

Abstract
Knowledge graphs, powerful tools that explicitly transfer knowledge to machines, have significantly advanced new knowledge inferences. Discovering unknown relationships between diseases and genes/proteins in biomedical knowledge graphs can lead to the identification of disease development mechanisms and new treatment targets. Generating high‐quality representations of biomedical entities is essential for successfully predicting disease‐gene/protein associations. We developed a computational model that predicts disease‐gene/protein associations using the Precision Medicine Knowledge Graph, a biomedical knowledge graph. Embeddings of biomedical entities were generated using two different methods—a large language model (LLM) and the knowledge graph embedding (KGE) algorithm. The LLM utilizes information obtained from massive amounts of text data, whereas the KGE algorithm relies on graph structures. We developed a disease‐gene/protein association prediction model, “Cross‐Modal Embedding Integrator (CMEI),” by integrating embeddings from different modalities using a multi‐head attention mechanism. The area under the receiver operating characteristic curve of CMEI was 0.9662 (± 0.0002) in predicting disease‐gene/protein associations. In conclusion, we developed a computational model that effectively predicts disease‐gene/protein associations. CMEI may contribute to the identification of disease development mechanisms and new treatment targets.
Keywords: disease, gene, knowledge graph embedding, large language model, multi‐head attention, protein
Abbreviations
- AUC
area under the receiver operating characteristic curve
- AUPR
area under the precision‐recall curve
- CMEI
cross‐modal embedding integrator
- GDF1
growth differentiation factor 1
- GNN
graph neural network
- HolE
holographic embeddings
- IL6
interleukin 6
- KGE
knowledge graph embedding
- LLM
large language model
- lncRNA
long non‐coding RNA
- miRNA
microRNA
- MLP
multilayer perceptron
- PPI
protein–protein interaction
- PrimeKG
precision medicine knowledge graph
1. INTRODUCTION
Discovering unknown associations between diseases and genes/proteins can reveal novel mechanisms of disease development and potential therapeutic targets. 1 Therefore, it is a critical starting point for drug development; however, it requires expensive and time‐consuming biological experiments. 2 , 3 A computational model that predicts unknown associations between diseases and genes/proteins can aid the development of new drugs.
Knowledge graphs express knowledge as “head entity, relation, and tail entity,” which explicitly transfer knowledge to machines. 4 They have led to major advances in inferring new knowledge and have found utility in various fields. A biomedical knowledge graph comprises entities such as diseases, genes, proteins, drugs, and biological processes, and their relationships. Knowledge that is yet to be discovered remains a missing link in knowledge graphs. New knowledge can be created by identifying the missing links. For instance, identifying unknown relationships between drugs and diseases may facilitate drug repurposing. 5 Discovering unknown associations between diseases and genes/proteins may help in drug discovery. 6 Biomedical knowledge graphs could be a good database for identifying unknown associations between diseases and genes/proteins.
Inferring new knowledge requires representations that preserve the meaning of each entity and the relationships between them. Generating high‐quality representations of biomedical entities is essential for successfully performing downstream tasks, such as link prediction and node classification. The representations of biomedical entities can be generated using the graph structure, including the relationships between biomedical entities. Various knowledge graph embedding (KGE) algorithms have been developed. 7 , 8 , 9 Among these, DistMult represents relational embeddings as diagonal matrices, which facilitates the learning by reducing the parameter space. 7 , 10 Holographic embeddings (HolE) utilize circular correlation to acquire various interactions. 8 , 10 RotatE considers the relation from head entity to tail entity as rotation and represents relations and entities to the complex latent space. 9 , 10 Several models for predicting the association between biomedical entities have been developed using the KGEs. 11 , 12 Another approach, graph neural networks (GNNs), has been used for developing disease‐gene/protein association prediction models. 13 , 14 , 15 Han et al. utilized GNN and matrix factorization to identify the associations between diseases and genes. 13 Cinaglia et al. suggested a disease‐gene association prediction model consisting of an encoder and a decoder using GNN. 14 As these methods obtain information about biomedical entities based on the graph structures, a graph database must be established before developing the prediction model. Recently, a large‐scale biomedical knowledge graph has been generated. 16 , 17 , 18 , 19 , 20 This may further facilitate the development of a computational model for predicting disease‐gene/protein associations.
In addition to the graph structures, biomedical entities contain considerable amounts of information. Therefore, a better representation may be generated by utilizing additional information that reflects the characteristics of biomedical entities. Zhou et al. showed that in predicting circular RNA‐microRNA (miRNA) interactions, better predictive performance can be achieved by adding features reflecting entity characteristics to the features obtained from the network structure. 21 Accordingly, a better performance may be achieved in predicting the disease‐gene/protein associations by adding information that can reflect the disease and gene/protein characteristics to the features obtained from the graph structure.
Recently, with the rapid development of large language models (LLMs), such as ChatGPT, 22 , 23 LLMs have played a role in complementing knowledge graphs. In LLM‐augmented knowledge graphs, the LLM plays a role in generating embeddings, structuring knowledge graphs, and generating text from knowledge graphs, 24 thereby reducing its limitations and increasing usefulness. In the node classification task using graph datasets, it has been reported that embeddings obtained by the LLM can be used as the initial embeddings of the nodes in the GNN to improve performance. 25 Therefore, leveraging information from two different modalities—information from an LLM, which is obtained from massive text data, and information from the graph structure—may contribute to a better performance. We developed a disease‐gene/protein association prediction model, “Cross‐Modal Embedding Integrator (CMEI),” which integrated the embeddings, obtained using an LLM 26 and the KGE algorithm, 7 , 8 , 9 via a multi‐head attention mechanism. 27 CMEI integrated various embeddings with appropriate weights using a multi‐head attention mechanism and showed favorable performance.
2. MATERIALS AND METHODS
2.1. Knowledge graph
Prediction models were developed with Precision Medicine Knowledge Graph (PrimeKG), 19 , 20 which is available in Harvard Dataverse at https://doi.org/10.7910/DVN/IXA7BM. 19 , 20 PrimeKG 19 , 20 is a heterogeneous graph and has the following 10 node types: gene/protein (n = 27 671), drug (n = 7957), effect/phenotype (n = 15 311), disease (n = 17 079), biological process (n = 28 642), molecular function (n = 11 169), cellular component (n = 4176), exposure (n = 818), pathway (n = 2498), and anatomy (n = 14 035). A total of 26 types of relationships between nodes were as follows: disease‐gene/protein, gene/protein‐effect/phenotype, gene/protein‐anatomy, gene/protein‐biological process, gene/protein‐cellular component, gene/protein‐molecular function, gene/protein‐pathway, drug‐disease, drug‐gene/protein, drug‐effect/phenotype, disease‐effect/phenotype, exposure‐disease, exposure‐gene/protein, exposure‐biological process, exposure‐cellular component, exposure‐molecular function, gene/protein‐gene/protein, drug–drug, disease‐disease, effect/phenotype‐effect/phenotype, anatomy‐anatomy, exposure‐exposure, biological process‐biological process, cellular component‐cellular component, molecular function‐molecular function, and pathway‐pathway. Among these, the relationships between drugs and genes/proteins were subdivided into carriers, enzymes, targets, and transporters; however, all these were considered to have the same relationships in the present study. The relationships between drugs and diseases were divided into indications, contraindications, and off‐label use, of which only indications were used in this study. Relationships between diseases and phenotypes were divided into positive and negative relationships, and only positive relationships were used in the current study. The relationships between anatomies and genes/proteins were divided into present and absent, and only present relationships were used in this study.
2.2. Construction of dataset
A total of 80 411 edges between diseases and genes/proteins were randomly assigned to four groups—message passing dataset (n = 45 031), training dataset (n = 19 298), validation dataset (n = 8041), and testing dataset (n = 8041) using “RandomLinkSplit” of PyTorch Geometric. 28 Other edges, except for edges between diseases and genes/proteins, were assigned to the message‐passing dataset. The training, validation, and testing datasets consisted only of edges between diseases and genes/proteins. Some edges between diseases and genes/proteins were adjusted to ensure that there were no isolation nodes in the message‐passing dataset. The number of edges in each dataset remained the same after the adjustment. The ratio of the number of edges between diseases and genes/proteins belonging to the message passing and training datasets was 7:3. The validation and test datasets included 10% of the total edges between the diseases and genes/proteins.
The training, validation, and testing datasets require negative pairs for model learning and evaluation, which in this study were disease and gene/protein combinations with no edges. Negative pairs were generated through the following process: pairs were generated by combining diseases and genes/proteins from the dataset and pairs with edges removed. Finally, from the remaining ones, some pairs were selected and used as negative pairs. However, the frequencies of diseases and genes/proteins in the positive pairs comprising the edges of the training, validation, and testing datasets were uneven. For example, “hereditary breast‐ovarian cancer syndrome” was included in 280 edges, whereas “asthma” was included in 16 edges of the training datasets. Among the genes/proteins, “interleukin 6 (IL6)” was included in 63 edges of the training datasets, whereas “growth differentiation factor 1 (GDF1)” was included in only 3 edges. This indicates that if the disease and gene/protein pairs are randomly selected to form negative pairs, biased predictions may occur because of differences in disease and gene/protein frequencies between the positive and negative pairs. To avoid this, if the frequency of diseases and genes/proteins in positive pairs constituting the entire edge of the training, validation, and testing dataset or negative pairs in the whole dataset were 20 or more, negative pairs were constructed to ensure that the frequency ratio of disease or gene/protein between the positive and negative pairs was more than 0.5 and less than 2. This rule also applies to positive pairs that constitute the edges of the training dataset as well as to negative pairs of the training dataset. Thus, 59 359 negative pairs were constructed. Based on the edge ratio, 32 377 negative pairs were assigned to the training dataset, whereas 13 491 pairs each were assigned to the validation and testing datasets. Finally, the training dataset comprised 19 298 positive and 32 377 negative pairs, whereas the validation and testing datasets comprised 8041 positive and 13 491 negative pairs.
Embeddings by the KGE algorithm were generated using the message‐passing dataset. Prediction models using the KGE algorithm or LLM embeddings were developed using the training and validation datasets. The performances of the prediction models were evaluated using the testing dataset. The message‐passing edges used to develop a GNN‐based prediction model were constructed using the message‐passing, training, and validation datasets. The message‐passing edges during the training process were constructed using the message‐passing datasets. The message‐passing edges during the validation process were constructed using the message passing and training datasets. The message passing edges during the testing process were constructed using the message passing, training, and validation datasets. The training, validation, and testing datasets were used as supervision edges during the training, validation, and testing process, respectively.
2.3. Development of the prediction model
We developed CMEI, a prediction model integrating embeddings obtained using both the LLM 26 and KGE algorithm 7 , 8 , 9 via a multi‐head attention mechanism. 27 In addition to CMEI, we further developed a prediction model in the following three ways for model performance comparison:
Using only embeddings obtained via the KGE algorithms 7 , 8 , 9
Using only embeddings obtained via the LLM 26
Using embeddings obtained via the LLM 26 as the initial embeddings of the nodes in GNN 14 , 25
The embedding model of OpenAI 26 was used to obtain the LLM embeddings. DistMult, 7 HolE, 8 and RotatE 9 were used as the KGE algorithms. Experiments were conducted on five different seeds for representative models.
2.3.1. Integrating the embeddings generated by the KGE algorithms and LLM using a multi‐head attention mechanism
Embeddings reflecting the graph structure were generated using the following representative KGE algorithms: DistMult, 7 HolE, 8 and RotatE. 9 The PyKEEN package was used to generate the embeddings. 29 The embedding dimension was set as 1536 to match the embedding model of OpenAI 26 and was generated by changing the number of epochs by 50 from 200 to 300. Prediction models for associations between diseases and genes/proteins were developed using the generated embeddings. Input data for developing prediction models, that is, disease‐gene/protein pairs, were obtained by combining disease and gene/protein embeddings generated by the KGE algorithms via a vector operation—concatenation, element‐wise averaging, or element‐wise product. 30 The prediction model comprised two or three multilayer perceptron (MLP) layers. 31 , 32 The models were developed by modifying the model structure and adjusting the hyperparameters. Their performance was assessed using the area under the receiver operating characteristic curve (AUC), area under the precision‐recall curve (AUPR), accuracy, recall, precision, specificity, and the F1 score.
For each KGE algorithm, the prediction model with the best AUC value in the validation dataset was selected as the final model. The performance of the final model was assessed using the testing dataset. Their performance was compared with that of CMEI. The embeddings generated by the KGE algorithm, which exhibited the best AUC value in the testing dataset, were used to develop CMEI.
Next, the LLM embeddings were obtained using the embedding model of OpenAI, “text‐embedding‐ada‐002.” 26 A combination of two words, the type and name of the biomedical entity, was used as the input. For instance, for “osteogenesis imperfecta,” the embedding was obtained by entering “disease, osteogenesis imperfecta.” The number of embedding dimensions was 1536. As mentioned earlier, pairs of disease and gene/protein embeddings were combined using a vector operation 30 and passed through two or three MLP layers. 31 , 32 The associations between diseases and genes/proteins were predicted. The performance of the model was evaluated using the AUC, AUPR, accuracy, recall, precision, specificity, and the F1 score. The performance of the final model established using the LLM embeddings was compared with that of CMEI. These embeddings were also used for the development of CMEI.
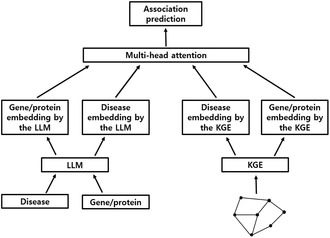
CMEI was developed by integrating the embeddings generated by the selected KGE algorithm 7 with those from the OpenAI 26 embedding model using a multi‐head attention mechanism (Figure 1). 27 The following embeddings were sequentially entered into the prediction model: gene/protein embedding by the LLM, disease embedding by the LLM, disease embedding by the KGE, and gene/protein embedding by the KGE. The prediction model was developed in two ways: adding a classification (CLS) token 33 as the first sequence or not. The embeddings were processed using the prediction module, including the multi‐head attention layer, 27 , 34 skip connection, 35 layer normalization, 36 , 37 feed‐forward layer, 31 , 32 skip connection, 35 and layer normalization. 36 , 37 One or two prediction modules were used. Learnable positional embeddings were applied. 27 , 33 The prediction value was obtained from the CLS token using an MLP layer 31 , 32 when the CLS token was used. The prediction value was obtained from the last output sequence when the CLS token was not used. The models were developed by varying the input order of embeddings, model structure, and hyperparameters. The input order of the embeddings, structure, and hyperparameters of the final model was determined based on the AUC value in the validation dataset. The performance of the final model was evaluated using the test dataset. PyTorch frameworks 38 was used to develop the prediction model. The “MultiheadAttention” 27 , 34 and “Linear” 31 , 32 of PyTorch were used as multi‐head attention and MLP layers, respectively. The “Adam” of PyTorch 39 , 40 was used as the optimizer, and the “Dropout” of PyTorch 41 , 42 was used for regularization. The learning rate was reduced by a factor of 0.1 if the AUC value did not improve in the validation dataset during 30 epochs using the “ReduceLROnPlateau” of PyTorch. 43 Learning was stopped if the AUC value in the validation dataset did not improve for 60 consecutive epochs.
FIGURE 1.
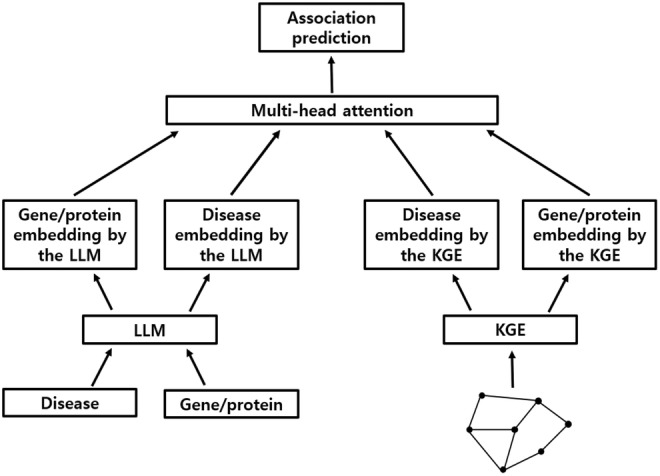
Structure of CMEI. CMEI, Cross‐Modal Embedding Integrator; LLM, large language model; KGE, knowledge graph embedding.
2.3.2. GNN‐based model established using embeddings generated by the LLM
To evaluate the performance of CMEI, a GNN‐based prediction model consisting of an encoder and a decoder was developed. 14 , 25 In the encoder, the embeddings generated by the LLM were used as the initial node embeddings. The initial embeddings were processed through GNN layers 44 , 45 to capture the graph structure. In the decoder, the associations between diseases and genes/proteins were predicted using the embeddings obtained from the encoder. The embeddings of the diseases and genes/proteins were combined using a vector operation—concatenation, element‐wise averaging, or element‐wise product. 30 The prediction value was obtained by passing them through the MLP layers. 31 , 46 The models were developed by changing the model structure and hyperparameters. The structure and hyperparameters of the final GNN‐based model were determined based on the AUC values of the validation dataset. The performance of the final model was evaluated using the test dataset. The PyTorch 38 and PyTorch Geometric 47 frameworks were used to develop the prediction model. The “SAGEConv” 44 , 45 and “Linear” 31 , 46 of PyTorch Geometric were used as GNN and MLP layers, respectively. The “Adam” of PyTorch 39 , 40 was used as the optimizer, and the “Dropout” of PyTorch 41 , 42 was used for regularization. A minibatch was not utilized. The learning rate was reduced by a factor of 0.1 if the AUC value did not improve in the validation dataset during 30 epochs using the “ReduceLROnPlateau” of PyTorch. 43 Learning was stopped if the AUC value in the validation dataset did not improve for 60 consecutive epochs.
2.4. Case study
Case studies have been conducted on breast, lung, colorectal, and prostate cancers, hepatocellular carcinoma, schizophrenia, anxiety disorders, and neurotic disorders. CMEI was applied to the testing dataset, and 30 genes/proteins predicted to be associated with each disease were selected based on the highest probability. The number of genes/proteins not identified in the knowledge graph used as the dataset was checked.
2.5. Ablation study
The ablation study was conducted such that the following four types of embedding were not used to develop prediction models individually: gene/protein embedding by the LLM, disease embedding by the LLM, disease embedding by the KGE, and gene/protein embedding by the KGE. Using the three types of embeddings, a prediction model was developed with the same structure as that of the final model, CMEI. The importance of each embedding in predicting the disease‐gene/protein association was evaluated by comparing the performances of all models.
3. RESULTS
3.1. Performance of the prediction models
Figure 2 and Table 1 present information about the performances of the prediction models for the testing dataset. The prediction models were established using only embeddings obtained by DistMult, 7 HolE, 8 and RotatE 9 performed the best when they consisted of two MLP layers 31 , 32 (size of each output sample: 1024 and 1) and used concatenations of disease and gene/protein embeddings 30 as the input data. Among KGE algorithms, DistMult 7 performed the best. Embeddings generated by DistMult 7 were used to develop CMEI. The prediction models established using only embeddings obtained by the LLM 26 performed the best when they consisted of two MLP layers 31 , 32 (size of each output sample: 1024 and 1) and used concatenations of disease and gene/protein embeddings 30 as the input data. GNN‐based models performed the best when they consisted of three GNN 44 , 45 (size of output sample: 256, 128, and 128) and four MLP 31 , 46 (size of output sample: 1024, 512, 256, and 1) layers and used element‐wise products of disease and gene/protein embeddings 14 , 30 as input data.
FIGURE 2.
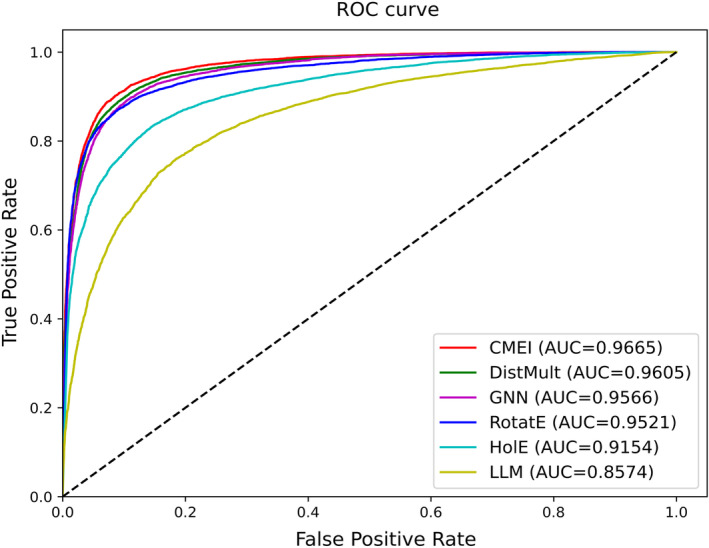
ROC curves of CMEI and baseline prediction models. Results of the representative models among the five trials performed by changing the seed number. DistMult, Rotate, HolE, and LLM refer to the prediction models established using the embeddings obtained from DistMult, RotatE, HolE, and the LLM, respectively. GNN refers to the GNN‐based model. CMEI, Cross‐Modal Embedding Integrator; GNN, graph neural network; HolE, holographic embeddings; LLM, large language model; ROC, receiver operating characteristic.
TABLE 1.
Results of the prediction models.
| CMEI | Prediction model established using the embeddings obtained from DistMult. 7 | Prediction model established using the embeddings obtained from HolE. 8 | Prediction model established using the embeddings obtained from RotatE. 9 | Prediction model established using the embeddings obtained from the LLM. 28 | GNN‐based model | |
|---|---|---|---|---|---|---|
| AUC | 0.9662 ± 0.0002 | 0.9604 ± 0.0001 | 0.9149 ± 0.0004 | 0.9518 ± 0.0002 | 0.8560 ± 0.0016 | 0.9545 ± 0.0014 |
| AUPR | 0.9469 ± 0.0007 | 0.9377 ± 0.0004 | 0.8836 ± 0.0009 | 0.9324 ± 0.0005 | 0.7983 ± 0.0025 | 0.9286 ± 0.0027 |
| F1 score | 0.8799 ± 0.0010 | 0.8716 ± 0.0014 | 0.8010 ± 0.0014 | 0.8606 ± 0.0005 | 0.7169 ± 0.0007 | 0.8602 ± 0.0029 |
| Accuracy | 0.9080 ± 0.0011 | 0.9043 ± 0.0010 | 0.8518 ± 0.0006 | 0.8964 ± 0.0004 | 0.8004 ± 0.0014 | 0.8944 ± 0.0021 |
| Recall | 0.9030 ± 0.0056 | 0.8699 ± 0.0026 | 0.7987 ± 0.0042 | 0.8564 ± 0.0042 | 0.6770 ± 0.0037 | 0.8696 ± 0.0078 |
| Precision | 0.8581 ± 0.0059 | 0.8734 ± 0.0025 | 0.8033 ± 0.0018 | 0.8648 ± 0.0035 | 0.7620 ± 0.0052 | 0.8510 ± 0.0062 |
| Specificity | 0.9110 ± 0.0048 | 0.9248 ± 0.0018 | 0.8834 ± 0.0019 | 0.9202 ± 0.0028 | 0.8739 ± 0.0042 | 0.9092 ± 0.0050 |
Note: Experiments were conducted using five different seeds. The results are reported as average ± standard deviation.
Abbreviations: AUC, area under the receiver operating characteristic curve; AUPR, area under the precision‐recall curve; CMEI, Cross‐Modal Embedding Integrator; GNN, graph neural network; HolE, Holographic embeddings; LLM, large language model.
The final structure of CMEI is shown in Figure 1 (Data S1). Embedding was entered into the prediction model in the following order: gene/protein embedding by the LLM, disease embedding by the LLM, disease embedding by the KGE, and gene/protein embedding by the KGE. The CLS token was not used. CMEI has one prediction module, including a multi‐head attention layer, 27 , 34 skip connection, 35 layer normalization, 36 , 37 feed‐forward layer, 31 , 32 skip connection, 35 and layer normalization. 36 , 37 An initial learning rate of 0.001 and minibatch size of 32 were used. The AUC value of CMEI for predicting disease‐gene/protein associations in the testing dataset was 0.9662 (± 0.0002). Among the prediction models, CMEI showed the best AUC value.
3.2. Case studies
Genes/proteins associated with breast, lung, colorectal, and prostate cancers, hepatocellular carcinoma, schizophrenia, anxiety disorders, and neurotic disorders were predicted using the testing dataset (Table 2). In breast cancer, hepatocellular carcinoma, and anxiety disorders, all the top 30 genes/proteins were identified in the knowledge graph used as the dataset. In lung cancer, colorectal cancer, prostate cancer, schizophrenia, and neurotic disorder, two, two, one, one, and one of the top 30 genes/proteins, respectively, were not identified in the knowledge graph used as the dataset.
TABLE 2.
Results of case studies using CMEI.
| Breast cancer | Lung cancer | Colorectal cancer | Prostate cancer | Hepatocellular carcinoma | Schizophrenia | Anxiety disorder | Neurotic disorder |
|---|---|---|---|---|---|---|---|
| PACC1 | RPL13A | ZNF569 | MMP9 | FABP5 | ACHE | HCRT | HTR2C |
| TFAP2D | MIR222 | ZNF442 | BCAS1 | FAM180A | HTR1A | HTR2A | HTR7 |
| LBX1 | CRP | TNS4 | NCOA7 | ANGPTL6 | HTR2C | GABRA6 | GRIA3 |
| C1orf87 | TP73 | LRRC47 | HAO1 | IRF2 | NCAM1 | HTR7 | GLO1 |
| COL19A1 | TYMS | PTPRU | SHBG | RNF157 | MTHFR | GRIK3 | TAC1 |
| ACCS | IL1B | ZNF480 | ALOX5 | LCAT | GRIA1 | CPLX1 | GRPR |
| FAM217B | PYCARD | JAKMIP2 | TLR4 | PYGL | GABBR1 | ADRA2A | OXT |
| PCDHGB6 | STN1 | ACTL9 | ENPP5 | TCF19 | HCRTR1 | MAOB | GRIN2A |
| CEP85L | TFRC | ALOX12B | ETV4 | BMPER | SST | GRIA1 | ADCY5 |
| MIA2 | NEK2 | TYMP | ARG2 | CLEC4G | ERVW‐4 | NEFM | GAP43 |
| PADI3 | EGFR | SH3TC1 | GSTM1 | MPO | SNCB | ADCY5 | PCLO |
| TAFA4 | MYC | IPP | TGFBR2 | CENPW | GRM4 | GDNF | CLOCK |
| KRTAP10‐8 | ZNF765* | CACUL1 | HRAS | MIR885 | APOE | CSMD2 | HSPB3 |
| AHSA2P | MIR19A | DMRTA1 | HMOX1 | TGM3 | GRM8 | PDE4D | SGCE |
| SCGB3A2 | MIR144 | SLC22A9 | ETV5 | MRO | TET1 | CDH7 | DBH |
| TLL1 | TNFSF10 | CHRM5* | RNF130 | CDCA8 | NRGN | CMYA5 | SOD1 |
| MFAP5 | H2BC4 | TNF | FOXA1 | PTTG1 | PDE4B | TG | MAPK8 |
| TBC1D9B | TP63 | SFRP2 | CLDN9 | CYP2C8 | NRXN1 | GAL | REN |
| ANKEF1 | RAF1 | GRID1 | IVL* | CCN1 | SLC29A1* | CCL24 | PDE4A |
| NID2 | RPS6KA6 | IFNG | ASZ1 | ACLY | TF | MAGI2 | RNF123 |
| LRRC37A | BAP1 | PAIP2 | TOM1L1 | HSD3B2 | ESR2 | RELN | GNB3 |
| WARS1 | DNMT3A | PTGS2 | SERPINB10 | MYBL2 | CAV1 | GAD1 | NRXN1 |
| MALAT1 | EEF2 | TCERG1L | CDKN1B | GPX3 | ATF4 | MMP8 | GSTT1 |
| DNAH9 | ERBB3 | ARHGEF10L | TMSB4X | IGF2BP3 | DISC1 | SOD1 | DGKB |
| FBXO8 | GSTM2 | SLC5A8 | BIRC5 | IL2 | DLGAP2 | PRL | BCL2 |
| ARAP3 | MLH1 | KRT71* | GSTK1 | HPSE | NCAN | MS | SFRP1 |
| MIR10A | RPL27A | NQO1 | HDAC6 | NNMT | HSPA1L | NRG1 | WFS1 |
| C16orf58 | FST* | GUCY1A1 | SLC7A1 | AKR1C2 | HDAC2 | MAPT | VEGFA |
| NOA1 | JAG1 | AKR1B10 | RPN2 | ATM | CHAT | DUSP1 | ERRFI1* |
| KIAA1324 | MIR155 | FPGS | ZFP36L2 | TP53 | ACP1 | IL18 | AKT1 |
Note: Asterisks in bold font indicate genes/proteins not identified in the knowledge graph used as the dataset.
Abbreviation: CMEI, Cross‐Modal Embedding Integrator.
3.3. Ablation study
The results of the ablation study are presented in Table 3. Excluding gene/protein embeddings by KGE resulted in the most significant deterioration in the performance of the model, while excluding gene/protein embeddings by LLM caused the least deterioration.
TABLE 3.
Results of the ablation study.
| Excluded embedding | Gene/protein embedding by the LLM | Disease embedding by the LLM | Disease embedding by the KGE | Gene/protein embedding by the KGE |
|---|---|---|---|---|
| AUC | 0.9653 ± 0.0002 | 0.9633 ± 0.0002 | 0.9520 ± 0.0005 | 0.8794 ± 0.0031 |
| AUPR | 0.9455 ± 0.0008 | 0.9404 ± 0.0004 | 0.9261 ± 0.0008 | 0.8236 ± 0.0052 |
| F1 score | 0.8774 ± 0.0026 | 0.8739 ± 0.0034 | 0.8552 ± 0.0018 | 0.7432 ± 0.0032 |
| Accuracy | 0.9051 ± 0.0032 | 0.9018 ± 0.0047 | 0.8903 ± 0.0016 | 0.8110 ± 0.0044 |
| Recall | 0.9096 ± 0.0115 | 0.9105 ± 0.0203 | 0.8672 ± 0.0098 | 0.7325 ± 0.0097 |
| Precision | 0.8477 ± 0.0139 | 0.8408 ± 0.0218 | 0.8437 ± 0.0088 | 0.7545 ± 0.0132 |
| Specificity | 0.9023 ± 0.0116 | 0.8966 ± 0.0190 | 0.9041 ± 0.0072 | 0.8577 ± 0.0119 |
Note: The results were reported as average ± standard deviation.
Abbreviations: AUC, area under the receiver operating characteristic curve; AUPR, area under the precision‐recall curve; GNN, graph neural network; LLM, large language model.
4. DISCUSSION
Interaction prediction research has been conducted in various fields of computational biology, including the prediction of various biological associations such as long non‐coding RNA (lncRNA)–miRNA interactions, 48 lncRNA‐protein interactions, 49 metabolite‐disease associations, 50 , 51 and drug‐toxicity associations. 52 , 53 , 54 Analyzing interactions among various biomedical entities, such as genes, diseases, drugs, and metabolites, is pivotal in medicine, as it contributes to a systemic understanding of diseases and facilitates the development of innovative therapeutic strategies. Recent research has increasingly focused on depicting these interactions within networks and analyzing them to uncover previously unknown interactions. For instance, Hulovatyy et al. predicted links by calculating the number of shared neighboring nodes or common pathways. 55 However, this approach has the disadvantage of showing significant performance variations depending on the dataset owing to the noisy nature of biomedical graphs. This method has evolved into a more advanced technique that utilizes diffusion kernels to model the interactions between individual entities, thereby enhancing our understanding of the representation of each node. Notably, research suggests that genes contributing to the same phenotype are likely to interact. 56 Furthermore, Kovács et al. have explored the possibility of predicting protein interactions through their similarity with connected nodes. 57 Recently, the prediction of miRNA‐disease associations has emerged as a vibrant area of graph‐based link prediction research employing large‐scale heterogeneous networks comprising miRNAs and diseases. 58 , 59 Additionally, various methodologies have been employed to predict disease‐associated proteins. For example, extracting disease‐related graph structures from protein–protein interaction (PPI) networks has proven to be a promising approach. 60 , 61 Chen et al. leveraged the PPI, expression data, subcellular localization, and orthology information to develop transfer neural networks to predict disease‐associated proteins. 62 Nevertheless, although these studies offer valuable insights, they are limited by their dependence on PPI networks.
While several studies have sought to represent entities through the various interrelationships among them, recently developed LLMs have made significant strides in comprehending the meanings of individual entities within vast contexts across diverse fields of expertise. Models such as ChatGPT 22 , 23 encode the meaning of each text into a representation vector endowed with common knowledge and robust semantic comprehension. Attempts have been made to leverage this ability to effectively capture the characteristics of each text to improve the representation of nodes in GNNs. 25 , 63 One fundamental approach utilizes embedding vectors derived by LLMs in the initial node embedding, constituting a form of feature‐level enhancement. 25 Another strategy is to use text‐to‐text LLM to generate additional texts with more profound and richer information, thereby creating relationships between these texts and the original ones. 25 , 63 This enriches the graph being learned, making it more comprehensive. LLM‐derived feature embeddings are widely used to learn from tabular data. Approaches such as TabLLM 64 and LIFT 65 aim to leverage LLMs to convert column features into embeddings for each row by utilizing the context awareness inherent in LLMs for tabular data learning. Models like CAAFE 66 utilize LLMs to generate new features with a high correlation with labels from column names. Additionally, TransTab proposed a method to convert each column name into an embedding vector using word embeddings. 67
Embeddings using the KGE algorithm include information on the relationships between biomedical entities. Embeddings generated by the LLM include comprehensive common knowledge from the literature. We integrated embeddings from two different modalities, graph structure and text, using a multi‐head attention mechanism. 27 This approach allowed the effective integration of these two types of information with appropriate weights and excellent performance was achieved. Additionally, we employed embeddings generated by the LLM as initial node embeddings for GNN to integrate the data. 25 We attempted to add the graph structure information by learning the embeddings obtained by the LLM through GNN. However, CMEI integrating the two types of information using a multi‐head attention mechanism 27 showed a better performance than the GNN‐based model. Additionally, a prediction model was developed using only embeddings based on the KGE algorithm or LLM. CMEI performed better than the other models. When comparing the performances of the models developed using only KGE embeddings versus only LLM embeddings, the model established using the KGE embeddings performed better. This result suggests that information from biomedical entities may be extracted more efficiently by the KGE algorithm than by the LLM. The importance of each embedding was evaluated using an ablation study. When the gene/protein embeddings obtained by KGE were excluded, the performance of the model deteriorated the most. Conversely, excluding gene/protein embeddings derived from the LLM led to the least deterioration in the performance of the model. This may be attributed to the fact that information about genes/proteins may be more specialized in biomedical fields than information about diseases; thus, they might be better extracted by the KGE algorithm than by the general‐purpose LLM.
We developed a prediction model by integrating the embeddings extracted from the two modalities. In this study, the embedding model of OpenAI, 26 a general‐purpose LLM, was used. Better predictions may be possible using LLMs specialized for biomedical tasks. However, further studies are needed to confirm this hypothesis. Case studies on several diseases have been conducted. Most genes/proteins predicted to be associated with each disease in CMEI were identified in the knowledge graph used as the dataset. However, there were several genes/proteins whose associations with the diseases were not identified in the knowledge graph used as the dataset. It may be possible that the associations between these genes/proteins and diseases have yet to be discovered. This suggestion by CMEI can be the starting point for the identification of disease development mechanisms and new treatment targets. Our study has limitations in making inferences regarding diseases and genes/proteins that are not included in the PrimeKG. 19 , 20 However, as PrimeKG 19 , 20 contains information about many diseases and genes/proteins, identifying the missing links between these diseases and genes/proteins will contribute significantly to discovering new treatment targets and identifying disease development mechanisms.
Comprehensive information about biomedical entities is crucial for predicting their associations. We obtained a wide range of information from various relationships using large biomedical knowledge graphs. Additionally, information regarding biomedical entities available in a wide range of literature was obtained using the LLM. Subsequently, a multi‐head attention mechanism integrated this information with appropriate weights to ensure an excellent prediction performance. Further studies using CMEI to identify the missing links between diseases and genes/proteins may contribute to the development of novel drugs.
AUTHOR CONTRIBUTIONS
Munyoung Chang: Conceptualization, methodology, software, validation, formal analysis, investigation, data curation, writing—original draft, writing—review and editing, visualization, supervision, project administration, funding acquisition. Junyong Ahn: Conceptualization, methodology, software, investigation, writing—original draft, writing—review and editing. Bong Gyun Kang: Conceptualization, methodology, software, investigation, writing—original draft, writing—review and editing. Sungroh Yoon: Conceptualization, methodology, software, formal analysis, investigation, resources, writing—original draft, writing—review and editing, supervision, project administration, funding acquisition.
FUNDING INFORMATION
This work was supported by the BK21 FOUR program of the Education and Research Program for Future ICT Pioneers, Seoul National University in 2024, the National Research Foundation of Korea grants funded by the Korean government (Ministry of Science and ICT, no. 2022R1A3B1077720), Institute of Information & Communications Technology Planning & Evaluation (IITP) grant funded by the Korea government (MSIT) [NO.RS‐2021‐II211343, Artificial Intelligence Graduate School Program (Seoul National University)], and AI‐Bio Research Grant through Seoul National University.
CONFLICT OF INTEREST STATEMENT
All authors declare that they have no known competing interests.
Supporting information
Data S1: Source code for Cross‐Modal Embedding Integrator. A, B, C, and D in the source code refer to gene/protein embedding by the LLM, disease embedding by the LLM, disease embedding by the KGE, and gene/protein embedding by the KGE, respectively. LLM, large language model; KGE, knowledge graph embedding.
Chang M, Ahn J, Kang BG, Yoon S. Cross‐modal embedding integrator for disease‐gene/protein association prediction using a multi‐head attention mechanism. Pharmacol Res Perspect. 2024;12:e70034. doi: 10.1002/prp2.70034
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available in Harvard Dataverse at https://doi.org/10.7910/DVN/IXA7BM. 19 , 20
REFERENCES
- 1. Paliwal S, de Giorgio A, Neil D, Michel JB, Lacoste AM. Preclinical validation of therapeutic targets predicted by tensor factorization on heterogeneous graphs. Sci Rep. 2020;10:18250. doi: 10.1038/s41598-020-74922-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yoon S, Nguyen HCT, Yoo YJ, et al. Efficient pathway enrichment and network analysis of GWAS summary data using GSA‐SNP2. Nucleic Acids Res. 2018;46:e60. doi: 10.1093/nar/gky175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ganegoda GU, Sheng Y, Wang J. ProSim: a method for prioritizing disease genes based on protein proximity and disease similarity. Biomed Res Int. 2015;2015:213750. doi: 10.1155/2015/213750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Peng C, Xia F, Naseriparsa M, Osborne F. Knowledge graphs: opportunities and challenges. Artif Intell Rev. 2023;56:1‐32. doi: 10.1007/s10462-023-10465-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Malas TB, Vlietstra WJ, Kudrin R. Drug prioritization using the semantic properties of a knowledge graph. Sci Rep. 2019;9:6281. doi: 10.1038/s41598-019-42806-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bonner S, Barrett IP, Ye C, et al. A review of biomedical datasets relating to drug discovery: a knowledge graph perspective. Brief Bioinform. 2022;23(6):bbac404. doi: 10.1093/bib/bbac404 [DOI] [PubMed] [Google Scholar]
- 7. Yang B, Yih W‐t, He X, Gao J, Deng L. Embedding entities and relations for learning and inference in knowledge bases. ICLR 2015. 2015.
- 8. Nickel M, Rosasco L, Poggio T. Holographic Embeddings of Knowledge Graphs. AAAI'16: Proceedings of the Thirtieth AAAI Conference on Artificial Intelligence 2016. 1955–1961.
- 9. Sun Z, Deng Z‐H, Nie J‐Y, Tang J. RotatE: Knowledge Graph Embedding by Relational Rotation in Complex Space. ICLR 2019. 2019.
- 10. Rossi A, Barbosa D, Firmani D, Matinata A, Merialdo P. Knowledge graph embedding for link prediction: a comparative analysis. ACM Trans Knowl Discov Data (TKDD). 2020;15:1‐49. [Google Scholar]
- 11. Mohamed SK, Nováček V, Nounu A. Discovering protein drug targets using knowledge graph embeddings. Bioinformatics. 2020;36:603‐610. doi: 10.1093/bioinformatics/btz600 [DOI] [PubMed] [Google Scholar]
- 12. Gualdi F, Oliva B, Piñero J. Predicting gene disease associations with knowledge graph embeddings for diseases with curtailed information. NAR Genom Bioinform. 2024;6:lqae049. doi: 10.1093/nargab/lqae049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Han P, Yang P, Zhao P, et al. GCN‐MF: disease‐Gene Association identification by graph convolutional networks and matrix factorization. KDD'19: Proceedings of the 25th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining 2019. 705–713.
- 14. Cinaglia P, Cannataro M. Identifying candidate gene‐disease associations via graph neural networks. Entropy (Basel). 2023;25(6):909. doi: 10.3390/e25060909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mastropietro A, De Carlo G, Anagnostopoulos A. XGDAG: explainable gene‐disease associations via graph neural networks. Bioinformatics. 2023;39(8):btad482. doi: 10.1093/bioinformatics/btad482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Himmelstein DS, Lizee A, Hessler C, et al. Systematic integration of biomedical knowledge prioritizes drugs for repurposing. elife. 2017;6:e26726. doi: 10.7554/eLife.26726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Walsh B, Mohamed SK, Nováček V. BioKG: A Knowledge Graph for Relational Learning On Biological Data. CIKM'20: Proceedings of the 29th ACM International Conference on Information & Knowledge Management 2020. 3173–3180.
- 18. Zheng S, Rao J, Song Y, et al. PharmKG: a dedicated knowledge graph benchmark for bomedical data mining. Brief Bioinform. 2021;22(4):bbaa344. doi: 10.1093/bib/bbaa344 [DOI] [PubMed] [Google Scholar]
- 19. Chandak P, Huang K, Zitnik M. Building a knowledge graph to enable precision medicine. Sci Data. 2023;10:67. doi: 10.1038/s41597-023-01960-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chandak P. PrimeKG; Harvard Dataverse. V2 2022. doi: 10.7910/DVN/IXA7BM [DOI]
- 21. Zhou J, Wang X, Niu R, Shang X, Wen J. Predicting circRNA‐miRNA interactions utilizing transformer‐based RNA sequential learning and high‐order proximity preserved embedding. iScience. 2024;27:108592. doi: 10.1016/j.isci.2023.108592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. OpenAI . Introducing ChatGPT. Accessed March 2, 2024. https://openai.com/index/chatgpt
- 23. OpenAI . Gpt‐4 Technical Report. arXiv. 2303:08774 [cs.CL].
- 24. Melnyk I, Dognin P, Das P. Grapher: Multi‐stage knowledge graph construction using pretrained language models. NeurIPS 2021 Workshop on Deep Generative Models and Downstream Applications 2021.
- 25. Chen Z, Mao H, Li H, et al. Exploring the potential of large language models (LLMs) in learning on graphs. NeurIPS 2023 New Frontiers in Graph Learning Workshop.
- 26. OpenAI . Embedding models. Accessed March 2, 2024. https://platform.openai.com/docs/guides/embeddings
- 27. Vaswani A, Shazeer N, Parmar N, et al. 31st Conference on Neural Information Processing Systems. 2017.
- 28. PyTorch Geometric Team . RandomLinkSplit. Accessed March 2, 2024. https://pytorch‐geometric.readthedocs.io/en/stable/generated/torch_geometric.transforms.RandomLinkSplit.html
- 29. PyKEEN . Pipeline. Accessed July 11, 2024. https://pykeen.readthedocs.io/en/stable/api/pykeen.pipeline.pipeline.html#pykeen.pipeline.pipeline
- 30. Nunes S, Sousa RT, Pesquita C. Multi‐domain knowledge graph embeddings for gene‐disease association prediction. J Biomed Semantics. 2023;14(1):11. doi: 10.1186/s13326-023-00291-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rumelhart D, Hinton G, Williams R. Learning representations by back‐propagating errors. Nature. 1986;323:533‐536. doi: 10.1038/323533a0 [DOI] [Google Scholar]
- 32. PyTorch . Linear. Accessed July 25, 2024.https://pytorch.org/docs/stable/generated/torch.nn.Linear.html
- 33. Devlin J, Chang M‐W, Lee K, Toutanova K. BERT: Pre‐Training of Deep Bidirectional Transformers for Language Understanding. 2019;arXiv 1810.04805v2 [cs.CL]. doi: 10.48550/arXiv.1810.04805 [DOI]
- 34. PyTorch . Multihead Attention. Accessed July 25, 2024. https://pytorch.org/docs/stable/generated/torch.nn.MultiheadAttention.html
- 35. He K, Zhang X, Ren S, Sun J, Deep Residual Learning for Image Recognition . Proceedings of the ieee conference on computer vision and pattern recognition (CVPR). 2016.
- 36. Ba JL, Kiros JR, Hinton GE. LayerNormalization. 2016;arXiv 1607:06450 [stat.ML]. doi: 10.48550/arXiv.1607.06450 [DOI]
- 37. PyTorch . LayerNorm. Accessed July 25, 2024. https://pytorch.org/docs/stable/generated/torch.nn.LayerNorm.html
- 38. PyTorch . PyTorch. Accessed March 2, 2024. https://pytorch.org
- 39. Kingma DP, Ba J. Adam: a method for stochastic optimization. 2014;arXiv 1412:6980 [cs.LG]. doi: 10.48550/arXiv.1412.6980 [DOI]
- 40. PyTorch . ADAM. Accessed March 2, 2024. https://pytorch.org/docs/stable/generated/torch.optim.Adam.html
- 41. Hinton GE, Srivastava N, Krizhevsky A, Sutskever I, Salakhutdinov RR. Improving neural networks by preventing co‐adaptation of feature detectors. 2012;arXiv 1207:0580 [cs.NE]. doi: 10.48550/arXiv.1207.0580 [DOI]
- 42. PyTorch . DROPOUT. Accessed March 2, 2024. https://pytorch.org/docs/stable/generated/torch.nn.Dropout.html
- 43. PyTorch . REDUCELRONPLATEAU. Accessed March 2, 2024. https://pytorch.org/docs/stable/generated/torch.optim.lr_scheduler.ReduceLROnPlateau.html
- 44. Hamilton WL, Ying R, Leskovec J. Inductive Representation Learning on Large Graphs. 31st Conference on Neural Information Processing Systems (NIPS 2017) 2017. 1025–1035.
- 45. PyTorch Geometric Team . conv.SAGEConv. Accessed March 2, 2024. https://pytorch‐geometric.readthedocs.io/en/stable/generated/torch_geometric.nn.conv.SAGEConv.html#torch_geometric.nn.conv.SAGEConv
- 46. PyTorch Geometric Team . Linear. Accessed March 2, 2024. https://pytorch‐geometric.readthedocs.io/en/stable/modules/nn.html
- 47. PyTorch Geometric Team . PyG Documentation. Accessed March 2, 2024. https://pytorch‐geometric.readthedocs.io/en/stable/index.html
- 48. Wang W, Zhang L, Sun J, Zhao Q, Shuai J. Predicting the potential human lncRNA‐miRNA interactions based on graph convolution network with conditional random field. Brief Bioinform. 2022;23(6):bbac463. doi: 10.1093/bib/bbac463 [DOI] [PubMed] [Google Scholar]
- 49. Zhao J, Sun J, Shuai SC, Zhao Q, Shuai J. Predicting potential interactions between lncRNAs and proteins via combined graph auto‐encoder methods. Brief Bioinform. 2023;24(1):bbac527. doi: 10.1093/bib/bbac527 [DOI] [PubMed] [Google Scholar]
- 50. Sun F, Sun J, Zhao Q. A deep learning method for predicting metabolite‐disease associations via graph neural network. Brief Bioinform. 2022;23(4):bbac266. doi: 10.1093/bib/bbac266 [DOI] [PubMed] [Google Scholar]
- 51. Gao H, Sun J, Wang Y, et al. Predicting metabolite‐disease associations based on auto‐encoder and non‐negative matrix factorization. Brief Bioinform. 2023;24(5):bbad259. doi: 10.1093/bib/bbad259 [DOI] [PubMed] [Google Scholar]
- 52. Wang T, Sun J, Zhao Q. Investigating cardiotoxicity related with hERG channel blockers using molecular fingerprints and graph attention mechanism. Comput Biol Med. 2023;153:106464. doi: 10.1016/j.compbiomed.2022.106464 [DOI] [PubMed] [Google Scholar]
- 53. Chen Z, Zhang L, Sun J, Meng R, Yin S, Zhao Q. DCAMCP: a deep learning model based on capsule network and attention mechanism for molecular carcinogenicity prediction. J Cell Mol Med. 2023;27:3117‐3126. doi: 10.1111/jcmm.17889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wang J, Zhang L, Sun J, et al. Predicting drug‐induced liver injury using graph attention mechanism and molecular fingerprints. Methods. 2024;221:18‐26. doi: 10.1016/j.ymeth.2023.11.014 [DOI] [PubMed] [Google Scholar]
- 55. Hulovatyy Y, Solava RW, Milenković T. Revealing missing parts of the interactome via link prediction. PLoS One. 2014;9:e90073. doi: 10.1371/journal.pone.0090073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cowen L, Ideker T, Raphael BJ, Sharan R. Network propagation: a universal amplifier of genetic associations. Nat Rev Genet. 2017;18:551‐562. doi: 10.1038/nrg.2017.38 [DOI] [PubMed] [Google Scholar]
- 57. Kovács IA, Luck K, Spirohn K, et al. Network‐based prediction of protein interactions. Nat Commun. 2019;10:1240. doi: 10.1038/s41467-019-09177-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Liu M, Yang J, Wang J, Deng L. Predicting miRNA‐disease associations using a hybrid feature representation in the heterogeneous network. BMC Med Genet. 2020;13:153. doi: 10.1186/s12920-020-00783-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yan C, Duan G, Li N, Zhang L, Wu FX, Wang J. PDMDA: predicting deep‐level miRNA‐disease associations with graph neural networks and sequence features. Bioinformatics. 2022;38:2226‐2234. doi: 10.1093/bioinformatics/btac077 [DOI] [PubMed] [Google Scholar]
- 60. Hu K, Hu J‐B, Tang L, et al. Predicting disease‐related genes by path structure and community structure in protein–protein networks. J Stat Mech. 2018;2018:100001. doi: 10.1088/1742-5468/aae02b [DOI] [Google Scholar]
- 61. Yang L, Zhao X, Tang X. Predicting disease‐related proteins based on clique backbone in protein‐protein interaction network. Int J Biol Sci. 2014;10:677‐688. doi: 10.7150/ijbs.8430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chen S, Huang C, Wang L, Zhou S. A disease‐related essential protein prediction model based on the transfer neural network. Front Genet. 2022;13:1087294. doi: 10.3389/fgene.2022.1087294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. He X, Bresson X, Laurent T, Perold A, LeCun Y, Hooi B. Harnessing Explanations: LLM‐to‐LM Interpreter for Enhanced Text‐Attributed Graph Representation Learning. The Twelfth International Conference on Learning Representations (ICLR 2024) 2024.
- 64. Hegselmann S, Buendia A, Lang H, Agrawal M, Jiang X, Sontag D. TabLLM: Few‐Shot Classification of Tabular Data with Large Language Models. Proceedings of the 26th International Conference on Artificial Intelligence and Statistics 2023. 206:5549–5581.
- 65. Dinh T, Zeng Y, Zhang R, et al. LIFT: Language‐Interfaced Fine‐Tuning for Non‐Language Machine Learning Tasks. 36th Conference on Neural Information Processing Systems (NeurIPS 2022) 2022.
- 66. Hollmann N, Müller S, Hutter F. Large Language Models for Automated Data Science: Introducing CAAFE for Context‐Aware Automated Feature Engineering. 37th Conference on Neural Information Processing Systems (NeurIPS 2023) 2023.
- 67. Wang Z, Sun J. TransTab: Learning Transferable Tabular Transformers across Tables. 36th Conference on Neural Information Processing Systems (NeurIPS 2022) 2022.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1: Source code for Cross‐Modal Embedding Integrator. A, B, C, and D in the source code refer to gene/protein embedding by the LLM, disease embedding by the LLM, disease embedding by the KGE, and gene/protein embedding by the KGE, respectively. LLM, large language model; KGE, knowledge graph embedding.
Data Availability Statement
The data that support the findings of this study are available in Harvard Dataverse at https://doi.org/10.7910/DVN/IXA7BM. 19 , 20