any thiol:disulfide exchange does. However, oxidation of substrates by DsbA is highly accelerated by the fact that Cys30 has a very low pKa, making it an excellent leaving group, and by the instability of the active-site disulfide bond, which drives DsbA toward the reduction of this tense bond. Taken together, cysteine pKa and conformational tension make DsbA an excellent oxidant able to catalyze thiol:disulfide exchange reaction at rates that are about a million times faster than noncatalyzed reactions (8).
Another key residue important in the function of DsbA is Pro151 (Fig. 2). This proline is one of the most conserved residues in Trx-fold proteins (31) and is often found in the cis conformation instead of the highly favorable trans conformation that most prolines adopt within proteins (54). In most thioredoxin structures, this cis-proline is found in close proximity to the active site, facing the redox-active cysteines. Mutations in Pro151 to alanine destabilize DsbA significantly (55), resulting in alterations in the structure of the active site (56). Mutation of Pro151 to serine, histidine, asparagine, tryptophan, or glycine results in accumulation of DsbA-DsbB mixed-disulfide complexes (57), while a change to threonine results in accumulation of DsbA-substrate mixed-disulfide complexes (58). Purification of these complexes resulted in the identification of numerous in vivo substrates of DsbA in E. coli. The Pro151 residue appears to be critical in resolving disulfide-bonded DsbA complexes. Another residue that may play a critical role is the one immediately before the conserved proline. Ren et al. (59, 60) showed that this residue in Francisella tularensis DsbA strongly modifies both the redox properties of Trx-fold proteins and their ability to interact with substrates.
The dsbA gene is part of a two-gene operon with an upstream gene called srkA (yihE) for “stress response kinase A.” There appear to be two promoters: a distal promoter upstream of the srkA gene and a proximal promoter within the 3′ end of this gene. Transcription from the proximal promoter is constitutive at low levels (61). Under normal laboratory growth conditions, dsbA is expressed constitutively in the periplasm at a level of about 850 molecules per cell (62). The expression of dsbA is increased in response to cell envelope protein folding defects. This increase is dependent on positive regulation by the Cpx two-component system (63, 64). DsbA is also induced at high pH in both aerobic and anaerobic conditions (65).
Due to the significant role disulfide bonds play in the folding and/or stability of numerous periplasmic proteins, mutations in DsbA and its homologues in other Gram-negative bacteria result in a myriad of phenotypes. Bacteria lacking a functional copy of dsbA are viable but have increased sensitivity to reduced dithiothreitol, benzylpenicillin (66), Cd2+, Hg2+ (67), and Zn2+ (68). These mutants are also defective in intracellular survival (69), infection (70, 71), competence (72), biofilm formation (73), pertussis toxin (74), fimbriae (75), and pilus production (76). Interestingly, DsbA mutants are also resistant to lysis by Lyz endolysin of bacteriophage P1 because its proper folding and activity rely on the formation of three disulfides (77). Thus far, more than two dozen periplasmic proteins in E. coli have been shown to be dependent on DsbA for their correct folding (Agp, AppA, ArtJ, Bla, DegP, DppA, FlgI, GltI, GltX, HisJ, Imp, LivJ, LivK, MepA, OmpA, OppA, PhoA, RcsF, Rna, STa, STb, UgpB, YbeJ, YbjP, YcdO, YedD, YggN, YodA, and ZnuA).
Neither DsbA nor its partner, DsbB (see below), are essential for aerobic growth of E. coli (20), despite the fact that two proteins which are essential have disulfide bonds that are required for either their stability, FtsN (78), or folding, LptD (79). The pathway is essential for anaerobic growth, which also requires these two proteins, suggesting that aerobic growth may be dependent on background oxidation sufficient for the activity of at least those two proteins (79). An explanation involving an alternative pathway is possible. Such a pathway can be constructed by export of the cytosolic reductase Trx1 to the periplasm (80) or overexpression of the periplasmic rhodanese (PspE), which partially restores disulfide bond formation in a dsbA-null strain (81). However, no such pathway has been found in wild-type E. coli.
DsbB
The C30-C33 disulfide bond of DsbA becomes reduced during its interaction with substrates and must be reoxidized to regain oxidase activity. It is reoxidized by DsbB, which transfers the electron pair it extracts from DsbA to the electron transport chain via quinones, to ubiquinone during aerobic growth or to menaquinone when growing anaerobically. DsbB was discovered in the same screen as DsbA (82) and at about the same time using other approaches (66, 83). DsbB is an integral membrane protein of 176 amino acids with 4 transmembrane helices and 2 pairs of active-site Cys residues at positions 41 to 44 in the first periplasmic loop and 104 to 130 in the second (Fig. 4). As shown in Fig. 5 (based on Fig. 8 in reference 84), a de novo disulfide bond is formed between the cysteine residues at C41 and C44 of DsbB via a transient charge transfer complex formed between C44 of DsbB and a quinone. It has also been demonstrated that the Arg48 in DsbB plays a critical role in this charge transfer (84–86). Subsequent transfer of this bond to reduced DsbA by disulfide exchange involves disulfide bonds between C41 and C130 of DsbB and an intermolecular disulfide between C104 of DsbB and C30 of DsbA. Two conformationally distinct pathways differing in the order of formation of these two bonds may both be involved (84).
Figure 4.
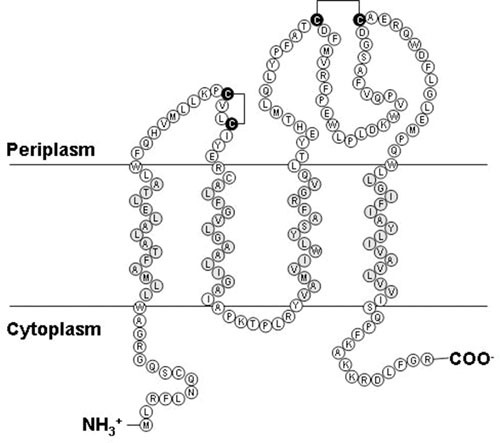
Topology of DsbB. The topology of DsbB based on alkaline phosphatase fusion studies (188). The active site cysteines are shown in their oxidized states, and the putative transmembrane domain amino acids are highlighted.
Figure 5.
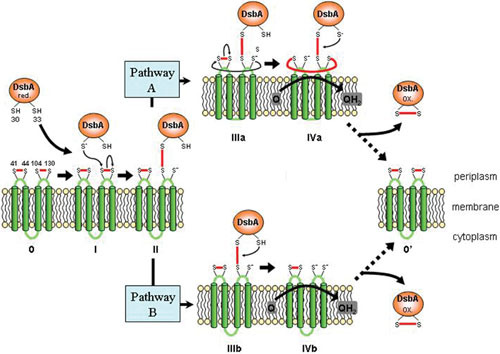
Mechanism of DsbA reoxidation by DsbB. A reduced DsbA interacts with oxidized DsbB, resulting in the reoxidation of DsbA and reduction of DsbB. The DsbA-DsbB complex is formed via a disulfide bond between C33 of DsbA and C104 of the second periplasmic loop of DsbB. The resolution of this complex is believed to occur through two pathways. In pathway A, a disulfide bond is formed between the first and second periplasmic loop, which is resolved by the oxidation of DsbB by quinones. In pathway B, the DsbA-DsbB complex is resolved by quinones without the interaction of the first periplasmic loop. Figure based on Fig. 8 in reference 84.
Diversity of Disulfide Bond Formation Pathways in Bacteria
Bioinformatic analysis (87) suggests that there is an unexpected diversity in the mechanisms of disulfide bond formation in bacteria. Strict anaerobes and obligate intracellular bacteria which do not live in oxidizing environments do not generally oxidize cysteines of exported proteins. Among bacteria predicted to generally oxidize cysteines of exported proteins, some aerobic and facultative bacteria have DsbA-DsbB oxidative pathways that are homologous to that of E. coli, notably most aerobic proteobacteria and members of the phylum Deinococcus-Thermus. Others, including most Actinobacteria, Cyanobacteria (including chloroplasts), aerobic Deltaproteobacteria, and some Spirochetes, have a homologue of the metazoan protein vitamin K epoxide reductase instead of DsbB. Subsequent work has shown that the vitamin K epoxide reductase of Mycobacterium tuberculosis together with a linked Gram-positive type DsbA (see below) constitute an essential disulfide bond formation pathway in that organism (88) and is a prototype for a second type of disulfide bond formation pathway found in a wide variety of organisms. M. tuberculosis vitamin K epoxide reductase is not homologous to DsbB but is also capable of functional replacement of DsbB in E. coli (87). It has a transmembrane architecture that is reminiscent of that of DsbB (89), and mutational analysis supports strong mechanistic similarity (90).
Structural analysis of homologues of DsbA with greater than 35% sequence identity suggests division into two main classes on the basis of the topology of their beta strands (91, 92). These classes roughly correspond with the phylogenetic distinction between Gram-negative (class I) and Gram-positive (class II) DsbAs. Class I can be further subdivided into the subclasses Ia with a long continuous groove adjacent to the active site cysteines and Ib with a smaller binding pocket. Class II DsbAs have various other features in this region. Many of these homologous pathways have been reviewed recently (9).
Homologues of DsbA with even less homology are also widespread. For example, Salmonella enterica serovar Typhimurium contains, in addition to a DsbA and a DsbB, a second pair of homologues in DsbL and DsbI (with, respectively, 27% and 30% homology to DsbA and DsbB) together in an operon with an aryl sulfate-sulfotransferase, assT, which is dependent on oxidation by DsbL/I for activity. A similar DsbL/DsbI pair was found in uropathogenic E. coli strain CFT073, which is also highly divergent from DsbA/B (19/24% sequence identity) but clearly paralogous in function (93, 94). DsbL is more oxidizing than DsbA (redox potential of –95 mV) and differs in the substrate-binding groove harboring basic residues instead of the hydrophobic patch of DsbA, suggesting narrower substrate specificity. It is one of the few genes specific to the uropathogenic strains and is involved in antibiotic resistance (93). DsbL and DsbI are also key components of the disulfide bond formation pathway of S. enterica serovar Typhimurium, where they play a role in general disulfide bond formation as well as ensuring complete oxidation of the linked AssT (95). Similar DsbL/I homologues are found in many proteobacteria both with and without associated assT genes, suggesting that horizontal gene transfer may have been involved (87).
ISOMERIZATION OF MISMATCHED DISULFIDES AND PROTECTION OF LONELY CYSTEINES
DsbC
About two-thirds of the E. coli periplasmic proteins whose structure has been determined have one or two disulfide bonds, and these bonds are usually formed between cysteines that are close in the primary structure. This correlates with the assumption that DsbA has no substrate specificity and may simply react with the first cysteine of the polypeptide chain to cross into the periplasm and join it into a disulfide bond with the next cysteine to appear. The indiscriminate oxidation of cysteines by DsbA is demonstrated by the introduction of nonnative disulfide bonds in secreted β-galactosidase (LacZ) (96). Further evidence of this lack of selectivity for substrates by DsbA is provided by DsbA acting on nonnatural substrates such as the cysteine-containing Bacteroides β-lactamase, which is not ordinarily translocated into the E. coli periplasm but acquires nonnative disulfide bonds that interfere with their folding and thus with their activity when secreted to the periplasm via a peptide signal (20, 97). However, a few cases of periplasmic proteins are known whose structure have one or more nonconsecutive disulfide bonds, such as phytase (AppA, four disulfide bonds), a murein endopeptidase (MepA, three disulfide bonds), and periplasmic RNA degrading enzyme (RNase I, four disulfide bonds). The formation of a disulfide bond between nonconsecutive cysteines poses a challenge to the linear mechanism of oxidative folding catalyzed by DsbA. E. coli, and other bacteria express a protein disulfide isomerase (DsbC) that is necessary to correct the “errors” that are made by DsbA. DsbC acts by promoting rearrangement of disulfide bonds, allowing generation of the proper array in the final folded product (98, 99). In concordance, the folding of each of the periplasmic proteins with the nonconsecutive disulfide bonds mentioned above (AppA, MepA, and RNase I) shows strong dependence on DsbC (100, 101).
There is a fairly strict correlation between native disulfide bond patterns and DsbC dependence; proteins that contain nonconsecutive disulfide bonds in their final structure are dependent on DsbC for proper folding, while proteins that should be made with consecutive disulfide bonds show very little or no dependence on DsbC (100–103). For example, two nearly identical proteins of E. coli, phytase and glucose-1-phosphatase, differ in that the former contains one nonconsecutive disulfide bond, while the latter contains only consecutive disulfide bonds. Phytase is dependent on DsbC for assembly into an active enzyme, while glucose-1-phosphatase is not (100). Introduction of a nonconsecutive disulfide bond into glucose-1-phosphatase results in a DsbC-dependent enzyme, and removing the nonconsecutive disulfide bond from phytase makes its folding DsbC independent.
DsbC is a 236-amino acid homodimer directed to the periplasm by a 20-amino acid-long amino-terminal signal peptide. Each monomer of DsbC is composed of a dimerization domain linked to a Trx-fold domain by a short α-helix (Fig. 6) (104–106). The Trx-fold domain contains four cysteines: Cys141 and Cys163 form a structural disulfide that is essential for the folding and stability of DsbC (107), while Cys98 and Cys101 form the redox-active pair, located in the conserved position into the Trx-fold (103). The redox-active site cysteine pairs are maintained in a reduced state by the inner-membrane protein, DsbD (see below). The dimerization of the two monomers of DsbC results in the formation of an uncharged hydrophobic pocket (Fig. 6) that has been postulated to be responsible for the recognition of misfolded proteins due to hydrophobic interactions. Since misoxidized proteins will most likely be misfolded, the chaperone properties of DsbC would allow it to recognize and bind to those proteins that contain incorrect disulfide bonds (108). The in vitro chaperone activity of DsbC is independent of its redox-active cysteines but is dependent on the dimerization domain (109).
Figure 6.
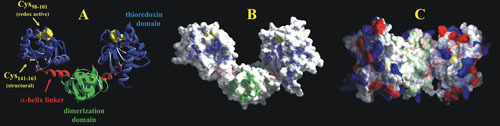
Domain organization of DsbC. (A) The crystal structure of the homodimer DsbC showing the two domains (thioredoxin in blue and dimerization in green) separated by the short α-helix linker in red (PDB accession number 1EEJ). Redox-active cysteines (C98 to C101) are represented as yellow spheres, and the structural disulfide bond (C141 to C163) is indicated. (B) The molecular surface of DsbC is superimposed, visualizing the pocket formed by the dimerization of DsbC. (C) Top-down view of DsbC displaying the noncharged pocket devoid of acidic (red) and basic (blue) amino acid residues.
While it seems likely that the chaperone properties of DsbC allow it to interact with misfolded proteins, the complete in vivo mechanism of disulfide bond isomerization remains to be elucidated. The first step is the attack on the incorrect disulfide bond exposed in the misfolded protein by the amino-terminal cysteine of the DsbC active site, resulting in the formation of a mixed disulfide. Two mechanisms have been proposed for the resolution of this intermediate. In the reducing model, the DsbC-substrate mixed-disulfide complex is resolved by the carboxyl-terminal Cys101 of the DsbC active site, resulting in the reduction of the substrate and the oxidation of DsbC. Further steps to generate the correct disulfide bonds would require one or more new cycles of oxidation by DsbA, and DsbC needs to be recycled back to its reduced state by DsbD. Supporting this mechanism, DsbC has been characterized in vitro to be an efficient reductase (110), and a periplasmic Trx-fold containing protein from Bacteroides fragilis known as TrxP could complement dsbC-null strains via its reductase activity, despite having no detectable isomerase activity (111). A second model, called the isomerase model, has been proposed for the reduction of the intermolecular disulfide. The mixed-disulfide bonded complex between DsbC and its substrate undergoes thiol:disulfide exchange reactions within the substrate, resulting in a new disulfide. This intra-thiol:disulfide exchange reaction can proceed until the substrate protein is in its native oxidized state, with the final reaction resulting in the resolution of DsbC in its reduced state and the substrate in its oxidized state. Disulfide bond isomerization of a substrate by DsbC does not result in any net gain or loss of electrons. Thus, at the end of the isomerization reaction, DsbC remains in its reduced form. The existence of a system devoted to maintain DsbC in its reduced state suggests that the reducing model for DsbC is more likely. However, DsbC may also be oxidized by oxidants in the milieu, suggesting that a role for DsbD may be to maintain DsbC in its reduced state to perform its action through electron-neutral isomerization (112, 113).
A new role for DsbC in the recycling of oxidized periplasmic proteins that contain odd or unique reactive cysteines was proposed. Because they are not involved in the formation of a disulfide, single cysteine residues can be oxidized to sulfenic acid (Cys-SOH). The sulfenic acid form of cysteine is highly reactive and prone to react with oxidants of thiols forming higher oxidized species (sulfinic [Cys-SO2H] or sulfonic [Cys-SO3H]) or disulfides, respectively, leading to inactivation or misfolding of the oxidized protein (114, 115). DsbC (and in some cases DsbG; see below) was shown to react with sulfenic acids on the l,d-transpeptidase YbiS (116), l-arabinose-binding protein AraF (117), and thiosulfate sulfur transferase PspE (81), resulting in mixed disulfides that lead to the oxidation of DsbC and release of reduced substrate. Oxidized DsbC is reduced by its cognate reductase DsbD (see below). DsbC has also been shown to play a major role in the insertion of proteins in the outer membrane. For example, the folding and function of RcsF, an outer membrane lipoprotein involved in the biofilm formation membrane (118), and LtpD, a protein essential for lipopolysaccharide insertion in the outer membrane (119), depend on both DsbA and DsbC.
DsbC is the most versatile of the Dsb members. While its biological function is allegedly to isomerize disulfides, DsbC can efficiently reduce insulin in vitro with kinetics similar to thioredoxin (110) or act as an oxidase in vivo when the interaction with the reducing DsbD is removed (103). The versatility of DsbC is believed to be essential for reducing mismatched disulfide bonds but raises questions about how reductases, isomerases, and oxidases are maintained in the needed redox state in the periplasm. Since the in vitro properties of the active site cysteines and the Trx-fold of DsbC are similar to those of DsbA (120), the lack of oxidation of DsbC by DsbB is intriguing. The most widely accepted hypothesis is the steric hindrances between DsbB and DsbC dimeric V-shaped structures. Modeling of DsbC-DsbB interactions based on the DsbA-DsbB cocrystal (121, 122) revealed that while one monomer of DsbC can interact with DsbB, the other monomer of DsbC clashes with the inner membrane. This model is supported by the fact that mutations that disrupt the dimerization of DsbC, resulting in periplasmic DsbC monomers, can efficiently interact with DsbB (123). These mutant DsbC monomers are now capable of acting as general oxidases and can functionally complement DsbA, acting as oxidases that are reoxidized by DsbB (123). In addition, chimeras of DsbC where the thioredoxin domain was replaced with DsbA, Trx1, or the Trx-fold redox-active domain “a” of the eukaryotic isomerase PDI promoted isomerization of misoxidized substrates in vivo and displayed chaperone activity in vitro at varying levels (110, 124). Moreover, when PspE (see “DsbA” above) is overexpressed in a dsbA-null background, the restoration of disulfide-bond capacity depends on DsbC reacting with oxidized PspE (81). The steric hinderances between DsbB and DsbC can also be alleviated by mutations in DsbB. Mutations in DsbB that disrupt its association with the inner membrane permit DsbB to oxidize DsbC, again resulting in oxidized DsbC to functionally complement DsbA (125).
The separation of the DsbA/B oxidative pathway from the reductive DsbD/C pathway within the periplasmic compartment is of paramount importance to maintaining DsbA in its oxidized state while maintaining DsbC in its reduced state (102, 126, 127). Intriguingly, even though DsbA (–120 mV) and DsbC (–129 mV) have similar redox potentials, they do not equilibrate. (50). While this reaction is thermodynamically possible, it has been shown to be too slow to be biologically relevant (100 M–1s–1 [120]), suggesting that both proteins have structural features that isolate them from reacting with each other.
Another molecule that needs to be considered to understand the redox biology of the periplasm is glutathione (GSH). While GSH plays an active role in the redox reactions within the endoplasmic reticulum (128), little is known about its potential role in bacterial periplasm. GSH is actively exported to the periplasm via CydCD (129), and recombinant glutaredoxin 1 can support GSH-dependent disulfide bond formation in the periplasm (130). However, a possible role for GSH in the mechanism of Dsb-dependent folding still remains to be determined, since the kinetics for the reaction between DsbC and GSSG (30 to 60 M–1s–1 [108, 120]) is outcompeted by the reaction with DsbD by 105 orders of magnitude (131, 132). However, DsbA oxidase activity is inhibited by GSH (133), probably because its reduction by GSH is significantly faster (103 M–1s–1 [53]), and may be biologically relevant considering that the periplasmic concentrations of GSH are estimated to be in the low millimolar range (129). Suggesting a possible connection between GSH and Dsb proteins in the periplasm, a new Dsb member, DsbM, was described in Pseudomonas aeruginosa which has structural similarity to DsbA and, to a lesser extent, to the Trx-domain of DsbC (134). While DsbM is cytoplasmic, it was reported to have a GSH-dependent reductase activity against an oxidized OxyR mechanism of direct regulation of OxyR by reduction (135, 136), suggesting that well-known Dsb proteins such as DsbA and DsbC may have GSH-dependent mechanisms that are still undisclosed.
The dsbC gene is in a bi-cistronic operon with the downstream gene recJ, which encodes a 5′→3′ exonuclease. Promoter analysis of dsbC shows putative CpxR binding sites, indicating regulation by the Cpx two-component stress response, similar to that of DsbA. DsbC transcription is also under the control of the σE pathway (137), which responds to cell envelope stress and relays the signal via a regulated cascade of intermembrane proteolysis. The cytoplasmic RNase E also regulates dsbC mRNA transcript levels. RNase E is an essential endoribonuclease that degrades mRNAs and assists the maturation of a variety of rRNAs and tRNAs in E. coli. Mutations in rne, which encodes RNase E, result in an increased half-life of dsbC mRNA, leading to increases in disulfide bond isomerase activity in E. coli (138).
DsbG
DsbG was discovered in 1997 by screening for mutants which conferred sensitivity or resistance to dithiothreitol when overexpressed (139). It was quickly recognized that DsbG is related to DsbC (24% identity) and may provide additional thiol:disulfide isomerase activity. DsbG is a homodimer with 231 amino acids in its mature form. The crystal structure reveals a dimeric structure similar to DsbC but with some interesting differences (140). The α-helix linker of DsbG is 2.5 turns longer, making the hydrophobic pocket of DsbG significantly larger. Furthermore, unlike the uncharged pocket of DsbC, there are seven negatively charged amino acids within the pocket of DsbG and three in the dimerization domain, suggesting a different substrate specificity and activity between DsbC and DsbG. In vitro, DsbG cannot reduce insulin but does promote refolding of substrate proteins, presumably by the same mechanism as DsbC (141). Multicopy expression of DsbG complements a dsbC– mutant and promotes isomerization and proper assembly of bovine pancreatic trypsin inhibitor in vivo. The in vivo function of DsbG is dependent on DsbD, which reduces the oxidized form of DsbG as it does with DsbC (142).
Oxidoreductases that lack the resolving cysteine in their active sites have poor kinetics in resolving from their mixed-disulfide bonded substrates. This biochemical property was utilized to capture the substrates of DsbG. In coimmune precipitation of DsbG active-site mutants that are not able to resolve the mixed disulfide with its substrate, Depuydt et al. (116) identified three periplasmic l,d-transpeptidases—YbiS, ErfK, and YnhG—as substrates of DsbG. These transpeptidases catalyze the cross-linking of peptidoglycan in a reaction that depends on a single essential and conserved cysteine in the peptidase active site. Those cysteines can be oxidized to Cys-SOH in the periplasm, leading to the inactivation of the peptidase. DsbG reacts with the sulfenic form of YbiS, forming a mixed-disulfide intermediate that resolves releasing reduced YbiS and oxidized DsbG, which is subsequently reduced by DsbD (116).
DsbG and AhpC (a cytoplasmic peroxiredoxin) are located adjacent to each other on the E. coli chromosome and are transcribed in opposite directions. Both are regulated by the oxidative stress regulator, OxyR. OxyR is a cytoplasmic Lys-R type transcriptional regulator which is activated upon having its cysteines oxidized to form a disulfide bond. Footprinting analyses have confirmed the binding of oxidized OxyR 54 bp upstream of the dsbG gene (143). However, in vitro analysis demonstrated that it was the ahpCF-proximal binding site 238 bp upstream of dsbG which was responsible for the upregulation of both dsbG and ahpCF transcripts. The evidence that only oxidized OxyR was capable of binding upstream of dsbG along with the fact that dsbG transcripts were detected with primer extension analysis only after the induction of oxidative stress indicates that DsbG is induced under conditions of oxidative stress.
Chaperone activities for DsbC and DsbG have been demonstrated in vitro using classical chaperone function assays, which monitor their ability to prevent aggregation and assist with refolding of denatured model substrates (109, 141, 144). Moreover, a recent publication demonstrated that DsbG also has cysteine-independent chaperone activity in vivo, in a fashion similar to Spy (144), adding this protein to the list of chaperones that contribute to the folding of periplasmic proteins (145). This ability to chaperone protein folding is thought to be important primarily for the function of DsbC and DsbG as disulfide bond isomerases, because proteins containing incorrect disulfide bonds are likely to become misfolded. The ability to break and reform bonds must therefore be intimately connected to the refolding of denatured protein, preferably alongside the ability to specifically recognize nonnative structures (146). Despite this connection between their two functions, DsbC and DsbG have low selectivity between misfolded proteins containing oxidized cysteines and other misfolded proteins that lack them (109, 141). They are thus capable of providing general protection against folding stress within the periplasm.
The structures of DsbC (105) and DsbG (140) demonstrate unequivocally that both proteins are evolutionarily related, sharing a V-like structure with N-terminal dimerization domains required for the chaperone activity and C-terminal catalytic domains at the prongs of the V fork. The major difference between DsbC and DsbG is that the latter is larger, with an extended α-helical connection region altering the relative orientations of the C-terminal domains. For both proteins, the substrate-binding sites are thought to reside in hydrophobic patches located on the inside of the C-terminal domains and linkers, with the distance between the two binding sites able to change due to flexibility in the hinge region. However, in DsbG there are some additional acidic, negatively charged residues within the substrate-binding region, perhaps conferring a substrate specificity different from that of DsbC (140).
DsbD
To be active as isomerases, DsbC and DsbG require their redox-active cysteines to be maintained in their reduced states (103). This reaction is performed by the inner-membrane protein DsbD, a 546-amino acid membrane protein whose primary function is to connect cytoplasmic thioredoxins to DsbC (for reviews, see 147 and 148). DsbD is the largest and most complex protein of the Dsb family, formed by three domains named α, β, and γ (Fig. 7). The amino-terminal α domain has an immunoglobulin (Ig)-like fold and contains two of the six essential cysteines, Cys103 and Cys109 (149); the central (β) domain has eight transmembrane segments and two essential cysteines: Cys163 in the first transmembrane segment and Cys285 in the fourth (150). The central membrane-embedded β domain of DsbD is the only domain for which a structure is yet to be determined. Finally, the carboxyterminal γ domain has a Trx-fold and the last of the essential cysteines displayed in the unusual Trx-like active site CVAC (Cys461 and 464) (Fig. 8) (132, 151).
Figure 7.
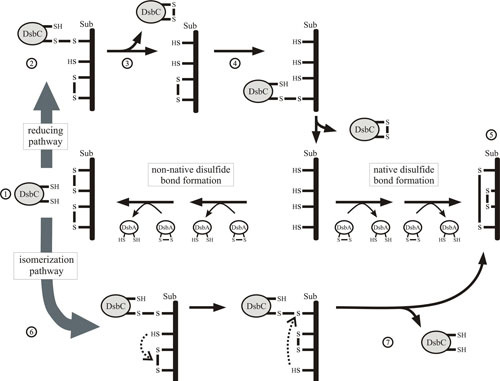
Proposed mechanism of isomerization by DsbC. For clarity, only a monomer of DsbC is shown. Reduced active DsbC recognizes misoxidized substrate (1) and forms a mixed-disulfide bonded complex. This complex (2) can be resolved by the reduction of the disulfide bond in the substrate, resulting in the oxidation of DsbC (3). A secondary cycle of reduction is necessary for the substrate to be fully reduced (4), allowing DsbA to reoxidize the substrate (5). Alternatively, the disulfide bonds in the complex can be shuffled (6) by the isomerase action of DsbC, resulting in native disulfide bonded substrate and reduced DsbC (7).
Figure 8.
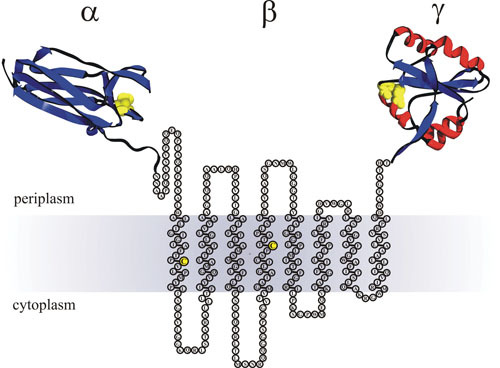
Domain organization of DsbD. The predicted membrane topology of DsbD from the web-based program Phobius (http://phobius.binf.ku.dk). The immunoglobin-like α domain is crystallized (PDB accession number 1L6P) devoid of its signal peptide from the amino acids Arg8 to Asn125. The amino acids of the β domain from Asn126 to Thr422 are depicted as circles. The redox-active cysteines (C163 to C285) are highlighted as yellow circles. The thioredoxin-like γ domain from Ala423 to Pro546 is crystallized (PDB accession number 1UC7). Active site cysteines in the crystal structures are shown as yellow spheres (α domain C103 to C109 and β domain C461 to C464). The membrane is shaded gray.
The three redox centers of DsbD are essential for its electron transfer activity, but the domains do not need to be linked, because they can efficiently reduce DsbC and DsbG when split (152, 153). Based on cumulative in vivo and in vitro data, the mechanism of DsbD can be described as a cascade of thiol:disulfide exchanges that initiate from the cytosolic pool of NADPH, ultimately terminating in the periplasm (131, 132, 150, 152–158). The first step in the reduction of the disulfide pair in the central transmembrane domain of DsbD is catalyzed by cytosolic Trx1. This connection between a cytosolic reductase and an integral membrane protein was demonstrated by the isolation of heterodimers formed between Cys163 of the β domain and active-site Cys32 of Trx1 (152). After donating its electrons, Trx1 is released in its oxidized state and DsbDβ disulfide is reduced. The next step is the attack of Cys163 of the β domain on the oxidized Trx-fold-containing active site of the γ domain. It is important to remember that Trx1 is cytoplasmic, while the γ domain resides in the periplasm, suggesting that the cysteines in the β domain are accessible from both sides of the membrane. How this single intramembrane disulfide embedded in a constrained environment interacts with targets on both sides of the membrane and 50 to 60 Å apart is not completely understood, but the most plausible explanation for this mechanism is that reduction of transmembrane disulfide triggers a conformational change that exposes those cysteines to the periplasmic γ domain (154, 159). A set of conserved prolines was suggested to play a role in this redox-dependent mechanism (160). Additional models have proposed a funnel-like opening and closing of DsbD, allowing for the interaction of the periplasmic γ domain with the redox-active membrane embedded cysteines in the β domain (148).
Following the thiol:disulfide exchange between Trx1 and the β domain, the interdomain disulfide between the β and γ domains is resolved by the attack of Cys285, resulting in the reoxidation of the β domain and release of the reduced Trx-fold γ domain. In the final step, Cys461 from the Trx-fold γ domain attacks the disulfide on the Ig-fold α domain formed by Cys103 and Cys109. An intermediate between Cys461 and Cys109 was isolated, suggesting large conformational changes that bring the γ and α domains together (132). The thiol:disulfide exchange between γ and α is the rate-limiting step of the full cycle, with a reaction constant of 23 s–1 (131). Once the α domain is reduced by the γ domain, it can interact with its oxidized substrates (see below) via Cys109 (151, 152, 161, 162).
The three domains of DsbD have redox potentials that are practically identical (about –240 mV), intermediate between Trx1 (–270 mV) and DsbC (–130 mV). This suggests that the flow of electrons toward DsbC is thermodynamically favorable but does not indicate why these electrons flow following the concerted order β → γ → α or why DsbC is not directly reduced by DsbDγ. The reaction between the γ and α domains and how DsbC is reduced by DsbDα have been thoroughly characterized in vitro. The reduction of DsbC by the DsbDα domain has been characterized to have extremely fast kinetics (4 × 106 M–1s–1), a million times faster than the reduction by DsbDγ (57 M–1s–1) (132). This is an unusual reaction where a dithiol on an Ig-fold domain reduces a disulfide on a Trx-fold protein (DsbC, DsbG, CcmG). Based on Cys to Ala mutation of the two essential cysteines in the α domain that resulted in complete destabilization of the Ig-fold (152), it has been proposed that this reaction is driven by the chaperone activity of DsbC, which recognizes the partially unfolded DsbDα domain and docks in, allowing the thiol:disulfide exchange to take place (151).
Besides the reduction of DsbC, DsbD provides electrons to a variety of redox pathways in the periplasm; particularly relevant is the membrane-anchored protein CcmG (132, 163), which is involved in cytochrome-c maturation (164). A homologous DsbD found in proteobacteria and Chlamydia (ScsB) can also participate in the antioxidant defense, providing electrons for a peroxidase system formed by a thioredoxin-like protein, TlpA, and a peroxiredoxin, PprX (165).
ENGINEERING E. coli FOR DISULFIDE BOND FORMATION
The pharmaceutical and biotechnological industries are extremely interested in disulfide-bonded proteins. Most eukaryotic cell surface and secreted proteins are rich in disulfide bonds due to the increased stability they confer, making these proteins attractive candidates for therapeutics (also known as biopharmaceuticals). For example, the most profitable biopharmaceutical is the monoclonal antibody against human tumor necrosis factor-α (Humira, adalimumab) with nine disulfide bonds essential for its folding and function (166). Indeed, antibodies represent the fastest-growing category of biopharmaceutical due to their specificity-favorable pharmacokinetic profiles within the human body. The production of antibodies for therapeutic applications is a well-established pipeline dominated by the use of mammalian cells (mostly Chinese hamster ovary cells) or mouse hybridomas. However, even in these eukaryotic systems, disulfide mismatching is a major source of undesired heterogeneity on monoclonal antibodies (167).
Though E. coli is currently not as established as Chinese hamster ovary or hybridoma cell lines for the production of therapeutic IgG, it is slowly becoming a more common host for the production of antibodies. It can potentially combine the power of prokaryotic hosts as protein factories with the versatility of the molecular biology tools available for screening and selection. Considering that the disulfide-producing machinery is located in the periplasm, most of the antibodies and antibody fragments produced on E. coli have been targeted to that compartment (168, 169). However, translocation of the target protein across the inner membrane to the periplasm can be problematic and may require extensive optimization of both the expression conditions and the targeting signal sequence. Furthermore, the lack of ATP in the periplasm makes it an energy-poor environment for proteins that require ATP-dependent chaperones for their folding. The cytoplasm is therefore a more suitable compartment for high-yielding protein production. It also obviates the problems of crossing the membrane and is rich in ATP, chaperones, and folding factors.
However, production of disulfide-rich proteins on E. coli cytoplasm is hampered by the presence of very active reducing systems formed by the thioredoxin reductase (trxB) and GSH reductase (gor) systems. Deletion of trxB and gor is lethal, and a suppressor screen for the double mutant ΔtrxB Δgor lethality generated a strain in which a mutation in a thiol-dependent peroxidase (ahpC) gained the ability to reduce Grx1, restoring reducing power to the cell (170). The absence of the most important reducing pathways makes the cytosol of this strain less reductive and suitable for expression of disulfide-rich protein, leading to its commercialization under the name Origami (Novagen). Our lab engineered ΔtrxB Δgor, adding a signal sequenceless DsbC that is constitutively expressed in the cytoplasm and enhances the production of disulfide-rich proteins (46). Those cells have been commercialized under the name SHuffle by New England Biolabs. SHuffle cells are the first example of how the Dsb system can be engineered for biotechnological applications. Using SHuffle cells, our group was able to express various antibody fragments and full-length antibodies in the cytoplasm (171), as well as isotopically labeled antibodies, by expressing them in minimal media (172). DsbB has also been engineered to improve protein production in the cytoplasm. Inversion of topology of DsbB allows the reoxidation of a cytosolic DsbA and improves the formation of disulfide-containing protein in the cytosol in E. coli even in the presence of the reducing system (173). Along the same line, solubilizing DsbB in the cytosol by fusion to a truncated version of human apolipoprotein A-I, together with cytosol expression of DsbA, also increases the expression yield of protein that requires disulfide bonds for its activity (174).
THE EXPANDING Dsb FAMILY: FUTURE DIRECTIONS AND UNANSWERED QUESTIONS
Although great progress has been achieved in the past 30-plus years in furthering our understanding of how enzymes form, break, and shuffle disulfide bonds, there remain, as always, many unanswered questions. The in vivo mechanism of how DsbC recognizes misoxidized proteins and returns them to their correctly oxidized states remains to be understood. In fact, although DsbC has been shown to be an effective chaperone in vitro, this ability of DsbC to assist in the folding of proteins in a redox-independent manner has yet to be shown. Similarly, the exact mechanism of how electrons are transferred across the inner membrane without any proton leakage via cysteine-mediated electron relay between DsbD and the cytosolic Trx1 is not fully understood.
In parallel, scientists have started to expand their research into oxidative folding of proteins in clinically relevant strains, such as Mycobacterium (15, 175, 176), Pseudomonas (136, 177, 178), Helicobacter (179, 180), and Vibrio (181, 182). The discovery of Dsb machinery in these clinically relevant strains has resulted in the discovery of potential biopharmaceuticals with the ability to target and inhibit disulfide bond formation in pathogenic strains (183–185). The targeting of Dsb machinery also addresses the need for new approaches to overcome the problem of rapidly developing antibiotic resistance. This new class of antibacterial drug does not necessarily kill bacteria or inhibit their growth but instead combats disease by targeting the pathogen’s capacity to make virulence factors (186, 187).
Contributor Information
Bruno Manta, New England Biolabs, Ipswich, MA 01938.
Dana Boyd, Department of Microbiology and Molecular Genetics, Harvard Medical School, Boston, MA 02115.
Mehmet Berkmen, New England Biolabs, Ipswich, MA 01938.
James M. Slauch, The School of Molecular and Cellular Biology, University of Illinois at Urbana-Champaign, Urbana, IL
Michael Ehrmann, Center for Medical Biotechnology (ZMB), University of Duisburg-Essen, Essen, Germany.
REFERENCES
- 1.Fass D. 2012. Disulfide bonding in protein biophysics. Annu Rev Biophys 41:63–79. [PubMed] [DOI] [PubMed] [Google Scholar]
- 2.Fass D, Thorpe C. 2018. Chemistry and enzymology of disulfide cross-linking in proteins. Chem Rev 118:1169–1198. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sideris DP, Tokatlidis K. 2010. Oxidative protein folding in the mitochondrial intermembrane space. Antioxid Redox Signal 13:1189–1204. [PubMed] [DOI] [PubMed] [Google Scholar]
- 4.Laurindo FR, Pescatore LA, Fernandes DC. 2012. Protein disulfide isomerase in redox cell signaling and homeostasis. Free Radic Biol Med 52:1954–1969. [PubMed] [DOI] [PubMed] [Google Scholar]
- 5.Putker M, Vos HR, Dansen TB. 2014. Intermolecular disulfide-dependent redox signalling. Biochem Soc Trans 42:971–978. [PubMed] [DOI] [PubMed] [Google Scholar]
- 6.Mallick P, Boutz DR, Eisenberg D, Yeates TO. 2002. Genomic evidence that the intracellular proteins of archaeal microbes contain disulfide bonds. Proc Natl Acad Sci U S A 99:9679–9684. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Senkevich TG, White CL, Koonin EV, Moss B. 2002. Complete pathway for protein disulfide bond formation encoded by poxviruses. Proc Natl Acad Sci U S A 99:6667–6672. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nagy P. 2013. Kinetics and mechanisms of thiol-disulfide exchange covering direct substitution and thiol oxidation-mediated pathways. Antioxid Redox Signal 18:1623–1641. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Landeta C, Boyd D, Beckwith J. 2018. Disulfide bond formation in prokaryotes. Nat Microbiol 3:270–280. [PubMed] [DOI] [PubMed] [Google Scholar]
- 10.Depuydt M, Messens J, Collet JF. 2011. How proteins form disulfide bonds. Antioxid Redox Signal 15:49–66. [PubMed] [DOI] [PubMed] [Google Scholar]
- 11.Pedone E, Limauro D, Bartolucci S. 2008. The machinery for oxidative protein folding in thermophiles. Antioxid Redox Signal 10:157–169. [PubMed] [DOI] [PubMed] [Google Scholar]
- 12.Denoncin K, Collet JF. 2013. Disulfide bond formation in the bacterial periplasm: major achievements and challenges ahead. Antioxid Redox Signal 19:63–71. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bocian-Ostrzycka KM, Grzeszczuk MJ, Dziewit L, Jagusztyn-Krynicka EK. 2015. Diversity of the Epsilonproteobacteria Dsb (disulfide bond) systems. Front Microbiol 6:570. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hatahet F, Boyd D, Beckwith J. 2014. Disulfide bond formation in prokaryotes: history, diversity and design. Biochim Biophys Acta 1844:1402–1414. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reardon-Robinson ME, Ton-That H. 2015. Disulfide-bond-forming pathways in Gram-positive bacteria. J Bacteriol 198:746–754. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meyer AJ, Riemer J, Rouhier N. 2018. Oxidative protein folding: state-of-the-art and current avenues of research in plants. New Phytol. 10.1111/nph.15436. [DOI] [PubMed] [Google Scholar]
- 17.Anfinsen CB. 1973. Principles that govern the folding of protein chains. Science 181:223–230. [PubMed] [DOI] [PubMed] [Google Scholar]
- 18.Anfinsen CB, Haber E. 1961. Studies on the reduction and re-formation of protein disulfide bonds. J Biol Chem 236:1361–1363. [PubMed] [PubMed] [Google Scholar]
- 19.Anfinsen CB, Haber E, Sela M, White FH Jr. 1961. The kinetics of formation of native ribonuclease during oxidation of the reduced polypeptide chain. Proc Natl Acad Sci U S A 47:1309–1314. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bardwell JC, McGovern K, Beckwith J. 1991. Identification of a protein required for disulfide bond formation in vivo. Cell 67:581–589. [DOI] [PubMed] [Google Scholar]
- 21.Beckwith J. 2007. What lies beyond Uranus? Preconceptions, ignorance, serendipity and suppressors in the search for biology’s secrets. Genetics 176:733–740. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cranford-Smith T, Huber D. 2018. The way is the goal: how SecA transports proteins across the cytoplasmic membrane in bacteria. FEMS Microbiol Lett 365:fny093. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crane JM, Randall LL. 21 November 2017, posting date. The Sec system: protein export in Escherichia coli. Ecosal Plus 2017. 10.1128/ecosalplus.ESP-0002-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Josefsson LG, Randall LL. 1981. Different exported proteins in E. coli show differences in the temporal mode of processing in vivo. Cell 25:151–157. [DOI] [PubMed] [Google Scholar]
- 25.Steinberg R, Knüpffer L, Origi A, Asti R, Koch HG. 2018. Co-translational protein targeting in bacteria. FEMS Microbiol Lett 365:fny095. [PubMed] [DOI] [PubMed] [Google Scholar]
- 26.Mas G, Hiller S. 2018. Conformational plasticity of molecular chaperones involved in periplasmic and outer membrane protein folding. FEMS Microbiol Lett 365:fny121. [PubMed] [DOI] [PubMed] [Google Scholar]
- 27.Sone M, Kishigami S, Yoshihisa T, Ito K. 1997. Roles of disulfide bonds in bacterial alkaline phosphatase. J Biol Chem 272:6174–6178. [PubMed] [DOI] [PubMed] [Google Scholar]
- 28.Kadokura H, Beckwith J. 2009. Detecting folding intermediates of a protein as it passes through the bacterial translocation channel. Cell 138:1164–1173. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schierle CF, Berkmen M, Huber D, Kumamoto C, Boyd D, Beckwith J. 2003. The DsbA signal sequence directs efficient, cotranslational export of passenger proteins to the Escherichia coli periplasm via the signal recognition particle pathway. J Bacteriol 185:5706–5713. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huber D, Boyd D, Xia Y, Olma MH, Gerstein M, Beckwith J. 2005. Use of thioredoxin as a reporter to identify a subset of Escherichia coli signal sequences that promote signal recognition particle-dependent translocation. J Bacteriol 187:2983–2991. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Atkinson HJ, Babbitt PC. 2009. An atlas of the thioredoxin fold class reveals the complexity of function-enabling adaptations. PLOS Comput Biol 5:e1000541. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martin JL, Bardwell JC, Kuriyan J. 1993. Crystal structure of the DsbA protein required for disulphide bond formation in vivo. Nature 365:464–468. [PubMed] [DOI] [PubMed] [Google Scholar]
- 33.Guddat LW, Bardwell JC, Martin JL. 1998. Crystal structures of reduced and oxidized DsbA: investigation of domain motion and thiolate stabilization. Structure 6:757–767. [DOI] [PubMed] [Google Scholar]
- 34.Schirra HJ, Renner C, Czisch M, Huber-Wunderlich M, Holak TA, Glockshuber R. 1998. Structure of reduced DsbA from Escherichia coli in solution. Biochemistry 37:6263–6276. [PubMed] [DOI] [PubMed] [Google Scholar]
- 35.Frech C, Wunderlich M, Glockshuber R, Schmid FX. 1996. Preferential binding of an unfolded protein to DsbA. EMBO J 15:392–398. [PubMed] [PMC free article] [PubMed] [Google Scholar]
- 36.Guddat LW, Bardwell JC, Zander T, Martin JL. 1997. The uncharged surface features surrounding the active site of Escherichia coli DsbA are conserved and are implicated in peptide binding. Protein Sci 6:1148–1156. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vinci F, Couprie J, Pucci P, Quéméneur E, Moutiez M. 2002. Description of the topographical changes associated to the different stages of the DsbA catalytic cycle. Protein Sci 11:1600–1612. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ondo-Mbele E, Vivès C, Koné A, Serre L. 2005. Intriguing conformation changes associated with the trans/cis isomerization of a prolyl residue in the active site of the DsbA C33A mutant. J Mol Biol 347:555–563. [PubMed] [DOI] [PubMed] [Google Scholar]
- 39.Kurth F, Duprez W, Premkumar L, Schembri MA, Fairlie DP, Martin JL. 2014. Crystal structure of the dithiol oxidase DsbA enzyme from proteus mirabilis bound non-covalently to an active site peptide ligand. J Biol Chem 289:19810–19822. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Flohé L. 2013. The fairytale of the GSSG/GSH redox potential. Biochim Biophys Acta 1830:3139–3142. [PubMed] [DOI] [PubMed] [Google Scholar]
- 41.Mössner E, Huber-Wunderlich M, Rietsch A, Beckwith J, Glockshuber R, Aslund F. 1999. Importance of redox potential for the in vivo function of the cytoplasmic disulfide reductant thioredoxin from Escherichia coli. J Biol Chem 274:25254–25259. [PubMed] [DOI] [PubMed] [Google Scholar]
- 42.Aslund F, Berndt KD, Holmgren A. 1997. Redox potentials of glutaredoxins and other thiol-disulfide oxidoreductases of the thioredoxin superfamily determined by direct protein-protein redox equilibria. J Biol Chem 272:30780–30786. [PubMed] [DOI] [PubMed] [Google Scholar]
- 43.Bessette PH, Aslund F, Beckwith J, Georgiou G. 1999. Efficient folding of proteins with multiple disulfide bonds in the Escherichia coli cytoplasm. Proc Natl Acad Sci U S A 96:13703–13708. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stewart EJ, Aslund F, Beckwith J. 1998. Disulfide bond formation in the Escherichia coli cytoplasm: an in vivo role reversal for the thioredoxins. EMBO J 17:5543–5550. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.García-Santamarina S, Boronat S, Calvo IA, Rodríguez-Gabriel M, Ayté J, Molina H, Hidalgo E. 2013. Is oxidized thioredoxin a major trigger for cysteine oxidation? Clues from a redox proteomics approach. Antioxid Redox Signal 18:1549–1556. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lobstein J, Emrich CA, Jeans C, Faulkner M, Riggs P, Berkmen M. 2012. SHuffle, a novel Escherichia coli protein expression strain capable of correctly folding disulfide bonded proteins in its cytoplasm. Microb Cell Fact 11:56. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nelson JW, Creighton TE. 1994. Reactivity and ionization of the active site cysteine residues of DsbA, a protein required for disulfide bond formation in vivo. Biochemistry 33:5974–5983. [DOI] [PubMed] [Google Scholar]
- 48.Gane PJ, Freedman RB, Warwicker J. 1995. A molecular model for the redox potential difference between thioredoxin and DsbA, based on electrostatics calculations. J Mol Biol 249:376–387. [PubMed] [DOI] [PubMed] [Google Scholar]
- 49.Warwicker J. 1998. Modeling charge interactions and redox properties in DsbA. J Biol Chem 273:2501–2504. [PubMed] [DOI] [PubMed] [Google Scholar]
- 50.Bessette PH, Qiu J, Bardwell JC, Swartz JR, Georgiou G. 2001. Effect of sequences of the active-site dipeptides of DsbA and DsbC on in vivo folding of multidisulfide proteins in Escherichia coli. J Bacteriol 183:980–988. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wunderlich M, Glockshuber R. 1993. Redox properties of protein disulfide isomerase (DsbA) from Escherichia coli. Protein Sci 2:717–726. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wunderlich M, Jaenicke R, Glockshuber R. 1993. The redox properties of protein disulfide isomerase (DsbA) of Escherichia coli result from a tense conformation of its oxidized form. J Mol Biol 233:559–566. [PubMed] [DOI] [PubMed] [Google Scholar]
- 53.Zapun A, Bardwell JC, Creighton TE. 1993. The reactive and destabilizing disulfide bond of DsbA, a protein required for protein disulfide bond formation in vivo. Biochemistry 32:5083–5092. [PubMed] [DOI] [PubMed] [Google Scholar]
- 54.Martin JL. 1995. Thioredoxin: a fold for all reasons. Structure 3:245–250. [DOI] [PubMed] [Google Scholar]
- 55.Moutiez M, Burova TV, Haertlé T, Quéméneur E. 1999. On the non-respect of the thermodynamic cycle by DsbA variants. Protein Sci 8:106–112. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Charbonnier JB, Belin P, Moutiez M, Stura EA, Quéméneur E. 1999. On the role of the cis-proline residue in the active site of DsbA. Protein Sci 8:96–105. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kadokura H, Nichols L II, Beckwith J. 2005. Mutational alterations of the key cis proline residue that cause accumulation of enzymatic reaction intermediates of DsbA, a member of the thioredoxin superfamily. J Bacteriol 187:1519–1522. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kadokura H, Tian H, Zander T, Bardwell JC, Beckwith J. 2004. Snapshots of DsbA in action: detection of proteins in the process of oxidative folding. Science 303:534–537. [PubMed] [DOI] [PubMed] [Google Scholar]
- 59.Ren G, Champion MM, Huntley JF. 2014. Identification of disulfide bond isomerase substrates reveals bacterial virulence factors. Mol Microbiol 94:926–944. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ren G, Stephan D, Xu Z, Zheng Y, Tang D, Harrison RS, Kurz M, Jarrott R, Shouldice SR, Hiniker A, Martin JL, Heras B, Bardwell JC. 2009. Properties of the thioredoxin fold superfamily are modulated by a single amino acid residue. J Biol Chem 284:10150–10159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Belin P, Boquet PL. 1994. The Escherichia coli dsbA gene is partly transcribed from the promoter of a weakly expressed upstream gene. Microbiology 140:3337–3348. [DOI] [PubMed] [Google Scholar]
- 62.Akiyama Y, Kamitani S, Kusukawa N, Ito K. 1992. In vitro catalysis of oxidative folding of disulfide-bonded proteins by the Escherichia coli dsbA ( ppfA) gene product. J Biol Chem 267:22440–22445. [PubMed] [Google Scholar]
- 63.Danese PN, Silhavy TJ. 1997. The sigma(E) and the Cpx signal transduction systems control the synthesis of periplasmic protein-folding enzymes in Escherichia coli. Genes Dev 11:1183–1193. [DOI] [PubMed] [Google Scholar]
- 64.Pogliano J, Lynch AS, Belin D, Lin EC, Beckwith J. 1997. Regulation of Escherichia coli cell envelope proteins involved in protein folding and degradation by the Cpx two-component system. Genes Dev 11:1169–1182. [DOI] [PubMed] [Google Scholar]
- 65.Yohannes E, Barnhart DM, Slonczewski JL. 2004. pH-dependent catabolic protein expression during anaerobic growth of Escherichia coli K-12. J Bacteriol 186:192–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Missiakas D, Georgopoulos C, Raina S. 1993. Identification and characterization of the Escherichia coli gene dsbB, whose product is involved in the formation of disulfide bonds in vivo. Proc Natl Acad Sci U S A 90:7084–7088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stafford SJ, Humphreys DP, Lund PA. 1999. Mutations in dsbA and dsbB, but not dsbC, lead to an enhanced sensitivity of Escherichia coli to Hg2+ and Cd2+. FEMS Microbiol Lett 174:179–184. [DOI] [PubMed] [Google Scholar]
- 68.Hayashi S, Abe M, Kimoto M, Furukawa S, Nakazawa T. 2000. The dsbA- dsbB disulfide bond formation system of Burkholderia cepacia is involved in the production of protease and alkaline phosphatase, motility, metal resistance, and multi-drug resistance. Microbiol Immunol 44:41–50. [DOI] [PubMed] [Google Scholar]
- 69.Yu J, Edwards-Jones B, Neyrolles O, Kroll JS. 2000. Key role for DsbA in cell-to-cell spread of Shigella flexneri, permitting secretion of Ipa proteins into interepithelial protrusions. Infect Immun 68:6449–6456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gonzalez MD, Lichtensteiger CA, Vimr ER. 2001. Adaptation of signature-tagged mutagenesis to Escherichia coli K1 and the infant-rat model of invasive disease. FEMS Microbiol Lett 198:125–128. [DOI] [PubMed] [Google Scholar]
- 71.Peek JA, Taylor RK. 1992. Characterization of a periplasmic thiol:disulfide interchange protein required for the functional maturation of secreted virulence factors of Vibrio cholerae. Proc Natl Acad Sci U S A 89:6210–6214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tomb JF. 1992. A periplasmic protein disulfide oxidoreductase is required for transformation of Haemophilus influenzae Rd. Proc Natl Acad Sci U S A 89:10252–10256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Genevaux P, Bauda P, DuBow MS, Oudega B. 1999. Identification of Tn10 insertions in the dsbA gene affecting Escherichia coli biofilm formation. FEMS Microbiol Lett 173:403–409. [DOI] [PubMed] [Google Scholar]
- 74.Stenson TH, Weiss AA. 2002. DsbA and DsbC are required for secretion of pertussis toxin by Bordetella pertussis. Infect Immun 70:2297–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bouwman CW, Kohli M, Killoran A, Touchie GA, Kadner RJ, Martin NL. 2003. Characterization of SrgA, a Salmonella enterica serovar Typhimurium virulence plasmid-encoded paralogue of the disulfide oxidoreductase DsbA, essential for biogenesis of plasmid-encoded fimbriae. J Bacteriol 185:991–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jacob-Dubuisson F, Pinkner J, Xu Z, Striker R, Padmanhaban A, Hultgren SJ. 1994. PapD chaperone function in pilus biogenesis depends on oxidant and chaperone-like activities of DsbA. Proc Natl Acad Sci U S A 91:11552–11556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xu M, Struck DK, Deaton J, Wang IN, Young R. 2004. A signal-arrest-release sequence mediates export and control of the phage P1 endolysin. Proc Natl Acad Sci U S A 101:6415–6420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Meehan BM, Landeta C, Boyd D, Beckwith J. 2017. The essential cell division protein FtsN contains a critical disulfide bond in a non-essential domain. Mol Microbiol 103:413–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Meehan BM, Landeta C, Boyd D, Beckwith J. 2017. The disulfide bond formation pathway is essential for anaerobic growth of Escherichia coli. J Bacteriol 199:e00120-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Debarbieux L, Beckwith J. 1998. The reductive enzyme thioredoxin 1 acts as an oxidant when it is exported to the Escherichia coli periplasm. Proc Natl Acad Sci U S A 95:10751–10756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chng SS, Dutton RJ, Denoncin K, Vertommen D, Collet JF, Kadokura H, Beckwith J. 2012. Overexpression of the rhodanese PspE, a single cysteine-containing protein, restores disulphide bond formation to an Escherichia coli strain lacking DsbA. Mol Microbiol 85:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bardwell JC, Lee JO, Jander G, Martin N, Belin D, Beckwith J. 1993. A pathway for disulfide bond formation in vivo. Proc Natl Acad Sci U S A 90:1038–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dailey FE, Berg HC. 1993. Mutants in disulfide bond formation that disrupt flagellar assembly in Escherichia coli. Proc Natl Acad Sci U S A 90:1043–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Inaba K, Murakami S, Nakagawa A, Iida H, Kinjo M, Ito K, Suzuki M. 2009. Dynamic nature of disulphide bond formation catalysts revealed by crystal structures of DsbB. EMBO J 28:779–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Inaba K, Takahashi YH, Ito K, Hayashi S. 2006. Critical role of a thiolate-quinone charge transfer complex and its adduct form in de novo disulfide bond generation by DsbB. Proc Natl Acad Sci U S A 103:287–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Malojcić G, Owen RL, Grimshaw JP, Glockshuber R. 2008. Preparation and structure of the charge-transfer intermediate of the transmembrane redox catalyst DsbB. FEBS Lett 582:3301–3307. [DOI] [PubMed] [Google Scholar]
- 87.Dutton RJ, Boyd D, Berkmen M, Beckwith J. 2008. Bacterial species exhibit diversity in their mechanisms and capacity for protein disulfide bond formation. Proc Natl Acad Sci U S A 105:11933–11938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ke N, Landeta C, Wang X, Boyd D, Eser M, Beckwith J. 2018. Identification of the thioredoxin partner of VKOR in mycobacterial disulfide bond formation. J Bacteriol 200:e00137-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li W, Schulman S, Dutton RJ, Boyd D, Beckwith J, Rapoport TA. 2010. Structure of a bacterial homologue of vitamin K epoxide reductase. Nature 463:507–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang X, Dutton RJ, Beckwith J, Boyd D. 2011. Membrane topology and mutational analysis of Mycobacterium tuberculosis VKOR, a protein involved in disulfide bond formation and a homologue of human vitamin K epoxide reductase. Antioxid Redox Signal 14:1413–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kurth F, Rimmer K, Premkumar L, Mohanty B, Duprez W, Halili MA, Shouldice SR, Heras B, Fairlie DP, Scanlon MJ, Martin JL. 2013. Comparative sequence, structure and redox analyses of Klebsiella pneumoniae DsbA show that anti-virulence target DsbA enzymes fall into distinct classes. PLoS One 8:e80210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.McMahon RM, Premkumar L, Martin JL. 2014. Four structural subclasses of the antivirulence drug target disulfide oxidoreductase DsbA provide a platform for design of subclass-specific inhibitors. Biochim Biophys Acta 1844:1391–1401. [DOI] [PubMed] [Google Scholar]
- 93.Grimshaw JP, Stirnimann CU, Brozzo MS, Malojcic G, Grütter MG, Capitani G, Glockshuber R. 2008. DsbL and DsbI form a specific dithiol oxidase system for periplasmic arylsulfate sulfotransferase in uropathogenic Escherichia coli. J Mol Biol 380:667–680. [DOI] [PubMed] [Google Scholar]
- 94.Narzi D, Siu SW, Stirnimann CU, Grimshaw JP, Glockshuber R, Capitani G, Böckmann RA. 2008. Evidence for proton shuffling in a thioredoxin-like protein during catalysis. J Mol Biol 382:978–986. [DOI] [PubMed] [Google Scholar]
- 95.Lin D, Kim B, Slauch JM. 2009. DsbL and DsbI contribute to periplasmic disulfide bond formation in Salmonella enterica serovar Typhimurium. Microbiology 155:4014–4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dwyer RS, Malinverni JC, Boyd D, Beckwith J, Silhavy TJ. 2014. Folding LacZ in the periplasm of Escherichia coli. J Bacteriol 196:3343–3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Alksne LE, Keeney D, Rasmussen BA. 1995. A mutation in either dsbA or dsbB, a gene encoding a component of a periplasmic disulfide bond-catalyzing system, is required for high-level expression of the Bacteroides fragilis metallo-beta-lactamase, CcrA, in Escherichia coli. J Bacteriol 177:462–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Missiakas D, Georgopoulos C, Raina S. 1994. The Escherichia coli dsbC ( xprA) gene encodes a periplasmic protein involved in disulfide bond formation. EMBO J 13:2013–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shevchik VE, Condemine G, Robert-Baudouy J. 1994. Characterization of DsbC, a periplasmic protein of Erwinia chrysanthemi and Escherichia coli with disulfide isomerase activity. EMBO J 13:2007–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Berkmen M, Boyd D, Beckwith J. 2005. The nonconsecutive disulfide bond of Escherichia coli phytase (AppA) renders it dependent on the protein-disulfide isomerase, DsbC. J Biol Chem 280:11387–11394. [DOI] [PubMed] [Google Scholar]
- 101.Hiniker A, Bardwell JC. 2004. In vivo substrate specificity of periplasmic disulfide oxidoreductases. J Biol Chem 279:12967–12973. [PubMed] [DOI] [PubMed] [Google Scholar]
- 102.Joly JC, Swartz JR. 1997. In vitro and in vivo redox states of the Escherichia coli periplasmic oxidoreductases DsbA and DsbC. Biochemistry 36:10067–10072. [PubMed] [DOI] [PubMed] [Google Scholar]
- 103.Rietsch A, Bessette P, Georgiou G, Beckwith J. 1997. Reduction of the periplasmic disulfide bond isomerase, DsbC, occurs by passage of electrons from cytoplasmic thioredoxin. J Bacteriol 179:6602–6608. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sun XX, Wang CC. 2000. The N-terminal sequence (residues 1-65) is essential for dimerization, activities, and peptide binding of Escherichia coli DsbC. J Biol Chem 275:22743–22749. [PubMed] [DOI] [PubMed] [Google Scholar]
- 105.McCarthy AA, Haebel PW, Törrönen A, Rybin V, Baker EN, Metcalf P. 2000. Crystal structure of the protein disulfide bond isomerase, DsbC, from Escherichia coli. Nat Struct Biol 7:196–199. [PubMed] [DOI] [PubMed] [Google Scholar]
- 106.Zhang M, Monzingo AF, Segatori L, Georgiou G, Robertus JD. 2004. Structure of DsbC from Haemophilus influenzae. Acta Crystallogr D Biol Crystallogr 60:1512–1518. [PubMed] [DOI] [PubMed] [Google Scholar]
- 107.Liu X, Wang CC. 2001. Disulfide-dependent folding and export of Escherichia coli DsbC. J Biol Chem 276:1146–1151. [PubMed] [DOI] [PubMed] [Google Scholar]
- 108.Darby NJ, Raina S, Creighton TE. 1998. Contributions of substrate binding to the catalytic activity of DsbC. Biochemistry 37:783–791. [PubMed] [DOI] [PubMed] [Google Scholar]
- 109.Chen J, Song JL, Zhang S, Wang Y, Cui DF, Wang CC. 1999. Chaperone activity of DsbC. J Biol Chem 274:19601–19605. [PubMed] [DOI] [PubMed] [Google Scholar]
- 110.Zhao Z, Peng Y, Hao SF, Zeng ZH, Wang CC. 2003. Dimerization by domain hybridization bestows chaperone and isomerase activities. J Biol Chem 278:43292–43298. [PubMed] [DOI] [PubMed] [Google Scholar]
- 111.Shouldice SR, Cho SH, Boyd D, Heras B, Eser M, Beckwith J, Riggs P, Martin JL, Berkmen M. 2010. In vivo oxidative protein folding can be facilitated by oxidation-reduction cycling. Mol Microbiol 75:13–28. [PubMed] [DOI] [PubMed] [Google Scholar]
- 112.Collet JF, Bardwell JC. 2002. Oxidative protein folding in bacteria. Mol Microbiol 44:1–8. [DOI] [PubMed] [Google Scholar]
- 113.Walker KW, Gilbert HF. 1997. Scanning and escape during protein-disulfide isomerase-assisted protein folding. J Biol Chem 272:8845–8848. [PubMed] [DOI] [PubMed] [Google Scholar]
- 114.Heppner DE, Janssen-Heininger YMW, van der Vliet A. 2017. The role of sulfenic acids in cellular redox signaling: reconciling chemical kinetics and molecular detection strategies. Arch Biochem Biophys 616:40–46. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Claiborne A, Miller H, Parsonage D, Ross RP. 1993. Protein-sulfenic acid stabilization and function in enzyme catalysis and gene regulation. FASEB J 7:1483–1490. [PubMed] [DOI] [PubMed] [Google Scholar]
- 116.Depuydt M, Leonard SE, Vertommen D, Denoncin K, Morsomme P, Wahni K, Messens J, Carroll KS, Collet JF. 2009. A periplasmic reducing system protects single cysteine residues from oxidation. Science 326:1109–1111. [PubMed] [DOI] [PubMed] [Google Scholar]
- 117.Denoncin K, Vertommen D, Arts IS, Goemans CV, Rahuel-Clermont S, Messens J, Collet JF. 2014. A new role for Escherichia coli DsbC protein in protection against oxidative stress. J Biol Chem 289:12356–12364. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Leverrier P, Declercq JP, Denoncin K, Vertommen D, Hiniker A, Cho SH, Collet JF. 2011. Crystal structure of the outer membrane protein RcsF, a new substrate for the periplasmic protein-disulfide isomerase DsbC. J Biol Chem 286:16734–16742. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Denoncin K, Vertommen D, Paek E, Collet JF. 2010. The protein-disulfide isomerase DsbC cooperates with SurA and DsbA in the assembly of the essential β-barrel protein LptD. J Biol Chem 285:29425–29433. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zapun A, Missiakas D, Raina S, Creighton TE. 1995. Structural and functional characterization of DsbC, a protein involved in disulfide bond formation in Escherichia coli. Biochemistry 34:5075–5089. [PubMed] [DOI] [PubMed] [Google Scholar]
- 121.Inaba K, Murakami S, Suzuki M, Nakagawa A, Yamashita E, Okada K, Ito K. 2006. Crystal structure of the DsbB-DsbA complex reveals a mechanism of disulfide bond generation. Cell 127:789–801. [PubMed] [DOI] [PubMed] [Google Scholar]
- 122.Inaba K, Ito K. 2008. Structure and mechanisms of the DsbB-DsbA disulfide bond generation machine. Biochim Biophys Acta 1783:520–529. [PubMed] [DOI] [PubMed] [Google Scholar]
- 123.Bader MW, Hiniker A, Regeimbal J, Goldstone D, Haebel PW, Riemer J, Metcalf P, Bardwell JC. 2001. Turning a disulfide isomerase into an oxidase: DsbC mutants that imitate DsbA. EMBO J 20:1555–1562. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Segatori L, Paukstelis PJ, Gilbert HF, Georgiou G. 2004. Engineered DsbC chimeras catalyze both protein oxidation and disulfide-bond isomerization in Escherichia coli: reconciling two competing pathways. Proc Natl Acad Sci U S A 101:10018–10023. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pan JL, Sliskovic I, Bardwell JC. 2008. Mutants in DsbB that appear to redirect oxidation through the disulfide isomerization pathway. J Mol Biol 377:1433–1442. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Denoncin K, Nicolaes V, Cho SH, Leverrier P, Collet JF. 2013. Protein disulfide bond formation in the periplasm: determination of the in vivo redox state of cysteine residues. Methods Mol Biol 966:325–336. [PubMed] [DOI] [PubMed] [Google Scholar]
- 127.Kishigami S, Akiyama Y, Ito K. 1995. Redox states of DsbA in the periplasm of Escherichia coli. FEBS Lett 364:55–58. [DOI] [PubMed] [Google Scholar]
- 128.Delaunay-Moisan A, Ponsero A, Toledano MB. 2017. Reexamining the function of glutathione in oxidative protein folding and secretion. Antioxid Redox Signal 27:1178–1199. [PubMed] [DOI] [PubMed] [Google Scholar]
- 129.Smirnova G, Muzyka N, Oktyabrsky O. 2012. Transmembrane glutathione cycling in growing Escherichia coli cells. Microbiol Res 167:166–172. [PubMed] [DOI] [PubMed] [Google Scholar]
- 130.Eser M, Masip L, Kadokura H, Georgiou G, Beckwith J. 2009. Disulfide bond formation by exported glutaredoxin indicates glutathione’s presence in the E. coli periplasm. Proc Natl Acad Sci U S A 106:1572–1577. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Rozhkova A, Glockshuber R. 2007. Kinetics of the intramolecular disulfide exchange between the periplasmic domains of DsbD. J Mol Biol 367:1162–1170. [PubMed] [DOI] [PubMed] [Google Scholar]
- 132.Rozhkova A, Stirnimann CU, Frei P, Grauschopf U, Brunisholz R, Grütter MG, Capitani G, Glockshuber R. 2004. Structural basis and kinetics of inter- and intramolecular disulfide exchange in the redox catalyst DsbD. EMBO J 23:1709–1719. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wunderlich M, Glockshuber R. 1993. In vivo control of redox potential during protein folding catalyzed by bacterial protein disulfide-isomerase (DsbA). J Biol Chem 268:24547–24550. [PubMed] [PubMed] [Google Scholar]
- 134.Wang X, Li M, Liu L, Mou R, Zhang X, Bai Y, Xu H, Qiao M. 2012. DsbM, a novel disulfide oxidoreductase affects aminoglycoside resistance in Pseudomonas aeruginosa by OxyR-regulated response. J Microbiol 50:932–938. [PubMed] [DOI] [PubMed] [Google Scholar]
- 135.Li M, Guan X, Wang X, Xu H, Bai Y, Zhang X, Qiao M. 2014. DsbM affects aminoglycoside resistance in Pseudomonas aeruginosa by the reduction of OxyR. FEMS Microbiol Lett 352:184–189. [PubMed] [DOI] [PubMed] [Google Scholar]
- 136.Jo I, Park N, Chung IY, Cho YH, Ha NC. 2016. Crystal structures of the disulfide reductase DsbM from Pseudomonas aeruginosa. Acta Crystallogr D Struct Biol 72:1100–1109. [PubMed] [DOI] [PubMed] [Google Scholar]
- 137.Dartigalongue C, Missiakas D, Raina S. 2001. Characterization of the Escherichia coli sigma E regulon. J Biol Chem 276:20866–20875. [PubMed] [DOI] [PubMed] [Google Scholar]
- 138.Zhan X, Gao J, Jain C, Cieslewicz MJ, Swartz JR, Georgiou G. 2004. Genetic analysis of disulfide isomerization in Escherichia coli: expression of DsbC is modulated by RNase E-dependent mRNA processing. J Bacteriol 186:654–660. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Andersen CL, Matthey-Dupraz A, Missiakas D, Raina S. 1997. A new Escherichia coli gene, dsbG, encodes a periplasmic protein involved in disulphide bond formation, required for recycling DsbA/DsbB and DsbC redox proteins. Mol Microbiol 26:121–132. [PubMed] [DOI] [PubMed] [Google Scholar]
- 140.Heras B, Edeling MA, Schirra HJ, Raina S, Martin JL. 2004. Crystal structures of the DsbG disulfide isomerase reveal an unstable disulfide. Proc Natl Acad Sci U S A 101:8876–8881. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Shao F, Bader MW, Jakob U, Bardwell JC. 2000. DsbG, a protein disulfide isomerase with chaperone activity. J Biol Chem 275:13349–13352. [PubMed] [DOI] [PubMed] [Google Scholar]
- 142.Bessette PH, Cotto JJ, Gilbert HF, Georgiou G. 1999. In vivo and in vitro function of the Escherichia coli periplasmic cysteine oxidoreductase DsbG. J Biol Chem 274:7784–7792. [PubMed] [DOI] [PubMed] [Google Scholar]
- 143.Zheng M, Wang X, Doan B, Lewis KA, Schneider TD, Storz G. 2001. Computation-directed identification of OxyR DNA binding sites in Escherichia coli. J Bacteriol 183:4571–4579. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kouidmi I, Alvarez L, Collet JF, Cava F, Paradis-Bleau C. 2018. The chaperone activities of DsbG and Spy restore peptidoglycan biosynthesis in the elyC mutant by preventing envelope protein aggregation. J Bacteriol 200:e00245-18. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Stull F, Betton JM, Bardwell JCA. 30 April 2018, posting date. Periplasmic chaperones and prolyl isomerases. Ecosal Plus 2018 10.1128/ecosalplus.ESP-0005-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Nakamoto H, Bardwell JC. 2004. Catalysis of disulfide bond formation and isomerization in the Escherichia coli periplasm. Biochim Biophys Acta 1694:111–119. [PubMed] [DOI] [PubMed] [Google Scholar]
- 147.Cho SH, Collet JF. 2013. Many roles of the bacterial envelope reducing pathways. Antioxid Redox Signal 18:1690–1698. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Porat A, Cho SH, Beckwith J. 2004. The unusual transmembrane electron transporter DsbD and its homologues: a bacterial family of disulfide reductases. Res Microbiol 155:617–622. [PubMed] [DOI] [PubMed] [Google Scholar]
- 149.Goulding CW, Sawaya MR, Parseghian A, Lim V, Eisenberg D, Missiakas D. 2002. Thiol-disulfide exchange in an immunoglobulin-like fold: structure of the N-terminal domain of DsbD. Biochemistry 41:6920–6927. [PubMed] [DOI] [PubMed] [Google Scholar]
- 150.Chung J, Chen T, Missiakas D. 2000. Transfer of electrons across the cytoplasmic membrane by DsbD, a membrane protein involved in thiol-disulphide exchange and protein folding in the bacterial periplasm. Mol Microbiol 35:1099–1109. [PubMed] [DOI] [PubMed] [Google Scholar]
- 151.Haebel PW, Goldstone D, Katzen F, Beckwith J, Metcalf P. 2002. The disulfide bond isomerase DsbC is activated by an immunoglobulin-fold thiol oxidoreductase: crystal structure of the DsbC-DsbDalpha complex. EMBO J 21:4774–4784. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Katzen F, Beckwith J. 2000. Transmembrane electron transfer by the membrane protein DsbD occurs via a disulfide bond cascade. Cell 103:769–779. [DOI] [PubMed] [Google Scholar]
- 153.Krupp R, Chan C, Missiakas D. 2001. DsbD-catalyzed transport of electrons across the membrane of Escherichia coli. J Biol Chem 276:3696–3701. [PubMed] [DOI] [PubMed] [Google Scholar]
- 154.Cho SH, Beckwith J. 2009. Two snapshots of electron transport across the membrane: insights into the structure and function of DsbD. J Biol Chem 284:11416–11424. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Cho SH, Porat A, Ye J, Beckwith J. 2007. Redox-active cysteines of a membrane electron transporter DsbD show dual compartment accessibility. EMBO J 26:3509–3520. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Katzen F, Deshmukh M, Daldal F, Beckwith J. 2002. Evolutionary domain fusion expanded the substrate specificity of the transmembrane electron transporter DsbD. EMBO J 21:3960–3969. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kim JH, Kim SJ, Jeong DG, Son JH, Ryu SE. 2003. Crystal structure of DsbDgamma reveals the mechanism of redox potential shift and substrate specificity(1). FEBS Lett 543:164–169. [DOI] [PubMed] [Google Scholar]
- 158.Rozhkova A, Glockshuber R. 2008. Thermodynamic aspects of DsbD-mediated electron transport. J Mol Biol 380:783–788. [PubMed] [DOI] [PubMed] [Google Scholar]
- 159.Hiniker A, Vertommen D, Bardwell JC, Collet JF. 2006. Evidence for conformational changes within DsbD: possible role for membrane-embedded proline residues. J Bacteriol 188:7317–7320. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Cho SH, Beckwith J. 2006. Mutations of the membrane-bound disulfide reductase DsbD that block electron transfer steps from cytoplasm to periplasm in Escherichia coli. J Bacteriol 188:5066–5076. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Goldstone D, Haebel PW, Katzen F, Bader MW, Bardwell JC, Beckwith J, Metcalf P. 2001. DsbC activation by the N-terminal domain of DsbD. Proc Natl Acad Sci U S A 98:9551–9556. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Haebel PW, Wichman S, Goldstone D, Metcalf P. 2001. Crystallization and initial crystallographic analysis of the disulfide bond isomerase DsbC in complex with the alpha domain of the electron transporter DsbD. J Struct Biol 136:162–166. [PubMed] [DOI] [PubMed] [Google Scholar]
- 163.Stirnimann CU, Rozhkova A, Grauschopf U, Grütter MG, Glockshuber R, Capitani G. 2005. Structural basis and kinetics of DsbD-dependent cytochrome c maturation. Structure 13:985–993. [DOI] [PubMed] [Google Scholar]
- 164.Crooke H, Cole J. 1995. The biogenesis of c-type cytochromes in Escherichia coli requires a membrane-bound protein, DipZ, with a protein disulphide isomerase-like domain. Mol Microbiol 15:1139–1150. [DOI] [PubMed] [Google Scholar]
- 165.Cho SH, Parsonage D, Thurston C, Dutton RJ, Poole LB, Collet JF, Beckwith J. 2012. A new family of membrane electron transporters and its substrates, including a new cell envelope peroxiredoxin, reveal a broadened reductive capacity of the oxidative bacterial cell envelope. MBio 3:e00291-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Zhang L, Chou CP, Moo-Young M. 2011. Disulfide bond formation and its impact on the biological activity and stability of recombinant therapeutic proteins produced by Escherichia coli expression system. Biotechnol Adv 29:923–929. [DOI] [PubMed] [Google Scholar]
- 167.Wang X, Das TK, Singh SK, Kumar S. 2009. Potential aggregation prone regions in biotherapeutics: a survey of commercial monoclonal antibodies. MAbs 1:254–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Lee YJ, Lee DH, Jeong KJ. 2014. Enhanced production of human full-length immunoglobulin G1 in the periplasm of Escherichia coli. Appl Microbiol Biotechnol 98:1237–1246. [DOI] [PubMed] [Google Scholar]
- 169.Zhou Y, Liu P, Gan Y, Sandoval W, Katakam AK, Reichelt M, Rangell L, Reilly D. 2016. Enhancing full-length antibody production by signal peptide engineering. Microb Cell Fact 15:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Ritz D, Lim J, Reynolds CM, Poole LB, Beckwith J. 2001. Conversion of a peroxiredoxin into a disulfide reductase by a triplet repeat expansion. Science 294:158–160. [DOI] [PubMed] [Google Scholar]
- 171.Robinson MP, Ke N, Lobstein J, Peterson C, Szkodny A, Mansell TJ, Tuckey C, Riggs PD, Colussi PA, Noren CJ, Taron CH, DeLisa MP, Berkmen M. 2015. Efficient expression of full-length antibodies in the cytoplasm of engineered bacteria. Nat Commun 6:8072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Reddy PT, Brinson RG, Hoopes JT, McClung C, Ke N, Kashi L, Berkmen M, Kelman Z. 2018. Platform development for expression and purification of stable isotope labeled monoclonal antibodies in Escherichia coli. MAbs 10:992–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Hatahet F, Ruddock LW. 2013. Topological plasticity of enzymes involved in disulfide bond formation allows catalysis in either the periplasm or the cytoplasm. J Mol Biol 425:3268–3276. [DOI] [PubMed] [Google Scholar]
- 174.Mizrachi D, Robinson MP, Ren G, Ke N, Berkmen M, DeLisa MP. 2017. A water-soluble DsbB variant that catalyzes disulfide-bond formation in vivo. Nat Chem Biol 13:1022–1028. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 175.Chim N, Harmston CA, Guzman DJ, Goulding CW. 2013. Structural and biochemical characterization of the essential DsbA-like disulfide bond forming protein from Mycobacterium tuberculosis. BMC Struct Biol 13:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Ke N, Landeta C, Wang X, Boyd D, Eser M, Beckwith J. 2018. Identification of the thioredoxin partner of vitamin K epoxide reductase in mycobacterial disulfide bond formation. J Bacteriol 200:e00137-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Urban A, Leipelt M, Eggert T, Jaeger KE. 2001. DsbA and DsbC affect extracellular enzyme formation in Pseudomonas aeruginosa. J Bacteriol 183:587–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Arts IS, Ball G, Leverrier P, Garvis S, Nicolaes V, Vertommen D, Ize B, Tamu Dufe V, Messens J, Voulhoux R, Collet JF. 2013. Dissecting the machinery that introduces disulfide bonds in Pseudomonas aeruginosa. MBio 4:e00912-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Lester J, Kichler S, Oickle B, Fairweather S, Oberc A, Chahal J, Ratnayake D, Creuzenet C. 2015. Characterization of Helicobacter pylori HP0231 (DsbK): role in disulfide bond formation, redox homeostasis and production of Helicobacter cystein-rich protein HcpE. Mol Microbiol 96:110–133. [DOI] [PubMed] [Google Scholar]
- 180.Grzeszczuk MJ, Bąk A, Banaś AM, Urbanowicz P, Dunin-Horkawicz S, Gieldon A, Czaplewski C, Liwo A, Jagusztyn-Krynicka EK. 2018. Impact of selected amino acids of HP0377 (Helicobacter pylori thiol oxidoreductase) on its functioning as a CcmG (cytochrome c maturation) protein and Dsb (disulfide bond) isomerase. PLoS One 13:e0195358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Fengler VH, Boritsch EC, Tutz S, Seper A, Ebner H, Roier S, Schild S, Reidl J. 2012. Disulfide bond formation and ToxR activity in Vibrio cholerae. PLoS One 7:e47756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Yu J, McLaughlin S, Freedman RB, Hirst TR. 1993. Cloning and active site mutagenesis of Vibrio cholerae DsbA, a periplasmic enzyme that catalyzes disulfide bond formation. J Biol Chem 268:4326–4330. [PubMed] [Google Scholar]
- 183.Dutton RJ, Wayman A, Wei JR, Rubin EJ, Beckwith J, Boyd D. 2010. Inhibition of bacterial disulfide bond formation by the anticoagulant warfarin. Proc Natl Acad Sci U S A 107:297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Totsika M, Vagenas D, Paxman JJ, Wang G, Dhouib R, Sharma P, Martin JL, Scanlon MJ, Heras B. 2018. Inhibition of diverse DsbA enzymes in multi-DsbA encoding pathogens. Antioxid Redox Signal 29:653–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Mohanty B, Rimmer K, McMahon RM, Headey SJ, Vazirani M, Shouldice SR, Coinçon M, Tay S, Morton CJ, Simpson JS, Martin JL, Scanlon MJ. 2017. Fragment library screening identifies hits that bind to the non-catalytic surface of Pseudomonas aeruginosa DsbA1. PLoS One 12:e0173436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Heras B, Scanlon MJ, Martin JL. 2015. Targeting virulence not viability in the search for future antibacterials. Br J Clin Pharmacol 79:208–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Heras B, Shouldice SR, Totsika M, Scanlon MJ, Schembri MA, Martin JL. 2009. DSB proteins and bacterial pathogenicity. Nat Rev Microbiol 7:215–225. [DOI] [PubMed] [Google Scholar]
- 188.Jander G, Martin NL, Beckwith J. 1994. Two cysteines in each periplasmic domain of the membrane protein DsbB are required for its function in protein disulfide bond formation. EMBO J 13:5121–5127. [DOI] [PMC free article] [PubMed] [Google Scholar]