

Abstract
Adoptive cell therapy using chimeric antigen receptor (CAR) T cells has proven to be lifesaving for many cancer patients. However, its therapeutic efficacy has been limited in solid tumors. One key factor for this is cancer-associated fibroblasts (CAFs) that modulate the tumor microenvironment (TME) to inhibit T cell infiltration and induce “T cell dysfunction.” Additionally, the sparsity of tumor-specific antigens (TSA) and expression of CAR-directed tumor-associated antigens (TAA) on normal tissues often results in “on-target off-tumor” cytotoxicity, raising safety concerns. Using TALEN-mediated gene editing, we present here an innovative CAR T cell engineering strategy to overcome these challenges. Our allogeneic "Smart CAR T cells” are designed to express a constitutive CAR, targeting FAP+ CAFs in solid tumors. Additionally, a second CAR targeting a TAA such as mesothelin is specifically integrated at a TCR signaling-inducible locus like PDCD1. FAPCAR-mediated CAF targeting induces expression of the mesothelin CAR, establishing an IF/THEN-gated circuit sensitive to dual antigen sensing. Using this approach, we observe enhanced anti-tumor cytotoxicity, while limiting "on-target off-tumor" toxicity. Our study thus demonstrates TALEN-mediated gene editing capabilities for design of allogeneic IF/THEN-gated dual CAR T cells that efficiently target immunotherapy-recalcitrant solid tumors while mitigating potential safety risks, encouraging clinical development of this strategy.
Keywords: CAR T cell, immunotherapy, solid tumors, gene editing, CAR logic gate, on-target off-tumor toxicity, cell therapy, TALEN, allogeneic
Graphical abstract
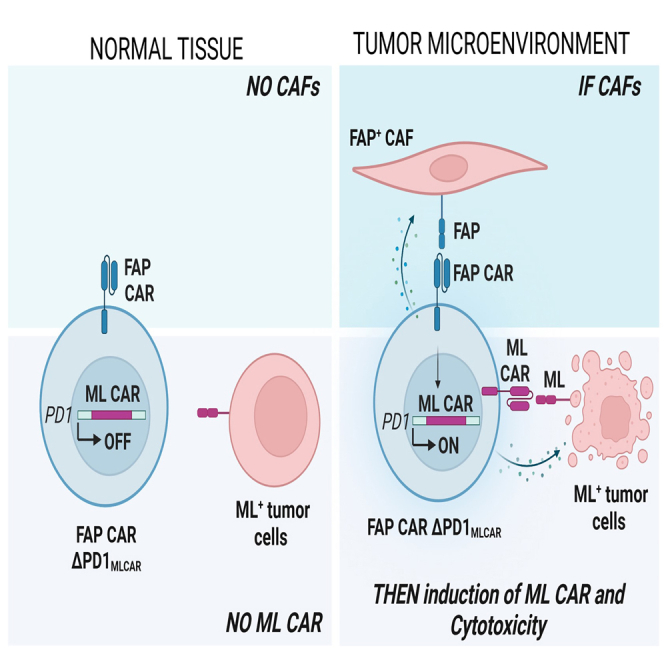
This study uses TALEN-mediated engineering of "Smart CAR T cells” constitutively expressing CAF-targeting FAPCAR and a mesothelin-targeting CAR integrated at TCR signaling-inducible PDCD1 locus. FAPCAR-mediated CAF targeting induces expression of the mesothelin CAR, establishing an IF/THEN-gated circuit sensitive to dual antigen sensing. These cells display enhanced anti-tumor cytotoxicity without "on-target off-tumor" toxicity.
Introduction
In recent years, autologous chimeric antigen receptor (CAR) T cell therapy1 has revolutionized the treatment of hematologic malignancies, offering new hope to patients with diseases like leukemia, lymphoma, and multiple myeloma.2,3,4,5 However, the application of CAR T cell therapy in solid tumors has proven to be more challenging. Solid tumors present unique obstacles, including tumor antigen heterogeneity, a hostile tumor microenvironment that favors immune escape, and limitations in CAR T cell persistence and infiltration.6,7,8 Consequently, results of CAR T cell trials targeting widely expressed tumor antigens such as mesothelin (ML), GPC3, and mucin-1 (MUC1) in a broad range of cancers have so far been disappointing.8,9
Unlike most hematological malignancies, most solid tumors evolve a complex tumor microenvironment (TME) characterized by desmoplasia and a heterotypic immune profile that together ablate CAR T cell infiltration and persistence while promoting T cell exhaustion.6,7,8,9,10,11 Moreover, while the TME obstructs CAR T cell function, the tumor cells themselves pose their own challenges for CAR target selection. Apart from a few surface neoantigens that are unique to tumor cells, called tumor-specific antigens (TSAs), most other antigens are overexpressed on tumor cells, while also expressed at albeit lower levels on healthy tissues.6,12,13 These tumor-associated antigens (TAAs) pose a critical safety concern for the application of CAR T cell therapy against them since they pose the risk of serious “on-target off-tumor” toxicities.8,9 Furthermore, CAR-mediated targeting of a single tumor antigen is susceptible to partial response and frequent relapse due to the clonal heterogeneity of these tumors, wherein different cellular sub-populations express distinct surface antigens.6,7
Despite these challenges, considerable efforts are being made to advance CAR T cell therapy for solid tumors. Technological advances in genetic modifications have enabled innovative approaches for CAR T cell engineering aimed at circumventing the obstacles described above. These include incorporation of novel co-stimulatory domains to promote T cell persistence and evade dysfunction.14,15,16,17,18 Additionally, development of “armored” CAR T cells that incorporate transgenic “payloads” that enhance CAR T cell anti-tumor function.18,19,20 Prominent among these are fourth-generation armored CAR T cells that can stably or inducibly secrete cytokines to create an immune-reactive TME.20 We have previously described a TALEN-mediated genetic network repurposing strategy that enables CAR-activation-dependent secretion of the interleukin (IL)-12 cytokine.21 These allogeneic armored CAR T cells demonstrate increased CAR T cell cytotoxicity and extend survival of hematological tumor-bearing mice.
Here we repurpose the endogenous components of T cell biology by using TALEN-mediated gene editing for generation of non-alloreactive, PD-1-inactivated T cells constitutively expressing a TME-targeting CAR, which upon activation in the tumor milieu induces expression of a TAA-targeting CAR. Our tailored dual inducible UCAR T cells thus single-handedly incorporate a three-pronged approach for effective and safe solid tumor targeting by (1) attenuating TME-mediated CAR T cell tumor exclusion, (2) direct tumor cell killing, and (3) circumventing “on-target, off-tumor” toxicities.
Results
FAP+ cancer-associated fibroblasts localize to the solid tumor microenvironment and inhibit T cell infiltration and anti-tumor cytotoxicity of CAR T cells
A specialized population of cells termed cancer-associated fibroblasts or CAFs are critical contributors to immune suppression and T cell exclusion in most solid tumors.22 TME CAFs evolve from quiescent resident fibroblasts in response to activating cues from transformed cells. In turn, the CAF secretome, which includes a plethora of cytokines and extracellular matrix proteins, manifests as physical and chemical barriers that can inhibit intra-tumoral T cell infiltration and anti-tumor activity.23,24,25,26,27 Thus, targeting CAFs is a promising avenue for enabling tumor infiltration and cytotoxicity of CAR T cells and is an active area of investigation.28,29,30,31,32,33,34
To assess the potential benefit of CAF targeting for enabling CAR T cell activity in solid tumors, we established a three-dimensional spheroid model that can physiologically proximate tumor architecture and tumor-supportive CAF properties. We confirmed that spheroids derived from two triple-negative breast cancer (TNBC) cell lines HCC70 and MDA-MB-231 co-cultured with two different TNBC patient-derived primary CAF cell lines displayed significant inhibition of intra-spheroid T cell infiltration when incubated with activated primary human T cells, recapitulating “cold” tumor properties (Figures 1A, 1B, and S1A).11 Furthermore, anti-tumor cytotoxicity of non-alloreactive TRACKO mesothelin CAR T (TRACKO; MLCAR T) cells was significantly attenuated against mesothelin-positive MDA-MB-231 spheroids when co-cultured with patient-derived CAFs (Figures 1C, 1D, S1B, and S1C). Overall, our results validate the role of CAFs in inhibiting T cell infiltration and anti-tumor cytotoxicity in a therapeutically pertinent tumor model that can be used further to assess the effect of CAF depletion on CAR T cell efficacy.
Figure 1.

Tumor localized FAP+ CAFs inhibit CAR T cell intra-tumoral infiltration and anti-tumor cytotoxicity
(A) Schematic illustrating T cell infiltration assay in tumor spheroids plated with or without CAFs. (B) Graph depicting flow cytometry quantitation of T cells infiltrated per tumor spheroids with or without CAFs, represented as percentage of T cell input. Bars show means ± SD, n = 3; p values determined by Student t test (two-tailed, unpaired). ns, not significant, ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001. (C) Schematic of TRACKO ML CAR T cell cytotoxicity assay against tumor spheroids of TNBC cell line MDA-MB-231-Luc alone or co-cultured with TNBC-derived CAFs. (D) Bar graph representing percentage MDA-MB-231-Luc tumor cell survival post cytotoxicity assay outlined in (C), at Effector:Target ratio = 5:1. Bars show means ± SD, n = 3; p values determined by Student t test (two-tailed, unpaired). ns, not significant, ∗p ≤ 0.05, ∗∗p ≤ 0.01. (E) Immunohistochemistry for detection of human FAP protein in tissue microarray of patient samples from different cancers. (F) Heatmap depicting percentage positive area stained for human FAP protein in immunohistochemical analysis of healthy human tissue microarray.
Since CAFs are specialized cells that differentiate specifically in an inflamed TME,22 they are characterized by a unique surfaceome that lends itself to precise targeting by CAR T cells. We recently described the development and efficacy of one such allogeneic CAR T cell candidate (FAP UCAR T cells)35 that targets a CAF surface protein called fibroblast activation protein-α or FAP.23,36 The FAP CAR used in this study is specific to human FAP protein and does not cross-react with mouse FAP.35 To assess whether FAP targeting by CAR T cells can be tumor-specific, we determined the distribution of FAP protein expression by analyzing tissue microarrays of patient tumor biopsies as well as healthy organ tissues using immunohistochemistry. Consistent with previous observations, FAP was prominently expressed in the stroma of various tumor types (Figures 1E and S1C; Table S1).28,30,34,36 On the other hand, FAP expression was largely undetectable in most of the 30 healthy tissue types assayed (Figures 1F and S1D; Table S2). In some instances, such as skeletal muscle, lung, and thyroid, FAP expression was heterogeneous between tissue samples collected from different donors, which could be due to undiagnosed inflammation, a condition that can also activate resident fibroblasts to express FAP.30 The placenta, stomach body, and adrenal glands were the only three tissues where FAP protein was detected in all three healthy donors. Most remarkably though, even where detected, FAP expression was limited to less than a quarter of each sample area, indicating the sparsity of FAP+ cells in healthy peripheral tissues. Overall, our results confirm low basal expression of FAP protein on a sub-set of healthy peripheral tissues. Combined with an encouraging safety profile of FAP CAR T cell therapy as determined in a clinical trial wherein three patients were treated with a low dose of FAP CAR T at a local FAP+ mesothelioma site,37 FAP can be a promising targetable tumor antigen that merits further efficacy and safety assessment. It is necessary to point out though that exceptions to this could be inflamed tissue during wound healing or fibrosis—a parameter to include during patient enrollment when considering FAP targeting as a therapeutic approach.30
Engineering non-alloreactive, checkpoint-evasive dual inducible CAR T cells via multiplex gene editing
In our previous study, we demonstrated the efficacy of a therapeutic strategy wherein pre-treatment of orthotopic TNBC-bearing mice with FAP CAR T cells targeting patient-derived CAFs increased the infiltration and anti-tumor cytotoxicity of subsequent ML CAR T cell therapy.35 While the combination CAR T cell therapy successfully led to tumor regression, the relatively promiscuous expression of mesothelin on cells in the pleura, pericardium, peritoneum, and sheath (in males) still poses the risk of ML CAR T cell-mediated “on-target, off-tumor” toxicity, especially at efficacious doses.38,39 Case in point are results of a recent clinical trial that reported severe pulmonary toxicity following ML CAR T cell infusion in the high-dose patient cohort.40
Given our observations on significant tumor localized expression of FAP and the high efficacy of combined FAP CAR T cell and ML CAR T cell treatment,35 we repurposed the TCR signaling pathway to devise an inducible IF/THEN logic gate where constitutive FAP CAR expression induces expression of the ML CAR strictly in the tumor milieu for resultant dual CAR anti-tumor cytotoxic output (Figure 2A). To do so, we used a previously described lentiviral construct for stable expression of FAP CAR.35 Additionally, we generated an AAV6 promoter-less donor template designed to integrate an ML CAR35 at the PDCD1 locus by homologous recombination. This template contains a 2A self-cleaving element upstream of the ML CAR expression cassette and 300 base pair (bp) homology arms specific for the PDCD1 locus. TALEN-mediated targeted integration of the donor template at the PDCD1 locus disrupts production of the functional PD-1 protein while simultaneously using its reading frame and regulatory elements for expression of the ML CAR. Furthermore, the ML CAR transgene donor template incorporates an EF1α promoter-driven non-signaling truncated (Δ)LNGFR transgene after the ML CAR expression cassette (PDCD1ML CAR;EF1αΔLNGFR) (Figure S2A). Since ΔLNGFR expression is independent of the PDCD1 promoter activity, all T cells with the integration of this donor template will constitutively express the ΔLNGFR surface protein. Consequently, we can use the GMP compliant ΔLNGFR enrichment process using magnetic activated cell sorting (MACS), enabling simultaneous co-enrichment of PDCD1ML CAR+ T cells.
Figure 2.

TALEN and AAV-mediated multiplex genome editing generates universal, PD-1-resistant dual inducible CAR T cells against solid tumor targets
(A) Schematic depicting IF/THEN-gate logic strategy wherein activation of constitutive expression of FAP CAR specifically in TME induces activation of ML CAR integrated through TALEN-mediated targeted disruption at the PDCD1 locus, resulting in enhanced tumor localized dual FAP CAR and ML CAR anti-tumor activity. (B) Pictorial representation of universal dual inducible CAR T cell developed by TALEN-mediated multiplex editing of TRAC and PDCD1 gene loci to downregulate surface TCRα/β and PD-1 expression, lentiviral FAP CAR random integration for stable surface expression AAV6 DNA repair matrix-mediated disruptive integration of ML CAR at PDCD1 gene locus, for inducible expression downstream of FAP CAR activation. (C) Flow cytometry plots showing frequency of CAR expression among viable engineered T cells. (D) Flow cytometry plots showing frequency of TCRα/β (−)/PD-1 (−) viable engineered T cells post activation with PMA/Ionomycin. (E) Graph representing quantitation of percentage of FAP CAR positive viable T cells, as determined in (C). Bars show the means ± SD, n = 3 donors. (F) Graph representing quantitation of percentage of TCRα/β (−)/PD-1 (−) viable T cells, as determined in (D). Bars show the means ± SD, n = 3 donors. (G) Graph representing quantitation of percentage of ML CAR allelic insertion at PDCD1 locus in positive viable T cells post ΔLNGFR enrichment, as determined by droplet digital PCR (ddPCR). Bars show the means ± SD, n = 3 donors.
Human primary T cells from healthy donors were transduced and TALEN-transfected as outlined in Figure S2B. The engineering strategy includes (1) lentiviral transduction to constitutively express the FAP CAR to target FAP+ CAFs in the TME; (2) two TALEN-mediated knockouts: TRAC, to prevent graft-versus-host disease, and PDCD1 to inactivate expression of the immune checkpoint protein PD-1, thereby conferring resistance to PD1-mediated immune suppression; (3) AAV-mediated disruptive integration at the TALEN-targeted PDCD1 locus of the ML CAR transgene for inducible expression in response to FAP CAR activation; (4) MACS-mediated ΔLNGFR positive selection for enrichment of PDCD1ML CAR;EF1αΔLNGFR template-integrated cells; and finally (5) G-REX T cell expansion. Successful enrichment of ΔLNGFR+ T cells was confirmed by flow cytometry (Figure S2C). Robust transduction of the FAP CAR (Figure 2B) was confirmed by flow cytometry (Figures 2C and 2E), and efficient TALEN gene editing of TRAC and PDCD1 loci was obtained, as assessed by site-directed DNA sequencing (Figure S2D) as well as flow cytometry post PMA/Ionomycin treatment-induced PDCD1 activation (Figures 2D and 2F). Moreover, we successfully enriched for PDCD1ML CAR+ T cells, attaining upwards of 75% ML CAR allelic integration at the PDCD1 locus (Figure 2G). The editing strategy employed here has been previously characterized by us extensively,21 with cytogenetic and genomic characterization of TRACKOPDCD1transgene T cells revealing no significant genomic instability events such as off-site cleavage or chromosomal translocations.
Henceforth for the sake of clarity, TRAC knockout T cells will be referred to as universal T cells (UT). The dual inducible CAR T cells expressing constitutive FAP CAR and PDCD1-integrated ML CAR will be referred to as FAPCAR_ΔPD1MLCAR UT cells. Reference controls for the study accounting for potential effects of PDCD1 knockout alone, stable FAP CAR expression alone, or for activation-independent ML CAR expression from PDCD1 (leakiness) will be referred to as ΔPD1 UT cells, FAPCAR_ΔPD1 UT cells, and ΔPD1MLCAR UT cells, respectively.
At the end of expansion, the engineered FAPCAR_ΔPD1MLCAR UT cells were characterized by flow cytometry to determine the composition of their immune sub-populations. As is expected, CD4+/CD8+ was variable between donors, with one outlier composed predominantly of CD4+ T cells while the other two with ∼1:1 CD4+/CD8+ T cell ratio (Figure S2E). Interestingly, the CAR+ population was strongly skewed toward a CD62L+ naive or stem-like population (Figure S2F), a positive indicator of anti-tumor efficacy.21
Specificity and sensitivity of FAP CAR synthetic circuit to regulate ML CAR expression and activity
To assess and validate specificity of FAP CAR-induced PDCD1-integrated ML CAR expression, we incubated the engineered UT cells on FAP protein-coated plates. FAPCAR_ΔPD1MLCAR UT cells alone significantly expressed ML CAR upon FAP CAR activation, as determined by flow cytometry (Figures 3A and 3B). Notably, no significant ML CAR expression was detected in either non-activated cells or in FAP protein-activated ΔPD1MLCAR UT cells, confirming rigorous regulation by the FAP CAR with little to no detectable leakiness from the PDCD1 locus. We further monitored the kinetics of ML CAR expression upon sustained exposure to the FAP protein and observed early temporal upregulation of the ML CAR as the FAP CAR interacted with its cognate ligand and was internalized, peaking at 48 h, and then gradually decaying over time as surface FAP CAR expression is restored (Figures 3C and S3A). No significant ML CAR expression was observed over this time course for control ΔPD1MLCAR UT cells exposed to the FAP protein in a similar manner (Figures S3B and S3C). The ML CAR expression kinetics observed thus indicates tight upregulation upon FAP CAR activation and subsequent downregulation to baseline as the signal decays.
Figure 3.

FAP CAR activation-dependent stringent regulation of ML CAR expression and activity in multiplex engineered FAPCAR_ΔPD1ML CAR UT cells
(A) Flow cytometry plots depicting FAP CAR and ML CAR expression in indicated UT cell groups, with or without human FAP (FAP) protein-mediated FAP CAR activation for 48 h. (B) Graph representing quantitation of percentage of ML CAR-positive viable T cells, as determined in (A). Bars show the means ± SD, n = 3 donors; p values determined by Student t test (two-tailed, unpaired). ns, not significant, ∗p ≤ 0.05. (C) Graph depicting kinetics of FAP CAR and ML CAR expression following FAP protein-mediated activation of FAP CAR on FAPCAR_ΔPD1ML CAR UT cells at 0 h. Each data point represents mean ± SD, n = 3 donors. (D) Graph representing time course of FAP CAR and MLCAR expression upon FAP CAR stimulation and withdrawal of stimulus (FAP protein) from FAPCAR_ΔPD1ML CAR UT cells. Each data point represents mean ± SD, n = 3 donors. (E) Schematic for assessing ML CAR cytotoxicity of indicated UT cells against ML+FAP− NCI-H226-LUC tumor cells upon FAP CAR stimulation with FAP for 3 days, and subsequent withdrawal of stimulus (FAP protein) for 4 days. (F) Bar graph representing percentage ML+FAP− NCI-H226-LUC tumor cell killing at different time points defined in (E). Cytotoxicity was measured 24 h post incubation of UT cells taken from indicated time points in (E) with target cells at Effector:Target ratio = 1:1. Bars show the means ± SD, n = 2 independent experiments; p values determined by Student t test (two-tailed, unpaired). ns, not significant, ∗p ≤ 0.05.
To test the agility of the system to respond to changes in the activating cue, in this case the FAP protein, we performed an assay wherein the engineered T cells were incubated on FAP-coated plates for 2 days and then replated without FAP for 4 days. This was repeated in tandem once, and surface ML CAR expression was measured by flow cytometry (Figures 3D, S3D, and S3E). As evident in Figure 3D, the engineered ML CAR-inducible circuit is highly responsive to FAP CAR activation and can alternate between an “ON” or “OFF” state, subject to FAP availability. This can be of great interest for alleviating “on-target, off-tumor” cytotoxicity in instances where these cells circulate out of an FAP+ML+ tumor milieu into FAP−ML+ peripheral sites. Yet again, no ML CAR expression was detected on either FAP-activated ΔPD1MLCAR UT cells or non-activated FAPCAR_ΔPD1MLCAR UT cells (Figures S3F and S3G) in the same assay, validating rigorous FAP CAR-specific induction.
Finally, we evaluated the cytotoxic activity of FAP CAR-induced ML CAR, as outlined in Figure 3E. FAPCAR_ΔPD1MLCAR UT cells activated with FAP protein for 48 h displayed significant cytotoxicity against the ML+ mesothelioma cell line NCI-H226 (Figures 3F and S3H). As a control, we compared the cytotoxic activity of activated FAPCAR_ΔPD1MLCAR UT cells with constitutive MLCAR_ΔPD1 UT cells. Forty-eight hours post FAP activation of FAPCAR_ΔPD1MLCAR UT cells, the percentage of MLCAR+ cells was comparable between the two cell populations and anti-NCI-H226 cytotoxicity of activated FAPCAR_ΔPD1MLCAR UT cells was not significantly different from that of constitutive MLCAR_ΔPD1 UT cells (Figure S3I). Furthermore, this cytotoxic activity dissipated upon removal of the FAP CAR activation signal.
We also determined the effect of persistent antigen stimulation of both FAPCAR and MLCAR simultaneously on potential exhaustion of FAPCAR_ΔPD1MLCAR UT cells. Mock transduced and FAPCAR_ΔPD1MLCAR UT cells were serially stimulated with FAP-transduced ML+ HCC70 cells over a period of 10 days (Figure S3J). As shown in Figure S3K, while LAG3, TIGIT, and TIM3 were strongly induced in the beginning, persistent upregulation of the monitored exhaustion markers was not observed in a majority of the cells by the end of the assay. However, as mentioned previously, since exhaustion of T cells in solid tumors can be induced by components of the TME, such a possibility cannot be ruled out.
Overall, our results validate the stringent and agile regulation and function of the inducible ML CAR, tailored to respond to FAP CAR activity in our gated dual CAR T cells.
FAP CAR-dependent ML CAR induction begets enhanced T cell cytotoxicity specifically directed against FAP+ tumor targets
To scrutinize the functional specificity and sensitivity of our inducible system, wherein integrated FAP and ML CAR killing should only transpire upon FAP CAR engagement (Figure 4A), we transduced ML+ mesothelioma cell line NCI-H226 with FAP-containing lentiviral particles to generate tumor pools with variable proportion of FAP+ cells (Figures S4A and S4B). These cells were used as targets in an in vitro cytotoxic assay, outlined in Figure 4B. MLCAR_ΔPD1 UT cells were engineered to use as positive controls for cytotoxicity (Figure S4C). Since all target cells are ML positive, MLCAR_ΔPD1 UT cells displayed nearly 100% cytotoxicity of all target cell groups, validating ML CAR activity (Figure 4C) in this system. No cytotoxicity of FAPCAR_ΔPD1ML CAR UT cells was observed against FAP(−) H226 target cells, which cannot activate the FAP CAR, indicating stringent regulation of ML CAR expression by FAP CAR, with no detectable leakiness. Interestingly, no significant cytotoxicity was detected with either FAPCAR_ΔPD1 or FAPCAR_ΔPD1ML CAR UT cells against FAP(LOW) H226 target cells, relative to both ΔPD1 UT and UT cell controls. This indicates that a baseline proportion of FAP+ cells is necessary in the target cell population to initiate the tailored signaling cascade, an observation that is significant considering the low proportion of FAP+ cells detected in our tissue microarray analysis of healthy donor tissues (Figures 1F and S1D). Most importantly, we observed nearly 100% cytotoxic activity of FAPCAR_ΔPD1MLCAR UT cells against both FAP(MED) and FAP(HIGH) target cells, which was significantly greater than the FAPCAR_ΔPD1 UT. This is only attainable by dual FAP CAR and ML CAR cytotoxicity against the target cells, thus validating the high activity and specificity of our dual inducible FAPCAR_ΔPD1MLCAR UT cells.
Figure 4.

FAPCAR_ΔPD1ML CAR UT cells display efficient FAP CAR activation-dependent dual CAR tumor killing in vitro
(A) Pictorial representation of strategy designed to test activity and sensitivity of the IF/THEN logic gate in FAPCAR_ΔPD1ML CAR UT cells. As depicted, tumor cells with no FAP expression should not induce FAP CAR-mediated ML CAR expression and should therefore survive, despite being ML positive. On the other hand, ML+FAP+ double-positive cells should induce FAP CAR-mediated ML CAR expression, resulting in dual CAR activity and enhanced killing of target cells. (B) Schematic of engineered UT cell cytotoxicity assay against tumor spheroids NCI-H226-Luc cells with or without varying proportions of FAP+ cells, co-incubated for 72 h at E:T = 2.5:1. (C) Heatmap representing percentage target tumor cell cytotoxicity when treated with indicated UT groups, as outlined in (B). Brackets indicate statistical comparison of cytotoxic activity between different target cell groups, n = 2 independent experiments, two donors per experiment, three technical replicates per donor per experiment; p values determined by Student t test (two-tailed, unpaired). ns, not significant, ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001. (D) Schematic of engineered UT cell cytotoxicity assay against 3D spheroids of TNBC cell line HCC70-NanoLuc alone or co-cultured with TNBC-derived CAFs at 1:2 ratio. Effector and target spheroids co-incubated for 72 h at 5:1 ratio. (E) Bar graph representing percentage of HCC70-GFP tumor cell cytotoxicity post cytotoxicity assay outlined in (D). Data representative of n = 2 independent experiments, two donors per experiment, three technical replicates per donor per experiment. Bars show the means ± SD; p values determined by Student t test (two-tailed, unpaired). ns, not significant, ∗p ≤ 0.05, ∗∗p ≤ 0.01. (F) Bar graph representing IFNγ release in cytotoxicity assay outlined in (D), as measured by ELISA in co-culture supernatant on day 6. Data representative of n = 2 independent experiments, two donors per experiment, three technical replicates per donor per experiment. Bars show the means ± SD; p values determined by Student t test (two-tailed, unpaired). ns, not significant, ∗p ≤ 0.05.
The CAR T cell engineering strategy described thus far is aimed at creating an IF/THEN logic gate, wherein dual cytotoxic activity of FAP and ML CAR against CAFs and tumor cells respectively should result in superior tumor killing35 while staying restricted to the TME. To test this hypothesis, we used our ex vivo heterotypic TNBC spheroid model composed of the mesothelin-expressing TNBC cell line NCI-HCC70 expressing a Nanoluciferase reporter (HCC70-NanoLuc) (Figure S4D) and TNBC patient-derived CAFs, co-cultured at a 1:2 ratio. Homotypic TNBC spheroids composed of HCC70-NanoLuc cells alone were used as controls in a CAR T cytotoxicity assay outlined in Figure 4E. Reminiscent of our results in Figure 1D with the MDA-MB231 tumor spheroids, while HCC70-GFP alone spheroids were vulnerable to MLCAR_ΔPD1 UT cell cytotoxicity, CAF addition to these tumor spheroids significantly decreased MLCAR_ΔPD UT cell killing, as measured by both Nanoluciferase activity of surviving target cells and interferon (IFN)γ release (Figures 4E and 4F). No cytotoxic activity against the HCC70-NanoLuc homotypic spheroids was observed with either FAPCAR_ΔPD1 UT or FAPCAR_ΔPD1MLCAR UT cells, once again reiterating FAP-specific activation of ML CAR in the latter. Above all, while FAPCAR_ΔPD1 CAR T cells did display some cytotoxicity against the HCC70-NanoLuc + CAF spheroids, likely due to bystander killing of HCC7-NanoLuc cells upon CAF cytotoxicity, maximum significant HCC70-NanoLuc + CAF spheroid killing was attained by FAPCAR_ΔPD1MLCAR UT cells, substantiating the superior anti-tumor activity of our designed dual CAR AND gate in a physiologically relevant tumor context. Like the NCI-H226 cytotoxicity assay (Figures 4B and 4C), we also titrated the level of FAP in this model by generating HCC70-GFP and CAF co-culture spheroids at 1:1 and 2:1 ratio (Figure S4E). While similar results were obtained as in Figure 4E with the HCC70-GFP (+) CAF spheroids at 1:1 ratio, no FAPCAR cytotoxicity was observed with either FAPCAR_ΔPD1 UT or FAPCAR_ΔPD1MLCAR UT cells against HCC70-GFP (+) CAF spheroids at 2:1 ratio, reiterating a baseline FAP expression/availability required to activate FAPCAR activity and signaling. Overall, our ex vivo results clearly demonstrate the enhanced anti-tumor activity of our dual inducible CAR T cell engineering strategy, while limiting “on-target, off-tumor” toxicity.
FAPCAR_PD1MLCAR UT cells effectively control CAF+ tumors with no detectable “on-target, off-tumor” toxicity
To assess the robustness of our in vitro results under more physiologically pertinent conditions, we used two different mouse tumor models that would allow us to evaluate respectively the “on-target, off-tumor” toxicity and the tumoricidal activity of our FAPCAR_ΔPD1MLCAR UT cells. First, we developed a murine bilateral tumor model wherein 100% ML+ NCI-H226 tumor cells were implanted subcutaneously on the left flank and 100% ML+ 40% FAP+ NCI-H226 (H226FAP40%) tumor cells were implanted subcutaneously on the right flank of the same NSG mouse. Thus, while the right flank H226FAP40% engraft represents a tumor site, the left flank H226 engraft is illustrative of “off-tumor” peripheral tissue expressing the ML antigen. These bilateral tumor-bearing mice were treated with UCAR T cells and monitored as outlined in Figure 5A. One week post UCAR T cell treatment, three mice from each cohort were humanely euthanized and analyzed by flow cytometry. Control ΔPD1 and ΔPD1MLCAR UT cells, as measured by presence of human CD45 (hCD45)+ cells, were nearly undetectable in all tumors, suggesting no activation-dependent expansion (Figures 5B and S5A). Interestingly, maximum intra-tumoral accumulation of both FAPCAR_ΔPD1 and FAPCAR_ΔPD1MLCAR UT cells was detected only in right flank H226-FAP40% tumors, indicative of FAP CAR-mediated T cell expansion. Further analysis of these populations revealed downregulation of surface FAP CAR levels only in right flank H226-FAP40% tumors, once again indicative of CAR internalization post activation (Figures 5C and S5B). Most notable though was the induction of surface ML CAR in FAPCAR_ΔPD1MLCAR UT cells in the H226-FAP40% alone, validating in vivo FAP CAR-specific upregulation of ML CAR (Figure 5D). Significant T cell accumulation was also detected in FAP− left flank tumors treated with either FAPCAR_ΔPD1 or FAPCAR_ΔPD1MLCAR UT cells, likely due to recirculation of expanded cells from the FAP+ flank tumors. This is supported by the fact that mice bearing FAP− H226 tumors alone did not show this accumulation following treatment with FAPCAR_ΔPD1 UT cells (Figures S5C–S5E).
Figure 5.

FAPCAR_ΔPD1ML CAR UT cells efficiently target FAP+ML+ tumors and impede CAF+ tumor growth with no “off-target” toxicity in vivo
(A) Schematic of UCAR T cell treatment and analysis of bilateral subcutaneous tumors implanted in NSG mice. Data representative of two independently conducted studies, each with different T cell donors. (B) Bar graph representing quantitation of total number of hCD45+ cells per gram of tumors from mice treated with indicated UT cells 7 days post administration, as determined by flow cytometry. p values determined by Student t test (two-tailed, paired), n = 3 mice per cohort. ns, not significant, ∗p ≤ 0.05, ∗∗p ≤ 0.01. (C) Bar graph representing percentage of FAP CAR+ cells among total hCD45+ cells in tumors from mice treated with indicated UT cells 7 days post administration, as determined by flow cytometry. p values determined by Student t test (two-tailed, paired), n = 3 mice per cohort. ns, not significant, ∗p ≤ 0.05, ∗∗p ≤ 0.01. (D) Bar graph representing percentage of ML CAR+ cells among total hCD45+ cells in tumors from mice treated with indicated UT cells 7 days post administration, as determined by flow cytometry. p values determined by Student t test (two-tailed, paired), n = 3 mice per cohort. ns, not significant, ∗p ≤ 0.05. (E) Top panel: Graph representing growth kinetics of subcutaneous left flank NCI-H226 tumors of remaining mice treated as indicated over time until study endpoint. p values determined by Student t test (two-tailed, unpaired), n = 5 mice per cohort. ns, not significant. Bottom panel: Graph representing growth kinetics of subcutaneous right flank NCI-H226-FAP40% tumors of remaining mice treated as indicated over time until study endpoint. p values determined by Student t test (two-tailed, unpaired), n = 5 mice per cohort. ns, not significant, ∗∗p ≤ 0.01. (F) Schematic of UT cell treatment and analysis of orthotopic TNBC tumors co-implanted with patient-derived CAFs in NSG mouse mammary fat pad. Data representative of two independently conducted studies, each with different T cell donors. (G) Graph representing growth kinetics of orthotopic TNBC tumors in mice treated as indicated over time. p values determined by Student t test (two-tailed, unpaired), n = 3–5 mice per cohort. ns, not significant, ∗p ≤ 0.05. (H) Kaplan-Meier curve for survival analysis of orthotopic TNBC tumor-implanted NSG mice treated as indicated (n = 3–5 per cohort). p values determined by log rank (Mantel-Cox) test. ns, not significant, ∗p ≤ 0.05.
These observed CAR expression profiles functionally correlated with effects on tumor growth monitored throughout the course of the study significantly. First, no differences were detected in left flank H226 tumor growth between the different treatment groups, indicating the absence of “on-target, off-tumor” toxicity (Figure 5E, top panel). Furthermore, significant reduction in growth was only observed for H226-FAP40% tumors when treated with FAPCAR_ΔPD1MLCAR UT cells (Figure 5E, bottom panel; Figure S5F), relative to both other treatment groups as well as the left flank H226 tumors within the same mice. At the study endpoint, all mice were humanely euthanized, and tumors were excised for further analysis. FAPCAR_ΔPD1MLCAR UT cell treatment significantly reduced H226-FAP40% tumor weight, relative to their H226 tumor counterpart (Figure S5G). Since all engrafted tumor cells expressed a GFP reporter, all excised tumors were also analyzed by immunohistochemistry for FAP and GFP expression. FAP+ cells were cleared in the right flank H226-FAP40% tumors by both FAPCAR_ΔPD1 and FAPCAR_ΔPD1MLCAR UT cells, validating FAP CAR cytotoxic activity (Figure S5H). Interestingly, while GFP+ area was the same for all left flank H226 tumors, right flank H226-FAP40% tumors treated with FAPCAR_PD1MLCAR UT cells displayed a necrotic center and had the least GFP+ area (Figure S5I), validating maximum tumor cell killing in this group. Overall, our results confirm maximal anti-tumor activity of the FAPCAR_ΔPD1MLCAR UT cells with no “on-target, off-tumor” toxicity observed.
Finally, to gauge and compare the activity of the dual inducible FAPCAR_ΔPD1MLCAR UT cells against FAPCAR_ΔPD1 or MLCAR_ΔPD1 UT cells alone in a physiologically relevant model, we orthotopically co-implanted HCC70 tumor cells with TNBC patient-derived CAFs in the mammary fat pad of NSG mice. Tumor-bearing mice cohorts were then treated with the different UCAR T cells and monitored for tumor growth as outlined in Figure 5F. The engrafted tumors displayed aggressive growth, reaching endpoint tumor burden within weeks of implantation. Nevertheless, FAPCAR_ΔPD1MLCAR UT cell treatment was still able to significantly decrease tumor growth (Figure 5G) and increase overall survival (Figure 5H), relative to all other treatment groups.
Altogether, our dual inducible UCAR T cell strategy, targeted at eliminating both tumor cells and CAFs demonstrates enhanced anti-tumor activity accompanied by a superior safety profile.
Discussion
Antigenic complexities and an immune evasive, hostile niche render most solid tumors recalcitrant to successful adoptive cell therapy. Advances in gene editing technologies has enabled ingenious strategies devised to empower next-generation CAR T cells that can combat these solid tumor challenges. In this study, our approach aims at appropriating the TCR signaling pathway to create a synthetic gene circuit aimed at CAR-induced expression of a second CAR while simultaneously deactivating the PD-1 checkpoint pathway. We employed a stably expressed CAR targeting FAP protein to induce the expression of a TAA CAR targeting ML. The stringent expression regulation of ML CAR by FAP CAR activity ensures that in the absence of FAP, the ML CAR is not expressed or promptly downregulated and no longer functional at a non-tumor site, as evidenced by both our in vitro and in vivo results. Particularly, in our bilateral tumor mouse model, control UT and PD1KO UT did not accumulate in the left flank FAP− H226 implants due to lack of FAP CAR-mediated expansion. It thus stands to reason that the FAPCAR_ΔPD1 and FAPCAR_ΔPD1MLCAR UT cells accumulated at this FAP− site because of recirculation from the right flank H226FAP40% tumors, where these cells did expand in response to FAP CAR stimulation. Yet, while the FAPCAR_ΔPD1MLCAR UT cells in the right flank H226FAP40% tumors expressed surface ML CAR, no ML CAR expression was detected on these cells in the left flank H226 tumors, and no subsequent regression of these tumors was observed. We can thus safely surmise that in peripheral FAP− sites, expression of the inducible ML CAR is rapidly downregulated and cannot illicit “on-target, off-tumor” toxicity.
Dual CAR T cell development holds great promise for solid tumors, especially since it can address the challenges of tumor heterogeneity and low antigen abundance.41 In our current study, we go one step further and intertwine dual CAR strategy with TME remodeling and tumor-sensing circuitry to create IF/THEN logic-gated bivalent CAR T cells that target different tumor components while circumventing “on-target, off-tumor” cytotoxicity. Using physiologically relevant in vitro and in vivo models, we demonstrate how simultaneous FAP CAR-mediated targeting of CAF creates a CAR T cell permissive TME wherein the ML CAR can now access and kill tumor cells. Co-targeting of the TME and tumor cells thus results in enhanced tumor control, relative to either single CAR T cell individually. Furthermore, the fine-tuning of ML CAR induction through FAP antigen abundance, as observed both in vitro by the absence of ML CAR cytotoxicity in FAP(LOW) target cells and in vivo by the downregulation of ML CAR expression and loss of activity of FAPCAR_ΔPD1 MLCAR UT cells in H226(FAP−) tumors validates the precise and potent control of the tumor, with no observed “off-tumor” toxicity.
Functionally, our approach manifests as an AND gate wherein our multi-antigenic CAR T cells demonstrate superior anti-tumor toxicity by targeting two tumor antigens, relative to single antigen-targeted CAR T cells. Other “AND” logic gate CAR circuits described in literature include the dual signaling strategy described by Lanitis et al. that physically separated the CD3ζ and co-stimulatory CD28 domains into two separate CARs with different antigen specificity,42 the recently described LINK-CAR wherein the CD3ζ and co-stimulatory domains are replaced with LAT and SLP6 for minimal “on-target, off-tumor” toxicity43 and the synNotch receptor-CAR circuit that uses a tumor-specific “priming signal” for flexible and precise CAR induction at the tumor site.44,45 While the latter also uses a tumor-antigen-specific sensor to induce expression of an anti-TAA CAR, this sensor acts solely as a transcription activator for the CAR. In contrast, our strategy combines the tumor-sensing module with its own CAR activity, resulting in expression of two CARs targeting two different antigens. Furthermore, our dual inducible CAR T cells can target tumor cells that express the priming antigen alone, as opposed to the SynNotch-CAR cells where absence of the priming antigen would result in no anti-tumor toxicity. Furthermore, even upon depletion of the TSA+ sub-population, in this case the primary FAP+ CAF cells, the gated-CAR once induced should be able to sustain its own expression driven by the TAA+ cells in the tumor milieu. An exception here could be CAF− micrometastatic niches that like the FAP− H226 tumors in our study (Figures 5B and 5D) could be infiltrated by the expanded dual inducible CAR T cells, but would escape clearance since the MLCAR expression is downregulated upon exiting the FAP+ML+ primary tumor site.
Given the heterogeneous antigen expression profile of different solid tumors and its evolution over time, this can provide a distinct advantage during the therapeutic window. As illustrated by our in vitro and in vivo CAF plus tumor models, our dual inducible CAR T cells can exhibit cytotoxic activity in trans, targeting two different single antigen-expressing cells. We can thus target different tumor-supportive cell types within the TME, adopting a more comprehensive approach for solid tumor targeting. Finally, by integrating our CAR-inducible strategy into the endogenous gene regulation network, we bypass the use of potentially immunogenic non-human transcription factors and their cognate binding sequences. Overall, our current study decisively illustrates the feasibility and distinct advantages of our strategy for safe and effective targeting of solid tumors. Further refinement of the approach will now be aimed at testing different co-stimulatory domain combinations for CAR design as well as exploring additional CAR-inducible gene loci for fine-tuning sensitivity to antigen abundance and to synergistically improve activity and persistence restricted to the tumor milieu.
Our proposed strategy can be extended to any relevant combination of TSA- and TAA-targeted CARs, expressed either in cis on the same target cells or in trans on different but proximal target cells, enabling targeting of a vast array of heterogeneous cancers. TALEN-mediated TRAC knockout enables us to engineer universal “off-the-shelf” CAR T cells, which are increasingly demonstrating several advantages over the autologous approach, including superior manufacturing efficiency with low cost, production time, and stocking ability that can enable rapid “bench-to-bedside,” as well as the ability to use healthy donor T cells with higher potency and fitness relative to T cells of patients who may have undergone several lines of treatment.46,47 Furthermore, since TALEN editing can easily allow for precise integration of the CAR transgene both in a disruptive or non-disruptive manner, we can multiplex our gene editing tools to enable addition of several useful attributes in our CAR T cells. This includes disruptive CAR integration at gene loci whose activity promotes T cell exhaustion or dysfunction, as well as targeted integration of bicistronic transgenes at the inducible locus. These can be integration cassettes that include more than one CAR with different co-stimulatory domains and antigen specificity to generate multivalent CAR T cells with potent activity, or even a CAR and an immune stimulatory cytokine such as IL-12 or IL-15 to boost activity and persistence and counter the immunosuppressive TME.
Overall, our work illustrates the therapeutic potential of an IF/THEN logic-gated dual CAR T cell engineering strategy that can integrate tumor-specific cues to output expansive tumoricidal activity against solid tumors with no detectable “off-target” toxicity.
Materials and methods
Primary cells, cell lines, and cell culture
Cryopreserved human peripheral blood mononuclear cells (PBMCs) were acquired from ALLCELLS (# PB006F). PBMCs were cultured in CTS OpTmizer media (obtained from Gibco, # A1048501), containing IL-2 (Miltenyi Biotec, # 130-097-748), human serum AB (Seralab, # GEM-100-318), and CTS Immune Cell SR (Gibco, # A2596101). Human T cell TransAct (Miltenyi Biotec t# 130-111-160) was used to activate T cells. PBMCs were cryopreserved in 90% albumin/10% DMSO.
HCC70-GFP-Nanoluciferase were engineered from HCC70 cells (ATCC, # CRL-2315) using an in-house rLV encoding NanoLuc_T2A_EGFP construct and AMSbio (# LVP323-PBS), respectively, using the manufacturer’s protocols. MDA-MB-231-GFP-Luciferase and NCI-H226-GFP-Luciferase were engineered from MDA-MB-231 cells (ATCC, #HTB-26) and NCI-H226 cells (ATCC, #CRL-5826) were generated by transducing the cells with a lentivirus encoding EF1a-Luciferase-2A-GFP (Neo; Amsbio #LVP438). TNBC patient-derived CAFs were obtained from BioIVT (cancer fibroblasts #HPCCAFBRTNB-05). All cell lines and CAFs were maintained in DMEM supplemented with 10% heat-inactivated FBS in 5% CO2 at 37°C. HCC70-GFP-Nanoluciferase-FAP and NCI-H226-GFP-Luciferase-FAP cells were generated by transducing HCC70-GFP-Nanoluciferase and NCI-H226-GFP-Luciferase cells respectively with human FAP lentivirus (#LVP1307, GenTarget Inc).
Spheroid T cell infiltration assay
To assess T cell infiltration in tumor cell spheroids, on day 0, 104 MDA-MB-231-Luciferase cells with or without CAFs at 1:2 ratio were seeded on low adherence 96-well round bottom plates (Thermo Fisher # 174925), in DMEM+10%FBS media. On day 4, T cells were added to the spheroids at tumor cell:T cell ratio of 1:1. Cocultures were cultivated at 37oC overnight. The next day, supernatant from wells was aspirated and spheroids were washed twice with PBS to remove any surface T cells. Spheroids were then dissociated using 0.25% Trypsin (Gibco) and single cells were suspended in full DMEM media, pelleted down, and stained by flow cytometry for CD8. Spheroid infiltrated T cells were calculated as percentage of input T cells.
UCAR T cell generation and expansion
Briefly, PBMCs were thawed, washed, resuspended, and cultivated in CTS OpTmizer complete media (reconstituted CTS OpTmizer, 5% human AB serum, 20 ng/mL IL-2). One day later, the cells were activated with Human T cell TransAct (25 μL of beads/106 CD3-positive cells) and transduced with recombinant lentiviral vectors (Flash Therapeutics) (Table S1) at a multiplicity of infection (MOI) of 15 for FAP CAR and 10 for Mesothelin CAR T cells, in retronectin-coated culture vessels (Takara Bio USA Inc, #T100B). Cells were transduced at a concentration of 2 × 106 cells/mL in full media with TransAct and cultured at 37oC in the presence of 5% CO2 for 3 days. The cells were then split into fresh complete media and transfected the next day according to the following procedure. On the day of transfection, the cells were washed twice in Cytoporation buffer T (BTX Harvard Apparatus, Holliston, Massachusetts), and resuspended at a final concentration of 28 × 106 cells/mL in the same solution. The cellular suspension (5 × 106 cells) was mixed with 5 μg mRNA encoding each TRAC TALEN arm (Trilink BioTechnology) and 15 μg of mRNA encoding each arm of PDCD1 TALEN (Trilink BioTechnology) in a final volume of 200 μL (Table S2). The cellular suspension was transfected in 0.4-cm gap cuvettes using Pulse Agile technology. The electroporation consisted of two 0.1-mS pulses at 2,000 V/cm followed by four 0.2-mS pulses at 325 V/cm. Electroporated cells were transferred to a 12-well plate containing 2 mL of prewarmed OpTmizer media (supplemented with human serum and IL-2) and incubated at 37°C for 15 min. The cells were then concentrated to 8E6 cells/mL in 250 μL of the same media in the presence of AAV6 particles (MOI = 1E5 vg/cells) comprising the donor matrices (Figure S2) in 48-well regular treated plates. After 2 h of culture at 30°C, 250 μL of OpTmizer media supplemented with 5% AB serum and 20 ng/mL IL-2 was added to the cell suspension, and the mix was incubated for 24 h under the same culture conditions. One day later, the cells were seeded at a density of 106 cells/mL in complete OpTmizer media and cultivated at 37°C in the presence of 5% CO2. On day 8 post thawing, the cells were resuspended in fresh complete medium supplemented with 20 ng/mL IL-2 and 5% CTS Immune Cell SR. The cells were seeded in GREX6 (Wilson Wolf, #80240M) at 0.125 × 106 cell/mL and cultivated in the same media according to the manufacturer’s guidelines.
Genomic DNA extraction
Cells were harvested and washed once with PBS. Genomic DNA extraction was performed using Mag-Bind Blood & Tissue DNA HDQ kits (Omega Bio-Tek) following the manufacturer’s instructions.
Targeted PCR and NGS
To assess TALEN-mediated TRAC and PDCD1 knockout on the genomic level, 100 ng genomic DNA from engineered UCART cells was used per reaction in a 50 mL reaction with Phusion High-Fidelity PCR Master Mix (NEB). The PCR condition was set to 1 cycle of 30 s at 98°C; 30 cycles of 10 s at 98°C, 30 s at 60°C, 30 s at 72°C; 1 cycle of 5 min at 72°C; hold at 4°C. The PCR product was then purified with Omega NGS beads (1:1.2 ratio) and eluted into 30 mL of 10 mM Tris buffer pH7.4. The second PCR that incorporates NGS indices was then performed on the purified product from the first PCR. Fifteen microliters of the first PCR product was set in a 50-mL reaction with Phusion High-Fidelity PCR Master Mix (NEB). The PCR condition was set to 1 cycle of 30 s at 98°C; 8 cycles of 10 s at 98°C, 30 s at 62°C, 30 s at 72°C; 1 cycle of 5 min at 72°C; hold at 4°C. Purified PCR products were sequenced on MiSeq (Illumina) on a 2 × 250 V2 cartridge.
In vitro FAP CAR activation assay
Human FAP (FAP) protein was obtained from LakePharma. One milliliter of FAP protein at 2 μg/mL concentration in PBS was added to each well of a 24-well tissue culture-treated plate. The plate was incubated at 37oC for 2 h in a humidified incubator with 5% CO2. Thereafter, the protein suspension was aspirated, and the wells were washed twice with 1 mL of PBS. After the last wash, PBS was aspirated and 1E6 FAP CAR+ or control UT cells were added to the FAP-coated wells in 1 mL complete OpTmizer media. Plates were incubated at 37oC in a humidified incubator with 5% CO2 for the duration of the experiment. At pre-determined intervals, 50 μL of cells was taken and analyzed by flow cytometry for FAP CAR and ML CAR expression. For the resting period, cells were taken from FAP-coated plates, spun down at 300 × g, and the pellet was resuspended at 1E6 cells/mL in complete OpTmizer media. This cell suspension was now plated in non-FAP-coated 24-well plates and incubated as above.
In vitro serial stimulation assay
To assess the exhaustion phenotype of FAPCAR_ΔPD1MLCAR UT cells persistently stimulated to FAP and mesothelin protein, 105 FAP-transduced HCC70 target tumor cells were plated on a 12-well plate; 5 × 105 mock transduced and transfected T cells or FAPCAR_ΔPD1MLCAR UT cells were added to the target cell well and incubated at 37oC. On day 3, 104 T cells from this co-incubation were stained and analyzed by flow cytometry. The remaining cells were transferred to a fresh well of target tumor cells. This was repeated for three rounds over a period of 10 days. T cells at every round were analyzed for exhaustion markers using NovoCyte Penteon flow cytometer (Agilent), and data were analyzed using NovoExpress V.1.5.6, respectively.
In vitro T cell cytotoxicity assays
To assess UCAR T cell cytotoxicity against tumor cell spheroids, on day 0, 104 NCI-H226-Luciferase cells or MDA-MB-231-Luciferase cells with or without CAFs at 1:2 ratio were seeded on low adherence 96-well round bottom plates (Thermo Fisher # 174925), in DMEM+10%FBS media. On day 3, UCAR T cells were added to the spheroids at tumor cell: CAR+ T cell ratio of 1:5 or 1:2.5, respectively. Cocultures were cultivated at 37°C for 3 days. On the day of measuring cytotoxicity, ONE-Glo reagent was prepared following the manufacturer’s instructions (Promega, #E6120). And 50 μL of ONE-Glo reagent was mixed at a 1:1 ratio with target:effector cells in a white, flat-bottom plate. Plate was incubated for 3 min at room temperature and luminescence was measured in a FLUOstar Omega microplate reader (BMG Labtech).
The following calculations were used to estimate the percentage of lysis:
To assess UCAR T cell cytotoxicity against Tumor-CAF spheroids, on day 0, 104 triple-negative breast tumor cells HCC70-Nanoluciferase cells were seeded either alone or with TNBC-derived CAFs at a 1:2 ratio on low adherence 96-well round bottom plates (Thermo Fisher # 174925), in DMEM+10%FBS media. Tumor cells and CAFs were given 3 days to organize themselves into spheroids. On day 3, UCAR T cells were added to the spheroids at tumor cell:CAR+ T cell ratio of 1:5 and co-incubated for 72 h at 37oC. On day of measuring cytotoxicity, supernatants from wells were aspirated and 100 μL of 0.26% Triton X-100 lysis buffer was added to each well. Plate was incubated for 10 min, vortexing every 5 min for 30 s- 1 min at maximum power. Subsequently, the plate was spun down at 1,500 rpm for 3 min and 25 μL surviving cell lysate was mixed at 1:1 ration with Nano-Glo buffer (Promega #N1110) in a white, flat-bottom plate. Plate was incubated for 3 min at room temperature and luminescence was measured in a FLUOstar Omega microplate reader (BMG Labtech).
The following calculations were used to estimate the percentage of lysis:
IFNγ secretion assay
The levels of INFγ were evaluated in supernatants obtained from cytotoxicity assays using the Human IFN-Gamma Quantikine Kit (R&D Systems, SIF50) following the manufacturer’s instructions. As positive control, CAR T cells were activated with Ionomycin and PMA).
Flow cytometry
For in vitro cell cultures, cells in a U-bottom 96-well plate were spun down and washed with PBS (150 μL/well) at 300 × g for 2 min. Prior to surface staining, cells were stained with Fixable Viability Dye eFluor 450 or eFluor 780 (eBiosciences) according to the manufacturer’s instructions. The cells were then stained with antibodies diluted in FACS buffer (2% FBS + 5 mM EDTA + 0.05% azide in PBS, 50 μL/well) for at least 30 min in the dark at 4°C. Cells were washed with PBS (150 μL/well), spun at 300 × g for 2 min, and resuspended in fix buffer (4% paraformaldehyde in PBS, 100 μL/well). Sample collection was performed on a FACSCanto II cytometer (BD) or NovoCyte Penteon flow cytometer (Agilent), and data were analyzed using FlowJo V.10.6.1 (Treestar) or NovoExpress V.1.5.6, respectively.
For tumor samples, tumor tissue was chopped finely with a razor in 5 mL Accutase (Biolegend, #4232201) and incubated at 37oC water bath for 30 min. Digested tumor suspension was passed through a 100-μm strainer (Corning) and filtrate was spun at 300 × g for 10 min. Cell pellet was subsequently stained for flow cytometry as described above. Staining was performed with the antibody panels outlined in Table 1.
Table 1.
Antibody panel for flow cytometry staining
| UCAR T phenotype | |||
| FAP protein | Fluorescein (FITC) | AcroBiosystems | FAP-HF263 |
| Mesothelin protein | R-Phycoerythrin (PE) | AcroBiosystems | MSN-HP2H8 |
| TCRα/β | Allophycocyanin (APC) | Miltenyi Biotec | 130-098-859 |
| PD-1 | BV421 | BioLegend | 329920 |
| Tumor analysis | |||
| Antigen | Fluorochrome | Company | |
| hCD45 | R-Phycoerythrin (PE) | Miltenyi | 130-110-632 |
| mCD45 | Brilliant Violet-421 (BV421) | BD Biosciences | 560501 |
| FAP | Alexa Fluor 488 | R&D Systems | MAB9727-100 |
| hMesothelin | Allophycocyanin (APC) | R&D Systems | FAB32652P |
| hCD4 | PE-Vio770 | Miltenyi | 130-113-227 |
| hCD8 | Brilliant Violet 510 | BD Bioscience | 563919 |
| hCD62L | R-Phycoerythrin (PE) | Miltenyi | 130-110-632 |
| hCD45RA | Brilliant Violet 421 | BD Bioscience | 562885 |
| hCD45RO | Allophycocyanin (APC) | Miltenyi | 130-095-460 |
| LAG3 | PerCP-eFluor-710 | Thermo Fisher | 46-2239-42 |
| CTLA-4 | Allophycocyanin (APC) | Biolegend | 349907 |
| TIM3 | Brilliant Violet 711 | BD Bioscience | 565567 |
| TIGIT | PE/Cyanine7 | Biolegend | 372714 |
Mice and animal procedures
All procedures involving animals were approved by The Mispro Institutional Animal Care and Use Committee and were performed in accordance with the guidelines of the PHS (Public Health Service) Policy on Humane Care and Use of Laboratory Animals, OLAW (Office of Laboratory Animal Welfare), and the USDA (United States Department of Agriculture) AWA (Animal Welfare Act). Experimental/control animals were co-housed.
All experiments were performed on 8-week-old, female NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice obtained from the Jackson Laboratory (Stock # 005557). Animals were housed in an SPF animal facility. Mouse room light cycles were on a 12-h on/off (on from 6 a.m. to 6 p.m. and off from 6 p.m. to 6 a.m.), temperature reading was maintained between 68 and 79 °F, and humidity between 30% and 70%.
Bilateral subcutaneous tumor model
Immunodeficient NSG mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ, the Jackson Laboratory), were received and acclimatized. NSG mice (7–8 weeks old, female) were then injected with 5 × 106 NCI-H226 tumor cells in left flank and 5 × 106 NCI-H226-FAP40% cells in right flank, as a suspension in PBS+matrigel (1:1; Corning, CB-40234C) subcutaneously. Mice were randomized after day 6 based on average tumor growth that was measured using digital calipers. Next day, mice were adoptively intravenously transferred with 15 × 106 CAR+ UT cells or control UT cells in 100-μL PBS. The mice were monitored for health and weighed at least once weekly. On day 7 post CAR T cell administration, three mice from each treatment cohort were humanely euthanized using CO2 asphyxiation followed by cervical dislocation, and tumors were harvested for flow cytometry and immunohistochemistry (IHC) analysis. The remaining mice were monitored as described above until study endpoint at day 28 post CAR T cell administration. Disease progression was monitored on a weekly basis by measuring tumor dimensions using digital calipers and tumor volume was calculated using the formula [length × (width)2/2].
Orthotopic triple-negative breast cancer model
For in vivo triple-negative breast cancer modeling and CAR T treatment, 2 × 106 HCC70-GFP cells mixed with 5 × 105 TNBC-derived CAFs in 50 μL of ice-cold PBS:Matrigel (1:1) were injected into the mammary fat pad of 8-week-old, female NSG mice. Mice were randomly enrolled into the study once tumor volume reached ∼150 mm3. For CAR T cell treatment, tumor-bearing mice received a single-dose treatment of 15 × 106 CAR+ UT or mock cells in 100 μL of PBS via intravenous injection. The mice were monitored for health, weighed at least once weekly, and followed to measure survival. Disease progression was monitored on a weekly basis by measuring tumor dimensions using digital calipers and tumor volume was calculated using the formula [length × (width)2/2]. Humane endpoint criteria for tumor models were (1) weight loss greater than or equal to 20% from baseline; (2) abnormal gait, paralysis, or inability to ambulate properly; (3) respiratory distress/labored breathing; (4) lethargy or persistent recumbency; and (5) loss of righting reflex or other abnormal neurological behaviors. The method for euthanasia was CO2 asphyxiation followed by cervical dislocation to ensure death.
Histology and IHC
Human tumor tissue and healthy tissue microarray slides were obtained from Novus Biologicals (NBP2-30234, NBP2-78082, NBP2-30232, NBP2-30233, NBP2-30189). Mouse tumors were fixed in 10% buffered formalin (Thermo Fisher Scientific) overnight and moved to 70% ethanol thereafter. Paraffin embedding, tissue sectioning, H&E staining, IHC, and Trichrome staining was performed by HistoWiz Inc. (histowiz.com) using a standard operating procedure and fully automated workflow. Samples were processed, embedded in paraffin, and sectioned at 4 μm. IHC was performed on a Bond Rx autostainer (Leica Biosystems) with heat-induced epitope retrieval (pH6 or pH9) using standard protocols. The bond polymer refine detection (Leica Biosystems) was used according to the manufacturer’s protocol. After staining, sections were dehydrated and film coverslipped using a TissueTek-Prisma and Coverslipper (Sakura). Whole-slide scanning (40x) was performed on an Aperio AT2 (Leica Biosystems). For IHC, antibodies used were α-GFP (#ab183734, Abcam, 1:100) and α-FAP (#ab227703, Abcam, 1:100). Percentage FAP+ area was quantitation of using QuPath software. A grid was applied to the TMA to extract the individual TMA cores and the percentage of pixels positive for the FAP stain was calculated for each TMA core.
Data and code availability
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Acknowledgments
We would like to thank the clinical, translational, and process and analytical development teams at Cellectis for their support through this work. We would also like to thank Kathryn Newhall, Mark Frattini, Valerie Meyrial, and Fabien Delacote for reviewing the manuscript and providing valuable advice, as well as the Cellectis publications team for their input. All funding was provided by Cellectis. All illustrations were generated using BioRender.
Author contributions
S.Das, J.V., P.D., and L.P. conceived the study. A.J., J.V., L.P., and S.Das designed the experiments. S.Dharani, H.C., J.P.F., and S.Das performed the experiments. S.Dharani, H.C., J.P.F., and S.Das analyzed experimental data. S.Das, J.V., A.J., P.D., and L.P. drafted, edited, and reviewed the manuscript.
Declaration of interests
S.Dharani, H.C., A.J., J.V., P.D., L.P., and S.Das are current employees and equity holders at Cellectis. TALEN is a Cellectis patented technology. J.P.F. is a former Cellectis employee and is a current employee of Bristol Myers Squibb.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2024.08.018.
Supplemental information
References
- 1.Imai C., Mihara K., Andreansky M., Nicholson I.C., Pui C.H., Geiger T.L., Campana D. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 2004;18:676–684. doi: 10.1038/sj.leu.2403302. [DOI] [PubMed] [Google Scholar]
- 2.Brentjens R.J., Rivière I., Park J.H., Davila M.L., Wang X., Stefanski J., Taylor C., Yeh R., Bartido S., Borquez-Ojeda O., et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817–4828. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Westin J.R., Kersten M.J., Salles G., Abramson J.S., Schuster S.J., Locke F.L., Andreadis C. Efficacy and safety of CD19-directed CAR-T cell therapies in patients with relapsed/refractory aggressive b-cell lymphomas: Observations from the JULIET, ZUMA-1, and TRANSCEND trials. Am. J. Hematol. 2021;96:1295–1312. doi: 10.1002/ajh.26301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li D., Li X., Zhou W.L., Huang Y., Liang X., Jiang L., Yang X., Sun J., Li Z., Han W.D., Wang W. Genetically engineered T cells for cancer immunotherapy. Signal Transduct. Targeted Ther. 2019;4:35. doi: 10.1038/s41392-019-0070-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schuster S.J., Svoboda J., Chong E.A., Nasta S.D., Mato A.R., Anak Ö., Brogdon J.L., Pruteanu-Malinici I., Bhoj V., Landsburg D., et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N. Engl. J. Med. 2017;377:2545–2554. doi: 10.1056/NEJMoa1708566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mirzaei H.R., Rodriguez A., Shepphird J., Brown C.E., Badie B. Chimeric antigen receptors T cell therapy in solid tumor: Challenges and clinical applications. Front. Immunol. 2017;8:1850. doi: 10.3389/fimmu.2017.01850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miao L., Zhang Z., Ren Z., Tang F., Li Y. Obstacles and coping strategies of CAR-T cell immunotherapy in solid tumors. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.687822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen K., Wang S., Qi D., Ma P., Fang Y., Jiang N., Wu E., Li N. Clinical Investigations of CAR-T Cell Therapy for Solid Tumors. Front. Immunol. 2022;13 doi: 10.3389/fimmu.2022.896685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Secondino S., Canino C., Alaimo D., Muzzana M., Galli G., Borgetto S., Basso S., Bagnarino J., Pulvirenti C., Comoli P., Pedrazzoli P. Clinical trials of cellular therapies in solid tumors. Cancers. 2023;15:3667. doi: 10.3390/cancers15143667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hou A.J., Chen L.C., Chen Y.Y. Navigating CAR-T cells through the solid-tumour microenvironment. Nat. Rev. Drug Discov. 2021;20:531–550. doi: 10.1038/s41573-021-00189-2. [DOI] [PubMed] [Google Scholar]
- 11.Bonaventura P., Shekarian T., Alcazer V., Valladeau-Guilemond J., Valsesia-Wittmann S., Amigorena S., Caux C., Depil S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019;10:168. doi: 10.3389/fimmu.2019.00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Merlotti A., Sadacca B., Arribas Y.A., Ngoma M., Burbage M., Goudot C., Houy A., Rocañín-Arjó A., Lalanne A., Seguin-Givelet A., et al. Noncanonical splicing junctions between exons and transposable elements represent a source of immunogenic recurrent neo-antigens in patients with lung cancer. Sci. Immunol. 2023;8 doi: 10.1126/sciimmunol.abm6359. [DOI] [PubMed] [Google Scholar]
- 13.Burbage M., Rocañín-Arjó A., Baudon B., Arribas Y.A., Merlotti A., Rookhuizen D.C., Heurtebise-Chrétien S., Ye M., Houy A., Burgdorf N., et al. Epigenetically controlled tumor antigens derived from splice junctions between exons and transposable elements. Sci. Immunol. 2023;8 doi: 10.1126/sciimmunol.abm6360. [DOI] [PubMed] [Google Scholar]
- 14.Li Y., Rezvani K., Rafei H. Next-generation chimeric antigen receptors for T- and natural killer-cell therapies against cancer. Immunol. Rev. 2023;320:217–235. doi: 10.1111/imr.13255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goodman D.B., Azimi C.S., Kearns K., Talbot A., Garakani K., Garcia J., Patel N., Hwang B., Lee D., Park E., et al. Pooled screening of CAR T cells identifies diverse immune signaling domains for next-generation immunotherapies. Sci. Transl. Med. 2022;14 doi: 10.1126/scitranslmed.abm1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hamieh M., Mansilla-Soto J., Rivière I., Sadelain M. Programming CAR T cell tumor recognition: tuned antigen sensing and logic gating. Cancer Discov. 2023;13:829–843. doi: 10.1158/2159-8290.CD-23-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harrison A.J., Du X., von Scheidt B., Kershaw M.H., Slaney C.Y. Enhancing co-stimulation of CAR-T cells to improve treatment outcomes in solid cancers. Immunothr Adv. 2021;1 doi: 10.1093/immadv/ltab016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giardano Attianese G.M.P., Ash S., Irving M. Coengineering specificity, safety, and function into T cells for cancer immunotherapy. Immunol. Rev. 2023;320:166–198. doi: 10.1111/imr.13252. [DOI] [PubMed] [Google Scholar]
- 19.Hawkins E.R., D’Souza R.R., Klampatsa A. Armored CAR T-cells: the next chapter in T-cell cancer immunotherapy. Biologics. 2021;15:95–105. doi: 10.2147/BTT.S291768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chmielewski M., Abken H. TRUCKs: the fourth generation of CARs. Expert Opin. Biol. Ther. 2015;15:1145–1154. doi: 10.1517/14712598.2015.1046430. [DOI] [PubMed] [Google Scholar]
- 21.Sachdeva M., Busser B.W., Temburni S., Jahangiri B., Gautron A.S., Maréchal A., Juillerat A., Williams A., Depil S., Duchateau P., et al. Repurposing endogenous immune pathways to tailor and control chimeric antigen receptor T cell functionality. Nat. Commun. 2019;10:5100. doi: 10.1038/s41467-019-13088-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Räsänen K., Vaheri A. Activation of fibroblasts in cancer stroma. Exp. Cell Res. 2010;316:2713–2722. doi: 10.1016/j.yexcr.2010.04.032. [DOI] [PubMed] [Google Scholar]
- 23.Kraman M., Bambrough P.J., Arnold J.N., Roberts E.W., Magiera L., Jones J.O., Gopinathan A., Tuveson D.A., Fearon D.T. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science. 2010;330:827–830. doi: 10.1126/science.1195300. [DOI] [PubMed] [Google Scholar]
- 24.De P., Aske J., Dey N. Cancer-Associated Fibroblast Functions as a Road-Block in Cancer Therapy. Cancers (Basel) 2021;13:5246. doi: 10.3390/cancers13205246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jenkins L., Jungwirth U., Avgustinova A., Iravani M., Mills A., Haider S., Harper J., Isacke C.M. Cancer-Associated Fibroblasts Suppress CD8+ T-cell Infiltration and Confer Resistance to Immune-Checkpoint Blockade. Cancer Res. 2022;16:2904–2917. doi: 10.1158/0008-5472.CAN-21-4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bougherara H., Mansuet-Lupo A., Alifano M., Ngô C., Damotte D., Le Frère-Belda M.A., Donnadieu E., Peranzoni E. Real-Time Imaging of Resident T Cells in Human Lung and Ovarian Carcinomas Reveals How Different Tumor Microenvironments Control T Lymphocyte Migration. Front. Immunol. 2015;6:500. doi: 10.3389/fimmu.2015.00500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koppensteiner L., Mathieson L., O'Connor R.A., Akram A.R. Cancer Associated Fibroblasts - An Impediment to Effective Anti-Cancer T Cell Immunity. Front. Immunol. 2022;13 doi: 10.3389/fimmu.2022.887380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xin L., Gao J., Zheng Z., Chen Y., Lv S., Zhao Z., Yu C., Yang X., Zhang R. Fibroblast Activation Protein-α as a Target in the Bench-to-Bedside Diagnosis and Treatment of Tumors: A Narrative Review. Front. Oncol. 2021;11 doi: 10.3389/fonc.2021.648187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Loeffler M., Krüger J.A., Niethammer A.G., Reisfeld R.A. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J. Clin. Invest. 2006;116:1955–1962. doi: 10.1172/JCI26532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee J., Fassnacht M., Nair S., Boczkowski D., Gilboa E. Tumor immunotherapy targeting fibroblast activation protein, a product expressed in tumor-associated fibroblasts. Cancer Res. 2005;65:11156–11163. doi: 10.1158/0008-5472. [DOI] [PubMed] [Google Scholar]
- 31.Ostermann E., Garin-Chesa P., Heider K.H., Kalat M., Lamche H., Puri C., Kerjaschki D., Rettig W.J., Adolf G.R. Effective immunoconjugate therapy in cancer models targeting a serine protease of tumor fibroblasts. Clin. Cancer Res. 2008;14:4584–4592. doi: 10.1158/1078-0432.CCR-07-5211. [DOI] [PubMed] [Google Scholar]
- 32.Lindner T., Giesel F.L., Kratochwil C., Serfling S.E. Radioligands Targeting Fibroblast Activation Protein (FAP) Cancers (Basel) 2021;13:5744. doi: 10.3390/cancers13225744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Das S., Shapiro B., Vucic E.A., Vogt S., Bar-Sagi D. Tumor Cell–Derived IL1β Promotes Desmoplasia and Immune Suppression in Pancreatic CancerIL1β Promotes Immune Suppression in Pancreatic Cancer. Cancer Res. 2020;80:1088–1101. doi: 10.1158/0008-5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lo A., Wang L.C.S., Scholler J., Monslow J., Avery D., Newick K., O'Brien S., Evans R.A., Bajor D.J., Clendenin C., et al. Tumor-Promoting Desmoplasia Is Disrupted by Depleting FAP-Expressing Stromal Cells. Cancer Res. 2015;75:2800–2810. doi: 10.1158/0008-5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Das S., Valton J., Duchateau P., Poirot L. Stromal depletion by TALEN-edited universal hypoimmunogenic FAP-CAR T cells enables infiltration and anti-tumor cytotoxicity of tumor antigen-targeted CAR-T immunotherapy. Front. Immunol. 2023;14 doi: 10.3389/fimmu.2023.1172681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garin-Chesa P., Old L.J., Rettig W.J. Cell surface glycoprotein of reactive stromal fibroblasts as a potential antibody target in human epithelial cancers. Proc. Natl. Acad. Sci. USA. 1990;87:7235–7239. doi: 10.1073/pnas.87.18.7235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hiltbrunner S., Britschgi C., Schuberth P., Bankel L., Nguyen-Kim T.D.L., Gulati P., Weder W., Opitz I., Lauk O., Caviezel C., et al. Local delivery of CAR T cells targeting fibroblast activation protein is safe in patients with pleural mesothelioma: first report of FAPME, a phase I clinical trial. Ann. Oncol. 2021;32:120–121. doi: 10.1016/j.annonc.2020.10.474. [DOI] [PubMed] [Google Scholar]
- 38.Lv J., Li P. Mesothelin as a biomarker for targeted therapy. Biomark. Res. 2019;7:18. doi: 10.1186/s40364-019-0169-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhai X., Mao L., Wu M., Liu J., Yu S. Challenges of anti-mesothelin CAR-T-cell therapy. Cancers. 2023;15:1357. doi: 10.3390/cancers15051357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haas A.R., Golden R.J., Litzky L.A., Engels B., Zhao L., Xu F., Taraszka J.A., Ramones M., Granda B., Chang W.J., et al. Two cases of severe pulmonary toxicity from highly active mesothelin-directed CAR T cells. Mol. Ther. 2023;31:2309–2325. doi: 10.1016/j.ymthe.2023.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Han X., Wang Y., Wei J., Han W. Multi-antigen-targeted chimeric antigen receptor T cells for cancer therapy. J. Hematol. Oncol. 2019;12:128. doi: 10.1186/s13045-019-0813-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lanitis E., Poussin M., Klattenhoff A.W., Song D., Sandaltzopoulos R., June C.H., Powell D.J., Jr. Chimeric antigen receptor T cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol. Res. 2013;1:43–53. doi: 10.1158/2326-6066.CIR-13-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tousley A.M., Rotiroti M.C., Labanieh L., Rysavy L.W., Kim W.J., Lareau C., Sotillo E., Weber E.W., Rietberg S.P., Dalton G.N., et al. Co-opting signalling molecules enables logic-gated control of CAR T cells. Nature. 2023;615:507–516. doi: 10.1038/s41586-023-05778-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roybal K.T., Rupp L.J., Morsut L., Walker W.J., McNally K.A., Park J.S., Lim W.A. Precision Tumor Recognition by T Cells with Combinatorial Antigen-Sensing Circuits. Cell. 2016;164:770–779. doi: 10.1016/j.cell.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Srivastava S., Salter A.I., Liggitt D., Yechan-Gunja S., Sarvothama M., Cooper K., Smythe K.S., Dudakov J.A., Pierce R.H., Rader C., Riddell S.R. Logic-Gated ROR1 Chimeric Antigen Receptor Expression Rescues T Cell-Mediated Toxicity to Normal Tissues and Enables Selective Tumor Targeting. Cancer Cell. 2019;201:489–503.e8. doi: 10.1016/j.ccell.2019.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martínez Bedoya D., Dutoit V., Migliorini D. Allogeneic CAR T cells: an alternative to overcome challenges of CAR T cell therapy in glioblastoma. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.640082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Metelo A.M., Jozwik A., Luong L.A., Dominey-Foy D., Graham C., Attwood C., Inam S., Dunlop A., Sanchez K., Cuthill K., et al. Allogeneic anti-BCMA CAR T cells are superior to multiple myeloma-derived CAR T cells in preclinical studies and may be combined with gamma secretase inhibitors. Cancer Res. Commun. 2022;2:158–171. doi: 10.1158/2767-9764.CRC-21-0157. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.