

Abstract
Prostate cancer (PC) is a leading cause of cancer-related deaths in men worldwide. Interleukin-30 (IL-30) is a PC progression driver, and its suppression would be strategic for fighting metastatic disease. Biocompatible lipid nanoparticles (NPs) were loaded with CRISPR-Cas9gRNA to delete the human IL30 (hIL30) gene and functionalized with anti-PSCA-Abs (Cas9hIL30-PSCA NPs). Efficiency of the NPs in targeting IL-30 and the metastatic potential of PC cells was examined in vivo in xenograft models of lung metastasis, and in vitro by using two organ-on-chip (2-OC)-containing 3D spheroids of IL30+ PC-endothelial cell co-cultures in circuit with either lung-mimicking spheroids or bone marrow (BM)-niche-mimicking scaffolds. Cas9hIL30-PSCA NPs demonstrated circulation stability, genome editing efficiency, without off-target effects and organ toxicity. Intravenous injection of three doses/13 days, or five doses/20 days, of NPs in mice bearing circulating PC cells and tumor microemboli substantially hindered lung metastasization. Cas9hIL30-PSCA NPs inhibited PC cell proliferation and expression of IL-30 and metastasis drivers, such as CXCR2, CXCR4, IGF1, L1CAM, METAP2, MMP2, and TNFSF10, whereas CDH1 was upregulated. PC-Lung and PC-BM 2-OCs revealed that Cas9hIL30-PSCA NPs suppressed PC cell release of CXCL2/GROβ, which was associated with intra-metastatic myeloid cell infiltrates, and of DKK1, OPG, and IL-6, which boosted endothelial network formation and cancer cell migration. Development of a patient-tailored nanoplatform for selective CRISPR-mediated IL-30 gene deletion is a clinically valuable tool against PC progression.
Keywords: prostate cancer, interleukins, CRISPR-Cas9, metastasis, immunoliposomes, nanotechnology
Graphical abstract
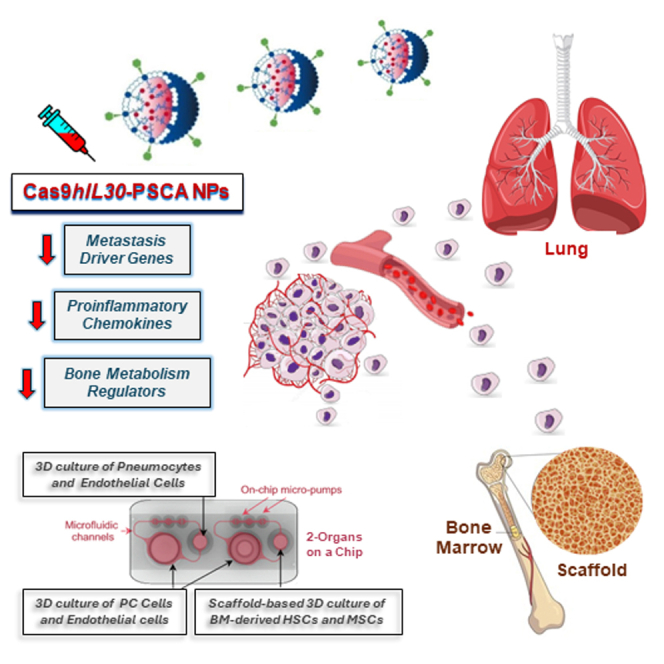
Fieni and colleagues demonstrated that the administration of lipid nanoparticles loaded with CRISPR-Cas9gRNA targeting human IL30 (hIL30) gene, in mice bearing circulating hIL30+ PC cells and microemboli, inhibited cancer cell proliferation and metastasis driver gene expression and downmodulated pro-inflammatory and bone marrow metabolism regulators, ultimately hindering lung and bone marrow metastasis.
Introduction
Prostate cancer (PC) is the leading cancer diagnosis among men and the second most common diagnosis overall.1 Although mortality has declined due to early screening and improved treatment of the primary tumor,2 metastatic disease, which may be detected at the first clinical observation or be diagnosed years after prostatectomy, is currently incurable, and represents a global public health challenge. Furthermore, its incidence is expected to increase due to the worldwide population aging.3
Metastasis is a lethal dynamic process consisting of five major steps: (1) acquisition of invasion and migration capabilities by cancer cells, (2) their intravasation and (3) survival into the bloodstream or lymphatic vessels, (4) trans-endothelial migration and extravasation at the secondary sites, and finally (5) colonization of target organs.4 Each phase requires cancer cells to overcome harsh physicochemical and microenvironmental conditions, and their survival in the bloodstream, where hemodynamic shear forces, immune stresses, and collision with red blood cells seriously threaten their vitality, constitutes an extremely difficult event, which is essential to secondary site colonization.5 In this contest, a primary role has been established for cancer cell-derived growth or immunoregulatory factors.6,7
Recently, an immunoregulatory mediator, known as interleukin-30 (IL-30),8,9 has emerged as a critical regulator of PC onset and behavior.10,11,12 It can be expressed, as a membrane-anchored cytokine, by cancer cells,13,14,15 or released in the microenvironment by activated myeloid-derived cells (MDCs). IL-30 silencing, or deletion, results in a considerable inhibition of PC growth and progression, associated with a substantial remodeling of the tumor’s genetic and microenvironmental programs.15,16 However, the implication of IL-30 in the different stages of the metastasis cascade it is not yet fully understood.
Here, we wondered whether IL-30 may be involved in the survival of PC cells spread throughout the bloodstream, their extravasation and colonization of the main target organs, and, therefore, whether its selective suppression, by using an IL-30-targeting CRISPR-Cas9-gRNA complex delivered at the sites of disseminated PC cells by anti-prostate stem cell antigen (PSCA) antibody (Ab)-guided nanocarriers, may impact the onset and development of PC metastasis.
CRISPR-associated nuclease 9 (Cas9) is an efficient RNA-guided genome editing tool with great potential for clinical application,17 including targeted cancer therapy, which offers several advantages compared with conventional methods, such as cost-effectiveness, flexibility, and simplicity to use.18
PSCA is a cell surface antigen barely detected in the basal cells of the normal prostate and overexpressed in more than 80% of PCs. Its expression level correlates with tumor stage, grade and androgen independence and may be a useful molecular target in advanced PC.19
By using a cutting-edge tool, such as the two organ-on-chip (2-OC) technology,20 which recapitulates the dynamic in vivo conditions of (1) the primary tumor and (2) the major metastatic sites (such as bone marrow [BM] and lungs21), connected by a pulsatile and continuous perfusion system, mimicking blood stream shear stress, as well as in vivo models of metastasis, we highlighted the biomolecular pathways triggered by IL-30 on the path to PC metastasis and determined the therapeutic value of active IL-30 targeting at the sites of metastatic colonization.
A major challenge was the identification of the key mechanisms underlying IL-30 regulation of the PC progression programs, with the final goal of developing a molecularly targeted nanoplatform to prevent metastasis, while minimizing side effects and improving the well-being especially of the frail and elderly patients.
Results
Production of biocompatible nanoliposomes loaded with CRISPR-Cas9gRNA targeting the IL30 gene in PC cells
Cationic lipid nanoparticles (NPs) coated with PEG were synthesized using a microfluidic device (Dolomite Microfluidics), with lipid compounds approved for medical use by both the European Medicines Agency (EMA) and the Food and Drug Administration (FDA).
NPs were either unloaded (empty), and used as control, or loaded with CRISPR-Cas9gRNA-hIL30 complex (Cas9hIL30) at a 1:3 flow rate ratio (lipids/CRISPR-Cas9gRNA-hIL30) and exhibited an entrapment efficiency of 60%. For active targeting of PC cell-anchored IL-30, NPs were then conjugated, via an aldehyde-maleimide reaction, on the PEG-CHO derivatives of the their external bilayer, with specific Abs that recognize and bind to the PSCA (Figure 1A), a prostate-specific cell surface glycosylphosphatidylinositol-linked marker, which is overexpressed in more than 80% of prostate tumors,22 to generate empty- and Cas9hIL30-loaded immunoliposomes, hereinafter referred as empty-PSCA NPs and Cas9hIL30-PSCA NPs, respectively.
Figure 1.

Physical characterization of immunoliposomes, binding to and taken up by prostate cancer cells
(A) Therapeutic nanoplatform consisting of spherical immunoliposomes of less than 1 μm in diameter, loaded with CRISPR-Cas9gRNA targeting the human IL30 gene, and functionalized, via an aldehyde-maleimide reaction that links the Fc domain of the anti-PSCA Abs on the PEG derivatives present on the external bilayer of the nanoparticles. (B) Flow cytometric assessment of the specific binding of anti-hPSCA-conjugated/rhodamine-labeled nanoliposomes (RhB-hPSCA-NPs) to the surface of DU145 (top right) and PC3 (bottom right) cells, compared with unconjugated/rhodamine-labeled nanoliposomes (RhB-NPs) (top left and bottom left). Blue areas, anti-hPSCA conjugated or unconjugated NPs; red areas, isotype controls. Experiments were performed in triplicate. (C and D) Dynamic laser light scattering analysis of the zeta potential of empty-PSCA NP (20.41 ± 3.16 mV) (C) and of the zeta potential of Cas9hIL30-PSCA NP (5.05 ± 1.60 mV) (D). (E–H) Transmission electron microscopy (TEM) images show that NPs consist of spherical vesicles homogeneous in size and shape (E), which are quickly taken up and endocytosed by PC (DU145) cells when conjugated with anti-PSCA Abs (Cas9hIL30-PSCA NPs) (F). (G) A magnification of endocytosed NPs compared with unconjugated NPs (Cas9hIL30 NPs). (H) Nanoparticles are indicated by arrows. N, nuclei; PM, plasma membrane. Ultrastructural images of NP-treated PC3 cells are comparable with those of NP-treated DU145 cells. (I) On-target and off-target characterization of CRISPR-Cas9gRNA-mediated hIL30 editing, delivered by immunoliposomes in vitro. Average frequency of CRISPR-Cas9-induced variants in the IL30 gene (editing efficiency or on-target effects [ONTs]) and in off-target sites (Off-target effects [OFTs]) in DU145 and PC3 cell lines treated with Cas9hIL30-PSCA NPs. The frequency of variants (ONTs and OFTs) in cells treated with PBS or empty-hPSCA NPs (controls) was <0.1%. Experiments were performed in triplicate. (J and K) On-target and off-target characterization of CRISPR-Cas9gRNA-mediated hIL30 editing delivered by immunoliposomes in vivo. Average frequency of CRISPR-Cas9-induced variants in the IL30 gene (editing efficiency or ONTs) and in OFTs in the indicated organs of DU145 (J) and PC3 (K) metastases-bearing NSG mice treated with Cas9hIL30-PSCA NPs. The frequency of variants (ONTs and OFTs) in organs of mice treated with PBS or empty-hPSCA NPs (controls) was <0.1%. (L and M) Serum (L) and pH (M) stability of the Cas9hIL30-PSCA NPs measured at different time points over a 24h period. Experiments were performed in triplicate. (N) Pharmacokinetics of free Cas9 and Cas9hIL30 NPs, conjugated or unconjugated with anti-hPSCA Abs in DU145 metastasis-bearing NSG mice. % ID/g, percentage of total injected dose per weight. ANOVA, p < 0.001. ∗p < 0.01, Tukey HSD test versus free Cas9.
PSCA is expressed by both PC3 and DU145 cells, which also express membrane-bound IL-30. These cell lines, which are representative of AR−CD44+PSA−CgA+NSE+ and AR+CK8/14+PSA+ metastatic PCs, respectively,15,23,24 were used in this study. Binding specificity of anti-PSCA conjugated/rhodamine (RhB)-labeled NPs, RhB-PSCA-NPs, versus unconjugated RhB-NPs, to the surface of PC cells was assessed by flow cytometry (Figure 1B).
Average particle size and zeta electromotive force determination by dynamic laser light scattering demonstrated that empty-PSCA NPs and Cas9hIL30-PSCA NPs were submicron nanoparticles (Figures 1C and 1D; Table S1). Both immunoliposomes consisted of spherical electron-dense vesicles that were homogeneous in size and shape, as demonstrated by transmission electron microscopy (TEM) (Figure 1E), which also showed faster uptake and more efficient endocytosis by PC cells of Ab-conjugated Cas9hIL30-PSCA NPs compared with unconjugated Cas9hIL30 NPs, as inferred from the ultrastructural images captured 2 h after treating the tumor cells (Figures 1F–1H). Similar vesicles were absent, both nearby and inside the untreated or PBS-treated cells (Figure S1).
To assess the potential genotoxicity of the CRISPR-Cas9gRNA-hIL30 complex due to non-specific cleavage of the genome,25 whole-genome sequencing of the off-target regions (Table S2) for the selected single-guide RNA was performed in both PC3 and DU145 cell lines, and in the tissues from different organs of NSG mice treated with Cas9hIL30-PSCA NPs (1 mg/mL, 48 h) or with PBS. The frequency of variants of the off-target regions detected in the cell cultures (Figure 1I) in the micrometastases obtained from lung microdissection and in the organs of mice (Figures 1J and 1K) treated with Cas9hIL30-PSCA NPs was comparable with that observed, respectively, in cancer cell cultures or in metastasis and organs of mice treated with PBS (controls) (<0.1%), which demonstrated the specificity of the genome editing.
Serum and pH stability of immunoliposomes was assessed by measuring changes over time in their size in medium containing 10% FBS (Figure 1L) and at different pH values (Figure 1M). The size of the immunoliposomes was constant over 24 h, and over a wide pH range, suggesting their suitability for drug delivery.
The release rate of Cas9 by Cas9hIL30-loaded Ab-conjugated and -unconjugated NPs, as measured at 30 min, was 27% and 30%, respectively, which complies with the liposome formulation guidelines, in the international pharmacopoeia (https://digicollections.net/phint/2020/index.html#d/b.1) that recommends a release rate <40% in 0.5 h.
Since our focus was the treatment of disseminated tumor cells and tumor microemboli, the pharmacokinetics, biodistribution, and cancer cell uptake of NPs were assessed in mice bearing disseminated PSCA+ cancer cells and tumor microemboli 3 days after intravenous (i.v.) inoculation of DU145 or PC3 cells (Figure 2A). Pharmacokinetics of Cas9hIL30-PSCA NPs demonstrated a half-life (t1/2) of 1 h, which was significantly longer than that of free Cas9, which was 5 min (ANOVA, p < 0.001) (Figure 1N) and comparable with the half-life of unconjugated Cas9hIL30 NPs (1 h), suggesting that the lipidic nanocarrier protects the Cas9gRNA-hIL30 complex from enzymatic degradation. Pharmacokinetics were similar between mice bearing DU145 cell-derived metastases and mice bearing PC3 cell-derived metastases.
Figure 2.

Treatment of lung PC microemboli with immunoliposomes carrying Cas9gRNA-hIL30
(A) Hematoxylin and eosin (H&E) (a) and immunohistochemical staining for PSCA (b) of DU145 tumor cell clusters observed in the lung of NSG mice 3 days after cancer cell inoculation into the dorsal tail vein (intravenously [i.v.]). Similar images were obtained from PC3 tumor cell clusters. Magnification: ×400. Scale bars, 40 μm. (B and C) Confocal microscopy images of lungs from NSG mice bearing GFP-labeled DU145 tumor microemboli (developed 3 days after i.v. cancer cell injection), after 15 min (B, top pictures) and 1 h (C) from i.v. inoculation of (red-labeled) RhB-hPSCA-NPs. The signal intensity starts to decline 10 min after inoculation in the case of unconjugated RhB-NPs (B, bottom). Similar results were obtained from NSG mice, bearing GFP-labeled PC3 tumor microemboli inoculated with RhB-hPSCA-NPs or RhB-NPs. DAPI, DNA-stained nuclei. Magnification: ×400. Scale bars, 40 μm. (D and E) Quantification, by LSC, of the NP uptake in DU145 (D, green bars) and PC3 (E, blue bars) tumor microemboli developed in NSG mice. The uptake by tumor cell clusters of the RhB-NPs (light green or blue) and RhB-hPSCA-NPs (dark green or blue) was expressed as the mean percentage ± SD of RhB+GFP+ cells/total number of GFP+cells. ∗p < 0.01, Student’s t-test versus RhB-NPs at the same time point. (F–H) TEM images of tumor cell (DU145) clusters within the lung revealed that the NP penetration and internalization in metastatic PC cells is negligible for unconjugated NPs (F), which were frequently found among cancer cells (arrows), and very efficient for anti-PSCA Ab-conjugated NPs (G and H, endocytosed NPs are indicated by arrows). Ultrastructural images of lung clusters of PC3 cells, after NPs administration, were comparable with those of DU145 cell clusters. N, nuclei; PM, plasma membrane. (I) Viability of DU145 (green bars) and PC3 (blue bars) cells after 48 h incubation with different concentrations (0.2, 0.4, 1 mg/mL) of empty-hPSCA NPs versus PBS-treated cells. ANOVA, p > 0.05. Results obtained from untreated cells were comparable with those from PBS-treated cells. Experiments were performed in triplicate. (J) Viability of DU145 (green bars) and PC3 (blue bars) cells after 72 h incubation with 1 mg/mL of Cas9hIL30-PSCA NPs versus PBS-treated and IL30KO cells. ANOVA: p < 0.001. ∗p < 0.01, Tukey HSD test versus PBS-treated cells. #p < 0.01, Tukey HSD test versus 1 mg/mL-treated, and PBS-treated cells. Results from PBS-treated cells were comparable with those obtained from empty-hPSCA NP-treated and untreated cells. Experiments were performed in triplicate. (K) Human metastasis PCR array. Fold differences of the mRNAs of metastasis-related genes between IL30KO-DU145 and control NTgRNA-treated DU145 cells (light green bars) or PC3 cells (light blue bars), and between Cas9hIL30-PSCA NP-treated DU145 cells and control empty-hPSCA NP-treated DU145 cells (dark green bars) or PC3 cells (dark blue bars). A significant threshold of a 2-fold change in gene expression corresponded to p < 0.001. Only genes with a fold change >2 are shown. Experiments were performed in duplicate. The dashed lines represent the 2-fold change cutoff. (L) Schedules of five (top gray arrow) or three (bottom gray arrow) treatments with Cas9gRNA-hIL30-loaded immunoliposomes administered to NSG mice bearing single or small clusters of PC3 (fuchsia stained) or DU145 (orange stained) cells (which constitutively express membrane-anchored IL-30) in the lung circulation. The mice were treated with a biweekly dose of immunoliposomes (250 μL dose, with 50 μg/mL of Cas9 and 20 mg/mL of lipids concentration), starting from the third day after the intravenous administration of 3 × 105 cancer cells. Treatment administration was stopped 20 (5 treatments) or 13 (3 treatments) days later. (M) Mean number of lung metastasis developed in NSG mice, after i.v. injection of IL30KO or wild-type DU145 cells, and 5 treatments with PBS, empty-hPSCA NPs, or Cas9hIL30-PSCA NPs. ANOVA: p < 0.01. ∗p < 0.01, Tukey HSD test compared with DU145 tumor microemboli treated with PBS or empty-hPSCA NPs. (N) Mean number of lung metastases developed in NSG mice after i.v. injection of IL30KO or wild-type PC3 cells, and 5 treatments with PBS, or empty-hPSCA NPs, or Cas9hIL30-PSCA NPs. ANOVA: p < 0.0001. ∗p < 0.01, Tukey HSD test compared with PC3 tumor microemboli treated with PBS or empty-hPSCA NPs.
To confirm that the lipid shell protects the CRISPR-Cas9-gRNA complex from enzymatic degradation, nanoparticle suspensions were incubated overnight with or without proteinase K (50 μg/μL).26 After proteinase K removal and cleavage with dimethyl sulfoxide (DMSO), the ELISA assay revealed that the amounts of encapsulated CRISPR-Cas9-gRNA complex were comparable between proteinase K-treated and -untreated NPs, thus demonstrating the suitability of the nanoparticles in protecting their load.
Nanoparticle biodistribution and co-localization with GFP-marked lung tumor microemboli was assessed by laser scanning confocal (LSC) microscopy. LSC images of lungs from mice bearing GFP-labeled DU145 or PC3 cell microemboli, which were i.v. inoculated with RhB-labeled NPs unconjugated or conjugated with anti-PSCA Abs (namely RhB-NPs and RhB-PSCA-NPs, respectively), showed a distinct RhB fluorescence signal 5 min after inoculation of both RhB-PSCA-NPs and RhB-NPs. In mice inoculated with RhB-NPs, the signal intensity declined 10 min later and was nearly lost after 30 min. By contrast, inoculation of RhB-PSCA-NPs generated a robust signal 10 min after injection, which peaked an hour later (Figures 2B–2E). At this time point, ultrastructural images of tumor cell clusters entrapped in the lung revealed that the uptake and internalization in metastatic PC cells were negligible for unconjugated NPs (Figure 2F) and very efficient for anti-PSCA Ab-conjugated NPs (Figures 2G and 2H). Similar vesicles, formed by electron-dense membranes, were never found nearby or in the cytoplasm of neoplastic cells lodged in the lungs of PBS-treated animals (Figure S2), excluding that they may be of cellular origin.
To assess the uptake of NPs by different organs, LSC microscopy analyses of liver, kidneys, lungs, spleen, and prostate were performed at specific time points (5, 10, 30 , and 90 min) after RhB-PSCA-NPs or RhB-NPs injection. The signal intensity from the uptake of both NPs, progressively increased in the liver and the kidneys, the organs primarily involved in liposome metabolism and clearance27 and reached its peak 1 h later. At 30 and 90 min post-injection, the signal intensity from these organs remained higher than the signals from the lungs, the spleen, and prostate, but significantly lower than the signals from lung metastases of mice inoculated with RhB-PSCA-NPs (Figures S3 and S4).
The results from both confocal and electron microscopy analysis steered us to use anti-PSCA Ab-conjugated nanoparticles to perform subsequent in vitro and in vivo experiments.
Cas9gRNA-hIL30-releasing immunoliposomes inhibit PC cell proliferation and regulate a wide range of metastasis driver genes
Exposure of DU145 and PC3 cells to increasing concentrations of empty-PSCA NPs was ineffective and comparable with that of untreated or PBS-treated cells, demonstrating the lack of toxicity of nanocarriers in vitro (Figure 2I). By contrast, treatment with Cas9hIL30-PSCA NPs gave rise to a progressive inhibition of cancer cell proliferation in the same concentration range (Figure 2J). Before testing the impact of Cas9gRNA-hIL30-loaded immunoliposomes in vivo, in metastatic disease, we investigated whether they could affect the gene expression profile of PC cells, specifically the expression of metastasis drivers, which was also analyzed, for comparative studies, in IL30KO-DU145 and IL30KO-PC3 cells, obtained via classic CRISPR-Cas9 editing of the IL30 gene (i.e., lipid transfection or lipofection) in wild-type DU145 e PC3 cells.15
Expression of the tumor suppressor and antimigratory gene CDH1/E-Cadh28 was upregulated both in DU145 and PC3 cells treated with Cas9hIL30-PSCA NPs compared with controls treated with empty-PSCA NPs, and in IL30KO-DU145 and IL30KO-PC3 cells compared with their respective wild-type clones. By contrast, the expression of chemokine receptors CXCR2 and CXCR4, which regulate different cellular processes including chemotactic cell migration,29,30 was substantially downmodulated in both PC cell lines treated with Cas9hIL30-PSCA NPs and in IL30KO-DU145 and IL30KO-PC3 cells (Figure 2K).
Expression of IGF1, which has been shown to have autocrine growth factor activity and to promote epithelial-to-mesenchymal transition (EMT),31,32 together with the expression of METAP2 (methionine aminopeptidase 2), which has been associated with biochemical PC recurrence,33 and MMP2 (matrix metalloproteinase 2), which has been associated with PC progression and reduced disease-free survival,34,35 were strongly downregulated in both PC cell lines by treatment with immunoliposomes, as well as in IL30KO-DU145 and IL30KO-PC3 cells compared with wild-type clones. PC cell expression of L1CAM, associated with tumor aggressiveness and bone metastasis,36 and a member of the TNF superfamily, TNFSF10/TRAIL, which induces apoptosis and regulates inflammation and metastasis,37 was also substantially inhibited by treatment with Cas9hIL30-PSCA NPs, as well as following IL-30 gene knockout in IL30KO-DU145 and IL30KO-PC3 cells.
While upregulated in both IL30KO-DU145 and IL30KO-PC3 cells, the expression of the metastasis suppressor CD82/KAI138,39 was efficiently upregulated by immunoliposome treatment only in PC3 cells. By contrast, the expression of the metastasis suppressor GNRH140 of the pro-invasive and pro-angiogenic endoglycosidases, HPSE,41 and pro-angiogenic growth factor, VEGF42 were all downmodulated in both IL30KO-DU145 and IL30KO-PC3 cells, whereas they were efficiently suppressed by treatment only in PC3 cells (Figure 2K).
Immunoliposome delivery of CRISPR-Cas9-targeting IL30 in PSCA+PC cells spread through the pulmonary circulation effectively inhibits their metastasization
To determine whether suppression of IL-30 expression in PC cells, which have already spread throughout the bloodstream, could affect their survival in the circulation, or their extravasation and colonization to the lung, starting 3 days after i.v. inoculation, when single or minimal clusters of PSCA+IL30+PC cells were trapped in the pulmonary capillaries (Figure 2Aa,b), mice underwent biweekly administrations of IL-30 gene-targeting immunoliposomes (250 μL dose, with a 20 mg/mL lipid concentration, and loaded with 50 μg/mL of Cas9) according to a schedule of five or three, treatments, as shown in Figure 2L. Intravenously administered immunoliposomes were efficiently taken up by single or small clusters of cancer cells trapped into the lung capillaries, as shown by TEM (Figures 2F–2H).
To compare the effects on the metastatic process of the immunoliposome-based delivery of CRISPR-Cas9gRNA-hIL30 complex versus classic IL-30 gene editing in PC cells, which generated IL30KO-DU145 and IL30KO-PC3 cells, in addition to three groups of NSG (NOD scid gamma) mice i.v. inoculated with wild-type DU145 or PC3 cells, and then treated with Cas9hIL30-PSCA NPs, empty-hPSCA NPs, or PBS; two groups of NSG mice inoculated with IL30KO-DU145 or IL30KO-PC3 cells and then treated with PBS were also included in this study.
Twenty days after cancer cell inoculation, i.e., 48 h after the last treatment, animals were euthanized, and autopsy and histopathological examinations of the lungs and other organs (liver, kidneys, spleen, prostate, heart) were performed.
Administration of the five-treatment schedule of Cas9hIL30-PSCA NP in mice bearing pulmonary microemboli of wild-type DU145 or PC3 cells led to a consistent reduction in the number of pulmonary micrometastases compared with the treatment with empty-PSCA NPs or PBS (Figures 2M and 2N). The results were confirmed even with the shorter schedule of three treatments (Figure S5), which indicates rapid-acting therapeutic efficacy.
Notably, the reduction of metastases was comparable with that obtained, in the same time frame, in mice that were inoculated with IL30KO-DU145 or IL30KO-PC3 cells versus mice receiving wild-type control cells (Figures 2M and 2N).
Immunopathological analyses revealed that lung micrometastasis from Cas9hIL30-PSCA NP-treated mice lacked IL-30 expression and had reduced proliferative activity compared with metastasis from control mice (Figure 3A; Table 1). Remarkably, the immunoliposome-mediated regulation of metastasis driver genes observed in vitro in PC cells, was largely confirmed in vivo by the immunohistochemical staining of tumor micrometastasis from animals undergoing IL-30-targeting treatment, showing a substantial downmodulation of CXCR2, CXCR4, IGF1, METAP-2, MMP2, VEGF-A, and TNFSF10/TRAIL, whereas expression of CDH1/E-Cadh was upregulated (Figures 3A and S6).
Figure 3.

Immunopathology of lung metastases treated with immunoliposomes and assessment of the treatment in biomimetic PC-lung or PC-BM 2-OCs
(A) Lung metastasis, developed after i.v. inoculation of DU145 cells, from empty-hPSCA NP-treated mice express IL-30 (a) show a robust proliferation (b), strong expression of CXCR4 (c), IGF1 (d), METAP-2 (e), and weak CDH1 expression (f). By contrast, the few metastases developed in Cas9hIL30-PSCA NP-treated animals lack IL-30 expression (g) and show a low cancer cell proliferation (h) and a weak expression of CXCR4 (i), IGF1 (j), and METAP-2 (k), while the expression of CDH1 was strengthened (l). Immunopathological features of lungs from mice injected with wild-type DU145 cells and treated with PBS were comparable with those of lungs from empty-hPSCA-treated mice. Results from mice bearing lung metastasis developed after i.v. inoculation of PC3 cells were comparable with those obtained from mice bearing lung metastasis developed after i.v. inoculation of DU145 cells. Magnification: ×400. Scale bars, 40 μm. (B) The prostate, liver, kidneys, spleen, and heart of NSG mice bearing DU145 tumor microemboli in their lungs, and then treated with Cas9hIL30-PSCA NPs (a–e), are free of signs of tissue or cell damage, and their histologic features are fully comparable with that of organs of NSG mice treated with empty-hPSCA NPs (f–j). Similar results were obtained from the histopathological analyses of the organs of PBS-treated mice. Histologic features of the organs of immunoliposome-treated and control NSG mice bearing PC3 lung tumor microemboli were comparable with those of treated and control mice bearing DU145 tumor microemboli. Magnification: ×400. Scale bars, 40 μm. (C) Microfluidic bioreactor, HUMIMIC Chip2 (a), which housed in one compartment the spheroid co-culture of PC and endothelial cells, (b) interconnected, via microfluidic channels, to a second compartment containing 3D spheroid co-culture of pneumocytes and endothelial cells, to mimic the lung, or a ceramic scaffold, comprising BM-derived MSCs and CD34+ MSCs, to mimic the BM niche. Micropumps, generating a pulsatile flow, adjusted by a control unit (c), ensured the dynamic circulation, between the two compartments of the culture medium. (D) Confocal microscopy of 3D PC-EC spheroids containing GFP-labeled DU145 cells treated with PBS or with RhB-labeled Cas9hIL30-PSCA NPs, showing the progressive uptake of red-labeled NPs by green-labeled PC cells. DAPI, DNA-stained nuclei. Magnification: ×200. Scale bars, 60 μm. (E–G) Ultrastructural images of 3D (DU145 cell containing) spheroids from the 2-OC platform untreated (E) or treated with Cas9hIL30-PSCA NPs (F and G), demonstrating that immunoliposomes (arrows) are efficiently taken up and internalized by DU145 (F) or PC3 (G) cells. N, nuclei; PM, plasma membrane.
Table 1.
Quantitation of PC cell proliferation, and of granulocytes and myeloid-derived cells infiltrating lung metastases
| Marker (%) | PBS | Empty-PSCA NPs | Cas9hIL30-PSCA NPs | IL30KO | ANOVA p valuec | |
|---|---|---|---|---|---|---|
| DU145 | Ki67a | 77.6 ± 7.5 | 78.0 ± 8.2 | 56.0 ± 7.2d | 52.2 ± 6.4d | <0.001 |
| Ly6Gb | 17.5 ± 5.5 | 18.1 ± 4.7 | 6.4 ± 2.9d | 5.2 ± 2.4d | <0.001 | |
| Gr-1/CD11bb | 20.0 ± 5.8 | 22.5 ± 6.2 | 7.0 ± 3.2d | 5.7 ± 2.0d | <0.001 | |
| PC3 | Ki67a | 86.5 ± 7.6 | 88.0 ± 7.3 | 64.0 ± 6.8d | 60.3 ± 6.2d | <0.001 |
| Ly6Gb | 18.3 ± 6.0 | 20.0 ± 5.2 | 9.0 ± 3.3d | 7.4 ± 2.3d | <0.001 | |
| Gr-1/CD11bb | 21.0 ± 5.5 | 24.1 ± 6.0 | 8.2 ± 2.6d | 6.5 ± 2.7d | <0.001 |
Gr-1+/CD11b+ cells, myeloid-derived cells; Ki67, cell proliferation marker; Ly6G+ cells, granulocytes.
Proliferation index was assessed by light microscopy at ×400 in an 85,431.59 μm2 field with Qwin image analysis software (version 2.7). Results are expressed as mean percentage of Ki67-positive cells/number of total cells.
Results are expressed as the mean percentage of area covered by Ly6G+ or Gr-1+/CD11b+ cells/total neoplastic area.
One-way ANOVA for comparisons between all groups.
p < 0.01 Tukey’s HSD test compared cells treated with PBS or empty-hPSCA NPs.
The absence of histologically evident alterations in the organs of treated animals compared with controls (Figure 3B), together with the lack of significant increases in the level of biomarkers of inflammation or organ damage, as assessed by hematological analyses (Table S3), confirmed that in vivo the immunoliposomes showed no obvious toxicity.
IL30 gene-targeting immunoliposomes hinder PC cell colonization to the lungs and downregulate CXCL2/GROβ release in a biomimetic PC-lung on a chip platform
The relationship between PC and endothelial cells (ECs) is essential in regulating cancer cell extravasation and colonization to the secondary sites,43 and IL-30 has been demonstrated to be deeply involved in the PC-EC crosstalk.16 Therefore, the mechanisms underlying the antimetastatic effects of immunoliposome-mediated delivery of Cas9gRNA-hIL30 to disseminated PC cells was further investigated by using a microfluidic bioreactor, HUMIMIC Chip2 (TissUse, Berlin, Germany) (Figure 3Ca), which housed in one compartment the 3D spheroid co-culture of PC cells and endothelium, arranged in microvessel networks (compartment A, Figure 3Cb), interconnected via microfluidic channels to a second compartment containing the 3D spheroid co-culture of microvascular network and pneumocytes with type-I-like properties (expression of caveolin-1, absence of surfactant protein C) arranged into alveolar-like structures to mimic the lung (compartment B, Figure 3Cb). Micropumps, generating a pulsatile flow, adjusted by a control unit (Figure 3Cc) ensured the dynamic circulation of the culture medium between the two compartments, and allowed cell migration events to be investigated in a biomimetic condition.
Treatments with IL-30-targeting immunoliposomes were applied to the circulating culture medium according to the same timeline of the five-treatment schedule used in vivo.
LSC microscopy and TEM images confirmed that the immunoliposomes penetrated PC-EC spheroids and were efficiently taken up by PC cells (Figures 3D–3G).
LSC microscopy and automated image analyses revealed that the five-treatment schedule led to a consistent reduction in the number of PC cells that colonized lung spheroids (ANOVA, p < 0.01) compared with controls (Figures 4A and 4Ba,b), as confirmed by flow cytometry analyses (Figure 4Bc, d). Moreover, the viability of both PC cells and ECs forming the tumor spheroids was substantially inhibited (Figures 4C–4F), whereas apoptotic events were unchanged, as demonstrated by Ki67 and annexin V staining (Figure S7), respectively, followed by flow cytometric analyses. Analysis of the culture supernatant, by the LegendPlex MultiAnalyte Flow Assay Kit Human Proinflammatory Chemokines, to assess whether IL-30 inhibition in PC cells affected the release of inflammatory mediators functional to metastatization, revealed a consistent reduction in the content of the neutrophil- and myeloid-derived cell chemoattractant CXCL2/GROβ44 in both DU145 and PC3 spheroids in the 2-OC platforms, starting after the third administration of IL-30-targeting immunoliposomes (Figures 4G–4H). ELISA assay of the supernatant of the single cellular components of the PC-lung on a chip platform, in static culture conditions, determined their production of CXCL2 and, more importantly, established that treatment with Cas9hIL30-PSCA NPs inhibited CXCL2 production and release in both PC cell lines, while the production by endothelia and pneumocytes was essentially unchanged (Figure 4I).
Figure 4.

Confocal microscopy and cytofluorimetric analyses of PC spheroids and PC cell migration into lung spheroids from the 2-OC, after immunoliposome treatments
(A) Confocal microscopy images of 3D lung spheroids containing red-labeled (LuminiCell Tracker 670) pneumocytes and metastasizing green-labeled (LuminiCell Tracker 540) PC cells (DU145), taken from the PC lung on a chip platform, show a consistent reduction in the number of PC cells colonizing the lung spheroids after treatment with 5 doses of immunoliposomes (a) compared with controls (b). Magnification: ×200. Scale bars, 60 μm. (B) Histograms representing the quantization of DU145 (a and c) and PC3 (b and d) cells colonizing the lung spheroids in the 2-OC platform after 5 doses of immunoliposome treatment, as assessed by LSC microscopy images (a and b) and by flow cytometry analyses (c and d). The automated quantization of the number of DU145 (a) or PC3 (b) cells (visualized with the LSM 800 confocal microscope, Zeiss, Oberkochen, Germany; RRID: SCR_015963) that colonized the lung spheroids were performed using Zen software (Zeiss). Four to six high-power fields were analyzed for each well and two optical sections per well were evaluated. Results are expressed as mean ± SD of GFP-labeled cells per field. (a) DU145 cells, ANOVA: p < 0.0022. ∗p < 0.01, Tukey HSD test versus spheroids treated with PBS or empty-hPSCA NPs. (b) PC3 cells, ANOVA: p = 0.0051. ∗p < 0.05, Tukey HSD test versus spheroids treated with PBS or empty-hPSCA NPs. (c) Flow cytometry assessment of DU145 cells colonizing lung spheroids. ANOVA: p = 0.0022. ∗p < 0.01, Tukey HSD test versus treatment with PBS or empty-hPSCA NPs. (d) Flow cytometry assessment of PC3 cells colonizing lung spheroids. ANOVA: p = 0.0064. ∗p < 0.05, Tukey HSD test versus treatment with PBS or empty-hPSCA NPs. (C) Cytofluorimetric images of Ki67+ DU145 cells (a) and Ki67+ HUVECs (b) forming tumor spheroids in the PC lung on a chip after the treatment with five doses of immunoliposomes. Blue areas, isotype controls; red areas, specific Abs. The image is representative of a triplicate experiment. (D) Cytofluorimetric images of Ki67+ PC3 cells (a) and Ki67+ HUVECs (b) forming tumor spheroids in the PC lung on a chip after treatment with five doses of immunoliposomes. Blue areas, isotype controls; red areas, specific Abs. The image is representative of a triplicate experiment. (E) Flow cytometric analyses of Ki67+ DU145 cells (a) and Ki67+ HUVECs (b) isolated from PC spheroids of PC lung on a chip treated with five doses of immunoliposomes. MFI ratios were calculated by dividing the MFI of Ki67+ cell population by the MFI of the negative/isotype control. (a) DU145 cells, ANOVA: p = 0.0044. ∗p < 0.05, Tukey HSD test versus spheroids treated with PBS or empty-hPSCA NPs. (b) HUVECs, ANOVA: p = 0.0018. ∗p < 0.01, Tukey HSD test versus spheroids treated with PBS or empty-hPSCA NPs. (F) Flow cytometric analyses of Ki67+ PC3 cells (a) and Ki67+ HUVECs (b) isolated from PC spheroids of PC lung on a chip treated with five doses of immunoliposomes. MFI ratios were calculated by dividing the MFI of the Ki67+ cell population by the MFI of the negative/isotype control. (a) PC3 cells, ANOVA: p = 0.0005. ∗p < 0.01, Tukey HSD test versus spheroids treated with PBS or empty-hPSCA NPs. (b) HUVECs, ANOVA: p = 0.0046. ∗p < 0.05, Tukey HSD test versus spheroids treated with PBS or empty-hPSCA NPs. (G and H) Quantification of CXCL2/GRO-β in the supernatant collected from the PC spheroid lung on a chip containing DU145 (G) or PC3 (H) cells using LEGENDplex flow cytometry-based immunoassay. ANOVA: p < 0.0001. ∗p < 0.01, Tukey HSD test compared with PBS and empty-hPSCA-NP. #p < 0.01, Tukey HSD test compared with PBS and empty-hPSCA-NP. §p < 0.01, Tukey HSD test compared with T0. T0, the beginning of the experiment. T3, day 10 of the experiment, i.e., after the third treatment with NPs. T5, day 20 and final day of the experiment, i.e., after the fifth treatment with NPs. (I) ELISA assay of GRO-β release by DU145 and PC3 cells, alveolar cell type I cells, HUVECs, MSCs, and CD34+ cells, after treatment with PBS, empty-hPSCA, or Cas9hIL30-PSCA. ANOVA: p < 0.001. ∗p < 0.01, Tukey HSD test compared with PBS and empty-hPSCA. Experiments were performed in triplicate. (J) Immunohistochemical features of DU145 lung metastasis after the treatment with the five-dose schedule of immunoliposomes showing a distinct downmodulation of CXCL2 expression and reduction of Ly6G+ granulocyte and Gr-1+/CD11b+ myeloid cell infiltrates when compared with metastasis from control mice. Magnification: ×400. Scale bars, 40 μm.
Interestingly, immunohistochemistry revealed that lung metastases from the treated mice had low to absent CXCL2 expression and lacked the Ly6G+ granulocyte and Gr-1+/CD11b+ MDC (Table 1) infiltrate, which was detectable in the metastases of animals treated with empty-hPSCA NPs or PBS (Figure 4J).
IL30 gene-targeting immunoliposomes hinder PC cell colonization to the BM and downregulate cancer cell release of OPG, DKK1, and IL-6 in a biomimetic PC-BM on a chip platform
Bones are the most common site of PC metastasis, occurring in 85%–90% of patients with metastatic disease.45 Therefore, assessment of whether IL-30-targeting nanotherapy might interfere with the process of bone metastasis is relevant. This issue was addressed by using 3D spheroids containing DU145 or PC3 cells and endothelial networks, which were interconnected by a common pulsatile flow with a (hydroxyapatite-coated zirconium oxide) 3D ceramic scaffold, comprising BM-derived mesenchymal stem cells (MSCs) (CD105+, CD166+, CD44+, CD90+, and CD73+) and CD34+ multipotent hematopoietic stem and progenitor cells, which overall mimics the BM niche. The timeline of the five-administration schedule of Cas9hIL30-PSCA NPs was applied to the circulating culture medium and PC cell spread within the ceramic cavities of the BM-like niche was assessed 48 h after the last treatment. LSC microscopy and computerized image analysis, together with flow cytometry data, demonstrated that colonization of the BM scaffold by both DU145 and PC3 cells was consistently reduced by treatment with IL-30-targeting immunoliposomes compared with treatment with empty-hPSCA NPs or PBS (Figures 5A–5C). Moreover, as observed in the PC-lung on a chip platform, the viability of both PC cells and ECs present in the tumor spheroids was reduced since Ki67+ stained cells decreased (Figures 5D–5G), although apoptosis was unchanged, and assessment of pro-inflammatory chemokines in the common culture supernatant confirmed the consistent downregulation of CXCL2/GROβ after the third and fifth immunoliposome treatment (Figures 5H and 5I). Analysis with the LegendPlex MultiAnalyte Flow Assay of Human Bone Metabolism regulating factors, of the culture supernatant of PC-BM on a chip, revealed that the IL-30-targeting treatment determined a substantial reduction in the content of DKK1, OPG, and IL-6, important regulators of oncogenesis and metastasis in the bone tissue microenvironment,46,47,48,49 albeit to a different extent between the PC3- and DU145-containing platforms, starting from the third administration of IL-30-targeting immunoliposomes (Figures 5J–5O). ELISA assays of the supernatant of the individual cellular components of the PC-BM on a chip platform established that treatment with Cas9hIL30-PSCA NPs substantially reduced the release of CXCL2, DKK1, OPG, and IL-6 by both DU145 and PC3 cells, while the production of all these mediators by ECs, MSCs, or HSCs, was unaltered (Figures 5P–5R).
Figure 5.

Confocal microscopy and cytofluorimetric analyses of PC spheroids and PC cell colonization of the BM scaffolds in the 2-OC after immunoliposome treatment
(A) Confocal microscopy image of the penetration of Cas9hIL30-PSCA RhB-labeled immunoliposomes (a) into GFP-labeled DU145 cells (b) forming the spheroid taken from the PC-BM 2-OC platform. Similar images were obtained from confocal analysis of spheroids containing PC3 cells and treated with immunoliposomes. (c) DAPI, DNA-stained nuclei. (d) Merge images showing, in yellow, the NP uptake by PC cells. Magnification: ×400. Scale bars, 40 μm. (B) Confocal microscopy image of the BM scaffold coated with red-stained MSCs, and colonized by green-stained DU145 cells, migrated from the spheroids contained in the 2-OC, and treated with Cas9hIL30-hPSCA NPs or Empty-PSCA NPs. Similar images were obtained from confocal analysis of spheroids containing PC3 cells and treated with immunoliposomes. DAPI, DNA-stained nuclei. Magnification: ×400. Scale bars, 40 μm. (C) Histograms representing the quantization of DU145 (a and c) and PC3 (b and d) cells, which colonized the BM scaffolds in the 2-OC platform after 5 treatments with immunoliposomes, as assessed by LSC microscopy images (a and b) and by flow cytometry analyses (c and d). The automated quantization of the number of DU145 (a) or PC3 (b) cells (visualized with the LSM 800 confocal microscope, Zeiss, Oberkochen, Germany; RRID: SCR_015963) that colonized BM scaffolds were performed using Zen software (Zeiss). Four to six high-power fields were analyzed for each well and two optical sections per well were evaluated. Results are expressed as mean ± SD of GFP-labeled cells per field. (a) DU145 cells, ANOVA: p < 0.001. ∗p < 0.01, Tukey HSD test versus spheroids treated with PBS or empty-hPSCA NPs. (b) PC3 cells, ANOVA: p < 0.001. ∗p < 0.01, Tukey HSD test versus spheroids treated with PBS or empty-hPSCA NPs. (c) Flow cytometry assessment of DU145 cells colonizing the BM scaffold. ANOVA: p < 0.001. ∗p < 0.01, Tukey HSD test versus treatment with PBS or empty-hPSCA NPs. (d) Flow cytometry assessment of PC3 cells colonizing the BM scaffold. ANOVA: p = 0.001. ∗p < 0.01, Tukey HSD test versus treatment with PBS or empty-hPSCA NPs. (D) Cytofluorimetric images of Ki67+ DU145 cells (a) and Ki67+ HUVECs (b) forming tumor spheroids in the PC-BM on a chip, after the treatment with five doses of immunoliposomes. Blue areas, isotype controls; red areas, specific Abs. The image is representative of a triplicate experiment. (E) Cytofluorimetric images of Ki67+ PC3 cells (a) and Ki67+ HUVECs (b) forming tumor spheroids in the PC-BM on a chip, after the treatment with five doses of immunoliposomes. Blue areas, isotype controls; red areas, specific Abs. The image is representative of a triplicate experiment. (F) Flow cytometric analyses of Ki67+ DU145 cells and Ki67+ HUVECs isolated from PC spheroids of PC-BM on a chip treated with the five doses of immunoliposomes. MFI ratios were calculated by dividing the MFI of Ki67+ cell population by the MFI of the negative/isotype control. DU145 cells, ANOVA: p = 0.0044. ∗p < 0.05, Tukey HSD test versus spheroids treated with PBS or empty-hPSCA NPs. HUVECs, ANOVA: p = 0.0018. ∗p < 0.01, Tukey HSD test versus spheroids treated with PBS or empty-hPSCA NPs. (G) Flow cytometric analyses of Ki67+ PC3 cells and Ki67+ HUVECs , isolated from PC spheroids of PC-BM on a chip, treated with five doses of immunoliposomes. MFI ratios were calculated by dividing the MFI of the Ki67+ cell population by the MFI of the negative/isotype control. PC3 cells, ANOVA: p = 0.0005. ∗p < 0.01, Tukey HSD test versus spheroids treated with PBS or empty-hPSCA NPs. HUVECs, ANOVA: p = 0.0046. ∗p < 0.05, Tukey HSD test versus spheroids treated with PBS or empty-hPSCA NPs. (H and I) Quantification of CXCL2/GROβ, in the supernatant collected from PC-BM 2-OC, containing DU145 (H) or PC3 (I) cells, using LEGENDplex flow cytometry-based immunoassay. (H) ANOVA: p < 0.001. ∗p < 0.01, Tukey HSD test compared with PBS and empty-hPSCA. #p < 0.01, Tukey HSD test compared with PBS and empty-hPSCA. §p < 0.01, Tukey HSD test compared with T0. (I) ANOVA: p < 0.001. ∗p < 0.01, Tukey HSD test compared with PBS and empty-hPSCA. #p < 0.01, Tukey HSD test compared with PBS and empty-hPSCA. §p < 0.01, Tukey HSD test compared with T0. (J and K) Quantification of DKK1 in the supernatant collected from PC-BM 2-OC, containing DU145 (J) or PC3 (K) cells, using LEGENDplex flow cytometry-based immunoassay. (J) ANOVA: p < 0.01. ∗p < 0.01, Tukey HSD test compared with PBS and empty-hPSCA-NPs. §p < 0.01, Tukey HSD test compared with T0. (K) ANOVA: p < 0.0001. ∗p < 0.01, Tukey HSD test compared with PBS and empty-hPSCA-NPs. #p < 0.01, Tukey HSD test compared with PBS and empty-hPSCA-NPs. §p < 0.01, Tukey HSD test compared with T0. (L and M) Quantification of OPG, in the supernatant collected from PC-BM 2-OC, containing DU145 (L) or PC3 (M) cells using LEGENDplex flow cytometry-based immunoassay. (L) ANOVA: p < 0.0001. ∗p < 0.01, Tukey HSD test compared with PBS and empty-hPSCA-NPs. #p < 0.01, Tukey HSD test compared with PBS and empty-hPSCA-NPs. §p < 0.01, Tukey HSD test compared with T0. (M) ANOVA: p < 0.0001. ∗p < 0.01, Tukey HSD test compared with PBS and empty-hPSCA-NPs. #p < 0.01, Tukey HSD test compared with PBS and empty-hPSCA-NPs. §p < 0.01, Tukey HSD test compared with T0. (N and O) Quantification of IL-6, in the supernatant collected from PC-BM 2-OC, containing DU145 (N) or PC3 (O) cells, using LEGENDplex flow cytometry-based immunoassay. (N) ANOVA: p < 0.0001. ∗p < 0.01, Tukey HSD test compared with PBS and empty-hPSCA-NPs. #p < 0.01, Tukey HSD test compared with PBS and empty-hPSCA-NPs. §p < 0.01, Tukey HSD test compared with T0. (O) ANOVA: p < 0.01. ∗p < 0.01, Tukey HSD test compared with PBS and empty-hPSCA-NPs. §p < 0.01, Tukey HSD test compared with T0. (P) ELISA assay of DKK1 release by DU145 and PC3 cells, HUVECs, MSCs, and CD34+ cells, after treatment with PBS, empty-hPSCA or Cas9hIL30-PSCA. ANOVA: p < 0.001. ∗p < 0.01, Tukey HSD test compared with PBS and empty-hPSCA. Experiments were performed in triplicate. (Q) ELISA assay of OPG release by DU145 and PC3 cells, HUVECs, MSCs, and CD34+ cells after treatment with PBS, empty-hPSCA, or Cas9hIL30-PSCA. ANOVA: p < 0.001. ∗p < 0.01, Tukey HSD test compared with PBS and empty-hPSCA. The columns corresponding to OPG production by MSCs were truncated at 25 pg/mL to improve readability. Experiments were performed in triplicate. (R) ELISA assay of IL-6 release by DU145 and PC3 cells, HUVECs, MSCs, and CD34+ cells after treatment with PBS, empty-hPSCA, or Cas9hIL30-PSCA. ANOVA: p < 0.001. ∗p < 0.01, Tukey HSD test compared with PBS and empty-hPSCA. Experiments were performed in triplicate.
DKK1, OPG, and IL-6 promote invasiveness and EMT in PC cells and stimulate EC activation and vascular network formation
Inhibition of the release, from PC cells, of regulators of bone metabolism following treatment with IL-30-targeting immunoliposomes, prompted us to investigate their involvement in the metastatic potential of PC cells. Treatment of both DU145 and PC3 cells with recombinant (r) DKK1, rOPG, or rIL6 revealed a significant proliferative response, which was also observed in ECs (Figures 6A–6C), hinting at their role in promoting PC growth, as well as angiogenesis.50,51,52,53 Furthermore, treatment with rDKK1, rOPG, and rIL6 substantially (Student’s t-test, p < 0.05) increased the invasion and migration ability of both PC cell lines (Figures 6D–6F) and, accordingly, all three mediators fostered PC cell expression of the major EMT transition genes, as assessed by transcriptional analyses.
Figure 6.

Effects of the second-level mediators suppressed in PC cells by IL-30 targeting with immunoliposomes on the viability and phenotype of PC and endothelial cells
(A–C) MTT assay of DU145, or PC3, and ECs (HUVECs) treated with (50 ng/mL – 48 h) rhDKK1 (A), rhOPG (B), rhIL6 (C). ∗p < 0.05, Student’s t test versus untreated control (CTRL) cells. (D–F) Migration and invasion assays of DU145 and PC3 cells in response to stimulation with (50 ng/mL – 48 h) of rDKK1 (D), rOPG (E), or rIL6 (F). ∗p < 0.05, Student’s t-test versus untreated control (CTRL) cells. (G–I) Fold differences of the mRNAs of epithelial-to-mesenchymal transition-related genes between DU145 (blue bars), or PC3 (green bars), cells untreated or treated with (50 ng/mL) rhDKK1(G), rhOPG (H), or rhIL6 (I). A significant threshold of a 2-fold change in gene expression corresponded to p < 0.001. Only genes with a fold change >2 are shown. Experiments were performed in duplicate. (J–L) Fold differences of the mRNAs of regulatory genes of EC activation, proliferation, and sprouting between ECs untreated or treated with (50 ng/mL) rDKK1 (J), rOPG (K), or rIL6 (L). A significant threshold of a 2-fold change in gene expression corresponded to p < 0.001. Only genes with a fold change >2 are shown. Experiments were performed in duplicate. (M) Mean number of endothelial tubes formed by HUVECs cultured on Matrigel-coated slides and stimulated with rhDKK1, rhIL6, rhOPG, or left untreated (PBS). Results are expressed as mean ± SD of tubes/field (4×). ANOVA: p < 0.0001. ∗p < 0.01, Tukey HSD test compared with untreated cells (PBS). Experiments were performed in triplicate. (N) Mean number of capillary meshes formed by HUVECs, cultured on Matrigel-coated slides, and stimulated with rhDKK1, rhIL6, rhOPG, or left untreated (PBS). Results are expressed as mean ± SD of meshes/field (4×). ANOVA: p < 0.0001. ∗p < 0.01, Tukey HSD test compared with untreated cells (PBS). Experiments were performed in triplicate. (O) Mean number of vascular nodes formed by HUVECs, cultured on Matrigel-coated slides, and stimulated with rhDKK1, rhIL6, rhOPG, or left untreated (PBS). Results are expressed as mean ± SD of nodes/field (4×). ANOVA: p < 0.0001. ∗p < 0.01, Tukey HSD test compared with untreated cells (PBS). Experiments were performed in triplicate. (P) Mean number of tube junctions formed by HUVECs cultured on Matrigel-coated slides and stimulated with rhDKK1, rhIL6, rhOPG, or left untreated (PBS). Results are expressed as mean ± SD of junctions/field (4×). ANOVA: p < 0.0001. ∗p < 0.01, Tukey HSD test compared with untreated cells (PBS). Experiments were performed in triplicate. (Q) Analyses of the tube-forming capabilities of HUVECs untreated (a–c) and treated with rDKK1 (d–f), rOPG (g–i), and rIL6 (j–l) was performed using the Angiogenesis analyzer plug-in of the ImageJ software, as described in the materials and methods. Magnification: ×4. Scale bars, 100 μm. (R) Flow cytometry assessment of DU145 cells colonizing the bone marrow scaffold after treatment of spheroids contained in the PC-BM 2-OC platform with anti-DKK1-neutralizing Abs. ANOVA: p = 0.001. ∗p < 0.01, Tukey HSD test versus spheroids treated with PBS or empty-hPSCA NPs. ∗∗p < 0.01, Tukey HSD test versus spheroids treated with PBS, empty-hPSCA NPs, or Cas9hIL30-PSCA-NPs. (S) Flow cytometry assessment of DU145 cells colonizing the bone marrow scaffold after treatment of spheroids contained in the PC-BM 2-OC platform with anti-OPG-neutralizing Abs, ANOVA: p = 0.001. ∗p < 0.01, Tukey HSD test versus spheroids treated with PBS or empty-hPSCA NPs. ∗∗p < 0.01, Tukey HSD test versus spheroids treated with PBS, empty-hPSCA NPs, or Cas9hIL30-PSCA-NPs. (T) Flow cytometry assessment of DU145 cells colonizing the bone marrow scaffold after treatment of spheroids contained in the PC-BM 2-OC platform with anti-IL-6-neutralizing Abs. ANOVA: p = 0.001. ∗p < 0.01, Tukey HSD test versus spheroids treated with PBS or empty-hPSCA NPs. ∗∗p < 0.01, Tukey HSD test versus spheroids treated with PBS, empty-hPSCA NPs, or Cas9hIL30-PSCA-NPs.
Treatment with rDKK1 upregulated the expression of AKT1, ITGB1, BMP7, NOTCH1, SNAI1, SNAI2, TGFB1, TWIST2, and VIM1, while it downregulated WNT11A in both DU145 cells and PC3 cells (Figure 6G). Recombinant OPG increased the expression of BMP7 and NOTCH1 in both PC cell lines (Figure 6H), while treatment with rIL6, also upregulated the expression of SNAI1 and SNAI2 (Figure 6I).
Interestingly, the EC gene expression profile was also remodeled toward an activated and proangiogenic phenotype following stimulation with the second-level mediators of IL-30, since treatment with rDKK1 upregulated VEGFR1, MMP14, ACKR3, and CCR2. Treatment with rOPG upregulated EC expression of ANG, PDGFA, ACKR3, and DLL4, whereas treatment with rIL6 upregulated MMP9, CCR2, and JAG1. All three second-level mediators of IL-30 upregulated the expression of CCL2, FGF1, FGF2, and ACKR1/DARC, while they downregulated the expression of TGFB1 and CXCR4 (Figures 6J–6L).
Next, we wondered whether the three IL-30-inducible mediators could also regulate EC assembly into 3D vascular networks. To address this issue, EC tube formation assay was performed by treating the ECs, seeded onto Matrigel, with rDKK1, rOPG, or rIL6 (50 ng/mL) for 18 h. Quantification of tube network formation, performed with the angiogenesis analyzer plug-in from the ImageJ software, demonstrated that treatment with rDKK1, rOPG, and rIL6 leads to a significant (ANOVA: p < 0.0001) increase in the number of tubes, meshes, junctions, and nodes per field (Figures 6M–6P), which overall indicate an implementation of the microvessel network induced by tumor-derived mediators that are suppressed by IL-30 targeting (Figure 6Q).
Finally, to determine the impact of second-level mediators in the process of BM colonization by PC cells, we repeated the metastasis experiment in the PC-BM 2-OC platforms containing DU145 or PC3 cells in the presence of neutralizing anti-DKK1, anti-OPG, and anti-IL6 Abs. Administration of neutralizing Abs, starting 48 h from the experimental setup, as described in the materials and methods, significantly inhibited, but did not abrogate, the colonization of the BM scaffold by both DU145 and PC3 cells (Figures 6R–6T), which substantiated the implication of DKK1, OPG, and IL-6, whose PC cell release is suppressed by IL-30-targeting NPs in IL-30-driven PC progression.
Discussion
Bone and lung metastases are the leading cause of death in patients with PC since effective therapy is still lacking. The development of new strategies for the treatment or prevention of cancer cell spread and colonization to secondary organs represents one of the key challenges in oncology.54 Understanding the cellular and molecular mechanisms driving disease progression will yield the necessary information to address this issue.55 IL-30 has recently emerged as a PC driver gene that acts as an autocrine growth factor in both stem and non-stem cancer cells and as an upstream regulator of PC progression programs,10,11,12 whereas in a paracrine way it reshapes the tumor microenvironment (TME) by promoting myeloid-derived immune cell recruitment and EC activation and angiogenetic reprogramming.14,15,16
Here, we demonstrate the antimetastatic efficacy of CRISPR-Cas9 targeting of the IL30 gene,15 delivered by submicron liposomes to PC cells and tumor microemboli trapped within the lung microvascular network. These nanovectors, which are suitable for large-scale production with high-throughput microfluidic technology,56 were functionalized with Abs that, through binding to surface tumor antigens, favored NP uptake and internalization in PC cells. Expression of PSCA, which involves most PCs, and is positively related to androgen independence and metastatic disease,57 is absent in the majority of normal tissues and barely detected by single-cell RNA-seq in the prostatic, salivary, respiratory, and gastrointestinal epithelium,58 where IL-30 expression is missing.59 Therefore, potential on-target toxicities of Cas9hIL30-PSCA NPs in humans are reduced, while their significant editing efficiency, which ranges from 45% to 61% in the lung metastasis of the NP-treated animals, highlights the effectiveness of this nanotherapeutic strategy.
Although the percentage of cells successfully edited by CRISPR-Cas9gRNA can vary widely based on several factors, including cell cycle phase,60 the genetic modifications introduced are permanent and passed on to daughter cells during cell division leading to long-lasting genetic changes. However, to keep unedited clones under control, repeating treatment cycles are needed to ensure long-lasting therapeutic effects.61
Both treatment schedules of five and even three administrations of NPs, starting from the onset of tumor microemboli in the pulmonary capillaries, substantially hindered metastasis development, as observed in untreated mice bearing tumor pulmonary microemboli of IL30KO-PC cells that have been generated by direct CRISPR-Cas9-mediated gene deletion.15 The safety of the proposed IL-30 gene-targeting nanoplatform is demonstrated by the absence of histological and ultrastructural signs of damage in association with the absence of alteration in blood levels of organ damage biomarkers. The absence of evident inflammatory responses, as assessed by testing immunoliposomes in fully immunocompetent animals, suggests the lack of immunogenicity of the treatment, which is essential for its eligibility for the clinical setting.62
The antimetastatic effects of the nanotherapy result from: (1) the direct action of IL-30 genomic editing in PC cells, which leads to the inhibition of their proliferation and to a crackdown of their metastasis gene expression program, highlighted by the upregulation of CDH128 and downregulation of invasion and migration driver genes, such as CXCR2,30 CXCR4,28 IGF1,31,32 METAP2,33 MMP2,34,35 L1CAM,36 and TNFSF10/TRAIL,37 and (2) the abrogation of IL-30-dependent downstream soluble mediators of PC progression and EC activation and proliferation, such as CXCL2/GROβ,43 DKK1,46,47 OPG,48,52 and IL-6,49,53 whose involvement emerged with the use of PC-Lung and PC-BM 2-OC platforms and from the assessment, in the 2-OC culture supernatant, of chemokines and BM metabolism regulating factors.
While in vivo xenograft models of experimental metastasis allow for experimentation with patient-derived cancer cell lines, they have the limitation of lacking cell-mediated immunity. The involvement of innate immune cells in IL-30-driven PC progression, which was possible to explore in this model, is strongly suggested by the reduction of the intra-metastatic influx of granulocytes and MDCs following IL-30-targeting nanotherapy, and associated inhibition of PC cell expression of CXCL2/GROβ, a powerful chemoattractant for these immune cells,44 which could enable tumor invasiveness through a variety of mechanisms.63
Second-level mediators, DKK1, OPG, and IL-6, whose tumor release is regulated by IL-30, demonstrate a crucial role in strengthening the metastatic phenotype of PC cells by promoting their EMT and migratory ability, but also by reshaping the transcriptional profile of the ECs, and finally by fostering vascular branching and angiogenesis.64 The significant reduction of the antimetastatic efficacy of IL-30-targeting nanotherapy, due to anti-DKK1, OPG, and IL-6 Abs included in PC-BM 2-OC platform, confirms the deep implication of DKK1, OPG, and IL-6 in the dynamics of the IL-30-driven PC metastatic process.
Before IL-30-targeting nanotherapy can be a candidate for clinical trials, important steps must be taken. It is essential to develop a standardized and low-cost patient selection protocol to quickly determine tumor expression levels of IL-30 and identify subjects eligible for treatment. It would be of clinical significance to test the efficacy of treatment in patient-derived 3D organoid cultures containing the PC immune cell background,65 which would help in selecting patients who may benefit from the treatment. Overcoming the immunosuppression orchestrated by MDCs in the PC microenvironment66 by using the IL-30-targeting nanoplatform could implement the poor effects thus far obtained with immunotherapy in the treatment of advanced disease.67,68 Testing a combined treatment, tailored on the immunological profile of the patient’s tumor, will be the next step to work on.
Nanoparticle-based delivery has several advantages, including the ability to load and protect a variety of drugs to control drug release, to improve drug pharmacokinetics and biodistribution, and site- or cell-specific targeting. CRISPR-Cas9 is the most developed and widely used tool for current genome editing strategies69 since it is highly specific, efficient, and versatile,70 and it has already entered clinical trials for the treatment of different human diseases, including cancer.71
Therefore, the development of a biocompatible immunoliposome-based delivery system for selective CRISPR-Cas9 genome editing of the IL-30 gene in disseminated PC cells and micrometastasis can provide a modern and patient-tailored weapon against PC progression and metastatic spread.
Materials and methods
Cell lines
-
(1)
Human PC cell lines derived from the metastases of high-grade PCs, DU145 cells, endowed with a CK8/14+AR+PSA+ phenotype15,23 and castration-resistant PC3 cells, endowed with a CD44+AR−PSA–CgA+NSE+ neuroendocrine phenotype,24 were purchased from the American Type Culture Collection (ATCC) (Manassas, VA). DU145 cell line was cultured in RPM1 1640 (no. 15-040-CV, Corning, Manassas, VA) with 10% FCS (Seromed, Biochrom KG, Berlin, Germany). PC3 cell line was cultured in F12 Ham’s (no. 10-080-CV, Corning) with 10% FCS (Seromed, Biochrom KG).
-
(2)
Human alveolar type I epithelial cell line, CI-hAELVi, immortalized by using the CI-SCREEN technology72 and characterized by Kuehn et al.73 was purchased from InSCREENeX (no. INS-CI-1015, Braunschweig, Germany). CI-hAELVi were cultured in huAEC Medium plus supplement kit (INS-ME-1013, InSCREENeX) on T-75 flasks coated with huAEC Coating Solution (INS-SU-1018, InSCREENeX).
-
(3)
Human umbilical vein endothelial cells (HUVECs) (no. PCS-100–010) were purchased from the ATCC and cultivated in Vascular Cell Basal Medium (no. PCS-100-030; ATCC) plus Endothelial Cell Growth Kit-VEGF (no. PCS-100–041, ATCC).
-
(4)
Bone marrow CD34+hematopoietic stem and progenitor cells, isolated from BM mononuclear cells, were purchased from Lonza (no. 2M-101, Morrisville, NC) and cultured in StemSpan SFEM II medium (no. 09655, STEMCELL Technologies, Vancouver, Canada) supplemented with StemSpan CD34+ Expansion Supplement (no. 02691, STEMCELL Technologies).
-
(5)
Human MSCs (hMSCs), isolated from human tissue, were purchased from Lonza (PT-2501), and cultured in Mesencult-ACF Plus Medium Kit (05445, STEMCELL Technologies).
All cell lines were authenticated by short tandem repeat profile analysis and confirmed mycoplasma-free by PCR analysis and passaged for fewer than 6 months after resuscitation.
Preparation of PC and lung spheroids
PC spheroid (PC cell aggregates and vascular channels)
To mimic the in vivo tumor microenvironment, PC spheroids containing PC cells DU145 or PC3, in co-culture with HUVECs, were set up. DU145 and/or PC3 cells and HUVECs were seeded in 96-well U-bottom ultra-low-attachment plate (no. 174925; Thermo Fisher Scientific, Waltham, MA), in a 1:3 ratio, respectively, for 24 h to allow spheroid formation. After 24 h the spheroids were transferred with a sterile Transfer Pipet (no. 204-1S, Thermo Fisher Scientific) into a 24-well ultra-low attachment plate (no. 174930, Thermo Fisher Scientific) for 48 h. After 48 h, the spheroids were transferred into the 24-well compartment of the 2-OC (HUMIMIC Chip2, 24-well; TissUse). Depending on the experimental set up, the PC spheroids were cultured in cell culture medium consisting in Stem-Span-AOF medium containing 10 ng/mL thrombopoietin (no. 300-18; Thermo Fisher Scientific), 25 ng/mL Fms-related tyrosine kinase 3 ligand (no. 300-19, Thermo Fisher Scientific), and Vascular Cell Basal Medium (no. PCS-100–030, ATCC), supplemented with Endothelial Cell Growth Kit-VEGF (no. PCS-100–041, ATCC), in a 1:1 ratio, for the study of BM metastasis, and in huAEC Medium, plus supplement kit (no. INS-ME-1013, InSCREENeX), and Vascular Cell Basal Medium (no. PCS-100–030, ATCC) supplemented with Endothelial Cell Growth Kit-VEGF (no. PCS-100–041, ATCC), in a 1:1 ratio, for the study of Lung metastasis.
Lung spheroid (alveolar acini and vascular channels)
To simulate the lung tissue, lung spheroids, consisting of CI-hAELVi, in co-culture with HUVECs, were prepared. CI-hAELVi and HUVECs were seeded in 96-well U-bottom ultra-low-attachment plates (no. 174925, Thermo Fisher Scientific), in a 1:2 ratio respectively, for 24 h, to allow spheroid formation. After 24 h, the spheroids were transferred, with a sterile Transfer Pipet (no. 204-1S, Thermo Fisher Scientific), into a 24-well ultra-low attachment plate (no. 174930, Thermo Fisher Scientific) for 48 h. After 48 h, the spheroids were transferred in the 96-well compartment of the 2-OC. The lung spheroids were cultured in huAEC Medium plus supplements (no. INS-ME-1013, InSCREENeX) and Vascular Cell Basal Medium (no. PCS-100–030, ATCC) supplemented with Endothelial Cell Growth Kit-VEGF (no. PCS-100–041, ATCC) in a 1:1 ratio.
3D models of BM niche
3D models of the BM were prepared according to TissUse’s protocols.74 hMSCs were cultured in static, T-175 flasks using the Mesencult-ACF Plus Medium for 7–10 days (no. 05445, STEMCELL Technologies). They were then seeded on Sponceram cylinders made of hydroxyapatite-coated zirconium oxide ceramic scaffold, and cultured for 7–10 days in Mesencult-ACF Plus Medium. On day 1 of the 2-OC experiment, the hMSCs containing scaffold were seeded with BM CD34+ hematopoietic stem and progenitor cells and inserted into the 96-well compartment of the 2-OC.
Incorporation of PC and lung spheroids or BM scaffold into 2-OC
The 2-OC used consisted of a 24-well compartment connected, in circuit, by microfluidic channels to a 96-well compartment. On each chip, two circuits were present. PC spheroids were placed inside an 8 μm culture plate insert (no. PI8P01250, Merck, Burlington, MA) present in the 24-well compartments of each circuit. Lung spheroids and BM scaffolds were loaded into the 96-well compartments of each circuit.
Flow cytometry-based multiplex immunoassays
To investigate potential TME mediators released in the “microenvironment” of the 2-OC, and to see if any of these mediators were affected by the treatment with the NPs, supernatants from the chips were collected at the beginning of the experiments (T0), after the third treatment with NPs (T3, day 8 of the experiment), and after the fifth treatment with NPs (T5, day 15 and final day of the experiment). Flow cytometry-based multiplex immunoassays, Human Bone Metabolism Panel version 2 (no. B741362, BioLegend, San Diego), which allows the detection of 13 molecules (OPG, OPN, PDGF-BB, ALPL, ACP5, Leptin, TNFSF11, TNF-α, IL-6, PTH, IL-1β, BMP-2, and DKK-1) and Human Proinflammatory Chemokine Panel 2 (no. 741158, BioLegend), which allows the detection of 12 molecules (CCL1, CCL13, CCL22, CCL7, CCL24, CCL19, CXCL13, CCL8, CX3CL1, CXCL12, CXCL2, and CCL18) were used. Cell culture supernatants were processed and acquired on a BD Scientific FACSCanto Flow Cytometer (RRID: SCR_018055) according to the manufacturer’s protocol. Flow cytometry data files were then analyzed using the LEGENDplex Data Analysis Software Suite provided on the manufacturer’s website.
ELISA
To determine the source, or sources, of the mediators found in the supernatant of the 2-OCs, ELISAs detecting OPG (no. ab100617, Abcam, Cambridge, UK), DKK1 (no. EHDKK1, Thermo Fisher Scientific), IL-6 (no. EH2IL6, Thermo Fisher Scientific), and CXCL2 (no. ab184862, Abcam) were used. Each of the cell lines present in the chips were seeded in a six-well plate, in single and/or in co-culture, in the same ratios and culture media used in the 2-OC, faithfully replicating the culture conditions used in the chips. The supernatants from the single and co-cultures were collected after 48 h and analyzed according to the manufacturer’s protocols.
CRISPR-Cas9-mediated IL30 gene knockout of PC cells
The CRISPR-Cas9 technology was used to generate IL30 gene knockout (IL30KO) in both DU145 and PC3 cells, and abrogation of IL-30 expression was validated by western blotting, as we described.15
Flow cytometry and Ab conjugation efficiency
PSCA expression on DU145 and PC3, and conjugation efficiency between the anti-PSCA Ab and the aldheyde-modified 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-(amino(polyethylene glycol)-2000) (DSPE-PEG2000) lipid, present on the external layer of the NP, were assessed by flow cytometry, as described in the supplemental materials and methods.
Synthesis of NPs
Cationic lipid nanocomplexes coated with PEG (NP) were synthesized using a Dolomite Microfluidics device (Royston, UK), which enables the standardized production of NPs (low polydispersion index, in the range of 0.06–0.25), high encapsulation efficiency, and overcomes the need for high temperatures during component assembly, which have proven detrimental to the stability of the Cas9-gRNA complex. Lipids used to produce the NPs are as follows:
-
(1)
1,2-Dioleoyl-3-trimethylammonium-propane (DOTAP) (Avanti Polar Lipids, Alabaster, AL), a cationic lipid used to obtain optimal loading efficiency of the Cas9-gRNA complex, which is negatively charged.75,76 DOTAP also function as PEG linker.
-
(2)
Fusogenic lipids, specifically, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) (Avanti Polar Lipids), useful to foster nanoparticle entry into the targeted cells.77
-
(3)
Stealth lipids, specifically DSPE-PEG2000 (Avanti Polar Lipids), used to increase circulation time and to prevent opsonization and subsequent phagocytosis by macrophages.78
-
(4)
DSPE-PEG2000, aldheyde/maleimide-modified PEG lipids, used for Ab conjugation.79
-
(5)
Cholesterol (Sigma-Aldrich, St. Louis, MO), used to provide structural stability to the lipid bilayer.80
All of the components used are approved for clinical use by the EMA and by the FDA.
To produce empty NPs, lipids (DOPE, DOTAP, CHOL, DSPEpeg2000, and DSPE-PEG-CHO) (Avanti Polar Lipids) were dissolved in ethanol (at molar ratios of 3.45:3.45:2.1:0.75:0.25, respectively) and then mixed in a micromixer chip with PBS at a 1:3 flow rate ratio (lipids/PBS). Then, nanoliposomes were dialyzed with Slide-A-Lyzer Dialysis Cassettes, 20K MWCO (no. 66003, Thermo Fisher Scientific), against PBS, to remove ethanol. Finally, the nanoparticle suspension was concentrated with Pierce Protein Concentrator PES, 100K MWCO (no. 88523X4, Thermo Fisher Scientific) to reach a final lipid concentration of 10 mg/mL.
To prepare RhB-labeled nanoliposomes, Lissamine Rhodamine B 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine, triethylammonium salt (no. L1392, Thermo Fisher Scientific) was added to the lipid mixture, at a 0.1% molar ratio.
To produce core-encapsulated (CE) gadolinium (Gd) nanoliposomes, CE-Gd-NPs, lipids were dissolved in ethanol and then mixed, in a micromixer chip, with a gadoteric acid (Gd-DOTA; no. G360000, SimSon Pharma, Mumbai, India) solution at a 1:3 flow rate ratio (lipids/Gd-DOTA). After mixing, unencapsulated Gd was removed by dialysis with Slide-A-Lyzer Dialysis Cassettes, 20K MWCO (Thermo Fisher Scientific). Then, the nanoparticle suspension was concentrated with Pierce Protein Concentrator PES, 100K MWCO (Thermo Fisher Scientific) to reach a final lipid concentration of 10 mg/mL. Lastly, nanoliposomes were conjugated with anti-PSCA Abs (no. PA5-65080, Thermo Fisher Scientific) and dialyzed with a 300K MWCO membrane (no. 131456T, Thermo Fisher Scientific) to remove unbound Abs.
Synthesis of the CRISPR-Cas9hIL30 NP and functionalization with anti-PSCA Abs
To produce CRISPR-Cas 9hIL30 NPs, lipids were dissolved in ethanol and then mixed, in a micromixer chip, with CRISPR-Cas9gRNA-hIL30 complex solution, at a 1:3 flow rate ratio (lipids/CRISPR-Cas9gRNA-hIL30). Then, the liposomes were dialyzed against PBS, at a pH of 6.5, with a 300K MWCO membrane (Thermo Fisher Scientific) to remove unencapsulated CRISPR-Cas9gRNA-hIL30 complex. The nanoparticle suspension was concentrated with Pierce Protein Concentrator PES, 100K MWCO (Thermo Fisher Scientific), to reach a final lipid concentration of 10 mg/mL.
Subsequently, anti-PSCA-specific Abs (Thermo Fisher Scientific cat. no. PA5-65080, RRID: AB_2662130) were conjugated on DSPE PEG Aldehyde (no. PG2-ALDS-2k, Nanocs, New York, NY) of the external bilayer of the pre-formed nanoliposomes to obtain CRISPR-Cas9hIL30-PSCA NP, referred to as Cas9hIL30-PSCA NP, using a displacement reaction mediated by sodium cyanoborohydride (no. 156159, Merck, Darmstadt, Germany). The nanoliposomes were dialyzed against PBS, at a pH 7.4, with a 300K MWCO membrane (Thermo Fisher Scientific) to remove unbound Abs and excess of sodium cyanoborohydride.
Physical characterization of NPs
The nanoparticles were characterized as follows.
Laser particle size analysis and zeta electromotive force analysis
To determine particle size and zeta electromotive force, empty NPs and CRISPR-Cas9gRNA-hIL30 NPs were dispersed in double-distilled water (to a final volume of 2 mL) and particle size and zeta electromotive force were measured using a 90Plus/BI-MAS ZetaPlus multiangle particle size analyzer (Brookhaven Instruments, Holtsville, NY).
Serum stability of the CRISPR-Cas9gRNA-IL30 NPs
To determine the serum stability of CRISPR-Cas9gRNA-hIL30 NPs, they were resuspended in medium containing 10% fetal calf serum, and the size of the nanoliposomes was measured every 6 h for 24 h using a 90Plus/BI-MAS ZetaPlus multiangle particle size analyzer (Brookhaven Instruments).
Resistance of CRISPR-Cas9gRNA-hIL30 NPs to enzymatic digestion
To confirm that the lipid shell protects the Cas9-gRNA complex from enzymatic degradation, 50 μg/μL proteinase K was added to the nanoparticle suspension (20 mg/mL lipid concentration), which was then incubated overnight at 37°C. Then, the nanoliposomes were dialyzed to remove the proteinase K and cleaved in DMSO. Subsequently, Cas9 quantification was performed by ELISA assay (Cas9 ELISA Kit, no. PRB-5079, Cell Biolabs, San Diego, CA) and compared with that obtained from an untreated nanoparticle suspension. The experiments were performed in triplicate.
Stability of CRISPR-Cas9gRNA-hIL30 NPs to different external pH values
The CRISPR-Cas9gRNA-hIL30 NPs were resuspended in a medium containing 50% fetal calf serum at pH 2.5, 6.5, or 9, and mixed by vortexing. The resuspended solution was placed on a horizontal shaker (70 rpm, 37°C ± 1°C) and, at fixed time intervals (0, 2, 4, 6, 12, and 24 h), the particle size of the nanoliposome was measured by using a 90Plus/BI-MAS ZetaPlus multiangle particle size analyzer (Brookhaven Instruments).
Morphological analyses of CRISPR-Cas9gRNA-hIL30 NPs by TEM
Size and shape of the nanoparticles were characterized by TEM. empty nanoliposomes conjugated with anti-PSCA Abs (empty-PSCA NPs) and CRISPR-Cas9gRNA-hIL30 NPs, conjugated with anti-PSCA Abs (Cas9IL30-PSCA NPs) were incubated in fresh culture medium, at 1:25, 1:50, and 1:100 dilutions, for 3 h. Then, the NPs were fixed in cacodylate-buffered 2.5% glutaraldehyde, post-fixed in osmium tetroxide, and embedded in Epon 812. Ultrathin sections were stained with uranyl acetate-lead citrate and analyzed using a Philips CM10 and a Fei-Philips Morgagni 268D transmission electron microscope (Philips, Eindhoven, NL).
Encapsulation efficiency of the CRISPR-Cas9hIL30 NPs
The Cas9hIL30 NP was cleaved in DMSO and diluted in PBS (pH 7.4). The resulting mixture was then centrifuged in a high-speed refrigerated centrifuge at 15,000 rpm for 1 h, and Cas9quantification was performed by ELISA assay (Cas9ELISA Kit, no. PRB-5079, Cell Biolabs) on the supernatant. Blank control was prepared by the same method, using the supernatant from the empty NPs. The encapsulation efficiency was calculated according to the following formula: encapsulation rate (%) = [(A2 –A 1)/A2] × 100%, where A1 was the amount of Cas9 measured in the supernatant and A2 was the initial amount of Cas9 used to prepare the Cas9hIL30 NP.
Release rate of the CRISPR-Cas9hIL30 NP
The Cas9hIL30 NP suspension was dialyzed against PBS, as described above, to remove unencapsulated CRISPR-Cas9gRNA-hIL30 complex. Subsequently, at scheduled time points (0, 5, 10, 20, 30 min and 1, 2, 3, 4, 5, 6, 7, 8, 10, 12, 24, and 36 h), an aliquot of the preparation was centrifuged at 15,000 rpm, the supernatant was discarded and the pellet, treated with DMSO, was used for Cas9 quantification (Cas9 ELISA Kit, Cell Biolabs). The Cas9 release rate was calculated according to the following formula: release rate (%) = 100 – [A1/A2 × 100], where A1 was the amount of Cas9 measured at the different time points and A2 was the initial amount of Cas9 measured at 0 min. The release rate of Cas9 by Cas9hIL30-loaded Ab-conjugated or unconjugated NPs was <40% at 30 min, which meets the liposome formulation guidelines in the international pharmacopoeia (https://digicollections.net/phint/2020/index.html#d/b.1) for less than 40% release in 0.5 h.
Editing quantification and analysis of the off-target cleavage in NP-treated PC cells in vitro and in the organs and lung metastasis of NP-treated mice in vivo
The two Trueguide synthetic gRNAs (sgRNAs) that we used for IL30 gene editing (no. CRISPR947272_SGM, ACCAGCTTGAACCAGGAGCA and no. CRISPR947284_SGM, AACCTCGGAGAGCAGCTTCC), were designed and synthesized by Thermo Fisher Scientific using a validated proprietary algorithm that predicts high-efficiency editing.
For editing quantification and evaluation of off-target cleavage events for the selected sgRNAs (Table S2), the whole genome obtained from PC cell cultures, treated or not with Cas9IL30-PSCA NxPs, and from lung metastases, prostate, lungs, liver, kidneys, and spleen of three PC3, or DU145 metastases-bearing mice, treated with one injection of Cas9IL30-PSCA NxPs, and three untreated mice, was sequenced by Lexogen (Wien, Austria), using the Illumina Platform NovaSeq 6000 System and Dragen software. The reads were aligned to the reference genomes and the percentages of sequence reads with insertions or deletions (variants) over the total number of sequence reads were calculated. The average frequency of variants induced in the IL-30 gene represented the on-target effects (editing efficiency), whereas the average frequency of variants induced in off-target sites represented the off-target effects.
The editing efficiency in lung metastases of NP-treated and untreated mice was assessed after isolating the metastatic cells from the lung tissue by laser capture microdissection (LCM), using the P.A.L.M. Micro Beam System (P.A.L.M. Microlaser Technologies, Bernried, Germany), as we described previously.81 Two 10-μm frozen sections were obtained from each lung sample. Tissue sections were mounted on PEN membrane-covered slides (P.A.L.M.), thawed at room temperature, and immersed in cold acetone (5 min). All reagents were prepared using Ultrapure DNAse/RNAse-Free distilled water (Invitrogen, Paisley, UK). Immediately after hematoxylin and eosin staining, tissue sections were used for LCM. Metastasis and tumor microemboli were selected from each section, then about 500–1,000 tumor cells were cut and catapulted intact into the cap of an LPC-Microfuge Tube (P.A.L.M.), and genomic DNA was immediately extracted and processed for whole genome sequencing according to the protocol reported.82
TEM analyses of PC spheroids incorporated in the 2-OC platform
The intracellular uptake of NPs by the 3D PC spheroid models composed of co-cultures of DU145, or PC3, and ECs was analyzed by TEM, as reported in the supplemental materials and methods.
LSC analyses of PC and lung spheroids, and BM scaffold incorporated in the 2-OC platform
To analyze the effects of NPs on the metastatic properties of PC cells, before preparation of PC and lung spheroids and 3D models of BM niche, PC3 and DU145 cells were stained with the LuminiCell Tracker 540 (Cell Labeling Kit, no. SCT010), whereas CI-hAELVi and hMSCs were stained with the LuminiCell Tracker 670 (Cell Labeling Kit, no. SCT011), both from Merck, according to manufacturer’s protocol.
At the end of the experiment, PC and lung spheroids and BM scaffolds were visualized under an LSM 800 confocal microscope (Zeiss, Oberkochen, Germany, RRID: SCR_015963) and the resulting images were evaluated and processed using ZEN Microscopy Software (Zeiss, RRID: SCR_013672).
Assessment of PC and EC viability in PC spheroids and analysis of PC cell colonization of lung spheroids or BM scaffolds in the 2-OC platform
Viability and apoptosis of tumor and ECs, and the number of tumor cells (GFP+) within the BM scaffolds or lung spheroids in the 2-OC platforms, were quantified by flow cytometry, as described in the supplemental materials and methods.
PCR array and real-time RT-PCR
PCR array and real-time RT-PCR were performed as described in the supplemental materials and methods, using the RT2 Profiler PCR Array Human Tumor Metastasis (no. PAHS-028Z, QIAGEN, Hilden, Germany) and selected primers specific for EMT genes and genes involved in ECs activation, proliferation, and sprouting (Table S4).
Biosafety and distribution of NPs in vivo
-
(1)
To measure markers of organ toxicity in serum, blood samples from mice treated with PBS, naked Cas9hIL30 complex, empty-PSCA NPs, Cas9hIL30 NPs, or Cas9hIL30-PSCA NPs were collected 21 days after the start of treatment, from the venous sinus, using a capillary tube. Aspartate aminotransferase (AST), alanine aminotransferase (ALT), blood urea nitrogen (BUN), creatinine (Cr), cardiac troponin-1 (cTnI), creatine kinase (CK), and lactate dehydrogenase (LDH) were measured in the sera using Mouse AST ELISA Kit (ab263882), Mouse ALT ELISA Kit (ab282882), Creatinine Assay Kit (ab65340), Creatine Kinase Activity Assay Kit (ab155901), and LDH Assay Kit (ab102526) (all from Abcam), and Urea Nitrogen (BUN) Colorimetric Detection Kit (Thermo Fisher Scientific) and Mouse Troponin I ELISA Kit (Novus Biologicals, Centennial, CO), according to the manufacturers’ protocols.
-
(2)
To assess the immunogenicity of NPs, four groups of five fully immunocompetent BALB/c male mice were i.v. injected with PBS, or naked Cas9hIL30 complex, or empty-PSCA NPs or Cas9hIL30-PSCA NPs. Blood samples were collected24 h post injection to measure the serum levels of TNF-α and IL-6 using the Mouse TNF-alpha Quantikine ELISA Kit (no. MTA00B, R&D Systems, Minneapolis, MN) and the IL-6 Mouse ELISA Kit (no. KMC0061, Thermo Fisher Scientific) according to the manufacturers’ protocol.
-
(3)
Histopathological and ultrastructural analyses of the intracellular uptake and toxicity of NP in vivo. After autopsy, tissue samples from organs (lungs, heart, liver, kidneys, prostate, and spleen) of NP-treated and control mice were collected. For the histological analyses, tissue samples were fixed in 4% formalin, embedded in paraffin, sectioned at 4 μm and stained with hematoxylin and eosin.
For ultrastructural analyses with TEM, small tissue samples (1 mm3) from liver, kidneys, and from the inner areas and edges of the tumor xenografts from NP-treated and control mice, were fixed in cacodylate-buffered 2.5% glutaraldehyde for 24 h at 4°C, post-fixed in 1% osmium tetroxide for 2 h, dehydrated with graded acetones, and embedded in Epon 812 (Electron Microscopy Sciences). Ultrathin sections (0.5–1 μm thick) were stained with 1% methylene blue and used to select suitable areas of ultrastructural sectioning. Finally, ultrathin sections (60 nm thick) were collected on copper grids, counterstained with uranyl-acetate and lead citrate, and examined with a Philips CM10 (TEM) and/or Fei-Philips Morgagni 268D transmission electron microscopes (FEI, Eindhoven, the Netherlands).
-
(4)
For LSC microscopy analyses of the uptake of NPs by lung metastases, lung parenchyma, prostate, spleen, liver, and kidneys, two groups of 24 NSG male mice were i.v. injected, in the tail vein, with GFP-labeled DU145 or with PC3 cells. After 10 days, each group of mice was divided into two groups of 12 mice, one was i.v. injected with RhB-labeled NPs (RhB-NP) and the other with RhB-labeled NPs conjugated with anti-PSCA Abs (RhB-PSCA-NP). Each group of 12 mice was then divided into 4 groups of 3 mice, which were sacrificed at different time points (5, 15, 30, 60, and 90 min) after the administration of NPs. LSC images of tumor emboli, lung parenchyma, prostate, spleen, liver, and kidneys were captured under an LSM 800 confocal microscope (Zeiss). To quantify the uptake of NPs by the lung metastases, the fluorescence intensity and distribution of RhB-NPs, RhB-PSCA-NPs, and GFP+ tumor cells were evaluated using ZEN Microscopy Software (Zeiss, RRID: SCR_013672), and the uptake of the NPs was expressed as the mean percentage ± SD of RhB+GFP+ cells/total number of GFP+ cells. The fluorescence intensity and distribution of RhB-NPs or RhB-PSCA-NPs in the lung parenchyma were visualized after staining of the alveolar cells with rabbit anti-EpCam antibody (Thermo Fisher Scientific cat. no. MA5-35283, RRID: AB_2849185), followed by goat anti-rabbit IgG Alexa Fluor 488-conjugated antibody (Thermo Fisher Scientific, cat. no. A32731, RRID: AB_2633280), and the uptake of the NPs was expressed as the mean percentage ± SD of RhB+EpCam+ cells/total number of EpCam+ cells. The fluorescence intensity and distribution of NPs in prostate, spleen, liver, and kidneys were visualized after staining with Alexa Fluor 488 Phalloidin (Thermo Fisher Scientific, cat. no. A12379) and the uptake of the NPs was expressed as the mean percentage ± SD of RhB+Phalloidin+ cells/total number of Phalloidin+ cells. PBS-treated GFP+ metastases-bearing mice were used as a negative control. Three sections per sample were analyzed and six to eight high-power fields were evaluated for each section.
Pharmacokinetic studies
For pharmacokinetic studies, three groups of five BALB/c mice were i.v. injected in the tail vein with 3 × 105 wild-type DU145 cells, and three groups of five BALB/c mice were i.v. injected with 3 × 105 wild-type PC3 cells. After 12 days, mice were i.v. treated with Cas9hIL30 NPs conjugated or unconjugated with anti-PSCA Abs, or free Cas9 (in PBS), at a dose of 1.5 mg of Cas9 equiv/kg. Subsequently, blood was collected at pre-determined time intervals (5, 15, and 30 min, or 1, 2, 4, 6, 8, 10, and 12 h) for ELISA assay (Cas9 ELISA Kit, no. PRB-5079, Cell Biolabs).
In vivo testing of CRISPR-Cas9gRNA-hIL30 NP treatment in the experimental models of PC metastasis
Eight-week-old NSG male mice were purchased from Charles River (Wilmington, MA) and were housed under high barrier conditions according to the Jackson Laboratory’s guidelines (https://www.jax.org/jax-mice-and-services/find-and-order-jax-mice/nsg-portfolio/housing-and-breeding-considerations-for-nsg-mice) in the animal facility of the Center for Advanced Studies and Technology at the "G. d'Annunzio" University of Chieti-Pescara.
To study the effects of IL-30 target treatment on the onset and development of lung metastases, 3 groups of 10, 8-week-old, NSG male mice were i.v. injected in the tail vein with 3 × 105 wild-type (CTRL) DU145 or PC3 cells.
Mice were treated with Cas9hIL30-PSCA NPs, empty-PSCA NPs, or PBS, starting from 72 h after the i.v. inoculation of PC cells, and twice a week thereafter, with a five or three administration schedule, as represented in Figure 2L. An additional two groups of 10, 8-week-old, NSG male mice were i.v. injected with non-targeting guide RNA-transfected (NTgRNA) or IL-30 knockout (IL30KO) DU145 or PC3 cells, and left untreated. Animal health and well-being were monitored daily. Forty-eight hours after the last treatment administration, mice were euthanized and autopsy was performed, followed by the collection of lungs and tissue sampling of the different organs (heart, liver, kidneys, spleen, and prostate) for histopathological and ultrastructural analyses.
Power analysis and sample size calculation
An overall sample size of 10 mice per group allowed the detection of a statistically significant difference in lung metastasis, between five groups (ANOVA), with an 80% power, at a 0.05 significance level (G∗Power, RRID: SCR_013726). Animal procedures were performed in accordance with the European Community and ARRIVE guidelines and were approved by the Institutional Animal Care Committee of “G. d’Annunzio” University and by the Italian Ministry of Health (authorization no. 892/2018-PR).
Histology, immunohistochemistry, and immunofluorescence for LSC microscopy
Histology, immunohistochemistry (performed with the Abs listed in the Table S5), assessment of proliferation indices, and evaluation of immune cell content and cytokine expression in metastasis, were performed as described in the supplemental materials and methods.
Statistics
For in vitro and in vivo studies, between-group differences were assessed by Student’s t-test, or ANOVA, followed by Tukey HSD test. All statistical tests were evaluated at an α level of 0.05, using Stata, version 13 (StataCorp, College Station, TX; RRID: SCR_012763). Results were expressed as mean ± standard deviation and Prism 6 (GraphPad Software, La Jolla, CA) was used to perform all statistical analyses.
Data and code availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Acknowledgments
The research leading to these results has received funding from AIRC under IG 2019 – ID. 23264 project – P.I. Di Carlo Emma, and MUR (Ministero dell'Università e della Ricerca) Programma Operativo Nazionale Ricerca e Innovazione/National Operational Program for Research and Innovation 2014–2020 (PON R&I) to Carlo Sorrentino.
Author contributions
E.D.C. conceived the study, interpreted the data, wrote the manuscript, and is responsible for the overall content. C.F. and S.L.C. performed the experiments and data analyses. C.S. and S.M. performed the experiments. L.V.L. performed the electron microscopy. P.L. and S.V. performed the flow cytometry analyses. All authors reviewed and approved the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2024.09.011.
Supplemental information
References
- 1.Sung H., Ferlay J., Siegel R.L., Laversanne M., Soerjomataram I., Jemal A., Bray F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Belkahla S., Nahvi I., Biswas S., Nahvi I., Ben Amor N. Advances and development of prostate cancer, treatment, and strategies: A systemic review. Front. Cell Dev. Biol. 2022;10 doi: 10.3389/fcell.2022.991330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Al-Mansour Z., Pang L., Bathini V. Novel Cancer Therapeutics in Geriatrics: What is Unique to the Aging Patient? Drugs Aging. 2019;36:1–11. doi: 10.1007/s40266-018-0619-2. [DOI] [PubMed] [Google Scholar]
- 4.Gerstberger S., Jiang Q., Ganesh K. Metastasis. Cell. 2023;186:1564–1579. doi: 10.1016/j.cell.2023.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hapach L.A., Mosier J.A., Wang W., Reinhart-King C.A. Engineered models to parse apart the metastatic cascade. NPJ Precis. Oncol. 2019;3:20. doi: 10.1038/s41698-019-0092-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bruni D., Angell H.K., Galon J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat. Rev. Cancer. 2020;20:662–680. doi: 10.1038/s41568-020-0285-7. [DOI] [PubMed] [Google Scholar]
- 7.Briukhovetska D., Dörr J., Endres S., Libby P., Dinarello C.A., Kobold S. Interleukins in cancer: from biology to therapy. Nat. Rev. Cancer. 2021;21:481–499. doi: 10.1038/s41568-021-00363-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pflanz S., Timans J.C., Cheung J., Rosales R., Kanzler H., Gilbert J., Hibbert L., Churakova T., Travis M., Vaisberg E., et al. IL-27, a heterodimeric cytokine composed of EBI3 and p28 protein, induces proliferation of naive CD4(+) T cells. Immunity. 2002;16:779–790. doi: 10.1016/s1074-7613(02)00324-2. [DOI] [PubMed] [Google Scholar]
- 9.Garbers C., Spudy B., Aparicio-Siegmund S., Waetzig G.H., Sommer J., Hölscher C., Rose-John S., Grötzinger J., Lorenzen I., Scheller J. An interleukin-6 receptor-dependent molecular switch mediates signal transduction of the IL-27 cytokine subunit p28 (IL-30) via a gp130 protein receptor homodimer. J. Biol. Chem. 2013;288:4346–4354. doi: 10.1074/jbc.M112.432955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Di Meo S., Airoldi I., Sorrentino C., Zorzoli A., Esposito S., Di Carlo E. Interleukin-30 expression in prostate cancer and its draining lymph nodes correlates with advanced grade and stage. Clin. Cancer Res. 2014;20:585–594. doi: 10.1158/1078-0432.CCR-13-2240. [DOI] [PubMed] [Google Scholar]
- 11.Sorrentino C., Ciummo S.L., Cipollone G., Caputo S., Bellone M., Di Carlo E. Interleukin-30/IL27p28 Shapes Prostate Cancer Stem-like Cell Behavior and Is Critical for Tumor Onset and Metastasization. Cancer Res. 2018;78:2654–2668. doi: 10.1158/0008-5472.CAN-17-3117. [DOI] [PubMed] [Google Scholar]
- 12.Sorrentino C., Yin Z., Ciummo S., Lanuti P., Lu L.F., Marchisio M., Bellone M., Di Carlo E. Targeting Interleukin(IL)-30/IL-27p28 signaling in cancer stem-like cells and host environment synergistically inhibits prostate cancer growth and improves survival. J. Immunother. Cancer. 2019;7:201. doi: 10.1186/s40425-019-0668-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Müller S.I., Friedl A., Aschenbrenner I., Esser-von Bieren J., Zacharias M., Devergne O., Feige M.J. A folding switch regulates interleukin 27 biogenesis and secretion of its α-subunit as a cytokine. Proc. Natl. Acad. Sci. USA. 2019;116:1585–1590. doi: 10.1073/pnas.1816698116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sorrentino C., Ciummo S.L., D'Antonio L., Fieni C., Lanuti P., Turdo A., Todaro M., Di Carlo E. Interleukin-30 feeds breast cancer stem cells via CXCL10 and IL23 autocrine loops and shapes immune contexture and host outcome. J. Immunother. Cancer. 2021;9 doi: 10.1136/jitc-2021-002966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sorrentino C., D'Antonio L., Ciummo S.L., Fieni C., Landuzzi L., Ruzzi F., Vespa S., Lanuti P., Lotti L.V., Lollini P.L., Di Carlo E. CRISPR/Cas9-mediated deletion of Interleukin-30 suppresses IGF1 and CXCL5 and boosts SOCS3 reducing prostate cancer growth and mortality. J. Hematol. Oncol. 2022;15:145. doi: 10.1186/s13045-022-01357-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ciummo S.L., Sorrentino C., Fieni C., Di Carlo E. Interleukin-30 subverts prostate cancer-endothelium crosstalk by fostering angiogenesis and activating immunoregulatory and oncogenic signaling pathways. J. Exp. Clin. Cancer Res. 2023;42:336. doi: 10.1186/s13046-023-02902-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morshedzadeh F., Ghanei M., Lotfi M., Ghasemi M., Ahmadi M., Najari-Hanjani P., Sharif S., Mozaffari-Jovin S., Peymani M., Abbaszadegan M.R. An Update on the Application of CRISPR Technology in Clinical Practice. Mol. Biotechnol. 2024;66:179–197. doi: 10.1007/s12033-023-00724-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karimian A., Azizian K., Parsian H., Rafieian S., Shafiei-Irannejad V., Kheyrollah M., Yousefi M., Majidinia M., Yousefi B. CRISPR/Cas9 technology as a potent molecular tool for gene therapy. J. Cell Physiol. 2019;234:12267–12277. doi: 10.1002/jcp.27972. [DOI] [PubMed] [Google Scholar]
- 19.Gu Z., Thomas G., Yamashiro J., Shintaku I.P., Dorey F., Raitano A., Witte O.N., Said J.W., Loda M., Reiter R.E. Prostate stem cell antigen (PSCA) expression increases with high gleason score, advanced stage and bone metastasis in prostate cancer. Oncogene. 2000;19:1288–1296. doi: 10.1038/sj.onc.1203426. [DOI] [PubMed] [Google Scholar]
- 20.Ingber D.E. Human organs-on-chips for disease modelling, drug development and personalized medicine. Nat. Rev. Genet. 2022;23:467–491. doi: 10.1038/s41576-022-00466-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Manna F.L., Karkampouna S., Zoni E., De Menna M., Hensel J., Thalmann G.N., Kruithof-de Julio M. Metastases in Prostate Cancer. Cold Spring Harbor Perspect. Med. 2019;9 doi: 10.1101/cshperspect.a033688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reiter R.E., Gu Z., Watabe T., Thomas G., Szigeti K., Davis E., Wahl M., Nisitani S., Yamashiro J., Le Beau M.M., et al. Prostate stem cell antigen: a cell surface marker overexpressed in prostate cancer. Proc. Natl. Acad. Sci. USA. 1998;95:1735–1740. doi: 10.1073/pnas.95.4.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stone K.R., Mickey D.D., Wunderli H., Mickey G.H., Paulson D.F. Isolation of a human prostate carcinoma cell line (DU 145) Int. J. Cancer. 1978;21:274–281. doi: 10.1002/ijc.2910210305. [DOI] [PubMed] [Google Scholar]
- 24.Tai S., Sun Y., Squires J.M., Zhang H., Oh W.K., Liang C.Z., Huang J. PC3 is a cell line characteristic of prostatic small cell carcinoma. Prostate. 2011;71:1668–1679. doi: 10.1002/pros.21383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiong X., Chen M., Lim W.A., Zhao D., Qi L.S. CRISPR/Cas9 for Human Genome Engineering and Disease Research. Annu. Rev. Genomics Hum. Genet. 2016;17:131–154. doi: 10.1146/annurev-genom-083115-022258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blanken D., Foschepoth D., Serrão A.C., Danelon C. Genetically controlled membrane synthesis in liposomes. Nat. Commun. 2020;11:4317. doi: 10.1038/s41467-020-17863-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ghosh S. In: Nanomaterials Safety: Toxicity and Health Hazards. Gosh S., editor. De Gruyter; 2019. 9. Toxicity and effect of nanoparticles on liver and kidneys, and kidney disorders; pp. 207–214. [Google Scholar]
- 28.Deep G., Gangar S.C., Agarwal C., Agarwal R. Role of E-cadherin in antimigratory and antiinvasive efficacy of silibinin in prostate cancer cells. Cancer Prev. Res. 2011;4:1222–1232. doi: 10.1158/1940-6207.CAPR-10-0370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hughes C.E., Nibbs R.J.B. A guide to chemokines and their receptors. FEBS J. 2018;285:2944–2971. doi: 10.1111/febs.14466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mollica Poeta V., Massara M., Capucetti A., Bonecchi R. Chemokines and Chemokine Receptors: New Targets for Cancer Immunotherapy. Front. Immunol. 2019;10:379. doi: 10.3389/fimmu.2019.00379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Graham T.R., Zhau H.E., Odero-Marah V.A., Osunkoya A.O., Kimbro K.S., Tighiouart M., Liu T., Simons J.W., O’Regan R.M. Insulin-like growth factor-I-dependent up-regulation of ZEB1 drives epithelial-to-mesenchymal transition in human prostate cancer cells. Cancer Res. 2008;68:2479–2488. doi: 10.1158/0008-5472.CAN-07-2559. [DOI] [PubMed] [Google Scholar]
- 32.Liu G., Zhu M., Zhang M., Pan F. Emerging Role of IGF-1 in Prostate Cancer: A Promising Biomarker and Therapeutic Target. Cancers (Basel) 2023;15:1287. doi: 10.3390/cancers15041287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xie J., Rice M.A., Chen Z., Cheng Y., Hsu E.C., Chen M., Song G., Cui L., Zhou K., Castillo J.B., et al. In Vivo Imaging of Methionine Aminopeptidase II for Prostate Cancer Risk Stratification. Cancer Res. 2021;81:2510–2521. doi: 10.1158/0008-5472.CAN-20-2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ross J.S., Kaur P., Sheehan C.E., Fisher H.A.G., Kaufman R.A., Jr., Kallakury B.V.S. Prognostic significance of matrix metalloproteinase 2 and tissue inhibitor of metalloproteinase 2 expression in prostate cancer. Mod. Pathol. 2003;16:198–205. doi: 10.1097/01.MP.0000056984.62360.6C. [DOI] [PubMed] [Google Scholar]
- 35.Trudel D., Fradet Y., Meyer F., Harel F., Têtu B. Significance of MMP-2 expression in prostate cancer: an immunohistochemical study. Cancer Res. 2003;63:8511–8515. [PubMed] [Google Scholar]
- 36.Altevogt P., Doberstein K., Fogel M. L1CAM in human cancer. Int. J. Cancer. 2016;138:1565–1576. doi: 10.1002/ijc.29658. [DOI] [PubMed] [Google Scholar]
- 37.Oh Y.T., Sun S.Y. Regulation of Cancer Metastasis by TRAIL/Death Receptor Signaling. Biomolecules. 2021;11:499. doi: 10.3390/biom11040499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dong J.T., Lamb P.W., Rinker-Schaeffer C.W., Vukanovic J., Ichikawa T., Isaacs J.T., Barrett J.C. KAI1, a metastasis suppressor gene for prostate cancer on human chromosome 11p11.2. Science. 1995;268:884–886. doi: 10.1126/science.7754374. [DOI] [PubMed] [Google Scholar]
- 39.Yan W., Huang J., Zhang Q., Zhang J. Role of Metastasis Suppressor KAI1/CD82 in Different Cancers. J. Oncol. 2021;2021 doi: 10.1155/2021/9924473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gründker C., Emons G. The Role of Gonadotropin-Releasing Hormone in Cancer Cell Proliferation and Metastasis. Front. Endocrinol. 2017;8:187. doi: 10.3389/fendo.2017.00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Masola V., Zaza G., Gambaro G., Franchi M., Onisto M. Role of heparanase in tumor progression: Molecular aspects and therapeutic options. Semin. Cancer Biol. 2020;62:86–98. doi: 10.1016/j.semcancer.2019.07.014. [DOI] [PubMed] [Google Scholar]
- 42.Saharinen P., Eklund L., Pulkki K., Bono P., Alitalo K. VEGF and angiopoietin signaling in tumor angiogenesis and metastasis. Trends Mol. Med. 2011;17:347–362. doi: 10.1016/j.molmed.2011.01.015. [DOI] [PubMed] [Google Scholar]
- 43.Ribatti D., Pezzella F. Overview on the Different Patterns of Tumor Vascularization. Cells. 2021;10:639. doi: 10.3390/cells10030639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang H., Ye Y.L., Li M.X., Ye S.B., Huang W.R., Cai T.T., He J., Peng J.Y., Duan T.H., Cui J., et al. CXCL2/MIF-CXCR2 signaling promotes the recruitment of myeloid-derived suppressor cells and is correlated with prognosis in bladder cancer. Oncogene. 2017;36:2095–2104. doi: 10.1038/onc.2016.367. [DOI] [PubMed] [Google Scholar]
- 45.Bubendorf L., Schöpfer A., Wagner U., Sauter G., Moch H., Willi N., Gasser T.C., Mihatsch M.J. Metastatic patterns of prostate cancer: an autopsy study of 1,589 patients. Hum. Pathol. 2000;31:578–583. doi: 10.1053/hp.2000.6698. [DOI] [PubMed] [Google Scholar]
- 46.Thudi N.K., Martin C.K., Murahari S., Shu S.T., Lanigan L.G., Werbeck J.L., Keller E.T., McCauley L.K., Pinzone J.J., Rosol T.J. Dickkopf-1 (DKK-1) stimulated prostate cancer growth and metastasis and inhibited bone formation in osteoblastic bone metastases. Prostate. 2011;71:615–625. doi: 10.1002/pros.21277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pang H., Ma N., Shen W., Zhao Q., Wang J., Duan L., Chen W., Zhang N., Zhao Z., Liu L., Zhang H. Effects of DKK1 overexpression on bone metastasis of SBC-3 cells. Oncol. Lett. 2018;15:6739–6744. doi: 10.3892/ol.2018.8160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y., Liang J., Liu P., Wang Q., Liu L., Zhao H. The RANK/RANKL/OPG system and tumor bone metastasis: Potential mechanisms and therapeutic strategies. Front. Endocrinol. 2022;13 doi: 10.3389/fendo.2022.1063815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harmer D., Falank C., Reagan M.R. Interleukin-6 Interweaves the Bone Marrow Microenvironment, Bone Loss, and Multiple Myeloma. Front. Endocrinol. 2018;9:788. doi: 10.3389/fendo.2018.00788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Smadja D.M., d’Audigier C., Weiswald L.B., Badoual C., Dangles-Marie V., Mauge L., Evrard S., Laurendeau I., Lallemand F., Germain S., et al. The Wnt antagonist Dickkopf-1 increases endothelial progenitor cell angiogenic potential. Arterioscler. Thromb. Vasc. Biol. 2010;30:2544–2552. doi: 10.1161/ATVBAHA.110.213751. [DOI] [PubMed] [Google Scholar]
- 51.Choi S.H., Kim H., Lee H.G., Kim B.K., Park J.Y., Kim D.Y., Ahn S.H., Han K.H., Kim S.U. Dickkopf-1 induces angiogenesis via VEGF receptor 2 regulation independent of the Wnt signaling pathway. Oncotarget. 2017;8:58974–58984. doi: 10.18632/oncotarget.19769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rochette L., Meloux A., Rigal E., Zeller M., Cottin Y., Vergely C. The Role of Osteoprotegerin and Its Ligands in Vascular Function. Int. J. Mol. Sci. 2019;20:705. doi: 10.3390/ijms20030705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Middleton K., Jones J., Lwin Z., Coward J.I.G. Interleukin-6: an angiogenic target in solid tumours. Crit. Rev. Oncol. Hematol. 2014;89:129–139. doi: 10.1016/j.critrevonc.2013.08.004. [DOI] [PubMed] [Google Scholar]
- 54.Ganesh K., Massagué J. Targeting metastatic cancer. Nat. Med. 2021;27:34–44. doi: 10.1038/s41591-020-01195-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Massagué J., Batlle E., Gomis R.R. Understanding the molecular mechanisms driving metastasis. Mol. Oncol. 2017;11:3–4. doi: 10.1002/1878-0261.12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li X., Fan X., Li Z., Shi L., Liu J., Luo H., Wang L., Du X., Chen W., Guo J., et al. Application of Microfluidics in Drug Development from Traditional Medicine. Biosensors. 2022;12:870. doi: 10.3390/bios12100870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lam J.S., Yamashiro J., Shintaku I.P., Vessella R.L., Jenkins R.B., Horvath S., Said J.W., Reiter R.E. Prostate stem cell antigen is overexpressed in prostate cancer metastases. Clin. Cancer Res. 2005;11:2591–2596. doi: 10.1158/1078-0432.CCR-04-1842. [DOI] [PubMed] [Google Scholar]
- 58.The Human Protein Atlas. 2024. https://www.proteinatlas.org/ENSG00000167653-PSCA
- 59.The Human Protein Atlas. 2024. https://www.proteinatlas.org/ENSG00000197272-IL27
- 60.Li G., Yang X., Luo X., Wu Z., Yang H. Modulation of cell cycle increases CRISPR-mediated homology-directed DNA repair. Cell Biosci. 2023;13:215. doi: 10.1186/s13578-023-01159-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rosenblum D., Gutkin A., Kedmi R., Ramishetti S., Veiga N., Jacobi A.M., Schubert M.S., Friedmann-Morvinski D., Cohen Z.R., Behlke M.A., et al. CRISPR-Cas9 genome editing using targeted lipid nanoparticles for cancer therapy. Sci. Adv. 2020;6 doi: 10.1126/sciadv.abc9450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee Y., Jeong M., Park J., Jung H., Lee H. Immunogenicity of lipid nanoparticles and its impact on the efficacy of mRNA vaccines and therapeutics. Exp. Mol. Med. 2023;55:2085–2096. doi: 10.1038/s12276-023-01086-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Swierczak A., Pollard J.W. Myeloid Cells in Metastasis. Cold Spring Harbor Perspect. Med. 2020;10:a038026. doi: 10.1101/cshperspect.a038026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Al-Ostoot F.H., Salah S., Khamees H.A., Khanum S.A. Tumor angiogenesis: Current challenges and therapeutic opportunities. Cancer Treat. Res. Commun. 2021;28 doi: 10.1016/j.ctarc.2021.100422. [DOI] [PubMed] [Google Scholar]
- 65.Bogoslowski A., An M., Penninger J.M. Incorporating Immune Cells into Organoid Models: Essential for Studying Human Disease. Organoids. 2023;2:140–155. [Google Scholar]
- 66.Lopez-Bujanda Z., Drake C.G. Myeloid-derived cells in prostate cancer progression: phenotype and prospective therapies. J. Leukoc. Biol. 2017;102:393–406. doi: 10.1189/jlb.5VMR1116-491RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Guan X., Polesso F., Wang C., Sehrawat A., Hawkins R.M., Murray S.E., Thomas G.V., Caruso B., Thompson R.F., Wood M.A., et al. Androgen receptor activity in T cells limits checkpoint blockade efficacy. Nature. 2022;606:791–796. doi: 10.1038/s41586-022-04522-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.King A. Could immunotherapy finally break through in prostate cancer? Nature. 2022;609:S42–S44. doi: 10.1038/d41586-022-02861-y. [DOI] [PubMed] [Google Scholar]
- 69.Knott G.J., Doudna J.A. CRISPR-Cas guides the future of genetic engineering. Science. 2018;361:866–869. doi: 10.1126/science.aat5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kaushik I., Ramachandran S., Srivastava S.K. CRISPR-Cas9: A multifaceted therapeutic strategy for cancer treatment. Semin. Cell Dev. Biol. 2019;96:4–12. doi: 10.1016/j.semcdb.2019.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Innovative Genomics Institute. 2024. https://innovativegenomics.org/news/crispr-clinical-trials-2022
- 72.Lipps C., Klein F., Wahlicht T., Seiffert V., Butueva M., Zauers J., Truschel T., Luckner M., Köster M., MacLeod R., et al. Expansion of functional personalized cells with specific transgene combinations. Nat. Commun. 2018;9:994. doi: 10.1038/s41467-018-03408-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kuehn A., Kletting S., de Souza Carvalho-Wodarz C., Repnik U., Griffiths G., Fischer U., Meese E., Huwer H., Wirth D., May T., et al. Human alveolar epithelial cells expressing tight junctions to model the air-blood barrier. ALTEX. 2016;33:251–260. doi: 10.14573/altex.1511131. [DOI] [PubMed] [Google Scholar]
- 74.Sieber S., Wirth L., Cavak N., Koenigsmark M., Marx U., Lauster R., Rosowski M. Bone marrow-on-a-chip: Long-term culture of human haematopoietic stem cells in a three-dimensional microfluidic environment. J. Tissue Eng. Regen. Med. 2018;12:479–489. doi: 10.1002/term.2507. [DOI] [PubMed] [Google Scholar]
- 75.Marty R., N'soukpoé-Kossi C.N., Charbonneau D., Weinert C.M., Kreplak L., Tajmir-Riahi H.A. Structural analysis of DNA complexation with cationic lipids. Nucleic Acids Res. 2009;37:849–857. doi: 10.1093/nar/gkn1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sun W., Ji W., Hall J.M., Hu Q., Wang C., Beisel C.L., Gu Z. Self-assembled DNA nanoclews for the efficient delivery of CRISPR-Cas9 for genome editing. Angew. Chem. Int. Ed. Engl. 2015;54:12029–12033. doi: 10.1002/anie.201506030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kube S., Hersch N., Naumovska E., Gensch T., Hendriks J., Franzen A., Landvogt L., Siebrasse J.P., Kubitscheck U., Hoffmann B., et al. Fusogenic Liposomes as Nanocarriers for the Delivery of Intracellular Proteins. Langmuir. 2017;33:1051–1059. doi: 10.1021/acs.langmuir.6b04304. [DOI] [PubMed] [Google Scholar]
- 78.Immordino M.L., Dosio F., Cattel L. Stealth liposomes: review of the basic science, rationale, and clinical applications, existing and potential. Int. J. Nanomed. 2006;1:297–315. [PMC free article] [PubMed] [Google Scholar]
- 79.Sapra P., Allen T.M. Internalizing antibodies are necessary for improved therapeutic efficacy of antibody-targeted liposomal drugs. Cancer Res. 2002;62:7190–7194. [PubMed] [Google Scholar]
- 80.Kulkarni J.A., Witzigmann D., Leung J., Tam Y.Y.C., Cullis P.R. On the role of helper lipids in lipid nanoparticle formulations of siRNA. Nanoscale. 2019;11:21733–21739. doi: 10.1039/c9nr09347h. [DOI] [PubMed] [Google Scholar]
- 81.Sorrentino C., Di Carlo E. Expression of IL-32 in human lung cancer is related to the histotype and metastatic phenotype. Am. J. Respir. Crit. Care Med. 2009;180:769–779. doi: 10.1164/rccm.200903-0400OC. [DOI] [PubMed] [Google Scholar]
- 82.Ellis P., Moore L., Sanders M.A., Butler T.M., Brunner S.F., Lee-Six H., Osborne R., Farr B., Coorens T.H.H., Lawson A.R.J., et al. Reliable detection of somatic mutations in solid tissues by laser-capture microdissection and low-input DNA sequencing. Nat. Protoc. 2021;16:841–871. doi: 10.1038/s41596-020-00437-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.