

Abstract
Background
Interleukin-37 (IL-37) has anti-inflammatory properties in innate and adaptive immunity. Patients with multiple sclerosis (MS), an autoimmune inflammatory demyelinating disease of the central nervous system (CNS), have increased serum levels of IL-37. However, it is unknown whether IL-37 has an inhibitory effect on ongoing autoimmune neuroinflammation, thus offering a potential MS therapy.
Aim
Here, we examined the effect of IL-37 in an experimental autoimmune encephalomyelitis (EAE) model after disease onset to determine if it was protective.
Findings
IL-37-treated mice developed a less severe disease than control mice, with reduced demyelination as determined by increased expression of myelin basic protein. IL-37 suppressed inflammation by decreasing infiltration of CD4 + T cells into the CNS and increasing the frequency of regulatory T cells, while IL-10 expression by CD4 + T cells decreased over time in the CNS.
Conclusion
Our findings confirm the immunomodulatory role of IL-37 in CNS inflammation during ongoing disease, thus indicating the potential of IL-37 as an inhibitory reagent for MS therapy.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12974-024-03295-1.
Keywords: IL-37, Multiple sclerosis, IL-10, Inflammation, Experimental autoimmune encephalomyelitis (EAE)
Introduction
IL-37 is a newly described cytokine of the IL-1 family with anti-inflammatory properties thought to suppress ongoing innate and adoptive immune responses [1–3]. Although mice do not have a homolog for IL-37, the human protein is bioactive in mice [4, 5]. Indeed, experiments with a transgenic mouse line expressing the human IL-37 protein under the control of the Cytomegalovirus (CMV) promoter showed a dual signaling pathway by which IL-37 exerts its suppressive functions [1, 3]. IL-37 can intracellularly bind to SMAD3 and act as a nuclear transcription factor, or it can be processed and released to the extracellular space and act as a cytokine [1]. Extracellular IL-37 signals through IL-18Ra and SIGIRR (IL-1R8), which are expressed by myeloid and lymphoid cells [6, 7].
In EAE or MS, inflammatory leukocytes infiltrate the CNS and inflict damage upon the myelin sheaths, neurons, and oligodendrocytes by producing inflammatory mediators such as IFN-γ, IL-17, GM-CSF, Granzyme B, and TNF-α [8–11], . Thus, therapies targeting inflammatory mediators or stimulating production of anti-inflammatory cytokines are necessary to suppress neuroinflammation. Unfortunately, therapies that reduce local inflammation in the CNS to stop disease progression and restore tissue homeostasis are limited in efficacy. Recently, Sánchez-Fernández et al. revealed that IL-37 suppresses inflammation and protects myelin loss in experimental autoimmune encephalomyelitis (EAE), a mouse model of multiple sclerosis (MS), when the treatment was started before disease onset [12]. They demonstrated that the beneficial effects of IL-37 in EAE are dependent on its extracellular receptor complex IL-1R5/IL-1R8.
Infiltration of immune cells across the blood–brain barrier (BBB) leads to inflammation and demyelination in MS and EAE models. Given the anti-inflammatory effect of IL-37, we explored whether IL-37 can suppress inflammation caused by infiltrated immune cells. We showed that IL-37-treated mice developed a much less severe disease and higher expression of myelin basic protein (MBP) than control mice when IL-37 was injected at disease onset, with increasing the frequency of Treg cells and decreasing CD4 + T-cell-derived IL-10 in the CNS.
Materials and methods
Mice
Adult female C57BL/6 mice (8–10 weeks) purchased from Jackson Laboratory (Bar Harbor, ME, USA) were kept in clean cages in a controlled environment with 12 h/12 h of light/dark cycles throughout the experimental procedures.
Ethics declaration
Experimental protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of Thomas Jefferson University (Protocol # 21-02-340).
EAE induction
Mice were injected subcutaneously with 200 µL of an emulsion containing 200 µg of MOG35 − 55 peptide (MEVGWYRSPFSRVVHLYRNGK; Genscript, NJ, USA) and 200 µL of Complete Freund’s adjuvant supplemented with 10 mg/mL of heat-killed Mycobacterium tuberculosis H37Ra. At the time of immunization and 48 h later, mice were also injected intraperitoneally with 200 ng of pertussis toxin. Depending on the severity of the symptom, mice were scored using the following scale: 0 - No clinical symptoms; 0.5 - Partial paralysis of the tail or waddling gait; 1.0 - Full paralysis of the tail; 1.5 - Full paralysis of the tail and waddling gait; 2.0 - Partial paralysis in one leg; 2.5 - Partial paralysis of both legs or one leg paralyzed; 3.0 - Both legs paralyzed; 3.5 - Ascending paralysis; 4.0 - Paralysis above the hips; 4.5 - mouse being unable to right itself for 30 s; and 5.0 - Death. EAE onset was the first day mice presented the first symptom of disease (EAE score 0.5, or 1) post immunization (p.i.). Peak of disease was considered on the day when EAE scores were the highest (approx. day 19 p.i.). The day mice exhibited decreased EAE score (day 26 p.i.) was considered as chronic phase of the disease.
Treatments
Recombinant human IL-37b (rhIL-37b) (R&D Systems) was reconstituted in phosphate-buffered saline (PBS) at 200 µg/mL. Mice were treated intraperitoneally with 1 µg of rhIL-37b in 200 µl of PBS daily at disease onset until the end the study and control mice were treated with 200 µl of PBS.
Preparation of infiltrating MNCs from the CNS and spleen
Mice were euthanized and perfused with ice-cold PBS for removal of brain and spinal cord, which were mechanically dissociated and enzymatically digested with 700 µg/mL Liberase TL (Sigma-Aldrich, USA) at 37 ºC for 30 min. The digested CNS preparation was passed through a 70 μm cell strainer (Falcon, Tewksbury, MA) and washed with ice cold PBS. Cells were then enriched on a 30% Percoll gradient and re-suspended in IMDM/10% FBS. Regarding splenocytes, they were also passed through a 70 μm cell strainer and incubated with red-blood-cell lysis buffer for approximately 2 min. Harvested cells were then washed with ice-cold PBS prior to flow cytometry.
Flow cytometry
For cell surface markers, CNS and spleen cells were incubated with the following antibodies conjugated with their respective fluorochrome: CD49b-APC, CD45-APC/Cy7, CD4-Alexa fluor 700, CD11c-DAPI, CD45-Alexa fluor 700, CD4-BV711, CD11b-BV786, and CD11c-PE/Texas Red (Biolegend and BD Biosciences) in two different panels at the recommended dilution or FMO control Abs for 30 min on ice. For intracellular staining, cells were stimulated for 4 h with 50 ng/ml PMA (phorbol 12-myristate 13-acetate) and 500 ng/ml ionomycin (Sigma-Aldrich) in the presence of GolgiPlug (1:1000; BD Pharmigen). Cells were permeabilized with Fix and Perm Mediums (Thermo Fisher) and incubated with the following antibodies: IFN-γ-BV711, IL-17-BV786, TNF-α-PE, IL-10-PE/Cy7, GM-CSF-PE/Texas Red, IL-4-DAPI, Foxp3-PE, IL-10-PE/Cy7 from Biolegend, and BD Biosciences in two different panels. Dead Cell Stain Kit (ThermoFisher, L34959) was used to exclude dead cells. Flow cytometry was done using BD FACSAria Fusion flow cytometer (BD Biosciences), and data were analyzed using FlowJo (version 10).
Immunohistochemistry (IHC) and fluorescence image analysis
Mice were transcardially perfused with PBS; lumbar sections were collected and placed in 4% Paraformaldehyde (PFA) overnight. The tissues were then transferred to 70% ethanol embedded in paraffin and cut into slices of 4 microns thick. To perform IHC, paraffin was removed by passing through a series of xylene and alcohol, permeablized with Triton X-100 and blocked with 2% bovine serum albumin (BSA). Primary antibody for MBP (1:500 dilution; ThermoFisher, Cat# MA5-42370), was added, incubated overnight and next day incubated with secondary antibody (Goat Anti-Rabbit IgG H&L [FITC]; abcam, Cat# ab97050). DAPI was used to stain the nucleus, and the sections were mounted and imaged using a microscope fluorescent microscopy (Nikon Eclipse E600; Nikon, Melville, NY). MBP expression was expressed as mean fluorescence intensity (MFI) in whole sections in both groups.
Statistical analyses
Two-tailed Student’s t-test was used for the comparison between two groups. Comparison of three or more groups was performed with one-way ANOVA. Graphs and statistical analyses were made in GraphPad Prism version 10 software. Data are shown as mean ± standard error of the mean (SEM). Values of p < 0.05 were considered significant.
Results
IL-37 suppressed ongoing EAE following onset administration
The immunomodulatory effect of IL-37 in EAE is still poorly understood. To investigate whether IL-37 suppresses EAE at priming phase or during disease, we immunized C57BL/6 mice with MOG35-55 peptide and treated them with rhIL-37b. We first examined the effect of IL-37 administered via intraperitoneal (i.p.) injection during the priming phase of EAE at days 1, 4, and 7 p.i. (1 µg/dose). IL-37-treated mice initially developed milder disease than control mice, but the difference diminished after disease peak (Fig. 1A). Indeed, at the end of the follow-up, IL-37-treated mice showed slight reduction (~ 0.4 point [16%]) in EAE clinical score. We next explored the effect of IL-37 treatment started after disease onset, when clinical at 1, by administering IL-37 daily. The treatment reduced EAE severity at the peak and chronic phase (Fig. 1B-C). At the end of the follow-up, IL-37-treated mice showed ~ 1.0 point (27.0%) and ~ 1.62 point (46.5%) decreases in EAE clinical score at peak and chronic phases of disease, respectively. Our data demonstrate that significant differences in clinical scores were observed 7–10 days after rhIL-37b administration; therefore, the difference in mean clinical scores was only significant in the chronic phase (Fig. 1C), highlighting that rhIL-37b needs time to efficaciously suppress EAE. These results show that IL-37 administration ameliorated severity of ongoing EAE when treatment was started at disease onset. Consistent with decreased clinical scores, IL-37-treated EAE mice had a higher expression of MBP compared with PBS-treated mice (Fig. 2), indicating a reduced demyelination after IL-37 treatment. The histopathological analysis showed lower cell infiltration in the spinal cord of IL-37-treated EAE mice than of PBS-treated mice (Figure S1).
Fig. 1.

IL-37 treatment ameliorated EAE at both priming and ongoing phases. (A) C57BL/6 mice were immunized to induce EAE and treated i.p. with IL-37 (1 µg/ dose; n = 5) and PBS (n = 4) at days 1, 4, and 7 p.i. The clinical scores were evaluated daily for 21 days p.i. Mean clinical scores for IL-37-treated and PBS-treated groups were calculated as the sum of daily scores for each mouse divided by the number of days per group. (B-C) EAE mice were treated i.p. with 1 µg of rhIL-37b/ day when they showed the first symptoms of EAE. PBS was administered i.p. to the control group in the same way. Clinical scores were followed for 20 days p.i. (peak; n = 7 per group) (B) and 26 days p.i. (chronic; n = 4 per group) (C), and mean clinical scores were compared between IL-37-treated mice and control. Clinical scores were analyzed by multiple unpaired t-tests. Mean clinical scores were analyzed by Student’s t-test. Data shown as mean ± SEM. Values of p < 0.05 (*), p < 0.01 (**) and p < 0.001 (***) were considered significant. Ns: not significant
Fig. 2.

IL-37 enhanced MBP expression. (A) The lumbar section of the spinal cord was harvested from EAE mice treated with PBS (left) or rhIL-37b (middle) at the peak of disease (n = 4 mice in each group). Naïve mice (right) are illustrated as a control to show healthy spinal cord. (B) Mean fluorescent intensity of MBP expression in both groups was measured using FIJI software. Unpaired t-test was performed to analyze these two groups. Data shown as mean ± SEM. Values of p < 0.05 (*), p < 0.01 (**) and p < 0.001 (***) were considered significant. Ns: not significant
IL-37 mostly decreased infiltration of CD4 + T cells into CNS but increased frequency of Treg cells
We next explored the impact of IL-37 on infiltration of immune cells in the CNS. We enumerated immune cells including CD4 + T cells, DC, B cells, NK cells, monocyte/macrophages (Mo/Macrophages; Mφ; CD11b + CD45hi) and microglia (CD11b + CD45lo) in the CNS, and found decreased numbers of CD4 + T cells and microglia following IL-37 administration (Fig. 3A). We then analyzed CD4 + T cell subsets by enumerating T helper (TH)-1 cells (CD4 + IFN-γ + IL-17-), TH-2 cells (CD4 + IL-4+), TH-17 cells (CD4 + IL-17 + IFN-γ-), TH-17 + 1 cells (CD4 + IFN-γ + IL-17+), and Treg cells (CD4 + Foxp3+) in the CNS at disease peak and chronic phase. An increase in the frequency of Treg cells in IL-37-treated mice was observed at disease peak (Fig. 3B), but no difference in the chronic phase (Fig. 3C and D). When we statistically compared CD4 + TH-cells between peak and chronic phases, we found a significant reduction in the frequency of TH-1 cells at chronic compared to the peak of disease, and this reduction was much stronger when IL-37 was administrated (Fig. 4A). We did not find any significant difference in the frequency of other CD4 + TH cells (Fig. 4A), even in Treg cells (Fig. 4B) in the presence or absence of IL-37 administration during inflammation.
Fig. 3.

IL-37 reduced infiltration of immune cells into the CNS. Numbers of infiltrated immune cells in the CNS of IL-37- and PBS-treated mice at the peak (A) and chronic phase (C) of EAE. Proportions of TH subsets among CD4 + T cells isolated from the CNS of IL-37- and PBS-treated mice at peak (B) and chronic phase (D) of EAE. The number of immune cells (E) and CD4 + T cell subsets (D) in the spleen of IL-37- and PBS-treated mice at the peak of disease. Unpaired t-test (n = 7 per group in peak; n = 4 per group in chronic) was used to analyze the two groups. Data shown as mean ± SEM. Values of p < 0.05 (*), p < 0.01 (**) and p < 0.001 (***) were considered significant. Ns: not significant
Fig. 4.

IL-37 reduces proportion of TH-1 cells at chronic phase during CNS inflammation. (A) The frequencies of CD4 + T cells at EAE peak compared to chronic phase in IL-37-treated (blue) and PBS-treated mice (black). (B) The frequencies Treg cells at peak compared to chronic phase of EAE disease in IL-37-treated (blue) and PBS-treated mice (black). Analyses between these two groups were carried out by unpaired t-tests (n = 7 per group in peak; n = 4 per group in chronic). Data shown as mean ± SEM. Values of p < 0.05 (*), p < 0.01 (**) and p < 0.001 (***) were considered significant. Ns: not significant
We also investigated the effect of IL-37 treatment on splenic cells. We found greater numbers of all immune cell types analyzed in the spleens of IL-37-treated mice compared to control mice, suggesting that fewer immune cells left the spleen to infiltrate the CNS following IL-37 administration (Fig. 3E). Among CD4 + T cell subsets, IL-37 significantly increased the frequency of Treg cells in the spleen, similar to the CNS (Fig. 3F). These results indicate that IL-37 treatment decreased infiltration of CD4 + T cells into the CNS and increased the frequency of Treg cells in both the CNS and the spleen at the peak of EAE disease.
IL-37 treatment reduced IL-10 production by CD4 + T cells
We explored whether IL-37 affects cytokine production by CD4 + T cells in EAE mice. The levels of all inflammatory cytokines produced by gated CD4 + T cells, including IL-17, GM-CSF, IFN-γ, and TNF-α were decreased after IL-37 administration in the CNS, although this reduction was only significant in the TNF-α level (Fig. 5). A slight increase in IL-4 level in IL-37-treated mice was observed, although that was not significant (Fig. 5).
Fig. 5.
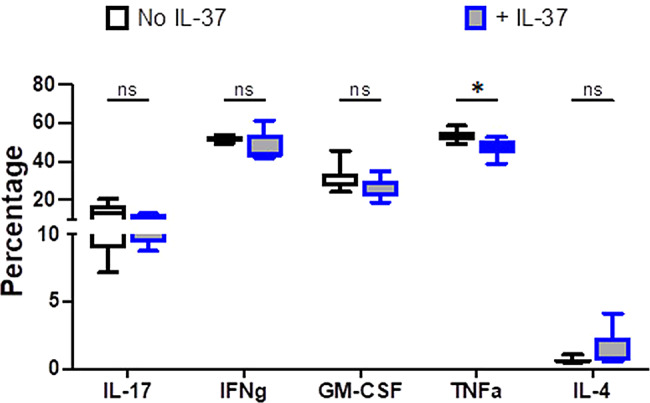
Production of inflammatory and anti-inflammatory cytokines in total CD4 + T cells of the CNS following IL-37 treatment. The frequencies of cytokine-producing CD4 + T cells in the CNS of IL-37- and PBS-treated mice at the peak of disease. Analyses between these two groups were carried out by unpaired t-tests (n = 7 per group). Data shown as mean ± SEM. Values of p < 0.05 (*), p < 0.01 (**) and p < 0.001 (***) were considered significant. Ns: not significant
T cells are the main source of IL-10 in vivo, regulating tissue homeostasis and immune reactions. Recently, it has been reported that CD4 + T cell-derived IL-10 promotes CNS inflammation in EAE mice by sustaining effector T cell survival [13]. We determined if IL-37 has an impact on IL-10 production by examining CD4 + T cells in the CNS. At the peak of EAE, IL-10 expression was comparable in all CD4 + T cell subsets between IL-37-treated mice and control (Fig. 6A and C); but IL-10 expression was significantly decreased in the chronic phase in both Treg and TH-17 cells following IL-37 treatment (Fig. 6B and C). When we statistically compared IL-10 expression by TH cells between peak and chronic phases, we observed significantly lower IL-10 expression in TH-17 cells at chronic compared to peak after IL-37 treatment, but IL-10 expression was comparable in other CD4 + TH cells (Fig. 7A). Significant augmentation of IL-10 expression was observed in Treg cells at the chronic phase compared to the peak of EAE without IL-37 treatment, while a similar level of IL-10 production was observed in both peak and chronic phases of IL-37-treated EAE mice. Interestingly, the percentage of IL-10 producing Tregs was significantly lower at the chronic phase in IL-37-treated mice than in control mice (Fig. 7B). Reduced IL-10 expression in TH-17 cells and suppression of increased IL-10 expression in Treg following IL-37 treatment highlight the inflammatory role of IL-10 at the chronic phase.
Fig. 6.

EAE suppression by IL-37 is mediated by reducing IL-10 producing CD4 + T cells of the CNS. The frequencies of IL-10 expression in different CD4 + T cell subsets from the CNS of IL-37-treated and PBS-treated EAE mice at the peak of disease (A) and chronic phase (B). Analyses between these two groups were carried out by unpaired t-tests (n = 7 per group in peak; n = 4 per group in chronic phase). (C) Representative flow cytometry dot-plots showing IL-10 expression in Treg and TH-17 cells from the CNS of PBS-treated and IL-37-treated EAE mice at disease peak and chronic phase. Data shown as mean ± SEM. Values of p < 0.05 (*), p < 0.01 (**) and p < 0.001 (***) were considered significant. Ns: not significant
Fig. 7.

IL-37 reduced frequency of IL-10 producing Treg cells during CNS inflammation. (A) The frequencies of IL-10 producing CD4 + T cells at EAE peak compared to chronic phase in IL-37-treated (blue) and PBS-treated mice (black). (B) The frequencies IL-10 producing Treg cells at peak compared to chronic of EAE disease in IL-37-treated (blue) and PBS-treated mice (black). Analyses between these two groups were carried out by unpaired t-tests (n = 7 per group in peak; n = 4 per group in chronic). Data shown as mean ± SEM. Values of p < 0.05 (*), p < 0.01 (**) and p < 0.001 (***) were considered significant. Ns: not significant
Discussion
In the present study, for the first time, we show that IL-37 treatment after disease onset effectively suppressed EAE, with increased number of Tregs in the CNS and reduced IL-10 production by CD4 + T cells. Transgenic expression of human IL-37, or treatment with IL-37 before disease onset, were reported to decrease neurological deficits and inflammation in the spinal cord of EAE mice, and beneficial effects of IL-37 in EAE relied upon IL-37 receptor (IL-1R5/IL-1R8) [12]. However, whether IL-37 inhibits ongoing disease and the mechanism whereby IL-37 ameliorates CNS autoimmunity has not been elucidated until now.
Few studies have assessed the role of IL-37 in MS. Some studies reported increased IL-37 levels in the serum of MS patients compared to healthy individuals [14, 15], although production of IL-37 from CD4 + T cells in MS patients was lower than in healthy controls [16]. MS patients with higher IL-37 levels had lower severity scores than patients with less IL-37 [16]. This suggests that IL-37 likely suppresses inflammation in MS, similar to its effect in EAE. However, investigation regarding the functional mechanism of IL-37 in EAE models remains to be clarified. IL-37 levels were found to increase at the peak and chronic phase of EAE [12], and significant reduction of clinical scores was observed over time. In our study, the greatest ameliorating effect of IL-37 was seen in the chronic phase. The reduced clinical scores in our IL-37-treated mice correlated with decreased infiltration of CD4 + T cells into the CNS, similar to the previous study [12]. Given that neuroinflammation in EAE/MS is driven by inflammatory leukocytes, including CD4 + T cells, infiltrating the CNS parenchyma [17, 18], decreased infiltrated CD4 + T cells into the CNS by IL-37 can be correlated with the suppression of CNS inflammation in EAE/MS. Significantly reduced levels of pro-inflammatory cytokines were observed at the early stage of EAE, e.g., day 3 after disease onset, in mice with constitutive transgenic expression of hIL-37, indicating its regulatory effect in the priming phase of EAE development [12]. In the present study, we confirmed the therapeutic effect of IL-37 treatment on T cell responses at the peak and chronic phase of EAE, which is more relevant to its clinical application. Further, while an increase in proportions of Treg cells was reported in the spinal cord of hIL-37tg transgenic mice [12]; we also observed this increase in the CNS following IL-37 administration, especially at the chronic phase. Reduced infiltration of inflammatory CD4 + T cells and increase in frequency of Treg cells in the CNS in the chronic phase of EAE highlight that IL-37 suppresses inflammation by switching the phenotype of CD4 + T cells in the CNS to regulatory phenotypes over time in EAE.
T cells are believed to be the main source of IL-10 in vivo, facilitating its delivery to inflammatory sites to regulate tissue homeostasis and immune reactions [19]. It has been recently reported that CD4 + T cell-derived IL-10 promoted CNS inflammation in MOG35 − 55-induced EAE, as mice with T cell-specific IL-10 knockout had reduced disease severity. However, IL-10 produced by B cell and myeloid cells had protective roles in EAE, highlighting that T cells are the key source of pro-inflammatory IL-10 in EAE [13]. The study defined that CD4 + T cell-derived IL-10 promotes effector T cell survival in the CNS, highlighting that CD4 + T cells are important as both a prominent source and target of pro-inflammatory IL-10 in vivo [13]. Given the immunomodulatory role of IL-37 in EAE, we explored whether IL-37 ameliorated EAE by reducing CD4 + T cell-derived IL-10 in an EAE model. Interestingly, we found a significant decrease in IL-10 production by CD4 + T cells, especially TH-17 and Treg cells, of IL-37-treated EAE mice. Suppression of IL-10 expression by IL-37 administration during inflammation indicates that IL-10 is probably an inflammatory cytokine in the CNS, consistent with the previous report [13]. Support for this hypothesis comes from a study by Liu et al., showing that IL-10 production in the CNS increased EAE severity by restraining the autoreactive T cells essential for immunopathogenesis [20]. Cannella et al. also showed that IL-10 had a worsening or no effect on EAE [21]. Sánchez-Fernández et al. found a slightly reduced IL-10 protein in the spinal cord of hIL-37tg mice relative to wild type mice at the peak of EAE; however, they did not characterized IL-10-producing CD4 + T cells in the CNS [12]. The deleterious effect of IL-10 in other autoimmune diseases, such as in a mouse model of systemic lupus erythematous, has also been reported, as IL-10 neutralization delayed disease onset [22]. Whether IL-10 has pro or anti-inflammatory role probably depends on the specific context, including the type of cells involved, the presence of other stimuli or factors, and the overall inflammatory environment. Therefore, the therapeutic approach using IL-10 as an anti-inflammatory agent has failed due to an insufficient understanding of complex IL-10 effects in immune system [23, 24]. The clear point derived from our findings is that IL-10 producing CD4 + T cells are probably inflammatory in the CNS, as reducing their presence ameliorates EAE scores.
Whether the suppressive feature of Treg is related to IL-10 is still controversial. Owing to the inhibitory feature of IL-10 on IL-2 and IFN-γ production by TH1 cells, IL-10 was initially considered an “anti-inflammatory cytokine.” However, it is now defined as an immunoregulatory cytokine with pleiotropic functions, exerting multiple and sometimes even opposite effects on immune cells [25], as IL-10 can potentiate killing capacity of effector CD8 + T cells [26]. The different cellular source of IL-10 and the location of the cells producing and responding to IL-10 and even timing of its secretion might determine suppressive vs. inflammatory activity of IL-10 [25], as opposite outcomes were found when EAE mice were treated with IL-10 short- or long-term [21, 27, 28].
In addition to IL-10 production, Treg cells exert their regulatory effects on immune responses by multiple other mechanisms, including secretion of suppressor cytokines (e.g., TGF-β1, IL-35), IL-2 consumption, cytolysis of targeted cells, and/or decreased costimulation and antigen presentation [29, 30]. For example, although Treg-derived TGF-β1 may not be essential for Treg suppressive function, this cytokine is crucial for Treg development in the thymus and periphery [31]. TGF-β1 also inhibits differentiation and effector functions of Th1 cells [32] and the MHC class II expression in antigen-presenting cells (APCs), thus inhibiting their functions [33]. Treg-derived IL-35 inhibits the differentiation of TH17 cells from naïve CD4 + T cells [34]. Tregs express high levels of cytotoxic T-lymphocyte-associated antigen (CTLA)-4, a negative regulator that, by binding CD80 and CD86 on APCs, inhibits their costimulatory signaling required for T cell activation [35]. Further, Tregs can stimulate dendritic cells to produce indoleamine 2,3-dioxygenase (IDO), an enzyme whose metabolites suppress T cell responses [36]. IL-37 has been shown to increase the suppressive activity of Treg cells in vitro by inducing their expression of CTLA-4 and Foxp3, as well as elevating production of TGF-β1, but not of IL-10 [37]. Nold et al. reported the similar findings, demonstrating that pro-inflammatory cytokines, including IL-1β, IL-6 and TNF, were increased in human PBMCs treated with siRNA against IL-37 without affecting IL-10 expression [1]. Regarding the effects of IL-37 independent of IL-10 in other autoimmune model, McNamee et al. demonstrated that IL-10 was not required for anti-inflammatory effects of IL-37 in colitis, as IL-10-receptor antibody blockade did not reverse IL-37-mediated protection [38]. These data, together with our findings, demonstrate a dispensable role of IL-10 production in IL-37-induced suppressive activity of Tregs. Further studies are needed to define regulatory mechanisms of IL-37-induced Tregs .
Conclusion
Our findings show the protective effect IL-37 in EAE model after disease onset, as IL-37-treated mice developed a less severe disease than control mice, with significantly reduced demyelination. IL-37 suppressed infiltration of CD4 + T cells into the CNS and increased the frequency of Tregs, while IL-10 expression by CD4 + T cells decreased over time in the CNS. Together, our findings confirm the immunomodulatory role of IL-37 in CNS inflammation during the ongoing stage, thus indicating the potential of IL-37 as a novel inhibitory reagent for MS therapy.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
This work was supported by grants from the National Institutes of Health (NIH, grant # 5R01AI156384). We thank Ms. Pamela Walter of TJU for editorial assistance.
Author contributions
RY performed the experiments, wrote the manuscript and analysed the data. HN, GA, MA, AMA and JA participated in data collection and contributed to flow cytometry experiments. BC and GXZ edited the manuscript. AR contributed to the conception and design of the study, revised the manuscript and supervised the project.
Data availability
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Nold MF, Nold-Petry CA, Zepp JA, Palmer BE, Bufler P, Dinarello CA. IL-37 is a fundamental inhibitor of innate immunity. Nat Immunol. 2010;11(11):1014–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tete S, Tripodi D, Rosati M, Conti F, Maccauro G, Saggini A, et al. IL-37 (IL-1F7) the newest anti-inflammatory cytokine which suppresses immune responses and inflammation. Int J Immunopathol Pharmacol. 2012;25(1):31–8. [DOI] [PubMed] [Google Scholar]
- 3.Li S, Amo-Aparicio J, Neff CP, Tengesdal IW, Azam T, Palmer BE, et al. Role for nuclear interleukin-37 in the suppression of innate immunity. Proc Natl Acad Sci U S A. 2019;116(10):4456–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cavalli G, Dinarello CA. Suppression of inflammation and acquired immunity by IL-37. Immunol Rev. 2018;281(1):179–90. [DOI] [PubMed] [Google Scholar]
- 5.Wang L, Quan Y, Yue Y, Heng X, Che F. Interleukin-37: a crucial cytokine with multiple roles in disease and potentially clinical therapy. Oncol Lett. 2018;15(4):4711–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nold-Petry CA, Lo CY, Rudloff I, Elgass KD, Li S, Gantier MP, et al. IL-37 requires the receptors IL-18Ralpha and IL-1R8 (SIGIRR) to carry out its multifaceted anti-inflammatory program upon innate signal transduction. Nat Immunol. 2015;16(4):354–65. [DOI] [PubMed] [Google Scholar]
- 7.Lunding L, Webering S, Vock C, Schroder A, Raedler D, Schaub B, et al. IL-37 requires IL-18Ralpha and SIGIRR/IL-1R8 to diminish allergic airway inflammation in mice. Allergy. 2015;70(4):366–73. [DOI] [PubMed] [Google Scholar]
- 8.El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol. 2011;12(6):568–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu D, Notarbartolo S, Croonenborghs T, Patel B, Cialic R, Yang TH, et al. Transcriptional signature of human pro-inflammatory TH17 cells identifies reduced IL10 gene expression in multiple sclerosis. Nat Commun. 2017;8(1):1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201(2):233–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421(6924):744–8. [DOI] [PubMed] [Google Scholar]
- 12.Sánchez-Fernández A, Zandee S, Amo-Aparicio J, Charabati M, Prat A, Garlanda C, et al. IL-37 exerts therapeutic effects in experimental autoimmune encephalomyelitis through the receptor complex IL-1R5/IL-1R8. Theranostics. 2021;11(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yogev N, Bedke T, Kobayashi Y, Brockmann L, Lukas D, Regen T, et al. CD4(+) T-cell-derived IL-10 promotes CNS inflammation in mice by sustaining effector T cell survival. Cell Rep. 2022;38(13):110565. [DOI] [PubMed] [Google Scholar]
- 14.Kouchaki E, Tamtaji OR, Dadgostar E, Karami M, Nikoueinejad H, Akbari H. Correlation of serum levels of IL-33, IL-37, Soluble Form of Vascular endothelial growth factor receptor 2 (VEGFR2), and circulatory frequency of VEGFR2-expressing cells with multiple sclerosis severity. Iran J Allergy Asthma Immunol. 2017;16(4):329–37. [PubMed] [Google Scholar]
- 15.Farrokhi M, Rezaei A, Amani-Beni A, Etemadifar M, Kouchaki E, Zahedi A. Increased serum level of IL-37 in patients with multiple sclerosis and neuromyelitis optica. Acta Neurol Belg. 2015;115(4):609–14. [DOI] [PubMed] [Google Scholar]
- 16.Cavalli E, Mazzon E, Basile MS, Mammana S, Pennisi M, Fagone P et al. In Silico and in vivo analysis of IL37 in multiple sclerosis reveals its probable homeostatic role on the clinical activity, disability, and treatment with Fingolimod. Molecules 2019;25(1). [DOI] [PMC free article] [PubMed]
- 17.Dobson R, Giovannoni G. Multiple sclerosis - a review. Eur J Neurol. 2019;26(1):27–40. [DOI] [PubMed] [Google Scholar]
- 18.Galli E, Hartmann FJ, Schreiner B, Ingelfinger F, Arvaniti E, Diebold M, et al. GM-CSF and CXCR4 define a T helper cell signature in multiple sclerosis. Nat Med. 2019;25(8):1290–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iyer SS, Cheng G. Role of interleukin 10 transcriptional regulation in inflammation and autoimmune disease. Crit Rev Immunol. 2012;32(1):23–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu X, Alli R, Steeves M, Nguyen P, Vogel P, Geiger TL. The T cell response to IL-10 alters cellular dynamics and paradoxically promotes central nervous system autoimmunity. J Immunol. 2012;189(2):669–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cannella B, Gao YL, Brosnan C, Raine CS. IL-10 fails to abrogate experimental autoimmune encephalomyelitis. J Neurosci Res. 1996;45(6):735–46. [DOI] [PubMed] [Google Scholar]
- 22.Godsell J, Rudloff I, Kandane-Rathnayake R, Hoi A, Nold MF, Morand EF, et al. Clinical associations of IL-10 and IL-37 in systemic lupus erythematosus. Sci Rep. 2016;6:34604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Garra A, Barrat FJ, Castro AG, Vicari A, Hawrylowicz C. Strategies for use of IL-10 or its antagonists in human disease. Immunol Rev. 2008;223:114–31. [DOI] [PubMed] [Google Scholar]
- 24.Saxena A, Khosraviani S, Noel S, Mohan D, Donner T, Hamad AR. Interleukin-10 paradox: a potent immunoregulatory cytokine that has been difficult to harness for immunotherapy. Cytokine. 2015;74(1):27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carlini V, Noonan DM, Abdalalem E, Goletti D, Sansone C, Calabrone L, et al. The multifaceted nature of IL-10: regulation, role in immunological homeostasis and its relevance to cancer, COVID-19 and post-COVID conditions. Front Immunol. 2023;14:1161067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Foulds KE, Rotte MJ, Seder RA. IL-10 is required for optimal CD8 T cell memory following Listeria monocytogenes infection. J Immunol. 2006;177(4):2565–74. [DOI] [PubMed] [Google Scholar]
- 27.Nagelkerken L, Blauw B, Tielemans M. IL-4 abrogates the inhibitory effect of IL-10 on the development of experimental allergic encephalomyelitis in SJL mice. Int Immunol. 1997;9(9):1243–51. [DOI] [PubMed] [Google Scholar]
- 28.Rott O, Fleischer B, Cash E. Interleukin-10 prevents experimental allergic encephalomyelitis in rats. Eur J Immunol. 1994;24(6):1434–40. [DOI] [PubMed] [Google Scholar]
- 29.Shevach EM. Mechanisms of foxp3 + T regulatory cell-mediated suppression. Immunity. 2009;30(5):636–45. [DOI] [PubMed] [Google Scholar]
- 30.Goldmann O, Nwofor OV, Chen Q, Medina E. Mechanisms underlying immunosuppression by regulatory cells. Front Immunol. 2024;15:1328193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moreau JM, Velegraki M, Bolyard C, Rosenblum MD, Li Z. Transforming growth factor-β1 in regulatory T cell biology. Sci Immunol. 2022;7(69):eabi4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol. 2006;24:99–146. [DOI] [PubMed] [Google Scholar]
- 33.Singh B, Krawetz MD, De Lima RM, Mukherjee R, Chaturvedi P, Lee-Chan E, et al. Role of TGF-β in self-peptide regulation of autoimmunity. Arch Immunol Ther Exp (Warsz). 2018;66(1):11–9. [DOI] [PubMed] [Google Scholar]
- 34.Niedbala W, Wei XQ, Cai B, Hueber AJ, Leung BP, McInnes IB, et al. IL-35 is a novel cytokine with therapeutic effects against collagen-induced arthritis through the expansion of regulatory T cells and suppression of Th17 cells. Eur J Immunol. 2007;37(11):3021–9. [DOI] [PubMed] [Google Scholar]
- 35.Hossen MM, Ma Y, Yin Z, Xia Y, Du J, Huang JY, et al. Current understanding of CTLA-4: from mechanism to autoimmune diseases. Front Immunol. 2023;14:1198365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Terness P, Bauer TM, Röse L, Dufter C, Watzlik A, Simon H, et al. Inhibition of allogeneic T cell proliferation by indoleamine 2,3-dioxygenase-expressing dendritic cells: mediation of suppression by tryptophan metabolites. J Exp Med. 2002;196(4):447–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang DW, Dong N, Wu Y, Zhu XM, Wang CT, Yao YM. Interleukin-37 enhances the suppressive activity of naturally occurring CD4(+)CD25(+) Regulatory T cells. Sci Rep. 2016;6:38955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McNamee EN, Masterson JC, Jedlicka P, McManus M, Grenz A, Collins CB, et al. Interleukin 37 expression protects mice from colitis. Proc Natl Acad Sci U S A. 2011;108(40):16711–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.