

Abstract
Objective:
A definite diagnosis of cerebral amyloid angiopathy (CAA), characterized by the accumulation of Amyloid-β in walls of cerebral small vessels, can only be obtained through pathological examination. A diagnosis of probable CAA during life relies on the presence of haemorrhagic markers, including lobar cerebral microbleeds (CMBs). The aim of this project was to study the histopathological correlates of lobar CMBs in false-positive CAA cases.
Methods:
In three patients who met criteria for probable CAA during life, but showed no CAA upon neuropathological examination, lobar CMBs were counted on ex vivo 3T MRI and on ex vivo 7T MRI. Areas with lobar CMBs were next sampled and cut in serial sections, on which the CMBs were then identified.
Results:
Collectively, there were 25 lobar CMBs on in vivo MRI and 22 on ex vivo 3T MRI of the analysed hemispheres. On ex vivo MRI we targeted 12 CMBs for sampling and definite histopathological correlates were retrieved for 9 of them of which seven were true CMBs. No CAA was found on any of the serial sections. The ‘culprit vessels’ associated with the true CMBs instead showed moderate to severe arteriolosclerosis. Furthermore, CMBs in false-positive CAA cases tended to be located more often in the juxtacortical or subcortical white matter, than in the cortical ribbon.
Interpretation:
These findings suggest that arteriolosclerosis can generate lobar CMBs and that more detailed investigations into the exact localization of CMBs with respect to the cortical ribbon could potentially aid the diagnosis of CAA during life.
Introduction
Cerebral amyloid angiopathy (CAA) is a condition pathologically defined by the accumulation of Amyloid-β (Aβ) in the walls of leptomeningeal and cortical vessels1. Like many neurodegenerative pathologies, a definite diagnosis can only be made at autopsy. A diagnosis of probable CAA can be made during life using the Boston criteria with high sensitivity and specificity. These criteria rely on typical hemorrhagic neuroimaging markers of the disease, such as cortical superficial siderosis (cSS), lobar intracerebral hemorrhage (ICH), and lobar cerebral microbleeds (CMBs)2. CMBs are defined as small round or ovoid hypointense foci, on T2*-weighted MRI sequences3. Patients >50 years of age, who present with clinical symptoms of CAA (i.e. symptomatic ICH, transient focal neurological episodes, subarachnoid hemorrhage, dementia/cognitive impairment) and with more than one strictly lobar CMB, meet criteria for a diagnosis of probable CAA during life2. A small number of individuals with strictly lobar CMBs and a diagnosis of probable CAA during life, do not show significant Aβ-accumulation in the cerebral vessels upon standard neuropathological examination2 (i.e. Vonsattel-grade ≥ 2 in full brain samples). The pathological substrate of lobar CMBs in these false-positive CAA cases is so far unknown. CAA can have a patchy distribution4 within the brain, so that it would be plausible to find it only in the vessels closely associated with the CMBs. CMBs have also been related to arteriolosclerosis in patients with hypertensive arteriopathy (HA)5. In these cases, the lesions are to be found mainly in deep brain regions, consistently with the thickening of the walls of the deep perforating vessels, which is associated with arteriolosclerosis6. However, arteriolosclerosis is not exclusively located in deep brain regions and often co-occurs with CAA7. CMBs are also a common finding in ageing8, Alzheimer’s disease (AD)9, and other neurodegenerative diseases10, meaning that other pathologies could play a role.
A detailed analysis of such cases might give insight into factors (e.g. morphology or localization of the CMBs) that can aid discrimination during life. This is of particular clinical relevance, considering that both the presence (and degree) of CAA pathology11 and the number of lobar CMBs have been related to risk of ICH12,13 and thus to the decision of administering anticoagulation.
The advent of MRI-guided targeted histopathological analysis in the past years14,15, has handed the community better tools to find pathological correlates of neuroimaging markers, which would otherwise remain elusive in standard neuropathological examination. In the case of CMBs, these have been identified as accumulation of extravasated erythrocytes (when acute) or as gold-brown hemosiderin deposits (when older)15, often accompanied by iron positive macrophages.
In the present study we used ex vivo MRI-guided sampling and serial sectioning to unravel the underlying histopathology of lobar CMBs in individuals who fulfilled criteria for probable CAA during life but did not show the pathological signatures of CAA upon routine neuropathological examination at autopsy. Moreover, we explored whether the localization of strictly lobar CMBs and positron emission topography (PET) imaging during life could discriminate between definite CAA and false-positive CAA cases.
Materials and Methods
Study design and case characteristics
The individuals included in the study, were part of an ongoing brain autopsy program that aims to evaluate the histopathology underlying MRI markers of CAA. The program was initiated in 2015 within the stroke research center at MGH and includes brain donations obtained from the MGH and outside hospitals16. Study approval was received from the MGH institutional review board (protocol number: 2021P001920), and informed consent was obtained from the next of kin or legal representative prior to autopsy according to the Declaration of Helsinki. Patients included in the study fulfilled the modified Boston criteria for probable CAA; they were > 55 years old at time of death, had multiple lobar CMBs or one lobar CMB and at least one area of cSS17. As per the clinical diagnostic criteria, possible alternative causes for lobar CMBs, such as antecedent head trauma or ischemic stroke, tumor of the central nervous system, vascular malformation, and vasculitis were excluded. After autopsy, one hemisphere underwent routine neuropathological examination by a board-approved neuropathologist, while the other and most intact hemisphere was fixed in 10% formalin for several weeks and underwent ex vivo 3T MRI scanning (Supplementary Figure 1). Then, the hemispheres were cut in 1 cm-thick coronal slabs and standardized histological examination was performed.
For this specific study, individuals who met criteria for probable CAA during life but showed no evidence of CAA upon standard histopathological examination (false-positive CAA cases) of their brains at autopsy were selected. Furthermore, cases with definite-CAA were chosen as a comparison group.
CMBs were detected on the ex vivo 3T MRI scan of the whole hemisphere. Additionally, for each false-positive case, the corresponding coronal slabs with CMBs were identified, based on anatomical landmarks (Figure 1). The selected tissue slabs were scanned again at higher resolution, using 7T MRI, which enabled a better localization of the CMBs. Finally, samples were taken from the presumed locations of the CMBs, they were embedded in paraffin, and serial sectioning was performed.
FIGURE 1:
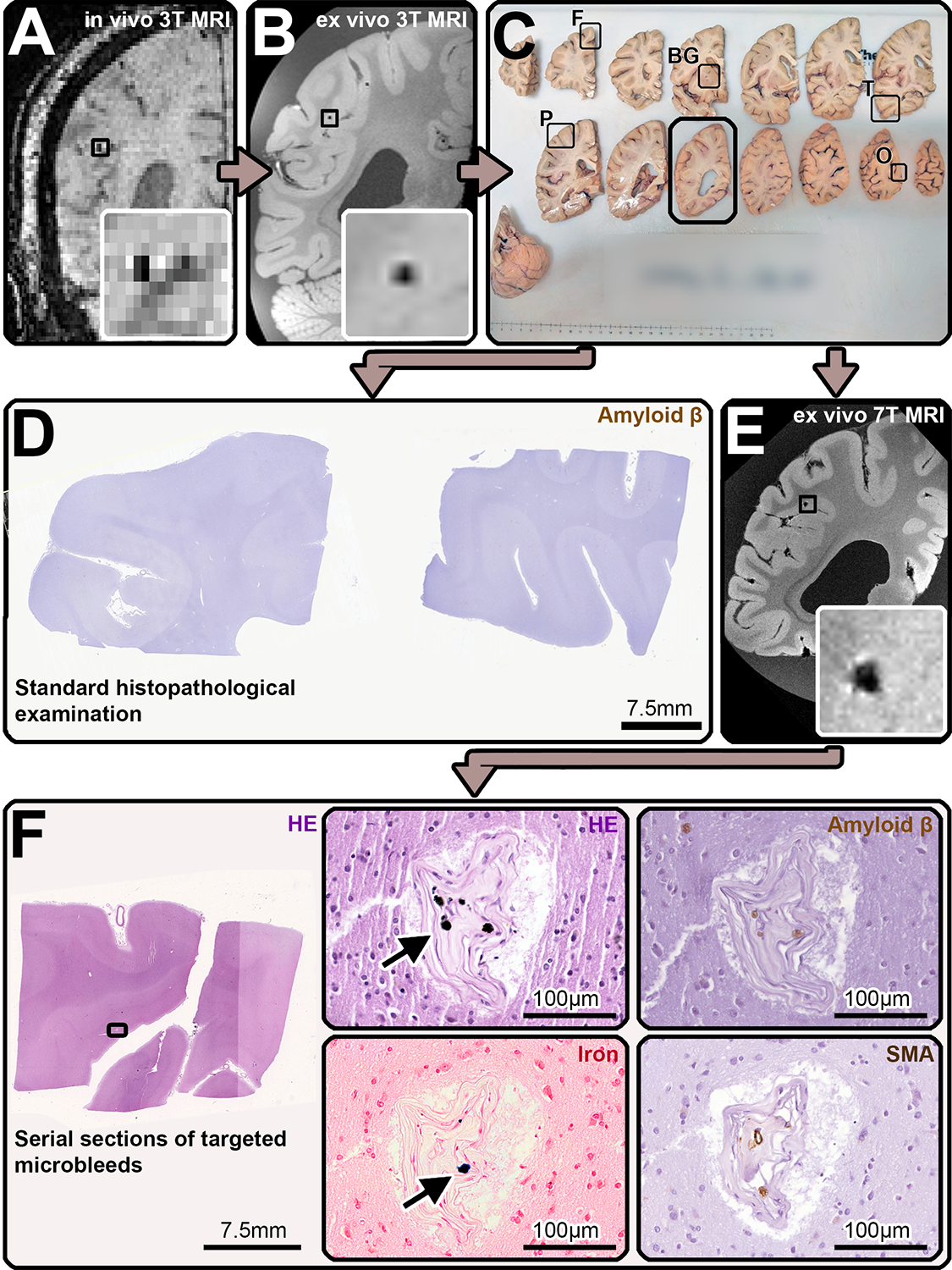
Study design. During life, the participants of this study underwent a research 3T MRI scan (A), which included a susceptibility weighted imaging (SWI) sequence, that allowed to identify cerebral microbleeds (CMBs) as hypointense round structures (inset). After death, the most intact hemisphere was donated to our research program, fixed in formalin, before undergoing ex vivo 3T MRI (B), including a spoiled gradient echo T2*-weighted (FLASH) on which hypointense round/oval lesions were identified as CMBs (inset). The hemisphere was afterwards cut in 1 cm thick coronal slabs (C) and samples were taken from standardized lobar regions (frontal (F), temporal (T), parietal (P), occipital (O) lobe) and from the basal ganglia (BG). These samples were embedded in paraffin and adjacent 6 μm-thick sections were cut and stained for standard histopathology, which involved Hematoxylin and Eosin (HE) and immunohistochemistry against Amyloid-β (Aβ), two examples displayed are from the frontal and parietal lobe (D)). Additionally, the tissue slabs that contained CMBs in the false-positive CAA cases were identified according to anatomical landmarks (black box in C) and scanned with an ultra-high resolution 7T MRI protocol that encompassed a FLASH sequence, on which the location of the CMBs was confirmed (E). Guided by the MRI, samples were cut to fit a standard tissue cassette, they were embedded in paraffin and underwent complete serial sectioning. Each 5th section was stained for HE and when a CMB correlate was found, adjacent sections were subjected to immunohistochemistry against Aβ, smooth muscle actin (SMA), and stained for Perls’ Prussian Blue to detect iron (F). When the presumed culprit vessel of the lesion was identified, it was traced, and adjacent sections were also stained for the above. In the here represented CMB, hemosiderin deposits (arrow in F, HE-enlargement) and iron (arrow in F, iron-enlargement) were observed predominantly within the occluded vessel, which likely represents an example of end-stage vessel remodeling. Neither Aβ (F, Aβ-enlargement), nor smooth muscle cells (F, SMA-enlargement) were seen along the course of the vessel.
In vivo 3T MRI
During life, scans were performed within the frame of a single-center longitudinal study, that included probable and possible CAA patients from a memory-clinic cohort18. All three false-positive cases underwent the same 3T MRI scan protocol (Magnetom Prisma-Fit, Siemens Healthineers, Erlangen, Germany), using a 32-channel head coil. The protocol included: a 3D T1-weighted sequence (repetition time [TR] 2300–2510 ms; echo time [TE] 1.69–2.98 ms; resolution 1 × 1 × 1 mm), a 3D fluid-attenuated inversion recovery sequence (FLAIR, TR 5000–6000 ms; TE 356–455 ms; resolution 1 × 1 × 1 mm), a susceptibility-weighted imaging sequence (SWI, TR 27–30 ms; TE 20 ms; slice thickness, 1.4 mm; in-plane resolution 0.9 × 0.9 mm), a 3D diffusion weighted imaging sequence (DWI, 60–64 directions; TR 8000–8040 ms; TE 82–84 ms; resolution 2 × 2 × 2 mm; b-value, 700 s/mm2), and a T2-weighted sequence (TR 7500 ms; TE 84 ms; slice thickness, 2 mm; in-plane resolution 0.5 × 0.5 mm).
Ex vivo 3T MRI
The details of the whole-hemisphere ex vivo 3T MRI scanning procedure have been previously described16,20,21. Briefly, hemispheres were vacuum sealed in a plastic bag containing periodate-lysine-paraformaldehyde, after eliminating air bubbles. Then, they were placed in the 32-channel head coil of a whole-body 3T MRI scanner (MAGNETOM Trio, Siemens Healthineers, Erlangen, Germany) and scanned overnight. The protocol included several sequences, including a T2-weighted turbo-spin echo (TSE) sequence (voxel size 0.5 × 0.5 × 0.5 mm; TE 61 ms; TR 1800 ms; flip angle 150°; total scan time ~ 3 hours), and a gradient-echo fast low angle shot (FLASH) sequence (voxel size 0.5 × 0.5 × 0.5 mm; TE-1 4.49 ms; TE-2 11.02 ms; TR 20 ms; flip angle 10°, 20°, 30°; receiver bandwidth 160 Hz/Px; 2 averages; total scan time ~ 2 hours), which was used in this study (Supplementary Figure 1).
Ex vivo 7T MRI
Individual formalin-fixed coronal tissue slabs of the false-positive CAA cases were scanned again with 7T MRI, in order to better identify the CMBs (Figure 1). Scans were acquired overnight on a whole-body 7T MRI scanner (MAGNETOM, Siemens Healthineers, Erlangen, Germany), with a custom built 32-channel head coil. The protocol included spoiled gradient echo T2*-weighted (FLASH) (voxel size 0.2 × 0.2 × 0.2 mm; TE 8.67 ms; TR 20 ms; flip angle 20°; receiver bandwidth 180 Hz/Px total scan time ~ 1 hour) and T2-weighted (TSE) (voxel size 0.5 × 0.5 × 0.5 mm; TE 54 ms; TR 1000 ms; flip angle 90°; receiver bandwidth 401 Hz/Px, total scan time ~ 1 hour) acquisitions.
MRI analysis
On in vivo and ex vivo MRI, CMBs were defined as round/ovoid foci < 10 mm, that appear hypointense on T2*-weighted imaging, according to established criteria3. Ex vivo T2*-weighted imaging is susceptible to air bubbles, which remain trapped on the surface of the brain and in the sulci and can mimic CMBs. Therefore, the T2-weighted images were used to discriminate between actual CMBs and the artefacts. CMBs were visually assessed by one expert observer (V.P., 7 years of experience) and consensus was then achieved with another expert (S.J.v.V., 10 years of experience). Each CMB was identified, when possible, in all imaging modalities (in vivo and ex vivo 3T MRI, ex vivo 7T MRI), guided by anatomical landmarks (Table 2). Furthermore, a third observer blinded to clinical information and pathological diagnosis (M.C.Z.Z., a neuroradiologist with 9 years of experience) classified the lobar CMBs on the ex vivo 3T MRI of the three false-positive CAA cases and in the three true-positive CAA cases as either located strictly in the cortex, in the juxtacortical WM or in the subcortical WM.
TABLE 2.
Histopathological findings of the MRI-observed CMBs and of their culprit vessel. CAA and arteriolosclerosis score were assessed at a whole section level, at the site of rupture of the culprit vessel, as well as up- and downstream of it. Key: not available (N/A); smooth muscle cells (SMC).
| MB | Detection on in vivo 3T MRI | Detection on ex vivo 3T MRI | Detection on ex vivo 7T MRI | Histological correlate | Location (histology) | Iron | CAA score (whole section/ culprit vessel at site of rupture/ culprit vessel down- and upstream) | Arteriolo-sclerosis score (whole section/ culprit vessel at site of rupture/ culprit vessel down- and upstream) | SMC |
|---|---|---|---|---|---|---|---|---|---|
| Case 1 – 1 | + | + | Not scanned | Hemosiderin/Erythrocytes | Subcortical | - | 0/0/0 | 1/1/1 | + |
| Case 1 – 2 | - | + | + | Microaneurysm | Juxtacortical | - | 0/0/0 | 2/3/3 | + |
| Case 1 – 3 | - | - | - | Hemosiderin/Erythrocytes | Subcortical | - | 0/0/0 | 2/3/3 | + |
| Case 2 – 1 | - | + | + | No correlate found | N/A | N/A | N/A | N/A | |
| Case 2 – 2 | - | + | + | No correlate found | N/A | N/A | N/A | N/A | |
| Case 2 – 3 | - | + | + | No correlate found | N/A | N/A | N/A | N/A | |
| Case 2 – 4 | + | + | + | Hemosiderin | Cortical | + | 0/0/0 | 3/3/3 | - |
| Case 2 – 5 | + | + | + | Hemosiderin | Juxtacortical | + | 0/0/0 | 3/3/3 | - |
| Case 2 – 6 | - | - | + | Hemosiderin | Juxtacortical | + | 0/0/0 | 3/3/3 | + |
| Case 2 – 7 | + | + | + | Hemosiderin | Cortical | + | 0/0/0 | 3/3/3 | + |
| Case 3 – 1 | - | + | + | Hemosiderin | Juxtacortical | + | 0/0/0 | 2/2/2 | + |
| Case 3 – 2 | - | + | + | Hemosiderin | Subcortical | - | 0/0/0 | 1/3/3 | + |
| Case 3 – 3 | - | - | - | Hemosiderin | Subcortical | - | 0/0/0 | 1/3/3 | N/A |
| Case 3 – 4 | - | - | + | Calcification | Subcortical | + | 0/0/0 | 1/3/3 | + |
PET Scans and analysis
PET scans were acquired on a Siemens/CTI (Knoxville, TN) ECAT HR+ (3-D mode, 63 image planes, 15.2-cm axial field of view, 5.6-mm transaxial resolution, 2.4-mm slice interval) at MGH. Immediately before the scan, 8.5 to 15 mCi of [11C]-Pittsburgh compound B was injected as a bolus followed by a 60- to 70-minute dynamic acquisition in 69 frames, as previously published22. The [11C]- PiB was prepared directly at MGH.
PET data were reconstructed, corrected for attenuation, and inspected to verify adequate count statistics and absence of significant head motion. Data processing was performed using FreeSurfer (version 6.0), as previously described23. The images were aligned to T1-weighted MR images and measurements were made in regions of interest (ROIs) defined by the Desikan-Killian atlas24. The specific binding of PiB was calculated using the Logan graphical method as distribution volume ratio (DVR), on data collected 40 to 60 minutes after injection, adopting the cerebellar cortex as a reference region25. We chose to evaluate the DVRs of a composite ROI consisting of the frontal, lateral temporal/parietal, and retrosplenial (FLR) cortex, because previous studies demonstrated an increased PiB retention in these brain areas in both AD and CAA26. To this aim, we adopted a cutoff score (FLR DVR 1.08), that had demonstrated good sensitivity to presence of Aβ23,27.
Histopathology
Samples were taken from predefined standard brain areas of the frontal, parietal, temporal and occipital cortex and from the BG area in all cases, as previously described28. All tissue blocks were placed in thin cassettes and embedded in paraffin. Six μm-thick sections were cut with a microtome and stained using hematoxylin & eosin (H&E), following standard histology protocols. Brightfield immunohistochemistry against Aβ (mouse, Agilent Technologies, Santa Clara, CA; 1:200, Cat# M0872) was performed on adjacent sections. Following a standardized immunohistochemistry protocol, sections were deparaffinized and rehydrated through xylene and graded ethanol series. Next, endogenous peroxidase was blocked with 3% H2O2 (20 min), followed by antigen retrieval using formic acid treatment (for Aβ, 5 min). Tissue was then blocked with normal horse or goat serum (1 hour) and incubated overnight with the primary antibody at 4°C. On the next day, the biotinylated mouse or rabbit secondary antibody was applied (1 hour), followed by a mixture of avidin (A) and biotinylated HRP (B) (Vectastain ABC kit, Vector laboratories, 30 min) and 3,3’-Diaminobenzidine (DAB, Vector laboratories). Negative controls were included by omitting the primary antibodies and showed no immunopositivity, while the presence of Aβ-plaques served as a positive control.
Further samples were taken from the three false-positive CAA cases, targeting several CMBs. These samples had the same dimensions as the ones described above to fit a tissue cassette. They were also embedded in paraffin and cut in serial sections at a microtome. Sections at the estimated lesion location were collected and each fifth section was stained with H&E and then matched with the ex vivo MRI scans to verify whether the targeted CMB (or another correlate for the hypointensity seen on MRI) was visible. If this was the case, immunohistochemistry was performed on directly adjacent sections for Aβ, following the protocol described above, and for smooth muscle actin (SMA) (mouse, Agilent Technologies, Santa Clara, CA; 1:250, Cat# M0851), using heated citrate buffer (pH 6.0) for the antigen retrieval step. Moreover, adjacent sections were stained for Perls’ Prussian blue using a standard protocol, to identify iron deposits. Briefly, sections were incubated with a 1:1 mixture of 5% hydrochloric acid and 5% potassium ferrocyanide (30 min), and counterstained with filtered neutral red (1 min).
Histopathological Image Analysis
Stained sections were scanned using a Hamamatsu NanoZoomer Digital Pathology (NDP)-whole slide scanner (C9600–12, Hamamatsu Photonics KK, Japan), with a 20x objective. Obtained high-resolution (457 nm/pixel; 55579 dpi) digital images were visualized using the NDP.view2 software (version 2.8.24). The samples taken from the predefined standard brain areas were scored for cortical and leptomeningeal CAA severity, which was assessed on Aβ-stained sections, using a 4-point scale; absent (0), scant Aβ deposition (1), some circumferential Aβ (2), and widespread circumferential Aβ (3), following proposed consensus criteria29. Parenchymal Aβ-plaque severity was evaluated using a 4-point scale; absent (0), mild (1), moderate (2), severe (3). CAA severity and Aβ-plaques were assessed by two independent raters (S.J.v.V. and V.P.), followed by a consensus rating to obtain a final score. Moreover, arteriolosclerosis in the juxtacortical WM of the sections and in the BG was assessed according to a 4-point scale (Grades 0–3)6, assigning to the section the highest grade reached by at least three arteries. Two independent raters (V.P. and C.A.A.) evaluated the sections and reached consensus to obtain a final score.
The H&E sections collected when targeting the CMBs were matched to the ex vivo MRIs and visually inspected. The positive retrieval of a CMB was defined as presence of erythrocyte extravasation (indicative of a recent hemorrhage) or blood-breakdown products (such as hemosiderin, indicative of subacute or old hemorrhages). Because vasculopathies (such as microaneurysms, or vessel calcifications) have been previously reported as CMB-mimics15, these were also scored. Positivity for iron was addressed on adjacent sections stained for Perls’ Prussian blue. When possible, the ‘culprit vessel’ of the hemorrhage was identified if extravasation from it or near the CMB could be observed. When necessary, the blocks were further cut to completely retrieve the CMB or the culprit vessel. When a CMB was found, adjacent Aβ-stained sections, were inspected for presence and grade of CAA at a whole section level, at the site of rupture, as well as up- and downstream of the culprit vessel. Similarly, arteriolosclerosis was assessed on adjacent HE-stained sections. Moreover, the integrity of smooth muscle cells in the culprit vessel was examined on the adjacent SMA-stained sections and recorded as either complete circumferential or not. Observations were carried out by V.P. and discussed with S.J.v.V.
Statistical Analysis
The difference in ratio of the sum of CMBs in the juxtacortical plus subcortical WM and total CMBs, for false-positive vs true-positive CAA cases was calculated using a two-tailed Mann Whitney-U test. Statistical analysis was performed in R, version 3.6.0 (R Foundation for Statistical Computing, Vienna, Austria; www.R-project.org) and significance was set at p < 0.05.
Results
False-positive CAA cases
Three individuals who met criteria for probable CAA during life but showed no evidence of CAA upon standard histopathological examination of their brains at autopsy, were selected for the present study (Table 1a). During life, these three patients all presented with cognitive complaints to the Memory Disorders Division of the Massachusetts General Hospital (MGH) and were enrolled in a single-center longitudinal memory-clinic cohort, as previously described in detail elsewhere18. They underwent a neurological clinical evaluation, neuropsychological examination, 3 tesla (T) research MRI (on which CMBs were detected), and a Pittsburgh Compound-B (PiB) positron emission tomography (PET) scan. Case 1 was an 87-year-old Caucasian female with 16 years of education, who presented with increasing memory loss, that begun approximately six years earlier. Brain MRI showed three lobar CMBs, periventricular white matter hyperintensities (WMH) and moderate burden of MRI-visible perivascular spaces (PVS) in the centrum semiovale (CSO) and basal ganglia (BG). The second patient (case 2) was an 80-year-old Caucasian male, with 19 years of education, who first presented with cognitive complaints that begun two to three years earlier. Baseline MRI showed lobar CMBs, two intragyral hemorrhages19, lobar lacunes and WMH in the posterior periventricular region and CSO. Severe bilateral hippocampal atrophy could also be observed. The third patient (case 3) was a 79-year-old female, who had 18 years of education and presented with cognitive impairment beginning two years earlier. On the brain MRI performed on the same day of the first visit, two lobar CMBs, mild WMH, moderate burden of MRI-visible PVS in the CSO and a lacune in the caudate nucleus were seen. With the exception of the third patient, who had an ocular transient ischemic attach after initial evaluation, no further neurological symptoms were documented.
TABLE 1a.
Characteristics of the false-positive CAA cases and MRI findings. Cortical superficial siderosis, WMH, lacunes, intragyral bleeds, PVS and infarcts were assessed on the last in vivo scan before death, according to the STRIVE criteria3 and proposed differentiation of WMH-patterns45. The latter include the multi-spot WMH pattern, which have recently been included as a supporting feature for the diagnosis of probable CAA according to the Boston criteria V2.0. Key: Basal ganglia (BG); Body-mass index (BMI); Cerebral Amyloid Angiopathy (CAA); Cerebral microbleeds (CMBs); Cerebral microinfarcts (CMIs); Centrum semiovale (CSO); Perivascular spaces (PVS); Intracerebral hemorrhage (ICH); white matter hyperintensities (WMH).
| Demographic information | Case 1 | Case 2 | Case 3 |
|---|---|---|---|
| CAA | Probable CAA | Probable CAA | Probable CAA |
| Age at death | 88 | 85 | 82 |
| Sex | Female | Male | Female |
| Hypertension | + | + | + |
| Diabetes mellitus II | + | - | + |
| Hyperlipidemia | + | + | + |
| BMI | 25.2 | 25.8 | 58 |
| Stroke/TIA | - | - | + |
| MRI findings | Case 1 | Case 2 | Case 3 |
| MRI – death interval (months) | 17 | 30 | 38 |
| No. lobar CMBs in vivo (total) | 3 | 20 | 2 |
| No. lobar CMBs in vivo/ex vivo (relevant hemisphere) | 2/5 | 12/13 | 1/4 |
| No. lobar CMBs ex vivo subcortical + juxtacortical/total | 2/5 | 8/13 | 2/4 |
| No. CMBs targeted for histopathology | 2 | 7 | 3 |
| No. correlates found on histopathology | 2 | 4 | 3 |
| No. of extra CMBs found on histopathology | 1 | 0 | 1 |
| Cortical Superficial Siderosis | No | No | No |
| WMH | Multi-spot WMH | Confluent WMH | Multi-spot WMH |
| Lacunes | 1 | 4 | 3 |
| Intragyral bleeds | 0 | 2 | 0 |
| ICH | 0 | 0 | 0 |
| MRI-visible PVS CSO | 3 | 1 | 0 |
| MRI-visible PVS BG | 1 | 2 | 0 |
| Ischemic macroinfarcts | 0 | 1 | 1 |
True-positive CAA cases
To assess differences in the localization of lobar CMBs between false-positive and true-positive CAA cases, we included three additional individuals from the same pathological cohort, who had comparable radiological characteristics (i.e. low number of CMBs). All cases fulfilled criteria for probable CAA during life, which was confirmed as definite CAA upon standard histopathological examination. Case 4 was a male who presented at age 68 at MGH with his second hemorrhagic stroke and deceased one year later. Case 5 was an 80-year-old female patient, who was evaluated at MGH because of memory impairment and diagnosed with probable CAA and dementia. She deceased at age 84. Case 6 was an 85-year-old male, who presented to an outside hospital, deceased the same year, and for which no further clinical information was available (Supplementary Table 1).
General Neuropathology Findings
No evidence of severe (i.e. grade >2) cortical or leptomeningeal CAA was found in any of the three false-positive CAA cases upon routine neuropathological analysis. Only a few sections from the predefined standard areas of the donated hemisphere showed mild CAA, in the form of grade 1 leptomeningeal (occipital cortex in case 1 and all areas in case 2) or cortical CAA (parietal cortex in case 3), or grade 2 leptomeningeal CAA (parietal cortex in case 3). Moderate arteriolosclerosis was found in all three cases (Table 1b).
Neuropathology of MRI-observed CMBs
Across the three false-positive CAA cases, 25 MRI-observed CMBs were found on in vivo MRI, 15 of which were located in the post-mortem hemisphere that was included in our study. On the ex vivo 3T MRI scans, a total of 22 CMBs could be identified on the donated hemispheres. Nine of these were precisely localized on 7T MRI, 2 additional smaller ones were seen only on 7T MRI and one CMB was not searched for at high resolution, because the localization was easy to determine using 3T MRI (Figure 2). These 12 7T MRI-observed CMBs were targeted for histopathological analysis and a correlate was found for 9 of them (Table 2): 7/9 were hemosiderin deposits, indicating older microhemorrhages, or erythrocytes, indicating a more recent hemorrhage. 2/9 proved to be mimics on histopathology (one was a microaneurysm and one a calcified vessel) (Table 2). Two additional CMBs were identified only on histopathology and had no MRI correlate, also retrospectively. Five CMBs and the calcification were positive for iron.
FIGURE 2:
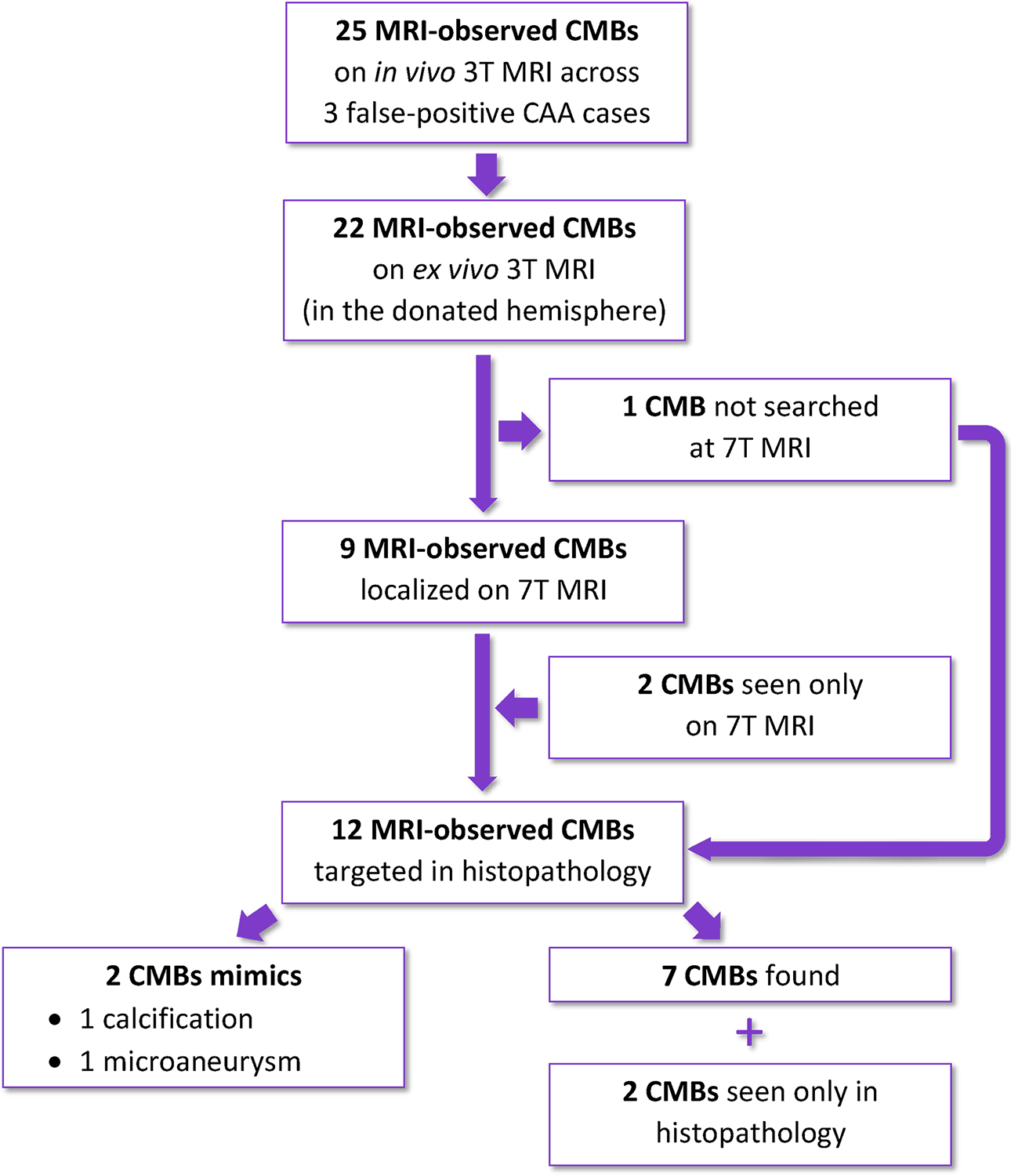
Flow chart exemplifying the number of lobar cerebral microbleeds (CMBs) analyzed in the three false-positive CAA cases.
Neuropathology of vessels associated with CMBs
The culprit vessel was identified for all histopathological CMBs. A total of 34 H&E-stained sections, and their adjacent sections stained for Aβ, iron and SMA, were examined in detail. In six sections, two CMBs were located on the same section. No CAA was found on any of the serial sections stained for Aβ (Figure 3), at the site of rupture of the culprit vessel, or up-/downstream of it. Conversely, 20/34 sections displayed moderate-to-severe arteriolosclerosis. In 8/9 CMBs and in 2/2 CMB-mimics (Figure 4) the culprit vessels had evidence of moderate-to-severe arteriolosclerosis at the rupture site (or site of most severe calcification or aneurysmal dilation), characterized by loss of smooth muscle cells with fewer nuclei visible within the wall, thickening of the vessel’s wall, and dysmorphic changes, associated with partial occlusion. The degree of arteriolosclerosis remained similar also when the vessel was traceable in adjacent sections (up- or downstream). A non-circumferential ring of SMA was visible only in two of the culprit vessels.
FIGURE 3:
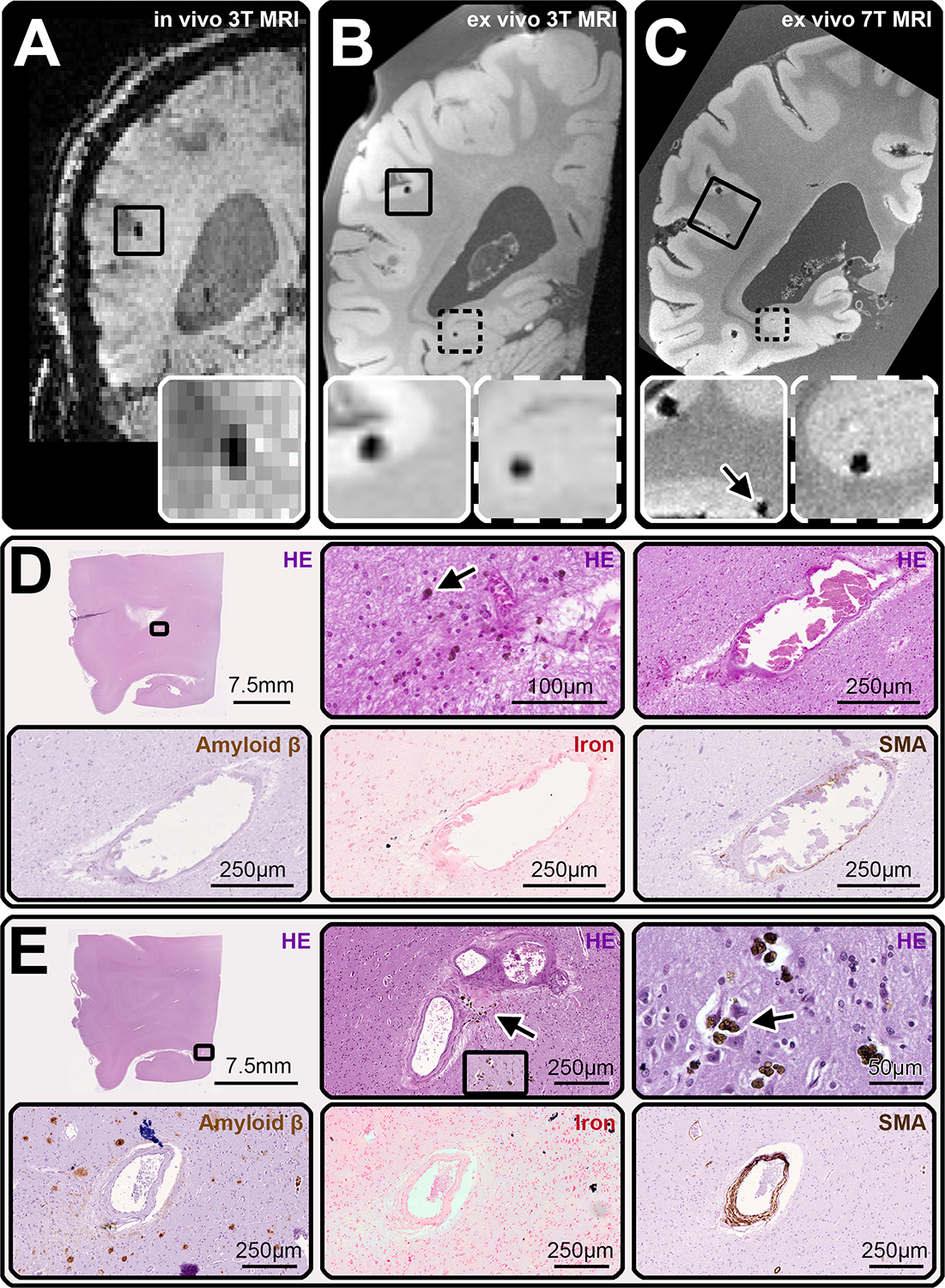
Examples of cerebral microbleeds (CMBs) identified on MRI and their pathological correlates. (A) displays an example of a coronal view of the susceptibility weighted imaging (SWI) scan of the in vivo 3T MRI of case 2, on which a CMB was identified (inset). The ex vivo 3T MRI (B), confirmed the presence of this first CMB (inset with continued line) and allowed to detect a second one (inset with broken line). On the ex vivo 7T MRI of the tissue slab (C) the localization of both CMBs was confirmed (the second CMB was visible on another plane) and a third CMB was observed in proximity to the first (arrow, inset with continued line). This area was sampled and cut in serial sections to retrieve both CMBs (D, E). Hemosiderin deposits (arrows in D and E, HE-enlargements) and iron (D and E, iron-enlargements) were seen at different depths as correlates for both CMBs. Moreover, the culprit vessels for each CMB were traced and showed thickening and loss of structure of the vessel’s wall and partial occlusion of the lumen, consistent with grade 3 arteriolosclerosis at the rupture site. There were no Aβ deposits in any of the vessels seen on the sections (D, E on Amyloid β). One of the culprit vessels had circumferential smooth muscle cells (E, SMA), while the other did not (D, SMA).
FIGURE 4:
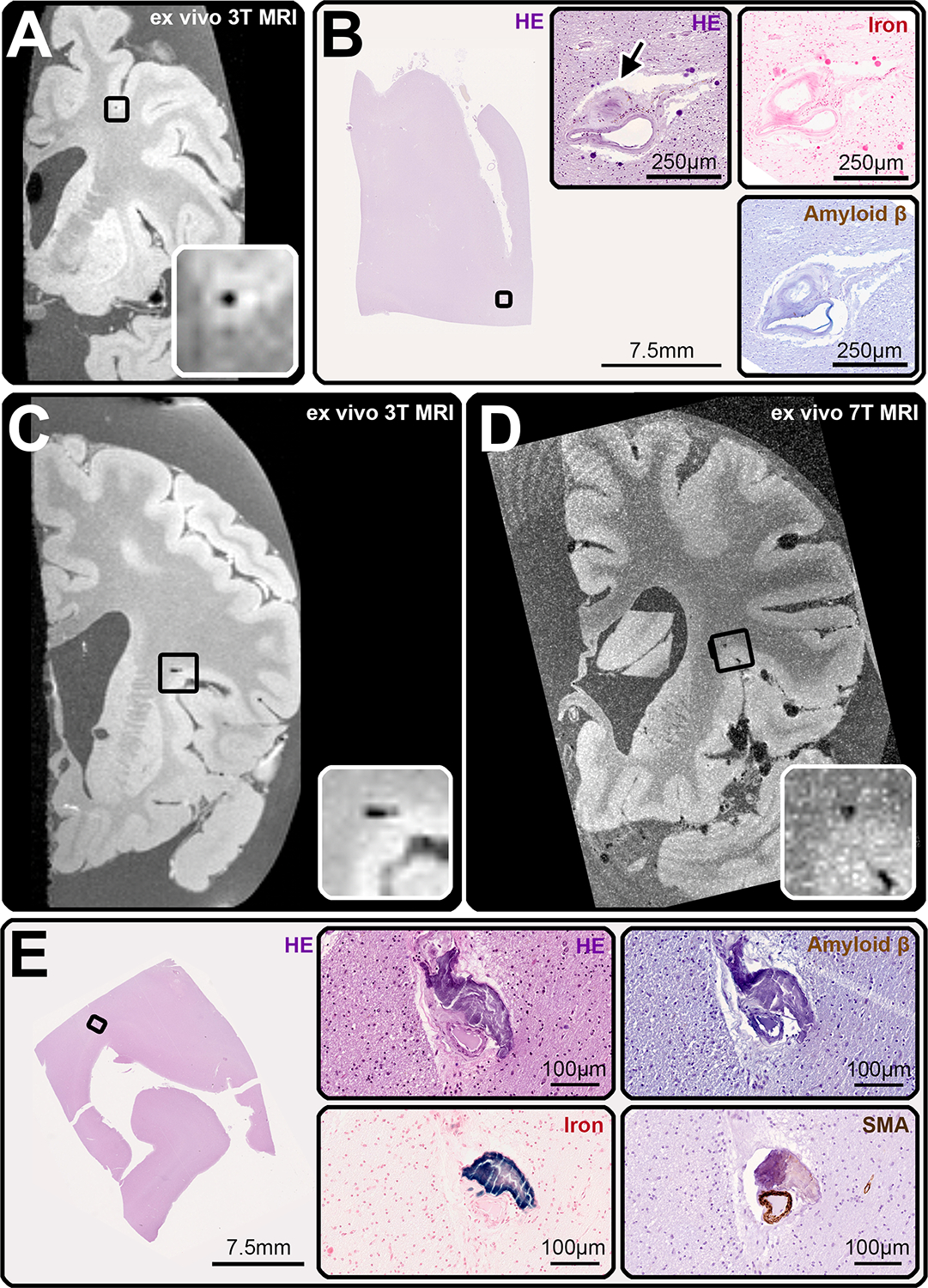
Examples of cerebral microbleed (CMB) – mimics. Two of the CMBs identified on MRI proved to be CMB-mimics upon histopathological analysis. The histological correlate of the first, which was observed on ex vivo 3T MRI in case 1 (A, inset), and ex vivo 7T MRI (not shown), was a partially occluded (arrow) microaneurysm with hemosiderin deposits within the vessel wall (B, HE), but no iron deposits (B, iron). Vascular Aβ was absent (B, Amyloid β). The smooth muscle cell layer was largely intact (not shown). The second CMB-mimic was found in case 3, also on ex vivo 3T (inset, C) and 7T MRI (inset, D). At the corresponding location on serial sectioning (E), we observed a calcified vessel (E, HE), which stained positively for iron (E, iron), had no Aβ deposits within the vessel wall (E, Amyloid β), nor circumferential smooth muscle cells (E, SMA).
Location of CMBs in false-positive CAA cases
It was noticed that the CMBs of false-positive CAA cases were mostly located in the juxtacortical or subcortical WM (7/9 pathologically confirmed CMBs) (Figure 5). This observation prompted a systematic comparison between the location of MRI-observed CMBs in false-positive and definite CAA cases, which was conducted on the 3T ex vivo MRI. The CMBs were classified as being located in the cortex, juxtacortical WM, or subcortical WM. The ratio between the sum of CMBs located in the juxtacortical WM and subcortical WM, to the total number of CMBs tended to be higher in the false-positive CAA cases (Median = 0.5) compared to the true-positive CAA cases (Median = 0; W = 9, p = 0.076).
FIGURE 5:
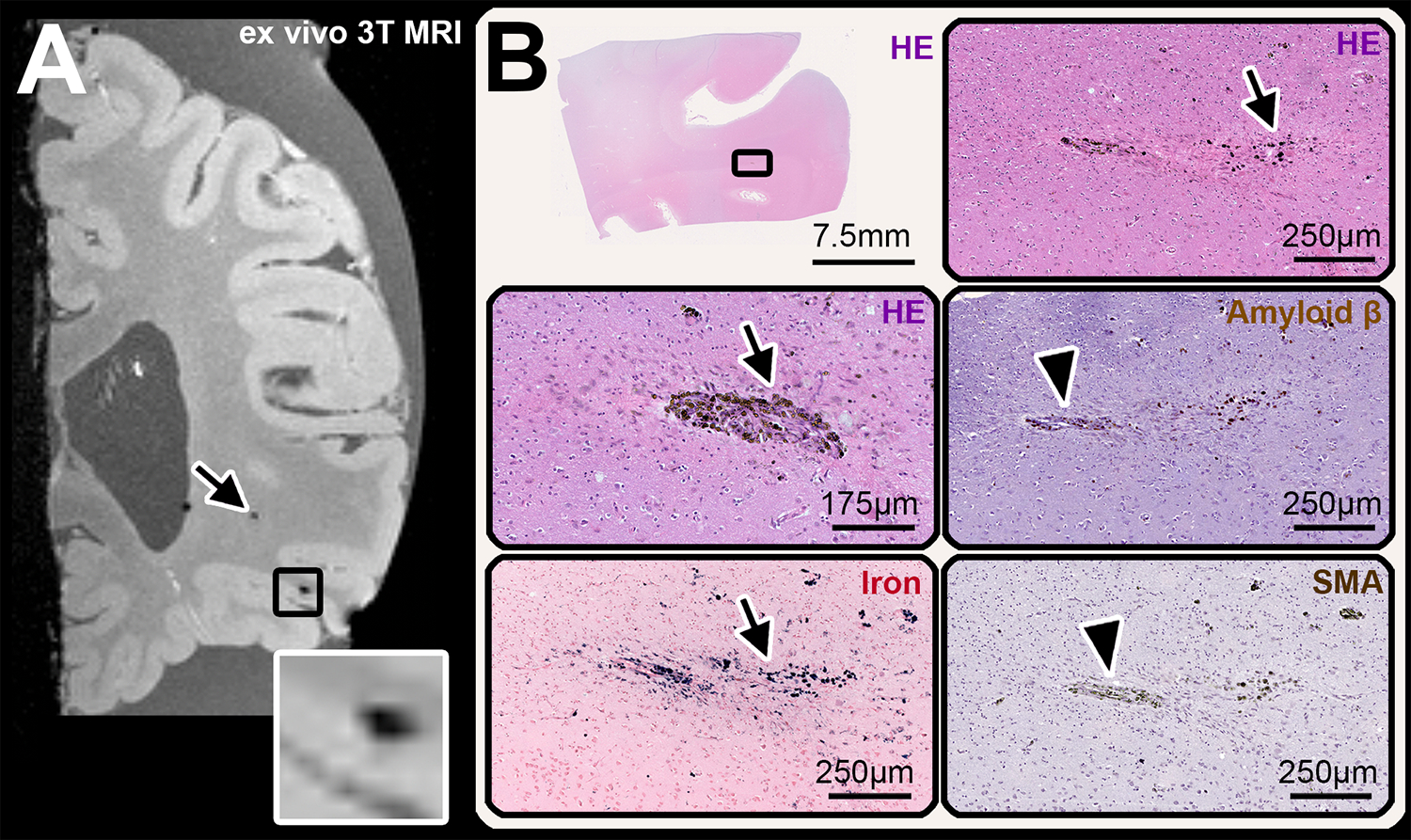
Example of cerebral microbleeds (CMB) identified in the subcortical and juxtacortical white matter (WM). (A) coronal view of the spoiled gradient echo T2*-weighted (FLASH) sequence at ex vivo 3T MRI of case 3, which shows a lobar subcortical CMB in the WM (arrow) and a juxtacortical CMB (inset). The juxtacortical CMB was found in the histopathological analysis (B) where hemosiderin deposits (B, HE) and iron (B, iron) were observed at different depths in serial sections. The presumed culprit vessel was negative for both Aβ (arrow in B, Amyloid β-enlargement) and smooth muscle cells (arrow in B, SMA-enlargement).
PiB-PET findings
Case 1 and 3 had a negative PiB PET scan (FLR DVR of 1.06 and 0.92 respectively). Case 2 had a positive scan (FLR DVR of 1.70), corresponding to the presence of parenchymal Aβ plaques on histopathology in this case (Figure 6).
FIGURE 6:
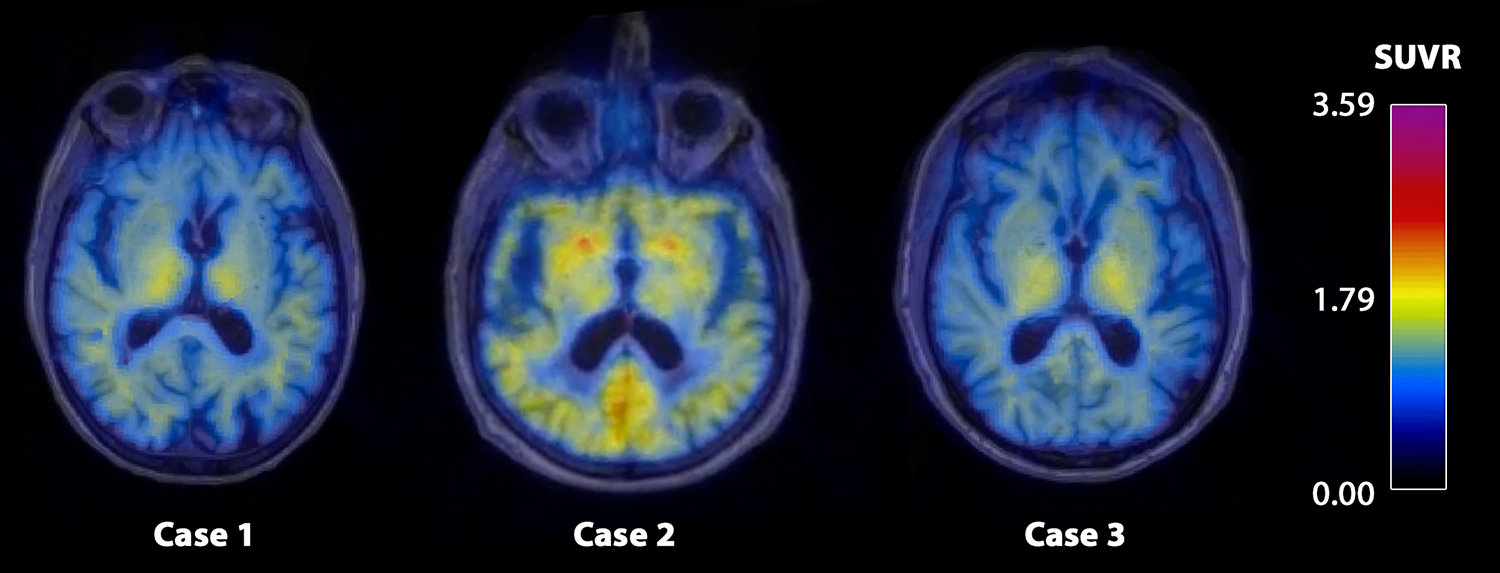
The figure displays the SUVR values of the Pittsburgh Compound-B PET in form of a heat map in the three false-positive CAA cases.
Discussion
Lobar CMBs are a typical marker of CAA, but they occur also in absence of this pathology. In three individuals who met Boston criteria for a clinical diagnosis of probable CAA during life but proved to have no CAA in their brains at autopsy, we performed ex vivo MRI-guided histological serial sectioning of lobar CMBs, with the aim of identifying the neuropathological correlates of these lesions. We found that the majority of MRI-observed CMBs had extravasation of erythrocytes (suggesting an acute hemorrhagic event) or blood-breakdown products (e.g. hemosiderin, revealing older hemorrhages) as pathological correlates. Two of 9 were CMB-mimics (one microaneurysm and one calcification), which is consistent with the previously described heterogeneity of the pathological correlates of lobar CMBs.30 Calcifications have been observed on in vivo imaging of small vessel disease patients31 and in hereditary Dutch-type CAA, where they were regarded as a marker of advanced pathology32.
No Aβ-depositions were found in any of the culprit vessels related to the CMBs or in any other vessels of the targeted sections, confirming that indeed CAA did not play a role in these lobar CMBs. Although it has been previously observed that the severity of CSVD-burden on MRI was lower in CAA cases without symptomatic ICH, compared to those with ICH, no differences have been reported in the number of CMBs on MRI between these two groups.33 Furthermore, the positive predictive value of probable CAA according to the modified Boston criteria was previously shown to be high (87.5%)34 in CAA patients without symptomatic ICH that came through a hospital-based memory clinic. Hence, the misclassification was likely not attributable to the absence of symptomatic ICH in these false-positive CAA cases. As per study design, inclusion in the MGH pathological cohort was based on the modified Boston criteria. Importantly, all cases included in the current study also met Boston V2.0 criteria for the diagnosis of probable CAA2. However, it is important to emphasize that cases 1 and 3 only had 2–3 lobar CMBs on in vivo MRI, and that in clinical practice they would possibly be considered as equivocal for a diagnosis of probable CAA.
Instead of CAA, all the identified culprit vessels had a moderate-to-severe degree of arteriolosclerosis, suggesting that this pathology might play a role not only in deep CMBs, but also in lobar CMBs. Moderate-to-severe arteriolosclerosis has indeed also been observed in the cortex and juxtacortical WM6. In fact, a co-occurrence of arteriolosclerosis and CAA was determined in 42% of cases with a lobar ICH35. In line with these previous observations, arteriolosclerosis is a frequent finding across our CAA pathological cohort. The co-occurrence of the two small vessel pathologies suggests shared pathophysiological pathways leading to hemorrhages, possibly through a process including vessel remodelling.14,36 Recent work from our group, using histopathological analysis of MRI-targeted CMBs in cases with advanced CAA has shown that cortical CMBs are the result of vascular remodeling, including hyaline thickening of the vessel wall, loss of smooth muscle cells, and loss of Aβ from affected vessels.14,28 While the exact mechanisms that trigger vessel remodeling and rupture in the context of advanced CAA remain unclear, it is possible that a comparable sequence of events may be implicated in the context of advanced arteriolosclerosis.
Relationships between CMBs and other neurodegenerative and neuroinflammatory pathologies have also been hypothesized10,37, though they have not been directly confirmed in neuropathology. Therefore, it cannot be excluded that the investigation of further pathological hallmarks could inform us on the genesis of these lesions. The contribution of globular glial tauopathy to hemorrhage, which was found in case 1, appears to be unlikely38,39.
Because of the clinical relevance that CAA burden has in predicting ICH risk during life, we explored possible characteristics that could aid the differentiation of false-positive CAA from true-positive CAA cases. Previous observations15 and a recent study on the validation of the Boston criteria V2.0 in a community-dwelling cohort (Zanon Zotin et al., under review)40 have pointed out that lobar CMBs in definite CAA cases are observed mainly within the cortical ribbon, suggesting potential discriminative power between definite and non-CAA cases. This is in line with the fact that CAA is a cortical pathology, which only very rarely affects subcortical areas. In our false-positive CAA cases, the CMBs were more often seen in the juxtacortical area (including the U-fibers) and in the subcortical WM. Furthermore, a trend towards higher ratio between juxtacortical/subcortical CMBs and total number of lobar CMBs was observed in the false-positive versus the true-positive CAA cases, suggesting that the localization of the CMBs might aid in the differentiation between pathologies, if replicated in future work. The use of PiB-PET imaging might further aid the differentiation between true- and false-positive CAA cases, as two out of three cases in this study had indeed a negative PiB-PET scan during life. That said, previous studies have suggested PiB-PET might not be sensitive enough to detect mild CAA41 and the co-occurrence of Aβ-plaques in the brain parenchyma may result in a positive scan, as was true for one of our cases42. As such, further research is needed to help determine the added value of PET imaging to aid diagnosis of CAA during life.
For a few of the CMBs seen on MRI, a histopathological correlate could not be retrieved, most likely because a mismatch between MRI and histopathology occurred, even though it cannot be excluded that pathologies we did not detect (e.g. thrombi) underlined some of the MRI-observed CMBs. Nonetheless, the combination of ex vivo 7T MRI and histopathology represents a strength of our study, which allowed to first localize and then characterize these minute lesions, which would have been missed on standard pathological examination. This exploratory study, based on a series of pathological cases, has some limitations. Firstly, only three false-positive CAA cases were available. Future case series should include a greater number of subjects, which will possibly disclose more heterogeneity within the pathological correlates of the lobar CMBs observed on MRI. At present, the qualitative nature of the study, together with its small sample size, hinders an analysis that accounts for the co-dependence by subject. Secondly, the cross-sectional nature of histopathological examination does not allow to infer the sequence of events that resulted in microhemorrhage. Thirdly, further pathological hallmarks of disease in the proximity of the lesions were not assessed. Systemic and local neuroinflammation and blood-brain-barrier leakage might for example play a role in the genesis of CMBs, as it has been suspected in multiple sclerosis43 and shown in CAA44. Future analyses should include the observation of reactive astrocytes and activated microglia.
Conclusion
This exploratory study aimed to investigate the pathological substrate of MRI-observed lobar CMBs in false-positive CAA cases. We confirmed that vascular Aβ accumulation was not present at the lesion site. Instead, the combination of ultra-high-resolution ex vivo MRI and serial sectioning gave insight in the likely role that arteriolosclerosis has in this context. Moreover, the observations made in this study, combined with those from a recent larger community-based study (Zanon Zotin et al., under review)40, suggest that the localization of CMBs strictly within the cortical ribbon rather than in the juxtacortical or subcortical WM, could represent an additional discriminative diagnostic factor during life. These observations are qualitative in nature and need to be confirmed in additional studies, ideally in population-based studies not subject to the selection bias present in this autopsy cohort, which is a highly selected group of clinical CAA patients.
Supplementary Material
Acknowledgments
We wish to thank the patients and their families for participating in the brain donation program. Furthermore, we wish to thank Dr. Romain Perbert for helpful discussions, Dr. Keith Johnson and Erin Chuba for providing the PiB-PET data, and the MGH Neuropathology core for the routine pathological analyses. This work was funded by the National Institutes of Health (AG059893 to S.J.v.V.) and the German Research Foundation (DFG) (454245528 to V.P.).
Footnotes
Potential conflicts of Interest
The authors declare that they have no conflict of interest.
Data Availability
Source data can be made available to collaborators upon reasonable request.
References
- 1.Charidimou A, Pantoni L, Love S. The concept of sporadic cerebral small vessel disease: A road map on key definitions and current concepts. Int. J. Stroke 2016;11(1):6–18. [DOI] [PubMed] [Google Scholar]
- 2.Charidimou A, Boulouis G, Frosch MP, et al. The Boston criteria version 2.0 for cerebral amyloid angiopathy: a multicentre, retrospective, MRI–neuropathology diagnostic accuracy study. Lancet Neurol. 2022;21(8):714–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wardlaw JM, Smith EE, Biessels GJ, et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. The Lancet Neurology 2013;12(8):822–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vinters HV. Cerebral amyloid angiopathy. A critical review. Stroke 1987;18(2):311–324. [DOI] [PubMed] [Google Scholar]
- 5.Fisher CM. Pathological observations in hypertensive cerebral hemorrhage. J. Neuropathol. Exp. Neurol 1971;30(3):536–550. [DOI] [PubMed] [Google Scholar]
- 6.Blevins BL, Vinters HV, Love S, et al. Brain arteriolosclerosis. Acta Neuropathol. 2021;141(1):1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brenowitz WD, Nelson PT, Besser LM, et al. Cerebral amyloid angiopathy and its co-occurrence with Alzheimer’s disease and other cerebrovascular neuropathologic changes. Neurobiol. Aging 2015;36(10):2702–2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vernooij MW, van der Lugt A, Ikram MA, et al. Prevalence and risk factors of cerebral microbleeds. Neurology 2008;70(14):1208–14. [DOI] [PubMed] [Google Scholar]
- 9.Cordonnier C, van der Flier WM. Brain microbleeds and Alzheimer’s disease: innocent observation or key player? Brain 2011;134(2):335–344. [DOI] [PubMed] [Google Scholar]
- 10.De Reuck J, Auger F, Durieux N, et al. The topography of cortical microbleeds in frontotemporal lobar degeneration: a post-mortem 7.0-tesla magnetic resonance study. Folia Neuropathol. 2016;54(2):149–155. [DOI] [PubMed] [Google Scholar]
- 11.Boulouis G, Charidimou A, Pasi M, et al. Hemorrhage recurrence risk factors in cerebral amyloid angiopathy: Comparative analysis of the overall small vessel disease severity score versus individual neuroimaging markers. J. Neurol. Sci 2017;380:64–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shoamanesh A, Charidimou A, Sharma M, Hart RG. Should Patients With Ischemic Stroke or Transient Ischemic Attack With Atrial Fibrillation and Microbleeds Be Anticoagulated? Stroke 2017;48(12):3408–3412. [DOI] [PubMed] [Google Scholar]
- 13.Shoamanesh A, Hart RG, Connolly SJ, et al. Microbleeds and the effect of anticoagulation in patients with embolic stroke of undetermined source: an exploratory analysis of the NAVIGATE ESUS randomized clinical trial. JAMA Neurol. 2021;78(1):11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jolink WMT, van Veluw SJ, Zwanenburg JJM, et al. Histopathology of Cerebral Microinfarcts and Microbleeds in Spontaneous Intracerebral Hemorrhage. Transl. Stroke Res 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Veluw SJ, Charidimou A, van der Kouwe AJ, et al. Microbleed and microinfarct detection in amyloid angiopathy: a high-resolution MRI-histopathology study. Brain 2016;139(12):3151–3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Veluw SJ, Reijmer YD, van der Kouwe AJ, et al. Histopathology of diffusion imaging abnormalities in cerebral amyloid angiopathy. Neurology 2019;92(9):933–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Linn J, Halpin A, Demaerel P, et al. Prevalence of superficial siderosis in patients with cerebral amyloid angiopathy. Neurology 2010;74(17):1346–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Raposo N, Zanon Zotin MC, Schoemaker D, et al. Peak Width of Skeletonized Mean Diffusivity as Neuroimaging Biomarker in Cerebral Amyloid Angiopathy. AJNR Am. J. Neuroradiol 2021;42(5):875–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koemans EA, Voigt S, Rasing I, et al. Striped occipital cortex and intragyral hemorrhage: Novel magnetic resonance imaging markers for cerebral amyloid angiopathy. Int. J. Stroke 2021;16(9):1031–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perosa V, Oltmer J, Munting LP, et al. Perivascular space dilation is associated with vascular amyloid-β accumulation in the overlying cortex. Acta Neuropathol. 2022;143(3):331–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Charidimou A, Perosa V, Frosch MP, et al. Neuropathological correlates of cortical superficial siderosis in cerebral amyloid angiopathy. Brain 2020;143(11):3343–3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Becker JA, Hedden T, Carmasin J, et al. Amyloid-β associated cortical thinning in clinically normal elderly. Ann. Neurol 2011;69(6):1032–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schoemaker D, Charidimou A, Zanon Zotin MC, et al. Association of Memory Impairment With Concomitant Tau Pathology in Patients With Cerebral Amyloid Angiopathy. Neurology 2021;96(15):1975–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Desikan RS, Cabral HJ, Hess CP, et al. Automated MRI measures identify individuals with mild cognitive impairment and Alzheimer’s disease. Brain 2009;132(8):2048–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Logan J, Fowler JS, Volkow ND, et al. Distribution volume ratios without blood sampling from graphical analysis of PET data. J. Cereb. Blood Flow Metab 1996;16(5):834–840. [DOI] [PubMed] [Google Scholar]
- 26.Johnson KA, Gregas M, Becker JA, et al. Imaging of amyloid burden and distribution in cerebral amyloid angiopathy. Ann. Neurol 2007;62(3):229–234. [DOI] [PubMed] [Google Scholar]
- 27.Villeneuve S, Rabinovici GD, Cohn-Sheehy BI, et al. Existing Pittsburgh Compound-B positron emission tomography thresholds are too high: statistical and pathological evaluation. Brain 2015;138:2020–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Veluw SJ, Scherlek AA, Freeze WM, et al. Different microvascular alterations underlie microbleeds and microinfarcts. Ann. Neurol 2019;86(2):279–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Love S, Chalmers K, Ince P, et al. Development, appraisal, validation and implementation of a consensus protocol for the assessment of cerebral amyloid angiopathy in post-mortem brain tissue. Am. J. Neurodegener. Dis 2014;3(1):19–32. [PMC free article] [PubMed] [Google Scholar]
- 30.van Veluw SJ, Biessels GJ, Klijn CJM, Rozemuller AJM. Heterogeneous histopathology of cortical microbleeds in cerebral amyloid angiopathy. Neurology 2016;86(9):867–871. [DOI] [PubMed] [Google Scholar]
- 31.Perosa V, Rotta J, Yakupov R, et al. Implications of quantitative susceptibility mapping at 7 Tesla MRI for microbleeds detection in cerebral small vessel disease. Front. Neurol Accepted for publication, 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bulk M, Moursel LG, van der Graaf LM, et al. Cerebral amyloid angiopathy with vascular iron accumulation and calcification. Stroke 2018;49(9):2081–2087. [DOI] [PubMed] [Google Scholar]
- 33.Charidimou A, Martinez-Ramirez S, Reijmer YD, et al. Total Magnetic Resonance Imaging Burden of Small Vessel Disease in Cerebral Amyloid Angiopathy: An Imaging-Pathologic Study of Concept Validation. JAMA Neurol. 2016;73(8):994–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martinez-Ramirez S, Romero J-R, Shoamanesh A, et al. Diagnostic value of lobar microbleeds in individuals without intracerebral hemorrhage. Alzheimers. Dement 2015;11(12):1480–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rodrigues MA, Samarasekera N, Lerpiniere C, et al. The Edinburgh CT and genetic diagnostic criteria for lobar intracerebral haemorrhage associated with cerebral amyloid angiopathy: model development and diagnostic test accuracy study. Lancet Neurol. 2018;17(3):232–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ritter MA, Droste DW, Hegedüs K, et al. Role of cerebral amyloid angiopathy in intracerebral hemorrhage in hypertensive patients. Neurology 2005;64(7):1233–1237. [DOI] [PubMed] [Google Scholar]
- 37.Pétrault M, Casolla B, Ouk T, et al. Cerebral microbleeds: Beyond the macroscope. Int. J. Stroke 2019;14(5):468–475. [DOI] [PubMed] [Google Scholar]
- 38.Ahmed Z, Bigio EH, Budka H, et al. Globular glial tauopathies (GGT): consensus recommendations. Acta Neuropathol. 2013;126(4):537–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Forrest SL, Kril JJ, Kovacs GG. Association Between Globular Glial Tauopathies and Frontotemporal Dementia—Expanding the Spectrum of Gliocentric Disorders: A Review. JAMA Neurol. 2021;78(8):1004–1014. [DOI] [PubMed] [Google Scholar]
- 40.Zanon Zotin MC, Makkinejad N, Schneider JA, et al. Application of the Boston criteria v2.0 for the diagnosis of cerebral amyloid angiopathy in a community-based sample. Ann. Neurol Under review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McCarter SJ, Lesnick TG, Lowe V, et al. Cerebral Amyloid Angiopathy Pathology and Its Association with Amyloid-β PET Signal. Neurology 2021;97(18):e1799–e1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ikonomovic MD, Buckley CJ, Abrahamson EE, et al. Post-mortem analyses of PiB and flutemetamol in diffuse and cored amyloid-β plaques in Alzheimer’s disease. Acta Neuropathol. 2020;140(4):463–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zivadinov R, Ramasamy DP, Benedict RRH, et al. Cerebral Microbleeds in Multiple Sclerosis Evaluated on Susceptibility-weighted Images and Quantitative Susceptibility Maps: A Case-Control Study. Radiology 2016;281(3):884–895. [DOI] [PubMed] [Google Scholar]
- 44.Kozberg MG, Yi I, Freeze WM, et al. Blood-brain barrier leakage and perivascular inflammation in cerebral amyloid angiopathy. Brain Commun 2022;4(5):245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Charidimou A, Boulouis G, Haley K, et al. White matter hyperintensity patterns in cerebral amyloid angiopathy and hypertensive arteriopathy. Neurology 2016;86(6):505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source data can be made available to collaborators upon reasonable request.