

RESUME
Introduction: Les syndromes hyper-IgE constituent un groupe d'erreurs innées de l'immunité, parfois syndromiques, caractérisés cliniquement par la triade classique : eczéma chronique, infections staphylococciques cutanées et/ou pulmonaires et des niveaux élevés d'IgE sériques (> 2000 UI/ ml ou > 10 x normale pour l’âge). Objectif: Rapporter les aspects cliniques et immunologiques de patients marocains présentant un HIES probable ou possible selon le score NIH-HIES. Méthodes: L'étude rétrospective couvrant la période de 1998 à 2023 a porté sur des patients marocains présentant des symptômes évocateurs de HIES et un score NIH ≥ 20. Résultats: La médiane d’âge à la présentation des signes cliniques était de 0,5 ans et la médiane d’âge au début des symptômes était de 5,5 ans. Les principaux symptômes inclus étaient l'eczéma (66%), les abcès cutanés (32,5%), les pneumonies (32,5%), les otites (20 %), les candidoses (19%), la diarrhée (12%), la dysmorphie faciale (10,3 %), les adénopathies (9,5 %), la dilatation bronchique (8%), les pneumatocèles (8%), la conjonctivite (7,1%), la rhinite (6,3%), le retard psychomoteur (5,6%), les fractures pathologiques (4%), la rétention des dents lactéales (4 %), le retard cognitif (3,2%). Conclusion: Cette étude représente la première description clinique d'une cohorte de patients marocains présentant un HIES selon les critères NIH. Bien que certaines orientations vers les gènes mutés aient été possibles sur la base du phénotype, une analyse moléculaire est nécessaire en raison du chevauchement des symptômes.
ABSTRACT
Introduction: Hyper-IgE syndrome is a group of inborn errors of immunity, some of which are syndromic, characterized clinically by the classic triad of chronic eczema, cutaneous and/or pulmonary staphylococcal infections and high serum IgE concentrations (> 2000 IU/ml or > 10 x normal for age). Aim: We report here the clinical and immunological aspects of Moroccan patients presenting probable or possible HIES according to NIH-HIES score. Methods: This retrospective study covers the period from 1998 to 2023 and includes Moroccan patients with a clinical presentation suggestive of HIES (skin and/or pulmonary infections, eczema, high IgE levels) and an NIH score ≥ 20. We attempted to classify the patients phenotypically according to the 2022 IUIS IEI Expert Committee classification. Results: Median age at symptom onset was 0.5 years and median age at diagnosis was 5.5 years. The main clinical signs were eczema (66%), skin abscesses (32.5%), pneumonia (32.5%), otitis (20%), mucocutaneous candidiasis (19%), diarrhea (12%), facial dysmorphism (10.3%), lymphadenopathy (9.5%), bronchial dilation (8%), pneumatoceles (8%), conjunctivitis (7.1%), rhinitis (6.3%), psychomotor delay (5.6%), pathological fractures (4%), retention of deciduous teeth (4%), cognitive delay (3.2%). Conclusion: This is the first clinical description of a cohort of Moroccan patients presenting HIES according to NIH criteria. Phenotype can sometimes orient towards identification of the mutated gene, but the overlapping clinical signs make molecular analysis necessary for genetic counseling and appropriate treatment.
Introduction
Hyper IgE syndrome (HIES) is a group of inborn errors of immunity (IEI, previously known as primary immunodeficiencies of PIDs).
HIES is a group 2b, combined associated or syndromic immunodeficiency.
It is rare and severe (OMIM #147060), and manifests early in life, with recurrent skin and/or pulmonary infections, eczema, and very high IgE levels (1).
HIES was first described in 1966 by Davis et al (2), paradoxically before the discovery of IgE antibodies in the late 1960s (3).
This multisystem syndrome has heterogeneous clinical manifestations and diverse genetic origins (4, 5 ).
Its classification system has been modified several times, with the exclusion of deficiencies of DOCK8 (dedicator of cytokinesis 8) and TYK2 (tyrosine kinase 2).
Indeed, DOCK8 deficiency was previously considered to be an autosomal recessive (AR) form of HIES but is now considered a combined immunodeficiency due to the predisposition to serious viral skin infections it confers, including Molluscum contagiosum, Herpes simplex and human papillomavirus infections (6), rarely noted in STAT3 deficiency as well as bone and connective tissue manifestations are less marked, or even non-existent in DOCK8 deficiency.
DOCK8 deficiency is no longer classified as HIES but still should be considered as a differential diagnosis with the STAT3-related forms (7 ).
Similarly, AR TYK2 deficiency was initially identified on the basis of its link to atypical mycobacterial disease, leading to its identification as a genetic predisposition to mycobacterial disease (Mendelian susceptibility to mycobacterial disease or MSMD) (8).
Several IEIs have recently been integrated into the HIES group: autosomal dominant (AD) defects of CARD11, ERBIN, TGFBR1, TGFBR2, AD and AR defects of IL6ST, and AR defects of ZNF341 and IL-6R(9).
According to the 2022 classification of the International Union of Immunological Societies (IUIS), HIES includes 11 pathological entities classified into two groups (4, 9) corresponding to autosomal dominant (AD-HIES) and autosomal recessive (AR-HIES) forms.
The principal genetic cause of this syndrome is AD STAT3 (signal transducer and activator of transcription 3) deficiency (OMIM #147060).
This form of the disease, also known as Job-Buckley Syndrome, is a multisystem disease.
In addition to the classic clinical triad of HIES, it includes non-immunological characteristics, such as facial dysmorphism, scoliosis, joint hyperlaxity, retention of deciduous teeth, a high-arched palate, and pathological fractures(10).
Seven other genetic deficiencies are known to result in clinical features partially or completely identical to those of STAT3 deficiency because they affect genes involved in the STAT3 signaling pathway (7).
These deficiencies are ZNF341 deficiency (AR HIES; Zinc finger protein 431)(11), IL-6R deficiency (AR HIES; interleukin 6 receptor)(12), IL6ST deficiency (AD and AR HIES; interleukin 6 signal transducer)(13), PGM3 deficiency (AR HIES; phosphoglucomutase 3) (14), and two other AD deficiencies of ERBIN (Erbb2-interacting protein) and TGFBR1 or TGFBR2 (Loeys-Dietz syndrome)(15, 16).
Another two deficiencies included in the HIES class of the IUIS do not present the non-immunological features of HIES: SPINK5 deficiency (Comel-Netherton syndrome; AR HIES; serine peptidase inhibitor Kazal type 5), which manifests as congenital ichthyosis and bamboo hair (17), and CARD11 (AD HIES) deficiency (Caspase recruitment domain family member 11), which manifests principally as atopy and susceptibility to a broad spectrum of infections (18).
In emerging countries like Morocco, it is difficult to obtain a genetic diagnosis of this disease.
It is therefore important to be able to make a reliable diagnosis based solely on the clinical and immunological phenotype.
We tested this approach with retrospective data from our cohort collected from the national reference registry of IEIs.
methods
We performed a retrospective study of patients with a strong suspicion of HIES followed by the Clinical Immunology Unit of the Pediatrics Department of Abderahim Harrouchi Children’s Hospital in Casablanca over a 25-year period extending from 1998 to 2023.
A form was completed for each patient, on which we recorded personal data (age, sex, family history, consanguinity, age at onset of clinical signs, and age at clinical diagnosis), clinical observations (cutaneous manifestations, pulmonary and skin infections, allergies, skeletal and connective tissue abnormalities, neurological abnormalities, autoimmunity) and the results of biological examinations (blood count, determinations of immunoglobulins IgM, G, A and E, lymphocyte subpopulations, and DHR test).
The data were collected from the patients' medical records and from the National Reference Registry for IEIs.
The ethics committee of the Faculty of Medicine and Pharmacy of Hassan II University approved this study, which was performed in accordance with the 2008 Helsinki Declaration.
An NIH-HIES score was calculated for all patients in this series for the purposes of prediagnosis.
This score was first proposed by Bodo Grimbacher in 1999 (19) and was subsequently developed by the American National Institutes of Health (NIH), using clinical and biological criteria based on IgE values (IU/ml), the number of eosinophils, the intensity of eczema, various suggestive infections (abscesses, pneumonia, pneumatocele bronchial dilation, candidiasis), and relatively specific nonimmunological signs, such as the retention of deciduous teeth, scoliosis, pathological fractures, characteristic facial features, a high-arched palate, and lymphomas.
An NIH-HIES score> 40 points indicates probable HIES, a score between 20 and 40 points is suggestive of HIES, and a score below 20 points excludes HIES.
The inclusion criteria were an NIH-HIES score ≥ 20, the classic clinical triad suggestive of HIES (recurrent skin and/or lung infections, eczema, and a high IgE level, > 10 times greater than the upper limit of the normal range for age), and a negative result in serological tests for HIV.
All patients with other types of IEIs associated with high IgE levels were excluded on the basis of clinical phenotype:Wiskott Aldrich syndrome (eczema, thrombocytopenia, autoimmunity and cancers), Omenn syndrome(neonatal generalized erythroderma, hypotrichosis, hepatosplenomegaly, lymphadenopathy), Di-George syndrome (cardiac malformation, hypoparathyroidism, hypoplasia), IPEX syndrome (enteropathy, polyendocrinopathy), and ARPC1B deficiency (hemorrhagic colitis, cutaneous vasculitis, thrombocytopenia ).
Statistical Analyses
All statistical analyses were performed with SPSS version 20 software.
Ethical Considerations
In September 2019, the Ethics Committee of the Faculty of Medicine and Pharmacy Hassan University approved the study under reference number 15/19 and written consent was obtained from the children’s parents or guardians after describing the study in detail
Results
After application of the inclusion and exclusion criteria,126 patients were included in the study.
Epidemiological characteristics
Age at presentation ranged from 3 months to 25 years.
There were slightly more male than female patients (58%), giving a sex ratio of 1.37.
Median age at symptom onset was 0.5 years [neonatal period - 15 years], and median age at clinical diagnosis was 5.5 years [3 months- 25 years].
Almost half (40.5%) the patients were born to consanguineous parents (51 patients), 24 patients (19%) reported similar cases within the family (siblings or other relatives), and 14 patients (11%) reported a history of death from the same cause within the family
Clinical findings
Almost all the patients had recurrent infections and allergies, with eczema in 66%, asthma in 25.4%, conjunctivitis in 7% and rhinitis in 6.3%.
Skin infections were identified in 39% of cases and manifested as skin abscesses (32.5%), mucocutaneous candidiasis (19%), and cold abscesses (10%).
Pulmonary infections were identified in 37.3% of cases, manifesting as pneumonia(32.5%), pneumatoceles (8%) and bronchial dilation (8%).
Ear, nose and throat (ENT) infections were observed in 27% of patients: otitis in 20%, sinusitis in 1.6%, two cases of dental abscess, and one case of parotid abscess.
Non-immunological manifestations were present in 16.6% of patients.
They included skeletal and connective tissue abnormalities (14.3%): facial dysmorphism in 10.3%, pathological fractures in 4%, retention of primary teeth in 4%, and ligamentous hyperlaxity and a high arched palate in 1.6%.
Scoliosis was detected in only one patient (0.8%).
Neurological abnormalities were highlighted in 6.3% of cases: psychomotor delay was detected in seven patients(5.6%), and cognitive delay in four patients (3.2%).
Finally, lymphadenopathy was recorded in 12 patients: inguinal(seven patients), cervical (four patients), axillary (three patients) and intraparotid (one patient).
Autoimmunity was observed in 1.6% of cases, in the form of a single case of lichenoid lupus erythematosus and one case of celiac disease.
Meningococcal meningitis was documented in 1.6% of cases.
One patient (0.8%) developed a non-Hodgkin's malignant lymphoma of the T phenotype, mycosis fungoides type.
Gastrointestinal manifestations were observed, including diarrhea (12%), splenomegaly (3.2%), rectal bleeding (2 cases), infectious colitis (one case), renal abscess (one case) and digestive bleeding (two cases).
Immunological findings
Cytobacteriological, skin (scales, pus) and ENT examinations detected several different microorganisms, including Gram+ Cocci and Gram+ and Gram- bacilli: Bacillus sp.(n=1), Citrobacter freudii (n=1), Streptococcus beta hemolytic group A (n=4), Staphylococcus aureus (n=20), Pseudomonas aeruginosa (n=2), Haemophilus influenzae (n=3), Proteus mirabilis (n=1), Proteus rettgeri (n=1), Streptococcus dysgalactiae (n=4), Candida parapsilosis (n=1), Candida dubliniensis (n=2), Enterobacter (n=1), Giardia intestinalis (n =1) and coagulase-positive Staphylococcus (n=1).
NIH-HIES score: An NIH-HIES score ≥ 20 points was required for inclusion and the mean score was 29, 6.
The patients were assigned to two groups on the basis of this score: a group of 15 patients (12%) with a score of at higher 40 points, indicating probable HIES, and a group of 111 patients (88.1%) with scores between 20 and 40 points, indicating possible HIES.
Mortality
Nine patients died (7.1%) during the study, at a median age of 5.9 years.
The causes of death were diverse: complications following respiratory infection (two cases), septicemia (one case), pulmonary hemoptysis (one case), peritonitis due to rupture of a spleen abscess (one case),and cardiorespiratory arrest (one case).
The cause of death was not reported for three patients.
The results are presented in Table 1.
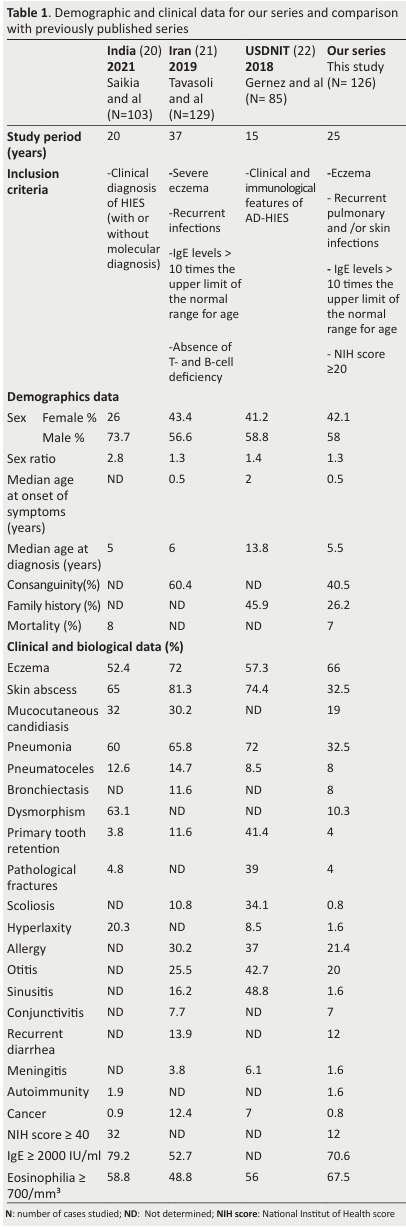
Table 1. Demographic and clinical data for our series and comparison with previously published series .
Disscussion
Until recently, IEIs were largely unknown in Morocco.
Since the creation, in 1995, of the first center specializing in the diagnosis and management of IEIs in Morocco (the Clinical Immunology Unit of Abderahim Harrouchi Ibn Rochd Children's Hospital in Casablanca), this discipline has greatly increased in importance in the country.
A few years after the creation of this first center, a large national network was established involving different specialists (pediatricians, dermatologists, neurologists, pulmonologists, psychologists) interested in IEIs working at different university hospitals, leading to the creation of the Moroccan Society for Primary Immunodeficiencies.
In this context, we investigated the particular features of HIES in Morocco.
This is the first retrospective study of HIES focusing exclusively on patients in Morocco.
We compared our cohort of 126 patients to three large published series: an Indian series (n = 103) (20), an Iranian series (n = 129) (21) in which genetic variants were studied in addition to clinical, epidemiological and immunological profiles, and a third series from USIDNET (n = 85) (22), which included patients with a phenotypic profile of AD HIES from different ethnic groups (25 US states and Quebec) for whom no molecular study was conducted.
In our series, the mean age at diagnosis was 6.9 years(median = 5.5 years), whereas the mean age at symptom onset was 1.2 years (median = 0.5 years).
This late diagnosis, with an interval between symptom onset and diagnosis of 5.7 years may be partly accounted for by the difficulty of diagnosis based solely on the classic triad of HIES, which overlaps with other diseases, without recourse to genetic studies, together with the moderate symptoms observed in patients of the ages recruited.
The lungs and skin are the most common sites of infection in patients with HIES (23).
Repeated infections were the most frequent clinical sign reported (75.4% of patients), followed by atopic diseases (73% of patients).
Pulmonary infections manifest as frequent, recurrent bacterial pneumonia, which can become complicated over time,developing into bronchiectasis or pneumatoceles (24).
In our series, such infections were present in 37.3% of patients, with pneumonia in 32.5% of cases, versus, 60% in India, 65.8% in Iran, and 72% USIDNET.
Pneumatoceles is a typical manifestation of the AD HIES form, especially that due to STAT3 (25) and IL6ST deficiencies (26, 27).
However, a significant number of patients with AR HIES forms and mutations of ZNF341 (11), IL6ST (13) or PGM3 (28) presenting pneumatoceles have been reported.
We identified pneumatoceles in 8% of patients, this value being similar to that reported for cohorts from India (12%), Iran (14.7%), and USIDNET (8.5%).
Bronchial dilation was found in 8% of our patients, a value close to that for the Iranian series (11.6%).
The patients in our series also presented skin infections in the form of abscesses (32.5%), albeit at a lower frequency than in the other cohorts considered here (65% in India,81.3% in Iran and 74.4% in USIDNET).
ENT infections should also be considered carefully, particularly if accompanied by severe eczema or growth retardation (28).
Recurrent or chronic ear infections were frequently reported in all types of HIES in several series (29–31).
Such infections were detected in 20% of our patients, versus 25.5% of Iranian patients and 42.7% of patients in the USIDNET series.
Eczema was the predominant clinical manifestation.
Typically, atopic dermatitis (AD) begins in infants with HIES during their first few months of life (32).
It is diagnosed clinically on the basis of a neonatal skin rash that develops into chronic pruritic eczema, characterized by an appearance and distribution different from that of classic AD.
Atopic dermatitis was observed in 66% of our patients ( 33), and its frequency, duration, and severity were highly variable.
Atopy was also present in the other series, with frequencies similar to that in our cohort: 72% in Iran (21), 57.3% in the USIDNET cohort (22) and 52.4% in India (20).
We identified allergies in 21.4% of our patients, versus 30.2% in India and 37% in the USIDNET) cohort, and 25.4% of our patients had asthma, versus 39% in the USIDNET cohort.
Other phenotypes were also recorded, such as meningitis, which was found in 1.6% of our patients, 3.8% of patients in Iran and 6.1% of patients in the USIDNET cohort, and autoimmunity, found in 1.6% of our patients and 1.9% of those in the Indian series.
Oral thrush, genital candidiasis, and onychomycosis are also common in HIES patients (34).
These conditions were identified in 20% of our patients, a frequency similar to those for the Indian (32%) and Iranian (30.5%) cohorts.
Gastrointestinal infections were less frequent than pulmonary and skin infections, but were nevertheless identified in our patients, manifesting as acute or chronic diarrhea in 12% of cases, a frequency close to that in the Iranian series (13.9%).
HIES is a group of heterogeneous, multisystemic immune disorders.
In addition to the classic triad found in all forms of HIES, other non-immunological features(skeletal, connective, dental, dysmorphic, neurological, or vascular abnormalities) may initially be absent or mild early in life, subsequently becoming more evident over time, which can be problematic for diagnosis.
Only 16.6% of our patients presented non-immunological abnormalities: a typical facial appearance for 10.3% of our patients, versus 63% in India.
Skeletal abnormalities were represented by scoliosis (0.8%), ligamentous hyperlaxity (1.6%), and pathological fractures (4%), all at frequencies lower than those in other series (35, 36).
We found dental abnormalities in 4% (5/126) of patients, with the retention of deciduous teeth, at a frequency similar to that in the Indian series (3.8%).
Characteristic facial features, musculoskeletal and dental abnormalities are classically observed in STAT3-HIES patients, and these features are, therefore, essential predictors for a diagnosis of AD HIES at an early stage of the disease(37)
However, some patients with deleterious ZNF341 or IL6ST or PGM3 mutations may have non-immunological clinical phenotypes similar to those observed in patients with STAT3 mutations(13).
Neurological abnormalities were identified in 6.3% of cases: psychomotor delay was detected in seven patients (5.6%), and cognitive delay in four patients (3.2%), suggesting a PGM3 deficiency.
In addition to significant immunodeficiencies, patients with PGM3 mutations also frequently exhibit dysmorphic features, skeletal anomalies, and various neurological abnormalities.
Another criterion to distinguish patients with biallelic PGM3 mutations from other HIES patients could be a reversed CD4/CD8 ratio, in addition to developmental delay(38).
Four of our patients had CD4+ lymphopenia but without any recorded neurological clinical manifestations
As its name suggests, high serum concentrations of total IgE are one of the main immunological characteristics of HIES.
IgE levels more than 10 times higher than the upper limit of the normal range for age were found in all of our patients; 70.6% (89/126) of our patients had serum IgE levels ≥ 2000 IU/ml, versus 79% (80/101) in the Indian series, and 53% in the Iranian series.
Hypereosinophilia was noted in 68% (85/126) of our patients, a frequency similar to that reported for other series.
An NIH-HIES score ≥40 was found in 12% (15/126) of the patients in our series, versus 32% (33/103) for the Iranian cohort, in which the mean NIH score was 29.6 [20 - 65],and 45.3 [10 - 69] for the Iranian series (n=129).
These differences can be explained by the choice of the age group studied, with certain symptoms, such as scoliosis, primary tooth retention and facial dysmorphism, detectable only in older patients.
Likewise, the number of infectious episodes or fractures increases with age, and it takes several years for their contribution to become evident; this results in higher NIH scores in series containing adults in addition to children (39,40).
Conclusion
We describe here, for the first time, the phenotypic profile of a large (n = 126) cohort of Moroccan patients presenting the classic triad of HIES with a high NIH-HIES score.
These patients have a broad clinical phenotype, with a wide range of immunological and extrahematopoietic abnormalities. HIES diagnosis on the basis of clinical and immunological findings alone is difficult.
Molecular analysis is required to identify the precise genetic cause of the disease.
References
- Grimbacher B, Holland SM, Gallin JI, Greenberg F, Hill SC, Malech HL, et al. Hyper-IgE Syndrome with Recurrent Infections — An Autosomal Dominant Multisystem Disorder. N Engl J Med. 1999 Mar 4;340(9):692–702. doi: 10.1056/NEJM199903043400904. [DOI] [PubMed] [Google Scholar]
- Davis SD, Schaller J, Wedgwood RJ. Job’s Syndrome. Recurrent, “cold”, staphylococcal abscesses. Lancet. 1966 May 7;1(7445):1013–1015. doi: 10.1016/s0140-6736(66)90119-x. [DOI] [PubMed] [Google Scholar]
- Bennich HH, Ishizaka K, Johansson SGO, Rowe DS, Stanworth DR, Terry WD. Immunoglobulin E. A new class of human immunoglobulin. Immunochemistry. 1968 Jul;5(4):327–328. doi: 10.1016/0019-2791(68)90128-6. [DOI] [PubMed] [Google Scholar]
- Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. Human Inborn Errors of Immunity: 2022 Update on the Classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol. 2022 Oct;42(7):1473–1507. doi: 10.1007/s10875-022-01289-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadil I, Ailal F, Béziat V, Casanova JL, Boisson B, Bousfiha AA. Hyperimmunoglobulinémie E et Déficits Immunitaires Héréditaires Hyperimmunoglobulinemia E and hereditary immune deficiencies. LA TUNISIE MEDICALE. 2023;101 [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, C Davis J, Lamborn I, Freeman A, Jing H, Favreau-Lessard A, et al. Combined Immunodeficiency Associated with DOCK8 Mutations. The New England journal of medicine. 2009 Sep 1;361:2046–2055. doi: 10.1056/NEJMoa0905506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadil I, Ben-Ali M, Jeddane L, Barbouche MR, Bousfiha AA. The Seven STAT3-Related Hyper-IgE Syndromes. J Clin Immunol. 2021 Aug;41(6):1384–1389. doi: 10.1007/s10875-021-01041-3. [DOI] [PubMed] [Google Scholar]
- Minegishi Y, Saito M, Morio T, Watanabe K, Agematsu K, Tsuchiya S. Human Tyrosine Kinase 2 Deficiency Reveals Its Requisite Roles in Multiple Cytokine Signals Involved in Innate and Acquired Immunity. Immunity. 2006 Nov;25(5):745–755. doi: 10.1016/j.immuni.2006.09.009. [DOI] [PubMed] [Google Scholar]
- Bousfiha A, Moundir A, Tangye SG, Picard C, Jeddane L, Al-Herz W. The 2022 Update of IUIS Phenotypical Classification for Human Inborn Errors of Immunity. J Clin Immunol. 2022 Oct;42(7):1508. doi: 10.1007/s10875-022-01352-z. [DOI] [PubMed] [Google Scholar]
- Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N. STAT3 Mutations in the Hyper-IgE Syndrome. N Engl J Med. 2007 Oct 18;357(16):1608–1619. doi: 10.1056/NEJMoa073687. [DOI] [PubMed] [Google Scholar]
- Béziat V, Li J, Lin JX, Ma CS, Li P, Bousfiha A. A recessive form of hyper-IgE syndrome by disruption of ZNF341-dependent STAT3 transcription and activity. Science Immunology. 2018 Jun 15;3(24):eaat4956. doi: 10.1126/sciimmunol.aat4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer S, Köstel Bal S, Egner W, Lango Allen H, Raza SI, Ma CA. Loss of the interleukin-6 receptor causes immunodeficiency, atopy, and abnormal inflammatory responses. J Exp Med. 2019 Sep 2;216(9):1986–1998. doi: 10.1084/jem.20190344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwerd T, Twigg SRF, Aschenbrenner D, Manrique S, Miller KA, Taylor IB. A biallelic mutation in IL6ST encoding the GP130 co-receptor causes immunodeficiency and craniosynostosis. J Exp Med. 2017 Sep 4;214(9):2547–2562. doi: 10.1084/jem.20161810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Yu X, Ichikawa M, Lyons JJ, Datta S, Lamborn IT. Autosomal recessive PGM3 mutations link glycosylation defects to atopy, immune deficiency, autoimmunity, and neurocognitive impairment. J Allergy Clin Immunol. 2014 May;133(5):1400–1409. doi: 10.1016/j.jaci.2014.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felgentreff K, Siepe M, Kotthoff S, von Kodolitsch Y, Schachtrup K, Notarangelo LD. Severe eczema and Hyper-IgE in Loeys Dietz-syndrome — Contribution to new findings of immune dysregulation in connective tissue disorders. Clinical Immunology. 2014 Jan 1;150(1):43–50. doi: 10.1016/j.clim.2013.11.008. [DOI] [PubMed] [Google Scholar]
- Lyons JJ, Liu Y, Ma CA, Yu X, O’Connell MP, Lawrence MG. ERBIN deficiency links STAT3 and TGF-β pathway defects with atopy in humans. J Exp Med. 2017 Mar 6;214(3):669–680. doi: 10.1084/jem.20161435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner ED, Hartl D, Rylaarsdam S, Young ML, Monaco-Shawver L, Kleiner G. Comèl-Netherton syndrome defined as primary immunodeficiency. Journal of Allergy and Clinical Immunology. 2009 Sep;124(3):536–543. doi: 10.1016/j.jaci.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorjbal B, Stinson JR, Ma CA, Weinreich MA, Miraghazadeh B, Hartberger JM. Hypomorphic caspase activation and recruitment domain 11 (CARD11) mutations associated with diverse immunologic phenotypes with or without atopic disease. Journal of Allergy and Clinical Immunology. 2019 Apr;143(4):1482–1495. doi: 10.1016/j.jaci.2018.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimbacher B, Schäffer AA, Holland SM, Davis J, Gallin JI, Malech HL. Genetic linkage of hyper-IgE syndrome to chromosome 4. Am J Hum Genet. 1999 Sep;65(3):735–744. doi: 10.1086/302547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saikia B, Rawat A, Minz RW, Suri D, Pandiarajan V, Jindal A. Clinical Profile of Hyper-IgE Syndrome in India. Front Immunol. 2021 Feb 26;12:626593. doi: 10.3389/fimmu.2021.626593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavassoli M, Abolhassani H, Yazdani R, Ghadami M, Azizi G, Abolrahim Poor Heravi S. The first cohort of Iranian patients with hyper immunoglobulin E syndrome: A long-term follow up and genetic analysis. Pediatric Allergy Immunology. 2019 Jun;30(4):469–478. doi: 10.1111/pai.13043. [DOI] [PubMed] [Google Scholar]
- Gernez Y, Freeman AF, Holland SM, Garabedian E, Patel NC, Puck JM. Autosomal Dominant Hyper-IgE Syndrome in the USIDNET Registry. The Journal of Allergy and Clinical Immunology: In Practice. 2018 May;6(3):996–1001. doi: 10.1016/j.jaip.2017.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimbacher B, Belohradsky BH, Holland SM. Immunoglobulin E in primary immunodeficiency diseases. Allergy. 2002 Nov;57(11):995–1007. doi: 10.1034/j.1398-9995.2002.02168.x. [DOI] [PubMed] [Google Scholar]
- Yong PF, Freeman AF, Engelhardt KR, Holland S, Puck JM, Grimbacher B. An update on the hyper-IgE syndromes. Arthritis Research & Therapy. 2012;14(6):228. doi: 10.1186/ar4069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogensen TH. STAT3 and the Hyper-IgE syndrome: Clinical presentation, genetic origin, pathogenesis, novel findings and remaining uncertainties. JAK-STAT. 2013 Apr;2(2):e23435. doi: 10.4161/jkst.23435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arlabosse T, Materna M, Riccio O, Schnider C, Angelini F, Perreau M, et al. New Dominant-Negative IL6ST Variants Expand the Immunological and Clinical Spectrum of GP130-Dependent Hyper IgE Syndrome. J Clin Immunol. 2023;43(7):1566–80. doi: 10.1007/s10875-023-01517-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Béziat V, Tavernier SJ, Chen YH, Ma CS, Materna M, Laurence A, et al. Dominant-negative mutations in human IL6ST underlie hyper-IgE syndrome. Journal of Experimental Medicine. 2020 Mar 24;217(6):e20191804. doi: 10.1084/jem.20191804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson NW, Hogan MB. Otitis media as a presenting complaint in childhood immunodeficiency diseases. Curr Allergy Asthma Rep. 2008 Nov;8(6):519–24. doi: 10.1007/s11882-008-0095-6. [DOI] [PubMed] [Google Scholar]
- Wu J, Chen J, Tian ZQ, Zhang H, Gong RL, Chen TX, et al. Clinical Manifestations and Genetic Analysis of 17 Patients with Autosomal Dominant Hyper-IgE Syndrome in Mainland China: New Reports and a Literature Review. Journal of Clinical Immunology. 2017 Feb;37(2):166–79. doi: 10.1007/s10875-017-0369-7. [DOI] [PubMed] [Google Scholar]
- Chandesris MO, Melki I, Natividad A, Puel A, Fieschi C, Yun L, et al. Autosomal Dominant STAT3 Deficiency and Hyper-IgE Syndrome: Molecular, Cellular, and Clinical Features From a French National Survey. Medicine (Baltimore) 2012 Jul;91(4):e1–19. doi: 10.1097/MD.0b013e31825f95b9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stray-Pedersen A, Backe PH, Sorte HS, Mørkrid L, Chokshi NY, Erichsen HC, et al. PGM3 Mutations Cause a Congenital Disorder of Glycosylation with Severe Immunodeficiency and Skeletal Dysplasia. The American Journal of Human Genetics. 2014 Jul;95(1):96–107. doi: 10.1016/j.ajhg.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gernez Y, Tsuang A, Smith TD, Shahjehan K, Hui Y, Maglione PJ, et al. Hemoptysis in a Patient with Elevated Immunoglobulin E. The Journal of Allergy and Clinical Immunology: In Practice. 2016 Nov;4(6):1054–58. doi: 10.1016/j.jaip.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Hong L, Chen TX. Clinical Manifestation of Hyper IgE Syndrome Including Otitis Media. Curr Allergy Asthma Rep. 2018 Oct;18(10):51. doi: 10.1007/s11882-018-0806-6. [DOI] [PubMed] [Google Scholar]
- Olaiwan A, Chandesris MO, Fraitag S, Lortholary O, Hermine O, Fischer A, et al. Cutaneous findings in sporadic and familial autosomal dominant hyper-IgE syndrome: A retrospective, single-center study of 21 patients diagnosed using molecular analysis. Journal of the American Academy of Dermatology. 2011 Dec;65(6):1167–72. doi: 10.1016/j.jaad.2010.09.714. [DOI] [PubMed] [Google Scholar]
- Schimke LF, Sawalle-Belohradsky J, Roesler J, Wollenberg A, Rack A, Borte M, et al. Diagnostic approach to the hyper-IgE syndromes: Immunologic and clinical key findings to differentiate hyper-IgE syndromes from atopic dermatitis. Journal of Allergy and Clinical Immunology. 2010 Sep;126(3):611–617. doi: 10.1016/j.jaci.2010.06.029. [DOI] [PubMed] [Google Scholar]
- Woellner C, Gertz EM, Schäffer AA, Lagos M, Perro M, Glocker EO, et al. Mutations in STAT3 and diagnostic guidelines for hyper IgE syndrome. Journal of Allergy and Clinical Immunology. 2010 Feb;125(2):424–432. doi: 10.1016/j.jaci.2009.10.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman AF, Holland SM. Clinical Manifestations of Hyper IgE Syndromes. Dis Markers. 2010;29(3–4):123–30. doi: 10.3233/DMA-2010-0734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Khemis L, Mekki N, Ben-Mustapha I, Rouault K, Mellouli F, Khemiri M, et al. A founder mutation underlies a severe form of phosphoglutamase 3 (PGM3) deficiency in Tunisian patients. Molecular Immunology. 2017 Oct;90:57–63. doi: 10.1016/j.molimm.2017.06.248. [DOI] [PubMed] [Google Scholar]
- Robinson WS, Arnold SR, Michael CF, Vickery JD, Schoumacher RA, Pivnick EK, et al. Case report of a young child with disseminated histoplasmosis and review of hyper immunoglobulin e syndrome (HIES). Clinical and Molecular Allergy. 2011;9(1):14. doi: 10.1186/1476-7961-9-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woellner C, Gertz EM, Schäffer AA, Lagos M, Perro M, Glocker EO, et al. Mutations in the signal transducer and activator of transcription 3 (STAT3) and diagnostic guidelines for the Hyper-IgE Syndrome. J Allergy Clin Immunol. 2010 Feb;125(2):424–432. doi: 10.1016/j.jaci.2009.10.059. [DOI] [PMC free article] [PubMed] [Google Scholar]