

Abstract
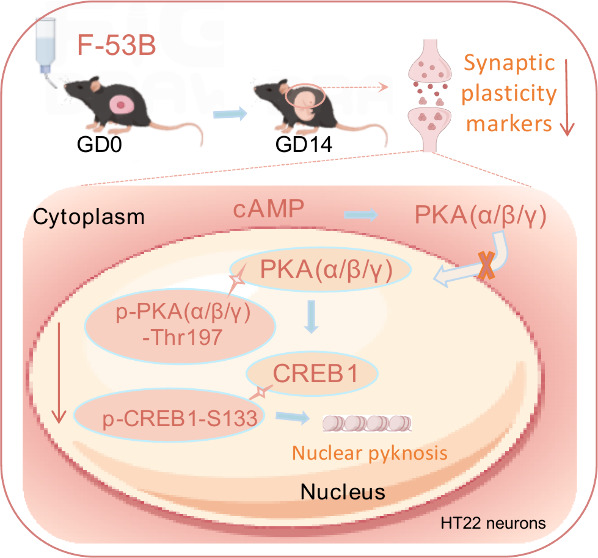
Chlorinated polyfluorinated ether sulfonate (F-53B), a chromium-fog depressant widely utilized as an alternative to perfluorooctanesulfonate, can transfer from mother to fetus. Recent research has demonstrated that prenatal exposure to F-53B results in synaptic damage in weaning mice. However, the mechanism underpinning F-53B-triggered synaptic damage during fetal development remains unclear. This study aims to investigate the role of the protein kinase A (PKA)/cAMP response element-binding protein (CREB) pathway, a crucial signaling mechanism known as “synaptic switch”, in the early neurotoxicity of F-53B exposure both in vivo and in vitro. Here, C57BL/6 fetal mice were subjected to exposure to F-53B (0, 4, and 40 μg/L) from gestation days (GD) 0 to 14 to evaluate nerve injury prior to delivery. HT22 neurons exposed to F-53B (0, 0.016, 0.08, 0.4, 2, and 10 μmol/L) for 24 h were utilized to elucidate the underlying mechanism. Our results demonstrated that F-53B significantly increased the fluorescence intensity of Nestin (a neural stem cell marker) in the fetal brain hippocampus (GD14). Subsequently, we found that F-53B downregulated the expression of synaptic plasticity markers (SYP, GAP43, and BDNF) in the fetal brain and HT22 neurons. Further molecular docking analysis revealed that F-53B fits into the ligand-binding pockets of PKA and CREB1. Results showed that F-53B inhibited the translocation of PKA protein from the cytoplasm to the neuronal nuclei and reduced the levels of PKA, CREB1, p-PKA(α/β/γ)-Thr197, and p-CREB1-S133 in the nucleus. Furthermore, the expression of synaptic plasticity markers altered by F-53B could be reversed by a PKA agonist and was intensified by a PKA antagonist. In summary, our findings suggest that intrauterine exposure to F-53B can weaken the expression of synaptic plasticity markers in the fetal brain, with this neurotoxicity being mediated by the cytoplasmic retention of PKA.
Keywords: F-53B, Synaptic plasticity, PKA/CREB signaling pathway, Early neurotoxicity, PKA cytoplasmic retention
1. Introduction
Chlorinated polyfluorinated ether sulfonate (F-53B), a chromium-fog depressant commonly utilized in the electroplating industry, has been considered a safer alternative to the prohibited chemical perfluorooctanesulfonate (PFOS) in China since the 1970s.1 However, recent studies have demonstrated that F-53B exhibits a higher bioconcentration potential and placental transfer coefficient than PFOS.2,3 Elevated levels of F-53B have been found in human cord blood, follicular fluid, and the placenta, posing a grave threat to offspring health.4,5 Furthermore, a recent finding indicates that the blood-brain barrier permeability of F-53B in humans is significantly higher than that of PFOS,6 eliciting significant concerns about its neurotoxicity. An epidemiological study has shown a negative correlation between F-53B levels in cord blood and the neurodevelopmental outcomes of 3-month-old children.7In vivo and in vitro studies have indicated that F-53B can impairs neurobehavior of mice offspring and zebrafish larvae8,9 and affects neural differentiation of mouse embryonic stem cells.10,11 While maternal F-53B exposure has been associated with adverse neurological outcomes in offspring, the underlying toxic mechanism remains to be elucidated.
Synaptic damage is regarded as a key event in offspring neurotoxicity following early life F-53B exposure. In our prior study, we observed altered neurotransmitter expressions that could mediate synaptic development for the first time in F-53B-exposed zebrafish before fertilization.12 Our subsequent research demonstrated that exposure to F-53B during pregnancy and lactation led to lower expression of hippocampal synaptic plasticity markers and memory impairment in weaning mice.9 These studies primarily focus on the effects of F-53B on postpartum offspring synapses. However, synaptic connectivity in the human brain initiates during the second trimester, as differentiating neurons extend axons and dendrites, with gradual maturation in the third trimester.13 The delayed fetal synaptic development during pregnancy may present challenges to remediation and lead to lasting effects on adult neurobehavior.14 For instance, intrauterine exposure to ethanol in fetal mice causes synaptic plasticity impairments during the embryonic stage, which persist and contribute to neurobehavioral deficits resembling “fetal alcohol spectrum disorder” in adult mice.15 However, a significant research gap remains in the elucidation of the effects and mechanisms of F-53B on prenatal offspring synapses.
The protein kinase A (PKA)/cAMP response element-binding protein (CREB) pathway, referred to as the “synaptic switch”,16 is acknowledged as a crucial mechanism by which common dietary supplements (eg., docosahexaenoic acid) for pregnant women and antipsychotic drugs (eg., desloratadine) safeguard synaptic and cognitive functions.17,18 PKA is capable of translocating to the nucleus, phosphorylating CREB, and activating gene promoters involved in synaptic development.19 Observations indicate that exposure to perfluorononanoic acid (PFNA) in zebrafish embryos results in the suppression of PKA/CREB pathway activity and the development of synaptic spines.20 Investigating this pathway could yield crucial insights into counteracting the early neurological damage induced by F-53B. However, studies on the effects of the PKA/CREB pathway on synapses affected by F-53B are currently lacking. We hypothesize that F-53B impairs synaptic development and function by suppressing the PKA/CREB pathway. Further in vitro studies are necessary to validate this hypothesis.
In this study, we provide novel evidence demonstrating that F-53B impairs the expressions of synaptic plasticity markers in fetal mice and pinpoint the PKA/CREB signaling pathway as the underlying mechanism. These findings hold significance in clarifying the impact of early F-53B exposure on the nervous system and potentially mitigating risks to fetus.
2. Materials and Methods
2.1. Chemicals
For mice, a stock solution of F-53B was formulated in 0.5% Tween 20 (Sigma-Aldrich, USA) and then diluted to the desired concentration with deionized water. For cells, F-53B, 8-bromo-cAMP sodium salt (a PKA agonist, HY-12306, MCE, USA), and H-89 dihydrochloride (a PKA antagonist, HY-15979A, MCE, USA) were prepared in dimethyl sulfoxide (DMSO) (Sigma-Aldrich, USA) at a concentration below 0.01% (v/v). All reagents employed were of either analytical or high-performance liquid chromatography (HPLC) grade.
2.2. Animal Experimental Design
C57BL/6 mice (n = 50, male: female = 1:1, aged 7–8 weeks) weighing an average of 20 g were procured from Guangdong Medical Laboratory Animal Center (Guangdong, China). Mice were housed in a temperature- and humidity-controlled room with a 12:12-h light-dark cycle and provided with specific pathogen-free (SPF)-grade foods. Following a one-week quarantine period, the mice were randomly paired in cages. Gestation day 0 (GD 0) was identified through vaginal smear microscopy. The drinking water for pregnant mice contained 0, 4, and 40 μg/L F-53B, and was made available ad libitum from GD0 to GD14. The dosage was converted from the F-53B concentrations in the general adult population (4.78 ng/mL) and occupational workers (51.5 ng/mL),21 as detailed in our previous study.9,22 At GD14, the mice were euthanized. The amniotic fluid, placenta, and fetal brain tissues were harvested and preserved at −80 °C. The organ coefficient of the fetal brain was calculated as the ratio of the organ weight (g) to the body weight (g).
2.3. Cell Culture and Treatment
Mouse hippocampal neuronal cells (HT22 cells) were purchased from the National Collection of Authenticated Cell Cultures (Shanghai, China). HT22 cells, between passages 4–10, were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Thermo Fisher Scientific Inc., USA), enriched with 10% FBS and 0.1% antibiotics (penicillin and streptomycin, P/S), in an environment of 37 °C with 5% CO2. A stock solution containing F-53B, a PKA agonist, and a PKA antagonist, all dissolved in DMSO, was introduced into DMEM devoid of FBS and subsequently incubated for 24 h.
2.4. Cell Proliferation Assay
Cytotoxicity evaluation was performed with the cell counting kit-8 (CCK8, Beyotime, China). HT22 cells were plated in 96-well plates (Corning, USA) to achieve approximately 80% confluency and subsequently exposed to escalating doses of F-53B (0, 0.025, 0.25, 1.25, 2.5, 5, 12.5, 25, 50, and 125 μmol/L) for 24 h. Following this, CCK-8 reagents were administered to each well and allowed to incubate for 2 h. Absorbance readings were taken at 450 nm using a microplate reader (Bio-Rad, USA).
2.5. Calculations for 50% Inhibitory Concentration (IC50) and Benchmark Dose (BMD)
The IC50 value was calculated using GraphPad Prism 8.0 software, which fitted the log-transformed dose of F-53B and the corresponding cell survival rates obtained from the CCK-8 assay to a four-parameter nonlinear curve model. The IC50 value is defined as the dose resulting in a cell survival rate of 50% on the model’s fitted curve. The BMD and its upper (BMDU) and lower (BMDL) limits at a 95% confidence interval were determined using Benchmark Dose Software (BMDS) Version 3.2 (https://www.epa.gov/bmds). Similar methodologies have been utilized in preceding studies.23
2.6. Cell Viability Assay
The effects of various doses of F-53B and their combined exposure with PKA agonist/antagonist on HT22 cells viability were evaluated using the lactate dehydrogenase release (LDH) kit (Beyotime, China). HT22 cells, maintained at 80% confluency, were exposed to F-53B for 24 h, with subsequent addition of LDH-release reagents to the culture medium, followed by an incubation period of 1 h. Following treatment, the mixture underwent centrifugation at 400 xg for 5 min. Subsequently, 120 μL of the supernatant from each well was combined with 60 μL of the LDH detection reagent. The absorbance level at 490 nm was quantified using a microplate reader (Bio-Rad, USA).
2.7. Immunofluorescent and Confocal Microscopy
The fetal brain, fixed in 4% paraformaldehyde for 24 h, underwent ethanol gradient elution and paraffin embedding. Subsequently, brain sections (measuring 8 μm in thickness) were subjected to treatment in a citrate buffer solution at 95 °C for 10 min. Similarly, HT22 cells were fixed in 4% paraformaldehyde for 30 min. The brain sections and HT22 cells underwent permeabilization for 30 min using 0.3% Triton X-100 and were subsequently blocked with 5% Bovine Serum Albumin (BSA) for 30 min at room temperature. Next, the samples were incubated with primary antibodies (Nestin, PKA α/β/γ, and SYP, 1:100) overnight at 4 °C, followed by treatment with secondary antibodies (goat-antirabbit IgG Alexa Fluor 566 and 488, 1:400, Abcam, UK) for 1 h at room temperature. Finally, the samples were incubated with an antifade mounting medium containing Diamidino-phenyl-indole (Beyotime, China) for 10 min and visualized with a laser scanning confocal microscope (Nikon, Japan).
2.8. Cell Apoptosis Assay
Cell apoptosis was assessed using the Annexin V-FITC/PI apoptosis kit (Beyotime, China) following the manufacturer’s instructions and analyzed with a CytoFLEX-2 Flow Cytometer (Beckman Coulter, USA). Additional methodological details are provided in the Supporting Information (Text S2).
2.9. RNA Extraction and Quantitative Real-Time PCR (qRT-PCR)
RNA was isolated from the fetal brain and HT22 cells using TRIzol Reagent (Invitrogen, USA) and subsequently transcribed into cDNA utilizing the ReverTra Ace qPCR RT Kit (TOYOBO, Japan). The remaining experimental procedures are delineated in the Supporting Information (Text S3) with comprehensive detail. Relative gene expression levels were determined using the 2–ΔΔCt method, with Gapdh serving as the reference gene. The mRNA expression levels of the PKA/CREB signaling pathway-related genes (Adcy1, cAMP, PKA, and CREB1) and synaptic plasticity-related genes (BDNF, SYP, PSD95, and GAP43) were meticulously analyzed. Specific primer sequences are detailed in the Supporting Information (Table S1).
2.10. Western Blot (WB)
Total proteins from the fetal brain and HT22 cells were extracted individually using a lysis buffer (Beyotime, China). Cytoplasmic and nuclear proteins from HT22 cells were extracted using the cytoplasmic and nuclear extraction kit (Beyotime, China). Further procedural details are available in the Supporting Information (Text S4). The specific antibodies used in the study included those for GAPDH (Proteintech, 10494–1-AP), Histone-H3 (Proteintech, 17168–1-AP), SYP (Abcam, ab52636), BDNF (Proteintech, 28205–1-AP), GAP-43 (Proteintech, 16971–1-AP), CREB1 (Proteintech, 12208–1-AP), CREB1 (phosphor Ser133) (Immunoway, YP0075), PKAα/β/γ (Immunoway, YT3749), PKAα/β/γ (phosphor Thr197) (Immunoway, YP0226), Caspase-3 (Abcam, ab184787), and Caspase-1 (Proteintech, 22915–1-AP).
2.11. Molecular Docking
The 3D structures of CREB1 (PD BID: 5ZKO), PKA (PDB ID: 2H77), 6:2 Cl-PFAES (Compound CID: 22568738), and 8:2 Cl-PFAES (Compound CID: 50851128) were employed in molecular docking analyses using AutoDock 1.5.7. The detailed process and parameters were performed as previously described with modifications,24 and are detailed in the Supporting Information (Text S5).
2.12. Statistical Analysis
Data analysis was performed utilizing IBM SPSS version 25.0 and GraphPad Prism 8.0. Variations between the F-53B-treated and control groups were assessed via one-way analysis of variance (ANOVA) followed by Tukey-HSD tests. All results are presented as the mean ± standard error of the mean (SEM), with statistical significance established at p < 0.05.
3. Results
3.1. Effects of F-53B on Synaptic Plasticity Markers in the Fetal Brain
The organ coefficient of the fetal brain showed no significant differences across the three groups (Table S2). In fetal brain sections exposed to F-53B, there was a marked increase in the fluorescence intensity of Nestin in the hippocampus compared to the control group (Figure 1A-B). Additionally, reductions in mRNA levels of SYP and BDNF by 0.66- and 0.74-fold, respectively, were observed at 40 μg/L F-53B (Figure 1C). In protein expression analyses, exposure to 4 and 40 μg/L F-53B resulted in a decrease in BDNF expression by 0.81- and 0.76-fold, respectively, and down-regulation of SYP by 0.70-fold was noted at 40 μg/L F-53B (Figure 1D-E).
Figure 1.

Effects of F-53B exposure on synaptic plasticity markers in fetal mice. (A-B) Representative photomicrographs of nestin in the fetal brain (n = 3). (C-E) The relative mRNA and protein levels of synaptic plasticity in the fetal brain (n = 3). Statistical significance is denoted by * for p < 0.05, ** for p < 0.01, and *** for p < 0.001.
3.2. Effects of F-53B on Cytotoxicity and Synaptic Plasticity Markers in HT22 Neurons
Initial exploration of the cytotoxic effects on HT22 neurons exposed to varying concentrations of F-53B (0- 125 μmol/L) revealed a dose-dependent decrease in cell survival rates (Figure 2A). Specifically, cell survival rate was greater than 75% below 5 μmol/L, dropped below 50% above 12.5 μmol/L, and approached 0% above 25 μmol/L (Figure 2A). To determine the IC50 and BMD values for F-53B on HT22 cells, we performed a logarithmic transformation of the doses and constructed a four-parameter nonlinear curve model. As shown in Figure 2B, the dose–response curves exhibited a sigmoidal shape, indicating increased cell inhibition rates with higher doses of F-53B. The IC50 and BMD values for F-53B effect on HT22 neurons were calculated to be 10.197 μmol/L and 1.957 μmol/L, respectively (Figure 2B). Based on the above data, we set 2 μmol/L and 10 μmol/L as the exposure doses of F-53B for subsequent experiments, and added three additional doses of 0.4 μmol/L, 0.08 μmol/L, and 0.016 μmol/L, following a 5-fold dilution series. Neurons exposed to F-53B at concentrations below 10 μmol/L maintained viability above 90%, whereas at 10 μmol/L, viability declined to below 70% (Figure 2C). Furthermore, a dose-dependent increase in apoptotic ratio was observed, with notable effects at concentrations of 0.4, 2, and 10 μmol/L F-53B, where the mean apoptosis rates were 7.32%, 8.45%, and 10.42%, respectively (Figure 2D-E). Similarly, the expression of the apoptotic protein Caspase-1 significantly increased at 2 and 10 μmol/L F-53B, with levels exceeding a 2-fold increase (Figure 2F-G).
Figure 2.

Effects of F-53B exposure on cytotoxicity and synaptic plasticity markers in HT22 neurons. (A) The cell survival rate of HT22 cells. (B) The fitting curves of HT22 cells for BMD and IC50 calculation of F-53B. (C) The cell viability of HT22 cells. (D-E) The apoptosis rate of HT22 cells. (F-G) The relative protein levels of apoptosis in HT22 cells. (H–I) The relative protein levels of synaptic plasticity in HT22 cells. (J-K) Representative photomicrographs of SYP levels and dendritic length in HT22 cells. Comparison with the control group is indicated by the symbol “#”. Statistical significance is denoted by * and # for p < 0.05, ** and ## for p < 0.01, and *** and ### for p < 0.001.
Significantly, marked reductions in protein levels of GAP43 (0.56-fold), SYP (0.53-fold), and BDNF (0.46-fold) were recorded at 10 μmol/L F-53B, with reductions in SYP (0.66-fold) levels also observed at 2 μmol/L (Figure 2H-I). Moreover, significant decreases in dendritic length, as well as in the fluorescence area and density of SYP, were observed in HT22 neurons exposed to 0.4, 2, and 10 μmol/L F-53B (Figure 2J-K).
3.3. Effects of F-53B on the PKA/CREB Pathway in the Fetal Brain and HT22 Neurons
As depicted in Figure 3, the primary constituents of F-53B (6:2 Cl-PFASE and 8:2 Cl-PFASE) were identified as interacting with crucial amino acid residues within the active sites of PKA and CREB1. In the structure of PKA, hydrogen bonds formed between 6:2 Cl-PFASE and Lysine (LYS)-189, as well as between 8:2 Cl-PFASE and LYS-217, LYS-192, and Asparagine (ASN)-216. In CREB1 docked complexes, 6:2 Cl-PFASE engaged in hydrogen bonding interactions with LYS-285, while 8:2 Cl-PFASE engaged with LYS-285, Arginine (ARG)-286, and Glutamic acid (GLU)-287.
Figure 3.

Molecular docking results of 6:2 Cl-PFAES and 8:2 Cl-PFAES with the PKA/CREB pathways. (A) 6:2 Cl-PFAES with PKA. (B) 8:2 Cl-PFAES with PKA. (C) 6:2 Cl-PFAES with CREB1. (D) 8:2 Cl-PFAES with CREB1.
Further investigation revealed that F-53B resulted in a reduction of mRNA expression of PKA and its upstream gene (cAMP) in the fetal brain at concentrations of 4 and 40 μg/L (Figure 4A). Neurons treated with 0.016–10 μmol/L of F-53B showed decreased expression of genes (Adcy1, PKA, and CREB1) and lowered cAMP levels in a dose-dependent manner (Figure 4B). Neurons exposed to F-53B and labeled with PKA(α/β/γ) demonstrated a noticeable ’cavity’ and a decrease in fluorescence area and density, suggesting a diminished nuclear localization of PKA(α/β/γ) following F-53B exposure (Figure 4C–D). Concurrently, HT22 neurons exposed to 0.4, 2, and 10 μmol/L of F-53B exhibited significant nuclear pyknosis (Figure 4C). Additionally, neurons treated with 2 and 10 μmol/L of F-53B showed reduced levels of PKA(α/β/γ), p-PKA(α/β/γ)-Thr197, and p-CREB1-Ser133 in the nucleus compared to the control group, with cytoplasmic levels remaining relatively unchanged (Figure 4F–I). These findings indicate that F-53B effectively sequesters PKA(α/β/γ) within the cytoplasm, inhibiting its nuclear translocation and consequently resulting in the downregulation of CREB1 and its phosphorylation within the nucleus.
Figure 4.

Effects of F-53B exposure on the PKA/CREB pathways in fetal mice and HT22 neurons. (A-B) The relative mRNA levels of the PKA/CREB pathways related genes in the fetal brain and HT22 neurons. (C-E) Representative photomicrographs of PKA levels and location in HT22 neurons. (F-I) The blots and diagrams of the PKA/CREB pathways related protein in the nucleus and cytoplasm of HT22 neurons. Comparison with the control group is indicated by the symbol “*”. Comparison with the 10 μmol/L F-53B-exposed group is indicated by the symbol “&”. * and & indicate p < 0.05, ** and && indicate p < 0.01, *** and &&& indicate p < 0.001.
3.4. Effects of F-53B on Synaptic Plasticity Markers Depend on the PKA/CREB Pathway
A PKA agonist and antagonist were introduced to investigate the impact of F-53B on the PKA/CREB signaling pathway and synaptic plasticity. Results indicate that a concentration of 100 μmol/L of the PKA agonist effectively enhanced mRNA levels of PKA, while 10 μmol/L of the PKA antagonist showed maximal inhibition (Figure 5A-B). Cell viability for treatments involving these doses of PKA agonist/antagonist in combination with F-53B (0.08, 0.4, and 2 μmol/L) exceeded 80%, except when coexposed to 2 μmol/L of F-53B with the PKA antagonist (Figure 5C). As shown in Figures 5D-F, nuclear protein expression levels of p-PKA(α/β/γ)-Thr197 and p-CREB1-Ser133 in neurons not exposed to F-53B increased in response to the PKA agonist and decreased with the PKA antagonist. In neurons exposed to F-53B, reduced levels of p-PKA(α/β/γ)-Thr197 and p-CREB1-Ser133 in the nucleus were reversed with the PKA agonist and further diminished by the PKA antagonist. Additionally, the PKA agonist was observed to restore the diminished total protein levels of SYP due to exposure to 0.04 and 2 μmol/L of F-53B. A similar trend was observed in the levels of BDNF and GAP43, albeit without reaching statistical significance (Figure 5G-J). These findings imply that F-53B-induced impairment of synaptic plasticity could be modulated through the PKA/CREB signaling pathway.
Figure 5.

Dependency of F-53B-declined synaptic proteins levels in HT22 neurons on the PKA/CREB pathway. (A) The survival rate of HT22 neurons treated with a PKA agonist and antagonist. (B) The relative mRNA levels of PKA in HT22 neurons treated with a PKA agonist and antagonist. (C) The survival rate of HT22 neurons treated with both a PKA agonist or antagonist and F-53B. (D-F) The nuclear protein blots and diagrams of the PKA/CREB pathways in HT22 neurons treated with both a PKA agonist or antagonist and F-53B. (G-J) The total protein blots and diagrams of the synaptic plasticity in HT22 neurons treated with both a PKA agonist or antagonist and F-53B. The “*” indicates a significant difference compared to the groups without F-53B, PKA agonist, and PKA antagonist. “*” and “&” indicate p < 0.05, “**” and “&&” indicate p < 0.01, “***” and “&&&” indicate p < 0.001.
4. Discussion
In this study, we investigated the role of the PKA/CREB pathway in the expression of synaptic plasticity markers caused by F-53B in fetal mice and HT22 neurons. Hindrances were observed in the translocation of PKA into neuronal nuclei, an effect attributed to F-53B. This led to reduced phosphorylation of PKA and CREB1 in neuronal nuclei. Furthermore, it was found that activation and inhibition of the PKA/CREB pathway could ameliorate and worsen the expressions of F-53B-altered synaptic plasticity proteins, respectively. These findings highlight the neurotoxicity risk associated with F-53B exposure during gestation and contribute to an enhanced understanding of the mechanisms underlying synaptic damage caused by F-53B.
This study revealed for the first time that prenatal exposure to F-53B adversely affects synaptic plasticity in the developing fetal brain. Previous studies have shown that intrauterine exposure to PFOS is associated with structural damage at the synaptic levels and down-regulation of genes such as Synapsin1/2/3 in the hippocampus of neonatal rats.25,26 Mice exposed to PFOS during lactation demonstrated abnormal synaptogenesis or impaired pre- and postsynaptic plasticity.27,28 Similarly, prenatal and postnatal exposure to PFOS can both disrupt synaptic transmission pathways in weaned rats.29 These findings underscore the health risks to offspring arising from maternal exposure to PFOS. However, an epidemiological study has shown that the maternal transfer coefficient of F-53B exceeds that of PFOS,3 implying that maternal F-53B may penetrate the placental barrier and negatively impact fetal health. This hypothesis is supported by our preceding research, which demonstrated memory impairment and lower expression of synaptic plasticity markers in the hippocampus of weaned mice exposed to F-53B during pregnancy and lactation.9 This study further explores the expression of synaptic plasticity markers in fetal mouse brains based on our previous animal models.9 It suggests that F-53B-induced changes in these markers could potentially affecting synaptic development before birth and have enduring effects into adulthood, impacting cognitive functions such as learning and memory. Additionally, our findings reveal a novel association between F-53B exposure and altered expression of neural stem cell markers in the developing hippocampus of fetal mice. This implies a role for F-53B in neural stem cell differentiation, corroborating previous in vitro studies.10,11
We focused on exploring how the PKA/CREB signaling pathway regulates the expression of synaptic plasticity markers altered by F-53B. We first predicted the interactions of the primary constituents (6:2 Cl-PFASE and 8:2 Cl-PFASE) of F-53B with PKA and CREB1 through molecular docking. LYS residues, which are positively charged, form hydrogen bonds and ionic interactions, contributing to protein–ligand stability and specificity. In our study, LYS-189, LYS-217, and LYS-192 in PKA, and LYS-285 in CREB1, formed hydrogen bonds with 6:2 Cl-PFASE and 8:2 Cl-PFASE through their amine groups interacting with the fluorinated and chlorinated segments. ASN, a polar amino acid, participates in hydrogen bonding due to its amide group. ASN-216 in PKA formed hydrogen bonds with 8:2 Cl-PFASE, enhancing binding specificity. ARG, another positively charged residue, engages in multiple hydrogen bonds and electrostatic interactions. ARG-286 in CREB1 interacted with 8:2 Cl-PFASE, where its guanidinium group increases binding affinity. GLU, a negatively charged residue, forms strong hydrogen bonds and salt bridges. GLU-287 in CREB1 formed hydrogen bonds with 8:2 Cl-PFASE, contributing to the protein–ligand complex’s stability. These interactions underline the importance of these residues in stabilizing and enhancing the specificity of the binding between PKA, CREB1, and the fluorinated and chlorinated segments of 6:2 Cl-PFASE and 8:2 Cl-PFASE. Previous studies have shown that PFNA and PFOS down-regulate genes within the PKA/CREB signaling pathway, resulting in aberrant synaptic development and epilepsy-like behavior in zebrafish embryos.20,30 Furthermore, the depressive-like behavior observed in mice was associated with PFOS-inhibited expressions of synaptic proteins in the hippocampus, modulated by the suppressed PKA/CREB signaling pathway.31 The termination of PKA/CREB signaling is due to the deactivation of nuclear PKA activity and the phosphorylation of CREB-S133. When nuclear PKA activity is suppressed, its ability to phosphorylate CREB-S133 is reduced, thereby weakening CREB’s capacity to bind to the cAMP response element (CRE) on DNA. This binding is essential for initiating the transcription of genes involved in synaptic growth, differentiation, and plasticity, including those coding for BDNF and other neurotrophins that support synaptic development, such as GAP43 and SYP.32,33 Our in vitro study addresses the gap in understanding how newer fluorinated compounds like F-53B affect critical signaling pathways. Specifically, we noted that F-53B prompted cytoplasmic sequestration of PKA and significantly reduced expression of PKA, CREB, and phosphorylated PKA-Thr197 and CREB-S133 in the nuclei of hippocampal neurons. Importantly, the downregulation of synaptic plasticity-related genes by F-53B could be reversed by the PKA agonist and was further inhibited by the PKA antagonist. Consistent interventions with the PKA agonist activated the PKA/CREB signaling pathway, thus leading to the restoration of synaptic protein levels in mice and hippocampal neurons.34,35 Furthermore, the PKA inhibitor was shown to disrupt long-term potentiation (LTP) of synaptic efficacy, while PKA activation was found to promote LTP.36 A study has presented evidence that F-53B significantly reduces the amplitude of LTP and field excitatory postsynaptic potential in adult rats.37 These findings emphasized the role of the PKA/CREB signaling pathway in synaptic markers expressions upon F-53B exposure.
This study presents several notable strengths. First, it highlights the impact of prenatal exposure to F-53B on synaptic plasticity markers in fetuses, underscoring the critical need to consider the intergenerational risks associated with this chemical in humans. Second, it provides the first exploration of the mechanism by which F-53B reduces levels of synaptic plasticity markers, specifically by blocking the entry of PKA into the neuronal nucleus. By elucidating the interaction between F-53B and the PKA/CREB signaling cascade, our study provides crucial insights into the broader implications of fluorinated compounds on neural health and synaptic integrity, and pave the way for further research into mitigating the neurotoxic impacts of F-53B. Third, the study offers critical toxicological data, including the IC50 and BMD for F-53B on neuronal cells, which are instrumental for future neurotoxicity research. However, it is crucial to acknowledge the limitations of this study, which necessitate further improvement. We have provided evidence of synaptic damage by F-53B only at the protein and mRNA levels, lacking electrophysiological or real-time imaging techniques to confirm the results. Moreover, the exposure doses for mice were limited to fewer than three groups, corresponding to internal exposure concentrations observed in both general and occupational populations. This limitation impedes the establishment of a clear dose–response relationship, an issue that will be addressed in future studies.
5. Conclusion
In conclusion, our research findings indicate that prenatal exposure to F-53B exerts detrimental effects on hippocampal neural stem cell differentiation and synaptic development in fetal mice. F-53B can bind to the active sites of PKA and CREB1, thereby inhibiting PKA nuclear entry and causing its cytoplasmic retention. This prevents the subsequent phosphorylation of nuclear proteins, leading to reduced expression of synaptic plasticity markers. The administration of a PKA agonist effectively reverses the reduced levels of syn aptic plasticity proteins induced by F-53B by activating the PKA/CREB signaling pathway. These results offer new insights into the underlying mechanisms behind F-53B-damaged synaptic plasticity. The safety supervision and risk assessment regarding F-53B in pregnant women and fetuses should be intensively enhanced in the future, especially focusing on exploring potential therapeutic interventions to mitigate the adverse effects of F-53B on neural development.
Acknowledgments
This study was supported by the National Science Foundation of China (No.82073503, 82173471, 82003409, 82103823), the Natural Science Foundation of Guangdong Province (No. 2021B1515020015, 2021A1515012212) and the Guangzhou Science and Technology Plan Project (No.2024A04J6476).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/envhealth.4c00098.
Additional experimental details, materials, and methods, including primer sequences used for qPCR in this study, and additional experimental results, including Organ coefficient of fetal mice (PDF)
Author Contributions
Shen-Pan Li: Conceptualization, Methodology, Software, Validation, Formal analysis, Investigation, Data curation, Writing - original draft, Writing - review and editing, Visualization. Hui-Xian Zeng: Writing - review &editing. Shuang-Jian Qin: Writing - review &editing. Qing–Qing Li: Methodology. Lu-Yin Wu: Methodology. Qi-Zhen Wu: Data curation. Li-Zi Lin: Supervision. Guang-Hui Dong: Supervision, Conceptualization, Funding acquisition. Xiao-Wen Zeng: Conceptualization, Funding acquisition, Resources, Supervision, Project administration.
This work has received approval for research ethics from the Biomedical Research Ethics Committee of the School of Public Health at Sun Yat-Sen University (Approval No. SYSU-IACUC-2022-001602) and a proof/certificate of approval is available upon request.
The authors declare no competing financial interest.
Supplementary Material
References
- He Y.; Lv D.; Li C.; Liu X.; Liu W.; Han W. Human exposure to F-53B in China and the evaluation of its potential toxicity: An overview. Environ. Int. 2022, 161, 107108 10.1016/j.envint.2022.107108. [DOI] [PubMed] [Google Scholar]
- Tu W.; Martinez R.; Navarro-Martin L.; Kostyniuk D. J.; Hum C.; Huang J.; et al. Bioconcentration and metabolic effects of emerging PFOS alternatives in developing zebrafish. Environ. Sci. Technol. 2019, 53 (22), 13427–13439. 10.1021/acs.est.9b03820. [DOI] [PubMed] [Google Scholar]
- Gao K.; Zhuang T.; Liu X.; Fu J.; Zhang J.; Fu J.; et al. Prenatal Exposure to Per- and Polyfluoroalkyl Substances (PFASs) and Association between the Placental Transfer Efficiencies and Dissociation Constant of Serum Proteins-PFAS Complexes. Environ. Sci. Technol. 2019, 53 (11), 6529–6538. 10.1021/acs.est.9b00715. [DOI] [PubMed] [Google Scholar]
- Kang Q.; Gao F.; Zhang X.; Wang L.; Liu J.; Fu M.; et al. Nontargeted identification of per- and polyfluoroalkyl substances in human follicular fluid and their blood-follicle transfer. Environ. Int. 2020, 139, 105686 10.1016/j.envint.2020.105686. [DOI] [PubMed] [Google Scholar]
- Bao J.; Shao L.-X.; Liu Y.; Cui S.-W.; Wang X.; et al. Target analysis and suspect screening of per-and polyfluoroalkyl substances in paired samples of maternal serum, umbilical cord serum, and placenta near fluorochemical plants in Fuxin, China. Chemosphere. 2022, 307 (pt1), 135731 10.1016/j.chemosphere.2022.135731. [DOI] [PubMed] [Google Scholar]
- Wang J.; Pan Y.; Cui Q.; Yao B.; Wang J.; Dai J. Penetration of PFASs Across the Blood Cerebrospinal Fluid Barrier and Its Determinants in Humans. Environ. Sci. Technol. 2018, 52 (22), 13553–13561. 10.1021/acs.est.8b04550. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Li Q.; Wang P.; Li J.; Zhao W.; Zhang L.; et al. Associations of prenatal PFAS exposure and early childhood neurodevelopment: Evidence from the Shanghai Maternal-Child Pairs Cohort. Environ. Int. 2023, 173, 107850 10.1016/j.envint.2023.107850. [DOI] [PubMed] [Google Scholar]
- Wu L.; Zeeshan M.; Dang Y.; Zhang Y.-T.; Liang L.-X.; Huang J.-W. Maternal transfer of F-53B inhibited neurobehavior in zebrafish offspring larvae and potential mechanisms: Dopaminergic dysfunction, eye development defects and disrupted calcium homeostasis. Sci. Total Environ. 2023, 894, 164838. 10.1016/j.scitotenv.2023.164838. [DOI] [PubMed] [Google Scholar]
- Liang L.-X.; Liang J.; Li Q.-Q.; Zeeshan M.; Zhang Z.; Jin N.; et al. Early life exposure to F-53B induces neurobehavioral changes in developing children and disturbs dopamine-dependent synaptic signaling in weaning mice. Environ. Int. 2023, 181, 108272–108272. 10.1016/j.envint.2023.108272. [DOI] [PubMed] [Google Scholar]
- Yin N.; Yang R.; Liang S.; Liang S.; Hu B.; Ruan T.; et al. Evaluation of the early developmental neural toxicity of F-53B, as compared to PFOS, with an in vitro mouse stem cell differentiation model. Chemosphere 2018, 204, 109–118. 10.1016/j.chemosphere.2018.04.011. [DOI] [PubMed] [Google Scholar]
- Qiu Y.; Gao M.; Cao T.; Wang J.; Luo M.; Liu S.; et al. PFOS and F-53B disrupted inner cell mass development in mouse preimplantation embryo. Chemosphere 2024, 349, 140948 10.1016/j.chemosphere.2023.140948. [DOI] [PubMed] [Google Scholar]
- Wu L.; Zeeshan M.; Dang Y.; Zhang Y.-T.; Liang L.-X.; Huang J.-W.; et al. Maternal transfer of F-53B inhibited neurobehavior in zebrafish offspring larvae and potential mechanisms: Dopaminergic dysfunction, eye development defects and disrupted calcium homeostasis. Sci. Total Environ. 2023, 894, 164838 10.1016/j.scitotenv.2023.164838. [DOI] [PubMed] [Google Scholar]
- Hall J.; Bray N. J. Schizophrenia Genomics: Convergence on Synaptic Development, Adult Synaptic Plasticity, or Both?. Biol. Psychiatry 2022, 91 (8), 709–717. 10.1016/j.biopsych.2021.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B.; Zhao T.; Chen X.-X.; Zhu Y.-Y.; Lu X.; Qian Q.-H.; et al. Gestational 1-nitropyrene exposure causes anxiety-like behavior partially by altering hippocampal epigenetic reprogramming of synaptic plasticity in male adult offspring. J. Hazard. Mater. 2023, 453, 131427 10.1016/j.jhazmat.2023.131427. [DOI] [PubMed] [Google Scholar]
- Subbanna S.; Basavarajappa B. A.-O. Binge-like Prenatal Ethanol Exposure Causes Impaired Cellular Differentiation in the Embryonic Forebrain and Synaptic and Behavioral Defects in Adult Mice. Brain. Sci. 2022, 12 (6), 793. 10.3390/brainsci12060793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M.; Gobert D.; Stern E.; Gamache K.; Colina R.; Cuello C.; et al. eIF2α phosphorylation bidirectionally regulates the switch from short- to long-term synaptic plasticity and memory. Cell. 2007, 129 (1), 195–206. 10.1016/j.cell.2007.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.-Y.; Huang B. X.; Spector A. A. Molecular and Signaling Mechanisms for Docosahexaenoic Acid-Derived Neurodevelopment and Neuroprotection. Int. J. Mol. Sci. 2022, 23 (9), 4635. 10.3390/ijms23094635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.; Zhang C.; Lv J.; Zhu X.; Jiang X.; Lu W.; et al. Antiallergic drug desloratadine as a selective antagonist of 5HT2A receptor ameliorates pathology of Alzheimer’s disease model mice by improving microglial dysfunction. Aging. Cell. 2021, 20 (1), e13286 10.1111/acel.13286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown-Leung J. M.; Cannon J. R. Neurotransmission Targets of Per- and Polyfluoroalkyl Substance Neurotoxicity: Mechanisms and Potential Implications for Adverse Neurological Outcomes. Chem. Res. Toxicol. 2022, 35 (8), 1312–1333. 10.1021/acs.chemrestox.2c00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.; Qiu W.; Li R.; Chen B.; Wu X.; Magnuson J. T.; et al. Perfluorononanoic Acid Induces Neurotoxicity via Synaptogenesis Signaling in Zebrafish. Environ. Sci. Technol. 2023, 57 (9), 3783–3793. 10.1021/acs.est.2c06739. [DOI] [PubMed] [Google Scholar]
- Shi Y.; Vestergren R.; Xu L.; Zhou Z.; Li C.; Liang Y.; et al. Human exposure and elimination kinetics of chlorinated polyfluoroalkyl ether sulfonic acids (Cl-PFESAs). Environ. Sci. Technol. 2016, 50 (5), 2396–2404. 10.1021/acs.est.5b05849. [DOI] [PubMed] [Google Scholar]
- Li S.; Wu L.; Zeng H.; Zhang J.; Qin S.; Liang L.-X.; et al. Hepatic injury and ileitis associated with gut microbiota dysbiosis in mice upon F-53B exposure. Environ. Res. 2024, 248, 118305 10.1016/j.envres.2024.118305. [DOI] [PubMed] [Google Scholar]
- Zhang S.; Chen K.; Li W.; Chai Y.; Zhu J.; Chu B. Varied thyroid disrupting effects of perfluorooctanoic acid (PFOA) and its novel alternatives hexafluoropropylene-oxide-dimer-acid (GenX) and ammonium 4,8-dioxa-3H-perfluorononanoate (ADONA) in vitro. Environ. Int. 2021, 156, 106745. 10.1016/j.envint.2021.106745. [DOI] [PubMed] [Google Scholar]
- Xin Y.; Ren X.-M.; Ruan T.; Li C.-H.; Guo L.-H.; Jiang G. Chlorinated polyfluoroalkylether sulfonates exhibit similar binding potency and activity to thyroid hormone transport proteins and nuclear receptors as perfluorooctanesulfonate. Environ. Sci. Technol. 2018, 52 (16), 9412–9418. 10.1021/acs.est.8b01494. [DOI] [PubMed] [Google Scholar]
- Zeng H. C.; Li Y. Y.; Li Y. y.; Zhang L.; Wang Y. J.; Chen J.; et al. Prenatal exposure to perfluorooctanesulfonate in rat resulted in long-lasting changes of expression of synapsins and synaptophysin. Synapse. 2011, 65 (3), 225–233. 10.1002/syn.20840. [DOI] [PubMed] [Google Scholar]
- Zeng H.-c.; Zhang L.; Li Y.-y.; Wang Y.-j.; Xia W.; Lin Y.; et al. Inflammation-like glial response in rat brain induced by prenatal PFOS exposure. Neurotoxicology 2011, 32 (1), 130–139. 10.1016/j.neuro.2010.10.001. [DOI] [PubMed] [Google Scholar]
- Ninomiya A.; Mshaty A.; Haijima A.; Yajima H.; Kokubo M.; Khairinisa M. A.; et al. The neurotoxic effect of lactational PFOS exposure on cerebellar functional development in male mice. Food. Chem. Toxicol. 2022, 159, 112751 10.1016/j.fct.2021.112751. [DOI] [PubMed] [Google Scholar]
- Johansson N.; Eriksson P.; Viberg H. Neonatal Exposure to PFOS and PFOA in Mice Results in Changes in Proteins which are Important for Neuronal Growth and Synaptogenesis in the Developing Brain. Toxicol. Sci. 2009, 108 (2), 412–418. 10.1093/toxsci/kfp029. [DOI] [PubMed] [Google Scholar]
- Wang F.; Liu W.; Jin Y.; Dai J.; Yu W.; Liu X.; et al. Transcriptional effects of prenatal and neonatal exposure to PFOS in developing rat brain. Environ. Sci. Technol. 2010, 44 (5), 1847–1853. 10.1021/es902799f. [DOI] [PubMed] [Google Scholar]
- Lee H.; Tran C. M.; Jeong S.; Kim S. S.; Bae M. A.; Kim K.-T. Seizurogenic effect of perfluorooctane sulfonate in zebrafish larvae. Neurotoxicology 2022, 93, 257–264. 10.1016/j.neuro.2022.10.007. [DOI] [PubMed] [Google Scholar]
- An Z.; Yang J.; Xiao F.; Lv J.; Xing X.; Liu H.; et al. Hippocampal Proteomics Reveals the Role of Glutamatergic Synapse Activation in the Depression Induced by Perfluorooctane Sulfonate. J. Agric. Food. Chem. 2023, 71 (20), 7866–7877. 10.1021/acs.jafc.3c01344. [DOI] [PubMed] [Google Scholar]
- Bedioune I.; Lefebvre F.; Lechene P.; Varin A.; Domergue V.; Kapiloff M. S.; et al. PDE4 and mAKAPβ are nodal organizers of β2-ARs nuclear PKA signalling in cardiac myocytes. Cardiovasc. Res. 2018, 114 (11), 1499–1511. 10.1093/cvr/cvy110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- More L.; Privitera L.; Perrett P.; Cooper D. D.; Bonnello M. V. G.; Arthur J. S. C.; et al. CREB serine 133 is necessary for spatial cognitive flexibility and long-term potentiation. Neuropharmacology 2022, 219, 109237 10.1016/j.neuropharm.2022.109237. [DOI] [PubMed] [Google Scholar]
- Ji M.; Zhang Z.; Gao F.; Yang S.; Wang J.; Wang X.; et al. Curculigoside rescues hippocampal synaptic deficits elicited by PTSD through activating cAMP-PKA signaling. Phytother. Res. 2023, 37 (2), 759–773. 10.1002/ptr.7658. [DOI] [PubMed] [Google Scholar]
- Malijauskaite S.; Sauer A. K.; Hickey S. E.; Franzoni M.; Grabrucker A. M.; McGourty K. Identification of the common neurobiological process disturbed in genetic and non-genetic models for autism spectrum disorders. Cell. Mol. Life. Sci. 2022, 79 (12), 589. 10.1007/s00018-022-04617-3. [DOI] [PubMed] [Google Scholar]
- Hashimotodani Y.; Nasrallah K.; Jensen K. R.; Chavez A. E.; Carrera D.; Castillo P. E. LTP at Hilar Mossy Cell-Dentate Granule Cell Synapses Modulates Dentate Gyrus Output by Increasing Excitation/Inhibition Balance. Neuron. 2017, 95 (4), 928. 10.1016/j.neuron.2017.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q.; Liu W.; Niu Q.; Wang Y.; Zhao H.; Zhang H.; et al. Effects of perfluorooctane sulfonate and its alternatives on long-term potentiation in the hippocampus CA1 region of adult rats in vivo. Toxicol. Res. (Camb) 2016, 5 (2), 539–546. 10.1039/C5TX00184F. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.