

Abstract
Acute myeloid leukemia (AML) derives from hematopoietic stem and progenitor cells (HSPCs). To date, no AML‐exclusive, non‐HSPC‐expressed cell‐surface target molecules for AML selective immunotherapy have been identified. Therefore, to still apply surface‐directed immunotherapy in this disease setting, time‐limited combined immune‐targeting of AML cells and healthy HSPCs, followed by hematopoietic stem cell transplantation (HSCT), might be a viable therapeutic approach. To explore this, we generated a recombinant single‐chain variable fragment‐based bispecific T‐cell engaging and activating antibody directed against CD3 on T‐cells and CD117, the surface receptor for stem cell factor, expressed by both AML cells and healthy HSPCs. Bispecific CD117xCD3 targeting induced lysis of CD117‐positive healthy human HSPCs, AML cell lines and patient‐derived AML blasts in the presence of T‐cells at subnanomolar concentrations in vitro. Furthermore, in immunocompromised mice, engrafted with human CD117‐expressing leukemia cells and human T‐cells, the bispecific molecule efficiently prevented leukemia growth in vivo. Additionally, in immunodeficient mice transplanted with healthy human HSPCs, the molecule decreased the number of CD117‐positive cells in vivo. Therefore, bispecific CD117xCD3 targeting might be developed clinically in order to reduce CD117‐expressing leukemia cells and HSPCs prior to HSCT.

INTRODUCTION
Hematopoietic stem and progenitor cells (HSPC) support lifelong hematopoiesis. 1 HSPC malignancies such as acute myeloid leukemia (AML), myelodysplastic neoplasia (MDS), and myeloproliferative neoplasia (MPN) derive from HSPCs by acquiring genetic alterations in a stepwise process. 2 , 3 Although some HSPC malignancies might present specific neoantigens in the context of their MHC, 4 , 5 there are currently no known broadly expressed surface antigens in HSPC malignancies that sufficiently distinguish healthy from malignant HSPCs. Thus, selective cell‐surface immune targeting of malignant HSPCs while sparing healthy, cell‐of‐origin HSPCs remains a yet unresolved challenge.
Indeed, in other hematologic malignancies such as B‐cell and plasma cell neoplasia, all currently successful clinical immunotherapeutic approaches, with either antibody constructs or chimeric antigen receptor (CAR) T‐cells, target cell‐surface antigens shared between neoplastic cells and their cells of origin. Targeting lineage‐specific B‐cell antigens, such as CD19, CD20, CD22, and CD79a, or plasma cell antigens, such as BCMA and GPRC5D, leads to healthy B‐ and plasma cell depletion, respectively. This collateral damage is clinically tolerated, and immunoglobulin substitution can temporarily compensate for B‐cell and plasma cell functions. 6 Furthermore, once immunotherapy against B‐cells and plasma cells is terminated, healthy target cells can re‐emerge from HSPCs.
We and others have hypothesized that selectively targeting a common surface antigen present on both neoplastic HSPCs and their healthy counterparts within a defined timeframe could be a viable approach. 7 It could potentially achieve the dual purpose of reducing or eliminating neoplastic cell growth and simultaneously freeing the HSPC niche, thereby serving as a preconditioning strategy. This, in turn would, upon termination of immunotherapy, enable subsequent healthy hematopoietic stem cell transplantation (HSCT) in order to rescue the hematopoietic system. 8 , 9 The target antigen for such an approach might be CD117 (c‐Kit), the healthy HSPC and HSPC malignancy expressed transmembrane tyrosine kinase and receptor for stem cell factor. 7 , 10 , 11 CD117 might be a particularly well‐suited target due to the in the hematopoietic‐system‐restricted expression in HSPCs and mast cells. 12 , 13 , 14 , 15 Additionally, CD117 signaling is vital for maintaining healthy HSPCs and is likely to be also important for the survival of malignant HSPC, possibly reducing the risk of immune escape by receptor loss under selective therapeutic pressure. 16 , 17
Bispecific antibodies or antibody‐derived constructs, capable of redirecting T‐cell activity in a TCR‐MHC‐independent manner against target cells, represent a potent class of antibody products that can achieve selective biocidal action in vivo. 18 , 19 , 20 T‐cell‐engaging bispecific antibodies (TCEs) can induce lysis of target cells at sub‐nanomolar concentrations in the presence of T‐cells. 21 The bispecific antibody product blinatumomab, which simultaneously binds human CD19 and CD3 antigens, is the first‐in‐class product to receive marketing authorization for the treatment of B‐cell precursor acute lymphoblastic leukemia [ALL; Kantarjian et al. 22 ]. Blinatumomab is a Bispecific T‐cell engager (BiTE™), a recombinant antibody‐construct produced by sequential fusion of two single‐chain Fv antibody fragments. 23 We reasoned that similar TCE binding to CD117 and CD3 might be an effective means to selectively eliminate CD117‐expressing healthy and malignant/diseased HSPCs within a controlled, limited timeframe prior to subsequent transplantation of allogeneic (or possibly genetically modified autologous) CD117‐expressing HSPCs. We here describe the generation and characterization of a tandem arrangement of two scFv antibody fragments, specific to CD117 and to CD3, resulting in a T‐cell engager (TCE), which we will subsequently call for simplicity “CD117xCD3 TCE.” We demonstrate that the novel TCE only activates T‐cells in target‐antigen presence and efficiently directs T‐cell biocidal activity toward CD117‐expressing human HSPCs, AML cell lines, and primary human AML cells in vitro and in xenogeneic immunodeficient mouse models in vivo.
MATERIALS AND METHODS
Generation of the CD117xCD3 bispecific T‐cell engaging and activating antibody‐construct
We constructed the CD117xCD3 TCE from the sequences of the anti‐human CD117 (c‐Kit) antibody 79D 24 and the anti‐human CD3 antibody OKT3 from blinatumomab. 23 It was genetically assembled by successive overlap PCR in the order VL79D‐Linker15aa‐VH79D‐Linker5aa‐VHOKT3‐Linker18aa‐VLOKT3 with or without a C‐terminal His6 tag, indicated as CD117xCD3 TCE His‐tag or CD117xCD3 TCE, respectively.
Protein expression and purification
We produced CD117xCD3 TCE His‐tag using transient gene expression (TGE) in Chinese hamster ovary cells (CHO‐S, RRID: CVCL_7183), and CD117xCD3 TCE using a stably transfected CHO clonal cell line. In both cases, we purified the product from the supernatant using protein A affinity chromatography and then exchanged it with phosphate buffer. The quality of the protein was assessed by SDS‐PAGE under nonreducing and reducing conditions and size exclusion chromatography using a Superdex 200 Increased 10/300GL column on an Äkta Pure FPLC system (both Cytiva). LC‐MS was performed on a Waters Xevo G2‐XS QTOF instrument coupled to a Waters Acquity UPLC H‐Class 147 System using a 2.1 × 50 mm Acquity BEH300 C4 1.7 μm column (Waters).
In vitro cytotoxicity assays
The leukemia cell lines (MOLM14, HL‐60, and Kasumi‐1) were maintained in the appropriate culture medium until the killing assay was established, whereas primary mononuclear cells (MNCs) or CD3+/CD19+ double‐depleted peripheral blood (PB) cells were thawed on the same day as the assay setup. Healthy donor apheresis samples and AML patient cells were obtained from the Department of Medical Oncology and Hematology Biobank, University Hospital Zürich, Zürich, Switzerland, with written informed patient consent. The study was conducted in accordance with the Declaration of Helsinki and approved by the Cantonal Ethics Board of Zürich, Switzerland (2009‐0062). Healthy donor bone marrow MNCs were purchased from STEMCELL (catalog no. 7001.2) and thawed on the day of the experiment. The flow cytometric cytotoxicity assay was adapted from previously published protocols 25 and was used to quantify T‐cell cytotoxicity. We co‐cultured T‐cells and target cells at the indicated effector‐to‐target ratios in T‐cell medium comprising advanced RPMI media supplemented with 10% FBS, penicillin/streptomycin (100 U/mL/100 µg/mL; Gibco, Thermo Fisher Scientific), and 1× Glutamax™ (Gibco, Thermo Fisher Scientific, catalog no. 35 050 061). CD117xCD3 TCE was diluted in the medium and added at the specified concentrations. At the indicated time points, cell lysis was assessed by flow cytometry using a BD LSR Fortessa II cell analyzer (Becton Dickinson). Antibodies used in this study are indicated in Supporting Information S1: Table 1. We calculated the percentage of specific cell lysis using the following formula 26 :
In vivo studies in xenogeneic mouse models
All procedures involving experimental animals were performed according to Swiss Animal Welfare Laws and Regulations (licenses 134/2022 and 004/2018). NSG (RRID: IMSR_JAX:005557) and 129/Sv (RRID: IMSR_JAX:009104) mice were purchased from Charles River (Germany) and maintained at our local animal facility. Healthy male and female mice 6–9 weeks old were selected for in vivo experiments. For pharmacokinetic studies, we intravenously (i.v.) administered 10–25 μg of bispecific antibody. We collected blood samples at the indicated time points to determine the antibody concentrations. For safety studies, we engrafted NSG mice with 1.4 × 107 peripheral blood mononuclear cells (PBMCs), and on the next day, 50 μg CD117xCD3 TCE was injected intraperitoneally (i.p.). Mouse bone marrow (BM), PB, and spleen were analyzed by flow cytometry for T‐cell activation, and serum was collected for cytokine analysis. For therapeutic studies, sub‐lethally irradiated NSG mice were intravenously injected with 105 MOLM14 CD117high GFP+Luc+ cells. The next day and on Day 7, 107 purified expanded T‐cells isolated from healthy donors or AML patients in remission were i.v. injected. Starting from Day 1 until the end of the experiment, we administered CD117xCD3 TCE i.p. (12.5 μg) twice daily every 12 h. Engraftment levels were monitored weekly using bioluminescence imaging (IVIS), and at the study endpoint, we performed flow cytometry analysis of the BM, PB, and spleen. BM cells from one femur were resuspended in 1 mL of FACS buffer in a single‐cell suspension, and 200 μL were analyzed by flow cytometry after the addition of counting beads (Biolegend). Single‐cell suspensions of spleens (1 mL) and 150 μL of blood were obtained after incubation in red blood cell lysis buffer (Biolegend). In therapeutic studies against primary AML, sublethally irradiated NSG mice received 5 × 106 CD3/CD19 double‐depleted AML blasts intravenously. After 7 days, 107 unexpanded healthy‐donor‐derived T‐cells were injected i.v., and mice received 12.5 μg CD117xCD3 TCE i.p. every 12 h. After 10 days, the experiment was terminated, and PB and BM were processed for flow cytometry as previously described. For xenograft models of healthy human hematopoiesis, adult donor‐mobilized PB CD34+ cells were engrafted in NSG mice, following established protocols. 27 Nine weeks postengraftment, as confirmed by PB flow cytometry, 107 T‐cells from the same apheresis donor were administered in the absence of preconditioning therapy, with or without CD117xCD3 TCE (12.5 μg b.i.d.). The study was terminated at Week 11 after 10 days of treatment, and PB and BM were analyzed by flow cytometry.
Statistical analysis
Unless otherwise noted, data are reported as mean ± standard deviation (SD). Statistical analyses were conducted using GraphPad Prism (version 10) using one‐way analysis of variance (ANOVA) with Šídák's multiple comparisons test or two‐way ANOVA followed by Tukey's post hoc test for multiple comparisons (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
RESULTS
CD117 antigen expression on primary AML samples at diagnosis and on healthy BM HSPCs and mast cells
We analyzed CD117 target antigen expression on the most recently diagnosed and coherently with the same diagnostic workflow evaluated AML patients (n = 69) at the Medical Oncology and Hematology department of our institution with the same clinical diagnostic analytic workflow. Background data on AML samples is provided in Supporting Information S1: Table 2. We further analyzed expression of CD117 on healthy bone marrow samples (n = 10) from our institution and on MOLM14 cells with the same diagnostic workflow. We present the respective data in Figure 1. Specifically, we show the representative flow cytometry analysis gating strategy for analyzing CD117 expression on healthy donor BM mast cells, B‐cell progenitors, and HSPCs by flow cytometry (Figure 1A) and the representative gating strategy to analyze CD117 expression on AML blasts from BM aspirate and PB samples (Figure 1B). Sequential gating includes time gating, doublet exclusion, and debris removal. CD15+ granulocytes and CD19+ cells are excluded before gating CD45dim blast cells based on CD45 expression and SSC. The CD45dim blast population was subsequently divided into two sub‐populations based on CD45 expression: CD45dim,higher and CD45dim,lower. The following figure panels (Figure 1C–I) illustrate comparative analyses of CD117 mean fluorescence intensity (MFI) on different cell types, beginning with a comparison of CD117 expression on PB versus BM AML blasts from different patients (total n = 69, PB n = 18; BM n = 51) in Figure 1C. We then examined CD117 expression by gating on the CD45 higher or lower blast populations (Figure 1D,E). In Figure 1F, AML CD117 expression is contrasted with healthy BM HSPCs and mast cells, as well as various AML cell lines with varying CD117 levels. Additionally, Figure 1G analyzes CD117 expression based on AML NPM1 and FLT3 mutational status, while Figure 1H relates it to the AML European LeukemiaNet (ELN 2017) classification. Finally, we correlate CD117 expression with the co‐analyzed surface antigens CD34, CD38, and CD33 (Figure 1I). In sum, the data shows broadly varying CD117 expression on blasts from AML patients, while the range of expression in healthy BM cells is rather small. The average expression of AML blasts and healthy HSPCs is similar and lies within the range of HL60 and MOLM14 low CD117 expressing cell lines. In the rather small AML cohort analyzed, we find no statistically significant association of CD117 expression with either NPM1/FLT3 mutational status or ELN 2017 classification. In the correlative analysis of surface marker expression, we find a weak but significant positive correlation between CD117 and CD34 and CD38 expression, while there is a weak negative correlation between CD117 and CD33 expression.
Figure 1.

CD117 antigen expression on primary AML samples at diagnosis, on healthy BM HSPCs and mast cells, and on CD117 expressing MOLM cells. (A) Representative gating strategy for analyzing CD117 expression on healthy donor BM mast cells, B‐cell progenitors, and HSPCs by flow cytometry. Data clean‐up includes time gating, doublet exclusion, and debris removal. A CD45/SSC plot is used to discriminate CD45dim cells. After excluding B‐cell progenitors (CD45dimCD19+CD34+/−), cells were further discriminated into CD45dimCD34+CD38− HSPCs and more differentiated CD45dimCD34+CD38+ HSPCs. Mast cell and HSPC gating presented as large dots. (B) Representative gating strategy to analyze CD117 expression on AML blasts from bone marrow (BM) aspirate (top) and peripheral blood (PB) samples (bottom) from two AML patients at diagnosis. Sequential gating includes time gating, doublet exclusion, and debris removal. CD15+ granulocytes and CD19+ cells are excluded before gating CD45dim blast cells based on CD45 expression and SSC. The CD45dim blast population was subsequently divided into two sub‐populations, based on CD45 expression: CD45dim,higher and CD45dim,lower. (C) CD117 median fluorescence intensity (MFI) with interquartile range on total CD45dim blasts in AML patient samples (total n = 69) collected from PB (n = 18) and BM (n = 51). Statistical analysis was conducted using an unpaired t‐test. (D) Median CD117 expression and interquartile range on CD45dim,higher and CD45dim,lower blast populations from AML patient samples. Statistical analysis was conducted using paired t‐test. (E) Within the same AML sample, CD117 MFI was compared between CD45dim,higher and CD45dim,lower populations. A total of 23 patients had rather higher CD117 MFI on CD45dim,lower cells (left), while 46 patients had higher or similar CD117 MFI on CD45dim,higher cells (right). (F) Median CD117 expression and interquartile range on malignant cells (Kasumi‐1, HL60 CD117 low, HL60 CD117 high, MOLM14 CD117 low, MOLM14 CD117 high, AML patient CD45dim blasts) and on healthy BM populations (B‐cell progenitors, CD34+CD38− HSPCs, CD34+CD38+ HSPCs and mast cells) (n = 11 healthy BM samples). Dashed horizontal lines indicate the CD117 MFI of MOLM14 CD117 low and MOLM14 CD117 high cells, measured using the same machine and settings as the patient samples and healthy donor BM samples. (G, H) CD117 MFI and interquartile range according to the mutational status of NPM1 and FLT3 in AML patient cells (G) and ELN Risk group 2017 (H). Statistical analysis was conducted using one‐way ANOVA with Tukey's multiple comparisons test. (I) Calculated degree of linear correlation (Pearson's r) from the log10 transformed MFI and its statistical probability (p‐value) to assess the relationship between the CD117 MFI and CD34 MFI (left), CD38 MFI (middle), and CD33 MFI (right) on total CD45dim blast populations from AML patients. (C–I) AML sample 1, used for both in vitro and in vivo experiments, was denoted in red.
Cloning, expression, and characterization of CD117xCD3 TCE
Figure 2A,B depicts the schematic structure of the vector and CD117xCD3 TCE, which was expressed in CHO cells. The chosen tandem scFv format featured a VL‐VH‐VH‐VL arrangement for the variable antibody domains. We used the 79D antibody for CD117 recognition, 24 while we chose the OKT3 moiety for CD3 targeting, as this scFv fragment has been successfully used for blinatumomab. 23 We used a short Gly4Ser linker to connect the two scFv fragments in order to yield a tandem scFv product with minimal amounts of oligomer (e.g., TandAb) formation. 28 The complete amino acid sequence is shown in Supporting Information S1: Figure 1A. The fusion protein was expressed in CHO cells by stable gene expression 29 and purified to homogeneity by Protein A chromatography, as the VH79D domain belongs to the VH3 family, followed by a hydroxyapatite polishing step. Mass spectrometric analysis revealed a main peak, whose mass (54,090 Da) was larger than the theoretical one (52,652 Da) due to predicted N‐linked glycosylation at position N154, confirmed by digestion with PNGase F enzyme (New England BioLabs), as illustrated in Figure 2C. A single band was visible by SDS‐PAGE electrophoresis (Figure 2D). The TCE was produced in various batches. Purity upon production was calculated to be >90% based on size‐exclusion chromatography analysis (representative example in Figure 2E). The CD117xCD3 TCE bound with high affinity to recombinant extracellular preparations of CD117 (Figure 2F) and CD3 (Figure 2G), as revealed by BIAcore analysis. 30 At the beginning of our studies, we generated an analogous TCE construct displaying a C‐terminal His‐tag by transient gene expression in CHO cells. In subsequent continuation of the project, we removed the His‐tag (as it was not essential for purification). We indicate throughout the manuscript in which experiments we used the TCE molecule with the His‐tag. For completeness, we show the characterization of the TCE with His‐tag in Supporting Information S1: Figure 1B–F. We tested the serum half‐life of CD117xCD3 TCE in mice upon i.v. injection, using two independent methods. We either injected the unmodified CD117xCD3 TCE or the 125I‐radiolabeled protein version. We then measured antibody levels in serum by ELISA (Figure 2H) or in whole blood by detecting the radioactivity with a gamma counter (Supporting Information S1: Figure 1G), respectively. Both methods revealed a serum half‐life of about 30 min (26.8 and 28 min, respectively). We also tested the serum half‐life of CD117xCD3 TCE upon i.p. injection (Figure 2I) as this application route in our hands is the most feasible for multiple repetitive injections in respective in vivo experiments. While the peak concentration occurred with delay and was lower compared to i.v. injection, the measured serum half‐life remained about the same.
Figure 2.

Cloning, expression, and characterization of the bispecific CD117xCD3 TCE. (A) Schematic representation of the arrangement of the CD117xCD3 TCE. The anti‐CD117 antibody 79D 24 was cloned in the chain order VL79D‐Linker‐VH79D and genetically fused to the anti‐CD3 antibody OKT3 in the order VHOKT3−VLOKT3 without a C‐terminal hexa histidine‐tag. (B) Schematic representation of the CD117xCD3 TCE. The N‐terminus of the fusion protein is indicated as N’. (C) Mass spectrometric analysis of the purified product, native and treated with PNGase, showed a product of 54,090 and 52,642 Da, indicating a single N‐glycosylation of 1448 Da, thus confirming the theoretical weight of 52,652 Da. (D) Purified CD117xCD3 TCE exhibited migration at the expected (monomeric) size of ~55 kDa in SDS‐PAGE gel. M, Marker, NR, non‐reducing conditions; R, reducing conditions. (E) Representative size exclusion chromatography profile of the CD117xCD3 TCE. (F) Binding of the CD117xCD3 TCE to recombinant human target antigen CD117 in a Biacore experiment at the indicated concentrations. (G) Binding of the CD117xCD3 TCE to recombinant human target antigen CD3 in a Biacore experiment at the indicated concentrations. (H, I) Quantitative pharmacokinetic study of CD117xCD3 TCE. Mice were injected i.v. (H) and i.p. (I) with 25 μg of CD117xCD3 TCE. Blood was taken at different time points and serum concentrations of CD117xCD3 TCE were measured by ELISA (mean ± SD from n = 2 mice per time‐point, analyzed in duplicate wells). Half‐life (t 1/2) and the highest concentration in blood (C max) are reported on the graph.
CD117xCD3 TCE induces T‐cell mediated lysis of CD117+ but not CD117− HeLa cells in vitro
To test and visualize if CD117xCD3 TCE recruits and activates T‐cells against CD117+ target cells, we artificially expressed the truncated form of CD117 on adherent HeLa cells and then co‐incubated them with T‐cells and CD117xCD3 TCE His‐tag. The representative 34.16 h time‐lapse video of CD117+ versus CD117− HeLa cells, co‐incubated with T‐cells and CD117xCD3 TCE His‐tag, demonstrated efficient T‐cell‐mediated lysis of CD117+ HeLa cells, while CD117− HeLa cells continued to grow without being lysed by T‐cells (Supporting Information S1: Video 1).
CD117xCD3 TCE activates T‐cells only in presence of CD117‐expressing target cells in vitro and in vivo
To first evaluate if CD117xCD3 TCE might induce T‐cell activation in the absence of CD117‐target cells, we incubated healthy donor‐derived PBMCs (which do not contain relevant amounts of CD117‐positive cells) with CD117xCD3 TCE for 72 h in the presence and absence of MOLM14 cells, either negative for CD117 or transduced to express CD117 at high levels (Figure 1F and Supporting Information S1: Figure 2). The ratio of PBMCs to MOLM14 target cells was set up as 1:1, leading to an effector T‐cell to MOLM14 target cell ratio of about 1:3 in disfavor of effector T‐cells (Figure 3A). To ensure correct read‐out with respect to CD117 flow‐cytometry detection, we excluded potential cross‐reactivity/hindrance of CD117 binding by the CD117xCD3 TCE (79D clone for CD117 binding) and the anti‐CD117 antibody used for CD117 detection by flow cytometry (104D2 clone) in Supporting Information S1: Figure 3. Over time, significant specific lysis of MOLM14 CD117+ cells was induced by the TCE, while this was not the case for MOLM14 CD117− cells (Figure 3B). This was paralleled by control of MOLM14 CD117+ cell growth in presence of the TCE, while both MOLM CD117+ cells in absence of TCE as well as MOLM14 CD117− cells (+/− TCE) continued to grow. T‐cells only expanded upon exposure to both MOLM14 CD117+ cells and TCE (Figure 3C). Similarly, only the presence of both MOLM14 CD117+ cells and TCE induced measurable IFN‐γ and IL‐2 cytokine release (Figure 3D) and T‐cell proliferation, as determined by FarRed dye dilution (Figure 3E). Furthermore, only presence of MOLM14 CD117+ cells and TCE induced full phenotypic T‐cell activation as determined by T‐cell surface expression of the activation markers CD69 and CD25 (Figure 3E,F).
Figure 3.

CD117xCD3 TCE elicits T‐cell proliferation, IFN‐γ and IL‐2 cytokine secretion, and immune‐phenotypic activation in presence but not in absence of CD117‐expressing target cells. (A) Graph depicting average absolute cell counts at the start (time zero) of the in vitro co‐culture experiment. Peripheral blood mononuclear cells (PBMCs) isolated from three healthy donors were combined with MOLM cells at a 1:1 ratio, resulting in a 1:3 between T‐cells within the PBMC fraction to MOLM cells. (B) Time‐dependent (24, 48, and 72 h) specific lysis of MOLM14 GFP+CD117high and control MOLM14 WT (CD117neg) cells, induced by addition of 1000 ng/mL CD117xCD3 TCE in combination with PBMCs. Data are shown as mean ± SD from duplicates of each donor. Statistical analysis was conducted using two‐way analysis of variance (ANOVA) with Tukey's multiple comparisons test; *p < 0.5, **p < 0.01. (C) Absolute count of the MOLM14 WT and MOLM14 GFP+CD117high cells (left panel) and T‐cells (right panel) in the presence and absence of TCE. Statistical analysis was conducted using two‐way ANOVA with Tukey's multiple comparisons test; *p < 0.05, **p < 0.01. (D) IFN‐γ and IL‐2 concentrations in the supernatants of co‐cultures at 48 h shown in (A–C). Data are shown as mean from duplicates of each donor ±SD. Statistical analysis conducted with two‐way ANOVA with Tukey's multiple comparison test; ****p < 0.0001. (E) Representative example of pre‐gated CD3+ T‐cell proliferation within the PBMC fraction at 48 h in the indicated conditions by means of CellVue Far‐Red dye dilution. (F) Representative flow cytometry plots at 48 h of co‐culture, gated on the CD4+ and CD8+ T‐cell subsets, showing the activation markers CD69 and CD25 expression on the respective T‐cell populations. (G) Quantification of T‐cell activation status at indicated time points (0, 24, 48, 72 h) highlighting the relative proportions of CD69+/− and CD25+/− CD4+ T‐cells and CD8+ T‐cells.
We also evaluated in vivo T‐cell activation in the absence of CD117 target cells, following administration of CD117xCD3 TCE, which is not cross‐reactive with the homolog mouse antigens. 24 , 31 The experimental outline is shown in Supporting Information S1: Figure 4. We injected NSG mice i.v. with 1.4 × 107 healthy donor PBMCs. The following day, mice were injected with either a bispecific antibody (50 µg, i.p.) or saline solution. Consistent with the results from the in vitro experiments, we observed a marginal increase in CD69 but no CD25 expression on human CD3‐positive cells upon CD117xCD3 TCE application, and no relevant cytokine release (Supporting Information S1: Figure 4B–D).
In summary, these experiments provide evidence that the newly generated CD117xCD3 TCE in combination with T‐cells leads to T‐cell activation and lysis of target‐antigen expressing cells. Importantly, the data also demonstrates that in absence of target antigen‐expressing cells, no relevant T‐cell activation and target cell lysis was observed (even in this allogeneic cell culture setting), indicating CD117 target‐antigen restricted activity of the CD117xCD3 TCE in this in vitro short‐term experimental setting. Furthermore, short‐term xenogeneic in vivo experiment adds additional evidence that T‐cell activation by CD117xCD3 TCE does not occur in absence of target antigen.
CD117xCD3 TCE elicits autologous T‐cell‐mediated lysis of CD117+ hematopoietic stem and progenitor cells in vitro
We next investigated the efficacy of CD117xCD3 TCE in eliciting autologous T‐cell‐mediated lysis of hematopoietic stem and progenitor cells by co‐incubating the TCE with bone marrow mononuclear cells from three healthy donors for up to 72 h (experimental outline given in Figure 4A). CD117xCD3 TCE efficiently induced T cell‐mediated depletion of HSPCs (identified by the HSC enrichment markers CD45dimCD34+CD117+), while sparing the CD117 negative populations (Figure 4B). Quantification of the specific lysis showed that 78.2% of target CD45dimCD34+CD117+ HSPCs were already lysed within 24 hours, reaching 94.8% by 72 h (Figure 4C). Additionally, supernatant analysis demonstrated a significant increase in IFN‐γ levels when cells were exposed to CD117xCD3 TCE (Figure 4D). The lysis of HSPCs and secretion of pro‐inflammatory cytokines correlated positively with T‐cell activation, evidenced by increased expression of activation markers CD69 and CD25 (Figure 4E). A similar experiment using mobilized HSPCs is shown in Supporting Information S1: Figure 5, which further confirms our findings.
Figure 4.

CD117xCD3 TCE mediates T‐cell cytotoxicity against healthy HSPCs. (A) Outline of the experimental setup. Human healthy‐donor‐derived BM mononuclear cells (MNCs) were purchased as frozen material and cultured in vitro in the presence or absence of CD117xCD3 TCE for up to 72 h. Panel created using Biorender.com. (B) Representative flow cytometry plots showing healthy donor bone marrow mononuclear cells, co‐cultured with and without 1000 ng/mL CD117xCD3 TCE after 48 h of culture. The gating strategies used to analyze target cells are denoted by the arrows, and the relative frequencies of cells within each plot are shown as percentages. (C) Quantification of percentage specific lysis of CD45dimCD34+CD117+ HSPCs at indicated time‐points. Three healthy bone marrow donors were plated in triplicate wells (mean ± SD). (D) Quantification of IFN‐γ in the supernatants at indicated time points. Data from three healthy donors (as in panel C), analysis performed in triplicate (mean ± SD). Statistical analysis was performed by one‐way ANOVA with Šidák's multiple comparisons test (*p < 0.05, **p < 0.01). (E) Representative flow cytometry plots showing healthy donor BM MNCs pre‐gated on CD45 high, co‐cultured with and without 1000 ng/mL CD117xCD3 TCE for 48 h. Arrows indicate the subsequent gating sequences for CD3+ effector cell analysis; the relative proportions of cells within a plot are indicated as percent.
CD117xCD3 TCE induces T‐cell mediated lysis of CD117+ AML cell lines in vitro
To evaluate the biocidal properties of CD117xCD3 TCE, we used three AML cell lines. We previously engineered the AML cell line HL60 to express the dual reporter, GFP‐Luciferase, and to display different levels of CD117 on its surface. 25 Similarly, we lentivirally transduced MOLM14 AML cells to express GFP‐luciferase and various levels of CD117 (Supporting Information S1: Figure 2A). Subsequently, we investigated the lytic activity of T‐cells elicited by CD117xCD3 TCE against MOLM14‐CD117high GFP+Luc+ cells at an effector‐to‐target ratio of 1:1 (E:T = 1:1). Cytotoxicity was exclusively observed when cells were co‐cultured with CD117xCD3 TCE as exemplified in the flow cytometry plots in Figure 5A. CD117xCD3 TCE induced phenotypic T‐cell activation, illustrated by CD69 and CD25 expression (Figure 5B), T‐cell mediated target cell lysis (Figure 5C), as well as IFN‐γ release in the culture supernatant (Figure 5D). Target cell lysis was positively correlated with CD117 target antigen expression on AML cell lines, with culture time, and with CD117xCD3 TCE concentration, reaching a maximum at 1000 ng/mL (Figure 5C,D). However, at the highest CD117xCD3 TCE concentration used (10,000 ng/mL), we measured a slight decrease of all induced activities, likely due to target antigen saturation on both effector and target cells by excess TCE, thus leading to loss of effective bridging activity, a phenomenon which has been described in multiple studies by others 32 , 33 and also by us. 34 Analysis of total numbers of target and effector cells revealed that, while target cells were already reduced compared to controls at 24 h, significant T‐cell proliferation was only observed at 72 h of culture (Figure 5E).
Figure 5.
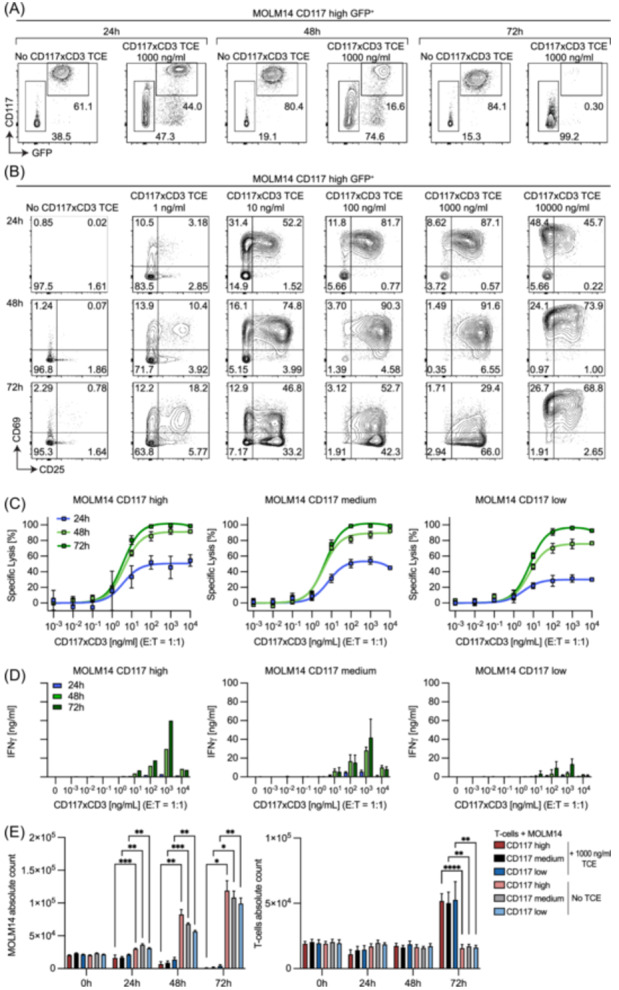
CD117xCD3 TCE elicits T‐cell cytotoxicity against human MOLM14 AML cells in a TCE concentration‐dependent, target antigen‐density‐dependent and time‐dependent manner. (A) Representative flow cytometry analyses at three subsequent time points (24, 48, 72 h), showing the relative proportions of MOLM14 CD117high GFP‐positive target cells and healthy donor‐derived T‐cells. Conditions with and without 1000 ng/mL CD117xCD3 TCE added into the co‐culture were compared as indicated. (B) Representative flow cytometry analyses of T‐cell activation at indicated time points (24, 48, 72 h), showing the relative proportions of CD69+/− and CD25+/− T‐cells (pre‐gated on CD3+ cells). Indicated concentrations of CD117xCD3 TCE were compared to conditions with no CD117xCD3 TCE added. (C) Quantification of percentage specific lysis of MOLM14 CD117high, CD117medium, and CD117low cells as a function of CD117xCD3 TCE concentration added to co‐cultures. Data from three T‐cell donors, experiment performed in duplicate wells (mean ± SD). (D) Quantification of IFN‐γ in the supernatants of co‐cultures from (C). (E) Absolute cell counts of MOLM14 cells (expressing various CD117 levels as indicated, left panel) and of effector T‐cells (right panel) in presence or absence of 1000 ng/mL TCE. Statistical analysis was conducted using two‐way ANOVA with Tukey's multiple comparisons test; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
When using CD117xCD3 TCE with and without terminal His‐tag we detected no difference in eliciting T‐cell‐mediated lysis against MOLM14‐CD117high cells as well as activating T‐cells at 24, 48, and 72 h (Supporting Information S1: Figure 6A,B). Expectedly, the efficacy of CD117xCD3 TCE was also dependent on the E:T ratio, as indicated in Supporting Information S1: Figure 7, where E:T ratios ranged from 1:20 to 20:1. To test complete tumor eradication, we performed long‐term in vitro co‐culture experiments over 120 h, and spiked T‐cells ± CD117xCD3 TCE for a second time at 72 h, which enhanced lytic activity against target cells (Supporting Information S1: Figure 8). To further test CD117xCD3 TCE in another AML cell line, we performed analogous experiments using HL60 cells, which we transduced to express CD117 (Supporting Information S1: Figure 9AB). 25 These experiments confirmed the dose‐response behavior, modulated according to antigen density as well as the E:T ratio. Interestingly, the addition of recombinant IL‐2 to the co‐culture as an exogenous T‐cell activation signal did not substantially enhance target cell lysis in this setting (Supporting Information S1: Figure 9C). Finally, we tested CD117xCD3 TCE‐mediated T‐cell activation and target cell lysis on a third AML cell line (Kasumi‐1), which endogenously expresses CD117. The in vitro results confirmed time and TCE concentration‐dependent target cell lysis (Supporting Information S1: Figure 10). In summary, these experiments indicate that CD117xCD3 TCE efficiently induces T‐cell mediated lysis of CD117+ AML cell lines in vitro.
Efficacy and dynamics of CD117xCD3 TCE and T‐cell versus direct anti‐CD117 CAR T‐cell response against target cells in vitro
We then compared the lytic efficacy of CD117xCD3 TCE in combination with T‐cells to the one of anti‐CD117 CAR T‐cells, previously described. 25 Lysis of MOLM14 CD117high cells was significantly more efficient after 24 h with anti‐CD117 CAR T cells, whereas after 48 h, lysis rates were comparable (Figure 6A). To study effector‐target‐cell interaction dynamics, we established time‐lapse imaging with CD117xCD3 TCE in combination with T‐cells and MACS‐purified anti‐CD117 CAR T‐cells, 25 both derived from the same T‐cell donor (Figure 6B). We were able to successfully track lysis of MOLM14 CD117high cells with anti‐CD117 CAR‐T cells as well as CD117xCD3 TCE in combination with T‐cells, while in the absence of CD117xCD3 TEA, no target cell lysis was observed (Figure 6C; Supporting Information S1: Video 2‐4). Quantification of >170 individual wells revealed almost complete attachment of effector cells to target cells within 24 h in both the direct anti‐CD117 CAR T‐cell and the CD117xCD3 TCE and T‐cell combination. However, the CD117xCD3 TCE and T‐cell combination showed faster recruitment to target cells compared to CAR T‐cells, possibly reflecting a quantitatively higher CD3 than CAR molecule expression in the respective effector cells in a fully saturated antigen setting (Figure 6D). Most importantly, after individual target cell attachment, anti‐CD117 CAR T‐cells lysed target cells more potently than CD117xCD3 TCE with T‐cells, and the lysis of multiple target cells was more frequently observed by anti‐CD117 CAR T‐cells compared to the CD117xCD3 TCE approach (Figure 6E, left panel). However, when analyzing only the successful killing events with either anti‐CD117 CAR T‐cells or CD117xCD3 TCE with T‐cells, we observed no differences in time from attachment to killing (Figure 6E, right panel), demonstrating comparable target‐cell lysis dynamics between anti‐CD117 CAR T‐cells and CD117xCD TCE with T‐cells. However, the efficacy of lysis at the single‐cell level was higher with anti‐CD117 CAR T‐cells compared with CD117xCD3 TCE and T‐cells, possibly due to the presence of co‐stimulatory molecule signaling in CAR T‐cells (4‐1BB), which is absent in the CD117xCD3 TCE and T‐cell setting.
Figure 6.

Comparison of lysis dynamics between anti‐CD117 CAR‐T cells and CD117xCD3 TCE and T‐cells using time‐lapse imaging. (A) Percentage of specific lysis of MOLM14 GFP+ CD117high cells after 24 and 48 h in co‐culture with anti‐CD117 CAR T‐cells or T‐cells from two matched donors plus 1000 ng/mL CD117xCD3 TCE. Cells were cocultured at an E:T = 1:1, two independent experiments from two CAR T‐cell and T‐cell matched healthy donors are shown. (B) Workflow of the time‐lapse imaging pipeline to study effector‐target cell interactions. Cells were co‐cultured at an E:T = 1:1 for up to 72 h on microgrids glued to chamber slides to allow for quantification of single attachment and lysis events. After meeting standard criteria (i.e., appropriate movie quality allowing for cell traceability and high cell viability of target and effector cells over the course of imaging) individual time‐lapse movies in which both target and effector cells were successfully plated in an E:T‐ratio of 3:1 to 1:3 were chosen for final analysis. Panel created using Biorender.com (C) Representative time‐lapse images of MOLM14 GFP+ CD117high cells (grey) co‐incubated with direct anti‐CD117 CAR T‐cells or T‐cells (effector cells in red) with CD117xCD3 TCE (1000 ng/mL). Target cell lysis was marked by propidium iodide (PI) influx (green). Time to PI influx was further divided into two components: the time to sustained effector‐target cell attachment and time from attachment to target cell lysis. (D) Effector‐target‐cell interactions were analyzed and quantified in >170 independent wells using time‐lapse imaging and experimental data were pooled from two different matched healthy donors. Co‐incubation of MOLM14 GFP+ CD117high cells with T‐cells and CD117xCD3 TCE (1000 ng/mL) led to similar attachment rates to target cells within 24 h when compared to anti‐CD117 CAR T‐cells (left panel). Tracking of successful attachment events showed significantly faster attachment to MOLM14 GFP+ CD117high cells with CD117xCD3 TCE (right panel). For the fraction of attached cells, Chi2‐test was used to determine p values; p values for time to attachment were determined by unpaired Student's t‐test; **p < 0.01. (E) After successful target‐cell engagement, target‐cell lysis was tracked in >150 individual wells. Anti‐CD117 CAR T‐cells lysed significantly more often when compared to CD117xCD3 TCE. In wells containing >1 target cell, anti‐CD117 CAR T‐cells significantly more frequently lysed multiple target cells when compared to CD117xCD3 TCE with T‐cells (left panel). Time from attachment to target‐cell lysis did not differ between both groups (right panel). For the fraction of lysed cells, Chi2‐test was used to determine p values; p value for time to killing was determined by unpaired Student's t‐test; ***p < 0.001; ****p < 0.0001.
T‐cells derived from AML patients in remission in combination with CD117xCD3 TCE induce efficient lysis of CD117 expressing MOLM14 cells and primary CD117 expressing human AML cells
We next asked the question if also AML patient‐derived T‐cells (similar to healthy donor‐derived T‐cells) are capable to induce AML cell line (MOLM14) or primary patient‐derived AML blast lysis. The expression of CD117 on six AML patient blasts and patients' characteristics used in this manuscript experiments are listed in Supporting Information S1: Figure 11A,B (AML blasts), and Supporting Information S1: Table 3 (AML patient T‐cells).
As demonstrated in Figure 7A–F, the observed lysis of MOLM14 CD117 high and MOLM14 CD117 low target cell lines was similarly efficient, independently of the healthy‐donor or AML‐patient derived effector T‐cells. As expected, the T‐cell expansion and IFN‐γ release to supernatants were lower when targeting MOLM14 CD117 low cells (Figure 7B,C,E,F). These observations were recapitulated when executing the same experimental setup against primary AML patient‐derived blast cells (Figure 7G–I), albeit the lytic activity was substantially lower compared to the lytic activity observed in MOLM14 cells, likely due to the lower CD117 expression of primary patient AML blasts. Further experiments testing CD117xCD3 TCE‐mediated healthy‐donor T‐cell or autologous T‐cell activation against patient AML cells are shown in Supporting Information S1: Figures 12 and 13.
Figure 7.

T‐cells derived from AML patients in first remission lyse MOLM14 cells and primary AML cells with similar efficacy as healthy donor T‐cells upon activation with CD117xCD3 TCE. (A) Percentage specific lysis of MOLM14 CD117high cells by T‐cells upon addition of 1000 ng/mL CD117xCD3 TCE at 24, 48, and 72 h. Cells were cocultured at an E:T = 1:1, and the experiment was performed in duplicate wells (mean ± SD). Data from three healthy‐donor‐derived T‐cells and five AML‐patient‐derived T‐cells isolated at first remission after AML diagnosis. (B) Absolute count of the target cells (left panel) and effector cells (right panel) in the presence and absence of TCE from the experiment shown in (A). (C) Quantification of IFN‐γ in the supernatant from experiments depicted in (A, B). (D–F) Same experimental setup and analysis as in (A–C) but with the use of MOLM CD117 low target cells. (G) Percentage specific lysis of CD45dimCD3− AML blasts from four AML patients (AML #1, 2, 3, 4) by T‐cells at indicated time‐points following the addition of 1000 ng/mL CD117xCD3 TCE. Cells from each AML patient were co‐cultured with T‐cells from healthy donors (n = 3) and from AML patients collected at first remission after AML diagnosis (n = 3). The experiment was performed in duplicate wells, and the data is presented as the mean of the T‐cell‐mediated lysis against the same AML patient blasts ± SD. (H) Absolute count of the target cells (left panel) and effector cells (right panel) in the presence and absence of TCE from the experiment shown in (G). (I) Quantification of IFN‐γ in the supernatant of experiment in (G, H). (A–I) Statistical analysis was conducted using two‐way ANOVA with Tukey's multiple comparison test; *p < 0.05; **p < 0.01; ***p < 0.001.
As biocidal activity increased as a function of time in most experiments, we thought to extend the observation time. We executed a long‐term in vitro assay for up to 192 h (similar to the MOLM14 cells in Supporting Information S1: Figure 8), spiking in T‐cells ± CD117xCD3 TCE a second time at 72 h to the co‐culture. A representative experiment demonstrating almost complete primary human AML cell elimination in the case of both the addition of a second dose of T‐cells and CD117xCD3 TCE is shown in Supporting Information S1: Figure 14.
In sum, these experiments demonstrate that both T‐cells from healthy donors as well as from AML patients in remission are capable of efficiently lysing MOLM14 cells as well as primary AML blasts (in autologous and allogeneic settings), when activated via CD117xCD3 TCE in vitro.
CD117xCD3 TCE leads to efficient T‐cell mediated AML cell depletion in vivo
In order to analyze CD117xCD3 TCE activity in vivo, we first established transplantation and growth kinetics of GFP+Luc+CD117high MOLM14 cells in sublethally irradiated (100 cGy) NSG mice (experimental setup shown in Supporting Information S1: Figure 15A). The analysis shows time‐dependent engraftment of 105 MOLM14 cells over 10 days by in vivo bioluminescence (Supporting Information S1: Figure 15B,C), absolute counts (Supporting Information S1: Figure 15D), and 3D microscopy (Supporting Information S1: Figure 15E). Based on this data, the doubling time of the MOLM14 cell line in vivo was calculated to be about 1.3 days.
Assuming that the CD117xCD3 TCE activity would likely be most efficient in a “minimal residual disease (MRD)” setting with a favorable E:T ratio, we tested the therapeutic activity starting at Day 1 after MOLM14 transplantation. The experimental setup is illustrated in Figure 8A. We intravenously injected 105 GFP+Luc+CD117high MOLM14 cells into sublethally irradiated (100 cGy) NSG mice. The following day, we administered 107 anti‐CD117 CAR T‐cells as positive control 25 or 107 T‐cells, with or without i.p. injection of 12.5 μg CD117xCD3 TCE every 12 h. Bioluminescence imaging revealed that MOLM14 cells were engrafted and expanded in all mice that received only MOLM14 or MOLM14 cells and T‐cells. In contrast, strong tumor growth inhibition was observed when mice were treated with either the combination of T‐cells and CD117xCD3 TCE or with CD117‐CAR T‐cells (Figure 8B,C). Data were further quantified by counting the fraction of GFP+ MOLM14 cells within the human CD45+ cell population isolated from the PB, spleen, and BM of a femur at terminal analysis (Figure 8D,E). We detected no statistically significant difference in the mice treated with either CD117xCD3 TCE plus T‐cells or CD117‐CAR T‐cells regarding both their flux signal at terminal analysis and the residual tumor cells detected by flow cytometry in the analyzed organs. Of note, as the CD117‐CAR T‐cell products used in the in vivo experiments were 60%–80% pure, their in vivo E:T ratio might have been somewhat lower compared to the E:T ratio of nontransduced T‐cells, activated via the CD117xCD3 TCE. However, this difference in E:T was possibly compensated by more efficient lytic activity of CAR T‐cells, which are enhanced by built‐in co‐stimulation, a feature absent in TCE‐activated T‐cells.
Figure 8.

T‐cells in combination with CD117xCD3 TCE elicit similar strong cytotoxicity against MOLM14 AML cells as CD117‐CAR T‐cells in a therapeutic, low disease‐burden setting in vivo in NSG mice. (A) Schematic outline of the experimental setup. NSG mice were sublethally irradiated (100 cGy) and i.v. injected with 105 GFP+Luc+CD117high MOLM14 cells. The following day, mice received either PBS, 107 anti‐CD117 CAR T‐cells, or 107 T‐cells i.v., the latter with or without 12.5 μg CD117xCD3 TCE i.p. injection every 12 h (n = 4). A second injection of 107 T‐cells was performed in T‐cell‐transferred mice on Day 7. (B) Bioluminescence imaging (BLI) of MOLM14 engraftment at Days 7, 11, 14, and 18 after transplantation in mice treated as indicated. (C) Quantification of the bioluminescence flux of whole‐body imaging of mice over at indicated time‐points. Statistical analysis was conducted using two‐way ANOVA with Tukey's multiple comparisons test (*p < 0.05, ***p < 0.001). (D) Absolute GFP+ MOLM14 cell count in blood, spleen, and bone marrow of one femur each at terminal analysis. Statistical analysis was performed by one‐way ANOVA with Tukey's multiple comparisons tests (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). (E) Representative flow cytometry plots of live cells from BM of indicated experimental groups.
To test if AML patient‐derived T‐cells, harvested at time of remission after induction therapy, are also able to induce CD117xCD3 TCE mediated lysis of GFP+Luc+CD117high MOLM14 cells in vivo, we repeated the same experimental setup, comparing patient‐derived T‐cells versus healthy‐donor T‐cells and healthy‐donor derived anti‐CD117 CAR T‐cells (Supporting Information S1: Figure 16A–D). The results indicate that that both T‐cells in combination with CD117xCD3 TCE (but not without it) are about equally efficient in inhibiting growth of GFP+Luc+CD117high MOLM14 target cells and that there is no relevant difference observed to direct anti‐CD117‐CAR T‐cell activity.
Additionally, we tested if the growth of primary patient‐derived AML cells engrafted into NSG mice can be inhibited by the addition of healthy‐donor T‐cells with or without the addition of CD117xCD3 TCE. Figure 9A–C shows the experimental setup and the results, indicating significant in vivo reduction of AML cells only in the presence of both T‐cells and CD117xCD3 TCE.
Figure 9.

T‐cells in combination with CD117xCD3 TCE elicit cytotoxicity against primary human AML cells in a murine xenograft model. (A) Schematic experimental setup. Sublethally (100 cGy) irradiated NSG mice were i.v. engrafted with 5 × 106 CD3/CD19 double‐depleted PB cells isolated from a primary AML patient (PID 1). Four days later, indicated mice were injected i.v. with PBS or 107 T‐cells. Subsequently, an indicated subgroup of mice was i.p. injected twice daily with 12.5 μg CD117xCD3 TCE until Day 15. (B) Absolute counts of AML cells (hCD45dim) in blood, spleen, and BM of a single femur per mouse in indicated groups at terminal analysis. Statistical analysis was conducted using one‐way ANOVA with Tukey's multiple comparisons test; *p < 0.05, **p < 0.01. (C) Representative flow cytometry plots of live cells from BM at terminal analysis for each experimental group. Gating sequence is indicated by arrows and percentages of cells in the respective plots are shown.
In summary, the here presented experiments document CD117xCD3 TCE and T‐cell (healthy donor and AML patient‐derived) mediated in vivo growth inhibition of GFP+Luc+CD117high MOLM14 and/or primary AML patient blasts.
CD117xCD3 TCE leads to efficient T‐cell mediated healthy human HSPC depletion in vivo
Finally, we tested the therapeutic activity of CD117xCD3 TCE against healthy human HSPCs in a xenograft mouse model. The experimental setup is depicted in Figure 10A. NSG mice were transplanted with mobilized peripheral blood healthy human donor CD34+ cells. After 7 weeks, engraftment was analyzed in blood of mice, showing multi‐lineage human hematopoietic development with T‐cells still lacking (Figure 10B). After 8 weeks, n = 2 mice were sacrificed and analyzed for engraftment in thymus and bone marrow, showing multi‐lineage human hematopoietic development, including double‐positive T‐cell development in the thymus (Figure 10C,D). Mice were then injected at Week 9 with human T‐cells from the same HSPC donor and were treated (or not) with CD117xCD3 TCE for 10 subsequent days b.i.d. as indicated. The subsequent terminal analysis is shown in Figure 10E,G. We observed significant CD3+ T‐cell expansion and CD19+ B‐cell reduction in peripheral blood and, more importantly, a significant reduction of CD117‐expressing HSPCs in the bone marrow. At the same time, the relative overall human CD45+ cell engraftment remained rather stable. Highly CD117‐expressing human mast cells in the bone marrow of NSG mice were detectable at low levels and those were further reduced in CD117xCD3 TCE‐treated animals, although, given the low numbers of mice analyzed, this reduction was not significant (Supporting Information S1: Figure 17A,B).
Figure 10.

CD117xCD3 TCE in combination with T‐cells mediate human CD117 + HSPCs lysis in humanized mice. (A) Schematic outline of the experimental setup. Sub‐lethally irradiated NSG mice (100 cGy) were engrafted i.v. with 2 × 106 CD34‐selected mobilized peripheral blood HSPCs (M‐PB HSPCs) from healthy donor 3. At Week 7 after cell inoculation, human chimerism was assessed in peripheral blood by flow cytometry. One week later, two representative cohort mice were terminally analyzed to assess human engraftment in the thymus and bone marrow. At Week 9, 107 T‐cells isolated from the M‐PB HSPC donor 3 were injected i.v. Subsequently, 12.5 μg CD117xCD3 TCE was administered i.p. every 12 h for 10 days. (B) Percentages of all human CD45+ cells (left) and human lineage antigen expression on cells (right) in peripheral blood 7 weeks after transplantation. Each symbol represents an individual mouse. (C) Human T‐cell analysis in the thymus from a representative mouse 8 weeks after transplantation. (D) Representative flow cytometry plots of BM cells isolated from a single femur of a mouse 8 weeks after transplantation. Sequential gating is indicated. (E) Representative flow cytometry plots of live cells from BM at terminal analysis for each experimental group. The sequence of gating is shown in the top row. (F) Percentages of lymphoid and myeloid cells in the peripheral blood of treated and untreated mice at terminal analysis. Statistical analysis was performed using one‐way ANOVA; ****p < 0.0001. (G) Percentages of all human CD45+ cells (left) in the bone marrow from a femur and absolute counts of lymphoid and myeloid cells (middle) and progenitors (right). Statistical analysis was performed using one‐way ANOVA; *p < 0.05, **p < 0.01.
In summary, this indicates a relative selective reduction of CD117 expressing human HSPCs, which, over the short treatment period of 10 days, did not yet substantially decrease HSPC offspring cells.
DISCUSSION
Here, we report on a novel single‐chain variable fragment‐based, bispecific, T‐cell engaging and activating antibody, which binds to human CD117, expressed on HSPCs and AML cells, and to CD3, expressed on T‐cells (named CD117xCD3 TCE). In vitro, CD117xCD3 TCE induced T‐cell mediated lysis of healthy HSPCs from human BM and PB (by activating the same‐donor BM and PB T‐cells) and of AML cell lines, expressing various levels of CD117 on their surface, in a concentration‐, antigen‐density‐, as well as time‐dependent manner. Also, the CD117xCD3 TCE induced T‐cell mediated lysis of primary human AML cells. In a xenogeneic in vivo setting in NSG mice, resembling a minimal measurable (or residual) disease situation, the CD117xCD3 TCE in combination with T‐cells prevented the outgrowth of a CD117‐positive AML cell line with comparable efficacy as direct anti‐CD117 CAR T‐cells. Similarly, CD117xCD3 TCE in combination with T‐cells prevented outgrowth of human primary AML in vivo in a patient‐derived xenotransplantation setting. Furthermore, CD117xCD3 TCE in combination with T‐cells induced depletion of human HSPCs in human CD34+ cell engrafted, immunodeficient mice. Of importance, upon activation with CD117xCD3 TCE, peripheral blood T‐cells from AML patients in remission were able to lyse AML cells with similar efficacy as healthy donor‐derived T‐cells.
Currently, clinically successful cell‐surface immune‐targeting approaches for hematologic malignancies do not discriminate between malignant cells and healthy cell‐of‐origin counterparts. In fact, all clinically approved monoclonal antibodies, bispecific T‐cell engaging and activating antibodies, and CAR T‐cells utilized against B‐ and plasma cell malignancies eliminate both neoplastic and healthy cells without discrimination. 35 , 36 While such an approach is clinically acceptable in B‐ and plasma cell neoplasia, a similar, nondiscriminating approach with respect to cell‐of‐origin in malignancies derived from HSPCs, such as AML, MDS, and MPN, will lead to bone marrow aplasia, which is incompatible with the long‐term survival of patients. Therefore, the immune targeting of HSPC malignancies is only possible with more elaborate solutions. Ideally, distinct cell surface antigens, exclusively expressed on malignant HSPCs, should be identified. These could be either MHC‐presented epitopes of mutated driver molecules (NPM‐1; van der Lee et al. 4 or some broadly expressed antigens, such as CD70, CLL‐1 (CD371), or SICLEC‐6, which might be preferentially expressed in more mature blasts in AML 37 , 38 , 39 , 40 or combinations thereof. 41 , 42 While identifying truly malignant HSPC‐specific target antigens remains an important goal, a pragmatic solution for the time being is to non‐selectively target healthy and malignant HSPC‐expressed common antigens, such as CD33, CD45, CD117, CD123, or CD135, and replace collaterally eliminated healthy HSPCs with new, autologous, or allogeneic HSPCs by transplantation. To prevent the incoming transplant from being attacked, immunotherapy needs to be stopped prior to HSPC transplantation or, as recently demonstrated, the transplant must be genetically engineered to resist immunotherapy. 43 , 44 , 45 , 46
In our proposed approach, we suggest targeting CD117, which is expressed on most, if not all, AML, MDS, MPN initiating, and maintaining cells as well as on healthy HSPCs, 7 , 17 albeit at varying expression levels. Immune‐targeting of CD117 with a monoclonal antibody to eliminate healthy HSPCs as a means of selective preconditioning for hematopoietic stem cell transplantation has been demonstrated in mouse models more than a decade ago. 10 , 47 , 48 , 49 This concept has been explored in the clinical setting of hematopoietic stem cell transplantation for immune deficiencies, with remarkable engraftment results. 50 However, targeting CD117 with a stem cell factor‐blocking antibody may not be sufficient to achieve full donor chimerism and this approach may not efficiently eliminate malignant HSPCs. 51 , 52 , 53 More efficient CD117‐expressing cell depletion might be achieved by antibody‐drug conjugates or by CAR T‐cells. Indeed, fertility‐preserving myeloablative conditioning has been recently demonstrated in a non‐human primate therapy model with a CD117 targeting antibody‐drug conjugate. 15 Also, a more effective targeting approach, utilizing CD117‐directed CAR T‐cells in both primary and humanized mouse models, has been previously explored by others and us. 25 , 54 , 55 , 56 Although CAR T‐cell mediated approaches seem highly efficient, they also require elaborate in vitro effector cell generation followed by in vivo effector cell termination to allow subsequent HSPC engraftment.
With the development of a short half‐life, bispecific CD117xCD3 TCE, we here provide a possible, non‐genotoxic “off the shelf” solution for engagement of a strong endogenous T‐cell effector response against malignant and healthy HSPCs, with the advantage of rapid termination of treatment within hours in case of occurrence of side effects (as e.g., cytokine release syndrome, or potential release of mast cell substrates upon T‐cell induced mast cell apoptosis). In addition, termination of therapy should theoretically allow for timely healthy HSPC transplantation without the risk of targeting incoming CD117‐expressing cells, a procedure, which needs to be tested in future research.
Besides the described potential advantages of the CD117xCD3 TCE, there might also be limitations. While we believe that most of the observed biologic activity of the TCE is due to binding of the monomer to both target antigens, followed by subsequent T‐cell activation, we cannot formally exclude the theoretical concern that a likely small portion of the efficacy is due to possible functional aggregate formation in the non‐monomeric fraction in vitro and in vivo. Further studies in context of a possible clinical development of the TCE will need to address and resolve this concern. More than hundred alternative formats for bispecific antibodies have been proposed, 18 and it is well possible that longer‐lived or different affinity or avidity products may be preferable for clinical applications. Furthermore, in contrast to second‐generation CAR T‐cells, bispecific TCEs, such as the here‐described CD117xCD3 TCE, lack the delivery of a second co‐stimulatory signal in addition to the CD3‐mediated TCR signal. This lack of co‐stimulation might lead to insufficient biocidal activity to fully eliminate malignant cells, a limitation that will possibly be overcome by delivering a co‐stimulatory signal via a second binder. 57 , 58 Both, the here used 79D anti‐CD117 binder and the OKT3 anti‐CD3 binder do not cross‐react with respective mouse homologues. In addition, anti‐CD3 antibody moieties commonly used for the generation of bispecific products (e.g., OKT3 and UCHT‐1) do not cross‐react with the CD3 antigen of Cynomolgus monkeys. This restricted antigen recognition pattern prevents the execution of full toxicology studies in mice and non‐human primates. However, precedents for OKT3‐based bispecific antibodies have been explored in clinical trials based on limited safety pharmacology investigations. 59 , 60 Alternatively, the minimum anticipated biological effect (MABEL) dose may be determined based on in vitro studies rather than findings in non‐human primates. 61 Novel anti‐CD3 antibodies that cross‐react with Cynomolgus monkey antigens may facilitate future product development. 62 , 63
In summary, we have generated and characterized a novel bispecific antibody, which in the presence of T‐cells is able to selectively eliminate CD117‐positive cells. Although bispecific TCE or CAR T‐cell therapies are currently mostly approved and applied in clinically measurable disease settings, we believe that their clinical efficacy will benefit from more favorable in vivo E:T ratios in minimal disease‐burden settings, as for example, suggested by recent studies on the clinical first in class drug blinatumomab. 64 , 65 From our perspective, the immediate clinical application of the CD117xCD3 TCE might be in a time‐limited setting prior to state‐of‐the‐art allogeneic HSCT in patients with high‐risk minimal measurable residual AML or MDS in first remission. This would allow for testing of safety, on‐target and off‐target toxicity, 7 , 17 and efficacy with respect to CD117‐positive cell reduction in the bone marrow prior to pre‐conditioning for HSCT. Further research is needed to investigate whether such an approach might also permit the reduction and replacement of cytotoxic chemotherapy in the pre‐conditioning approach for HSC niche clearance, allowing subsequent incoming donor HSC to engraft.
AUTHOR CONTRIBUTIONS
Laura Volta, Renier Myburgh, Dario Neri, Markus G. Manz were involved in conceptualization. Laura Volta, Renier Myburgh, Christian Koch, Philipp Kaufmann, Serena Fazio, Florin Schneiter, Paul Büschl, Stefan Balabanov, Timm Schroeder, Dario Neri, Markus G. Manz were involved in data curation and analysis. César Nombela‐Arrieta, Timm Schroeder, Dario Neri, Markus G. Manz were involved in funding acquisition. Laura Volta, Renier Myburgh, Mara Hofstetter, Christian Koch, Jonathan D. Kiefer, Celeste Gobbi, Francesco Manfredi, Kathrin Zimmermann, Philipp Kaufmann, Serena Fazio, Christian Pellegrino, Norman F. Russkamp, Danielle Villars, Mattia Matasci, Monique Maurer, Jan Mueller, Florin Schneiter, Paul Büschl, Niclas Harrer, Jacqueline Mock, Stefan Balabanov, César Nombela‐Arrieta, Timm Schroeder, Dario Neri, Markus G. Manz were involved in investigation. Laura Volta, Renier Myburgh, Christian Koch, Jonathan D. Kiefer, Serena Fazio, Monique Maurer, Florin Schneiter, César Nombela‐Arrieta, Timm Schroeder, Dario Neri, Markus G. Manz were involved in methodology. Dario Neri, Markus G. Manz were involved in project administration. Stefan Balabanov, César Nombela‐Arrieta, Timm Schroeder, Dario Neri, Markus G. Manz were involved in resources. Paul Büschl, César Nombela‐Arrieta, Timm Schroeder were involved in software. Stefan Balabanov, César Nombela‐Arrieta, Timm Schroeder, Dario Neri, Markus G. Manz were involved in Supervision. Stefan Balabanov, César Nombela‐Arrieta, Timm Schroeder, Dario Neri, Markus G. Manz were involved in validation. Laura Volta, Renier Myburgh, Mara Hofstetter, Christian Koch, Jonathan D. Kiefer, Celeste Gobbi, Kathrin Zimmermann, Philipp Kaufmann, Serena Fazio, Mattia Matasci were involved in visualization. Laura Volta, Renier Myburgh, Dario Neri, Markus G. Manz were involved in writing—original draft. Laura Volta, Renier Myburgh, Mara Hofstetter, Christian Koch, Jonathan D. Kiefer, Celeste Gobbi, Francesco Manfredi, Kathrin Zimmermann, Philipp Kaufmann, Serena Fazio, Christian Pellegrino, Norman F. Russkamp, Danielle Villars, Mattia Matasci, Monique Maurer, Jan Mueller, Florin Schneiter, Paul Büschl, Niclas Harrer, Jacqueline Mock, Stefan Balabanov, César Nombela‐Arrieta, Timm Schroeder, Dario Neri, Markus G. Manz were involved in writing—review and editing.
CONFLICT OF INTEREST STATEMENT
Jonathan D. Kiefer, Renier Myburgh, Dario Neri, and Markus G. Manz are inventors in patent applications that describe the anti‐CD117xCD3 TCE. The commercialization rights/plans for this patent are overseen and regulated by the Technology Transfer Office of the University of Zurich and ETH Zürich, which granted an exclusive license to ATLyphe (www.atlyphe.com). Renier Myburgh, Jonathan D. Kiefer, Norman F. Russkamp, Danielle Villars, Dario Neri/Philogen, and Markus G. Manz are shareholders of ATLyphe. All other authors declare no conflict of interest.
ETHICS STATEMENT
Patient and healthy donor data and cells were obtained from the Department of Medical Oncology and Hematology Biobank, University Hospital Zürich, Zürich, Switzerland, and written informed consent was obtained from all patients. This study was conducted in accordance with the Declaration of Helsinki and approved by the Cantonal Ethics Board of Zürich, Switzerland (2009‐0062).
FUNDING
Financial support from ETH Zürich, Swiss National Science Foundation (grant no. 310030_182003/1 and 310030_184747/1), the European Research Council under the European Union's Horizon 2020 Research and Innovation Program (grant agreement 670603), the University of Zurich (Clinical Research Priority Program “ImmunoCure” of the University of Zurich and University Research Priority Project Translational Cancer Research), Swiss Cancer Research (KFS‐3846‐02‐2016), ETH Lymphoma Challenge Grant, the Dr. Horst Böhlke Foundation, and a Pioneer Fellowship from the ETH Foundation are gratefully acknowledged.
Supporting information
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
ACKNOWLEDGMENTS
The authors thank Prof. Martin Ehrbar for the use of a Leica microscope, Angelina Oestmann for technical assistance with mouse colony management, and Nadia Keller, Asuka Frey, Dr. Dominik Schneidawind, and Dr. Adrian Bachofner for the organization and preparation of primary AML‐patient derived samples and apheresis samples. The authors also acknowledge Prof. Jörg Scheuermann, Dr. Andreas Gloger, and Dr. Louise Plais for their support with the LC‐MS analysis.
DATA AVAILABILITY STATEMENT
The datasets generated and/or analyzed during the current study are available from the corresponding author upon request.
REFERENCES
- 1. Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008;132:631‐644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730‐737. [DOI] [PubMed] [Google Scholar]
- 3. Trumpp A, Haas S. Cancer stem cells: the adventurous journey from hematopoietic to leukemic stem cells. Cell. 2022;185:1266‐1270. [DOI] [PubMed] [Google Scholar]
- 4. van der Lee DI, Reijmers RM, Honders MW, et al. Mutated nucleophosmin 1 as immunotherapy target in acute myeloid leukemia. J Clin Invest. 2019;129:774‐785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Xue S‐A, Gao L, Hart D, et al. Elimination of human leukemia cells in NOD/SCID mice by WT1‐TCR gene–transduced human T cells. Blood. 2005;106:3062‐3067. [DOI] [PubMed] [Google Scholar]
- 6. Cappell KM, Kochenderfer JN. Long‐term outcomes following CAR T cell therapy: what we know so far. Nat Rev Clin Oncol. 2023;20:359‐371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Russkamp NF, Myburgh R, Kiefer JD, Neri D, Manz MG. Anti‐CD117 immunotherapy to eliminate hematopoietic and leukemia stem cells. Exp Hematol. 2021;95:31‐45. [DOI] [PubMed] [Google Scholar]
- 8. Griffin JM, Healy FM, Dahal LN, Floisand Y, Woolley JF. Worked to the bone: antibody‐based conditioning as the future of transplant biology. J Hematol Oncol. 2022;15:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Saha A, Blazar BR. Antibody based conditioning for allogeneic hematopoietic stem cell transplantation. Front Immunol. 2022;13:1031334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Czechowicz A, Kraft D, Weissman IL, Bhattacharya D. Efficient transplantation via antibody‐based clearance of hematopoietic stem cell niches. Science. 2007;318:1296‐1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Manz MG, Russkamp NF. Selective CD117+ HSC exchange therapy. Blood. 2019;133:2007‐2009. [DOI] [PubMed] [Google Scholar]
- 12. Ogawa M, Matsuzaki Y, Nishikawa S, et al. Expression and function of c‐kit in hemopoietic progenitor cells. J Exp Med. 1991;174:63‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Broudy VC. Stem cell factor and hematopoiesis. Blood. 1997;90:1345‐1364. [PubMed] [Google Scholar]
- 14. Lennartsson J, Rönnstrand L. Stem cell factor receptor/c‐Kit: from basic science to clinical implications. Physiol Rev. 2012;92:1619‐1649. [DOI] [PubMed] [Google Scholar]
- 15. Uchida N, Stasula U, Demirci S, et al. Fertility‐preserving myeloablative conditioning using single‐dose CD117 antibody‐drug conjugate in a rhesus gene therapy model. Nat Commun. 2023;14:6291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature. 2012;481:457‐462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Foster B, Zaidi D, Young T, Mobley M, Kerr B. CD117/c‐kit in cancer stem cell‐mediated progression and therapeutic resistance. Biomedicines. 2018;6:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brinkmann U, Kontermann RE. The making of bispecific antibodies. Mabs. 2017;9:182‐212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Labrijn AF, Janmaat ML, Reichert JM, Parren PWHI. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discovery. 2019;18:585‐608. [DOI] [PubMed] [Google Scholar]
- 20. Klein C, Brinkmann U, Reichert JM, Kontermann RE. The present and future of bispecific antibodies for cancer therapy. Nat Rev Drug Discovery. 2024;23:301‐319. [DOI] [PubMed] [Google Scholar]
- 21. Dreier T, Lorenczewski G, Brandl C, et al. Extremely potent, rapid and costimulation‐independent cytotoxic T‐cell response against lymphoma cells catalyzed by a single‐chain bispecific antibody. Int J Cancer. 2002;100:690‐697. [DOI] [PubMed] [Google Scholar]
- 22. Kantarjian H, Stein A, Gökbuget N, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. 2017;376:836‐847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Löffler A, Kufer P, Lutterbüse R, et al. A recombinant bispecific single‐chain antibody, CD19 × CD3, induces rapid and high lymphoma‐directed cytotoxicity by unstimulated T lymphocytes. Blood. 2000;95:2098‐2103. [PubMed] [Google Scholar]
- 24. Reshetnyak AV, Nelson B, Shi X, et al. Structural basis for KIT receptor tyrosine kinase inhibition by antibodies targeting the D4 membrane‐proximal region. Proc Natl Acad Sci USA. 2013;110:17832‐17837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Myburgh R, Kiefer JD, Russkamp NF, et al. Anti‐human CD117 CAR T‐cells efficiently eliminate healthy and malignant CD117‐expressing hematopoietic cells. Leukemia. 2020;34:2688‐2703. [DOI] [PubMed] [Google Scholar]
- 26. Hoffmann P, Hofmeister R, Brischwein K, et al. Serial killing of tumor cells by cytotoxic T cells redirected with a CD19‐/CD3‐bispecific single‐chain antibody construct. Int J Cancer. 2005;115:98‐104. [DOI] [PubMed] [Google Scholar]
- 27. Saito Y, Ellegast JM, Rafiei A, et al. Peripheral blood CD34+ cells efficiently engraft human cytokine knock‐in mice. Blood. 2016;128:1829‐1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Reusch U, Burkhardt C, Fucek I, et al. A novel tetravalent bispecific TandAb (CD30/CD16A) efficiently recruits NK cells for the lysis of CD30+ tumor cells. Mabs. 2014;6:727‐738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zuberbühler K, Palumbo A, Bacci C, et al. A general method for the selection of high‐level scFv and IgG antibody expression by stably transfected mammalian cells. Protein Eng, Des Sel. 2009;22:169‐174. [DOI] [PubMed] [Google Scholar]
- 30. Kim KS, Sun Z‐YJ, Wagner G, Reinherz EL. Heterodimeric CD3ϵγ extracellular domain fragments: production, purification and structural analysis. J Mol Biol. 2000;302:899‐916. [DOI] [PubMed] [Google Scholar]
- 31. Zorn JA, Wheeler ML, Barnes RM, et al. Humanization of a strategic CD3 epitope enables evaluation of clinical T‐cell engagers in a fully immunocompetent in vivo model. Sci Rep. 2022;12:3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Betts A, van der Graaf PH. Mechanistic quantitative pharmacology strategies for the early clinical development of bispecific antibodies in oncology. Clin Pharm Ther. 2020;108:528‐541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu C, Zhou J, Kudlacek S, Qi T, Dunlap T, Cao Y. Population dynamics of immunological synapse formation induced by bispecific T cell engagers predict clinical pharmacodynamics and treatment resistance. eLife. 2023;12:e83659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pellegrino C, Favalli N, Sandholzer M, et al. Impact of ligand size and conjugation chemistry on the performance of universal chimeric antigen receptor T‐cells for tumor killing. Bioconjug Chem. 2020;31:1775‐1783. [DOI] [PubMed] [Google Scholar]
- 35. Labanieh L, Mackall CL. CAR immune cells: design principles, resistance and the next generation. Nature. 2023;614:635‐648. [DOI] [PubMed] [Google Scholar]
- 36. Lanier OL, Pérez‐Herrero E, Andrea APD, et al. Immunotherapy approaches for hematological cancers. iScience. 2022;25:105326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Riether C, Schürch CM, Bührer ED, et al. CD70/CD27 signaling promotes blast stemness and is a viable therapeutic target in acute myeloid leukemia. J Exp Med. 2017;214:359‐380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Riether C, Pabst T, Höpner S, et al. Targeting CD70 with cusatuzumab eliminates acute myeloid leukemia stem cells in patients treated with hypomethylating agents. Nat Med. 2020;26:1459‐1467. [DOI] [PubMed] [Google Scholar]
- 39. van Rhenen A, van Dongen GAMS, Kelder A, et al. The novel AML stem cell–associated antigen CLL‐1 aids in discrimination between normal and leukemic stem cells. Blood. 2007;110:2659‐2666. [DOI] [PubMed] [Google Scholar]
- 40. Jetani H, Navarro‐Bailón A, Maucher M, et al. Siglec‐6 is a novel target for CAR T‐cell therapy in acute myeloid leukemia. Blood. 2021;138:1830‐1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Haubner S, Mansilla‐Soto J, Nataraj S, et al. Cooperative CAR targeting to selectively eliminate AML and minimize escape. Cancer Cell. 2023;41:1871‐1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Atilla PA, McKenna MK, Watanabe N, Mamonkin M, Brenner MK, Atilla E. Combinatorial antigen targeting strategies for acute leukemia: application in myeloid malignancy. Cytotherapy. 2022;24:282‐290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Marone R, Landmann E, Devaux A, et al Epitope engineered human haematopoietic stem cells are shielded from CD123‐targeted immunotherapy. J Exp Med. 2023;220(12):e20231235. 10.1038/s41375-024-02409-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wellhausen N, O'Connell RP, Lesch S, et al. Epitope base editing CD45 in hematopoietic cells enables universal blood cancer immune therapy. Sci Transl Med. 2023;15(714):eadi1145. 10.1126/scitranslmed.adi1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Casirati G, Cosentino A, Mucci A, et al. Epitope editing enables targeted immunotherapy of acute myeloid leukaemia. Nature. 2023;621:404‐414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Volta L, Manz MG. Twisted: escape of epitope‐edited healthy cells from immune attack. J Exp Med. 2023;220:e20231635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xue X, Pech NK, Shelley WC, Srour EF, Yoder MC, Dinauer MC. Antibody targeting KIT as pretransplantation conditioning in immunocompetent mice. Blood. 2010;116:5419‐5422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pang WW, Czechowicz A, Logan AC, et al. Anti‐CD117 antibody depletes normal and myelodysplastic syndrome human hematopoietic stem cells in xenografted mice. Blood. 2019;133:2069‐2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kwon H‐S, Logan AC, Chhabra A, et al. Anti‐human CD117 antibody‐mediated bone marrow niche clearance in nonhuman primates and humanized NSG mice. Blood. 2019;133:2104‐2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Agarwal R, Weinberg KI, Kwon H‐S, et al. First report of non‐genotoxic conditioning with JSP191 (anti‐CD117) and hematopoietic stem cell transplantation in a newly diagnosed patient with severe combined immune deficiency. Blood. 2020;136:10. [Google Scholar]
- 51. Chhabra A, Ring AM, Weiskopf K, et al. Hematopoietic stem cell transplantation in immunocompetent hosts without radiation or chemotherapy. Sci Transl Med. 2016;8:351ra105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. George BM, Kao KS, Kwon H‐S, et al. Antibody conditioning enables mhc‐mismatched hematopoietic stem cell transplants and organ graft tolerance. Cell Stem Cell. 2019;25:185‐192.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bankova AK, Pang WW, Velasco BJ, Long‐Boyle JR, Shizuru JA. 5‐Azacytidine depletes HSCs and synergizes with an anti‐CD117 antibody to augment donor engraftment in immunocompetent mice. Blood Adv. 2021;5:3900‐3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Arai Y, Choi U, Corsino CI, et al. Myeloid conditioning with c‐kit‐targeted CAR‐T cells enables donor stem cell engraftment. Mol Ther. 2018;26:1181‐1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Magnani CF, Myburgh R, Brunn S, et al. Anti‐CD117 CAR T cells incorporating a safety switch eradicate human acute myeloid leukemia and hematopoietic stem cells. Mol Ther Oncolytics. 2023;30:56‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Volta L, Myburgh R, Pellegrino C, et al. Efficient combinatorial adaptor‐mediated targeting of acute myeloid leukemia with CAR T‐cells. Leukemia. 2024:1‐16. In press. 10.1038/s41375-024-02409-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dickinson M, Gritti G, Carlo‐Stella C, et al. Phase 1 study of CD19 targeted CD28 costimulatory agonist in combination with glofitamab to enhance t cell effector function in relapsed/refractory B cell lymphoma. Blood. 2022;140:3818‐3820. [Google Scholar]
- 58. Wei J, Montalvo‐Ortiz W, Yu L, et al. CD22‐targeted CD28 bispecific antibody enhances antitumor efficacy of odronextamab in refractory diffuse large B cell lymphoma models. Sci Transl Med. 2022;14:eabn1082. [DOI] [PubMed] [Google Scholar]
- 59. Klinger M, Benjamin J, Kischel R, Stienen S, Zugmaier G. Harnessing T cells to fight cancer with BiTE® antibody constructs – past developments and future directions. Immunol Rev. 2016;270:193‐208. [DOI] [PubMed] [Google Scholar]
- 60. Pishvaian M, Morse MA, McDevitt J, et al. Phase 1 dose escalation study of MEDI‐565, a bispecific T‐cell engager that targets human carcinoembryonic antigen, in patients with advanced gastrointestinal adenocarcinomas. Clin Colorectal Cancer. 2016;15:345‐351. [DOI] [PubMed] [Google Scholar]
- 61. Muller PY, Milton M, Lloyd P, Sims J, Brennan FR. The minimum anticipated biological effect level (MABEL) for selection of first human dose in clinical trials with monoclonal antibodies. Curr Opin Biotechnol. 2009;20:722‐729. [DOI] [PubMed] [Google Scholar]
- 62. Falchi L, Vardhana SA, Salles GA. Bispecific antibodies for the treatment of B‐cell lymphoma: promises, unknowns and opportunities. Blood. 2023;141:467‐480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Pillarisetti K, Powers G, Luistro L, et al. Teclistamab is an active T cell–redirecting bispecific antibody against B‐cell maturation antigen for multiple myeloma. Blood Adv. 2020;4:4538‐4549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gökbuget N, Dombret H, Bonifacio M, et al. Blinatumomab for minimal residual disease in adults with B‐cell precursor acute lymphoblastic leukemia. Blood. 2018;131:1522‐1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Litzow MR, Sun Z, Mattison RJ, et al. Blinatumomab for MRD‐negative acute lymphoblastic leukemia in adults. N Engl J Med. 2024;391:320‐333. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Data Availability Statement
The datasets generated and/or analyzed during the current study are available from the corresponding author upon request.