

Significance
Many patients with neovascular age-related macular degeneration (nvAMD) respond inadequately to therapies targeting vascular endothelial growth factor (VEGF). Following treatment of patients with nvAMD with anti-VEGF therapy, we report decreased expression of VEGF but increased expression of a second angiogenic mediator, angiopoietin-like 4 (ANGPTL4). Increased ANGPTL4 expression is a consequence of accumulation of hypoxia-inducible factor (HIF)-1α in response to VEGF/kinase insert domain receptor (KDR) inhibition in the retinal pigment epithelium. By preventing the increase in HIF-1α accumulation in response to anti-VEGF therapy, combining 32-134D with aflibercept was more effective for choroidal neovascularization (CNV) treatment in mice than either drug alone. This suggests that combining 32-134D with current therapies may overcome the inadequate response of patients with nvAMD to anti-VEGF monotherapy.
Keywords: age-related macular degeneration, choroidal neovascularization, hypoxia inducible factor, vascular endothelial growth factor, angiopoietin-like 4
Abstract
Neovascular age-related macular degeneration (nvAMD) is the leading cause of severe vision loss in the elderly in the developed world. While the introduction of therapies targeting vascular endothelial growth factor (VEGF) has provided the first opportunity to significantly improve vision in patients with nvAMD, many patients respond inadequately to current anti-VEGF therapies. It was recently demonstrated that expression of a second angiogenic mediator, angiopoietin-like 4 (ANGPTL4), synergizes with VEGF to promote choroidal neovascularization (CNV) in mice and correlates with reduced response to anti-VEGF therapy in patients with nvAMD. Here, we report that expression of ANGPTL4 in patients with nvAMD increases following treatment with anti-VEGF therapy and that this increase is dependent on accumulation of hypoxia-inducible factor (HIF)-1α in response to inhibition of VEGF/KDR signaling in the retinal pigment epithelium (RPE). We therefore explored HIF-1 inhibition with 32-134D, a recently developed pharmacologic HIF-inhibitor, for the treatment of nvAMD. 32-134D prevented the expression of both VEGF and ANGPTL4 and was at least as effective as aflibercept in treating CNV in mice. Moreover, by preventing the increase in HIF-1α accumulation in the RPE in response to anti-VEGF therapy, combining 32-134D with aflibercept was more effective than either drug alone for the treatment of CNV. Collectively, these results help explain why many patients with nvAMD respond inadequately to anti-VEGF therapy and suggest that the HIF inhibitor 32-134D will be an effective drug—alone or in combination with current anti-VEGF therapies—for the treatment of patients with this blinding disease.
Choroidal neovascularization (CNV), the growth of abnormal leaky blood vessels under and within the retina, is a hallmark of patients with neovascular (nv) or “wet” age-related macular degeneration (nvAMD). Untreated, CNV leads to rapid and often irreversible vision loss from leakage of fluid (causing macular edema), bleeding, and scarring (1, 2). A single angiogenic mediator, vascular endothelial growth factor (VEGF), has been shown to play a central role in the development of CNV. Expression of VEGF in the eyes of patients with nvAMD is regulated by the transcription factor, hypoxia-inducible factor (HIF)-1 (3). Increased expression of VEGF by HIF-1, in turn, promotes the growth of the abnormal leaky vessels that compromise vision in patients with nvAMD.
The introduction of therapies targeting VEGF (i.e., anti-VEGF therapies) has revolutionized the treatment of nvAMD with almost half of treated patients demonstrating a clinically significant improvement of their vision (4, 5). Nonetheless, most patients with nvAMD will demonstrate persistent intraretinal or subretinal fluid despite strict adherence to the recommended treatment regimens (6). Why many patients with nvAMD treated with anti-VEGF therapies fail to respond adequately to treatment (7) remains unclear.
Using a protocol designated treat-and-extend (TAE), pause and monitor (TEP/M), a hybrid of the TAE, and pro re nata (PRN) protocols, designed to wean patients with nvAMD off anti-VEGF therapy, it was reported that after 1 y of treatment, patients fall into subgroups based on their sensitivity to anti-VEGF therapy (8). While their response to anti-VEGF therapy did not correlate with the aqueous levels of VEGF, they did correlate with the aqueous levels of a second HIF-regulated angiogenic mediator, angiopoietin-like 4 (ANGPTL4) (9). Expression of ANGPTL4 synergized with VEGF to promote endothelial cell tubule formation in vitro and CNV in vivo (9). Accordingly, simultaneously targeting both ANGPTL4 and VEGF in the laser CNV mouse model was more effective than targeting either factor alone (9). These observations support a role for ANGPTL4 in the development of CNV and the resistance to anti-VEGF therapy in patients with nvAMD. Here, we use a combination of human samples, mouse models, and cell-based studies to explore the mechanism whereby ANGPTL4 expression influences the response of patients with nvAMD to anti-VEGF therapy and provide evidence that a recently developed HIF-inhibitor, 32-134D, in combination with anti-VEGF therapy, may be a more effective approach to treat patients with nvAMD than anti-VEGF therapy alone.
Results
Treatment of Patients with nvAMD with Anti-VEGF Therapy Results in a Countertherapeutic Increase in the Expression of ANGPTL4.
Aqueous levels of ANGPTL4—but not VEGF—have been reported to correlate inversely with the response of patients with nvAMD to treatment with anti-VEGF therapy: The higher the levels of ANGPTL4, the poorer the response of patients to anti-VEGF therapy (9, 10). To better understand the relationship between ANGPTL4 expression in patients with nvAMD and their response to anti-VEGF therapy, we compared the expression of VEGF and ANGPTL4 in the aqueous of patients with treated and untreated nvAMD as well as patients with non-neovascular (nnv) or “dry” AMD and non-AMD (control) patients (SI Appendix, Table S1). Close inspection of the scatter plot demonstrated that the levels of ANGPTL4 and VEGF in the aqueous fluid of patients with nvAMD could be used to distinguish them from patients with nnvAMD and from control patients (Fig. 1A and SI Appendix, Fig. S1). VEGF levels were markedly lower in the aqueous fluid from patients who underwent their first treatment with anti-VEGF therapy (either bevacizumab or aflibercept) 4 to 6 wk prior to sample collection (nvAMD first treatment; Fig. 1A; vertical arrow), as expected. Conversely, in patients who had a remote (>12 wk) history of receiving anti-VEGF therapy and evidence of recurrent CNV, the aqueous levels of VEGF were similar to untreated patients with nvAMD (Fig. 1A). By contrast, in patients with nvAMD who had a prior history of anti-VEGF therapy, the levels of ANGPTL4 were further increased both in patients with active CNV who had a recent (within 4 to 6 wk; nvAMD first treatment) or remote (>12 wk; nvAMD recurrent) history of treatment compared to untreated patients with nvAMD (nvAMD Untreated; Fig. 1A; horizontal arrows).
Fig. 1.
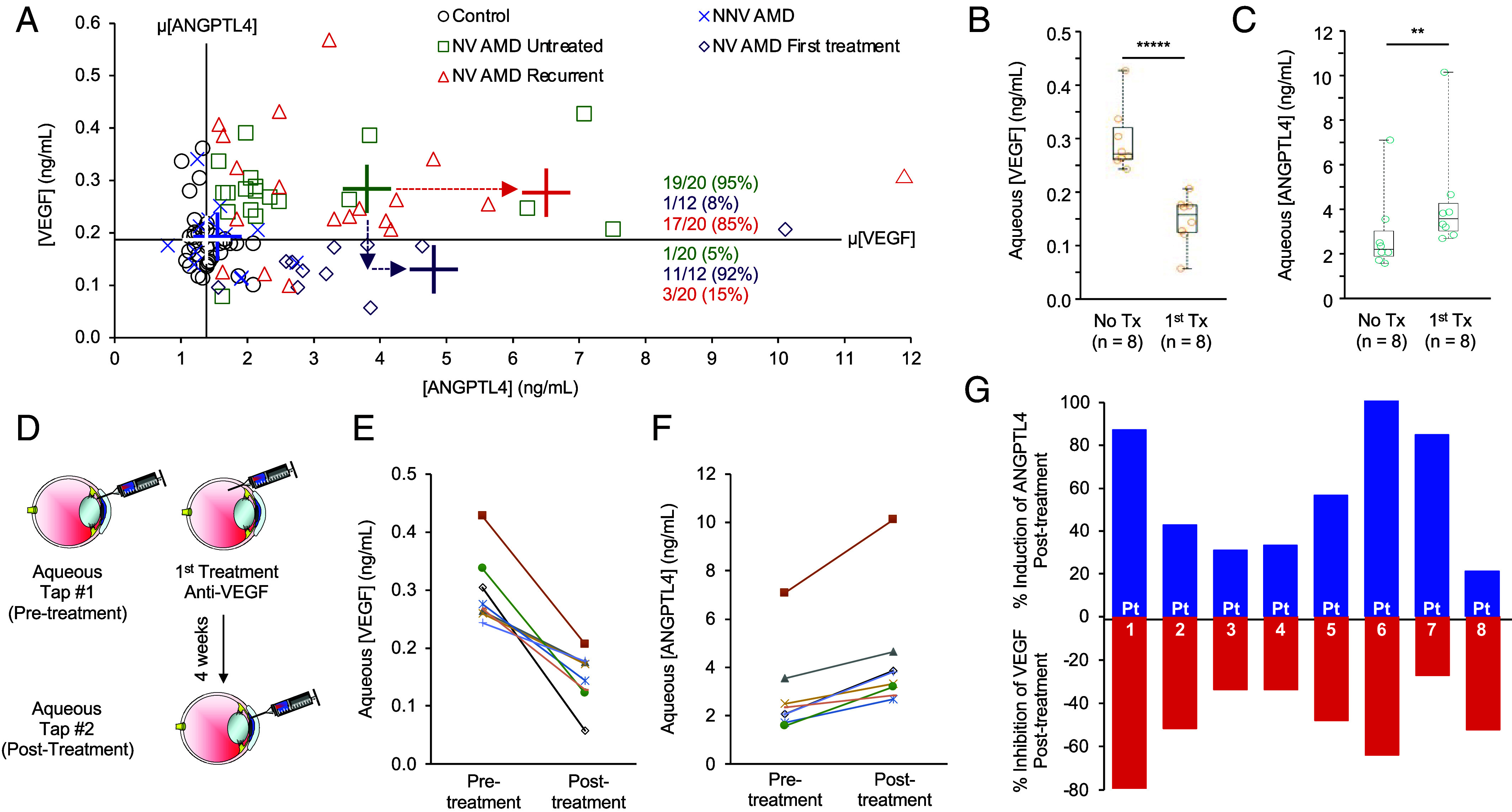
Aqueous levels of ANGPTL4 are increased in patients with nvAMD following treatment with anti-VEGF therapy. (A) Scatter plot comparing aqueous levels of VEGF and ANGPTL4 in NV AMD patients (untreated, following their first treatment or with recurrent CNV), compared to NNV AMD and non-AMD controls. Average (cross) VEGF (horizontal bar) and ANGPTL4 (vertical bar) levels for control (white), NN VAMD (blue), or NV AMD (untreated, green; first treatment, purple; or recurrent, red). The color matched number (percent) of patients with high VEGF/high ANGPTL4 or low VEGF/high ANGPTL4 are shown. Dashed arrows denote change in mean ANGPTL4 (horizontal) or VEGF (vertical) levels in untreated vs. first treatment or recurrent NV AMD. (B and C) Aqueous levels of VEGF (B) and ANGPTL4 (C) in NV AMD patients (treatment-naïve, No Tx; following one treatment, 1st Tx). (D) Schematic of aqueous fluid acquisition from NV AMD patients before and 4 wk after receiving a single treatment with anti-VEGF therapy. (E and F) Aqueous levels of VEGF (E) and ANGPTL4 (F) in patients with NV AMD prior to (pretreatment) and 4 wk following (posttreatment) their first anti-VEGF treatment. (G) Bar graph depicting the % change of aqueous ANGPTL4 (blue) or VEGF (red) in these patients. Statistical analyses were performed by two-tailed Student’s t test, *P < 0.05; **P < 0.01; *****P < 0.00001. NS, not significant.
To further explore the expression of ANGPTL4 following treatment with anti-VEGF therapy in patients with nvAMD, we examined its expression in the aqueous fluid from patients with nvAMD with no prior history of anti-VEGF therapy (nvAMD No Tx) or from a second group of patients with nvAMD who received their first treatment with anti-VEGF therapy within 4 to 6 wk of sample collection (nvAMD 1st Tx). We observed lower VEGF levels (Fig. 1B), but higher ANGPTL4 levels (Fig. 1C) in the aqueous fluid of patients with nvAMD who had recently been treated with anti-VEGF therapy compared to untreated patients.
We next analyzed aqueous fluid samples from 8 patients with newly diagnosed (i.e., treatment-naïve) nvAMD for the levels of VEGF and ANGPTL4 and then treated them with anti-VEGF therapy (Fig. 1D). When we reexamined aqueous levels of VEGF and ANGPTL4 in these patients 4 wk later, we observed a decrease in VEGF levels following treatment with anti-VEGF therapy in 8/8 patients with nvAMD (Fig. 1E). Conversely, we observed an increase in ANGPTL4 levels in 8/8 patients with nvAMD 4 wk after a single treatment with anti-VEGF therapy (Fig. 1F). Interestingly, the increase in ANGPTL4 mirrored the decrease in VEGF in 6/8 patients (Fig. 1G). Collectively, these results demonstrate that treatment of patients with nvAMD with anti-VEGF therapy results in a therapeutic decrease in VEGF, but a countertherapeutic increase in the expression of ANGPTL4, supporting a role for ANGPTL4 in limiting the treatment response of patients with nvAMD to anti-VEGF therapy.
Treatment with Anti-VEGF Therapy Results in Increased ANGPTL4 mRNA and Protein Expression in a Mouse Model for nvAMD.
We next employed a mouse model of CNV, in which a laser is used to rupture Bruch’s membrane (11), to explore the mechanism whereby treatment with anti-VEGF therapy results in an increase in ANGPTL4. The laser CNV model has proven to be a powerful tool to examine molecular events underlying the development of CNV (12) and the role of ANGPTL4 in its promotion (9). We treated mice with an intravitreal injection of aflibercept (which is effective in humans and mice) or vehicle control (PBS), 1 d following laser treatment (Fig. 2A) and examined the expression of ANGPTL4 protein; we used a low dose of aflibercept (300 ng) to allow for the development of the CNV lesion despite treatment. On day 3, when ANGPTL4 expression is not yet detected in the laser CNV model, we observed a marked increase in the expression of ANGPTL4 in the outer retina of CNV lesions of animals treated with aflibercept compared to vehicle control (Fig. 2B). By day 7, expression of ANGPTL4 is detected in the outer retina of CNV lesions but is further increased in animals treated with aflibercept (Fig. 2C), similar to what was observed in patients with nvAMD following treatment with anti-VEGF therapy. This correlated with an increase in Angptl4 mRNA expression in the RPE/choroid as measured by qPCR (Fig. 2D). These results were corroborated by RNAscope when mice were pretreated with a low dose of aflibercept (300 ng) 3 d prior to laser treatment to allow sufficient time to detect the accumulation of Angptl4 mRNA (Fig. 2E). Angptl4 mRNA was detected in the outer retina of CNV lesions in animals pretreated with aflibercept, but not with vehicle, on day 3 (Fig. 2F). By day 7, Angptl4 mRNA was observed in CNV lesions in animals pretreated with vehicle control but was increased in animals pretreated with aflibercept (Fig. 2G), similar to what was observed with ANGPTL4 protein. Collectively, these observations demonstrate that anti-VEGF therapy promotes increased ANGPTL4 mRNA and protein expression in a mouse model for CNV.
Fig. 2.
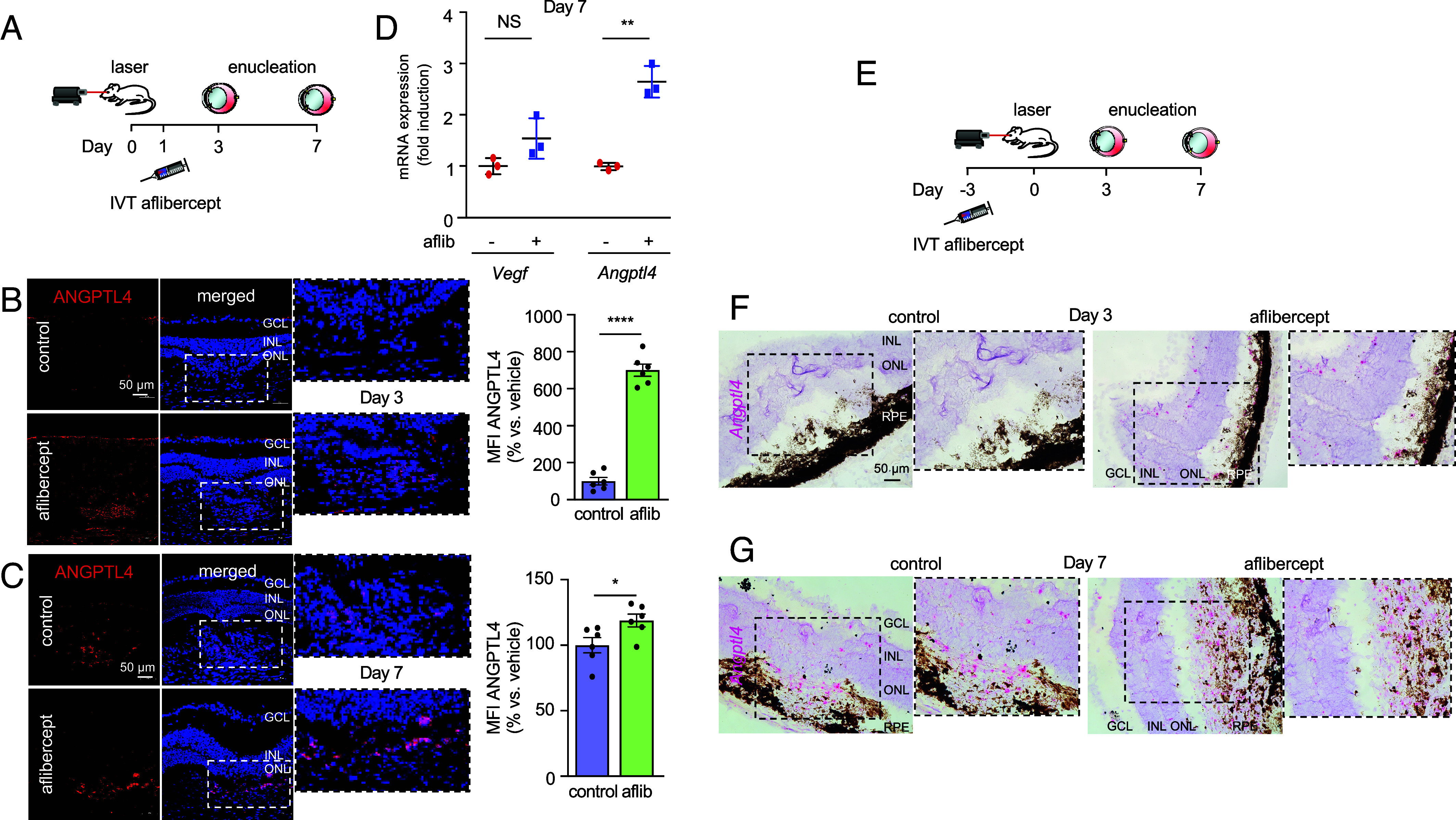
Anti-VEGF therapy increases ANGPTL4 expression in mice. (A) Schematic depicting laser CNV mice treated with an injection of aflibercept (300 ng) or PBS 1 d after laser treatment. (B and C) Representative image depicting expression of ANGPTL4 protein (immunofluorescence) within CNV lesions on day 3 (B) or 7 (C) (Left) with quantitation (Right). (D) Scatter plot demonstrating expression of Vegf and Angptl4 mRNA (qPCR) in RPE/choroid lysates on day 7. (E) Schematic depicting laser CNV mice treated with an injection of aflibercept (300 ng) or PBS 3 d prior to laser treatment. (F and G) Expression of Angptl4 mRNA (in situ hybridization) within CNV lesions on day 3 (F) or 7 (G). n = 3-6 animals per group. GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer; RPE, retinal pigment epithelium; MFI, mean fluorescence intensity; IVT, intravitreal; aflib, aflibercept. Data are shown as means ± SD. Statistical analyses were performed by two-tailed Student’s t test, *P < 0.05; **P < 0.01; ****P < 0.0001. NS, not significant.
Hypoxia Promotes the Accumulation of HIF-1α and VEGF in the Laser-Induced Mouse Model of CNV.
To further interrogate the mechanism whereby treatment of CNV with anti-VEGF therapy results in an increase in ANGPTL4 mRNA and protein expression, we shifted our attention upstream from ANGPTL4 to HIF-1α, the transcription factor that has been previously implicated in regulating ANGPLTL4 expression in ocular disease (13–22), including nvAMD (3, 18). To this end, we explored HIF-1α expression in the laser CNV mouse model. Following laser treatment, we observed an increase in outer retinal hypoxia, as measured using HypoxyProbe™ that peaked at day 1, and largely resolved by day 3 (SI Appendix, Fig. S2 A and B). This correlated with an increase in HIF-1α protein accumulation as early as day 1, which was still present at day 3 but was no longer detectable by day 7 (SI Appendix, Fig. 2C). Accumulation of HIF-1α protein, in turn, promoted VEGF mRNA and protein expression (SI Appendix, Fig. S2 D and E). Intraocular administration of the pharmacologic HIF inhibitor, acriflavine 1 d following laser treatment (23) (SI Appendix, Fig. S2F) resulted in a reduction in HIF-1α accumulation and, in turn, Vegf mRNA expression in the RPE/choroid (SI Appendix, Fig. S2 G and H), and was sufficient to reduce CNV lesion size to that observed following treatment with a high dose (800 ng) of aflibercept (SI Appendix, Fig. S2I). Collectively, these results demonstrate the critical role for HIF-1α in promoting VEGF expression and the development of CNV in the laser CNV model.
Accumulation of HIF-1α Following Treatment with Anti-VEGF Therapy Promotes ANGPTL4 Expression in Laser CNV Mice.
We next examined whether the expression of HIF-1α is influenced by treatment with aflibercept. Treatment with a moderate dose of aflibercept (400 ng), 1 d after laser treatment, resulted in an increase in HIF-1α accumulation 24 h later (SI Appendix, Fig. S3A) that localized to the outer retina of CNV lesions (SI Appendix, Fig. S3B). These results suggested that the increase in ANGPTL4 expression following treatment with anti-VEGF therapy may be due to an increase in HIF-1α accumulation.
To interrogate the contribution of HIF-1α accumulation to the increase in ANGPTL4 mRNA and protein expression following treatment with anti-VEGF therapy, we used mice that were heterozygous for a knockout allele at the Hif1a locus (Hif1a+/−) (24). We took advantage of prior observations that basal levels of HIF-1α are relatively normal in Hif1a+/− mice, whereas in response to ischemia, HIF-1α expression is largely unchanged in Hif1a+/− mice but potently stimulated in wild type (wt) littermate controls (25). Following laser treatment (SI Appendix, Fig. S3C), both HIF-1α protein expression in the RPE/choroid (day 2; SI Appendix, Fig. S3D) as well as VEGF and ANGPTL4 protein expression (day 7; SI Appendix, Fig. S3 E and F) were markedly reduced in Hif1a+/− mice compared to wt littermate controls. In wt littermate controls, intravitreal injection with a moderate dose of aflibercept (400 ng) 1 d after laser treatment stimulated an increase in HIF-1α protein accumulation at day 2 (SI Appendix, Fig. S3D). Conversely, in Hif1a+/− mice, intravitreal injection with aflibercept resulted in only a modest increase in HIF-1α accumulation compared to PBS control. Interestingly, while Vegf mRNA expression at day 7 was unchanged in wt mice following intravitreal injection with aflibercept, expression of Vegf mRNA in the RPE/choroid was decreased in Hif1a+/− mice (SI Appendix, Fig. S3G), suggesting that the increase in HIF-1α accumulation in response to inhibition of VEGF is required to maintain stable Vegf mRNA expression. Conversely, intraocular injection with aflibercept stimulated a marked increase in Angptl4 mRNA expression in the RPE/choroid in wt animals; this increase was not observed in Hif1a+/− mice (SI Appendix, Fig. S3H). Collectively, these results suggest that inhibition of VEGF with anti-VEGF therapy could result in a compensatory increase in HIF-1α accumulation and, in turn, ANGPTL4 expression in treated patients.
RPE Cells Up-Regulate HIF-1α Expression in the Laser CNV Mouse Model.
RPE cells have been reported to drive the expression of the angiogenic mediators that stimulate the development of CNV in patients with nvAMD (26). In patients geographic atrophy (GA), CNV develops in areas with preserved RPE surrounding the GA or within the spared foveal island (27), consistent with prior histopathological (28) and clinical studies (29, 30). These studies suggest that a viable RPE is required for the development of CNV. We therefore examined whether RPE cells contribute to the expression of HIF-1α in the laser CNV mouse model. On day 1 (Fig. 3A), we observed coexpression of the RPE-specific marker, RPE65 in cells expressing HIF-1α in the outer retina in mice following treatment with laser (Fig. 3 B–F). On day 3 (Fig. 3G), expression of HIF-1α in the outer retina increased, but remained localized primarily to the outer retina, and again included RPE65-expressing cells (Fig. 3 H–K). The diffuse expression of HIF-1α, predominantly in RPE65-expressing cells, was confirmed in CNV lesions on choroidal flat mounts at day 3 (Fig. 3 L–N). Expression of HIF-1α was no longer detected by day 7 following laser treatment (SI Appendix, Fig. S4). HIF-1α expression was not observed in the outer retina of adjacent (nonlasered) control tissue at day 1 or 3 (Fig. 3 B and H), nor in the outer retina of the contralateral (nonlasered) control eyes (SI Appendix, Fig. S5). Collectively, these results support a role for expression of HIF-1α in RPE cells in the development of CNV.
Fig. 3.
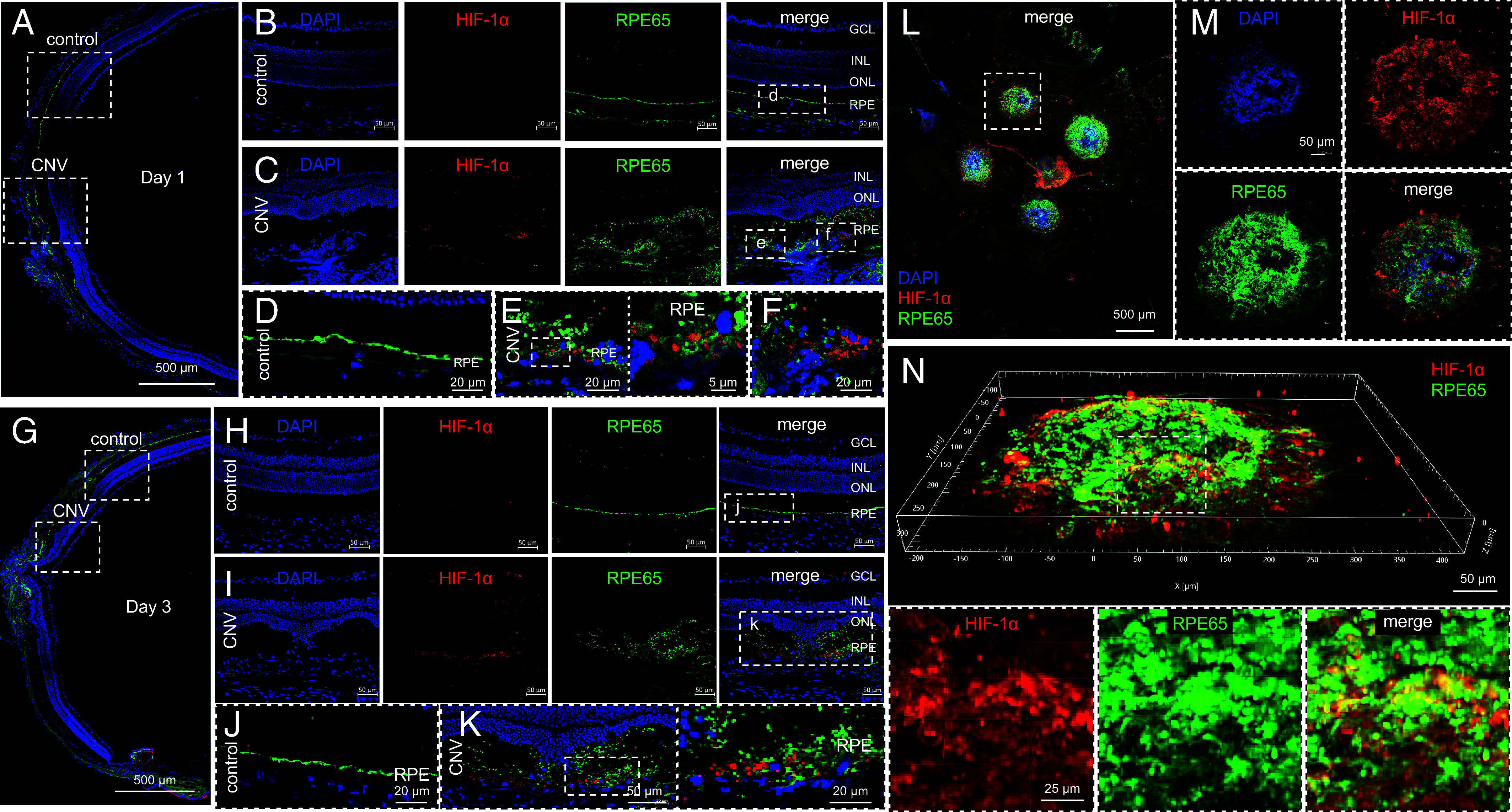
Expression of HIF-1α in RPE cells in laser CNV mice. (A–K) Representative image depicting expression of HIF-1α (red) and RPE65 (green) in laser CNV mice on day 1 (A–F) or day 3 (G–K). Adjacent (nonlasered) retina is shown as controls. (L–N) Representative image depicting expression of HIF-1α (red) and RPE65 (green) in flat mount (L and M) or Z-stack (N) from laser CNV mice on day 3. n = 4-6 animals per group. GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer; RPE, retinal pigment epithelium.
Increased HIF-1α Expressing RPE Cells in Laser CNV Mice Following Treatment with Aflibercept.
Administration of either aflibercept or ranibizumab to cocultures of RPE and endothelial cells has been reported to result in a dose–response increase in RPE cell viability and increased RPE cell migration and proliferation (31). We therefore next examined the response RPE cells following treatment with aflibercept in laser CNV mice. To ensure sufficient time for the RPE to respond to treatment with anti-VEGF therapy, we performed intraocular injections with aflibercept 3 d prior laser treatment (SI Appendix, Fig. S6A) and observed an increase in RPE65-expressing cells at day 1 (SI Appendix, Fig. 6 B and C). This was not observed in adjacent (nonlasered) control tissue (SI Appendix, Fig. S6D). These RPE cells demonstrated an increase in HIF-1α expression (Fig. 4 A–D).
Fig. 4.
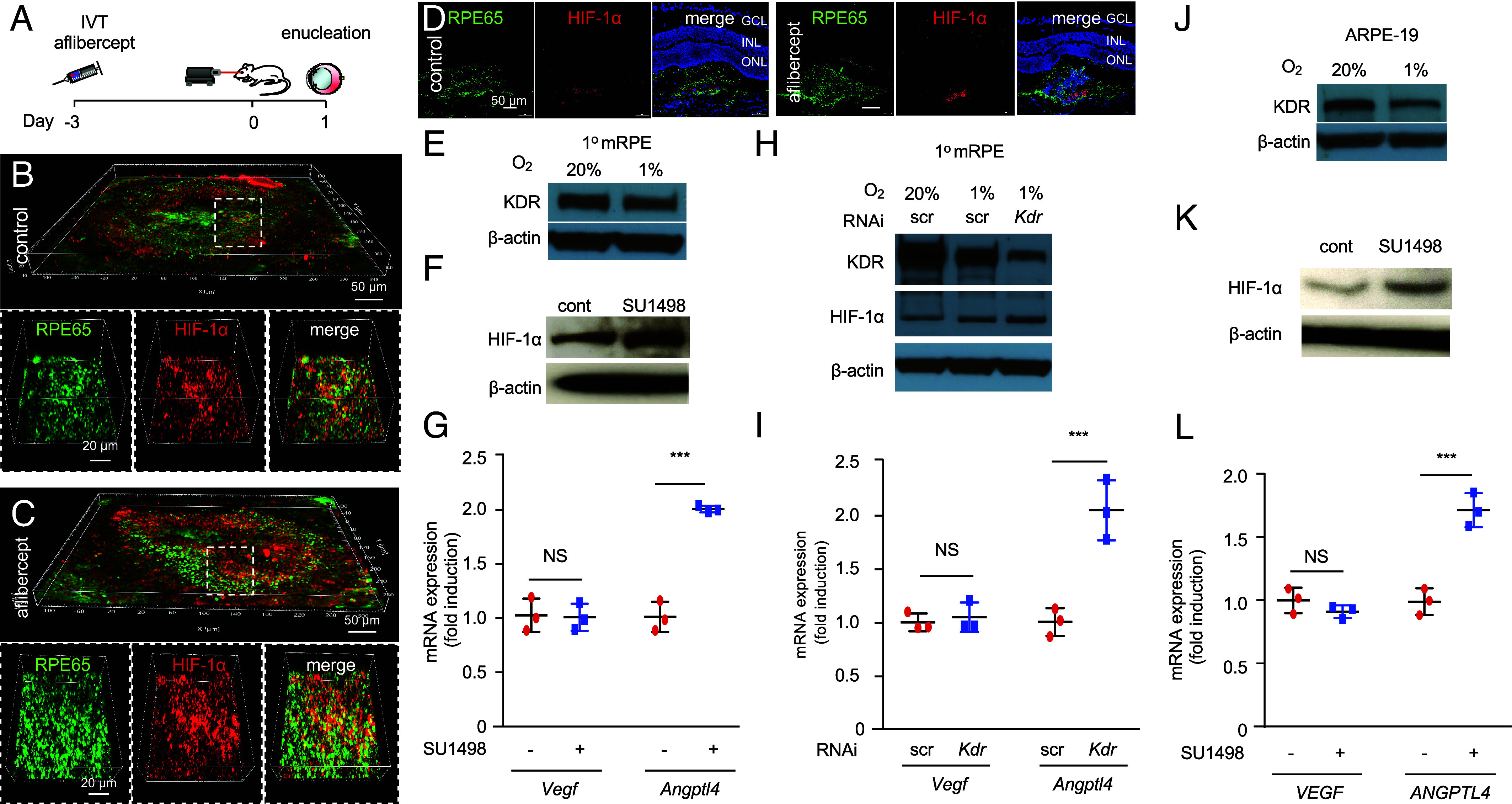
The VEGF/KDR axis regulates expression of HIF-1α expression in RPE cells. (A) Above, schematic depicting laser CNV mice treated with an injection of aflibercept (300 ng) or PBS 3 d before laser treatment. (B–D) Representative images depicting expression of the RPE cell-specific marker, RPE65 (green) and HIF-1α (red) in laser CNV mice on day 1 in flat mounts (B and C) and sections (D) after treatment with PBS or aflibercept. (E) Western blot depicting expression of KDR in primary mouse RPE cells (1° mRPE) cultured in normoxia (20% O2) or hypoxia (1% O2) for 24 h. (F and G) Effect of KDR inhibition using SU1498 (1 µM; 24 h) on HIF-1α accumulation (F) and Vegf and Angptl4 mRNA expression (qPCR; H) in 1° mRPE. Vehicle (DMSO) was used as a control. (H) Western blot depicting expression of KDR and HIF-1α protein in lysates from 1° mRPE cultured in 20% O2 or 1% O2 for 24 h in the presence of siRNA targeting Kdr or scrambled (scr) control for. (I) Vegf and Angptl4 mRNA expression (qPCR) in 1° mRPE in the presence of siRNA targeting Kdr or scrambled (scr) control. (J) Western blot depicting expression of KDR in ARPE-19 cells cultured in 20% or 1% O2 for 24 h. (K and L) Effect of KDR inhibition using SU1498 (1 µM; 24 h) vs. vehicle (DMSO) control on HIF-1α accumulation (F) and Vegf and Angptl4 mRNA expression (qPCR; H) in ARPE-19 cells. Data are shown as means ± SD. Statistical analyses were performed by two-tailed Student’s t test with Welch’s Correction, *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; NS, not significant.
RPE cells have been reported to express VEGF receptor 2 (KDR) (32) and that secreted VEGF may play a role as an autocrine survival for RPE in AMD (33, 34). Based on our observations, we postulated that expression of KDR on RPE cells may also allow these cells to detect (and respond to) reduced levels of angiogenic mediators critical for the survival of the underlying choriocapillaris. To interrogate the response of RPE cells to reduced VEGF/KDR signaling, we isolated primary RPE cells from mice. Expression of KDR in these cells was detected under physiologic (20% O2) culture conditions and was slightly reduced under hypoxic (1% O2) culture conditions (Fig. 4E). Inhibition of the KDR using the pharmacologic inhibitor, SU1498, resulted in an increase in HIF-1α protein expression (Fig. 4F) and, in turn, Angptl4 mRNA expression (Fig. 4G); expression of Vegf mRNA was not affected. These results were corroborated following knockdown of KDR expression in primary mouse RPE cells using RNA interference (Fig. 4 H and I), and in the immortalized human RPE cell line, ARPE-19 using the KDR inhibitor, SU1498 (Fig. 4 J–L). Collectively, these results support a role for the VEGF/KDR signaling axis in regulating HIF-1α protein expression (and the expression of HIF-regulated genes) in RPE cells.
RPE Cells Secrete ANGPTL4 and VEGF, but Not ANGPT2 or EPO.
HIF-1α regulates the expression of numerous angiogenic mediators, in addition to VEGF and ANGPTL4. We therefore set out to examine whether the increase in HIF-1α in response to anti-VEGF therapy resulted in an increase in other key HIF-regulated angiogenic mediators. Expression of the HIF-regulated angiogenic mediator ANGPT2 has been reported in surgically excised CNV membranes from patients with nvAMD (35). This has led to the development of therapies targeting ANGPT2 and its receptor, TIE2. Interestingly, unlike ANGPTL4, expression of ANGPT2 was not elevated in patients who had recently been treated with anti-VEGF therapy compared to untreated patients (SI Appendix, Fig. S7A). Similar results were observed for another HIF-regulated gene, erythropoietin (EPO; SI Appendix, Fig. S7B), which has also been previously implicated in the pathogenesis of nvAMD (36, 37).
We hypothesized that the reason why expression of ANGPTL4—but not ANGPT2 or EPO—was increased following treatment of nvAMD patients with anti-VEGF therapy could be a consequence of the ability of RPE cells to express only a subset of HIF-regulated angiogenic mediators. To interrogate this hypothesis, we examined the expression of HIF-regulated angiogenic factors in ARPE-19 cells cultured in hypoxia. Expression of VEGF, ANGPTL4, and ANGPT2 and EPO mRNA were all increased in ARPE-19 cells cultured in hypoxia (SI Appendix, Fig. S7 C–F). However, we observed secretion of VEGF and ANGPTL4, but not ANGPT2, by ARPE-19 cells cultured in hypoxia, consistent with prior studies demonstrating that ANGPT2 was expressed specifically by vascular endothelial cells (38). Similar results were obtained with EPO. These results help explain why expression of ANGPTL4—but not ANGPT2 and EPO—is increased in patients following treatment with anti-VEGF therapy.
Inhibition of HIF-1α with 32-134D Effectively Treats CNV Lesions in Mice.
Collectively, these studies identify a potential limitation of current anti-VEGF therapies for the treatment of patients with nvAMD. To avoid the countertherapeutic increase in HIF-1α and ANGPTL4 expression by RPE cells following treatment with anti-VEGF therapies, we set out to evaluate the therapeutic efficacy of targeting HIF-1α for the treatment of CNV. While preclinical studies of the pharmacologic HIF inhibitor, acriflavine (23), for the treatment of ocular vascular disease have been promising (39), recent studies demonstrate that its safety profile following intraocular administration makes it a less desirable candidate to translate to the clinic (22). Instead, we took advantage of a recently developed pharmacologic HIF inhibitor, 32-134D (40), which was reported to effectively inhibit HIF-1α and HIF-2α protein accumulation, resulting in a modest but broad reduction of dozens of HIF-regulated vasoactive mediators following intraocular administration at doses that are well tolerated in mouse models for diabetic eye disease (22). 32-134D has been shown to be more effective than aflibercept at preventing retinal neovascularization in a mouse model for ischemic retinal neovascularization (22). We first evaluated the efficacy of intraocular administration of 32-134D in the laser CNV model. Treatment with 32-134D (70 ng) on day 3 (to allow CNV lesion to develop prior to treatment) resulted in a marked reduction in the size of CNV lesion size in mice (Fig. 5A). Examination of the CNV lesion demonstrated a reduction in Vegf mRNA expression in the outer retina and RPE (Fig. 5B). This, in turn, resulted in a reduction of VEGF protein expression in the RPE and choroid as well as the neurosensory retina to the levels observed in nonlasered (control) animals (Fig. 5 C and D), similar to what was observed following treatment with aflibercept (SI Appendix, Fig. S8). While VEGF expression and CNV lesion size were both markedly reduced with treatment with 32-134D, VEGF expression was observed within the smaller CNV lesions, albeit significantly less than that observed in the untreated CNV lesions (Fig. 5E). Expression of ANGPTL4 following treatment with 32-134D was also markedly reduced even within these smaller CNV lesions (Fig. 5 F and G). These results suggest that 32-134D monotherapy, which reduces expression of both VEGF and ANGPTL4, could be an effective approach for the treatment of CNV.
Fig. 5.
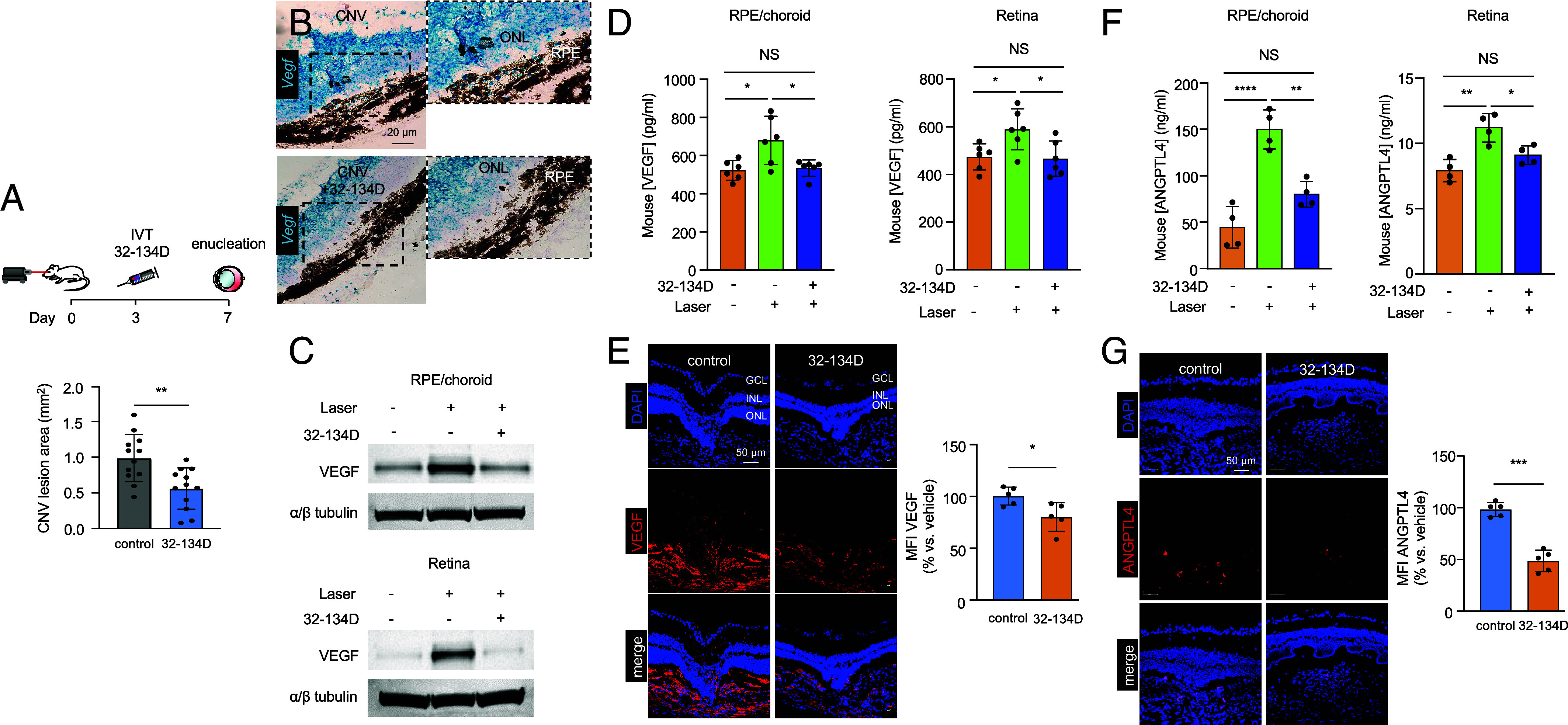
32-134D inhibits ANGPTL4 and VEGF expression and CNV lesion size in laser CNV mice. (A) Above, schematic depicting laser CNV in 13-to 15-wk-old mice treated with an injection of 32-134D (70 ng) or vehicle (DMSO; control) 3 d after laser treatment. Below, analysis of CNV lesion size on choroid flat mounts on day 7. (B) Expression of Vegf mRNA expression (in situ hybridization) within CNV lesions following treatment with 32-134D or vehicle. (C and D) Expression of VEGF (C, ELISA; D, WB) in RPE/choroid or neurosensory retina lysates from laser CNV mice treated with 32-134D or vehicle. (E–F) Expression of VEGF (E) or ANGPTL4 (F, ELISA; G) by immunofluorescence (Left) with quantitation (Right) on day 7 in laser CNV mice treated with 32-134D or vehicle. n = 5-6 animals. GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer; RPE, retinal pigment epithelium; MFI, mean fluorescence intensity; IVT, intravitreal. Data are shown as means ± SD. Statistical analyses were performed by two-tailed Student’s t test with Welch’s Correction (A, E, and G) or one-way ANOVA with Bonferroni’s multiple-comparison test (D and F). *P < 0.05; ** P < 0.01; ***P < 0.001; ****P < 0.0001; NS, not significant.
32-134D Prevents the Countertherapeutic Increase in HIF-1α Observed Following Treatment with Aflibercept.
We next set out to determine whether 32-134D could prevent the increase in HIF-1α observed following treatment with aflibercept. To examine the effect of 32-134D on the early expression of HIF-1α, we pretreated mice with 70 ng of 32-134D 1 d prior to laser treatment and then collected eyes 1 d after laser treatment (Fig. 6A) and examined HIF-1α accumulation. We observed a marked reduction in HIF-1α accumulation in the RPE/choroid, as well as the neurosensory retina in animals pretreated with 32-134D compared to vehicle control (Fig. 6 B and C). Pretreatment with a low dose of aflibercept (300 ng) 1 d prior to laser treatment resulted in augmentation of HIF-1α accumulation at day 1; this was prevented in animals pretreated with 32-134D (Fig. 6D). Interestingly, this was independent of the increase in RPE65-expressing cells, which was not affected by treatment with 32-134D (SI Appendix, Fig. S9 A and B). This effect was more notable when animals were pretreated with both 32-134D and aflibercept 3 d prior to laser treatment (Fig. 6 E–G and SI Appendix, Fig. S9 C and D). This suggests that the addition of 32-134D could prevent the countertherapeutic increase of HIF-1α accumulation (and ANGPTL4 expression) observed in patients treated with anti-VEGF therapy.
Fig. 6.
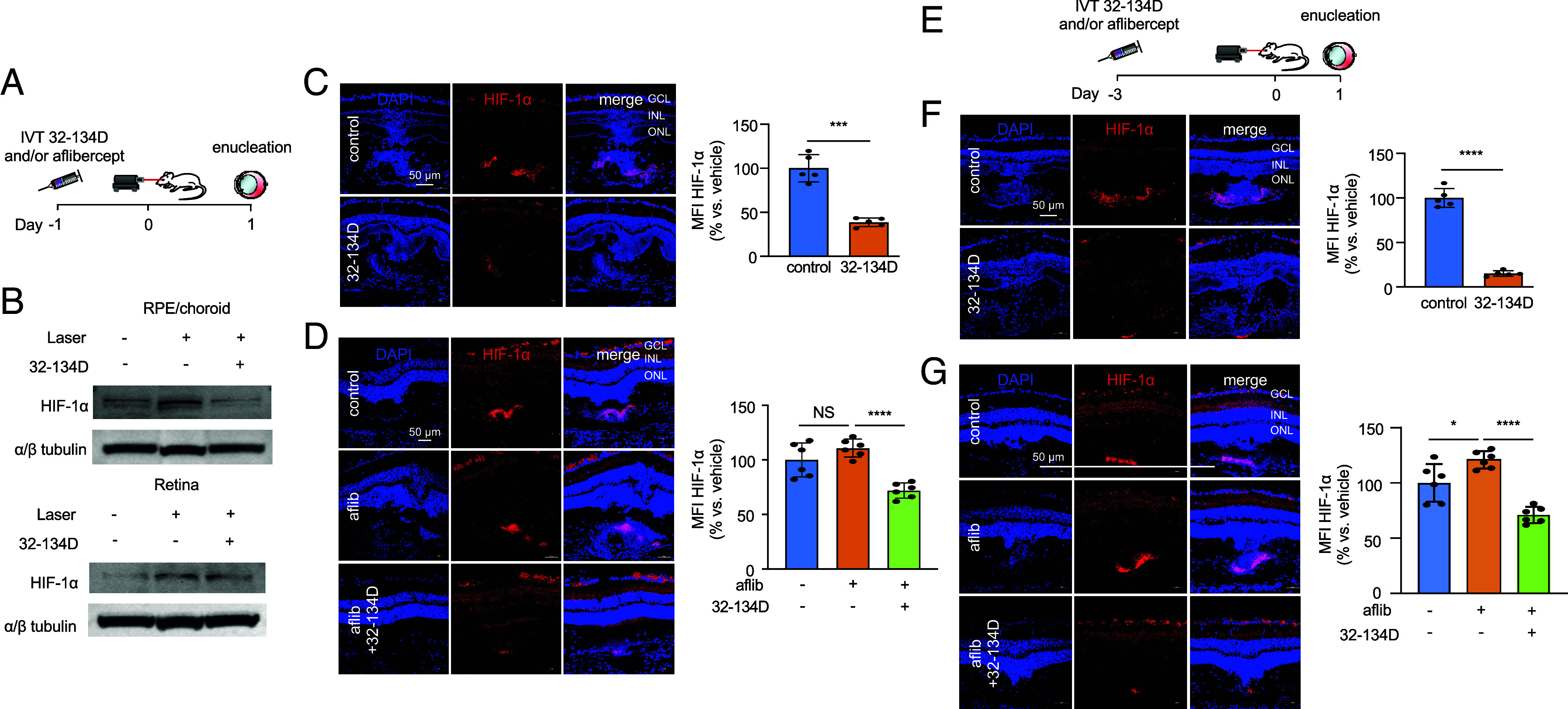
32-134D prevents aflibercept-induced increase in HIF-1α accumulation in laser CNV mice. (A) Schematic depicting laser CNV mice treated with an injection of aflibercept (300 ng) and/or 32-134D (70 ng) 1 d before laser treatment. (B) Expression of HIF-1α protein (WB) in RPE/choroid or neurosensory retina lysates from laser CNV mice treated with 32-134D or vehicle. (C and D) Expression of HIF-1α (immunofluorescence; Left) with quantitation (Right) within CNV lesions on day 1 in mice treated with 32-134D (C) or aflibercept and/or 32-134D (D). (E) Schematic depicting laser CNV mice treated with an injection of aflibercept (300 ng) and/or 32-134D (70 ng) 3 d before laser treatment. (F and G) Expression of HIF-1α (immunofluorescence; Left) with quantitation (Right) within CNV lesion on day 1 in mice treated with 32-134D (F) or aflibercept and/or 32-134D (G). n = 5-6 animals. GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer; IVT, intravitreal; aflib, aflibercept. Data are shown as means ± SD. Student’s t test with Welch’s Correction (C and F) and one-way ANOVA with Bonferroni’s multiple-comparison test (D and G). *P < 0.05; ***P < 0.001; ****P < 0.0001; NS, not significant.
Simultaneous Inhibition of Both HIF-1 and VEGF Is More Effective than Inhibition of Either Alone for the Treatment of CNV in Mice.
To determine the therapeutic potential of simultaneous inhibition of HIF-1 and VEGF for the treatment of CNV, we first performed a dose–response for aflibercept and 32-134D delivered on day 3 (Fig. 7A) to identify the threshold dose of each for the treatment of CNV. We observed that 300 ng of aflibercept and 30 ng of 32-134D (SI Appendix, Fig. S10) was sufficient to observe a small but significant reduction in CNV lesion size in treated animals. We then combined a subthreshold (100 ng) or threshold (300 ng) dose of aflibercept with a subthreshold (10 ng) or threshold (30 ng) dose of 32-134D (Fig. 7B) to determine whether targeting HIF-1α influences the response of CNV lesions size in mice treated with anti-VEGF therapy. There was a trend toward increased efficacy when combining a subthreshold dose of aflibercept (100 ng) and/or 32-134D (10 ng) with a subthreshold (Fig. 7B; highlighted in Fig. 7C) or threshold (Fig. 7B; highlighted in Fig. 7 D and E) dose of the other therapy; however, this did not reach statistical significance. When we combined a threshold dose of aflibercept (300 ng) with a threshold dose of 32-134D (30 ng), we observed a significant improvement in the efficacy of the combination of the two compared to either treatment alone (Fig. 7B; highlighted in Fig. 7F).
Fig. 7.
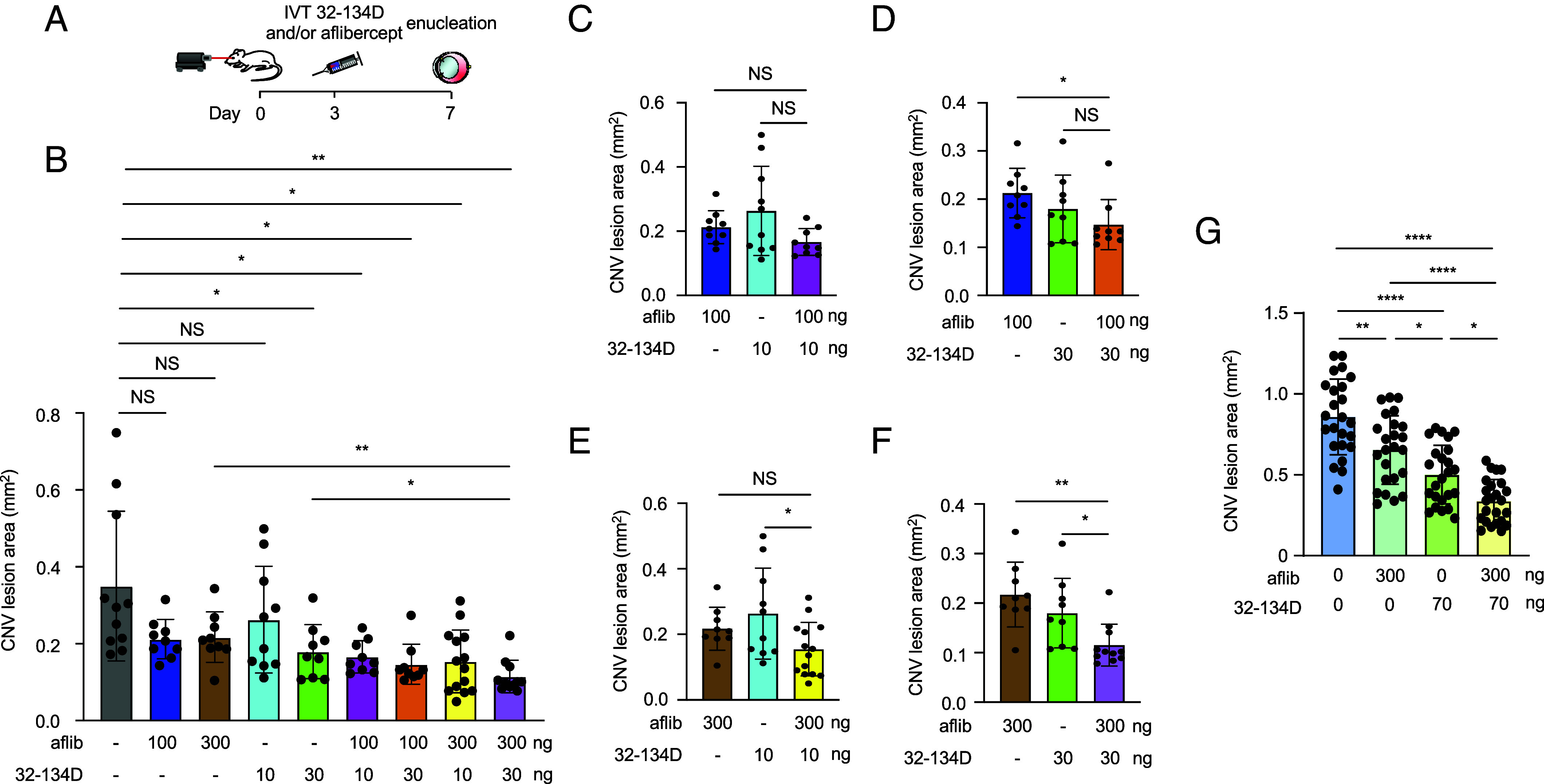
32-134D prevents the countertherapeutic increase in ANGPTL4 observed with aflibercept alone, thereby improving the efficacy of aflibercept. (A) Schematic depicting laser CNV mice treated with an injection of aflibercept and/or 32-134D 3 d after laser treatment. (B) Combined effect of a subthreshold or threshold doses of aflibercept and/or 32-134D on the CNV lesions size on day 7 in 9- to 12-wk-old mice. (C–F) Data from Panel B presented to highlight the effect of combining subthreshold and threshold doses of aflibercept and/or 32-134D. (G) Additive effect of a half maximal inhibitory concentration (IC50) of aflibercept and/or 32-134D on the CNV lesions size on day 7 in 13- to 15-wk-old mice. n = 4-5 animals. IVT, intravitreal; aflib, aflibercept. Data are shown as means ± SD. Statistical analyses were performed in a two-tailed Student’s t test with Welch’s Correction and one-way ANOVA with Bonferroni’s multiple-comparison test (B–G). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; ns, not significant.
We next examined whether the addition of 32-134D could improve upon an effective dose of aflibercept. To this end, we performed a dose–response of aflibercept and 32-134D to identify the dose that was 50% as effective as the maximal effective dose (i.e., the IC50) and determined that the IC50 for aflibercept was approximately 300 ng and for 32-134D was 70 ng (SI Appendix, Fig. S11). Combining 300 ng of aflibercept with 70 ng of 32-134D was more effective than either drug alone (Fig. 7G). Collectively, these results demonstrate that 32-134D is effective for the treatment of CNV in mice and provide the foundation for clinical trials assessing its efficacy when used alone or in combination with current therapies targeting VEGF for the treatment of patients with nvAMD.
Discussion
Results from multiple clinical trials consistently demonstrate that the majority of patients with nvAMD fail to achieve a clinically significant improvement in vision (i.e., an increase of 3 line or more on the ETDRS vision chart) despite monthly or bimonthly treatment over 2 y (5). Follow up studies on these clinical trials have further demonstrated that the vision gains experienced by patients in the initial 2 y are often lost over subsequent years despite continued treatment (6). While this may be due, in part, to inadequate treatment, these observations raise concerns that anti-VEGF monotherapy may not provide a long-term solution for the treatment of most patients with nvAMD.
There are also concerns that a subpopulation of patients with nvAMD are vulnerable to continued vision loss despite monthly treatment with anti-VEGF therapy. In a post hoc analysis of CATT, a subset of patients (approximately 6%) developed sustained vision loss despite ongoing treatment with anti-VEGF therapy (41). Another 10% of patients in CATT developed a transient, but significant decrease in visual acuity (i.e., a decrease of 3 line or more on the ETDRS vision chart; termed sporadic vision loss), despite treatment with anti-VEGF therapy (42). Analyses of the VIEW 1/2 and LUCAS trials similarly reported vision loss in some patients despite continued treatment during the trials (43, 44). Collectively, these studies highlight the importance of understanding why some patients respond inadequately, transiently, or not at all, despite treatment with therapies that effectively neutralize VEGF.
In this regard, it has previously been reported that expression of a second HIF-regulated vasoactive mediator, ANGPTL4, is expressed in the eyes of patients with nvAMD, and localizes to CNV lesions (9). In mice, ANGPTL4 synergized with VEGF to promote CNV, markedly increasing CNV lesions size and leakage, as assessed by fluorescein angiography (9). It was further observed that simultaneously targeting both ANGPTL4 and VEGF in the laser CNV mouse model was more effective than targeting either factor alone (9). These results suggest that the expression of ANGPTL4 may influence how patients with nvAMD will respond to anti-VEGF therapy.
Here, we report that aqueous fluid from patients with no history—or a remote history (i.e., >12 wk)—of anti-VEGF therapy fell into two categories: high levels of both VEGF and ANGPTL4 (90%) or low levels of VEGF and high levels of ANGPTL4 (10%). We speculate that the latter group could help identify the subpopulation of patients with nvAMD who are most vulnerable to continued vision loss despite treatment with anti-VEGF monotherapy. We further observed an increase in ANGPTL4 levels in patients following initiation of anti-VEGF therapy. This unexpected observation may further explain why some patients experience a return of fluid with deterioration in their vision following an initial response to treatment with anti-VEGF therapy (6).
Consistent with these observations, in patients with nvAMD, aqueous levels of ANGPTL4 at the time of treatment initiation correlated inversely with the response to anti-VEGF therapy at the end of 12 mo of treatment (9). In another study, aqueous levels of ANGPTL4 in patients with nvAMD were shown to correlate directly with the frequency of treatment with anti-VEGF therapy at 12 mo (10). Collectively, these studies provide a possible explanation for why patients with nvAMD respond inadequately to anti-VEGF therapy upon treatment initiation (i.e., in treatment-naïve eyes) as well as following an initial response to treatment (i.e., over time).
To provide a molecular explanation for these observations, we examined the expression of ANGPTL4 and VEGF in the laser CNV mouse model for nvAMD following treatment with anti-VEGF therapy (i.e., aflibercept). We observed an increase in ANGPTL4 protein expression following intraocular administration of aflibercept, consistent with our observations in patients in the clinic. We further observed that enhanced ANGPTL4 protein expression following treatment with anti-VEGF therapy was a consequence of an increase in Angptl4 mRNA expression and that this correlated with an increase in HIF-1α accumulation in the RPE. Prior work implicates the accumulation of HIF-1α in the RPE, caused by localized ischemia (45) and oxidative stress (3) as an early event in the development of CNV in patients with AMD. Preclinical studies lend additional support for a role for HIF-1 in the regulation of VEGF expression and the development of CNV in patients with nvAMD (9, 46–50). Our results, demonstrating that the use of anti-VEGF therapy may cause a countertherapeutic increase in HIF-1α (and, in turn, ANGPTL4) in RPE, could explain the diminished efficacy of anti-VEGF therapy monotherapy for the treatment of CNV in many patients with nvAMD.
We next explored whether a recently described HIF-1 inhibitor, 32-134D (40), would be an effective treatment for patients with nvAMD. Intraocular delivery of 32-134D effectively inhibits HIF-1α and HIF-2α protein accumulation, normalizes the expression of HIF-regulated vasoactive genes, and prevents the development of retinal NV and vascular hyperpermeability in mouse models of diabetic eye disease without affecting retinal histology or function (22). We therefore examined the efficacy of a single intraocular injection of 32-134D for the treatment of CNV in mice. 32-134D effectively prevented the increase in VEGF mRNA and protein in the laser CNV mouse model of nvAMD, similar to aflibercept. However, unlike with aflibercept, treatment with 32-134D did not result in a countertherapeutic increase in ANGPTL4 expression. Rather, expression of ANGPTL4 was also markedly diminished following treatment with 32-134D. Accordingly, treatment of mice with 32-134D effectively treated CNV in mice, demonstrating that 32-134D could be an effective monotherapy for the treatment of patients with nvAMD.
The observation that 32-134D effectively blocked HIF-1α accumulation as well as the expression of ANGPTL4 in the laser CNV model prompted us to explore whether the addition of 32-134D could prevent the countertherapeutic increase in HIF-1α (and, in turn, ANGPTL4) observed following treatment with aflibercept. We report that the addition of 32-134D to treatment with aflibercept effectively prevented the increase in both HIF-1α and ANGPTL4 and had more effective for the prevention of CNV in mice than either therapy alone, demonstrating the potential of simultaneous inhibition of HIF-1α and VEGF as a therapeutic approach for the treatment of CNV. Collectively, these studies provide the foundation for a clinical study assessing 32-134D, alone or in combination with anti-VEGF therapies, for the treatment of patients with nvAMD.
Materials and Methods
Mice.
All animal procedures were performed under the guidelines of the Johns Hopkins University School of Medicine Animal Care and Use Committee and the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research. Ten- to twelve-week-old pathogen-free female mice were used for all experiments. C57BL/6 mice and HIF-1α heterozygous mice were obtained from Jackson Laboratories (JAX).
Cell Culture and Reagents.
Cell culture of ARPE19 and primary mouse RPE cells was as previously described (3). Cell lines were routinely tested for Mycoplasma contamination by PCR. DMSO (472301-500ML) was purchased from Sigma-Aldrich. 4-(6-bromo-1H-indol-3-yl)-2-(7-bromo-1H-indol-3-yl)thiazole, designated 32-134D, was synthesized as previously described (40). Acriflavine and SU1498 were obtained from MilliporeSigma. Aflibercept was obtained from the Johns Hopkins University Pharmacy.
siRNA.
Predesigned control (scrambled or scr), Kdr siRNA sequences were obtained from Qiagen. Kdr (20 µM) and scr siRNA delivery was performed using HiPerFect (Qiagen).
Western Blot.
Antibodies are listed in SI Appendix, Table S2. Western blots were performed as previously described (3). Western blot scans are representative of at least 3 independent experiments.
Intraocular Injections.
Intraocular injections were performed as previously described (22).
Laser Treatment.
Laser CNV was used to rupture Bruch’s membrane and cause neovascularization as previously described (9). Four lesions were made using a diode laser photocoagulator (IRIS medical) and a slit lamp delivery system (532 nm wavelength, 300 mW power, 100 ms duration, and 100 μm spot size). Animals were killed at indicated time points and eyes were enucleated and further studies conducted of laser CNV lesions.
Immunofluorescence Assays.
All antibodies and dilutions are listed in SI Appendix, Table S2. Immunofluorescence in RPE/choroid flat mounts and cross sections of the eye were performed as previously described (9). Images were captured at high magnification (20×) with a Zeiss fluorescent microscope (Carl Zeiss Inc.), and the area of CNV lesions was calculated as mm2 by ImageJ software (NIH) by keeping the parameters the same for all the spots. 3D images of flat mounts of RPE/choroid were prepared using Imaris software. The quantitation of signal intensity in a region of interest (ROI) using the mean fluorescence intensity (MFI) method was calculated using ImageJ. Briefly, the original confocal image of a section and channel of interest to be quantified was selected. The ROI was selected by tracing the tissue using a high-resolution drawing, and the output from MFI was measured. MFI of the background from a rectangular area of the image that lacks tissue was also measured. The final MFI was the calculated by subtracting the background MFI from the ROI MFI. The percent final MFI of the sample compared to control was then calculated using Excel.
In Situ Hybridization.
RNA in situ hybridization was performed as previously described (21).
Reverse Transcription and Quantitative Real-Time PCR.
Primers are listed in SI Appendix, Table S3. Quantitative real-time PCR was performed as previously described (21).
Patient Samples.
All studies were in accordance with the Declaration of Helsinki. Institutional Review Board approval from the Johns Hopkins University School of Medicine was obtained for all patient tissue used in this study (NA_00075565). Consent was written and voluntary without stipend. All human samples used were deidentified prior to use. Aqueous samples were collected, and ELISA kits were performed as previously described (9). ELISAs are representative of at least three independent experiments.
Statistical Analysis.
Results are shown as a mean value ± SD from at least 3 independent experiments. Data from clinical samples are shown as mean ± SEM. Statistical analysis was done using Excel and Prism 9 software (GraphPad). Statistical differences between two or more heterogeneous groups were determined using the Kruskal–Wallis with Dunn’s multiple-comparison test, unpaired Student’s t test with Welch’s Correction, and the one-way or two-way ANOVA with Bonferroni’s multiple-comparison test. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. ns = not significant.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (PDF)
Acknowledgments
This work was supported by the NIH grants R01EY029750 to A.S., R01EY032104 to S.M. and A.S., and EY001765 (the Wilmer Core Grant for Vision Research, Microscopy and Imaging Core Module); the TEDCO Maryland Innovation Initiative to G.L.S. and A.S.; the Research to Prevent Blindness, Inc., Special Scholar Award to A.S., Sybil B. Harrington Stein Innovation Award to G.L.S., and an unrestricted grant to the Wilmer Eye Institute, Johns Hopkins School of Medicine; the Armstrong Family Foundation to G.L.S.; the C. Michael Armstrong Professorship and research funding from the Armstrong Family Foundation to G.L.S.; and the Branna and Irving Sisenwein Professorship in Ophthalmology to A.S. The funding organizations had no role in the design or conduct of this research.
Author contributions
A.S. designed research; D.S., E.L., Y.Q., K.J., M.R., C.G., A.D., E.M., and A.S. performed research; S.S., Y.H., and G.L.S. contributed new reagents/analytic tools; D.S., E.L., Y.Q., K.J., M.R., C.G., A.D., A.M., S.M., and A.S. analyzed data; D.S., E.L., Y.Q., A.M., G.L.S., and S.M. reviewed/edited manuscript; and A.S. wrote the paper.
Competing interests
G.L.S. and A.S. are co-founders of and hold equity in HIF Therapeutics, Inc. S.S., Y.H., G.L.S., and A.S. are inventors on provisional patent application PCT/US2022/039883. This arrangement has been reviewed and approved by the Johns Hopkins University in accordancewith its conflict of interest policies.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
All study data are included in the article and/or supporting information.
Supporting Information
References
- 1.Nguyen D. H., Luo J., Zhang K., Zhang M., Current therapeutic approaches in neovascular age-related macular degeneration. Discov. Med. 15, 343–348 (2013). [PubMed] [Google Scholar]
- 2.Jager R. D., Mieler W. F., Miller J. W., Age-related macular degeneration. N. Engl. J. Med. 358, 2606–2617 (2008). [DOI] [PubMed] [Google Scholar]
- 3.Babapoor-Farrokhran S., et al. , Pathologic vs. protective roles of hypoxia-inducible factor 1 in RPE and photoreceptors in wet vs. dry age-related macular degeneration. Proc. Natl. Acad. Sci. U.S.A. 120, e2302845120 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kovach J. L., Schwartz S. G., Flynn H. W. Jr., Scott I. U., Anti-VEGF treatment strategies for wet AMD. J. Ophthalmol. 2012, 786870 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Solomon S. D., Lindsley K., Vedula S. S., Krzystolik M. G., Hawkins B. S., Anti-vascular endothelial growth factor for neovascular age-related macular degeneration. Cochrane Database Syst. Rev. 3, CD005139 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maguire M. G., et al. , Comparison of Age-related Macular Degeneration Treatments Trials (CATT) Research Group, Five-year outcomes with anti-vascular endothelial growth factor treatment of neovascular age-related macular degeneration. Ophthalmology 123, 1751–1761 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hariprasad S. M., Morse L. S., Shapiro H., Wong P., Tuomi L., Fixed monthly versus less frequent ranibizumab dosing and predictors of visual response in exudative age-related macular degeneration. J. Ophthalmol. 2012, 690641 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cao X., et al. , Aqueous proteins help predict the response of patients with neovascular age-related macular degeneration to anti-VEGF therapy. J. Clin. Invest. 132, e144469 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qin Y., et al. , ANGPTL4 influences the therapeutic response of patients with neovascular age-related macular degeneration by promoting choroidal neovascularization. JCI Insight 7, e157896 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim J. H., Shin J. P., Kim I. T., Park D. H., Angiopoietin-like 4 correlates with response to intravitreal ranibizumab injections in neovascular age-related macular degeneration. Retina 38, 523–530 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Shah R. S., Soetikno B. T., Lajko M., Fawzi A. A., A Mouse Model for Laser-induced Choroidal Neovascularization. J. Vis. Exp. 106, e53502 (2015), 10.3791/53502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grossniklaus H. E., Kang S. J., Berglin L., Animal models of choroidal and retinal neovascularization. Prog. Retin. Eye Res. 29, 500–519 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Babapoor-Farrokhran S., et al. , Angiopoietin-like 4 is a potent angiogenic factor and a novel therapeutic target for patients with proliferative diabetic retinopathy. Proc. Natl. Acad. Sci. U.S.A. 112, E3030–E3039 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo C., et al. , HIF-1alpha accumulation in response to transient hypoglycemia may worsen diabetic eye disease. Cell Rep. 42, 111976 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu K., et al. , Hypoxia-inducible factor 1 upregulation of both VEGF and ANGPTL4 is required to promote the angiogenic phenotype in uveal melanoma. Oncotarget 7, 7816–7828 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meng Q., et al. , Hypoxia-inducible factor-dependent expression of angiopoietin-like 4 by conjunctival epithelial cells promotes the angiogenic phenotype of pterygia. Invest. Ophthalmol. Vis. Sci. 58, 4514–4523 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rodrigues M., et al. , Expression pattern of HIF-1alpha and VEGF supports circumferential application of scatter laser for proliferative sickle retinopathy. Invest. Ophthalmol. Vis. Sci. 57, 6739–6746 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sodhi A., et al. , Angiopoietin-like 4 binds neuropilins and cooperates with VEGF to induce diabetic macular edema. J. Clin. Invest. 129, 4593–4608 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sodhi A., Montaner S., Angiopoietin-like 4 as an emerging therapeutic target for diabetic eye disease. JAMA ophthalmol. 133, 1375–1376 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Xin X., et al. , Hypoxic retinal Muller cells promote vascular permeability by HIF-1-dependent up-regulation of angiopoietin-like 4. Proc. Natl. Acad. Sci. U.S.A. 110, E3425–E3434 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang J., et al. , HIF-1alpha and HIF-2alpha redundantly promote retinal neovascularization in patients with ischemic retinal disease. J. Clin. Invest. 131, e139202 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang J., et al. , Targeting hypoxia-inducible factors with 32–134D safely and effectively treats diabetic eye disease in mice. J. Clin. Invest. 133, e163290 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee K., et al. , Acriflavine inhibits HIF-1 dimerization, tumor growth, and vascularization. Proc. Natl. Acad. Sci. U.S.A. 106, 17910–17915 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 24.Yu A. Y., et al. , Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1alpha. J. Clin. Invest. 103, 691–696 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bosch-Marce M., et al. , Effects of aging and hypoxia-inducible factor-1 activity on angiogenic cell mobilization and recovery of perfusion after limb ischemia. Circ Res. 101, 1310–1318 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Bhutto I., Lutty G., Understanding age-related macular degeneration (AMD): Relationships between the photoreceptor/retinal pigment epithelium/Bruch’s membrane/choriocapillaris complex. Mol. Aspects Med. 33, 295–317 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sunness J. S., Gonzalez-Baron J., Bressler N. M., Hawkins B., Applegate C. A., The development of choroidal neovascularization in eyes with the geographic atrophy form of age-related macular degeneration. Ophthalmology 106, 910–919 (1999). [DOI] [PubMed] [Google Scholar]
- 28.Green W. R., Key S. N. III, Senile macular degeneration: A histopathologic study. Trans. Am. Ophthalmol. Soc. 75, 180–254 (1977). [PMC free article] [PubMed] [Google Scholar]
- 29.Sarks J. P., Sarks S. H., Killingsworth M. C., Evolution of geographic atrophy of the retinal pigment epithelium. Eye (Lond) 2, 552–577 (1988). [DOI] [PubMed] [Google Scholar]
- 30.Schatz H., McDonald H. R., Atrophic macular degeneration. Rate of spread of geographic atrophy and visual loss. Ophthalmology 96, 1541–1551 (1989). [DOI] [PubMed] [Google Scholar]
- 31.De Cilla S., et al. , Aflibercept and ranibizumab modulate retinal pigment epithelial cells function by acting on their cross talk with vascular endothelial cells. Cell Physiol. Biochem. 54, 161–179 (2020). [DOI] [PubMed] [Google Scholar]
- 32.Chen Y. S., et al. , Localisation of vascular endothelial growth factor and its receptors to cells of vascular and avascular epiretinal membranes. Br. J. Ophthalmol. 81, 919–926 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Byeon S. H., et al. , Vascular endothelial growth factor as an autocrine survival factor for retinal pigment epithelial cells under oxidative stress via the VEGF-R2/PI3K/Akt. Invest. Ophthalmol. Vis. Sci. 51, 1190–1197 (2010). [DOI] [PubMed] [Google Scholar]
- 34.Chu Y. K., Lee S. C., Byeon S. H., VEGF rescues cigarette smoking-induced human RPE cell death by increasing autophagic flux: Implications of the role of autophagy in advanced age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 54, 7329–7337 (2013). [DOI] [PubMed] [Google Scholar]
- 35.Hera R., et al. , Expression of VEGF and angiopoietins in subfoveal membranes from patients with age-related macular degeneration. Am. J. Ophthalmol. 139, 589–596 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Jonas J. B., Neumaier M., Erythropoietin levels in aqueous humour in eyes with exudative age-related macular degeneration and diabetic retinopathy. Clin. Exp. Ophthalmol. 35, 186–187 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Wang Z. Y., Zhao K. K., Song Z. M., Shen L. J., Qu J., Erythropoietin as a novel therapeutic agent for atrophic age-related macular degeneration. Med. Hypotheses 72, 448–450 (2009). [DOI] [PubMed] [Google Scholar]
- 38.Maisonpierre P. C., et al. , Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 277, 55–60 (1997). [DOI] [PubMed] [Google Scholar]
- 39.Zeng M., et al. , The HIF-1 antagonist acriflavine: Visualization in retina and suppression of ocular neovascularization. J. Mol. Med. (Berl) 95, 417–429 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Salman S., et al. , HIF inhibitor 32–134D eradicates murine hepatocellular carcinoma in combination with anti-PD1 therapy. J. Clin. Invest. 132, e156774 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ying G. S., et al. , Sustained visual acuity loss in the comparison of age-related macular degeneration treatments trials. JAMA Ophthalmol. 132, 915–921 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim B. J., et al. , Sporadic visual acuity loss in the comparison of age-related macular degeneration treatments trials (CATT). Am. J. Ophthalmol. 158, 128–135.e10 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Berg K., et al. , Ranibizumab or bevacizumab for neovascular age-related macular degeneration according to the Lucentis compared to Avastin study treat-and-extend protocol: Two-year results. Ophthalmology 123, 51–59 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Heier J. S., et al. , Intravitreal aflibercept (VEGF trap-eye) in wet age-related macular degeneration. Ophthalmology 119, 2537–2548 (2012). [DOI] [PubMed] [Google Scholar]
- 45.Arjamaa O., Nikinmaa M., Salminen A., Kaarniranta K., Regulatory role of HIF-1alpha in the pathogenesis of age-related macular degeneration (AMD). Ageing Res. Rev. 8, 349–358 (2009). [DOI] [PubMed] [Google Scholar]
- 46.Vadlapatla R. K., Vadlapudi A. D., Mitra A. K., Hypoxia-inducible factor-1 (HIF-1): A potential target for intervention in ocular neovascular diseases. Curr. Drug Targets 14, 919–935 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Inoue Y., et al. , Expression of hypoxia-inducible factor 1alpha and 2alpha in choroidal neovascular membranes associated with age-related macular degeneration. Br. J. Ophthalmol. 91, 1720–1721 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sheridan C. M., et al. , Expression of hypoxia-inducible factor-1alpha and -2alpha in human choroidal neovascular membranes. Graefes. Arch. Clin. Exp. Ophthalmol. 247, 1361–1367 (2009). [DOI] [PubMed] [Google Scholar]
- 49.Lin M., et al. , Impacts of hypoxia-inducible factor-1 knockout in the retinal pigment epithelium on choroidal neovascularization. Invest. Ophthalmol. Vis. Sci. 53, 6197–6206 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yoshida T., et al. , Digoxin inhibits retinal ischemia-induced HIF-1alpha expression and ocular neovascularization. FASEB J. 24, 1759–1767 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (PDF)
Data Availability Statement
All study data are included in the article and/or supporting information.