

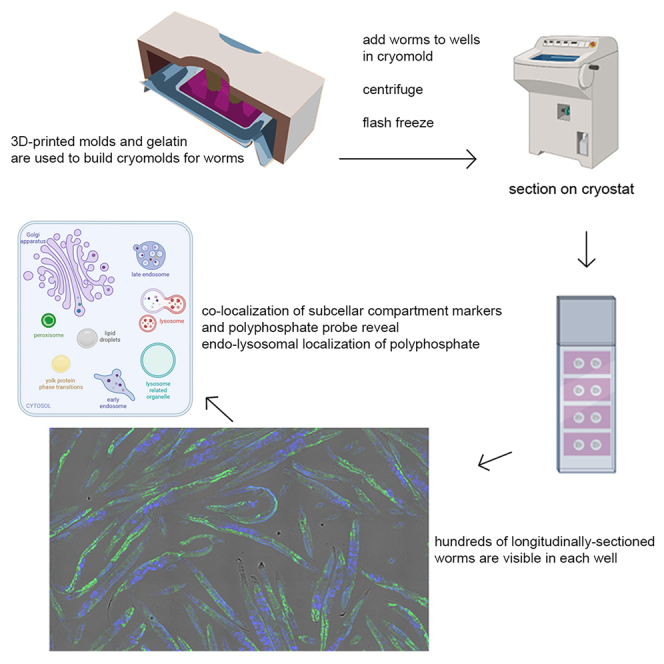
Summary
Polyphosphate (polyP) is a ubiquitous polyanion present throughout the tree of life. While polyP’s widely varied functions have been interrogated in single-celled organisms, little is known about the cellular distribution and function of polyP in multicellular organisms. To study polyP in metazoans, we developed the nematode Caenorhabditis elegans as a model system. We designed a high-throughput, longitudinal-orientation cryosectioning method that allowed us to scrutinize the intracellular localization of polyP in fixed C. elegans using fluorescent polyP probes and co-immunostaining targeting appropriate marker proteins. We discovered that the vast majority of polyP is localized within the endo-lysosomal compartments of the intestinal cells and is highly sensitive toward the disruption of endo-lysosomal compartment generation and food availability. This study lays the groundwork for further mechanistic research of polyPs in multicellular organisms and provides a reliable method for immunostaining hundreds of fixed worms in a single experiment.
Keywords: polyphosphate, Caenorhabditis elegans, cryosection, immunofluorescence staining, endosome, lysosome, tissue sectioning, freeze-crack, cuticle
Graphical abstract
Highlights
-
•
The cuticle of Caenorhabditis elegans has long hampered fluorescent staining approaches
-
•
This cryosection method allows staining of hundreds of worms in a single experiment
-
•
Staining for polyphosphate reveals its presence in endo-lysosomal compartments
-
•
Polyphosphate levels are sensitive to food availability
Motivation
Caenorhabditis elegans are a widely used system to study basic biological phenomena. However, studies requiring fluorescently tagged molecules are restricted to genetically manipulated organisms or low-throughput and disruptive staining techniques, in part due to the presence of a tough cuticle. Labeling polyphosphate with known probes is not compatible with either of these options in a high-throughput setting. Thus, we describe a method to cryosection worms longitudinally and en masse to allow for staining with fluorescent probes with minimal chemical perturbation of the samples.
Polyphosphate (polyP), a ubiquitous and highly conserved molecule, is present throughout all branches of life yet has mostly been studied in single cells. Quarles et al. describe a method to longitudinally section Caenorhabditis elegans, allowing high-throughput staining of worms using polyP-specific fluorescent probes. These studies establish C. elegans as a model for polyP research.
Introduction
Polyphosphate (polyP), a simple polyanion composed of orthophosphates linked by high-energy phosphoanhydride bonds, is ubiquitously present throughout the tree of life. Its known functions vary wildly, depending on the organism, tissue, and cellular context.1,2 PolyP is currently best understood in single-celled organisms,3 particularly bacteria and budding yeast. This is largely due to the fact that in these organisms, the enzymes that make polyP (i.e., bacterial polyphosphate kinase [PPK], yeast Vtc4) and break the polymer (bacterial and yeast exopolyphosphatase [PPX]) have been identified and allow for genetic interrogations of polyP functions in vivo.4 In contrast, the mammalian polyP synthesizing machinery remains unknown, and polyphosphatases of the Nudix protein family have only recently been discovered.5 Moreover, while it is well known that polyP fulfills a multitude of different functions in higher eukaryotic cells, ranging from blood clotting6 and neuronal glial signaling7 to neurodegenerative diseases8,9,10 and cancer,11,12 model systems to study polyP in a multicellular and multi-tissue context are still lacking. In fact, even extracting polyP from multicellular organisms remains a major challenge due to the orders-of-magnitude lower abundance of polyP in mammalian cells and tissues compared to the millimolar concentrations of polyP that accumulate in environmentally stressed bacteria13 or in the vacuole of Saccharomyces cerevisiae.14 Nevertheless, visualization of polyP has been made possible in fixed tissues by using a probe consisting of the polyP-binding domain of Escherichia coli PPX fused with EGFP or mCherry (EcPPXbd-EGFP and EcPPXbd-mCherry, respectively), which binds long-chain polyP specifically.15,16
Our primary goal for this study was to assess and visualize the amount and (sub)cellular distribution of polyP in a multicellular organism. To do this, we chose Caenorhabditis elegans for its relative anatomical simplicity, its cost effectiveness, the abundance of available transgenic strains, and the ease with which knockdown experiments can be conducted. However, staining of polyP with the EcPPXbd-EGFP probe posed a major challenge in C. elegans since antibody staining is notoriously difficult in whole worms.17 The primary challenge is the presence of their cuticle, a biological fortress that protects the worms from the insults of the outside world. To gain access to the inner tissues requires chemical or mechanical disruption,17 which is highly destructive to the structural components of the cell (chemical methods, dissection), only doable with very low throughput (dissection, freeze cracking), causes a high degree of technical error (dissection, freeze cracking), or requires chemicals that might compromise extraction and visualization efforts (chemical methods).18 Hence, immunofluorescence (IF) on fixed C. elegans using commercially available antibodies, of which there are few that are optimized for worm proteins, is rarely performed. To address this issue, we considered using cryosectioning,19 a method that is frequently performed with dissected tissues. The benefits of sectioning cryopreserved tissues include the low cost and technical ease of making the frozen blocks, the excellent antigenicity retained in the sections, and the possibility of long-term storage at −80°C. The challenge, however, lies in manipulating the position and orientation of small tissue sections or organisms such as worms within the same plane as the sectioning blade, given that wrong positioning will result in partial worm sections that make mapping that area back to the whole worm course-grained. We thus decided to develop a cryosectioning method using three-dimensional (3D)-printed molds that allow us to position hundreds of worms in the same plane, thus substantially increasing both the sample number and staining homogeneity. Our results indicate that C. elegans produce polyP endogenously, and, while polyP is contained in most tissues, the majority of worm polyP accumulates in endo-lysosomal vesicles in the intestine. Moreover, we demonstrate that this technique is a highly reliable tool for the immunostaining of hundreds of fixed worms in a single experiment, a useful addition to the C. elegans toolbox.
Results and discussion
Visualization of endogenous polyP in C. elegans
As a first approach to determine if C. elegans contain any visualizable polyP, we used a conventional dissection method in which the worm cuticle is nicked, allowing the extrusion of the gonad and intestine.20 We then stained the dissected worms with EcPPXbd-mCherry, which has been shown to specifically interact with polyP in fixed tissues.15 As shown in Figure 1A, we observed a clear mCherry signal in the intestine, providing the first evidence that polyP can indeed be detected within select tissues of C. elegans. However, since the food source of C. elegans is E. coli, which produces ample amounts of polyP, we wondered whether the intestinal polyP signal might originate from the ingested bacteria. To test this idea, we cultivated the worms on either wild-type (WT) E. coli MG1655 or the ppk deletion mutant, which fails to produce any polyP.21 We did not observe any obvious differences in the intensity of the EcPPXbd-mCherry signal in the intestines of worms grown on either of the bacteria, making us confident that C. elegans is indeed able to synthesize polyP (Figures 1B and S1A). Nonetheless, these studies also made us realize that the data analysis of these dissection images is extremely difficult due to the considerable variability in tissue integrity and completeness of the dissections. Moreover, by using DAPI co-staining, we noticed that even this highly penetrant stain only occasionally permeated tissues more than one or two cells deep into areas where the cuticle was damaged. We thus concluded that while this method allowed us to confirm the presence of endogenous polyP in C. elegans, it was insufficient to provide meaningful data about the subcellular distribution of polyP or its presence in parts of the worms from which the cuticle could not be removed.
Figure 1.

Visualization of C. elegans tissues reveals endogenous polyP
(A) A representative dissected adult N2 worm stained with EcPPXbd-mCherry and DAPI and the differential interference contrast (DIC) image, shown in overlay.
(B) Similar images to those in (A): intestinal and gonadal tissue dissected from N2 adults and stained with EcPPXbd-mCherry. The worms were raised on either wild-type MG1655 E. coli or a ppk deletion mutant. Bottom row: worms were stained with mCherry alone. Higher-resolution images are shown in Figure S1.
(C) Overview illustration of cryoblocking protocol.
(D) Representative image of adult worms stained with DAPI and EcPPXbd-EGFP showing full body lengths of sectioned adjacent worms.
(E) Left: Z-projections of three 10 μm sections of adult N2s stained with EcPPXbd-mCherry (pseudocolored red) and DAPI (pseudocolored teal) and fed either wild-type or ppk/ppx knockout OP50 bacteria. Right: graph of relative EcPPXbd-mCherry fluorescent signal in intestines of worms fed either strain of bacteria (mean +/− SD). All scale bars represent 200 μm.
Establishing a high-throughput cryosectioning technique for C. elegans
To improve the technical consistency in opening the cuticles and accessing all worm tissues as well as increasing the throughput and reproducibility of stained and imaged worms, we decided to develop a higher-throughput cryosectioning approach (Figures 1C and S2). Cryosectioning, which is employed in pathology labs around the world,19 involves the dissection of tissues, addition of a liquid cutting compound, arrangement of the tissue pieces into the preferred orientation using forceps, and flash freezing of the samples. However, the proper arrangements of worms in the same plane for cryosectioning, which is necessary to obtain full-length worm sections rather than circular or oval cross-sections, is technically very challenging and likely the major factor why this method has not yet been developed for C. elegans. Previous cryosectioning attempts in worms were restricted to individual nematodes and used to specifically isolate cross-sections for RNA isolation.22,23 To achieve the longitudinal alignment of several hundred worms in each sample, we designed and 3D-printed 2- to 18-well gelatin molds, which could be centrifuged to align the optimal cutting temperature (OCT; a freezing compound routinely used for preparing cryosamples)-treated worms at the bottom of the wells. After freezing the resulting blocks, we then generated sections using the cryostat (Figures 1C and S2). Since adult C. elegans are about 80 μm thick, taking several slices at 10–12 μm thickness allows for multiple sections of the same worms to be added to one or more slides. By using blocks with up to 18 wells, this setup allowed us to minimize variation in the staining from slide to slide by staining sections from the same population on separate slides, staining multiple populations on the same slide, or both. By applying this method and subsequently staining the sections using either EcPPXbd-EGFP or EcPPXbd-mCherry together with DAPI, we were able to image hundreds of worms in a single experiment at high resolution (Figure 1D). To quantitatively describe the potential effects of bacterial polyP on the EcPPXbd-mCherry signal in the worm intestines, we also compared worms fed on WT bacteria versus those lacking both ppx and ppk (OP50ΔPPKΔPPX) (Figure 1E). These experiments revealed a slight but reproducible decrease in the EcPPXbd-mCherry signal in the mutant OP50-fed worms, suggesting that while worms produce most of their own polyP, the food source can contribute to intestinal polyP levels. As before, we observed the strongest EcPPXbd-mCherry signal in the intestines of the worms. Importantly, this signal was completely lost upon pretreatment of the sections with S. cerevisiae PPX (ScPPX) and increased upon treatment with the endopolyphospatase DPP1, which cleaves polyP chains and thus increases the number of polyP termini, the site of polyP hydrolysis by PPX24 (Figures S3A and S3B). Combined treatment with both PPX and DPP1 re-reduced the signal although not to the full extent of the treatment with PPX alone. Since PPX is a processive enzyme with a chain length limit (∼15 residues),25 it is conceivable that DPP1 produces some polyP chains that are too short for PPX to be further hydrolyzed. Moreover, staining with mCherry alone only gave very faint non-specific background staining. These results not only demonstrated that we developed a tool that, for the first time, allows us and others to immunostain entire fixed C. elegans worms in a high-throughput manner but also provided further evidence that we can visualize endogenous polyP in a multicellular organism.
Tissue-wide and subcellular distribution of polyP in C. elegans
Now that we generated a technique that provided accessibility of all worm tissues to the stain, we took a more granular look at the polyP distribution in C. elegans (Figure 2A). While the most prominent polyP signal was found in the intestine, EcPPXbd-EGFP signals significantly above the EGFP background signal were distributed throughout the entire worm body (Figure 2A). Apart from the intestinal cells, we found polyP to be present in the gonad and to diffuse staining through the body wall and head. Due to the ease of separating their signals from surrounding tissues on the images, we analyzed three separate tissues—the head region, the gonad (pachytene and oocytes), and the intestinal cells (Figure 2B). The pachytene is an acellular space within the proximal gonad, chosen specifically because it had the brightest signal of the gonadal regions analyzed, with the least interference of signal from cellular spaces. As expected from the images, the head region and the gonadal areas showed much lower and more diffuse signals compared to the intestinal cells (Figure 2C), providing good evidence that the intestine indeed harbors the primary polyP stores in C. elegans. Since the brightest polyP signal was clearly in punctate structures in the gut, we focused on the intestinal polyP for the remainder of the study.
Figure 2.

C. elegans polyP accumulates in intestinal cells
Worm sections for tissue- or region-specific differences in PPXbd-EGFP fluorescence were analyzed.
(A) Representative N2 adults at day 1, 10 μm sections, stained with DAPI and EcPPXbd-EGFP, which is shown at two different intensity levels to visualize gut and non-gut signals.
(B) Schematic of regions analyzed. The pachytene is the non-cellular region of the proximal gonad. Oocytes: (late: 1st and 2nd oocytes counting from the proximal end of the gonad before the spermatheca valve; early: 4th and 5th positions).
(C) Graph of background-corrected EcPPXbd-EGFP fluorescence levels, normalized to mean intestinal signal within each worm (set to 100%), for each region in (A). Three replicate experiments are shown. Each point is one worm, and lines are mean ± SEM.
Intestinal polyP is most prominently located in endo-lysosome vesicles
Higher-resolution analysis of the stained worms revealed that the intestinal polyP signal appeared in round puncta, suggestive of its presence within distinct subcellular compartments. The ranges of sizes and shapes of the EcPPXbd-EGFP/mCherry puncta potentially fit several known organelles and liquid-liquid phase condensates known to be present in the C. elegans gut (Figure 3, middle). To characterize the subcellular localization of polyP in more detail, we either used publicly available chromosome-encoded fluorophore-protein fusion worm strains or exploited our new-found ability to use commercially available antibodies and dyes to stain subcellular organelles and liquid-liquid phase separations (LLPS) for which no fluorescently tagged worms strains were available. Our analysis of their potential co-localization with the EcPPXbd-EGFP or EcPPXbd-mCherry signal nicely illustrated that this cryosection technique is indeed a powerful method to detect, monitor, and quantify proteins and lipids in large numbers of fixed worms. We did not observe any co-localization of the EcPPXbd-mCherry signal with a fluorescently tagged Golgi membrane marker, which appears as a punctate structure in the C. elegans intestines,26 a peroxisome membrane marker, or the yolk protein marker vitulin-2 fused to GFP (Figure 3A). Moreover, by using an antibody against C. elegans rme-1, a marker for recycling endosomes, we also ruled out the presence of polyP in those organelles (Figure S4). To determine if early endosomes contained polyP, we used a rab-5::GFP worm strain, which appeared to have a small number of very large compartments labeled with EGFP, indicating that the strain has a compromised endo-lysosomal system. Still, we did not observe EcPPXbd-mCherry within these structures. We also did not observe any co-localization of EcPPXbd-EGFP with lipid droplets, which we visualized via oil red O staining (Figure S4). Instead, we found very clear co-localization with membrane markers of lysosomes (lmp-1::GFP), late endosomes (rab-7::GFP), and so called “gut granules” (glo-1::GFP) (Figure 3, right). Gut granules are lysosome-related organelles (LROs) specific to the worm intestine.27 In each of these vesicles, the EcPPXbd-EGFP signal was surrounded by membrane marker fluorescence, indicating that the polyP resides inside these vesicles. This result was particularly noteworthy because polyP has previously been observed to accumulate in cell-type-specific LROs, such as acidocalcisomes (also known as dense granules) in trypanosomes.3 V-ATPases in the acidocalcisome membrane have been suggested to be critical for polyP synthesis and transport in trypanosomes and other eukaryotes.28 C. elegans lysosomes, late endosomes, and LROs are highly dynamic compartments, which merge with each other and other vesicles, sharing membrane components in the process.29 Based on this reason, we are unable to conclusively state that polyP is only present in these specific compartments since other vesicles, such as the autophagosomes, share the same membrane markers, even if only temporarily. Nevertheless, these studies clearly demonstrated that polyP is a component of the endo-lysosomal system of vesicles. It is of note, however, that not every endo-lysosomally marked vesicle contained polyP and that not every polyP puncta co-localized with each specific membrane marker that we used. While we cannot rule out that other compartments outside of the endo-lysosomal system contain polyP, these results are consistent with polyP being spread throughout the endo-lysosomal system. In any case, however, these results suggested that polyP synthesis is likely coupled to the development of endo-lysosomal vesicles.
Figure 3.

EcPPXbd-mCherry-positive puncta are surrounded by membrane markers of endo-lysosomal vesicles
(A) Grayscale images of representative day 1 worm intestinal cells, expressing the listed markers of Golgi apparatus, peroxisomes, and yolk protein, stained with EcPPXbd-mCherry and overlays.
(B) Cartoon of various subcellular compartments in which EcPPXbd-mCherry co-localization was tested in this study prepared using BioRender (biorender.com).
(C) Same as in (A) but showing membrane marker signal surrounds EcPPXbd-mCherry signal for markers of endosomes, lysosomes, and lysosome-related organelles. Scale bar represents 8.5 μm.
Disrupting endo-lysosomal vesicle development reduces polyP abundance
To monitor whether the synthesis of polyP is connected to endo-lysosomal vesicle development, we obtained worms that carried a mutation in the membrane protein lmp-1. These mutant worms have a greatly reduced number and size of lysosomes.30 Indeed, cryosectioning and staining of the lmp-1 knockout (KO) worms with EcPPXbd-EGFP revealed a significant reduction in the number and size of the EcPPXbd-EGFP+ vesicles compared to age-matched WT N2 worms (Figures 4A–4D). In worms defective in functional glo-1,31 a protein that is involved in the formation of the gut granules, the dimmer and smaller vesicles were left intact, while the largest-diameter subset of EcPPXbd-EGFP+ vesicles disappeared (Figures 4A–4D). These results and the observation that lmp-1KO worms harbor the smallest population of polyP+ vesicles are consistent with previous research that showed that lmp-1 KO worms have a severely reduced population of lysosomes as well as a loss of downstream endo-lysosomal compartments, including gut granules.30 These results strongly suggest that polyP synthesis and its accumulation are coupled to the biogenesis of lysosomes in C. elegans. We also investigated the EcPPXbd-EGFP signal in the heads of the mutant worms and found no difference between N2, glo-1KO, and lmp-1KO worms (Figure S5). These results indicate that polyP levels in the head regions of the worms that are detectable with our EcPPXbd-EGFP probe are not dependent on intestine-specific lmp-1- or glo-1-containing organelles. The origin and maintenance of that pool of polyP requires further study.
Figure 4.

Gut EcPPXbd-EGFP signal is dependent on endo-lysosomal vesicle formation and food availability
(A) Representative images of N2, glo-1KO, and lmp-1KO day 1 adults stained with EcPPXbd-EGFP. Cuticle of worm is denoted by a dashed yellow line. Area used for fluorescence measurement in Figure 3B is shown with a white rectangle. Scale bar represents 20 μm.
(B) Relative fluorescence of EcPPXbd-EGFP signal in intestine. Each circle is one worm. Kruskal-Wallis test with Dunn's post hoc was used. Error bars are mean +/− SD.
(C) Quantification of diameter of EcPPXbd-EGFP-positive puncta in three representative images from 3 separate worms of intestinal cells. Bar = median. Each dot is one punctum.
(D) Graph of signal intensity of each puncta by its diameter. Each circle is one punctum.
(E) Quantification of EcPPXbd-EGFP before and after food removal and recovery. Each dot represents one worm. Kruskal-Wallis test with Dunn's post hoc was used. p value is shown for each comparison made. Error bars are mean +/− SEM.
(F) Relative fluorescence of EcPPXbd-EGFP signal in the heads of worms relative to the fed 4 h intestinal signal. Each circle is one worm. Kruskal-Wallis test with Dunn's post hoc was used. Lines are mean ± SEM. p value is shown for each comparison made. All comparisons between GFP only groups had p > 0.4.
Transient starvation depletes polyP stores from endo-lysosomal vesicles
PolyP has long been thought to act as an energy reservoir, allowing for the storage of high-energy phosphoanhydride bonds while keeping the osmotic burden at a minimum.32 We now wondered whether worms utilize the endo-lysosomal polyP stores as an energy source under food-poor conditions. To test this, we grew N2 worms to day 1 of adulthood and then transferred them onto fresh plates containing either OP50 WT bacteria or no bacteria. After 4 h of starvation, we either directly collected the worms for cryosectioning or transferred the starved worms onto OP50WT-containing plates for an additional 1, 2, 4, or 24 h before cryoblocking and analysis. As shown in Figure 4E, we found that the EcPPXbd-EGFP signal in the intestine dropped significantly within 4 h of food starvation. At this point, it is unclear whether polyP is converted back to ATP, transported elsewhere, or capped at the ends, which could render it invisible to EcPPXbd-EGFP. However, since the EcPPXbd-EGFP signal slowly returned upon the re-introduction of food, it is most likely that polyP stores are being depleted during starvation and filled up again as soon as food becomes re-available. We hypothesized that the depletion of polyP would occur throughout the entire worm tissues and, indeed, found a significant drop in polyP levels also in the head region of the worm, which was recovered upon the re-introduction of food (Figure 3F). The reduction in signal was not as dramatic as compared to the intestine, suggesting there is a difference in the dynamics of loss in this polyP pool.
In summary, our study adds a metazoan model to the polyP field, allowing the visualization and manipulation of endogenous polyP and forming the groundwork for future mechanistic studies on the cross-tissue control of polyP. At this point, we do not yet understand what is happening to the sources of polyP during starvation and how polyP stores are refilled upon food availability. Is polyP exported as a whole chain or broken down into orthophosphate units, and how are they exported from the vesicles? It may also be the case that the ends of the polyP chains are simply made unavailable to EcPPXbd-EGFP through some hitherto unknown binding event, which may render it invisible to our IF method.33 We do not know how polyP distributes in non-intestinal cells since all of our transgenic lines marking those intracellular compartments are intestine-specific. There are many potential areas of study now available, and we expect that our longitudinally-oriented cryosectioning method will open the door for more worm researchers to investigate polyP in this organism. Our method for sectioning hundreds of worms in different conditions simultaneously is a powerful tool for C. elegans research. For most protein localization work in C. elegans, researchers depended on the use of transgenic animals expressing fluorophore-fused native proteins. While this is a powerful system for studying proteins in live animals, it may alter the localization and activity of the fusion proteins. Moreover, each of these strains takes months to make, significantly increasing the costs of this research. Our method is inexpensive, fast, and highly reproducible. Moreover, since many of the established antibodies cross-react with worm proteins, we are convinced that this 3D-printed-well and cryosectioning method will be a welcome addition to the C. elegans toolbox.
Limitations of the study
We currently have no way of verifying that all of the polyP in the samples is retained after sample processing. Therefore, even if the fluorescent probe is detecting all available polyP, we cannot say for certain that all biologically important pools of polyP are visualizable using this method. It is unlikely that an abundant reservoir is missing due to the similar pattern of the EcPPXbd-mCherry signal in freeze-cracked versus cryosectioned worms.
When using the freeze-cracking method to prepare C. elegans samples for IF, the entirety of the worm is present on the slide. However, by using cryosectioning to bypass the cuticle, there will always be missing parts of each worm that is sectioned. Another limitation of sectioning is related to imaging: thinner sample sections (8–12 μm) will be easier to image clearly than thicker samples, which have more tissue to image through. Thicker samples will include more spatial information in the whole organism, which allows for easier identification of various tissues.
There are few probes for polyP in the literature. While the one we employed for this study is well verified as polyP-specific in mammalian cells,16 it is possible that the probe was unable to bind and fluoresce in all polyP pools in the samples. Further validation could include genetic manipulation of the C. elegans to express an PPX from bacteria to the gut granules specifically and observe a loss of EcPPXbd-EGFP staining. Alternatively, creating a non-binding version of this probe would be an excellent control for specificity in a C. elegans system.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ellen Quarles (equarles@gmail.com).
Materials availability
3D model files of the pronged molds and centrifuge adaptors are available in several formats. Contact Ellen Quarles for the files. If users are unable to print these files themselves, we recommend using a service such as Shapeways.com. Strains used in this paper are available upon request from the Jakob lab at the University of Michigan by contacting Ursula Jakob (ujakob@umich.edu).
Data and code availability
-
•
The data reported in this paper are available from the lead contact upon request.
-
•
This study does not report original code.
-
•
Any further information required to re-analyze the data reported in this paper is available from the lead contact upon request.
Acknowledgments
We thank Ken Wan for purifying the proteins, Traci Banjanin for preparing many of the materials used for the study, and James Bardwell (University of Michigan) for access to the Leica Thunder microscope. We also thank Gregg Sobocinski for his help and expertise with microscopy. We are thankful for the following funding: the Michigan Society of Fellows (E.Q.), a BrightFocus ADR Fellowship (A2019250F to B.O.), and a University of Michigan T32-AG000114 Career Training in the Biology of Aging Training Grant (to B.O.) Funding for the study came from National Institutes of Health grant GM122506 (to U.J.).
Author contributions
Conceptualization, E.Q. and U.J.; methodology, E.Q.; validation, E.Q., L.P., A.N., A.J., and J.W.; formal analysis, E.Q.; investigation, E.Q., B.O., A.R., L.P., A.N., A.J., and J.W.; resources, E.Q. and U.J.; writing – original draft, E.Q.; writing – review & editing, U.J.; visualization, E.Q., B.O., and A.R.; supervision, E.Q. and U.J.; funding acquisition, U.J.
Declaration of interests
The authors declare no competing interests.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Gelatin, bovine | Sigma-Aldrich | G9391-100G |
| Phosphate buffered saline (no Mg2+, no Ca2+) | Fisher Scientific | 70-011-044 |
| d-sucrose | Sigma-Aldrich | S9378 |
| Paraformaldehyde, 8% solution | Electron Microscopy Sciences | 157-8-100 |
| Triton X-100 | Sigma-Aldrich | T9284 |
| O.C.T. (Optical Cutting Compound) | Fisher Scientific | 23-730-571 |
| EcPPXbd-mCherry | This paper | |
| EcPPXbd-eGFP | This paper | |
| mCherry | This paper | |
| eGFP | This paper | |
| Oil Red O | Sigma-Aldrich | O9755 |
| Polypropylene glycol (100%) | MP Biomedicals, LLC | Catalog # 151957 |
| Food coloring | McCormick | Assorted Food Color and Egg Dye |
| Antibodies | ||
| Anti-LMP-1 | Developmental Studies Hybridoma Bank (https://dshb.biology.uiowa.edu/) LMP1 was deposited to the DSHB by Nonet, M.L./Hadwiger, G./Dour, S. |
LMP1, RRID: AB_2161795 |
| Anti-RME-1 | Developmental Studies Hybridoma Bank (https://dshb.biology.uiowa.edu/) RME1 was deposited to the DSHB by Nonet, M.L./Hadwiger, G./Dour, S. |
RME1, RRID: AB_10571460 |
| Experimental models: Organisms/strains | ||
| N2 (Bristol) | Caenorhabditis Genetics Center | N2 |
| Glo-1KO (glo-1(zu391)) | Caenorhabditis Genetics Center | JJ1271 |
| Lmp-1KO (lmp-1(nr2045)) | Caenorhabditis Genetics Center | PD4482 |
| Golgi apparatus membrane (vha-6p:mans:GFP + Cbr-unc-119(+)) | Caenorhabditis Genetics Center | RT1315 |
| Yolk (vit2:GFP) | Caenorhabditis Genetics Center | RT130 |
| peroxisome membrane (hjIs73 [vha-6p:GFP:daf-22 + C. briggsae unc-119(+)]) | Caenorhabditis Genetics Center | VS11 |
| lysosome related organelles (gut granules) (hjIs9 [ges-1p:glo-1:GFP + unc-119(+)]) | Caenorhabditis Genetics Center | VS17 |
| lysosome membrane (pwIs50 [lmp-1:GFP + Cbr-unc-119(+)]) | Caenorhabditis Genetics Center | RT258 |
| late endosome membrane (pwIs170 [vha6p:GFP:rab-7 + Cbr-unc-119(+)]) | Caenorhabditis Genetics Center | RT476 |
| Bacterial and virus strains | ||
| Escherichia coli OP50 | Caenorhabditis Genetics Center | Cat# OP50 |
| Escherichia coli OP50ΔPPKΔPPX | Datsenko and Wanner34 | OP50 ΔPPKΔPPX |
| Escherichia coli MG1655 | Blattner et al.35 | MG1655 (genotype: F− λ− rph-1 ilvG rfb-50) |
| Escherichia coli MG1655ΔPPK | Gray et al.21 | MG1655 Δppk-749 |
| Software and algorithms | ||
| FIJI (ImageJ) (RRID: SCR_003070) version 1.53c |
http://imagej.nih.gov/ij Schindelin et al.36 |
FIJI |
| GraphPad Prism v 9.5.1 (733) | GraphPad | Prism |
| Excel | Microsoft | Microsoft Office Professional Plus 2016 |
| LAS X version 3.3.0.16799 | Leica | LAS X |
| Other | ||
| Cellpath Stainless-steel Reusable Base Molds | Fisher Scientific | cat # 22-222-033 |
| 3D printed pronged molds (resin printed due to needing tight tolerances in shape) | This study | Contact Ellen Quarles for files |
| 3D printed centrifuge adaptors (filament printed due to larger size and strength needed under centrifugation) | This study | Contact Ellen Quarles for files |
| Wide-field microscope on SP8 base | Leica | Thunder |
| Cryostat | Leica | 3050s |
| SP8 confocal microscope (DMI8 base) | Leica | SP8 |
Experimental model and study participant details
Strains
All worm strains were obtained from the Caenorhabditis Genetics Center. Wild type bacterial strains (OP50 and MG1655) came from the Caenorhabditis Genetics Center. Introducing the null mutations in OP50 (OP50ΔppkΔppx) has been conducted as described by K. A. Datsenko, B. L. Wanner, 2000.34 Null mutation in MG1655 has been described in Gray et al., 2014.21
Culturing C. elegans
Worms were grown on nematode growth media (NGM) agar with appropriate antibiotics at 20°C. Plates were seeded with overnight cultures of OP50 or MG1655 bacteria, and worms were always kept in food-excess conditions unless otherwise stated. Age synchronization was accomplished with the bleach method (Wormbook.org, “Harvesting asynchronous culture and seeding synchronous cultures”). Hermaphrodites were used for all experiments. Worms were removed from plates by washing with PBS buffer.
Method details
Preparation of cryomolds
Gelatin solution (10% D-sucrose, 10% bovine gelatin, 1x PBS no calcium/magnesium, 1 drop per ∼10 mL, blue or green food coloring) was prepared and heated in a water bath at 37°C–42°C with occasional mixing until completely melted. Unused gelatin solution was stored at 4°C for up to two weeks. Into each metal cryomold (Cellpath Stainless-steel Reusable Base Molds, Fisher Scientific, cat # 22-222-033) the gelatin solution was pipetted until the mold was halfway filled. Immediately, the 3D printed pronged mold was added and centered, and the entire apparatus was kept level, and allowed to cool at 4°C until the gelatin had solidified. Bubbles were avoided, and removed by pipetting or running a flame briefly over the liquid. The 3D printed pronged molds were removed by gently rocking them and slowly pulling them away from the gelatin. These were then gently wiped clean and stored at room temp. The prepared cryomolds were either used immediately or stored at 4°C in a sealed plastic bag for up to one week to prevent drying.
Freezing worms in cryomolds
200 - 10,000 worms were washed off NGM plates using M9 media or PBS. Adult worms were allowed to settle by gravity for up to 2 min, then the supernatant was removed by decanting or pipetting. The pellet of worms was washed twice more with M9 or PBS. Then, 2 mL of 4% v/v paraformaldehyde (PFA) in PBS was added to the worm pellet. Worms were incubated at room temperature in the PFA solution for 10 min and gently mixed by tube inversion every 2–3 min. The PFA serves two purposes: to kill the worms, and to help maintain structural integrity of the worms throughout the cryosectioning process. PFA was removed, and the worm pellet was washed with 0.01% v/v Triton ×100 in PBS for 10 min. This step is necessary to maintain smooth cuticle structure during sectioning, and to help the worms not stick to the tube walls. Worms were centrifuged at 500∗g for 1 min at room temperature, and the supernatant was removed to allow easy pipette access to the pellet. Worms were then pipetted with a 1000 μL tip, with the end cut off by several mm to facilitate easy and gently moving of the worms, into one or more wells of the prepared cryomolds. The worms settle to the bottom of the cryomolds in seconds. Then, we used a 2–10 μL pipette tip to pipette away the supernatant from the cryomold wells using a stereoscope to help visualize the worms. This is made easier by holding the tip extremely close to the gelatin at the sides of the wells, close enough that the adult worms cannot get sucked into the tip. Once the supernatant was fully removed an embedding compound commonly used to support sectioning of frozen tissue (O.C.T., Optimal Cutting Compound, purchased from Fisher Scientific) was added to the wells, filling them approximately halfway. A 2–10 μL pipette tip was used to gently stir the worms around in the O.C.T. to ensure they were entirely covered by the compound, and that they were evenly distributed in the well. Care must be taken to not damage the gelatin mold in this step. Then, O.C.T. was added again to top off the wells, and to fill the cryoblocks with 0.5 cm or more height of O.C.T. over the gelatin layer. All blocks were weighed and balanced using O.C.T. The cryomolds were centrifuged in 3D printed (filament) centrifuge adaptors designed to hold 2 metal cryomolds each, sized to fit into standard plate holders. Centrifugation was done at 4°C or room temperature for 8 min at 1500∗g, in swinging plate holders. The blocks were then visually inspected to ensure all worms were only in the bottom of the wells, and evenly distributed within each well. If not, they were remixed, and re-balanced, and re-spun. Completed blocks were flash frozen in a dry ice-EtOH slurry. Frozen blocks were marked with permanent marker to delineate the orientation of the block for future cutting and removed from the metal molds by inverting them and hitting the metal molds against a napkin-covered countertop to dislodge the block. The blocks were wrapped in labeled paper and stored at −80C until sectioning.
Cryosectioning
Blocks were allowed to warm up to −20°C, and adhered to cryostat chucks with the non-gelatin side attached to the chuck by O.C.T. The food coloring in the gelatin allows easy visualization of the alignment of, and distance to, the white circles of the wells which become more apparent as further layers of gelatin are removed. This also allows for a clear reference of the angle of the cryosections and allows the user to correct that alignment before the bottom of the wells (where all the worms are present) gets sectioned. Blocks were cryosectioned on a Leica 3050s cryostat at 10–12 μm per slice. Thicker and thinner sections are also possible. Thicker sections allow for easier handling of the section, and more stainable/image-able tissue sections, but they have reduced image clarity due to the light refracting through the tissues when using standard confocal or wide field microscopy. Thinner sections give clearer fluorescent images but can be difficult to handle and reduce the z-depth of the tissue, which can be an issue when generating z stack images. Sections were dried immediately after being applied to the slides, on a slide warmer set to 30°C for 10–30 min. The slides were either stained the same day or the subsequent day after remaining in a dark box overnight at room temperature. Freezing the slides at −80°C before staining did not seem to affect EcPPXbd-eGFP signal intensity or distribution.
Purification of fluorescent probes, ScPPX and DPP1
The creation of the plasmids containing mCherry-EcPPXbd, eGFP-EcPPXbd and ScDPP1 was described previously.15,24,33 Purification of these and the HIS-tagged mCherry, eGFP, scPPX and ScDPP1 proteins was performed using a nickel-NTA column (Qiagen, Germany). In short, after 4-6h expression, E. coli strains transformed with each of the plasmids except ScDPP1 were lysed in lysis buffer (40 mM Tris, 10mM sodium phosphate buffer, 10% v/v glycerol, 10 mM MgCl2, pH 8.0) with cOmplete protease inhibitor cocktail, DNASE I, and RNase A (all from Milipore-Sigma). Samples were sonicated, centrifuged, and the supernatant was loaded onto HisTrap columns (Sigma-Aldrich). Proteins were eluted with imidazole, dialyzed against 20 mM NaP, 50 mM KCl, 10% v/v Glycerol, pH 7.5 and stored at −70 °C at 1 mg/mL. ScDDP1 protein was isolated by lysing cells in lysis buffer (50 mM sodium phosphate buffer, 500 mM NaCl, 10 mM imidazole, with 1 mg/mL lysozyme, 2 mM MgCl2, and 50 U/mL Benzonase). Cells were incubated on ice for 30 min, then sonicated and centrifuged to pellet cell debris. Lysates were loaded onto NiNTA columns and eluted with imidazole (pH 8.0). Protein was dialyzed against 20 mM Tris-HCl (pH 7.5), 50 mM KCl, 30% (v/v) glycerol, and stored at −70 °C at 1 mg/mL. The purity of the proteins was analyzed by reducing SDS PAGE followed by Coommassie staining (Figure S3C).
Staining and immunofluorescence
The wells of the cryosections are visible on the slide as they show up as clear circles against a colorful gelatin background. To track the samples, we used a permanent marker and labeled the positions on the side of the glass slide that did not contain the worms. On the same side of the slide as the worms, we used a PAP pen (Electron Microscopy Sciences, # 106-94-5) to corral the staining fluids away from the top label and bottom edge of the slides.
EcPPXbd-eGFP staining - Briefly, the sections were fixed with 4% v/v PFA in PBS for 10 min, followed by 3x washing with PBS for 5 min each. Permeabilization of the tissues was accomplished by incubation with 0.3% v/v Triton ×100 in PBS for 10 min (longer incubation times allowed DNA to leak out of nuclei, shorter times did not allow for complete EcPPXbd-eGFP staining in the intestine). Sections were washed three more times with PBS, and incubated in blocking buffer (2% BSA, 22.52 mg/mL glycine, 1x PBS) for 30–60 min. Blocking buffer containing either 10 μg/mL EcPPXbd-eGFP (or EcPPXbd-mCherry) or 10 μg/mL eGFP (or mCherry) was added to the sections, which were incubated for 4 h to overnight in a humid, dark chamber. The sections were subsequently washed with PBS three more times, and incubated for 10 min with 0.1 μg/mL DAPI in PBS with occasional gentle rocking. This was followed by two more washes with PBS. A 1.5 size coverglass (Corning) was mounted using Prolong Gold. Slides were sealed with clear nail polish (Sally Hansen Hard as Nails Xtreme Wear, Invisible) and stored at 4°C in the dark until imaging. All PBS-containing steps were conducted at room temperature, and with gentle rocking/mixing every 1–3 min, except for blocking and staining steps, which were allowed to remain still.
Oil Red O Staining - Freshly cryosectioned, air dried sections were washed with DI water two times, briefly. Polypropylene glycol (100%) was added for 2 min, then drained off the slides. The sections were then incubated in either 100% polypropylene glycol (controls) or Oil Red O working solution (0.5% Oil Red O in 100% polypropylene glycol, heated to 95C, filtered, and stored at room temperature) for 8 min at 60°C. Slides were destained in 85% polypropylene glycol 3 min at room temperature. Finally, sections were washed twice briefly in DI water. The sections were then stained with EcPPXbd-eGFP or GFP as above.
Antibody staining - Sections were treated as above for EcPPXbd-eGFP, with the following additions. For mouse anti-C. elegans RME-1 staining: After the blocking step, sections were incubated with 10 μg/mL anti-RME-1 or no primary antibody (with and without EcPPXbd-mCherry or mCherry) overnight in a dark, humid chamber at room temperature. Sections were washed with 1x PBS three times, then incubated with anti-mouse Alexa Fluor 488 secondary antibody, at 1:1000 dilution in blocking buffer, for 1 h at room temperature. Sections were then washed again with 1x PBS three times, and mounted as above. For mouse anti-C. elegans LMP-1 staining: Same as anti-RME-1 staining, except a heat-induced antigen retrieval (HIAR) step was added just prior to the blocking step. We accomplished HIAR by adding 10 mM sodium citrate, pH 7.0 pre-warmed to 95°C to the sections on a hot (95°C) block for 60 s. The slides were then immediately drained and room temperature 1x PBS was used to wash the slides twice. The rest of the protocol is as above for anti-RME-1.
Fluorescence microscopy
Widefield images were captured using a Leica THUNDER (widefield microscope) on a T8i inverted base, with an HC PL APO 40x/1.3 NA oil objective (catalog number 11506358). Leica Navigator software was used to run Tile Scan and stitching functions. THUNDER Instant Computational Clearing (ICC) method was run for background and blur reduction. Focusing over the tile scanned areas was achieved by adding Focus Points within the area to be scanned using Navigator. An LED8 was used to excite all fluorophores (DAPI, Ex: 395 nm, Em: 460/80 nm; EGFP, Ex: 488 nm, Em: 535/70 nm; mCherry, Ex: 555 nm, Em: 590/50 nm), and the CYR7101, DFT51010, and EMP_BF filter cubes were used for emission. Images were captured with an Andor Zyla sCMOS camera.
Confocal images were captured using a Leica SP8 scanning confocal microscope (Leica GmbH, Mannheim Germany) on a DMI8 microscope base, using LAS X software, and an HC PL APO 100x/1.40 oil objective (Leica 11506378). A 405 nm diode laser, and multi-line white light laser were used for excitation (set to 488 and 585 nm). PMT detectors were used for spectral detection (DAPI: 410–480 nm, EGFP: 495–560 nm, mCherry: 600–675 nm). A transmitted light PMT, with the 405 nm laser, was used to capture a brightfield image. Control images without antibodies or EcPPXbd-bound fluorophores are in Figures S1B–S1F.
Measurement of fluorescence intensity
Image analysis was performed using FIJI (ImageJ 1.53t, Java 1.8.0_172). For each image, a composite of the DAPI (for DNA) and brightfield channels was made and used to define regions of interest (ROIs) using a rectangle of defined width/height that would cover the width of the worms (150 px), and a height of 115 px. These were placed on worms along areas with visible gut cells covering the entire width of the worm, as identified by the nuclear shape and size. (Gut cell nuclei are much larger than gonadal nuclei.) The saved ROIs were then used to measure eGFP or mCherry fluorescence intensity on the appropriate tiff channels, and a large non-worm background ROI intensity was subtracted from each measurement per image. In worms in which multiple ROIs could be made, up to 3 were made and averaged.
Food removal and recovery
Age-synchronized D1 adult N2 worms, grown on OP50WT bacteria were washed with M9 media three times to remove bacteria. The worms were then re-distributed onto either NGM plates containing OP50WT or no bacteria. After 4 h at 20°C, fed and starved worms were cryoblocked. The remaining starved worms were transferred onto OP50WT-containing plates to recover from starvation for 1, 2, 4, or 24 h and then cryoblocked.
Enzyme digestion used in Figure S3
Sections were treated as for EcPPXbd-eGFP, with the following additions: After permeabilization with Triton X-100, sections were washed in 1x PBS, 3x, 5 min each. The sections then underwent Heat Induced Antigen Retrieval (HIAR) to allow for polyP-ScPPX1 interaction post fixation. To the sections, we added 10 mM sodium citrate, pH 7.0 pre-warmed to 95°C, on a hot (95°C) block for 60 s. The slides were then immediately drained and room temperature 1x PBS was used to wash the slides twice. Then the sections were washed 1x, 5 min at RT in ScPPX1 buffer (20 mM Tris-HCl pH 7.5, 5 mM MgCl2, 50 mM ammonium acetate). The ScPPX1 with heat-inactivated DDP1, DDP1 with heat-inactivated ScPPX1, ScPPX1 with DDP1, or both enzymes heat-inactivated were added to the sections at 5 ng/mL in ScPPX1 buffer. These were incubated in a dark, humid chamber at 37C for 2 h. Then the sections were washed 3x with 1x PBS, 5 min each, and the EcPPXbd-eGFP staining procedure was continued as described in the main text methods starting at the blocking step.
Quantification and statistical analysis
GraphPad Prism 9 was used to perform statistical analysis and to produce graphs. Specific tests and post-hoc analyses are described in figure legends. Unless otherwise stated, dots within graphs represent biological replicate experiments, and data are presented with a bar indicating the mean ± SEM. P-values are listed on the graphs for comparisons made. Kruskal-Wallis tests were used instead of ANOVA because at least one group in each comparison had unequal variance with the rest of the groups, and because some groups violated an assumption of a normal distribution. Number of worms used for measurements ranged from 20 to 50 per group.
Published: October 15, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2024.100879.
Supplemental information
References
- 1.Kornberg A., Rao N.N., Ault-Riché D. Inorganic Polyphosphate: A Molecule of Many Functions. Annu. Rev. Biochem. 1999;68:89–125. doi: 10.1146/annurev.biochem.68.1.89. [DOI] [PubMed] [Google Scholar]
- 2.Bowlin M.Q., Gray M.J. Inorganic polyphosphate in host and microbe biology. Trends Microbiol. 2021;29:1013–1023. doi: 10.1016/j.tim.2021.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Docampo R., Moreno S.N. Acidocalcisome: A Novel Ca2+ Storage Compartment in Trypanosomatids and Apicomplexan Parasites. Parasitol. Today. 1999;15:443–448. doi: 10.1016/s0169-4758(99)01531-8. [DOI] [PubMed] [Google Scholar]
- 4.Denoncourt A., Downey M. Model systems for studying polyphosphate biology: a focus on microorganisms. Curr. Genet. 2021;67:331–346. doi: 10.1007/s00294-020-01148-x. [DOI] [PubMed] [Google Scholar]
- 5.Samper-Martín B., Sarrias A., Lázaro B., Pérez-Montero M., Rodríguez-Rodríguez R., Ribeiro M.P.C., Bañón A., Wolfgeher D., Jessen H.J., Alsina B., et al. Polyphosphate degradation by Nudt3-Zn2+ mediates oxidative stress response. Cell Rep. 2021;37 doi: 10.1016/j.celrep.2021.110004. [DOI] [PubMed] [Google Scholar]
- 6.Baker C.J., Smith S.A., Morrissey J.H. Polyphosphate in thrombosis, hemostasis, and inflammation. Res. Pract. Thromb. Haemost. 2019;3:18–25. doi: 10.1002/rth2.12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holmström K.M., Marina N., Baev A.Y., Wood N.W., Gourine A.V., Abramov A.Y. Signalling properties of inorganic polyphosphate in the mammalian brain. Nat. Commun. 2013;4:1362. doi: 10.1038/ncomms2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lempart J., Jakob U. Role of Polyphosphate in Amyloidogenic Processes. Cold Spring Harb. Perspect. Biol. 2019;11:a034041. doi: 10.1101/cshperspect.a034041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arredondo C., Cefaliello C., Dyrda A., Jury N., Martinez P., Díaz I., Amaro A., Tran H., Morales D., Pertusa M., et al. Excessive release of inorganic polyphosphate by ALS/FTD astrocytes causes non-cell-autonomous toxicity to motoneurons. Neuron. 2022;110:1656–1670.e12. doi: 10.1016/j.neuron.2022.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Borden E.A., Furey M., Gattone N.J., Hambardikar V.D., Liang X.H., Scoma E.R., Abou Samra A., D-Gary L.R., Dennis D.J., Fricker D., et al. Is there a link between inorganic polyphosphate (polyP), mitochondria, and neurodegeneration? Pharmacol. Res. 2021;163 doi: 10.1016/j.phrs.2020.105211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shim Y.J., Chatterjee V., Swaidani S., Alluri R.K., Kundu S., Merkulova A., Angelini D., You D., Whitney S.A., Feener E.P., et al. Polyphosphate expression by cancer cell extracellular vesicles mediates binding of factor XII and contact activation. Blood Adv. 2021;5:4741–4751. doi: 10.1182/bloodadvances.2021005116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kulakovskaya E.V., Zemskova M.Y., Kulakovskaya T.V. Inorganic Polyphosphate and Cancer. Biochemistry. 2018;83:961–968. doi: 10.1134/s0006297918080072. [DOI] [PubMed] [Google Scholar]
- 13.Gray M.J. Inorganic Polyphosphate Accumulation in Escherichia coli Is Regulated by DksA but Not by (p)ppGpp. J. Bacteriol. 2019;201 doi: 10.1128/JB.00664-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Breus N.A., Ryazanova L.P., Suzina N.E., Kulakovskaya N.V., Valiakhmetov A.Y., Yashin V.A., Sorokin V.V., Kulaev I.S. Accumulation of inorganic polyphosphates in Saccharomyces cerevisiae under nitrogen deprivation: Stimulation by magnesium ions and peculiarities of localization. Microbiology. 2011;80:624–630. doi: 10.1134/s002626171105002x. [DOI] [PubMed] [Google Scholar]
- 15.Saito K., Ohtomo R., Kuga-Uetake Y., Aono T., Saito M. Direct labeling of polyphosphate at the ultrastructural level in Saccharomyces cerevisiae by using the affinity of the polyphosphate binding domain of Escherichia coli exopolyphosphatase. Appl. Environ. Microbiol. 2005;71:5692–5701. doi: 10.1128/AEM.71.10.5692-5701.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Borghi F., Azevedo C., Johnson E., Burden J.J., Saiardi A. A mammalian model reveals inorganic polyphosphate channeling into the nucleolus and induction of a hyper-condensate state. Cell Rep. Methods. 2024;4 doi: 10.1016/j.crmeth.2024.100814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duerr J.S. Antibody Staining in C. Elegans Using “Freeze-Cracking”. J. Vis. Exp. 2013 doi: 10.3791/50664-v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duerr J.S. Immunohistochemistry. WormBook: the online review of C. elegans biology. WormBook. 2006 doi: 10.1895/wormbook.1.105.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ross M.A., Kohut L., Loughran P.A. Cryosectioning. Curr. Protoc. 2022;2:e342. doi: 10.1002/cpz1.342. [DOI] [PubMed] [Google Scholar]
- 20.Crittenden S.L., Troemel E.R., Evans T.C., Kimble J. GLP-1 is localized to the mitotic region of the C. elegans germ line. Development. 1994;120:2901–2911. doi: 10.1242/dev.120.10.2901. [DOI] [PubMed] [Google Scholar]
- 21.Gray M.J., Wholey W.Y., Wagner N.O., Cremers C.M., Mueller-Schickert A., Hock N.T., Krieger A.G., Smith E.M., Bender R.A., Bardwell J.C.A., Jakob U. Polyphosphate Is a Primordial Chaperone. Mol. Cell. 2014;53:689–699. doi: 10.1016/j.molcel.2014.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ebbing A., Vértesy Á., Betist M.C., Spanjaard B., Junker J.P., Berezikov E., van Oudenaarden A., Korswagen H.C. Spatial Transcriptomics of C. elegans Males and Hermaphrodites Identifies Sex-Specific Differences in Gene Expression Patterns. Dev. Cell. 2018;47:801–813.e6. doi: 10.1016/j.devcel.2018.10.016. [DOI] [PubMed] [Google Scholar]
- 23.Rödelsperger C., Ebbing A., Sharma D.R., Okumura M., Sommer R.J., Korswagen H.C. Spatial Transcriptomics of Nematodes Identifies Sperm Cells as a Source of Genomic Novelty and Rapid Evolution. Mol. Biol. Evol. 2021;38:229–243. doi: 10.1093/molbev/msaa207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lonetti A., Szijgyarto Z., Bosch D., Loss O., Azevedo C., Saiardi A. Identification of an evolutionarily conserved family of inorganic polyphosphate endopolyphosphatases. J. Biol. Chem. 2011;286:31966–31974. doi: 10.1074/jbc.M111.266320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akiyama M., Crooke E., Kornberg A. An exopolyphosphatase of Escherichia coli. The enzyme and its ppx gene in a polyphosphate operon. J. Biol. Chem. 1993;268:633–639. [PubMed] [Google Scholar]
- 26.Sato M., Saegusa K., Sato K., Hara T., Harada A., Sato K. Caenorhabditis elegans SNAP-29 is required for organellar integrity of the endomembrane system and general exocytosis in intestinal epithelial cells. Mol. Biol. Cell. 2011;22:2579–2587. doi: 10.1091/mbc.E11-04-0279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morris C., Foster O.K., Handa S., Peloza K., Voss L., Somhegyi H., Jian Y., Vo M.V., Harp M., Rambo F.M., et al. Function and regulation of the Caenorhabditis elegans Rab32 family member GLO-1 in lysosome-related organelle biogenesis. PLoS Genet. 2018;14 doi: 10.1371/journal.pgen.1007772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lander N., Cordeiro C., Huang G., Docampo R. Polyphosphate and acidocalcisomes. Biochem. Soc. Trans. 2016;44:1–6. doi: 10.1042/BST20150193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Delahaye J.L., Foster O.K., Vine A., Saxton D.S., Curtin T.P., Somhegyi H., Salesky R., Hermann G.J. Caenorhabditis elegans HOPS and CCZ-1 mediate trafficking to lysosome-related organelles independently of RAB-7 and SAND-1. Mol. Biol. Cell. 2014;25:1073–1096. doi: 10.1091/mbc.E13-09-0521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kostich M., Fire A., Fambrough D.M. Identification and molecular-genetic characterization of a LAMP/CD68-like protein from Caenorhabditis elegans. J. Cell Sci. 2000;113:2595–2606. doi: 10.1242/jcs.113.14.2595. [DOI] [PubMed] [Google Scholar]
- 31.Hermann G.J., Schroeder L.K., Hieb C.A., Kershner A.M., Rabbitts B.M., Fonarev P., Grant B.D., Priess J.R. Genetic analysis of lysosomal trafficking in Caenorhabditis elegans. Mol. Biol. Cell. 2005;16:3273–3288. doi: 10.1091/mbc.e05-01-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xie L., Jakob U. Inorganic polyphosphate, a multifunctional polyanionic protein scaffold. J. Biol. Chem. 2019;294:2180–2190. doi: 10.1074/jbc.REV118.002808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xie L., Rajpurkar A., Quarles E., Taube N., Rai A.S., Erba J., Sliwinski B., Markowitz M., Jakob U., Knoefler D. Accumulation of Nucleolar Inorganic Polyphosphate Is a Cellular Response to Cisplatin-Induced Apoptosis. Front. Oncol. 2019;9:1410. doi: 10.3389/fonc.2019.01410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Datsenko K.A., Wanner B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blattner F.R., Plunkett G., 3rd, Bloch C.A., Perna N.T., Burland V., Riley M., Collado-Vides J., Glasner J.D., Rode C.K., Mayhew G.F., et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 36.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The data reported in this paper are available from the lead contact upon request.
-
•
This study does not report original code.
-
•
Any further information required to re-analyze the data reported in this paper is available from the lead contact upon request.