

Abstract
DNA from organelles called mitochondria is not inherited from the father. But mitochondrial RNAs that sense paternal diet and mitochondrial quality are delivered from sperm to egg, affecting offspring metabolism.
DNA found in organelles called mitochondria (mtDNA) is inherited exclusively from the mother, owing to mechanisms that include the elimination of mtDNA from sperm before fertilization1. However, various types of RNA carried by sperm can respond to environmental conditions and influence the traits (phenotypes) of offspring, including whether or not they develop metabolic disorders2–5. On page 720, Tomar et al.6 find that, in both mice and humans, specific mitochondrial RNAs in sperm act as sensors of paternal diet and mitochondrial quality. This potentially regulates offspring metabolism by affecting embryonic development.
Sperm cells first develop in the testes, then travel through a long, coiled tube called the epididymis, where they mature and are stored. The authors set out to dissect the distinct contributions of testicular and epididymal environments to the health of offspring. They did this by feeding two groups of male mice meticulously timed high-fat diets designed to emulate eating habits that produce obesity in humans. In one group, the mice were fed a high-fat diet for two weeks before mating with females, which meant that their epididymal sperm had been influenced by the diet. Around 30% of the male (but not female) offspring of these mice exhibited intolerance to glucose and resistance to insulin – traits that are associated with metabolic conditions such as type 2 diabetes in humans.
In the second group, males were fed a high-fat diet for two weeks, then allowed to empty mature sperm before being fed a normal diet for four weeks. This meant that, at the time offspring were conceived, only the fathers’ testicular sperm had been influenced by the high-fat diet. The offspring of these animals showed no signs of metabolic disorders. This highlights the sensitivity of epididymal sperm to the father’s diet, and its impact on offspring health.
To investigate the underlying mechanisms, the team analysed the small non-coding RNAs (sncRNAs) in sperm. These short RNA transcripts, which are not translated into proteins, are crucial for mediating the inheritance of phenotypes through epigenetic means – that is, without changes to the DNA sequence5. Using a conventional high-throughput sequencing technology called sncRNA-seq to identify and quantify small RNA molecules in a sample, the authors found that around one-quarter of mitochondrial sncRNAs were greatly upregulated in mice that were fed a high-fat diet. Among these mitochondrial small RNAs, those derived from transfer RNA (mt-tsRNA) or ribosomal RNA (mt-rsRNA) were especially affected. By contrast, tsRNAs and rsRNAs that were genomic (derived from the nuclear DNA rather than mitochondrial DNA) showed relatively small changes. Importantly, sperm from mice on a two-week high-fat diet followed by a four-week recovery on a normal diet showed minimal changes to sncRNAs.
Tomar and colleagues also looked at data derived from humans. They found that levels of mt-tsRNAs (but not genomic tsRNAs) in sperm correlate with paternal body mass index (BMI), and that paternal overweight is associated with metabolic disorders in their children. These data suggest that mitochondrial s ncRNAs in sperm act as molecular sensors that respond to metabolic challenges in both mice and humans. This might be because transcription of genomic DNA is inactive in mature sperm, whereas transcription of mtDNA remains active5.
What could be behind these observations? A plausible scenario is that a high-fat diet induces mitochondrial damage or dysfunction in sperm, resulting in compensatory increases to transcription of mtDNA so that the sperm cell can maintain energy and motility. However, this would also result in an abnormal accumulation of mitochondrial tRNA, ribosomal RNA and their fragments (mt-tsRNAs, mt-rsRNAs), disrupting the ‘sperm RNA code’5, which carries hereditary information that is crucial for the control of phenotypes in offspring. In other words, this scenario would suggest a trade-off between maintaining mitochondrial function and preserving the accuracy of sperm-borne RNA information. This idea aligns with previous reports that an acute high-sugar diet in humans can simultaneously increase sperm motility (possibly owing to enhanced mitochondrial activity) and elevate expression of mitochondrial sncRNA in sperm7,8.
Disturbing mitochondrial function through genetic means mirrored the effect of a high-fat diet. The authors found that when male mice carried mutations in genes (such as Mrpl23 and Ndufb8) that are important for mitochondrial function, they had high levels of mt-tsRNAs from the 5′ end (but not from the 3′ end) of mt-tRNAs in their sperm and gave rise to male offspring with metabolic disorders. This finding supports the idea that the signature of mt-tsRNAs acts as a ‘molecular readout’ of altered mitochondrial quality or function. Although sperm do not transmit mutated mtDNA to offspring, alterations in mitochondrial function – which are possibly signalled through mt-tRNAs or mt-tsRNAs, or both, and potentially through other information-carrying molecules – can profoundly affect offspring health.
To further investigate the relationship between sperm mitochondrial RNAs and offspring health, the team created hybrid mouse embryos through in vitro fertilization using sperm from male mice fed on a high-fat or low-fat diet, and oocytes (egg cells) from female mice belonging to a different genetic line. In this way, they could track the parental origin of mt-RNAs by looking at minor genetic variations, known as single nucleotide polymorphisms, that differ between the parents.
The authors used RNA sequencing to profile the cellular RNA transcripts (the transcriptome) in hybrid embryos at the two-cell stage. An analytical approach called principal component analysis, which enables plotting of the differences in gene expression between samples, revealed that embryos had different mitochondrial (but not genomic) transcriptomic profiles that were associated with the diet of the father. Around 30% of male embryos fathered by mice that were fed a high-fat diet had levels of paternal mt-tRNAs that were around 12 times higher than those in embryos from fathers on a low-fat diet. The rest of the male embryos and all female embryos had paternal mt-tRNAs that were around two to three times as high. Considerable transcriptional reprogramming was observed in the same 30% of male embryos, particularly in genes involved in oxidative phosphorylation – the pathway by which mitochondria produce the main molecule, ATP, that cells use for energy.
The fact that the 30% of male embryos with very high levels of mt-tRNAs coincided with the 30% of male offspring that had metabolic disorders is intriguing, because it hints at a connection between increased paternal mt-tRNAs (and possibly mt-tsRNAs) in embryos and metabolic outcomes in the offspring (Fig. 1).
Figure 1 |. RNAs in the mitochondria of sperm sense the father’s diet and influence offspring health.
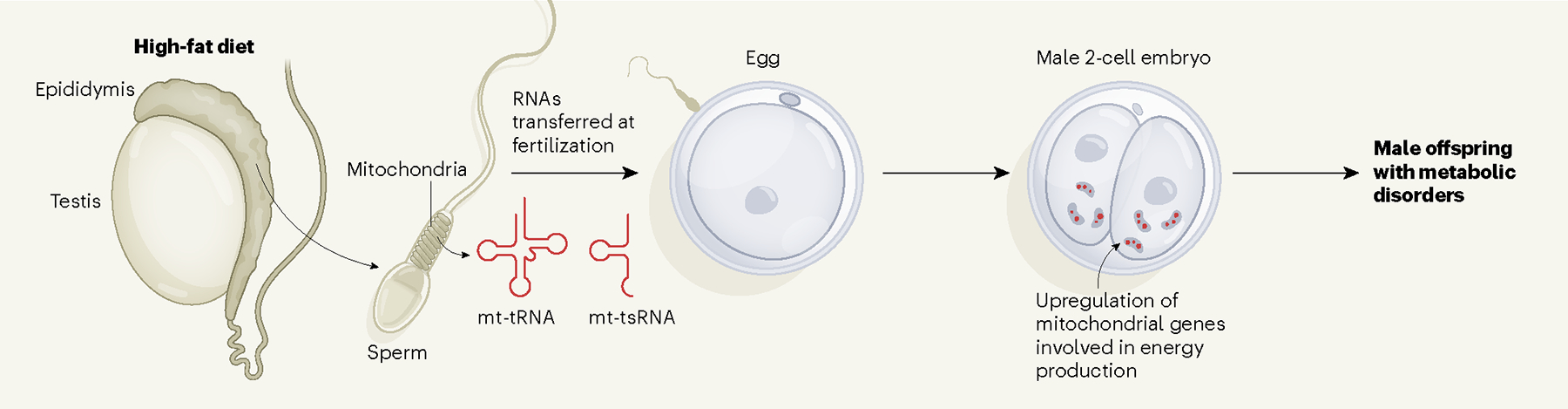
Sperm are made in the testes and mature in a structure called the epididymis. Tomar et al.6 found that feeding male mice a high-fat diet increases levels of small non-coding RNAs (sncRNAs) in the mitochondria of their epididymal sperm, possibly as a response to mitochondrial dysfunction. During fertilization, mitochondrial sncRNAs (mt-tRNAs and mt-tsRNAs) are delivered to the egg, resulting in a portion of embryos having high levels of these sncRNAs and showing an upregulation of genes involved in mitochondrial energy production. A similar proportion of male offspring of males that were fed a high-fat diet exhibited metabolic disorders, suggesting a link between the profiles of mitochondrial RNAs in the father’s sperm and the metabolic traits of the son. It is possible that changes in gene expression induced by mitochondrial sncRNAs could affect the differentiation of cells in the early embryo, influencing the growth ratio between the fetus and the placenta.
Despite the advances described in this study, many questions remain. In particular, how do RNAs from sperm affect embryonic development? Interestingly, oxidative phosphorylation and the functional state of mitochondria affect cell differentiation9. Therefore, altering either of these factors might bias embryonic-fate decisions. Such a decision might be whether a cell differentiates to become part of the inner cell mass (which forms the embryo) or part of the trophectoderm (which becomes supporting placental tissue). An imbalance in these cell types can lead to disproportional development of the embryo’s body compared with its placenta, potentially resulting in a birthweight that is either too high or too low – a risk factor for adult-onset metabolic disorders10.
The mechanisms that govern transcription in the mitochondria of sperm, and the enzymes needed for production of mitochondrial sncRNA, remain underexplored. Crucially, mt-tsRNAs and other sncRNAs harbour biochemical modifications that bias sequencing results gained from conventional sncRNA-seq11. Methods that have been developed in the past few years can bypass this challenge and provide deeper insights into the landscape of sncRNAs in sperm12.
The next challenge lies in deciphering the molecular mechanisms by which mt-tRNAs and mt-tsRNAs in sperm influence embryonic development. It is possible that this occurs not only through the typical means by which sncRNAs regulate gene expression – that is, by binding to RNA or DNA that has a complementary nucleic acid sequence – but also through dynamic changes in their 3D structures, and in their functional interactions with unexpected binding partners, such as proteins and metabolite molecules13,14. A deeper understanding of the sperm RNA code5 can help scientists and clinicians to make informed decisions that can predict, or even mitigate, the risk of metabolic disorders in offspring amid rising levels of obesity and diabetes worldwide.
Footnotes
The authors declare no competing interests.
References
- 1.Lee W et al. Nature Genet. 55, 1632–1639 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen Q et al. Science 351, 397–400 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Sharma U et al. Science 351, 391–396 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Y et al. Nature Cell Biol. 20, 535–540 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Y, Shi J, Rassoulzadegan M, Tuorto F & Chen Q Nature Rev. Endocrinol. 15, 489–498 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tomar A et al. Nature 630, 720–727 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nätt D et al. PLoS Biol. 17, e3000559 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ramesh R et al. Antioxid. Redox Signal. 38, 1167–1183 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang J et al. Cell Metab. 27, 332–338 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Fleming TP et al. Lancet 391, 1842–1852 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi J, Zhou T & Chen Q Nature Cell Biol. 24, 415–423 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shi J et al. Nature Cell Biol. 23, 424–436 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen Q & Zhou TJ Biol. Chem. 299, 105225 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuhle B, Chen Q & Schimmel P Mol. Cell 83, 3953–3971 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]