

Abstract
Mycobacterium tuberculosis complex (MTBC) is divided into several major phylogenetic lineages, with differential distribution globally. Using population-based data collected over a three year period, we performed 24-locus Mycobacterial Interspersed Repeat Unit – Variable Number Tandem Repeat (MIRU-VNTR) genotyping on all culture isolates from two districts of the country that differ in tuberculosis (TB) incidence (Gaborone, the capital, and Ghanzi in the Western Kalahari). The study objective was to characterize the molecular epidemiology of TB in these districts. Overall phylogenetic diversity mirrored that reported from neighboring Republic of South Africa, but differences in the two districts were marked. All four major lineages of M. tuberculosis were found in Gaborone, but only three of the four major lineages were found in Ghanzi. Strain diversity was lower in Ghanzi, with a large proportion (38%) of all isolates having an identical MIRU-VNTR result, compared to 6% of all isolates in Gaborone with the same MIRU-VNTR result. This study demonstrates localized differences in strain diversity by two districts in Botswana, and contributes to a growing characterization of MTBC diversity globally.
Keywords: Tuberculosis, Molecular epidemiology, Diversity, Lineage, Botswana
1. Introduction
Mycobacterium tuberculosis complex (MTBC) is divided into several major phylogenetic lineages that are differentially distributed globally. (Gagneux and Small, 2007) The phylogeographic population structure of MTBC has been suggested to reflect a combination of local pathogen adaptation to sympatric human populations and both historical and modern patterns of human migration. (Gagneux, 2018) Most disease in humans is caused by MTBC members M. tuberculosis sensu strico and Mycobacterim africanum; these members of MTBC can be divided into seven phylogenetic lineages (five M. tuberculosis sensu stricto lineages (lineages 1–4, 7) and two M. africanum lineages (lineages 5 and 6)). Euro-American/lineage 4 and East-Asian/lineage 2 are the most wide-spread globally, Indo-Oceanic/lineage 1 and East African-Indian/lineage 3 occur globally but are most prevalent in regions adjacent to the Indian Ocean, West African I/lineage 5 and West African II/lineage 6 occur predominately in West Africa, and lineage 7 is largely restricted to Ethiopia. (de Jong et al., 2010; Tessema et al., 2013) Based on available data on MTBC diversity in Africa, Euro-American lineage is predominant, and Indo-Oceanic and East African-Indian lineages are most commonly found in East Africa. East-Asian lineage is found widely in Africa but at a higher proportion in Southern Africa than elsewhere on the continent. The diversity of lineages varies across the continent as well, with South Africa described as having the highest diversity of strains based on available data. (Chihota et al., 2018; Mbugi et al., 2016)
Overall, the incidence rate of tuberculosis (TB) in Botswana was estimated at 414/100,000 population at the start of this study in 2013, but this varies widely within Botswana. (Global Tuberculosis Report, 2013) The Botswana National TB Program notified 7720 cases in 2013, with a notification rate of 343 per 100,000 population. (Botswana National Tuberculosis and Leprosy Program (BNTP), 2013–2014) Gaborone, the capital city with population of approximately 232,000 persons, is comprised primarily of Tswana, the dominant ethnic group in Botswana. Gaborone had a notification rate of 303/100,000 in 2013. (Botswana National Tuberculosis and Leprosy Program (BNTP) Combined Annual Report 2013 and 2014, 2013–2014; Population and housing census, 2011) By contrast, the rural district of Ghanzi, with population of approximately 43,000 persons, includes in large proportion of members of the indigenous San peoples, with 33.8% speaking the San language Sesarwa compared to 0.1% in Gaborone. (Population and housing census, 2011) Ghanzi historically had the highest TB notification rate in the country (663/100,000 population in 2013), a notification rate of over double that reported in Gaborone the same year. (Botswana National Tuberculosis and Leprosy Program (BNTP) Combined Annual Report 2013 and 2014, 2013–2014). At the start of this study in 2013, the HIV prevalence rate among the general population over age 18 months was 18.5%; the general population HIV prevalence rate in Gaborone was 17.0% and in Ghanzi was 17.1%. (Botswana AIDS Impact Survey IV, 2013; HIV estimates with uncertainty bounds 1990–2016, 2013)
We sought to determine the distribution of TB lineages in Botswana, and to understand in the molecular epidemiology of TB in two districts. We hypothesized that strain diversity would be greater in Gaborone and less in rural Ghanzi; Gaborone is a capital city and hypothesized to have more movement of people into the city with subsequent introduction of new strains.
2. Methods
2.1. Participant enrollment
Participants were enrolled in a prospective molecular epidemiology study – KOPANYO – in two districts of Botswana, Gaborone and Ghanzi, from September 2012–April 2016, as previously described. (Zetola et al., 2016)
2.2. Specimen collection and laboratory methods
Sputa were collected from all participants by spontaneous expectoration or, if that was not successful, by induction; other specimen types were accepted as well, if collected. Samples were decontaminated using the standard N-acetyl-L-cysteine/4% sodium hydroxide-sodium citrate (NALC/NaOH Na-citrate, final concentration of NaOH 1%) method. (PT Kent, 1985) Following centrifugation, specimens were resuspended in 50 ml sterile 0.067 phosphate buffer (pH 6.8) and 0.5 ml was inoculated into one Mycobacterial Growth Indicator Tube [(MGIT), BD, Franklin Lakes, NJ)]. MGIT cultures were incubated at 35–37 °C in the MGIT960™ instrument for up to 6 weeks. MGIT cultures flagged as positive were examined by microscopy using Ziehl-Neelsen staining to identify the presence of acid fast bacilli. SD Bioline TB Ag MPT64 Rapid test (Standard Diagnostics, Gyeonggi-do, Republic of Korea) was used to identify MTBC. Cultures positive for acid fast bacilli but with negative results for the SD Bioline TB Ag MPT64 Rapid test were considered to be non-tuberculous mycobacteria. Cultures with evidence of both mycobacterium species and other potential contaminating species were re-decontaminated using the standard method described above. Drug susceptibility testing (DST) for first-line anti-TB drugs was performed using MGIT DST. Susceptibility for isoniazid was set at 0.1 μg/ml and rifampin was set at 1.0 μg/ml. We used DST on LJ media in instances when MGIT DST was inconclusive.
One representative isolate for all participants with positive culture result was used for DNA preparation and subsequent genotyping by 24-locus Mycobacterial Interspersed Repeat Unit – Variable Number Tandem Repeat (MIRU-VNTR) typing performed at Genoscreen (Lille, France) using standardized methods. (Supply et al., 2006) DNA was prepared by crude extraction. (Mazars et al., 2001) Briefly, 1 ml of MGIT culture was centrifuged at 15,000 g × 15 min, the supernatant was discarded, pellets were resuspended in 200 μl of sterile nuclease-free water and incubated at 95 °C for 30 min. Cell debris was pelleted by centrifugation at 15,000 g × 1 min and the supernatant was used for genotyping.
2.3. Data analysis
MIRU-VNTR results with more than one copy number at one or more loci (“double alleles”) as seen with mixtures of different clonal sub-populations or strains, or with missing or indeterminate copy number at any locus, were considered to be non-interpretable for lineage assignment and were excluded from analysis. We built an unweighted pair group method with arithmetic mean (UPGMA) phylogenetic tree using Bionumerics v.7.6.2 (Austin, Texas) comparing study strains to a set of international reference strains (MIRU-VNTRplus). (Allix-Beguec et al., 2008) The MIRU-VNTRplus reference strain collection (n = 186) includes two M. africanum strains with double alleles; specifically, locus 2163b QUB11b has copy numbers 6 + 4 for strain 10,473/01 and copy numbers 6 + 3 for strain 5254/02. For these strains, we included both sub-strains as reference strains (e.g. for 10,473/01 we created two separate strains, one with 2163b QUB11b equal to 6 and one with 2163b QUB11b equal to 4 for use as reference strains), giving a total of 188 reference strains. For both reference and study strains, any strain with locus 580 MIRU04 coded as “3 s” was recoded to 23 for analysis using Bionumerics software; “s” indicates that only 3 repeats of 77 base pairs are present, and not the terminal repeat of 53 base pairs that is often found at this locus. (Supply et al., 2001; Supply et al., 2000)
For the purpose of assigning lineage to study strains, MIRU-VNTRplus lineage nomenclature (similar to that of Brudey, et al. with the exception of MIRU-VNTRplus names West African I (same as Brudey et al. AFRI2) and West African 2 (same as Brudey et al. AFRI1)) was first assigned the corresponding large sequence polymorhphism (LSP)-based nomenclature name (e.g. Euro-American/lineage 4, Indo-Oceanic/lineage 1, East African-Indian/lineage 3, East Asian/lineage 2). (Gagneux and Small, 2007; Brudey et al., 2006) Specifically, MIRU-VNTRplus lineages Cameroon, NEW-1, Ghana, TUR (Turkey), LAM (Latin American-Mediterranean), S, Haarlem, Uganda I, Uganda II, X, H37Rv, Ural were assigned as Euro-American/MTBC lineage 4; EAI (East African-Indian) was assigned as Indo-Oceanic/MTBC lineage 1; Beijing was assigned as East-Asian/MTBC lineage 2, and Delhi/CAS (Central Asian) was assigned as East African-Indian/MTBC lineage 3. MIRU-VNTRplus West African I corresponds to LSP-based name West African lineage I/MTBC lineage 5 and West African 2 corresponds to West African lineage II/MTBC lineage 6. (Niemann et al., 2016; Gagneux et al., 2006; Coscolla and Gagneux, 2014) Phylogenetic lineage of study strains was determined by visually comparing tree placement of study strains relative to reference strains and LSP-derived lineage names were assigned to study strains. (Gagneux and Small, 2007) Minimal spanning trees were generated for each district and for each lineage using all strains included in analysis from Gaborone and Ghanzi.
We compared proportions of participants living with HIV across lineages using a Chi square test and compared proportions of participants of with multi-drug resistant TB (MDR TB, defined as resistance to both isoniazid and rifampin) across lineages using Fisher’s exact test because 20% of cells had expected size < 5. Genotype clusters were defined as groups of two or more isolates with identical 24-locus MIRU-VNTR results. We compared the proportion of all isolates by lineage in each district. For each lineage, we compared by district the proportion that were clustered and the size of clusters (range and median with interquartile range).
HIV status was considered positive if a participant had documented HIV positive status prior to study enrollment or had a positive rapid test result at the enrollment; HIV status was considered negative based on a negative rapid test result at the time of enrollment or documented negative rapid test result within 3 months prior to enrollment.
2.4. Ethical approval
This study was approved by the USA Centers for Disease Control and Prevention (CDC) Institutional Review Board (IRB) (#6291), Health Research and Development Committee, Ministry of Health and Wellness, Botswana and the University of Pennsylvania IRBs. Participants provided written informed consent.
2.5. Funding
This study was funded by US National Institutes of Health grant number R01AI097045, by the U.S. Centers for Disease Control and Prevention, and by the President’s Emergency Plan for AIDS Relief (PEPFAR) through the Centers for Disease Control and Prevention. National Institute of Health did not participate in study design, data collection, or interpretation of results.
3. Results
3.1. Laboratory results – culture, genotype, specimen type
A total of 4331 participants were enrolled and of these, 3842 participants had smear or MGIT performed. A total of 59.2% (2162/3653) participants with culture results available had positive culture for MTBC and 8.2% (300/3653) had positive culture for non-tuberculous mycobacteria. MIRU-VNTR results were available for 2137 (98.8%) participants with MTBC cultures; of these, 213 were excluded from analysis: 122 were excluded because of the presence of double alleles for at least one locus, 82 were excluded because of indeterminate copy number for at least one locus, and 9 had both double and indeterminate alleles. A total of 90.0% (1924/2137) interpretable MIRU-VNTR genotype results, comprised of 686 unique genotypes, were included in analysis. Among 1918 participants with interpretable MIRU-VNTR genotype results and documented specimen type, specimen types included sputum 90.8% (1742/1918) and induced sputum 8.8% (169/1918); other specimen types accounted for the remaining 0.4% (7/1918).
3.2. Phylogenetic assignment results
Fig. 1 represents an unrooted UPGMA tree of study strains relative to international reference strains. Overall, Euro-American was the predominant lineage found in Botswana, representing 86.2% (1659/1924) of all isolates (Table 1). We found all four major LSP-defined M. tuberculosis lineages (Euro-American, Indo-Oceanic, East African-Indian, East Asian). Only one case (0.05%) of M. bovis was found, and no other members of the MTBC were found. A total of 36 strains, comprising 5 genotypes (234213321344351632225324, 23421432134435–1532228324, 234214321344351632225324, 23421432134435163–2227324, 234214321344351632228324, with allele order as specified in MIRU-VNTRplus), could not be readily classified based on the phylogenetic analysis described here and were classified as “Unknown.”
Fig. 1.
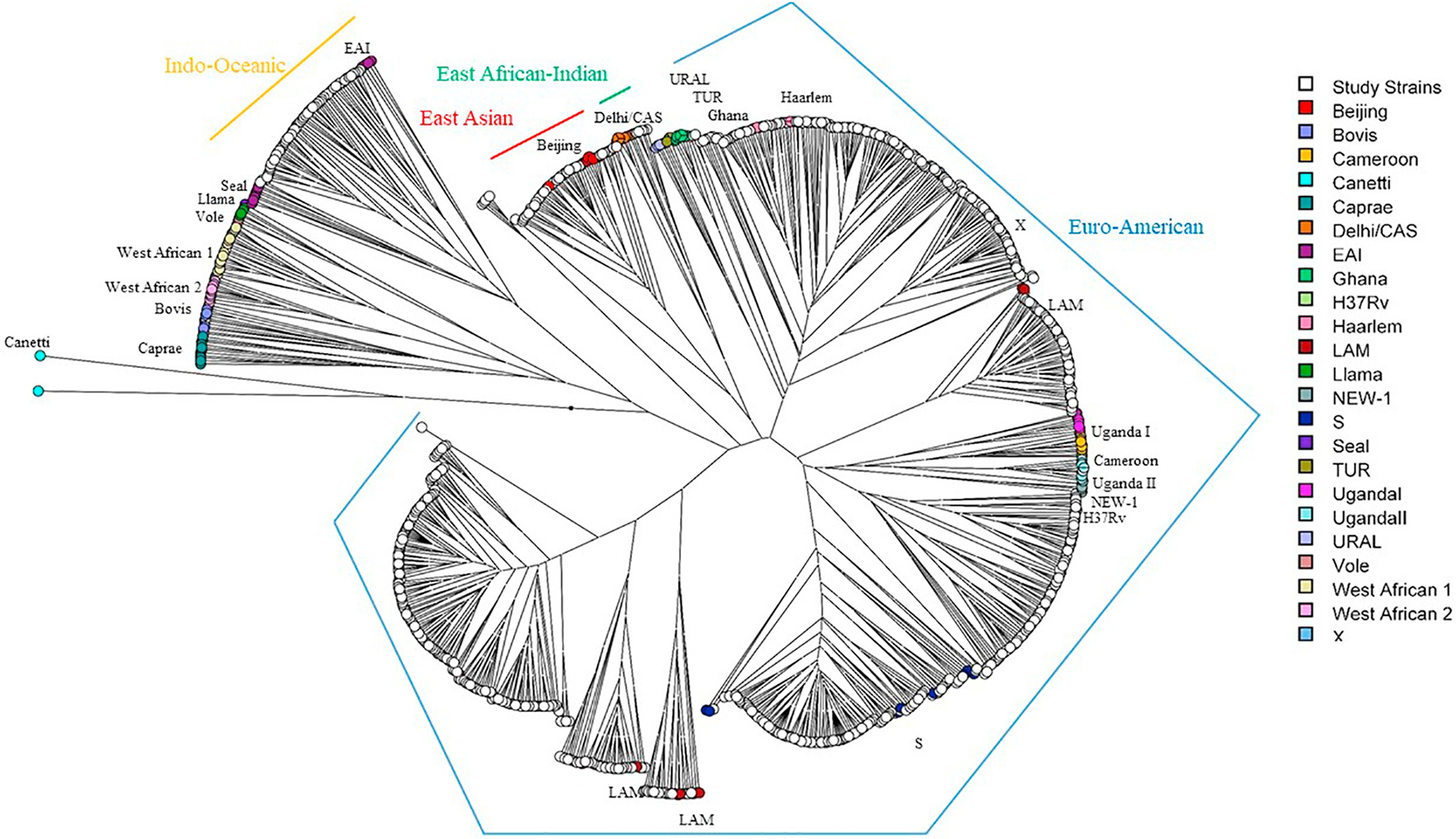
Unrooted UPGMA tree of study strains and MIRU-VNTRplus international reference strains. Reference strains are shown by color according to MIRU-VNTRplus lineage nomenclature; study strains are shown in white. Corresponding species are M. africanum (West African I and II), M. canettii (“Canetti” in the MIRU-VNTRplus reference set), M. caprae (“Caprae”), M. microti (“Vole”, “Llama”), M. pinnipedii (“Seal”), M. bovis (“Bovis”), and M. tuberculosis (all others). One representative of each genotype among study strains (n = 686) was used to determine phylogenetic relatedness to reference strains. Corresponding LSP-based nomenclature lineage names shown with colored lines.
Table 1.
Distribution of lineages by district of Botswana, September 2012–April 2016.
| Lineage | District | ||
|---|---|---|---|
| Gaborone | Ghanzi | Total | |
| Euro-American | 1277 (83.8) | 382 (95.5) | 1659 (86.2) |
| East Asian | 105 (6.9) | 13 (3.3) | 118 (6.1) |
| Indo-Oceanic | 94 (6.2) | 4 (1.0) | 98 (5.1) |
| East African-Indian | 12 (0.8) | 0 | 12 (0.6) |
| M. bovis | 1 (0.1) | 0 | 1 (0.1) |
| Unknown | 35 (2.3) | 1 (0.3) | 36 (1.9) |
| Total | 1524 | 400 | 1924 |
3.3. Strain-based results
Among the 686 genotypes identified in this study, 83.4% (572/686) were in Gaborone only, 8.0% (55/686) in Ghanzi only, and 8.6% (59/686) were in both Gaborone and Ghanzi; 63.6% (436/686) of the genotypes found were unique (isolated from only one participant) and 36.4% (250/686) were identified in 2 or more participants (clustered) at the national level. Among those 250 clusters, 69.6% (174/250) were among participants in Gaborone only, 6.8% (17/250) were among participants in Ghanzi only; 23.6% (59/250) of national-level clusters included participants from both Gaborone and Ghanzi. Among genotypes found in Gaborone only, 69.6% (398/572) were unique (found in one participant only), and among those found in Ghanzi only, 69.1% (38/55) were unique. Strains in Ghanzi comprised mainly a few large clusters and closely related strains (Fig. 2b). Although Euro-American is the predominant lineage in both districts, fewer sub-lineages were found in Ghanzi than in Gaborone. Euro-American strains in Ghanzi group in phylogenetic analysis primarily with just three sublineages (LAM, Haarlem, and S lineages by MIRU-VNTRplus spoligotype-based nomenclature). By contrast, Euro-American strains in Gaborone group in phylogenetic analysis with reference strains in the LAM, Haarlem, S, TUR, and Uganda II sublineages (Fig. 2a). While there was some overlap, the specific strains found in Ghanzi and Gaborone were largely distinct as seen in minimal spanning trees (Fig. 3). The amount of clustering across locations (i.e., clusters that included participants from both Ghanzi and Gaborone) also differed by lineage (Figs. 3 and 4 and Table 2). Only four Indo-Oceanic strains were found in Ghanzi and only one of these strains clustered with strains in Gaborone. Most East Asian strains found in Ghanzi clustered with strains in Gaborone. For Euro-American strains, the largest clusters were found almost exclusively in Ghanzi or in Gaborone, whereas some smaller clusters had more even distribution of strains from the two locations.
Fig. 2.
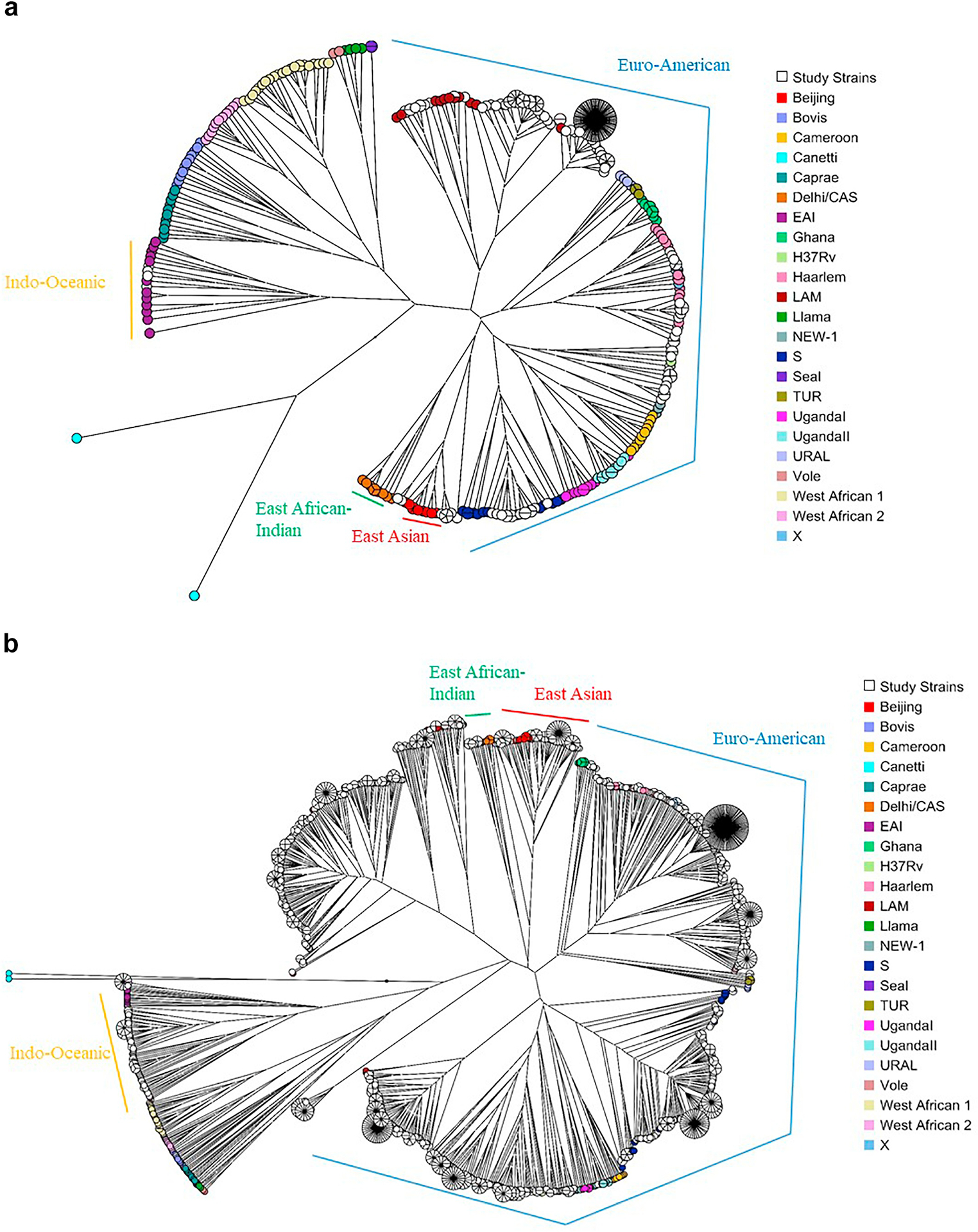
Strain diversity by district. Unrooted UPGMA trees of MIRU-VNTRplus reference strains and all study strains in (a) Gaborone and (b) Ghanzi are shown.
Fig. 3.
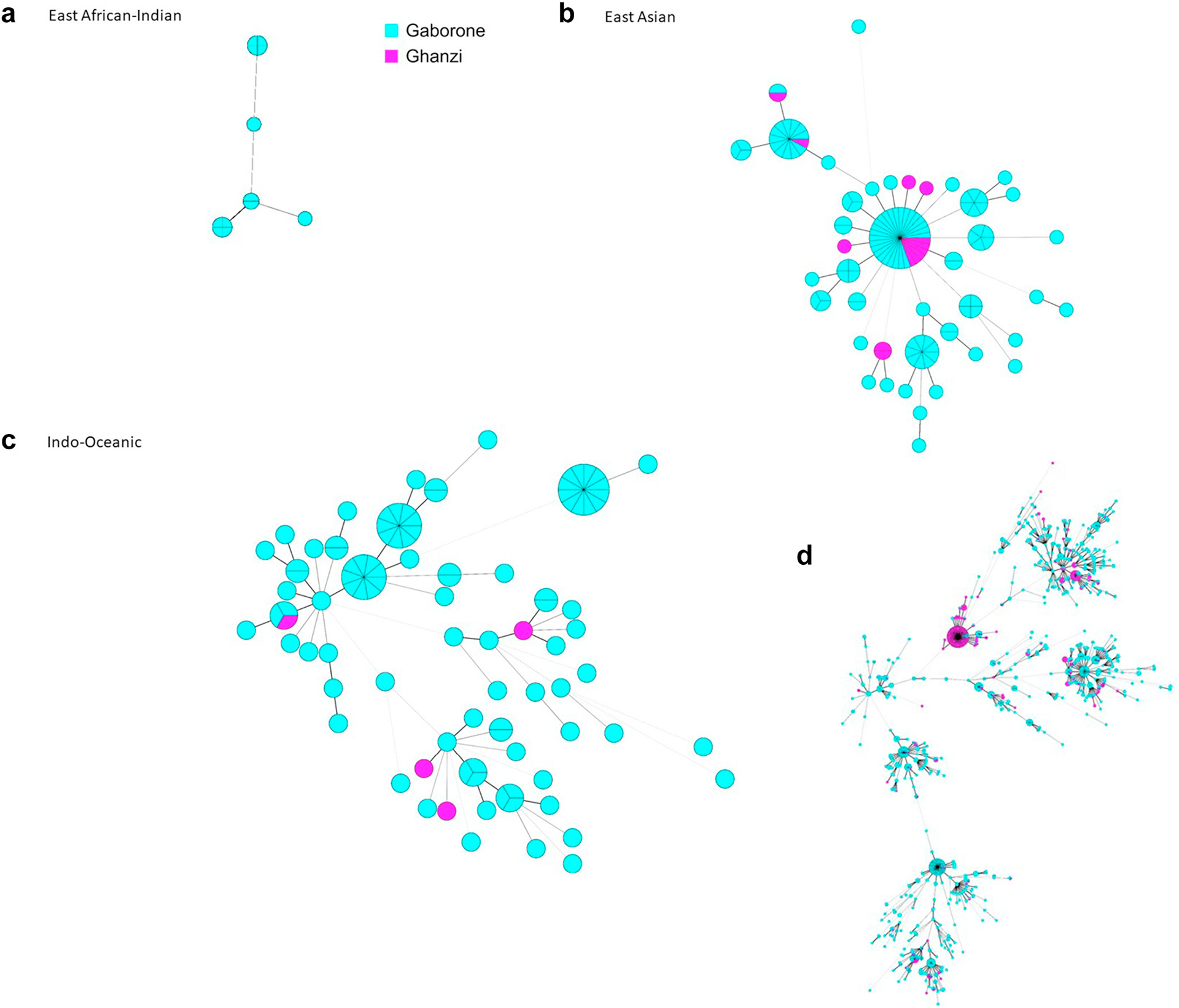
Minimal spanning trees of study strains by district and lineage. Minimal spanning trees for study strains with (A) East African-Indian lineage, (B) East Asian lineage, (C) Indo-Oceanic lineage, (D) Euro-American lineage. Each circle represents a single MIRU-VNTR pattern. The size each circle corresponds to the number of strains with the same genotype (indicated as ‘pieces’ of the circle). Lines between circles indicate genetic distance between genotypes, with shorter lines indicating more genetic similarity. Blue circles indicate patient-isolates from Gaborone; pink from Ghanzi.
Fig. 4.
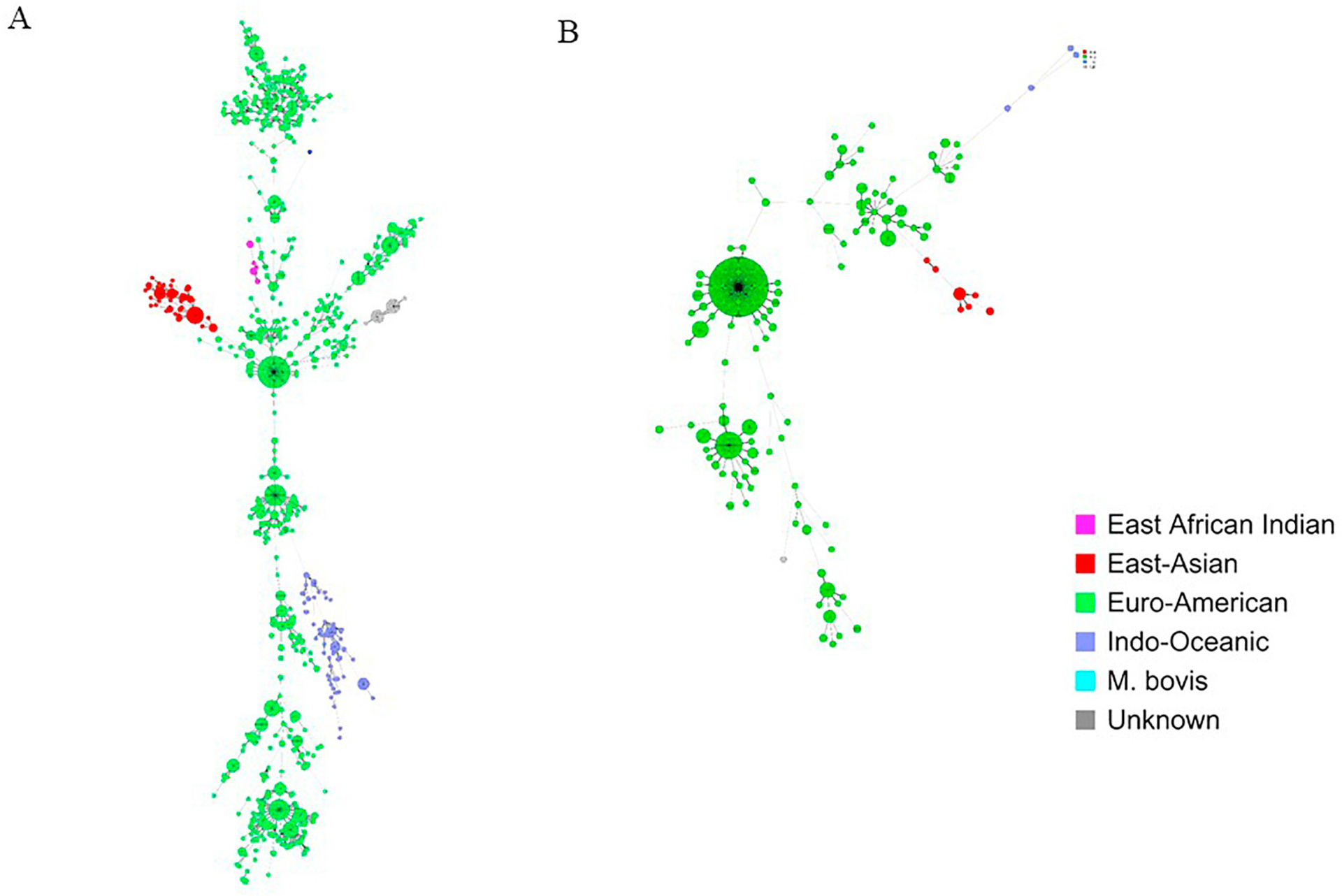
Minimal spanning trees of study strains in (a) Gaborone and (B) Ghanzi. Lineages are indicated by colors as specified in the key. Each circle represents a single MIRU-VNTR pattern. The size each circle corresponds to the number of strains with the same genotype (indicated as ‘pieces’ of the circle). Lines between circles indicate genetic distance between genotypes, with shorter lines indicating more genetic similarity.
Table 2.
Proportion of clustered strains and cluster sizes by lineage and location, September 2012–April 2016.
| Lineage | Location | Total Isolates | Total Clusters | Clustered Isolates n (%) |
Cluster Size Range | Cluster Size Median | Cluster Size IQR |
|---|---|---|---|---|---|---|---|
| Euro-American | Gaborone | 1277 | 184 | 934 (73.1) | 2–89 | 3 | 2–4 |
| Ghanzi | 382 | 41 | 321 (84.0) | 2–150 | 3 | 2–4 | |
| East Asian | Gaborone | 105 | 14 | 82 (78.1) | 2–25 | 3.5 | 2–6 |
| Ghanzi | 13 | 2 | 8 (61.5) | 2–6 | 4 | 2–6 | |
| Indo-Oceanic | Gaborone | 94 | 12 | 50 (53.2) | 2–12 | 2 | 2–6 |
| Ghanzi | 4 | 0 | 0 | – | – | – | |
| East African-Indian | Gaborone | 12 | 3 | 10 (83.3) | 2–4 | 4 | 2–4 |
| Ghanzi | 0 | 0 | – | – | – | – | |
| Unknown | Gaborone | 35 | 2 | 32 (91.4) | 15–17 | 16 | 15–17 |
| Ghanzi | 1 | 0 | 0 | – | – | – | |
| M. bovis | Gaborone | 1 | 0 | 0 | 1 | N/A | N/A |
| Ghanzi | 0 | 0 | – | N/A | N/A | N/A |
3.4. Case-based results
Among 1924 participants with an interpretable genotype result; 55.0% (1059) were male and median age was 33.1 years (IQR 26.3–40.9). A total of 1524 resided in Gaborone and 400 resided in Ghanzi. The distribution of patient-isolate lineage results by district are shown in Table 1. All four major lineages of M. tuberculosis sensu stricto were found among isolates in Gaborone, but among isolates from Ghanzi, only Euro-American, East Asian, and Indo-Oceanic lineage isolates were found. Euro-American lineage was the most common in both districts (83.8% (1277/1524) in Gaborone and 95.5% (382/400) in Ghanzi). No isolates of East African-Indian lineage or of M. bovis were found in Ghanzi.
A total of 1931 participants had known MDR status (first-line DST results available for both rifampin and isoniazid), and of these 3.4% (65/1931) had MDR TB. Among participants with interpretable genotype, 1747 had known MDR TB status and of these, 3.5% (61/1747) had MDR TB. The proportion of strains with MDR TB differed by lineage: 9.4% (8/85) Indo-Oceanic lineage strains, 8.0% (9/112) of East Asian lineage strains, and 2.9% (44/1505) of Euro-American lineage strains (Fisher’s exact test p ≤.0001); of 11 East African-Indian, 1 M. bovis and 33 unknown lineage isolates with DST results for rifampin and isoniazid, none had MDR TB (Table 3). Among participants with interpretable genotype, 1872 had known HIV status; of these participants, 1004 (53.6%) had known HIV positive status and 868 (46.4) had known HIV negative status. The proportion of participants with known HIV positive status differed among the main lineages of M. tuberculosis (Indo-Oceanic 67.4% (64/95), East African-Indian 66.7% [(8/12), East Asian 60.9% (70/115), Euro-American 52.1% (841/1613), Chi square p = .008)] (Table 3).
Table 3.
HIV and multidrug resistance status by lineage, September 2012–April 2016.
| HIV +a | Total | MDRb | Total | |
|---|---|---|---|---|
| n (%) | n (%) | |||
| Euro-American | 841 (52.1) | 1613 | 44 (2.9) | 1505 |
| East Asian | 70 (60.9) | 115 | 9 (8) | 112 |
| Indo-Oceanic | 64 (67.4) | 95 | 8 (9.4) | 85 |
| East African-Indian | 8 (66.7) | 12 | 0 | 11 |
| Unknown | 21 (58.3) | 36 | 0 | 33 |
| M. bovis | 0 | 1 | 0 | 1 |
Among participants interpretable 24-locus MIRU-VNTR result and aknown HIV status and bknown drug resistance testing result (positive of negative) for both rifampin and isoniazid.
In Ghanzi, 73.6% (281/382) of Euro-American lineage patient-isolates were the LAM sub-lineage. Among these, 150 patient-isolates (37.5% of all 400 strains in Ghanzi) were in a single cluster. By contrast, the single largest cluster in Gaborone comprised 89 Euro-American patient-isolates, accounting for 5.8% (89/1524) of all genotyped cases in Gaborone. District-specific clustering differed overall and by lineage. In Gaborone, 1108/1524 (72.7%) cases clustered with at least one other case in the district, and in Ghanzi 329/400 (82.3%) cases clustered. Among Euro-American strains, in Gaborone 73.1% (934/1277) clustered and in Ghanzi 84.0% (321/382) clustered whereas among East Asian strains, in Gaborone 77.4% (82/106) clustered and in Ghanzi 61.5% (8/13) clustered (Table 2).
3.5. Discussion
We found a variety of lineages in Botswana, including all 4 major global lineages of M. tuberculosis sensu stricto (i.e, Euro-American, East African-Indian, Indo-Oceanic, East Asian). We did not find any cases of M. africanum (Lineage 6; West African lineage I or West African lineage II) or Lineage 7, consistent with previous findings these lineages are restricted to specific geographic regions. (de Jong et al., 2010; Tessema et al., 2013)
Most isolates were readily categorized into major LSP-defined lineages; however, six 24-locus MIRU-VNTR patterns (among a total of 36 participants) did not fit straightforwardly into one of the main lineages based on phylogenetic analysis against an international reference set of strains. Different genotyping methods such as LSP analysis or comparison to a broader collection of reference strains might be informative for these isolates.
Compared to the distribution of lineages described for a number of countries in Africa, the overall strain diversity in Botswana was similar to neighboring Republic of South Africa. (Chihota et al., 2018; Mbugi et al., 2016) This may reflect the history of substantial cross-border movement between these countries for employment and commerce. (Crush et al., 2005) We did find that the proportion of strains with MDR differed by lineage, and that the proportion of MDR was the highest among Indo-Oceanic, followed by East Asian strains. Other reports have found East Asian lineage associated with higher rates of MDR and this lineage is suggested to have a higher propensity for development of MDR. (Ford et al., 2013) Notably, the proportion of participants with HIV-positive status was highest among persons with the least common M. tuberculosis sensu stricto lineages found in this study (i.e., Indo-Oceanic and East African-Indian). Reasons for the differences in HIV status by lineage are not known, but one possibility is that it could reflect the impact of immunosuppression on host susceptibility to strains of different lineages, consistent with the finding that host-pathogen associations can differ with HIV infection. (Fenner et al., 2013)
Interestingly, we found only one case of M. bovis despite the pre-ponderance of cattle in Botswana; this single case was found in the capital rather than in Ghanzi where cattle raising is an important component of the economy. TB in humans due to M. bovis is classically associated with extrapulmonary site of disease and with ingestion of unpasteurized milk products from infected cows. (Thoen et al., 2006) It is possible that our sample collection strategy with systematic collection only of sputum limited our ability to diagnose disease due to M. bovis. However, our findings would also be consistent with a low prevalence or absence of M. bovis reported for cattle in Botswana. (De Garine-Wichatitsky et al., 2013; Jori et al., 2013) M. bovis comprises a larger proportion of strains described for a number of African countries, though an understanding of the contribution of M. bovis to overall strain diversity are limited in many countries by small sample sizes that may not be representative of overall diversity. (Mbugi et al., 2016; Firdessa et al., 2013; Demay et al., 2012)
Phylogenetic diversity, both in terms of the number of strains and lineages observed, was greater in Gaborone as compared to Ghanzi. Regional variation in diversity of lineages is supported by another recent report in Botswana. (Mogashoa et al., 2019) In the report by Mogashoa et al., Euro-American/lineage 4 was the most common lineage followed by East Asian/lineage 2 and lineage 1; they also recovered one M. bovis isolate but in addition recovered one lineage 6 (M. africanum) isolate. Similar to our findings, they report very limited lineage diversity among isolates from South West Botswana, almost all of which were Euro-American/lineage 4; they also report greatest diversity of isolates from Southern Botswana, where Gaborone is located. Mogashoa et al. report a higher rate of drug resistance and of strain clustering; this may be due to selection bias of specimens for culture and differences in discriminating power of the geotyping methods used. Whereas we did find all 4 major global lineages of M. tuberculosis sensu stricto in Gaborone, we found only 3 of these lineages in Ghanzi and almost all strains were of the Euro-American lineage. Within the Euro-American lineage, diversity was also less in Ghanzi than in Gaborone, as evidenced by fewer sub-lineages represented, more clustered strains, and larger clusters of strains. These findings are consistent with our initial hypothesis that strain diversity would be less in Ghanzi than in Gaborone. (Chihota et al., 2018) The increased phylogenetic diversity of circulating strains in Gaborone would be consistent with a more ethnographically diverse and mobile human population in that metropolitan center, complemented by the importation of a diverse array of TB strains. The greater diversity of lineages found in Gaborone may reflect greater diversity in the ancestral region of origin of persons settling in Gaborone compared to Ghanzi. (Niemann et al., 2016). However, information on ethnicity was not collected as part of this study.
Similar findings to those in Ghanzi have been described among the indigenous Inuit populations of Canada, where the molecular epidemiology of TB is characterized by low strain diversity. (Nguyen et al., 2003; Lee et al., 2015) Such findings may indicate a founder effect with reactivation of a past epidemic strain or ongoing transmission, or both. However, because MIRU-VNTR genotyping only characterizes a small portion of the genome, apparently identical strains may overestimate direct lines of ongoing transmission due to insufficient resolution of the genotyping technique. The higher resolution genotyping method of whole genome sequencing (WGS) may be helpful in distinguishing between these possibilities. Among the Inuit, WGS suggested that low diversity was primarily the result of ongoing transmission. (Lee et al., 2015) In Ghanzi, determining the contribution of recent transmission versus reactivation of a endemic strain could help guide the prioritization of appropriate interventions, focused on prevention of re-activation of tuberculosis infection versus activities that interrupt transmission.
This study was limited to cases with positive cultures derived almost exclusively from respiratory specimens; however, genotyping was applied systematically to all cultured isolates during the study period and therefore bias in selection of strains was not likely. We used visual inspection of phylogenetic relatedness to a set of international reference strains classified by spoligotyping-based nomenclature to classify study strains by LSP-defined nomenclature. Although LSP testing was not performed directly on study isolates, concordance between LSP- and spoligotype/MIRU-VNTR-based typing for phylogenetic typing is high. (Kato-Maeda et al., 2011; Sola et al., 2003) Use of MIRU-VNTR typing will tend to over-estimate clustering rates; we would anticipate that higher resolution methods such as WGS would allow better understanding of diversity and transmission at the strain level. Genotypes with “double alleles”, which may represent infection with more than one strain, were excluded from analysis; however, we would not expect the likelihood of mixed infection to differ by phylogenetic lineage and therefore would not expect this exclusion to affect our overall conclusions on diversity of lineages in Botswana.
To our knowledge, this study provides the first systematic look at MTBC diversity in two regions of Botswana and contributes to the growing body of data regarding global mycobacterial diversity. Differences in strain-level diversity in two districts suggest important differences in introduction and propagation of TB in two districts of Botswana.
Acknowledgements
This publication made use of the MIRU-VNTRplus database website (http://www.miru-vntrplus.org/) developed by D. Harmsen, S. Nieman, P. Supply and T. Weniger. We thank Tsaone Tamuhla, Dimpho Otukile, Kitso Ramogale and Tlotlo Mogodi for laboratory technical work, Kyle Kingsley at Applied Maths for assistance with use of Bionumerics software, Lauren Cowan for assistance with construction of phylogenetic trees and Philip Supply for assistance with interpretation of Genoscreen genotyping result data and of MIRU-VNTRplus reference strain data.
Footnotes
Disclaimer
The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention (CDC) and other funding institutions.
References
- Allix-Beguec C, et al. , 2008. Evaluation and strategy for use of MIRU-VNTRplus, a multifunctional database for online analysis of genotyping data and phylogenetic identification of Mycobacterium tuberculosis complex isolates. J. Clin. Microbiol 46 (8), 2692–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botswana AIDS Impact Survey IV, 2013. Statistical Report. Republic of Botswana, Botswana. [Google Scholar]
- Botswana National Tuberculosis and Leprosy Program (BNTP), 2013–2014. Combined Annual Report 2013 and 2014. Botswana Ministry of Health, Republic of Botswana. [Google Scholar]
- Brudey K, et al. , 2006. Mycobacterium tuberculosis complex genetic diversity: mining the fourth international spoligotyping database (SpolDB4) for classification, population genetics and epidemiology. BMC Microbiol 6, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chihota VN, et al. , 2018. Geospatial distribution of Mycobacterium tuberculosis genotypes in Africa. PLoS One 13 (8), e0200632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coscolla M, Gagneux S, 2014. Consequences of genomic diversity in Mycobacterium tuberculosis. Semin. Immunol 26 (6), 431–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crush J, Williams V, Perberdy S, 2005. Migration in Southern Africa. In: Global Commission on International Migration. [Google Scholar]
- De Garine-Wichatitsky M, et al. , 2013. A review of bovine tuberculosis at the wildlife-livestock-human interface in sub-Saharan Africa. Epidemiol. Infect 141 (7), 1342–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong BC, Antonio M, Gagneux S, 2010. Mycobacterium africanum–review of an important cause of human tuberculosis in West Africa. PLoS Negl. Trop. Dis 4 (9), e744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demay C, et al. , 2012. SITVITWEB–a publicly available international multimarker database for studying Mycobacterium tuberculosis genetic diversity and molecular epidemiology. Infect. Genet. Evol 12 (4), 755–766. [DOI] [PubMed] [Google Scholar]
- Fenner L, et al. , 2013. HIV infection disrupts the sympatric host-pathogen relationship in human tuberculosis. PLoS Genet. 9 (3), e1003318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firdessa R, et al. , 2013. Mycobacterial lineages causing pulmonary and extrapulmonary tuberculosis, Ethiopia. Emerg. Infect. Dis 19 (3), 460–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CB, et al. , 2013. Mycobacterium tuberculosis mutation rate estimates from different lineages predict substantial differences in the emergence of drug-resistant tuberculosis. Nat. Genet 45 (7), 784–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagneux S, 2018. Ecology and evolution of Mycobacterium tuberculosis. Nat Rev Microbiol 16 (4), 202–213. [DOI] [PubMed] [Google Scholar]
- Gagneux S, Small PM, 2007. Global phylogeography of Mycobacterium tuberculosis and implications for tuberculosis product development. Lancet Infect. Dis 7 (5), 328–337. [DOI] [PubMed] [Google Scholar]
- Gagneux S, et al. , 2006. Variable host-pathogen compatibility in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A 103 (8), 2869–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HIV estimates with uncertainty bounds 1990–-2016, 2013. United Nations Programme on HIV/AIDS.
- Jori F, et al. , 2013. Preliminary assessment of bovine tuberculosis at the livestock/wildlife interface in two protected areas of northern Botswana. Transbound. Emerg. Dis 60 (Suppl. 1), 28–36. [DOI] [PubMed] [Google Scholar]
- Kato-Maeda M, et al. , 2011. Strain classification of Mycobacterium tuberculosis: congruence between large sequence polymorphisms and spoligotypes. Int J Tuberc Lung Dis 15 (1), 131–133. [PMC free article] [PubMed] [Google Scholar]
- Lee RS, et al. , 2015. Population genomics of Mycobacterium tuberculosis in the Inuit. Proc. Natl. Acad. Sci. U. S. A 112 (44), 13609–13614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazars E, et al. , 2001. High-resolution minisatellite-based typing as a portable approach to global analysis of Mycobacterium tuberculosis molecular epidemiology. Proc. Natl. Acad. Sci. U. S. A 98 (4), 1901–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbugi EV, et al. , 2016. Mapping of Mycobacterium tuberculosis complex genetic diversity profiles in Tanzania and Other African Countries. PLoS One 11 (5), e0154571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogashoa T, et al. , 2019. Genetic diversity of Mycobacterium tuberculosis strains circulating in Botswana. PLoS One 14 (5), e0216306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen D, et al. , 2003. Tuberculosis in the Inuit community of Quebec, Canada. Am. J. Respir. Crit. Care Med 168 (11), 1353–1357. [DOI] [PubMed] [Google Scholar]
- Niemann S, et al. , 2016. Impact of genetic diversity on the biology of Mycobacterium tuberculosis complex strains. Microbiol Spectr 4 (6). [DOI] [PubMed] [Google Scholar]
- Population and housing census, 2011. Analytical Report. 2014. Statistics Botswana, Gaborone, Botswana. [Google Scholar]
- Kent PT, K. G, 1985. Public Health Mycobacteriology: A Guide for the Level III Laboratory. U.S. Department of Health and Human Services, Public Health Service, Centers for Disease Control, Atlanta, Georgia. [Google Scholar]
- Sola C, et al. , 2003. Genotyping of the Mycobacterium tuberculosis complex using MIRUs: association with VNTR and spoligotyping for molecular epidemiology and evolutionary genetics. Infect. Genet. Evol 3 (2), 125–133. [DOI] [PubMed] [Google Scholar]
- Supply P, et al. , 2000. Variable human minisatellite-like regions in the Mycobacterium tuberculosis genome. Mol. Microbiol 36 (3), 762–771. [DOI] [PubMed] [Google Scholar]
- Supply P, et al. , 2001. Automated high-throughput genotyping for study of global epidemiology of Mycobacterium tuberculosis based on mycobacterial interspersed repetitive units. J. Clin. Microbiol 39 (10), 3563–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supply P, et al. , 2006. Proposal for standardization of optimized mycobacterial interspersed repetitive unit-variable-number tandem repeat typing of Mycobacterium tuberculosis. J. Clin. Microbiol 44 (12), 4498–4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessema B, et al. , 2013. Molecular epidemiology and transmission dynamics of Mycobacterium tuberculosis in Northwest Ethiopia: new phylogenetic lineages found in Northwest Ethiopia. BMC Infect. Dis 13, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoen C, Lobue P, de Kantor I, 2006. The importance of Mycobacterium bovis as a zoonosis. Vet. Microbiol 112 (2–4), 339–345. [DOI] [PubMed] [Google Scholar]
- WHO (Geneva: ). [Google Scholar]
- Zetola NM, et al. , 2016. Protocol for a population-based molecular epidemiology study of tuberculosis transmission in a high HIV-burden setting: the Botswana Kopanyo study. BMJ Open 6 (5), e010046. [DOI] [PMC free article] [PubMed] [Google Scholar]