

Summary
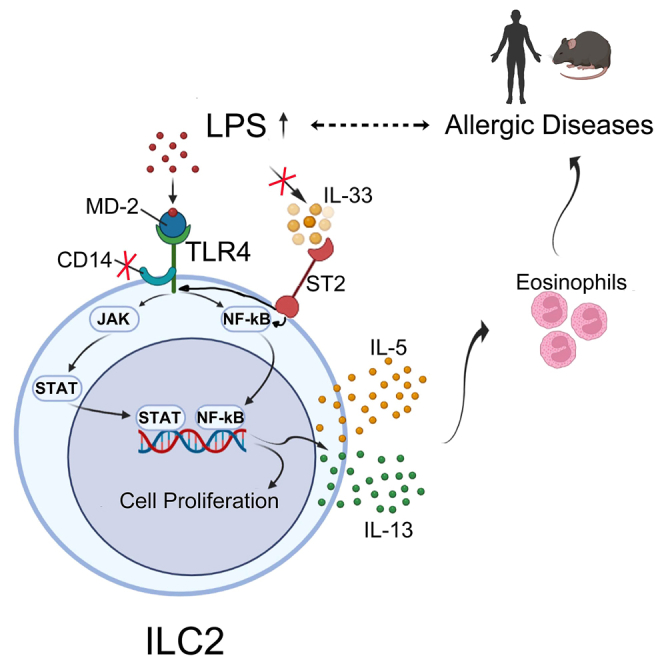
Group 2 innate lymphoid cells (ILC2s) are key players in type 2 immunity, but whether they can be directly activated by microbial ligands remain uncertain. In this study, we observed a positive correlation between blood endotoxin (LPS) levels and circulating ILC2s in allergic patients. In vitro, LPS robustly induced ILC2 proliferation and production of type 2 effector cytokines. RNA-seq revealed a type 2 immune-responsive profile in LPS-stimulated ILC2s. Notably, ILC2s lost their LPS-mediated growth and activation capacity when treated with TLR4 receptor antagonists and inhibitors of the NF-κB and JAK pathways, though this effect was not observed with IL-33 receptor blocking antibodies. Genetically, ILC2s from TLR4 knockout (KO) mice, but not from ST2 KO mice, were unresponsive to LPS. Collectively, these findings suggest a direct, non-canonical activation mechanism of ILC2s via the LPS-TLR4-NF-κB/JAK signaling axis.
Subject areas: Pathophysiology, Immunology, Cell biology
Graphical abstract
Highlights
-
•
LPS activates ILC2s via TLR4-NF-κB/JAK-STAT independent of IL-33-ST2 pathway
-
•
LPS alone can trigger the production of IL-5 and IL-13 by ILC2s
-
•
LPS promotes eosinophilic airway inflammation and associates with allergic diseases
Pathophysiology; Immunology; Cell biology
Introduction
Allergic disorders, including allergic rhinitis (AR), asthma, and atopic dermatitis (AD), are common diseases that affect more than 300 million people worldwide.1,2,3 Although these allergic diseases are traditionally characterized by an overzealous Th2-mediated inflammatory response, it has become increasingly appreciated in recent years that group 2 innate lymphoid cells (ILC2s) play an important role in the initiation and orchestration of type 2 immunopathologies.4,5 ILC2s are the innate counterparts of Th2 lymphocytes but lack rearranged antigen receptors.4,6,7,8 It has been reported that soluble factors are involved in orchestrating ILC2 responses, including cytokines, nutrients, hormones, and lipid mediators.9,10 However, the main direct activators of ILC2s are classical alarmin cytokines, such as IL-33, IL-25, and TSLP. Upon activation, ILC2s promptly produce large amounts of type 2 effector cytokines, such as IL-5 and IL-13, which drive the development of type 2 immunopathologies characterized by eosinophilia infiltration, airway remodeling, and mucus hypersecretion.11,12,13,14
Environmental allergens are complex and are often contaminated with microbial or parasitic products called pathogen-associated molecular patterns (PAMPs), such as lipopolysaccharides (LPS), which are also known as endotoxins. PAMPs stimulate innate immune cells by binding to and activating their corresponding pattern recognition receptors (PRRs). In addition to sensing PAMPs, PRRs can recognize self-ligands, such as damage-associated molecular patterns (DAMPs).15,16,17,18 Toll-like receptors (TLRs) were the first identified PRRs. All TLRs contain extracellular leucine-rich repeats (LRRs) for ligand binding and an intracellular Toll-interleukin-1 receptor (TIR) domain that recruits MyD88 and other adaptor proteins to activate signaling cascades, culminating in the production of various pro- or anti-inflammatory cytokines. To date, 10 TLRs (TLR1–10) have been identified in humans, and 12 (TLR1–9 and TLR11–13) have been identified in mice.19 TLR2 forms a heterodimer with TLR1, TLR6, or TLR10 to detect microbial lipopeptides and peptidoglycans. TLR4 detects bacterial LPS, whereas TLR5 recognizes bacterial flagellin. Nucleic acid-sensing TLRs include TLR3, TLR7/8, TLR9, and TLR13, which detect double-stranded RNA (e.g., poly[I:C]), R848 or single-stranded RNA (ssRNA), unmethylated CpG DNA (e.g., CpG-A), bacterial 23S rRNA and its derivative ISR23, a 13-nt sequence, respectively.20
The immune activation of PPRs by allergen-associated PAMPs and DAMPs is believed to be involved in the initiation of type 2 inflammation.21,22,23,24,25,26 Emerging evidence suggests that ILC2s express various PRRs, including TLRs and Nod2.27,28,29,30 Recent studies have demonstrated that microbial ligands, such as R848, ssRNA, and CpG-DNA, can activate the corresponding innate immune receptors to protect against eosinophilic airway diseases through the inhibition of ILC2 function,31,32,33 but these ligands go through the TLR-interferon signaling pathway in myeloid cells to indirectly regulate ILC2-driven type 2 inflammation. Although the role of PRRs in myeloid cells, such as monocytes and dendritic cells, in stimulating innate immune responses is well established, it remains largely unknown whether PPRs can act on ILC2s to regulate type 2 inflammatory responses. TLR4 was previously shown to play a role in regulating type 2 immune responses in mice34,35,36,37,38,39; however, whether TLR4 signaling directly regulates the function of ILC2s and its underlying mechanism remain to be explored.
In this study, we demonstrated that the abundance of LPS was positively correlated with the percentage of circulating ILC2s in patients with both AR and AD. Furthermore, among the various TLR ligands examined, only LPS robustly triggered ILC2s to expand and produce the type 2 effector cytokines IL-5 and IL-13. RNA-seq data revealed that the LPS-induced genes significantly overlapped with those induced by IL-33. However, the blockade of TLR4, NF-κB, and JAK signaling, not canonical IL-33 receptor signaling, completely abolished the activation of ILC2s stimulated by LPS. Moreover, LPS promotes ILC2-driven eosinophilic airway inflammation in vivo, which is alleviated in TLR4-deficient mice. These observations reveal a non-classical activation mode of ILC2s that is directly mediated by TLR4 signaling without the involvement of the IL-33-ST2 pathway, indicating that TLR4 signaling is a potential target for the treatment of microbial infection-associated inflammation mediated by ILC2.
Results
Endotoxin positively associates with ILC2s in peripheral blood samples of patients and can specifically activate cultured ILC2s in vitro
In the course of studying the ILC2 response in patients with AR and AD, we observed that the LPS concentration was positively associated with ILC2s in the plasma of patients with AR and AD, but not in healthy donors. The clinical information for these patients is presented in Tables S1 and S2. Remarkably, patients with AR and AD who had an elevated percentage of circulating ILC2s (Figures 1A and 1B) manifested higher amounts of LPS compared to healthy donors (Figure 1C). Furthermore, the concentration of LPS was positively correlated with the abundance of circulating ILC2s (Figures 1D and 1E). These clinical data suggest that elevated LPS levels are associated with an increased percentage of circulating ILC2s in patients with allergic inflammatory diseases.
Figure 1.

Endotoxin positively associates with ILC2s in peripheral blood samples of patients and can specifically activate cultured ILC2s in vitro
(A) FACS gating strategy. Human ILC2s from the peripheral blood of healthy donors, as well as patients with AR and AD, were stained with antibodies against lineage markers (CD45+Lin−CRTH2+CD127+).
(B) Percentages of human blood ILC2s were analyzed using FACS.
(C) Levels of blood LPS in healthy individuals and patients with AR and AD were measured by ELISA.
(D and E) The correlation analysis between the ILC2 percentage and LPS level in the peripheral blood of patients with AR (D) and AD (E) (unpaired t-test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
(F) Light microscopic images showing the growth of ILC2s. FACS-sorted human ILC2s were treated with various TLR ligands as indicated in a 96-well round-bottom plate for 5 days (1,000 cells were seeded in each well). Scale bar, 100 μm.
(G) The number of human ILC2s from each treatment was quantified using FACS. IL-5 (H) and IL-13 (I) levels from human ILC2s treated with the indicated TLR ligands were quantified by ELISA (two-way ANOVA, ∗∗p < 0.01, ∗∗∗∗p < 0.0001).
ILC2s are primarily activated by the alarmin cytokines IL-33, TSLP, or IL-25. However, whether a single TLR ligand is sufficient to activate ILC2s remains unclear. To address this issue, various TLR agonists were tested to activate in vitro-cultured human (CD45+Lin−CRTH2+CD127+) or mouse (CD45+Lin−T1/ST2+) ILC2s, which were originally isolated from human and mouse blood samples by fluorescence-activated cell sorting (FACS) (Figures S1A and S1B). The growth and cytokine production of ILC2s were analyzed by microscopy, FACS, and enzyme-linked immunosorbent assay (ELISA). Among the eight TLR ligands, LPS (Escherichia coli 0127:B8) was found to stimulate human ILC2s more potently than their murine counterparts, including bone marrow-ILC2s (BM-ILC2s) and lung-ILC2s, to proliferate (Figures 1F, 1G, S1C, S1D, S1G, and S1H) and produce the type 2 effector cytokines IL-5 and IL-13 (Figures 1H, 1I, S1E, S1F, S1I, and S1J). Interestingly, murine BM-ILC2s appear to be more sensitive to LPS than lung-ILC2s; therefore, BM-ILC2s were used for subsequent in vitro experiments. Taken together, these data suggest that LPS can directly stimulate ILC2s to mount a type 2 immune response.
LPS potently promotes the proliferation and cytokine production of ILC2s
We tested whether LPS activated ILC2s and determined the best activation mechanism. ILC2s harvested from human peripheral blood or cord blood, murine bone marrow, and murine lung tissue were cultured in vitro with or without LPS under various concentrations (0.01–100 μg/mL) for 3 or 5 days (this applies to all experiments if not specifically indicated). The number of human ILC2s and the secretion of type 2 cytokines, including IL-5 and IL-13, were promoted by the presence of LPS in a dose-dependent manner, and 10 μg/mL LPS was used thereafter in this study as the optimal activation dose (Figures 2A–2F). The promotion of cell growth was also measured by labeling with the cell proliferation markers Ki-67 and CSFE (Figures 2G–2I). Notably, LPS treatment did not compromise cell viability, implying negligible cytotoxic effects (Figure S2A). Similar observations were made in murine BM-ILC2s (Figures S2B–S2J) and murine lung ILC2s (data not shown). Taken together, these results indicate that LPS promotes ILC2 responses in vitro.
Figure 2.

LPS potently promotes the proliferation and cytokine production of ILC2s
(A) Light microscopic images showing the growth of human ILC2s treated with increasing doses of LPS, as indicated. Scale bar, 100 μm.
(B) The number of human ILC2s from each dose of LPS treatment was quantified by FACS analysis.
(C) Representative results of FACS showing the intracellular staining of human IL-5+IL-13+ ILC2s activated by increasing doses of LPS.
(D) The percentage of human ILC2s from each dose of LPS treatment was quantified using FACS. ELISA measuring the production of IL-5 (E) and IL-13 (F) by human ILC2s treated with increasing doses of LPS.
(G) Representative FACS images of Ki-67 staining showing the proliferation of LPS-treated human ILC2s.
(H) Quantification of Ki-67-positive cells.
(I) CSFE labeling for tracking the proliferation of human ILC2s treated with LPS or IL-33 for 3 or 5 days (two-way ANOVA, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). The data are representative from 2 experiments.
To further determine whether LPS from other gram-negative bacteria could stimulate ILC2s, we performed similar experiments using LPS extracted from E. coli 055:B5, Pseudomonas aeruginosa, and Salmonella enterica serotype typhimurium. The three LPS treatments similarly triggered the growth and cytokine production of human ILC2s at various concentrations (Figures 3A–3D). The murine BM-ILC2s exhibited similar responses (Figures S3A–S3D). Additionally, to avoid personal heterogeneity in human ILC2s, we obtained ILC2s from four healthy donors and cultured them in the presence of LPS or IL-33. LPS strongly enhanced the growth and cytokine production of ILC2s and significantly increased the effects of IL-33 in three of the donors (Figures 3E–3H), implying a synergistic effect between LPS and IL-33. Taken together, these data suggest that, at specific concentrations, LPS can directly activate both human and murine ILC2s to proliferate and induce the secretion of the type 2 effector cytokines IL-5 and IL-13.
Figure 3.

LPS from various Gram-negative bacterial species stimulates the growth and cytokine production of human ILC2s
(A) Light microscopic images showing the growth of human ILC2s treated with LPS isolated from E. coli 055:B5, P. aeruginosa, and S. typhimurium. Scale bar, 100 μm.
(B) The number of human ILC2s treated with various forms of LPS was quantified by FACS analysis. ELISA measuring the production of IL-5 (C) and IL-13 (D) by human ILC2s treated with the indicated LPS. Human ILC2s were derived from four individual donors as indicated.
(E) Light microscopic images showing the growth of human ILC2s. Scale bar, 100 μm.
(F) The number of human ILC2s was quantified by FACS analysis. IL-5 (G) and IL-13 (H) production were measured by ELISA (two-way ANOVA, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). Representative data from 1 experiment are shown here. Similar results were obtained from at least 3 experiments.
RNA-seq analysis reveals that LPS-induced genes in human ILC2s overlap significantly with those induced by IL-33
RNA-seq analysis was performed to obtain a global view of LPS-activated human ILC2s at the transcriptional level. To determine the best time point for sample collection, we examined the kinetics of the selected genes (IL-4, IL-5, IL-13, and TNF-α) induced by LPS in comparison with the IL-33 control using RT-qPCR. The peak levels of these genes appeared at 6 h in both treatments (Figure S4A). Therefore, we chose the 6-h samples to perform RNA-seq analysis of the differential expression of all genes (Figure 4). In the volcano plot analysis, 213 genes were upregulated, whereas 52 genes were downregulated compared to the control, which was also observed in the heatmap analysis (Figures 4A and 4B). Further cytokine analysis revealed that numerous type 2 effector cytokines, such as IL-4, IL-5, and IL-13, were induced, as highlighted in red (Figure 4C). To further define the functional connections between differentially expressed genes (DEGs), we performed Kyoto Encyclopedia of Genes and Genomes pathway enrichment and Gene Ontology Biological Process analyses. We discovered that genes from the LPS-treated samples were significantly enriched in biological processes highly related to immune modulation by cytokines, such as TNF and IL-17 (Figures 4D and S4B). By a direct comparison using gene set enrichment analysis (GSEA), the IL-33-upregulated gene set showed statistically significant concordance with the genes induced by LPS (Figure 4E), sharing more than 50% overlapped genes (141 genes) (Figure 4F). Taken together, these results reveal that LPS-induced genes overlap significantly with the gene signature induced by IL-33 in human ILC2s.
Figure 4.

RNA-seq analysis revealed that LPS-induced genes in human ILC2s overlap significantly with those induced by IL-33
Human ILC2s were stimulated by LPS in vitro for 6 h and then subjected to RNA-seq. Volcano plot (A) showing the upregulated and downregulated genes. Heatmap showing the DEGs (B), and the selected cytokine genes related to type 2 immunity are highlighted in red (C) in LPS-activated human ILC2s.
(D) Dot plot showing the enrichment analysis of Kyoto Encyclopedia of Genes and Genomes.
(E) GSEA analysis of DEGs upregulated by LPS and IL-33 in human ILC2s.
(F) Venn diagram depicting the overlapping and non-overlapping genes upregulated in human ILC2s stimulated by IL-2 and IL-7 with either LPS or IL-33, as compared to those treated with IL-2 and IL-7 alone.
LPS activates ILC2s via TLR4 without the involvement of the IL-33-ST2 pathway
Next, we investigated the involvement of TLR4- and ST2-mediated pathways in LPS-triggered signaling in human ILC2s. Although it has been reported that human ILC2s have a detectable level of TLR4 mRNA and are able to respond to a mixture of TLR-ligands,40 it remains to be determined whether LPS alone can activate human ILC2s via TLR4 signaling. To address this, we analyzed the mRNA and protein expression levels of TLR4 in ILC2s. Compared to other TLRs, TLR4 was highly expressed in untreated human ILC2s, indicating the importance of TLR4 signaling in this cell type (Figure 5A). In addition, the cell surface expression of TLR4, but not of CD14, was clearly detected by FACS staining (Figure 5B). Chemically, LPS contains three components: lipid A, an O-antigen, and a core oligosaccharide joined by a covalent bond. The lipid A domain is responsible for the toxicity of gram-negative bacteria and their binding to TLR4. To determine the specific role of the TLR4 receptor in the LPS-mediated activation of human ILC2s, we used a chemically synthesized TLR4 agonist, lipid A analog (CRX-527),41 and a natural antagonist, LPS-RS, to stimulate or block TLR4-specific signaling, respectively. Given that the CRX-527 solvent, DMSO, is likely to be toxic to cells at higher concentrations, DMSO alone at different concentrations was included as a control to rule out its potential confounding effects on ILC2s. CRX-527 at 0.1 and 1 μg/mL strongly promoted growth (Figures 5C and 5D) and the production of IL-5 and IL-13 (Figures 5E and 5F). Owing to the toxic effects of DMSO, CRX-527 completely lost its stimulatory effects at higher concentrations (10 or 100 μg/mL).
Figure 5.

LPS activates ILC2s via the TLR4 receptor without the involvement of the IL-33-ST2 pathway
(A) Heatmap showing the normalized mRNA levels of all 10 human TLRs in human ILC2s from two donors.
(B) FACS staining of TLR4 or CD14 proteins on the surface of human ILC2s. Light microscopic images showing the growth of human ILC2s treated with increasing doses of CRX-527, a lipid A analog, or its solvent DMSO (C). Scale bar, 100 μm.
(D) The number of human ILC2s was quantified by FACS analysis. IL-5 (E) and IL-13 (F) from human ILC2s were examined by ELISA. Light microscopic images showing human ILC2s exposed to increasing doses of LPS-RS (G). Scale bar, 100 μm.
(H) The number of human ILC2s was quantified using FACS. ELISA was used to measure the production of IL-5 (I) and IL-13 (J) from human ILC2s (unpaired t-test, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). Human ILC2s were treated by increasing doses of recombinant protein IL1RL1, and light microscopy (K) was used to observe their growth. Scale bar, 100 μm.
(L) FACS analysis showing the number of human ILC2s. ELISA measuring the production of IL-5 (M) and IL-13 (N) by human ILC2s (two-way ANOVA, ∗∗∗∗p < 0.0001). The data are representative from 2 experiments.
Next, we investigated whether LPS activity could be competitively blocked by LPS-RS, an LPS isolated from the photosynthetic bacterium Rhodobacter sphaeroides that is incapable of triggering TLR4 signaling.42 Remarkably, LPS-RS at 50 μg/mL or above almost completely inhibited the biological effects of LPS (Figures 5G–5J). It is worth mentioning that both LPS and LPS-RS dissolved in water. To demonstrate that TLR4 signaling is essential for LPS to directly activate ILC2s, we studied the LPS responsiveness of BM-ILC2s isolated from WT and TLR4−/− mice. As expected, we found that TLR4−/− ILC2s were completely unresponsive to LPS stimulation (Figures S5A–S5D).
To further address the possible involvement of the IL-33-ST2 pathway, we performed a competitive assay by adding increasing amounts of recombinant protein IL1RL1 to the ILC2 culture to sequester extracellular IL-33 in the media. Interestingly, while IL1RL1 at 1 and 10 μg/mL significantly blocked the effects of IL-33, it failed to affect the activity of LPS on human ILC2s (Figures 5K 5L, 5M, and 5N). To provide genetic evidence to rule out the IL-33-ST2 pathway in activating ILC2 in repose to LPS treatment, we have treated ST2 KO (Il1rl1−/−) and control WT mice with LPS and demonstrated that ILC2 cells from both conditions could be equally activated by LPS, indicating that LPS is activating ILC2 in an IL-33 or ST2 independent fashion (Figure S8). These results indicate that LPS activates human ILC2s via the TLR4 without the involvement of the IL-33-ST2 pathway.
NF-κB and JAK-STAT pathways are critical for LPS-induced activation of human ILC2s
The engagement of TLRs often triggers common signaling cascades, leading to the activation of a number of transcription pathways.43 We determined the pathways required for LPS to activate human ILC2s. To this end, we performed an additional RNA-seq analysis and found that the NF-κB and JAK-STAT signaling pathways were strongly associated with the function of LPS-activated human ILC2s (Figures 6A and 6B). We then aimed to determine whether NF-κB or JAK inhibitors could affect the growth and cytokine production of human ILC2s treated with LPS. As expected, NF-κB inhibition with Bay 11–7082 (1–100 μM) completely blocked LPS-mediated stimulating effects. A pan-JAK inhibitor also blocked all responses to LPS (Figures 6C–6F). Similarly, these two inhibitors blocked the IL-33-mediated effects on ILC2s (Figures 6G–6J). These data indicate that the NF-κB- and JAK-dependent pathways significantly contribute to the activation of human ILC2s.
Figure 6.

NF-κB and JAK-STAT pathways are critical for LPS-induced activation of human ILC2s
(A) Venn diagram showing the DEGs in the NF-κB pathway in human ILC2s stimulated by IL-2 and IL-7 with LPS, as compared to those treated with IL-2 and IL-7 alone.
(B) Venn diagram showing the DEGs in the JAK-STAT pathway in human ILC2s. Light microscopic images showing the growth of LPS-activated human ILC2s cultured under increasing concentrations of NF-κB and JAK inhibitors, or DMSO (C). Scale bar, 100 μm.
(D) FACS showing the number of LPS-activated human ILC2s. ELISA measuring the production of IL-5 (E) and IL-13 (F) by human ILC2s. Light microscopic images showing the growth of IL-33-activated human ILC2s treated with increasing concentrations of NF-κB and JAK inhibitors, or DMSO (G). Scale bar, 100 μm.
(H) FACS showing the number of IL-33-activated human ILC2s. ELISA measuring the production of IL-5 (I) and IL-13 (J) by human ILC2s (unpaired t-test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). Representative data from 1 experiment are shown here. Similar results were obtained from at least 3 experiments.
LPS promotes the activation of ILC2s and eosinophilic airway inflammation in vivo
Since we have shown that LPS is a potent ILC2 activator in vitro, we next evaluated whether LPS was able to elicit this function in vivo. To this end, we first used different doses of LPS to treat Rag1−/− mice via intravenous injection (Figure S6A). Consistent with our in vitro data, LPS induced the proliferation and activation of murine ILC2s in a dose-dependent manner, as measured by cell number, ST2 expression, and intracellular cytokine staining (Figures S6B–S6E). To demonstrate that TLR4 signaling is essential for LPS activation of ILC2s in vivo, we systemically delivered LPS into WT and TLR4−/− mice (Figure S6A) and found that LPS-induced growth, effector function, and ST2 expression of ILC2s were strictly dependent on TLR4 signaling (Figures S6F, S6D, S6H, and S6I).
The active role of LPS in ILC2s, both in vitro and in vivo, prompted us to explore its potential function in eosinophilic airway inflammation. To avoid the systemic effects of LPS in other tissues, intratracheal administration of LPS was used in a mouse model (Figure S7A). Our results showed that intratracheal administration of LPS induced ILC2 responses and promoted eosinophilic airway inflammation in WT mice and that the function of LPS was much weaker than that of IL-33 (Figures S7B–S7H).
To further mimic clinical situations and demonstrate the LPS function on human ILC2s in vivo, two humanized mouse models were developed, in which NSG or Rag2−/−γc−/− mice were reconstituted with human ILC2, as illustrated in Figure 7A. Similar models have recently been reported for IL-33 administration.27,44 Consistent with our in vitro data, the in vivo LPS stimulation led to the activation of the adoptively transferred human ILC2s, as shown by cell growth and type 2 cytokine production, which were measured by the cell number and intracellular cytokine staining (Figures 7B and 7C). Owing to a high level of protein sequence homology, human IL-5 could function as mouse IL-5 and was previously used to activate mouse eosinophils.45,46 Moreover, we also observed an increased number of mouse lung eosinophils in both NSG and Rag2−/−γc−/− mice (Figure 7D). These data indicate that LPS may play an important role in the regulation of ILC2-mediated inflammatory diseases.
Figure 7.

LPS induces human ILC2-mediated eosinophilic lung inflammation in humanized mouse models
(A) An experimental protocol for studying the activation of human ILC2s by LPS in two humanized mouse models.
(B) FACS showing the number of human ILC2s in the lungs of NSG or Rag2−/−γc−/− mice, which were reconstituted with human ILC2s and treated with or without LPS.
(C) As in B, the percentage of IL5+IL13+-double positive human ILC2s in NSG or Rag2−/−γc−/− mice was analyzed by intracellular staining.
(D) As in B, FACS showing the number of lung eosinophils in NSG or Rag2−/−γc−/− mice (unpaired t-test, ∗∗p < 0.01, ∗∗∗p < 0.001).
Discussion
In this study, we discovered a function of LPS-TLR4 signaling in ILC2s. Increased levels of circulating ILC2s appear to correlate well with elevated LPS levels in patients with AR and AD. Among the various TLR ligands, LPS activates the proliferation and function of ILC2. LPS extracted from multiple species of gram-negative bacteria can potently stimulate both human and murine ILC2s to proliferate and produce massive amounts of the type 2 effector cytokines IL-5 and IL-13. RNA-seq data revealed that LPS upregulated a large set of genes that significantly overlapped with those induced by IL-33. However, this direct effect on ILC2s by the LPS-TLR4 signaling axis does not require the classical IL-33-ST2 pathway but rather depends on the NF-κB and JAK pathways. Furthermore, in the lungs of WT and humanized mice, LPS promoted the proliferation of murine ILC2s and adoptively transferred human ILC2s in a TLR4-signaling-dependent manner. Collectively, these data suggest that LPS-TLR4 signaling directly functions in ILC2s to promote the proliferation and production of type 2 effector cytokines, independent of the endogenous IL-33-ST2 pathway.
ILC2s are activated by tissue-derived alarmin signaling upon exposure to environmental allergens. In recent years, accumulating evidence has suggested that diverse receptors expressed on the surface of ILC2s can either enhance or repress their activation and proliferation in response to various non-classical signals, hormones, regulatory cytokines, neuropeptides, and lipids.9,10,47 It was previously shown that human ILC2s express the mRNA of some TLRs (TLR1, TLR4, and TLR6) and can respond to a mixture of the three TLR ligands40; however, the TLR pathway that functions in human ILC2s remains unknown. Our findings show that the TLR4 signaling pathway plays an important role in the survival, proliferation, and production of type 2 cytokines by ILC2s. It has been reported that the co-expression of TLR4 and CD14 is required for cell type-specific responses to LPS, as well as for the maximal potency of TLR4-dependent cell activation by LPS, triggering downstream MyD88 and/or Toll/IL-1 receptor (TIR) domain-containing adaptor (TRIF) pathways to induce the secretion of pro-inflammatory factors and interferons. Moreover, CD14 is essential for efficient LPS delivery to the TLR4 receptor complex and its subsequent endocytosis, which is required for activating interferon regulatory factor 3 (IRF3)-mediated interferon responses through interactions with the adaptor molecules TRIF and TANK-binding kinase 1 (TBK1).48,49 In this study, we found that human ILC2s exhibited a high expression of TLR4 but lacked CD14. Moreover, the proliferation and activation of ILC2s induced by LPS or IL-33 were entirely dependent on the NF-κB and JAK pathways, which could be blocked by the pathway inhibitors. Thus, unlike other cell types, ILC2s are unable to activate the CD14-mediated signaling pathway because of their lack of CD14 expression.
It appears that mouse and human ILC2 cells had different capacities in response to LPS treatment (Figures 1F and S1C). Species difference in both innate and adaptive immunity including TLRs is common phenomenon.50 It is known that evolution across species can lead to substantial diversity in the TLR4’s affinity and specificity to its ligands, the TLR4 gene and cellular expression patterns and tissue distribution. The divergence in LPS responsiveness among species attributes to different levels of the effector cytokine production.51 In addition to TLR4, its signaling component MD-2 also displays some discrete structural differences between mouse and human. Lysines 122, 125, and 58 in human MD-2 contribute to the functional differences in response to the sensitivity to LPS between human and murine.52
Previous epidemiological and experimental studies have suggested that endotoxin-TLR4 signaling contributes to the development of type 2 inflammatory reactions.34,53,54,55,56 However, its underlying cellular and molecular mechanisms remain poorly understood. Natural allergens contain not only allergic proteins but also various PAMPs derived from bacteria, parasites, and viruses, which may stimulate innate immune sensors, such as TLRs, to participate in the regulation of type 2 inflammatory responses.21,22,23,24,25,26 More and more researchers have focused on the relationship between airway microbiota and allergic airway inflammation, and recent studies have shown that the presence of airway microbiota can significantly aggravate the response to allergic airway inflammation.57,58,59 In the current study, we demonstrated that bacterial endotoxins directly engage the TLR4 receptor expressed on the surface of ILC2s to robustly activate their proliferation and production of type 2 effector cytokines in vitro and in vivo, which leads to the recruitment of eosinophils in airway tissue. More importantly, our clinical data showed that the plasma concentrations of LPS in patients with AR and AD were higher than those of healthy donors and were positively correlated with the percentage of ILC2s. This indicates that the TLR4 signaling pathway may promote ILC2-mediated type 2 inflammatory conditions. However, a multicenter clinical study involving a large cohort of patients with allergic disorders, such as AR, asthma, and AD, is needed to further elucidate the relationship between LPS concentration and the incidence of these diseases. Besides its classical ligand, LPS, TLR4 can also be activated by various endogenous ligands, including DAMPs like HMGB1,60 hyaluronan,61 OxPAPC,62 surfactant protein A,63 S100 proteins,64 and HSP72.65,66 It has recently been reported that proteinase cleavage of fibrinogen can elicit allergic responses through the TLR4-mediated pathway.67 It would be interesting to evaluate whether self TLR4 ligands can activate human ILC2s in future studies.
ILC2s can communicate with various cell types through the surface expression of multiple receptors, which can bind to ligands present on other immune cells, such as dendritic cells and T or B cells.47,68,69 In our RNA-seq analysis, we found that LPS treatment alone was sufficient to induce the expression of many members of the TNF receptor superfamily and their ligands in ILC2s, which are known to provide key costimulatory signals to T and B cells. However, further research is required to understand the function and regulatory mechanisms of how these molecules on ILC2s interact with other immune cell types to regulate type 2 immunity.
Finally, it is important to note that the human ILC2 cells used in this study were expanded under standard culture conditions with IL-2 and IL-7. While these cells may differ biologically from ILC2 cells directly isolated from human PBMCs or cord blood mononuclear cells (CBMCs), our in vitro cultured ILC2 retained their original properties, as evidenced by their robust response to IL-33 stimulation, leading to the transcriptional and translational upregulation of type 2 response signature genes.
In conclusion, our findings provide evidence for an alternative mechanism of activating ILC2s via the LPS-TLR4 signaling axis without the involvement of the classical activation pathway mediated by the IL-33 receptor, IL1RL1 (also known as ST2). Considering the broad availability of TLR4 ligands from both foreign (e.g., bacterial infections) and endogenous sources, targeting the TLR4 signaling pathway could be developed as an alternative approach to alleviate ILC2-mediated type 2 inflammatory conditions.
Limitations of the study
The physiological role of endotoxin-activated ILC2 cells is not yet fully understood. The biological properties of human ILC2 cells may vary significantly between the in vitro conditions used in this study and those directly isolated from allergic patients who have elevated endotoxin levels in their blood. Therefore, it is crucial to compare the functions of ILC2 cells from both healthy individuals and patients in future research.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Xiao-Dong Li (lixiaodong@hotmail.com).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact with a completed Materials Transfer Agreement.
Data and code availability
-
•
Raw data in this study are available from Sequence Read Archive (SRA) of NCBI and accession number is listed in the key resources table. All data present in this study will be shared by the lead contact upon request.
•This paper does not report original code.
•Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Acknowledgments
We thank Ms. Karla Gorena, Mr. Sebastian Montagnino for technical assistance in flow cytometry and FACS sorting. We thank Drs. Zhao Lai, Yi Zou, and Yidong Chen for RNA-seq analysis and informatics assistance. X.Z., L.S., and Y.L. are supported by National Key Research and Development Program of China (No. 2023YFC2410204). L.S., X.Z., Y.L., and X.-D.L. are supported by National Natural Science Foundation of China (Nos. 82301286, 82073009, 81974424, and 82270022). L.S. is supported by China Postdoctoral Science Foundation (No. 2022M723557), Natural Science Foundation of Hunan (No. 2024JJ6629), Changsha Natural Science Foundation (No. kq2208390) and The Youth Science Foundation of Xiangya Hospital (No. 2022Q08). H.H.A. is supported by the Department of Clinical Laboratory Sciences, College of Applied Medical Sciences, Jouf University, Sakaka, Saudi Arabia. X.-D.L. is supported by the Max and Minnei Voelcker Fund and Guangzhou Medical University Startup Fund.
Author contributions
L.S. performed most experiments; L.S., H.H.A., Y.X., Y.Y., Y.G., J.W., D.P.C., Q.X., H.Z., Z.X., Y.S., N.X., W.J., Z.X., X.Z., Y.L., and X.-D.L. analyzed data; L.S., Y.L., and X.-D.L. planned, designed research. L.S., Y.L., and X.-D.L. wrote the manuscript; All authors discussed the results and participated in writing and commenting on the manuscript.
Declaration of interests
The authors declare no conflict of interest.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| FITC anti-human Lineage Cocktail | Biolegend | Cat#348801 |
| APC anti-human CD294 (CRTH2) Antibody | Biolegend | Cat#350110 |
| APC/Cyanine7 anti-human CD45 Antibody | Biolegend | Cat#368516 |
| PE anti-human CD127 | Biolegend | Cat#351340 |
| Biotin anti-mouse CD3ε (clone 145-2C11) | Biolegend | Cat#100304 |
| Biotin anti-mouse CD4 (clone GK1.5) | Biolegend | Cat#100404 |
| Biotin anti-mouse CD8α (clone 53-6.7) | Biolegend | Cat#100704 |
| Biotin anti-mouse CD11c (clone N418) | Biolegend | Cat#117304 |
| Biotin anti-mouse FceRIα (clone MAR-1) | Biolegend | Cat#134304 |
| Biotin anti-mouse NK1.1 (clone PK136) | Biolegend | Cat#108704 |
| Biotin anti-mouse CD19 (clone 6D5) | Biolegend | Cat#115504 |
| Biotin anti-mouse TER119 (clone TER-119) | BioLegend | Cat#116204 |
| Biotin anti-mouse CD5 (clone 53-7.3) | BioLegend | Cat#100604 |
| Biotin anti-mouse F4/80 (clone BM8.1) | Tonbo Biosciences | Cat#30-4801-U500 |
| Biotin anti-mouse Gr1 (clone RB6-8C5) | Tonbo Biosciences | Cat#30-5931-U500 |
| APC-conjugated streptavidin | BioLegend | Cat#405207 |
| PE-conjugated T1/ST2 (clone DIH9) | BioLegend | Cat#145304 |
| Anti-Mouse Siglec-F PE (clone E50-2440) | BD Biosciences | Cat#552126 |
| Anti-Mouse CD11c PE-Cy7 (clone N418) | Tonbo Biosciences | Cat#60-0114-U100 |
| Anti-Mouse CD45 APC-Cy7 (clone 30-F11) | BD Biosciences | Cat#561037 |
| PE anti-human Ki-67 Antibody | Biolegend | Cat#350504 |
| PE anti-mouse Ki-67 Antibody | Biolegend | Cat#652404 |
| Chemicals, peptides, and recombinant proteins | ||
| PamsCSK4 | InvivoGen | Cat#tlrl-pms |
| Poly(I:C) | InvivoGen | Cat#tlrl-pic |
| Flagellin | InvivoGen | Cat#tlrl-bsfla |
| R848 | InvivoGen | Cat#tlrl-r848-1 |
| CpG-A (ODN 2336, human)CpG-A (ODN 1585, mouse) | InvivoGen | Cat#tlrl-2336-1 Cat#tlrl-1585 |
| ISR23 | IDT | N/A |
| LPS from E. coli 0127:B8 LPS from E. coli 055:B5 LPS from P. aeruginosa 10 LPS from S. typhimurium | Sigma | Cat#L4516 Cat#L6529 Cat#L8643 Cat#L6143 |
| CRX-527 | InvivoGen | Cat#tlrl-crx-527 |
| LPS-RS Ultrapure | InvivoGen | Cat#tlrl-prslps |
| Human IL1RL1/ST2 protein | Sino Biologicl | Cat#13034-H08H |
| Bay 11-7082 | EMD Millipore | Cat#196870 |
| JAK inhibitor 1 | EMD Millipore | Cat#420099 |
| PMA and Brefeldin A | Sigma | Cat#P1585 Cat#B6542 |
| SPHERO AccuCount Fluorescent | Spherotech | Cat#ACFP-70-5 |
| Fixable Viability Dye eFluor 506 | Invitrogen | Cat#65-0866-14 |
| Recombinant Human IL-2 | PeproTech | Cat#200-02 |
| Recombinant Human IL-7 | PeproTech | Cat#200-07 |
| Recombinant Human IL-33 | PeproTech | Cat#200-33 |
| Recombinant Murine IL-2 | PeproTech | Cat#212-12 |
| Recombinant Murine IL-7 | PeproTech | Cat#217-17 |
| Recombinant Murine IL-33 | PeproTech | Cat#210-33 |
| CFSE Cell Division Tracker Kit | Biolegend | Cat#423801 |
| 1-Step™ TMB ELISA Substrate Solutions | Thermo Scientific | Cat#34028 |
| Cyto-Fast™ Fix/Perm Buffer Set | Biolegend | Cat#426803 |
| True-Nuclear™ Transcription Factor Buffer Set | Biolegend | Cat#4244011 |
| CFSE Cell Division Tracker Kit | Biolegend | Cat#423801 |
| Human IL-5 Uncoated ELISA Kit | Thermo Scientific | Cat#88-7056-88 |
| Human IL-13 Uncoated ELISA Kit | Thermo Scientific | Cat#88-7439-88 |
| Mouse IL-5 Uncoated ELISA Kit | Thermo Scientific | Cat#88-7054-88 |
| Mouse IL-13 Uncoated ELISA Kit | Thermo Scientific | Cat#88-7137-88 |
| Deposited data | ||
| RNA-seq raw data generated for this paper | Sequence Read Archive (SRA) of NCBI (https://trace.ncbi.nlm.nih.gov/Traces/?view=study&acc=SRP534046) | SRR30760103 SRR30760104 SRR30760105 SRR30760106 |
| Experimental models: Organisms/strains | ||
| Rag1−/− | Jackson Laboratory | Cat#002216 |
| Rag2−/−γc−/− | Jackson Laboratory | Cat#014593 |
| NSG | Jackson Laboratory | Cat#005557 |
| Il1rl1−/− | GemPharmatech | Cat#T005974 |
| Oligonucleotides | ||
| GAPDH | ATGACATCAAGAAGGTGGTG; CATACCAGGAAATGAGCTTG | N/A |
| IL-4 | ACTTTGAACAGCCTCACAGAG; TTGGAGGCAGCAAAGATGTC | N/A |
| IL-5 | AGCTGCCTACGTGTATGCCA; CAGGAACAGGAATCCTCAGA | N/A |
| IL-13 | TGAGGAGCTGGTCAACATCA; CAGGTTGATGCTCCATACCAT | N/A |
| TNF-α | CCTGGTATGAGCCCATCTATCTG; TAGTCGGGCCGATTGATCTC | N/A |
| Software and algorithms | ||
| Graphpad Prism 8 | Prism | N/A |
| FlowJo Version 9 | BD Biosciences | N/A |
Experimental model and study participant details
Mice
Rag1−/−, Rag2−/−γc−/− (Rag2tm1.1FlvIl2rgtm1.1Flv/J, 014593) and NSG (NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ, 005557) mice were purchased from Jackson Laboratory (USA). Il1rl1−/− mice were obtained from GemPharmatech. All animals were bred and maintained under specific pathogen-free conditions in the animal facility according to the experimental protocols approved by the Institutional Animal Care and Use Committee (IACUC# 2015055AR). ELISA to detect serum LPS in peripheral blood.
Human subjects/samples
Blood samples were collected from patients with AR and AD or from healthy donors in compliance with the guidelines of the Institutional Review Board of Xiangya Hospital at Central South University (No. 2023030472). PBMCs were first isolated from peripheral blood using Ficoll-Paque PLUS in a 15mL centrifuge tube. Cells were then washed and suspended with 2%FACS buffer (PBS with 2% FBS). After counting PBMCs with BIO-RAD cell counting slide and TC20 Automated Cell Counter, cells were pipetted into the well of a 96-well round bottom plate with 2∗10ˆ6 cells per well. Cells were then stained with Ghost DyeTM Violet 510, FITC anti-human Lineage Cocktail (CD3, CD14, CD16, CD19, CD20, CD56), APC anti-human CD294 (CRTH2), APC/Cyanine7 anti-human CD45, and PE anti-human CD127 (IL-7Rα). Human ILC2 cells were analyzed by flow cytometry. The level of serum LPS was analyzed by a highly sensitive ELISA kit according to the manufacture instruction (Cloud-Clone Corp.). Briefly, reagents A and B were sequentially applied. For detection, TMB substrate was added. The reaction was stopped by the stop solution provided by the kit. The OD value of each sample was read at 450nm.
Regents
The medium used was RPMI 1640 (Sigma-Aldrich), containing 10% fetal bovine serum (FBS; HyClone), 1% penicillin-streptomycin (Gibco), 1x GlutaMAX-I (Gibco), and 50 μM 2-Mercaptoethanol (Sigma-Aldrich). PamsCSK4, Poly(I:C), flagellin, R848, and CpG-A were purchased from InvivoGen. ISR23 was obtained from Integrated DNA Technologies (Beijing, China). LPS from E. coli 0127:B8, E. coli 055:B5, P. aeruginosa 10, and S. typhimurium were purchased from Sigma-Aldrich. Recombinant human and mouse cytokines IL-2, IL-7, and IL-33 (human and mouse) were obtained from PeproTech. The TLR4 agonist CRX-527 (tlrl-crx-527) and the TLR4 antagonist LPS-RS Ultrapure (tlrl-prslps) were obtained from InvivoGen. Human IL1RL1/ST2 protein (isoform a, His Tag; 13034-H08H) was purchased from Sino Biological. The NF-κB inhibitor Bay 11–7082 and JAK inhibitor 1 were both from EMD Millipore. PMA and Brefeldin A were purchased from Sigma-Aldrich and BioLegend, respectively.
Method details
ELISA for detecting serum LPS
Peripheral blood samples were collected from healthy donors and patients with AR or AD. Serum LPS levels were analyzed using a highly sensitive ELISA kit according to the manufacturer’s instructions (Cloud-Clone Corp.). For detection, TMB substrate was added. The reaction was stopped using the stop solution provided with the kit. The optical density (OD) of each sample was measured at 450 nm.
Isolation of ILC2s
Human ILC2s were isolated from the peripheral blood of healthy donors or umbilical cord blood samples from healthy full-term births at the Department of Obstetrics and Gynecology of UT Health San Antonio. All human samples were used in compliance with the UT Health San Antonio Institutional Review Board. PBMCs or cord blood mononuclear cells (CBMCs) were isolated from diluted blood (1:2) by density gradient centrifugation using density gradient medium (Histopaque, Sigma-Aldrich) and SepMateTM 50 mL tubes (STEMCELL Technologies).70 Cells were then washed once with dPBS-FBS buffer (dPBS, 3% FBS, 1 mM EDTA) and resuspended in dPBS-FBS, followed by staining with antibodies against CD45, lineage markers (CD3, CD14, CD16, CD19, CD20, and CD56), and the ILC2 markers CRTH2 and CD127 (all from BioLegend). Human ILC2s were sorted by the BD FACS Aria cell sorter as CD45+Lin−CRTH2+CD127+ cells. The purity of the sorted ILC2s was determined to be greater than 95%. Sorted human ILC2s were cultured and expanded in a medium supplemented with recombinant human (rh)-IL-2 and rh-IL-7 (all at 50 ng/mL) in 96-well round plates for 6 days before further experiments. In general, about 5,000 human ILC2s were obtained FACS-sorted from PBMCs or CBMCs isolated from 10 mL of peripheral blood or umbilical cord blood. About 300,000 ILC2 were generally obtained to perform following the in vitro ILC2 experiments.
Murine bone marrow or lung ILC2s were isolated from Rag1−/− mice treated with rm-IL-33 (250 ng/mouse) for 3 consecutive days and 2 days of rest before processing lung tissues for sorting ILC2s with a BD FACS Aria cell sorter. The criteria for identifying ILC2s included the lack of classical lineage markers (CD3ε, CD4, CD8α, CD11c, FceRIα, NK1.1, CD19, TER119, CD5, F4/80 and Gr-1) but expression of CD45 and T1/ST2 markers (all from BioLegend). The purity of the sorted ILC2s was greater than 95%. Sorted ILC2s were cultured and expanded in a medium supplemented with mouse IL-2 and IL-7 (all at 10 ng/mL) in 96-well round plates for 6 days before further experiments.
Culture and treatment of human and murine ILC2s
Sorted human ILC2s were cultured in the medium (200 μL) with or without rh-IL-2, rh-IL-7, and rh-IL-33 (all at 50 ng/mL) in 96-well round plates (500 or 1,000 cells/well) in a 37°C incubator with 5% CO2. The cells were treated with different concentrations of Pam3CSK4, Poly(I:C), LPS, flagellin, R848, CpG-A, and ISR23 for 3 or 5 days. The human ILC2s were treated with TLR4 agonist or antagonist, human IL1RL1/ST2 protein (isoform a, His Tag; added 1 h prior to the treatment with LPS or IL-33), or the NF-κB or JAK inhibitor for 5 days. After 3 days of treatment, the percentage of IL-5+IL13+ cells, expression of Ki-67, and cell death of ILC2s were analyzed by flow cytometry. After 5 days of treatment, the number and proliferation of ILC2s were analyzed using flow cytometry, and the levels of cytokines (IL-4, IL-5, and IL-13) in the supernatants were measured using ELISA.
Sorted murine bone marrow or lung ILC2s were cultured and treated with the same TLR ligands in 200 μL media with or without murine IL-2, IL-7, and IL-33 (all at 10 ng/mL) in 96-well round plates (1,000 cells/well) in a 37°C incubator with 5% CO2. The percentage of IL-5+IL13+ cells and the expression of Ki-67 in murine ILC2s were analyzed by flow cytometry 3 days later. The number and proliferation of ILC2s were analyzed by flow cytometry 5 days later, and the supernatants were collected for further detection of IL-5 and IL-13 by ELISA.
Flow cytometric analysis
Fc receptors were blocked with 2.4G2 hybridoma supernatant (generated in the laboratory). To detect early-stage apoptotic cells, Annexin V staining was performed according to the manufacturer’s protocol (eBioscience). Intranuclear staining of Ki-67 was performed with the True-Nuclear Transcription Factor Buffer Set (BioLegend) according to the manufacturer’s instructions. For intracellular cytokine staining, human or murine ILC2s were cultured and treated as indicated for 3 days, followed by incubation with Brefeldin A for 3 h. After surface staining, the cells were fixed and permeabilized with BioLegend Cytofix/Perm buffer and stained intracellularly with anti-human IL-5 and IL-13 or anti-mouse IL-5 and IL-13. For lung single-cell suspensions, 2 × 106 total live nucleated cells were stimulated in 200 μL media with Brefeldin A and PMA (phorbol 12-myristate 13-acetate; 30 ng/mL) at 37°C for 3 h. After surface staining, the cells were fixed, permeabilized, and intracellularly stained with anti-human IL-5 and IL-13 antibodies. Dead cells were stained with eFluor506 Fixable Viability Dye before fixation and permeabilization and excluded during the analysis. For TLR4 and CD14 expression in human ILC2s, cells were stained with anti-human TLR4 and anti-human CD14 surface markers.
Protein quantification in cell culture supernatants
Cytokines (IL-5 and IL-13) in the supernatants of human and mouse ILC2 cell cultures were analyzed using ELISA kits from Invitrogen. All final reactions were developed using TMB substrate (Thermo Scientific), stopped with sulfuric acid (0.16M), and the OD was measured at 450 nm.
CSFE staining
Human ILC2s were stained with 1 μM CSFE (CSFE cell division tracker kit, BioLegend) according to the manufacturer’s recommendations. This was followed by culture in the medium with or without rh-IL-2, rh-IL-7, rh-IL-33 (all at 50 ng/mL), or LPS (10 μg/mL) for 3 or 5 days in a 37°C incubator with 5% CO2. The proliferation of ILC2s was analyzed using flow cytometry.
Humanized mice
Purified human ILC2s were cultured with rh-IL-2 and rh-IL-7 (all at 50 ng/mL) for 6 days and then adoptively transferred to NSG mice or Rag−/−γc−/− mice (4 × 104 cells/mouse). Four hours after cell transfer, host mice were challenged with rh-IL-2 and rh-IL-7 (all 250 ng/mouse) in the absence or presence of LPS (5 μg/mouse) on day 0, 1, and 2, as shown in Figure 7A. On day 5, the mice were euthanized and the lungs were analyzed.
FACS analysis of lungs
Mice were euthanized at the indicated times, and the lung tissues were digested in 8 mL RPMI-1640 containing Liberase (50 μg/mL) and DNase I (1 μg/mL) for about 45 min at 37°C. The cell suspensions were filtered through 70 μm cell strainers and washed twice with RPMI-1640. Human ILC2s and murine eosinophils in the lungs were labeled with antibodies as indicated and mixed with counting beads for further FACS analysis on a BD Celesta cell analyzer. Flow cytometry data were analyzed using FlowJo software. The antibodies and reagents for FACS analysis are listed below: SPHERO AccuCount Fluorescent (ACFP-70-5, Spherotech), Anti-Human CD45 APC-Cy7 (HI30, BioLegend), Anti-Human CRTH2 APC (BM16, BioLegend), Anti-Human CD127 PE (A019D5, BioLegend), Anti-Mouse Siglec-F PE (E50-2440, BD Bioscience), Anti-Mouse CD11c PE-Cy7 (N418, TONBO bioscience), Anti-Mouse CD45 PerCP-Cy5.5 (30-F11, BioLegend), and Fixable Viability Dye eFluor 506 (Invitrogen).
RT-qPCR
Human ILC2s were treated with or without LPS (10 μg/mL) or rh-IL-33 (50 ng/mL) in 200 μL media with or without rh-IL-2 and rh-IL-7 (all 50 ng/mL) for 6 or 16 h. Total RNA was extracted using the TRIzol reagent (Life Technologies) according to the manufacturer’s instructions. RT-qPCR reactions were performed using the iScript cDNA synthesis kit and iQ SYBR Green Supermix (Bio-Rad). RT-qPCR was performed on a Bio-Rad CFX384 Touch Real-Time PCR Detection System using the human primers as listed in key resources table.
RNA isolation, RNA-seq, and bioinformatics
Purified human ILC2s (4 × 104) were cultured and stimulated with LPS (10 μg/mL) or rh-IL-33 protein (50 ng/mL) in the presence of rh-IL-2 and rh-IL-7 (all at 50 ng/mL) for 6 h. Total RNA was extracted using the RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions.
After the quality of the RNA samples was verified using an Agilent Bioanalyzer 2100 (Agilent), the RNA was further processed using an Illumina TruSeq RNA sample prep kit v2 (Illumina). Clusters were generated using the TruSeq Single-Read Cluster Generation Kit v3-cBot-HS on an Illumina cBot Cluster Generation Station. After quality control procedures, individual RNA-seq libraries were pooled based on their respective 6 bp index portions of the TruSeq adapters and sequenced at 50 bp/sequence using an Illumina HiSeq 3000 sequencer. The resulting reads were checked using a quality assurance pipeline and an initial genome alignment. After sequencing, demultiplexing with CASAVA was performed to generate a FASTQ file for each sample. All sequencing reads were aligned with the reference genome (GRCh38/hg38) using the HISAT2 default settings, yielding Bam files that were discarded using Picard and processed using HTSeq count to obtain counts for each gene. The RNA expression levels were determined using GENCODE annotation. Differential expression analysis was performed using the Deseq2 package in R post-normalization based on a Benjamini–Hochberg false discovery rate-corrected threshold for statistical significance of padj <0.05, or raw p-value <0.01. Transcript read counts were transformed into ln (x+1) used to generate heat maps using ClustVis. Volcano plots depicting log2-FoldChange and raw or adjusted p-values were generated in R. Venn diagrams were generated using the Venn Diagram package in R.
To investigate the biological pathways, DEGs were manually curated and compared to multiple public databases, including Gene Ontology and Kyoto Encyclopedia of Genes and Genomes, for enrichment analysis. For GSEA, an IL-33-upregulated gene set was identified from our RNASeq data. This gene set was obtained by comparing the sample treated with rh-IL-33 in the presence of rh-IL-2 and rh-IL-7 and the sample treated only with rh-IL-2 and rh-IL-7 stimulation. The threshold for statistical significance was a raw p-value <0.01 and log2-FoldChange >1. Samples with or without LPS in the presence of rh-IL-2 and rh-IL-7 were directly compared to this gene set to identify statistically significant concordance in expression using the GSEA algorithm.
Quantification and statistical analysis
Statistical analysis
Statistical analyses were performed using GraphPad Prism 8. For the comparison of two groups, p-values were determined using unpaired two-tailed Student’s t test. To compare more than two groups, two-way ANOVA was performed. p-values are indicated in the plots and figure legends. All error bars indicate standard errors of means (SEM). The number of patients and animals, and the number of repeating experiments have been indicated or stated in figure. p < 0.05 was considered statistically significant, while p ≥ 0.05 was not considered statistically significant (N.S.). Significance was indicated by asterisks: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Published: October 23, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.111240.
Contributor Information
Yong Liu, Email: liuyongent@csu.edu.cn.
Xiao-Dong Li, Email: lixiaodong@hotmail.com.
Supplemental information
References
- 1.Jackson K.D., Howie L.D., Akinbami L.J. NCHS Data Brief; 2013. Trends in Allergic Conditions Among Children: United States, 1997-2011; pp. 1–8.https://www.ncbi.nlm.nih.gov/pubmed/23742874 [PubMed] [Google Scholar]
- 2.Kay A.B. Allergy and allergic diseases. First of two parts. N. Engl. J. Med. 2001;344:30–37. doi: 10.1056/NEJM200101043440106. [DOI] [PubMed] [Google Scholar]
- 3.Kay A.B. Allergy and allergic diseases. Second of two parts. N. Engl. J. Med. 2001;344:109–113. doi: 10.1056/NEJM200101113440206. [DOI] [PubMed] [Google Scholar]
- 4.Vivier E., Artis D., Colonna M., Diefenbach A., Di Santo J.P., Eberl G., Koyasu S., Locksley R.M., McKenzie A.N.J., Mebius R.E., et al. Innate Lymphoid Cells: 10 Years On. Cell. 2018;174:1054–1066. doi: 10.1016/j.cell.2018.07.017. [DOI] [PubMed] [Google Scholar]
- 5.Jarick K.J., Topczewska P.M., Jakob M.O., Yano H., Arifuzzaman M., Gao X., Boulekou S., Stokic-Trtica V., Leclère P.S., Preußer A., et al. Non-redundant functions of group 2 innate lymphoid cells. Nature. 2022;611:794–800. doi: 10.1038/s41586-022-05395-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eberl G., Colonna M., Di Santo J.P., McKenzie A.N.J. Innate lymphoid cells. Innate lymphoid cells: a new paradigm in immunology. Science. 2015;348 doi: 10.1126/science.aaa6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Artis D., Spits H. The biology of innate lymphoid cells. Nature. 2015;517:293–301. doi: 10.1038/nature14189. [DOI] [PubMed] [Google Scholar]
- 8.Barlow J.L., McKenzie A.N.J. Innate Lymphoid Cells of the Lung. Annu. Rev. Physiol. 2019;81:429–452. doi: 10.1146/annurev-physiol-020518-114630. [DOI] [PubMed] [Google Scholar]
- 9.Cao Y., Li Y., Wang X., Liu S., Zhang Y., Liu G., Ye S., Zheng Y., Zhao J., Zhu X., et al. Dopamine inhibits group 2 innate lymphoid cell-driven allergic lung inflammation by dampening mitochondrial activity. Immunity. 2023;56:320–335.e9. doi: 10.1016/j.immuni.2022.12.017. [DOI] [PubMed] [Google Scholar]
- 10.Kabata H., Moro K., Koyasu S. The group 2 innate lymphoid cell (ILC2) regulatory network and its underlying mechanisms. Immunol. Rev. 2018;286:37–52. doi: 10.1111/imr.12706. [DOI] [PubMed] [Google Scholar]
- 11.Liew F.Y., Girard J.P., Turnquist H.R. Interleukin-33 in health and disease. Nat. Rev. Immunol. 2016;16:676–689. doi: 10.1038/nri.2016.95. [DOI] [PubMed] [Google Scholar]
- 12.Cayrol C., Girard J.P. Interleukin-33 (IL-33): A nuclear cytokine from the IL-1 family. Immunol. Rev. 2018;281:154–168. doi: 10.1111/imr.12619. [DOI] [PubMed] [Google Scholar]
- 13.Kubo M. Innate and adaptive type 2 immunity in lung allergic inflammation. Immunol. Rev. 2017;278:162–172. doi: 10.1111/imr.12557. [DOI] [PubMed] [Google Scholar]
- 14.Molofsky A.B., Savage A.K., Locksley R.M. Interleukin-33 in Tissue Homeostasis, Injury, and Inflammation. Immunity. 2015;42:1005–1019. doi: 10.1016/j.immuni.2015.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Akira S., Uematsu S., Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 16.Wu J., Chen Z.J. Innate immune sensing and signaling of cytosolic nucleic acids. Annu. Rev. Immunol. 2014;32:461–488. doi: 10.1146/annurev-immunol-032713-120156. [DOI] [PubMed] [Google Scholar]
- 17.Brubaker S.W., Bonham K.S., Zanoni I., Kagan J.C. Innate immune pattern recognition: a cell biological perspective. Annu. Rev. Immunol. 2015;33:257–290. doi: 10.1146/annurev-immunol-032414-112240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Man S.M., Karki R., Kanneganti T.D. AIM2 inflammasome in infection, cancer, and autoimmunity: Role in DNA sensing, inflammation, and innate immunity. Eur. J. Immunol. 2016;46:269–280. doi: 10.1002/eji.201545839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roach J.C., Glusman G., Rowen L., Kaur A., Purcell M.K., Smith K.D., Hood L.E., Aderem A. The evolution of vertebrate Toll-like receptors. Proc. Natl. Acad. Sci. USA. 2005;102:9577–9582. doi: 10.1073/pnas.0502272102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li X.D., Chen Z.J. Sequence specific detection of bacterial 23S ribosomal RNA by TLR13. Elife. 2012;1 doi: 10.7554/eLife.00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Locksley R.M. Asthma and allergic inflammation. Cell. 2010;140:777–783. doi: 10.1016/j.cell.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palm N.W., Rosenstein R.K., Medzhitov R. Allergic host defences. Nature. 2012;484:465–472. doi: 10.1038/nature11047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stewart G.A., Thompson P.J. The biochemistry of common aeroallergens. Clin. Exp. Allergy. 1996;26:1020–1044. https://www.ncbi.nlm.nih.gov/pubmed/8889258 [PubMed] [Google Scholar]
- 24.Van Dyken S.J., Mohapatra A., Nussbaum J.C., Molofsky A.B., Thornton E.E., Ziegler S.F., McKenzie A.N.J., Krummel M.F., Liang H.E., Locksley R.M. Chitin activates parallel immune modules that direct distinct inflammatory responses via innate lymphoid type 2 and gammadelta T cells. Immunity. 2014;40:414–424. doi: 10.1016/j.immuni.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Dyken S.J., Liang H.E., Naikawadi R.P., Woodruff P.G., Wolters P.J., Erle D.J., Locksley R.M. Spontaneous Chitin Accumulation in Airways and Age-Related Fibrotic Lung Disease. Cell. 2017;169:497–509.e13. doi: 10.1016/j.cell.2017.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van Dyken S.J., Garcia D., Porter P., Huang X., Quinlan P.J., Blanc P.D., Corry D.B., Locksley R.M. Fungal chitin from asthma-associated home environments induces eosinophilic lung infiltration. J. Immunol. 2011;187:2261–2267. doi: 10.4049/jimmunol.1100972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crellin N.K., Trifari S., Kaplan C.D., Satoh-Takayama N., Di Santo J.P., Spits H. Regulation of cytokine secretion in human CD127(+) LTi-like innate lymphoid cells by Toll-like receptor 2. Immunity. 2010;33:752–764. doi: 10.1016/j.immuni.2010.10.012. [DOI] [PubMed] [Google Scholar]
- 28.Hardman C.S., Chen Y.L., Salimi M., Nahler J., Corridoni D., Jagielowicz M., Fonseka C.L., Johnson D., Repapi E., Cousins D.J., et al. IL-6 effector function of group 2 innate lymphoid cells (ILC2) is NOD2 dependent. Sci. Immunol. 2021;6 doi: 10.1126/sciimmunol.abe5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hardman C.S., Chen Y.L., Salimi M., Jarrett R., Johnson D., Järvinen V.J., Owens R.J., Repapi E., Cousins D.J., Barlow J.L., et al. CD1a presentation of endogenous antigens by group 2 innate lymphoid cells. Sci. Immunol. 2017;2 doi: 10.1126/sciimmunol.aan5918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ishii T., Muroi M., Horiguchi K., Tanamoto K.I., Nagase T., Yamashita N. Activation through toll-like receptor 2 on group 2 innate lymphoid cells can induce asthmatic characteristics. Clin. Exp. Allergy. 2019;49:1624–1632. doi: 10.1111/cea.13490. [DOI] [PubMed] [Google Scholar]
- 31.Thio C.L.P., Lai A.C.Y., Chi P.Y., Webster G., Chang Y.J. Toll-like receptor 9-dependent interferon production prevents group 2 innate lymphoid cell-driven airway hyperreactivity. J. Allergy Clin. Immunol. 2019;144:682–697.e9. doi: 10.1016/j.jaci.2019.03.008. [DOI] [PubMed] [Google Scholar]
- 32.Maazi H., Banie H., Aleman Muench G.R., Patel N., Wang B., Sankaranarayanan I., Bhargava V., Sato T., Lewis G., Cesaroni M., et al. Activated plasmacytoid dendritic cells regulate type 2 innate lymphoid cell-mediated airway hyperreactivity. J. Allergy Clin. Immunol. 2018;141:893–905.e6. doi: 10.1016/j.jaci.2017.04.043. [DOI] [PubMed] [Google Scholar]
- 33.Sabatel C., Radermecker C., Fievez L., Paulissen G., Chakarov S., Fernandes C., Olivier S., Toussaint M., Pirottin D., Xiao X., et al. Exposure to Bacterial CpG DNA Protects from Airway Allergic Inflammation by Expanding Regulatory Lung Interstitial Macrophages. Immunity. 2017;46:457–473. doi: 10.1016/j.immuni.2017.02.016. [DOI] [PubMed] [Google Scholar]
- 34.Trompette A., Divanovic S., Visintin A., Blanchard C., Hegde R.S., Madan R., Thorne P.S., Wills-Karp M., Gioannini T.L., Weiss J.P., Karp C.L. Allergenicity resulting from functional mimicry of a Toll-like receptor complex protein. Nature. 2009;457:585–588. doi: 10.1038/nature07548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McAlees J.W., Whitehead G.S., Harley I.T.W., Cappelletti M., Rewerts C.L., Holdcroft A.M., Divanovic S., Wills-Karp M., Finkelman F.D., Karp C.L., Cook D.N. Distinct Tlr4-expressing cell compartments control neutrophilic and eosinophilic airway inflammation. Mucosal Immunol. 2015;8:863–873. doi: 10.1038/mi.2014.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eisenbarth S.C., Piggott D.A., Huleatt J.W., Visintin I., Herrick C.A., Bottomly K. Lipopolysaccharide-enhanced, toll-like receptor 4-dependent T helper cell type 2 responses to inhaled antigen. J. Exp. Med. 2002;196:1645–1651. doi: 10.1084/jem.20021340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deckers J., De Bosscher K., Lambrecht B.N., Hammad H. Interplay between barrier epithelial cells and dendritic cells in allergic sensitization through the lung and the skin. Immunol. Rev. 2017;278:131–144. doi: 10.1111/imr.12542. [DOI] [PubMed] [Google Scholar]
- 38.Hammad H., Lambrecht B.N. Barrier Epithelial Cells and the Control of Type 2 Immunity. Immunity. 2015;43:29–40. doi: 10.1016/j.immuni.2015.07.007. [DOI] [PubMed] [Google Scholar]
- 39.Hammad H., Chieppa M., Perros F., Willart M.A., Germain R.N., Lambrecht B.N. House dust mite allergen induces asthma via Toll-like receptor 4 triggering of airway structural cells. Nat. Med. 2009;15:410–416. doi: 10.1038/nm.1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maggi L., Montaini G., Mazzoni A., Rossettini B., Capone M., Rossi M.C., Santarlasci V., Liotta F., Rossi O., Gallo O., et al. Human circulating group 2 innate lymphoid cells can express CD154 and promote IgE production. J. Allergy Clin. Immunol. 2017;139:964–976.e4. doi: 10.1016/j.jaci.2016.06.032. [DOI] [PubMed] [Google Scholar]
- 41.Stover A.G., Da Silva Correia J., Evans J.T., Cluff C.W., Elliott M.W., Jeffery E.W., Johnson D.A., Lacy M.J., Baldridge J.R., Probst P., et al. Structure-activity relationship of synthetic toll-like receptor 4 agonists. J. Biol. Chem. 2004;279:4440–4449. doi: 10.1074/jbc.M310760200. [DOI] [PubMed] [Google Scholar]
- 42.Coats S.R., Pham T.T.T., Bainbridge B.W., Reife R.A., Darveau R.P. MD-2 mediates the ability of tetra-acylated and penta-acylated lipopolysaccharides to antagonize Escherichia coli lipopolysaccharide at the TLR4 signaling complex. J. Immunol. 2005;175:4490–4498. doi: 10.4049/jimmunol.175.7.4490. [DOI] [PubMed] [Google Scholar]
- 43.Kaisho T., Akira S. Toll-like receptor function and signaling. J. Allergy Clin. Immunol. 2006;117:979–988. doi: 10.1016/j.jaci.2006.02.023. quiz 988. [DOI] [PubMed] [Google Scholar]
- 44.Hurrell B.P., Galle-Treger L., Jahani P.S., Howard E., Helou D.G., Banie H., Soroosh P., Akbari O. TNFR2 Signaling Enhances ILC2 Survival, Function, and Induction of Airway Hyperreactivity. Cell Rep. 2019;29:4509–4524.e5. doi: 10.1016/j.celrep.2019.11.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ingley E., Cutler R.L., Fung M.C., Sanderson C.J., Young I.G. Production and purification of recombinant human interleukin-5 from yeast and baculovirus expression systems. Eur. J. Biochem. 1991;196:623–629. doi: 10.1111/j.1432-1033.1991.tb15858.x. [DOI] [PubMed] [Google Scholar]
- 46.Tavernier J., Cornelis S., Devos R., Guisez Y., Plaetinck G., Van der Heyden J. Structure/function analysis of human interleukin 5 and its receptor. Agents Actions Suppl. 1995;46:23–34. doi: 10.1007/978-3-0348-7276-8_3. [DOI] [PubMed] [Google Scholar]
- 47.Hurrell B.P., Shafiei Jahani P., Akbari O. Social Networking of Group Two Innate Lymphoid Cells in Allergy and Asthma. Front. Immunol. 2018;9:2694. doi: 10.3389/fimmu.2018.02694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zanoni I., Ostuni R., Marek L.R., Barresi S., Barbalat R., Barton G.M., Granucci F., Kagan J.C. CD14 controls the LPS-induced endocytosis of Toll-like receptor 4. Cell. 2011;147:868–880. doi: 10.1016/j.cell.2011.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beutler B., Rietschel E.T. Innate immune sensing and its roots: the story of endotoxin. Nat. Rev. Immunol. 2003;3:169–176. doi: 10.1038/nri1004. [DOI] [PubMed] [Google Scholar]
- 50.Mestas J., Hughes C.C.W. Of mice and not men: differences between mouse and human immunology. J. Immunol. 2004;172:2731–2738. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- 51.Vaure C., Liu Y. A comparative review of toll-like receptor 4 expression and functionality in different animal species. Front. Immunol. 2014;5:316. doi: 10.3389/fimmu.2014.00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vasl J., Oblak A., Gioannini T.L., Weiss J.P., Jerala R. Novel roles of lysines 122, 125, and 58 in functional differences between human and murine MD-2. J. Immunol. 2009;183:5138–5145. doi: 10.4049/jimmunol.0901544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Janeway C.A., Jr. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harbor Symp. Quant. Biol. 1989;54:1–13. doi: 10.1101/sqb.1989.054.01.003. http://www.ncbi.nlm.nih.gov/pubmed/2700931 [DOI] [PubMed] [Google Scholar]
- 54.Idzko M., Hammad H., van Nimwegen M., Kool M., Willart M.A.M., Muskens F., Hoogsteden H.C., Luttmann W., Ferrari D., Di Virgilio F., et al. Extracellular ATP triggers and maintains asthmatic airway inflammation by activating dendritic cells. Nat. Med. 2007;13:913–919. doi: 10.1038/nm1617. [DOI] [PubMed] [Google Scholar]
- 55.Kool M., Willart M.A.M., van Nimwegen M., Bergen I., Pouliot P., Virchow J.C., Rogers N., Osorio F., Reis e Sousa C., Hammad H., Lambrecht B.N. An unexpected role for uric acid as an inducer of T helper 2 cell immunity to inhaled antigens and inflammatory mediator of allergic asthma. Immunity. 2011;34:527–540. doi: 10.1016/j.immuni.2011.03.015. [DOI] [PubMed] [Google Scholar]
- 56.Schuijs M.J., Willart M.A., Vergote K., Gras D., Deswarte K., Ege M.J., Madeira F.B., Beyaert R., van Loo G., Bracher F., et al. Farm dust and endotoxin protect against allergy through A20 induction in lung epithelial cells. Science. 2015;349:1106–1110. doi: 10.1126/science.aac6623. [DOI] [PubMed] [Google Scholar]
- 57.Dang E.V., Lei S., Radkov A., Volk R.F., Zaro B.W., Madhani H.D. Secreted fungal virulence effector triggers allergic inflammation via TLR4. Nature. 2022;608:161–167. doi: 10.1038/s41586-022-05005-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Agaronyan K., Sharma L., Vaidyanathan B., Glenn K., Yu S., Annicelli C., Wiggen T.D., Penningroth M.R., Hunter R.C., Dela Cruz C.S., Medzhitov R. Tissue remodeling by an opportunistic pathogen triggers allergic inflammation. Immunity. 2022;55:895–911.e10. doi: 10.1016/j.immuni.2022.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Miao P., Jiang Y., Jian Y., Shi J., Liu Y., Piewngam P., Zheng Y., Cheung G.Y.C., Liu Q., Otto M., Li M. Exacerbation of allergic rhinitis by the commensal bacterium Streptococcus salivarius. Nat. Microbiol. 2023;8:218–230. doi: 10.1038/s41564-022-01301-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tang A.H., Brunn G.J., Cascalho M., Platt J.L. Pivotal advance: endogenous pathway to SIRS, sepsis, and related conditions. J. Leukoc. Biol. 2007;82:282–285. doi: 10.1189/jlb.1206752. [DOI] [PubMed] [Google Scholar]
- 61.Jiang D., Liang J., Fan J., Yu S., Chen S., Luo Y., Prestwich G.D., Mascarenhas M.M., Garg H.G., Quinn D.A., et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat. Med. 2005;11:1173–1179. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- 62.Imai Y., Kuba K., Neely G.G., Yaghubian-Malhami R., Perkmann T., van Loo G., Ermolaeva M., Veldhuizen R., Leung Y.H.C., Wang H., et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell. 2008;133:235–249. doi: 10.1016/j.cell.2008.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guillot L., Balloy V., McCormack F.X., Golenbock D.T., Chignard M., Si-Tahar M. Cutting edge: the immunostimulatory activity of the lung surfactant protein-A involves Toll-like receptor 4. J. Immunol. 2002;168:5989–5992. doi: 10.4049/jimmunol.168.12.5989. [DOI] [PubMed] [Google Scholar]
- 64.Vogl T., Pröpper C., Hartmann M., Strey A., Strupat K., van den Bos C., Sorg C., Roth J. S100A12 is expressed exclusively by granulocytes and acts independently from MRP8 and MRP14. J. Biol. Chem. 1999;274:25291–25296. doi: 10.1074/jbc.274.36.25291. [DOI] [PubMed] [Google Scholar]
- 65.Chase M.A., Wheeler D.S., Lierl K.M., Hughes V.S., Wong H.R., Page K. Hsp72 induces inflammation and regulates cytokine production in airway epithelium through a TLR4- and NF-kappaB-dependent mechanism. J. Immunol. 2007;179:6318–6324. doi: 10.4049/jimmunol.179.9.6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wheeler D.S., Chase M.A., Senft A.P., Poynter S.E., Wong H.R., Page K. Extracellular Hsp72, an endogenous DAMP, is released by virally infected airway epithelial cells and activates neutrophils via Toll-like receptor (TLR)-4. Respir. Res. 2009;10:31. doi: 10.1186/1465-9921-10-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Millien V.O., Lu W., Shaw J., Yuan X., Mak G., Roberts L., Song L.Z., Knight J.M., Creighton C.J., Luong A., et al. Cleavage of fibrinogen by proteinases elicits allergic responses through Toll-like receptor 4. Science. 2013;341:792–796. doi: 10.1126/science.1240342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Klose C.S.N., Artis D. Innate lymphoid cells control signaling circuits to regulate tissue-specific immunity. Cell Res. 2020;30:475–491. doi: 10.1038/s41422-020-0323-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gurram R.K., Zhu J. Orchestration between ILC2s and Th2 cells in shaping type 2 immune responses. Cell. Mol. Immunol. 2019;16:225–235. doi: 10.1038/s41423-019-0210-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Grievink H.W., Luisman T., Kluft C., Moerland M., Malone K.E. Comparison of Three Isolation Techniques for Human Peripheral Blood Mononuclear Cells: Cell Recovery and Viability, Population Composition, and Cell Functionality. Biopreserv. Biobank. 2016;14:410–415. doi: 10.1089/bio.2015.0104. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Raw data in this study are available from Sequence Read Archive (SRA) of NCBI and accession number is listed in the key resources table. All data present in this study will be shared by the lead contact upon request.
•This paper does not report original code.
•Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.