

Abstract
Aminoglycosides are cidal inhibitors of bacterial protein synthesis that have been utilized for the treatment of serious bacterial infections for almost 80 years. There have been approximately 15 members of this class approved worldwide for the treatment of a variety of infections, many serious and life threatening. While aminoglycoside use declined due to the introduction of other antibiotic classes such as cephalosporins, fluoroquinolones, and carbapenems, there has been a resurgence of interest in the class as multidrug-resistant pathogens have spread globally. Furthermore, aminoglycosides are recommended as part of combination therapy for empiric treatment of certain difficult-to-treat infections. The development of semisynthetic aminoglycosides designed to overcome common aminoglycoside resistance mechanisms, and the shift to once-daily dosing, has spurred renewed interest in the class. Plazomicin is the first new aminoglycoside to be approved by the FDA in nearly 40 years, marking the successful start of a new campaign to rejuvenate the class.
AMINOGLYCOSIDE HISTORY
Aminoglycoside antimicrobials were first discovered in the 1940s and originally isolated from actinomycetes. Streptomycin, isolated from Streptomyces griseus, was the first aminoglycoside introduced into clinical practice for the treatment of tuberculosis (1, 2). Selman Waksman (the first to coin the term “antibiotic”) won the Nobel Prize in 1952 for the discovery of streptomycin, along with Albert Schatz, who was eventually recognized as a co-discoverer. Since then, a number of aminoglycosides have been discovered as products from the Streptomyces group (“mycin” aminoglycosides, e.g., neomycin, kanamycin, tobramycin) or Micromonospora group (“micin” aminoglycosides, e.g., gentamicin, sisomicin) species, or developed through chemical modifications using existing aminoglycoside scaffolds (e.g., amikacin, netilmicin, arbekacin, plazomicin). Plazomicin is an aminoglycoside that was engineered to overcome aminoglycoside-modifying enzymes (AMEs), the most common aminoglycoside resistance mechanism in Enterobacteriaceae, and is the first aminoglycoside to be approved by the FDA (June 2018) since the approval of amikacin in 1981, marking the beginning of a class rejuvenation.
SPECTRUM OF AMINOGLYCOSIDE ACTIVITY
Aminoglycosides are rapidly bactericidal, concentration-dependent antibiotics with a broad spectrum of activity against Gram-negative and Gram-positive organisms, including those that are multidrug resistant (MDR). Overall, the class has potent activity against Enterobacteriaceae family members (3–7) and is less active against Pseudomonas aeruginosa and Acinetobacter spp. (4, 7, 8). Furthermore, aminoglycosides are also active against the biothreat pathogens Yersinia pestis, the causative agent of pneumonic plague (9, 10), and Francisella tularensis, the causative agent of tularemia (11, 12). Class members also have potent activity against some Gram-positive organisms such as Staphylococcus spp., including methicillin-resistant (MRSA) and vancomycin-intermediate and -resistant Staphylococcus aureus isolates (4) and certain Mycobacterium spp. (13, 14).
CLINICAL UTILITY OF AMINOGLYCOSIDES
Indications
Because of their broad spectrum, aminoglycosides have been used to treat a variety of serious, life-threatening infections and are commonly used as treatment for urinary tract infections, sepsis and neonatal sepsis, and pneumonia (15, 16). Aminoglycosides are administered for both empiric and directed therapy; and amikacin, gentamicin, and tobramycin are the most commonly utilized today. Specific indications for aminoglycoside therapy include amikacin and gentamicin administered intravenously for infections caused by MDR Gram-negative organisms. Gentamicin, in combination with ampicillin, is the most commonly used empiric therapy in neonatal sepsis (17). Inhalational tobramycin is utilized for the treatment of pneumonia and chronic P. aeruginosa infections in cystic fibrosis patients, while parental tobramycin is utilized for lower respiratory tract infections, osteomyelitis, and urinary tract infections. Additional uses include topical or oral neomycin for skin infections, oral paromomycin for amebic and parasitic infections, spectinomycin administered intramuscularly for the treatment of gonorrhea, and streptomycin, kanamycin, and amikacin administered intravenously or intramuscularly as second-line therapeutics for tuberculosis (15). In general, streptomycin, kanamycin, and netilmicin are infrequently used because of clinical resistance (15, 16).
Streptomycin and gentamicin are also considered first-line therapy for the treatment of infections caused by the biothreat pathogens Y. pestis and F. tularensis. Streptomycin has been the drug of choice for the treatment of bubonic, septicemic, and pneumonic plague caused by Y. pestis, but gentamicin has also been shown to be efficacious in the treatment of plague and is also recommended because of the limited supply of streptomycin (18, 19). Streptomycin is bactericidal against F. tularensis at concentrations achieved in humans and was the first antibiotic used for the treatment of tularemia; gentamicin is also an effective therapeutic option for tularemia and is suggested for use because of the supply limitations associated with streptomycin (20).
Associated Toxicities
Nephrotoxicity and ototoxicity are known safety concerns associated with the use of aminoglycosides. In addition, neuromuscular blockade associated with aminoglycosides has also been infrequently reported (21). Aminoglycosides are readily taken up and retained by proximal tubule cells in the kidney and cochlear cells in the ear and thus can be cytotoxic in these cell types (22–24). Although usually reversible, aminoglycoside-induced nephrotoxicity occurs in 5 to 15% of patients on therapy (24–26). However, when comparing incidences of nephrotoxicity, it is important to consider that the reported rate may be influenced by the dosing strategy (e.g., once versus multiple daily dosing, use of therapeutic drug management [TDM]), nephrotoxicity endpoint, frequency of renal function monitoring, and patient population (e.g., severity of illness, comorbidities) evaluated in the clinical trial. Clinically, aminoglycoside-induced nephrotoxicity may manifest as nonoliguric renal impairment accompanied by an increase in plasma creatinine, urea, and other metabolic products typically developing after several days to one week of therapy (25, 27, 28). Approximately 5% of the intravenously administered aminoglycoside dose is retained in the epithelial cells of proximal tubules after glomerular filtration where they accumulate within endosomal and lysosomal vacuoles (22, 29, 30). Aminoglycosides induce a variety of morphological and functional changes in these cells, contributing to injury of the kidney and reduced renal function (27). Strategies to reduce aminoglycoside-induced nephrotoxicity include short-duration, once-daily intravenous dosing, as is discussed in more detail below.
Aminoglycoside-induced ototoxicity can manifest as vestibulotoxicity (balance disorder) and/or cochleotoxicity (tinnitus and/or hearing loss) with intravenous aminoglycoside administration for multiple days, especially if multiple courses of treatment are administered (24, 31–33). Ototoxicity prevalence varies from 2 to 25% for cochleotoxicity and 1 to 10% for vestibulotoxicity (24, 31–33). Ototoxicity may be permanent, occurs in a dose-dependent manner, and is correlated with aminoglycoside blood concentration and duration of therapy; furthermore, patients may be genetically predisposed to ototoxicity via specific mitochondrial or nuclear genome mutations (33). Gentamicin and tobramycin are considered more vestibulotoxic, and amikacin, neomycin, and kanamycin are considered more cochleotoxic (24). As described below, proposed strategies to prevent aminoglycoside-induced ototoxicity include altering the aminoglycoside molecule to prevent aminoglycoside entry into hair cells and to interfere with other intracellular mechanisms that are attributed to aminoglycoside ototoxicity, such as the mitochondrial ribosome (24, 33).
Neuromuscular blockade associated with respiratory depression is linked with aminoglycoside use, although this is rarely observed in the clinic with only anecdotal case reports, often in the postoperative setting (21, 34, 35). Aminoglycosides induce neuromuscular blockade in animal models where it has been shown to be reversible with calcium administration (36–38). Neuromuscular blockade induced by aminoglycosides is hypothesized to be associated with high serum concentrations not typically seen in therapeutic dosing regimens, thus accounting for the rarity of this toxicity observed in clinical practice (34, 36).
Combination Therapy and Dosing Strategies
Aminoglycosides can be used as monotherapy but are also are commonly administered parentally as combination antibiotic therapy with the goals of broadening the spectrum of coverage in empiric therapy, accelerating pathogen clearance, preventing the development of resistance, and taking advantage of synergistic interactions between certain antibiotics, particularly with the β-lactam class (39, 40). Aminoglycosides are frequently used in combination with β-lactam antibiotics for empiric treatment of severe hospital-acquired infections, including hospital-acquired pneumonia, ventilator-associated pneumonia, and sepsis, when an MDR pathogen is suspected (41–43).
Short-duration, once daily dosing of aminoglycosides has been demonstrated to be less nephrotoxic than conventional multiple daily dosing regimens (16, 44). Furthermore, once daily dosing of gentamicin was shown to have no significant neuromuscular blocking effects on respiratory muscle (34). Higher doses given less often reduce the risk of toxicity while maintaining, and potentially enhancing, efficacy (45). As previously stated, aminoglycoside nephrotoxicity is linked to aminoglycoside uptake and retention by proximal renal tubular epithelial cells, where they cause cellular damage that leads to decreased renal function. However, this uptake is saturable, and studies have shown that less frequent aminoglycoside administration results in less aminoglycoside uptake and less nephrotoxicity (45). Additionally, once daily dosing provides a higher ratio of peak concentration of drug to minimum inhibitory concentration (MIC), which allows for rapid bacterial killing, and, as the serum concentration declines between dosing intervals, the kidney cells can recover, while the postantibiotic effect of aminoglycosides ensures that bacterial killing persists (46). Prolonged directed therapy is generally reserved for specific indications, including infections due to MDR pathogens. In these situations, TDM can be used, particularly in patients with increased risk of nephrotoxicity, so that aminoglycoside dose adjustments can be made based on serum concentrations (47).
AMINOGLYCOSIDE MECHANISM OF ACTION
Uptake
The primary mechanism of action of the aminoglycoside class is inhibition of key steps in bacterial protein synthesis (Fig. 1) (48, 49). Aminoglycosides are unique among other protein synthesis inhibitors in that they are bactericidal. All other classes of antibiotics that inhibit protein synthesis, including chloramphenicol, clindamycin, tetracyclines, macrolides, pleuromutilins, and oxazolidinones are bacteriostatic. The bactericidal activity of aminoglycosides is due in part to its method of uptake into the bacterial cell, which is described in more detail below (50–54). In addition, aminoglycosides typically produce a prolonged postantibiotic effect (55). The unique combination of the mechanism of aminoglycoside uptake and inhibition of bacterial protein synthesis lead to concentration-dependent bacterial cell killing.
Figure 1.
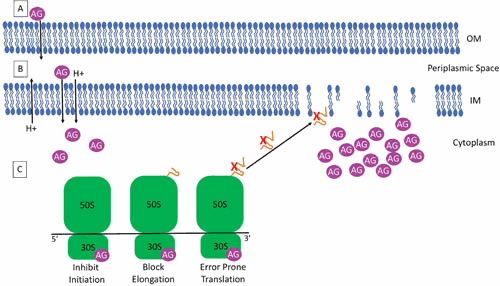
Mechanism of aminoglycoside uptake by Gram-negative bacterial cells and inhibition of protein synthesis. (A) Positively charged aminoglycosides (AG) enter the cell via electrostatic binding to the negatively charged components of the outer membrane (OM) including phospholipids and LPS in Gram-negative bacteria or teichoic acid in Gram-positive bacteria. This binding allows access of the AG to the periplasmic space. (B) A small number of AGs cross the inner membrane (IM) using the proton motive force and into the cytoplasm in an energy-dependent manner. (C) In the cytoplasm, AGs bind the 16S rRNA of the 30S ribosomal subunit where they inhibit initiation of translation, block elongation of translation, and induce error-prone translation. Mistranslated proteins are hypothesized to cause damage to the IM, facilitating AG entry into the cytoplasm.
Aminoglycosides are polycationic, hydrophobic molecules (typically with a molecular weight of ∼450 to 600), that enter bacterial cells in a three-stage self-promoted process (48, 50, 56). The first stage of aminoglycoside entry occurs via electrostatic binding of the positively charged aminoglycoside molecules to the negatively charged components on the bacterial cell surface and is termed the “ionic binding” phase (Fig. 1A). For Gram-negative bacteria, this includes binding to lipopolysaccharide (LPS) and phospholipids present in the outer membrane. For Gram-positive bacteria, which lack LPS, this involves binding to phospholipids and teichoic acid. The act of binding to these components results in displacement of divalent cations and increased permeability of the aminoglycoside molecules into the cells, thereby providing access to the periplasmic space (48, 50, 56).
The second phase of aminoglycoside uptake (energy-dependent phase I) requires the proton motive force and, as such, can be blocked by inhibitors of electron transport and oxidative phosphorylation (Fig. 1B) (48, 50, 56). This phase is characterized by the uptake of a small number of aminoglycoside molecules into the cytoplasm in an energy-dependent manner (56–60). These molecules begin to induce errors in protein synthesis by generating mistranslated proteins. Mistranslated proteins are hypothesized to cause damage to the cytoplasmic membrane creating nonspecific membrane channels, further facilitating aminoglycoside entry and additional cellular damage. The dependence on the electron transport chain can explain the reduced activity of aminoglycosides under anaerobic conditions and at low pH (50, 57).
The final energy-dependent stage of uptake (energy-dependent phase II) results in the rapid movement of aminoglycoside molecules into the cytoplasm (Fig. 1C) (48, 50, 56). As aminoglycoside molecules accumulate within the cell, inhibition of protein synthesis as well as mistranslation accelerates. This process results in the rapid, concentration-dependent bacterial killing observed with aminoglycosides (48, 50, 56, 61, 62).
Porins are involved in the uptake of several antibiotics (e.g., β-lactams, tetracycline, chloramphenicol, and fluoroquinolones), and loss of or functional changes in these porins leads to resistance, but the role that these channels play in aminoglycoside entry is still unclear (63–67). Porins, in particular, OmpF, are hypothesized to play a role in aminoglycoside entry and resistance by some investigators. Although most studies have focused on the role of the Escherichia coli major large porin OmpF (and homologs in other species), some studies have also implicated changes in expression of E. coli OmpC and homologs in the development of resistance (67, 68). Reduction in OmpF expression in E. coli has been linked to transient kanamycin resistance (69), and an E. coli mutant completely lacking OmpF was found to be resistant to gentamicin and kanamycin (70). Aminoglycosides have also been shown to diffuse efficiently through porins reconstituted in vesicle membranes (71). Taken together, these limited data suggest that OmpF, and potentially other porins, may play a role in aminoglycoside entry and that mutations altering porin expression may lead to altered aminoglycoside susceptibility.
Inhibition of Protein Synthesis
Bacterial protein synthesis involves numerous components and is composed of four key stages, “initiation, elongation, termination, and ribosome recycling” (72). In brief, the 30S and 50S ribosome subunits form the 70S ribosome, which binds to the messenger RNA (mRNA). Each subunit contains ribosomal RNAs (rRNA); the 30S contains the 16S rRNA, and the 50S contains the 32S and 5S rRNAs. The rRNAs play a role in the initiation and synthesis of proteins by acting as a scaffold. The 16S rRNA, in particular, contains a sequence complementary to the start codon at its 3′ end and thus can bring the ribosome and mRNA together to initiate protein synthesis (73).
The ribosome also possesses specific transfer RNA (tRNA) sites that facilitate the synthesis of the polypeptide chain as the ribosome moves along the mRNA. Each of these sites has particular roles; the aminoacyl-tRNA site (A-site), on the 16S rRNA, accepts new tRNAs, the peptidyl-tRNA site (P-site) houses the initiator tRNA and the subsequent growing polypeptide chain, and the E-site is the site at which the deacylated tRNA exits the ribosome (72, 74). At initiation, the start codon of the mRNA is located at the P-site with the initiating tRNA carrying the first amino acid of the polypeptide, typically a methionine. Aminoacyl-tRNAs are delivered to the A-site where codon-anticodon recognition and proofreading take place to ensure that the proper amino acid is delivered and added to the growing polypeptide chain (75). The polypeptide is formed between the nascent amino acid at the P-site and at the A-site via peptidyltransferase (74). The peptidyl-tRNA is deacylated and enters the E-site, while the aminoacyl-tRNA, containing the new polypeptide, moves into the P-site, allowing for a new aminoacyl-tRNA to deliver the next amino acid to the ribosome (74). Elongation of the polypeptide chain continues in this fashion as the ribosome moves along the mRNA until a stop codon is encountered; at this point, translation is terminated and the polypeptide is released.
Cocrystal structures show that aminoglycosides bind the bacterial ribosome at the 30S subunit with high affinity to the A-site on the 16S rRNA (76–78). The various members of the aminoglycoside class have different specificities for different regions on the A-site but all are thought to alter conformation. Binding to the 16S rRNA, in turn, impacts protein synthesis by inhibiting initiation, blocking elongation, and promoting misreading of the codon after delivery of the aminoacyl-tRNA, leading to error-prone translation after initiation. Furthermore, error-prone translation causes membrane damage, which is thought to also contribute to cell killing (Fig. 1C) (48, 51, 56, 62, 79–84).
Inhibition of translation has been demonstrated in vitro and in whole-cell experiments. Incorporation of 14C-valine into polypeptides was measured in an in vitro translation assay in the presence of spectinomycin using E. coli extracts from spectinomycin-susceptible and -resistant strains (79). Incorporation of valine was inhibited by spectinomycin in cell extracts from a spectinomycin-susceptible isolate but was unaffected in cell extracts from a spectinomycin-resistant isolate. Whole-cell experiments assessing incorporation of 35S-labeled amino acids into total cellular protein similarly showed dose-dependent decreases in translation with neomycin and paromomycin treatment (81).
In addition to inhibiting translation overall, aminoglycosides have been shown to promote mistranslation. Early work demonstrated that aminoglycosides act on the ribosome before the initiation of protein synthesis, leading to overall inhibition of translation, and they also act after initiation to promote codon misreading, resulting in error-prone protein synthesis (82, 83). Induction of misreading by streptomycin and other aminoglycosides was observed with chain-elongating noninitiating ribosomes (purified polysomes) that were limited for certain aminoacyl-tRNAs (85). In this work, it was shown that when the elongating ribosomes reached a codon for a missing aminoacyl-tRNA, the aminoglycosides promoted rapid substitution of another aminoacyl-tRNA, allowing for continued synthesis of the polypeptide. The degree of amino acid incorporation that was stimulated by the aminoglycosides varied with aminoglycoside concentration, providing additional evidence that aminoglycosides act on additional steps of translation beyond initiation.
AMINOGLYCOSIDE STRUCTURE GROUPS
The aminoglycoside class of antibiotics is composed of four different core structural groups (Fig. 2). In three of the four structure groups, an aminocyclitol 2-deoxystreptamine (DOS) ring is linked to amino sugars saturated with amino and hydroxyl substitutions. Molecules that contain a DOS ring are mono-substituted (e.g., apramycin, neamine), 4,5-disubstituted (e.g., neomycin B, paromomycin, and ribostamycin), and 4,6-disubstituted (e.g., amikacin, kanamycin, tobramycin, gentamicin, sisomicin, netilmicin, arbekacin, plazomicin); underlined molecules are depicted in Fig. 2 (86, 87). The fourth structural group contains a streptidine ring instead of a DOS ring (e.g., streptomycin) (86). While these four structural classes of aminoglycosides all confer bactericidal activity via protein synthesis inhibition, they differ in their ability to evade bacterial resistance mechanisms, specifically the AMEs and 16S rRNA methyltransferases (16S-RMTases).
Figure 2.
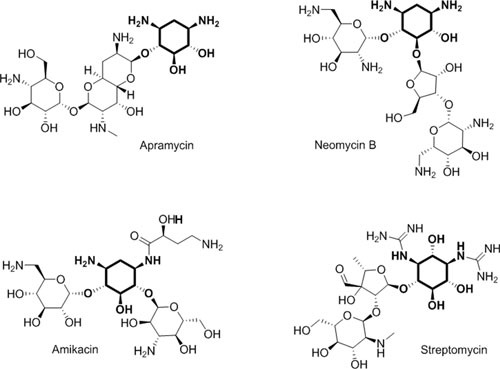
Four aminoglycoside structural groups. Structures of representative aminoglycosides, including the atypical aminoglycosides streptomycin and apramycin, 4,6-substituted amikacin and the 4,5-substituted neomycin B. The deoxystreptamine or streptidine rings are in bold. ©Cold Spring Harbor Press (171), used with permission.
AMINOGLYCOSIDE RESISTANCE MECHANISMS
A variety of aminoglycoside resistance mechanisms exist, including chemical modification of aminoglycosides by AMEs, target site modification via methylation (16S-RMTases) or via chromosomal mutation, and efflux and/or permeability mutations. AMEs and 16S-RMTases are thought to arise from organisms that produce aminoglycosides (e.g., Streptomyces spp., Micromonospora spp.) or via mutation, upregulation, or evolution of normal cellular genes (87, 88).
The aminoglycoside-producing Streptomyces spp., including S. fradiae (neomycin), S. ribosidificus (ribostamycin), and S. kanamyceticus (kanamycin), encode AMEs of the APH and AAC class, providing intrinsic protection from the aminoglycoside that the organism synthesizes (89). AMEs are also involved in normal cellular function, such as in the nonaminoglycoside producer, Providencia stuartii, which encodes a chromosomal AME AAC(2′)-Ia, whose primary function is to acetylate peptidoglycan during cell wall turnover in the cell wall biosynthesis pathway (88). The expression of this chromosomal AME is typically at a low level, dependent on environmental conditions, and is not inducible by aminoglycosides; thus, mutation for overexpression is required to result in aminoglycoside resistance. Furthermore, the phosphotransferases are distantly related to eukaryotic serine/threonine and tyrosine kinases, while the nucleotidyltransferase ANT(4′)-I is related to DNA polymerase β (90). Another example of intrinsic resistance in aminoglycoside producers is via the 16S-RMTases. Genes that encode this methyltransferase enzyme are found in the chromosomes of aminoglycoside-producing organisms such as the kgmA (kanamycin-gentamicin resistance methylation) gene in Monardella purpurea and the kamA (kanamycin-apramycin resistance methylation) gene in Streptomyces tenjimariensis (91).
Notably, in nonaminoglycoside producers, AMEs and 16S-RMTase mechanisms are predominantly acquired on mobile elements to confer aminoglycoside resistance (as described below). Chromosomal target site mutations are rare because of the multiple copies of the target 16S rRNA in most bacterial species, while mutations in efflux mechanisms and cell permeability, which reduce the intracellular concentration of aminoglycosides, are a resistance mechanism not unique to aminoglycosides. As is the case with all antibiotics, the development of bacterial resistance to each member of the aminoglycoside class followed its introduction. This section describes in more detail the mechanisms by which bacteria can acquire or develop resistance to aminoglycoside class members.
Acquired Resistance Mechanisms
Aminoglycoside-modifying enzymes
AMEs are the most common mechanism of resistance to aminoglycosides in clinical isolates (56, 92). AMEs are composed of three classes that render aminoglycosides inactive via acetylation (acetyltransferases; AAC), adenylation (nucleotidyltransferases; ANT), or phosphorylation (phosphotransferases; APH). The genes encoding these enzymes are frequently found on mobile elements (i.e., plasmids or transposons) that encode other antibiotic resistance genes, such as extended-spectrum β-lactamases (ESBLs) and carbapenemases, and thus are often found in MDR Gram-positive and Gram-negative bacteria (56). Notably, aminoglycoside-resistant clinical isolates often possess more than one AME gene; there have also been reports of AME genes encoded on the same plasmid as a 16S-RMTase gene (92–95). The three AME classes include multitudinous numbers of subclass enzymes, and hundreds have been reported, each varying slightly in its specificity for target aminoglycosides (56, 92). A depiction of each of the chemical modifications with example aminoglycosides is presented in Fig. 3.
Figure 3.
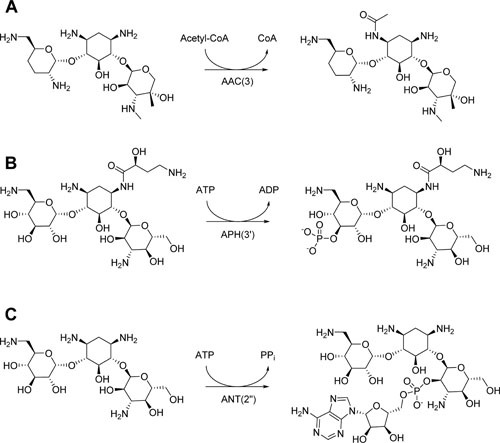
Chemical modification of aminoglycosides by aminoglycoside-modifying enzymes. (A) An example of chemical modification of gentamicin catalyzed by aminoglycoside acetyltransferase AAC(3). (B) An example of chemical modification of amikacin catalyzed by aminoglycoside phosphotransferase APH(3′). (C) An example of chemical modification of kanamycin catalyzed by the aminoglycoside nucleotidyltransferase ANT(2″). ©Cold Spring Harbor Press (171), used with permission.
As described above, the structure of aminoglycosides impacts their accessibility to certain AMEs. Over the years, a number of semisynthetic aminoglycoside molecules have been designed to evade these resistance mechanisms (Fig. 4). The first was dibekacin, which was synthesized from a kanamycin scaffold; arbekacin was subsequently synthesized from dibekacin, resulting in a molecule inaccessible to a number of important Gram-positive AMEs, including APH, ANT, and AAC class members (Fig. 4A) (96). Three aminoglycosides were developed by modification of existing aminoglycosides at the 1-NH group, specifically amikacin (from kanamycin; addition of (S)-4-amino-2-hydroxybutyrl group; Fig. 4B), isepamicin (from gentamicin B; addition of an (S)-3-amino-2-hydroxypropionyl group; Fig. 4C), and netilmicin (from sisomicin; addition of an ethyl group; Fig. 4D) (97). Plazomicin was also developed from sisomicin, which naturally lacks 3′- and 4′-OH groups, making it inaccessible to APH(3′) and ANT(4′) class enzymes (4). Modification of sisomicin via addition of a hydroxyethyl group at the 6′ position and a hydroxyl-aminobutyric acid (HABA) group at the N-1 position block clinically important AMEs, AAC(6′), and AAC(3), ANT(2″) and APH(2″), respectively, resulting in the ability of plazomicin to evade nearly every clinically relevant AME (Fig. 4D) (4, 98).
Figure 4.
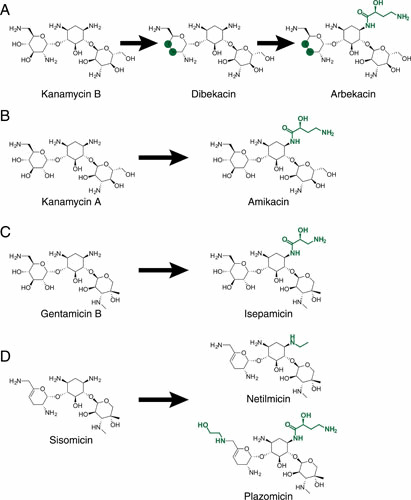
Semisynthetic aminoglycosides. (A) Kanamycin B scaffold with modification to dibekacin and subsequent modification to arbekacin. (B) Kanamycin A scaffold with modification to amikacin. (C) Gentamicin B scaffold with modification to isepamicin. (D) Sisomicin scaffold with modification to netilmicin and with modification to plazomicin. Green highlights indicate new chemical modifications; green circles indicate removal of hydroxyl groups.
Acetyltransferases
The AAC class of enzymes acetylates amino groups (-NH2) on aminoglycoside molecules (Fig. 3A). The enzymes are members of the GCN5-related N-acetyltransferase (GNAT) superfamily (99). There are four subclasses of this enzyme: AAC(1), (2′), (3), and (6′). Subtypes within the latter two subclasses (AAC(3) and AAC(6′)) are the most widespread enzymes, found in both Gram-positive and Gram-negative organisms. Depending on the specific variant, AAC(3) and AAC(6′) confer resistance to aminoglycosides including gentamicin, amikacin, and tobramycin; as described above, plazomicin maintains activity against isolates that encode these enzymes. A variant of AAC(6′)-Ib reported in 2007, AAC(6′)-Ib-cr, was shown to be capable of acetylating and inactivating fluoroquinolones (100).
The most prevalent AMEs reported in aminoglycoside-resistant Enterobacteriaceae spp. (via large [>1,000 clinical isolate] surveillance sample sizes) are AAC class enzymes, specifically AAC(6′)-Ib and its variant, AAC(6′)-Ib-cr, as well as AAC(3)-IIa (94, 101). These enzymes, along with AAC(3)-IVa, were also reported to be prevalent in carbapenem-resistant Enterobacteriaceae spp., predominantly Klebsiella pneumoniae (102, 103), demonstrating that aminoglycoside resistance is commonly observed with resistance to other classes of antibiotics.
The most common AAC class member observed in aminoglycoside-resistant clinical isolates of the Gram-negative glucose nonfermenter, P. aeruginosa, is also AAC(6′)-Ib, which is reported as a major contributor to amikacin and tobramycin resistance (93, 104, 105). Other AAC class members have also been reported in P. aeruginosa with less prevalence, including AAC(3)-Ia, AAC(3′)-II, and AAC(3)-IV (93, 104, 105). AAC(3)-Ia and AAC(6′)-Ib have also been reported in amikacin-resistant Acinetobacter baumannii clinical isolates (105, 106).
Phosphotransferases
The APH class of enzymes transfer a phosphate group from ATP to a hydroxyl (-OH) group on the aminoglycoside molecule (Fig. 3B). There are 7 subclasses of phosphotransferases, specifically APH(2″), (3′), (3″) (4), (6), (7″), and (9). Several of these classes of enzymes are not relevant to clinical aminoglycoside resistance and are utilized for molecular genetics and cloning purposes in the laboratory. These enzymes include APH(4), which only confers resistance to hygromycin; APH(6), which only confers resistance to streptomycin; APH(7″), which only confers resistance to hygromycin; and APH(9), which only confers resistance to spectinomycin (56). Furthermore, APH(3″), which only confers resistance to streptomycin, is limited in its distribution and was first reported on the chromosomes of S. griseus and Mycobacterium fortuitum and subsequently on mobile elements (56). However, some enzymes within this class are widespread mediators of clinical aminoglycoside resistance. Members of the APH(3′) class of enzymes confer resistance to amikacin, kanamycin, and neomycin in both Gram-negative and Gram-positive bacteria (4, 56, 107).
Aminoglycoside-resistant Gram-negative species encode a number of APH(3′) class members, including APH(3′)-VIa observed in Enterobacteriaceae spp. clinical isolates from Europe (101) and APH(3″)-Ia in carbapenem-resistant K. pneumoniae from the United States (103). Furthermore, APH(3′) class members are quite prevalent in the Gram-negative glucose nonfermenters. In P. aeruginosa, APH(3′) class members, including APH(3′)-Ia, APH(3′)-VI, and APH(3′)-IIb were each reported in individual studies as the most prevalent AMEs in MDR aminoglycoside-resistant P. aeruginosa clinical isolates from the United States, Korea, and Iran, respectively (93, 104, 105). Across each of these studies, the APH(3′) enzymes were also found present with additional AMEs, such as AAC(6)-I and APH(3′)-VI (93). Similarly, in A. baumannii, APH(3′)-VIa was the most prevalent AME reported in amikacin-resistant isolates from Iran (105), while APH(3′)-Ia was also reported as frequently detected in this same study, and was prevalent in isolates from China (106).
APH(3′)-IIIa is prevalent in Gram-positive organisms, including MRSA, and confers resistance to amikacin, isepamicin, and kanamycin, among other aminoglycosides (56, 95). In addition, APH(2″) enzymes confer resistance to gentamicin in Gram-positive organisms, including the variant APH(2″)-Id (or -IVa), which has only been reported in Enterococcus spp. (108), and the fused enzyme, AAC(6′)-Ie-APH(2″)-Ia, which is highly prevalent in Staphylococcus spp., including MRSA (95, 109, 110).
Adenyltransferases
The ANT class of enzymes transfers an adenosine monophosphate (AMP) group from ATP to a hydroxyl (-OH) group on the aminoglycoside molecule (Fig. 3C). Five subclasses of the enzyme exist: ANT(2″), (3″), (4′), (6), and (9) (56, 111). Members of the ANT class of enzymes (3″, 4′, 6, and 9) are predominantly found in Gram-positive organisms including Bacillus spp., Enterococcus spp., Staphylococcus spp., and Campylobacter spp. Specifically, the ANT(4′) and ANT(4″) are predominant in Enterococcus spp. and S. aureus, including MRSA, and confer resistance to amikacin, tobramycin, and isepamicin (56, 95). There have been two reports of ANT(4′)-IIa found in Pseudomonas spp. and Enterobacteriaceae (56).
The ANT(2″)-Ia enzyme, found in Gram-negative organisms only, is the most prevalent ANT enzyme. This AME has been reported in Enterobacteriaceae and P. aeruginosa at a lower frequency than observed for the AAC and APH class enzymes, but it is still an important contributor to aminoglycoside resistance (94, 102–104). Alternatively, in A. baumannii, ANT(2″)-Ia (as well as ANT(3′)-Ia) was reported to be prevalent in MDR clinical isolates (105, 106).
16S Ribosomal RNA methyltransferases
In addition to AME-mediated direct modification of aminoglycosides, modification of the aminoglycoside ribosomal target can confer aminoglycoside resistance. The 16S-RMTase enzymes methylate nucleotide residues on the 16S rRNA, thereby blocking aminoglycoside binding and conferring resistance (MICs ≥128 μg/ml). Notably, resistance to nearly all aminoglycosides is conferred by this mechanism (87, 91, 112, 113).
There are two classes of 16S-RMTase enzymes, those that methylate the N7 position of nucleotide G1405 and those that methylate the N1 position of nucleotide A1408 (49, 87, 91, 112, 114, 115). The enzymes that methylate G1405 are the most common plasmidic 16S-RMTase enzymes, with the first reported enzyme, RmtA, from a 1997 P. aeruginosa clinical isolate (116). Since then, a number of enzymes have been reported in a variety of Gram-negative species, including ArmA, RmtB1, RmtB2, RmtC, RmtD1, RmtD2, RmtE, RmtF, RmtG, and RmtH. These enzymes impact all 4,6-disubstituted aminoglycosides, including plazomicin, amikacin, gentamicin, and tobramycin (Table 1). In addition to rendering 4,6-disubstituted aminoglycosides inactive, NpmA (a member of the second class of 16S-RMTases) also renders 4,5-disubstituted and monosubstituted aminoglycosides, including neomycin and apramycin, inactive (Table 1). Clinical isolates with NpmA are rare, with only two reports currently in the literature (117, 118).
Table 1.
16S ribosomal RNA methyltransferase classesa
| Plasmidic 16S ribosomal RNA methyltransferase | Methylation Site | 4,6-DOS-linked aminoglycoside target(e.g., AMK, GEN, TOB, PLZ) | 4,5-DOS-linked aminoglycoside target(e.g., NEO) | Monosubstituted DOS-linked aminoglycoside target(e.g., APR) | No DOS ring(e.g., STR) |
|---|---|---|---|---|---|
| ArmA, RmtB, RmtB2, RmtC, RmtD1, RmtD2, RmtE, RmtF, RmtG, RmtH | N7 G1405 | Yesb | Noc | No | No |
| NpmA | N1 A1408 | Yes | Yes | Yes | No |
Abbreviations: DOS, 2-deoxystreptamine; AMK, amikacin; APR, apramycin; GEN, gentamicin; PLZ, plazomicin; STR, streptomycin; TOB, tobramycin.
No=16S rRNA methyltransferase does not methylate the respective aminoglycoside target and does not result in resistance.
Yes=16S rRNA methyltransferase methylates the respective aminoglycoside target, resulting in resistance.
Highly aminoglycoside-resistant A. baumannii are frequently associated with 16S-RMTases. The most prevalent 16S-RMTases in these organisms are ArmA and RmtB, and both are often associated with β-lactam resistance mechanisms, CTX-M, NDM, and OXA-23 (87, 106, 119–121). Although the first 16S-RMTase was found in a P. aeruginosa isolate, these enzymes are less prevalent in P. aeruginosa, but ArmA, RmtA, RmtB, and RmtD have also been described in aminoglycoside-resistant clinical isolates (87, 119, 120, 122).
The widest variety of 16S-RMTases are found in Enterobacteriaceae spp. (predominantly K. pneumoniae) with reports of ArmA, RmtB–RmtH, as well as the two reports of NpmA (87, 102). Similar to A. baumannii, the resistance mechanisms are often linked with resistance to other antibiotic classes, including resistance to β-lactams CTX-M, KPC, OXA, and plasmid-mediated quinolone resistance (QnrA and B) (87, 112, 118, 123–125). A high prevalence of 16S-RMTases among NDM producers exists in China and India (87, 112, 113). Overall, these enzymes are rarely reported in the United States. For instance, there were no 16S-RMTases found in a large set of Enterobacteriaceae spp. clinical isolates collected in 2016 from U.S. hospitals and surveyed as part of a global surveillance program (94). In Europe, the most recently reported rate was 1.4%, from a 2014 to 2015 global surveillance program of Enterobacteriaceae spp. (101).
Chromosomal Resistance
Target site mutations
Target site–based mutations are not typically observed with aminoglycosides because most species have multiple copies of rRNA-encoding genes, and resistance via this mechanism would thus require every copy of the gene to be mutated. Mycobacterium and Borrelia are unique genera with either a single copy of the entire ribosomal operon or a single copy of 16S rRNA, respectively (126, 127), and thus ribosomal mutations have conferred clinical resistance in these genera. In Mycobacterium tuberculosis, high-level resistance to streptomycin can result from mutations in the genes rrs and rpsL encoding two components of the ribosome, the 16S rRNA (128, 129) and the S12 protein (128, 130), respectively. High-level resistance to amikacin and other 2-deoxystreptamine aminoglycosides has also been observed in Mycobacterium abscessus and Mycobacterium chelonae clinical isolates due to mutations in the genes encoding 16S rRNA (131). In Borrelia burgdorferi, high-level resistance to spectinomycin and other aminoglycosides has been observed due to mutations in the 16S rRNA and the S12 protein (132); however, aminoglycosides are not commonly used to treat infections caused by this pathogen.
Efflux
Resistance nodulation division (RND) efflux systems play a major role in Gram-negative bacterial antimicrobial resistance (133, 134). The tripartite efflux systems consist of a transporter protein connected to a membrane fusion protein localized in the periplasmic space, which then connects the transporter to the outer membrane pore (OMP) to form a channel to actively export substrates, including antibiotics, via the proton motive force. As such, clinically relevant Gram-negative pathogens, such as P. aeruginosa, Acinetobacter spp., and Enterobacteriaceae, encode RND systems attributed to aminoglycoside resistance (133, 135–137). Furthermore, many efflux pumps are constitutively expressed, and as a result, some species exhibit intrinsic resistance to various antibiotics (see “Intrinsic Resistance”) (133, 134, 138). In addition, mutations in regulatory genes that derepress expression and/or result in overexpression of the pump can confer high-level antibiotic resistance.
The glucose nonfermenters, P. aeruginosa and Acinetobacter spp., contain important RND efflux systems that, when overexpressed, contribute to clinical resistance. The Mex (multiple efflux) XY-OprM system in P. aeruginosa initially contributes to low-level intrinsic resistance to aminoglycosides as well as other antibiotics such as tetracycline (139). Mutations in the mexZ repressor, which regulates expression of the mexXY operon, have been demonstrated to result in pan-aminoglycoside resistance in clinical isolates (122, 135). Additionally, mutations in other genes, such as parR, a member of the parRS two-component regulatory system, have been demonstrated to confer overexpression of mexXY (140). As a result, overexpression of this efflux system plays an important role in aminoglycoside resistance of P. aeruginosa clinical isolates, in particular, in chronic lung infections of cystic fibrosis patients (122, 141). Similarly, overexpression of the RND family Ade (Acinetobacter drug efflux) ABC efflux system via mutations in the AdeRS two-component regulator contributes to aminoglycoside resistance in clinical isolates of Acinetobacter spp. as well as to the resistance of diverse classes including fluoroquinolones, tetracyclines, β-lactams, and macrolides (136, 142, 143).
The AcrAD-TolC efflux system of E. coli and its homologs in Enterobacteriaceae spp. are also capable of exporting aminoglycosides as well as other antibiotics (92, 133, 137). Overall, clinical resistance to aminoglycosides via efflux pumps in Enterobacteriaceae spp. is rarely reported; this could hypothetically be due to the prominence of AMEs, which may reduce the selection pressure for efflux-mediated mechanisms. Alternatively, or in addition, efflux mutations may have a fitness cost that is different from AMEs (92, 137).
Permeability
Other antibiotic classes (e.g., β-lactams, fluoroquinolones, tetracyclines) traverse the bacterial membrane through porins, and, as a result, porin loss can confer resistance to these antibiotic classes. The limited data available suggest a potential role for porins in aminoglycoside entry and resistance, but this has not been definitively demonstrated in clinical isolates. Aminoglycosides have been shown to diffuse efficiently through porins in an in vitro system, and E. coli mutants lacking the major porin OmpF are resistant to aminoglycosides (69–71). E. coli OmpF mutants have also been found to have reduced survival compared to wild type, which suggests that these mutants may be rarely encountered in the clinic (144). It is likely that the prominence of plasmidic-based aminoglycoside resistance mechanisms, such as AMEs, negates the necessity for selection of porin-based mutations to confer aminoglycoside resistance in clinical pathogens.
Intrinsic Resistance
The aminoglycoside class lacks activity against obligate anaerobic bacteria (145). Uptake of aminoglycoside molecules requires active electron transport, and thus, under strict anaerobic conditions, aminoglycosides cannot be taken up by the cells (56, 57, 59, 60, 145). Several aerobic bacterial species are also intrinsically resistant to aminoglycosides, including the Gram-negative organisms Burkholderia cepacia complex, Stenotrophomonas maltophilia and Achromobacter xylosoxidans, and the Gram-positive organisms Enterococcus faecalis, Enterococcus faecium, and Enterococcus gallinarum/casseliflavus.
Intrinsic resistance is common across multiple drug classes in both B. cepacia complex, S. maltophilia, and A. xylosoxidans organisms; and there is substantial evidence to suggest that various efflux pumps are responsible for the observed resistance in these species (133, 146–149). For instance, the AmrAB-OprA RND efflux pump, an ortholog of the P. aeruginosa MexXY-OprM pump, has been shown to be a major contributor to intrinsic aminoglycoside resistance observed in Burkholderia pseudomallei, while the SmeJK pump is attributed to aminoglycoside resistance in S. maltophilia (133, 138). Additionally, AMEs, like the chromosomal AME aac(6′)-Iz found in Stenotrophomonas spp., can contribute to intrinsic resistance against some aminoglycosides (150). Likewise, in Enterococcus spp., intrinsic resistance is linked to expression of chromosomal AMEs including those encoded by the aac(6′)-Ii gene and the efmM gene, which encodes a 16S rRNA methyltransferase (151, 152).
REJUVENATION OF THE AMINOGLYCOSIDE CLASS
Plazomicin: A Recently Approved Aminoglycoside
Plazomicin is an aminoglycoside (as described above) that was approved in June 2018 by the FDA for use in patients 18 years of age or older for the treatment of complicated urinary tract infections (cUTIs), including pyelonephritis, caused by susceptible E. coli, K. pneumoniae, Proteus mirabilis, and Enterobacter cloacae. Approval was primarily based on results from the Phase 3 EPIC trial (NCT02486627), a randomized, multinational, double-blind, comparator-controlled noninferiority trial that evaluated the efficacy and safety of plazomicin, administered as a once daily 15 mg/kg intravenous (IV) infusion, versus meropenem in adult patients with cUTI, including acute pyelonephritis (AP) (153). The U.S. prescribing information indicates that plazomicin should be reserved for use in cUTI patients who have limited or no alternative treatment options (154). In vitro data for other aerobic Gram-negative bacteria, including Citrobacter spp., Klebsiella spp., Morganella morganii, Proteus vulgaris, Providencia stuartii, and Serratia marcescens is also noted as available, but that efficacy has not been established in infections caused by these bacteria in clinical trials.
Short-duration (30 minutes), once daily dosing of plazomicin 15 mg/kg administered by intravenous infusion is recommended based on creatinine clearance (CLCR) ≥ 90 ml/min; severity of infection and clinical status of the patient is to guide duration of therapy (154). Dose adjustments in patients with moderate or severe renal impairment are recommended based on estimated CLCR. Furthermore, trough-based TDM for patients with renal impairment (CLCR ≥15 and ≤90 ml/min) is recommended.
Plazomicin (15 mg/kg IV infusion once daily) was also studied in an open-label, comparator-controlled pathogen-focused study, the CARE trial (NCT01970371), in patients with serious infections caused by carbapenem-resistant Enterobacteriaceae (CRE). Two cohorts of patients were enrolled in this trial. Cohort 1 was a randomized, open-label, cohort comparing the efficacy and safety of plazomicin to that of colistin, each in combination with adjunctive meropenem or tigecycline, in adult patients with bloodstream infection (BSI), hospital-acquired bacterial pneumonia (HABP), or ventilator-associated bacterial pneumonia (VABP) due to CRE. Cohort 2 was an observational, single-arm cohort evaluating plazomicin-based therapy in patients with BSI, HABP/VABP, cUTI, or AP due to CRE who were not eligible for inclusion in the randomized cohort (155).
Aminoglycoside-associated toxicities including nephrotoxicity, ototoxicity, and neuromuscular blockade were monitored during these two phase 3 studies. In accordance with the aminoglycoside class, black box warnings are provided in regard to nephrotoxicity, ototoxicity, and neuromuscular blockade and the U.S. Prescribing Information (USPI) clinical trial experience for plazomicin reports observations of nephrotoxicity and ototoxicity (154). For more information, we direct the reader to refer to the full USPI for plazomicin (154). Further, details of both phase 3 studies are in preparation for future publications.
Design Strategies for Future Aminoglycosides
As described above, there have been many modification strategies discovered to overcome AME-mediated aminoglycoside resistance (156). Because of the success of these strategies, researchers have now turned to the more challenging task of identifying additional modifications that will reduce the nephrotoxic and ototoxic potential of this antibiotic class. If these two dosing-limiting toxicities could be reduced, then it is expected that higher exposures may be safely achieved, allowing for broader coverage against additional resistance mechanisms such as efflux upregulation or decreased membrane permeability. Furthermore, an improvement in safety would likely enable effective treatment of difficult-to-treat infections caused by P. aeruginosa and A. baumannii, in particular, because these organisms possess more challenging outer membrane barriers and more complex efflux machinery than do other species, such as those in the Enterobacteriaceae family.
The identification of aminoglycoside modifications that would reduce nephrotoxicity and ototoxicity is challenging because the atomic level interactions associated with the processes leading to these toxicities, which would be required to enable rational design, are not entirely understood. As described above, the toxicity caused by aminoglycosides is highly localized; the proximal tubule cells of the kidney and the hair cells of the cochlea are impacted, but the mechanism behind this is still unclear. Because the toxicities are highly localized, it is presumed that a specific molecular recognition event mediates the access of the aminoglycoside to the target cells. Several researchers have proposed that the receptor partnership of megalin and cubilin may control aminoglycoside accumulation in the target cells of both the proximal tubule cells and hair cells, but definitive experiments that rule out the potential role of other uptake mechanisms have not been exhaustively performed. Preventing entry into the specific cells associated with aminoglycoside toxicity would presumably be a viable strategy to reduce toxicity regardless of the downstream inner cellular distal and proximate causes of the toxicity.
In addition to preventing eukaryotic cell entry, many researchers also focus on addressing the more proximate cause of the nephrotoxicity, which is hypothesized to be aminoglycoside binding to the eukaryotic cytoplasmic and/or mitochondrial ribosome. The advantage of this approach is that the molecular details of these binding interactions are better understood and therefore more amenable to rational design of new aminoglycosides containing modifications that differentially effect eukaryotic versus prokaryotic ribosomal binding. Model systems of the aminoglycoside binding sites for the eukaryotic cytoplasmic and mitochondrial as well as the bacterial ribosome have been employed extensively to study these interactions (157–159). In addition, recent structures of more complete eukaryotic ribosomes promise researchers an even fuller view of how aminoglycosides interact with their target sites (160, 161). Such structures may allow for even more sophisticated design strategies to improve bacterial over eukaryotic ribosomal binding.
The most recent approaches toward improving the therapeutic window of aminoglycosides by either increasing antibacterial potency without changing toxicity or maintaining antibacterial potency while reducing toxicity are briefly described below. For earlier reviews on this topic, we direct the reader to the following references (162–164).
Perhaps the most exciting advancement is the discovery of 2-OH arbekacin (165). In this work, the authors installed a hydroxyl group onto the 2 position of the central ring of the arbekacin. This strategy was inspired by the observation that streptomycin, which also has a hydroxy group in analogous 2 position, demonstrates relatively low nephrotoxicity compared to other aminoglycosides. The new 2-OH arbekacin compound was assessed for in vitro microbiological activity against several species with and without the presence of AMEs. Potent antibacterial activity was observed for this 2-OH arbekacin derivative, and the activity was similar or improved relative to arbekacin. The arbekacin derivative and arbekacin were also tested side-by-side in a rat nephrotoxicity study, although the doses and duration used were not reported. Assuming the exposures were the same, 2-OH arbekacin displayed significantly lower levels of blood urea nitrogen and serum creatinine, biomarkers typically used as indicators of nephrotoxicity, when compared to arbekacin. Histopathological observations were consistent with the observed biomarker reduction showing marked findings for arbekacin but no findings for the 2-OH arbekacin compound. These encouraging results demonstrate that a relatively small change on the central ring of the 4,6-linked arbekacin aminoglycoside can significantly reduce nephrotoxicity without disrupting microbiological potency, thereby improving the therapeutic window of arbekacin.
Another recent publication examined the role of potentially nephroprotective modifications on the 4,6-linked aminoglycoside sisomicin (166). In this study, the authors report that ribosomal selectivity (i.e., the binding preference of the aminoglycoside for the bacterial ribosomal target over the eukaryotic cytoplasmic or mitochondrial ribosome) improved with the addition of a 6′-hydroxyethyl group on the sisomicin scaffold. In contrast, the authors report that the addition of the N1 HABA group to sisomicin has the opposite effect on ribosomal selectivity. Thus, the addition of both groups, which converts sisomicin to plazomicin, would result in a net effect of no change to the expected therapeutic window. The authors further speculate, based on ex vivo studies with mouse cochlear explants, that when both modifications are present on the sisomicin scaffold, the ototoxic potential of plazomicin will be similar to that of gentamicin. The extent to which the ex vivo model is predictive of outcomes in the standard preclinical guinea pig model of ototoxicity or of human clinical outcomes is currently unconfirmed, but data for plazomicin are discussed above.
As described earlier, one limitation of all known 4,6-linked aminoglycosides is that they are ineffective against bacteria encoding 16S-RMTases (Table 1). For this reason, some researchers are now turning to the 4,5-linked aminoglycosides, which are largely not affected by the methylation activity of nearly all 16S-RMTases (Table 1). Recent efforts have been made to make 4,5-linked aminoglycosides more selective for the bacterial ribosome. In one study, 4′-O modifications were installed on the 4,5-linked aminoglycoside paromomycin and in another study, N6′-hydroxyethyl and 4′-O-ethyl modifications were installed on the neomycin core (167, 168). In both cases, the modifications increased the ribosomal binding selectivity with little or no reduction in antibacterial potency (167). Whether the improvement in ribosomal binding selectivity in favor of binding the bacterial versus eukaryotic ribosomes translates to a significant reduction in in vivo nephrotoxicity and ototoxicity remains to be tested.
Research groups at Meiji Seika Pharma Co. have recently synthesized derivatives of apramycin, an atypical aminoglycoside that is also not affected by most 16S-RMTases (Table 1) (169, 170). The researchers found multiple modifications of apramycin at the 4″, 5, and 6 positions conferred enhanced potency against a panel of challenging Gram-positive and Gram-negative bacterial pathogens. One particular compound, TS3112, was reported to possess efficacy against 16S-RMTase-producing strains of K. pneumoniae, P. aeruginosa, and A. baumannii in a murine thigh infection model. In a 10-day repeat dose study in rats, TS3112 demonstrated a toxicity profile similar to gentamicin. Whether this compound or further modifications of this compound can be made to significantly improve the therapeutic window remains to be tested.
Finally, Achaogen is focused on the development of a 4,5-linked aminoglycoside capable of overcoming clinically relevant resistance mechanisms, including 16S-RMTases, for the potential of treating highly resistant Gram-negative pathogens including Enterobacteriaceae, A. baumannii, and P. aeruginosa. The development of a new synthetic route has enabled the exploration of further structure-toxicity relationships in this aminoglycoside class that were not previously accessible. The details of this synthetic approach, as well as results pertaining to the potential improvements to the therapeutic window of these new aminoglycosides, will appear in forthcoming publications.
CONCLUDING REMARKS
Aminoglycosides have been an important antibiotic class since their introduction in the 1940s. Their mechanism of action, resulting in rapid bactericidality and prolonged postantibiotic effect, is unique, because all other bacterial protein synthesis inhibitors are only bacteriostatic. Furthermore, the synergy observed when combined with other antibiotic classes, particularly the β-lactams, provides expanded utility for therapy in the face of serious infections due to MDR organisms. The recent resurgence of interest in the class provides an opportunity for the collection of contemporary data on clinical utility and optimized dosing, as well as a call to action to monitor aminoglycoside susceptibility and the prevalence of aminoglycoside resistance mechanisms in clinically important pathogens. Expansion of the aminoglycoside class, through thoughtful development, will help fill an important unmet need in the antimicrobial armamentarium against the ever-expanding spread of MDR pathogens, particularly those that encode resistance to last-line β-lactam-β-lactamase inhibitors, carbapenems, and polymyxins. The recent approval of plazomicin has the potential to pave the way for the development and approval of additional aminoglycosides and a resurgence of this historically important class of antibiotics.
Contributor Information
Alisa W. Serio, Achaogen Inc., South San Francisco, CA 94080
Tiffany Keepers, Achaogen Inc., South San Francisco, CA 94080.
Logan Andrews, Achaogen Inc., South San Francisco, CA 94080.
Kevin M. Krause, Achaogen Inc., South San Francisco, CA 94080
Karen Bush, Department of Biology, Indiana University, Bloomington, IN.
REFERENCES
- 1.Kresge N, Simoni RD, Hill RL. 2004. Selman Waksman: the father of antibiotics. J Biol Chem 279:e7. [Google Scholar]
- 2.Woodruff HB, Selman A. 2014. Selman A. Waksman, winner of the 1952 Nobel Prize for physiology or medicine. Appl Environ Microbiol 80:2–8. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ristuccia AM, Cunha BA. 1985. An overview of amikacin. Ther Drug Monit 7:12–25. [DOI] [PubMed] [Google Scholar]
- 4.Aggen JB, Armstrong ES, Goldblum AA, Dozzo P, Linsell MS, Gliedt MJ, Hildebrandt DJ, Feeney LA, Kubo A, Matias RD, Lopez S, Gomez M, Wlasichuk KB, Diokno R, Miller GH, Moser HE. 2010. Synthesis and spectrum of the neoglycoside ACHN-490. Antimicrob Agents Chemother 54:4636–4642. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Landman D, Babu E, Shah N, Kelly P, Bäcker M, Bratu S, Quale J. 2010. Activity of a novel aminoglycoside, ACHN-490, against clinical isolates of Escherichia coli and Klebsiella pneumoniae from New York City. J Antimicrob Chemother 65:2123–2127. [PubMed] [DOI] [PubMed] [Google Scholar]
- 6.Sorlozano A, Jimenez-Pacheco A, de Dios Luna Del Castillo J, Sampedro A, Martinez-Brocal A, Miranda-Casas C, Navarro-Marí JM, Gutiérrez-Fernández J. 2014. Evolution of the resistance to antibiotics of bacteria involved in urinary tract infections: a 7-year surveillance study. Am J Infect Control 42:1033–1038. [PubMed] [DOI] [PubMed] [Google Scholar]
- 7.Sader HS, Rhomberg PR, Farrell DJ, Jones RN. 2015. Arbekacin activity against contemporary clinical bacteria isolated from patients hospitalized with pneumonia. Antimicrob Agents Chemother 59:3263–3270. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Landman D, Kelly P, Bäcker M, Babu E, Shah N, Bratu S, Quale J. 2011. Antimicrobial activity of a novel aminoglycoside, ACHN-490, against Acinetobacter baumannii and Pseudomonas aeruginosa from New York City. J Antimicrob Chemother 66:332–334. [PubMed] [DOI] [PubMed] [Google Scholar]
- 9.Heine HS, Hershfield J, Marchand C, Miller L, Halasohoris S, Purcell BK, Worsham PL. 2015. In vitro antibiotic susceptibilities of Yersinia pestis determined by broth microdilution following CLSI methods. Antimicrob Agents Chemother 59:1919–1921. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mega WM, Doyle-Eisele M, Cass RT, Kostrub CF, Sherwood RL, Metz MA, Cirz RT. 2016. Plazomicin is effective in a non-human primate pneumonic plague model. Bioorg Med Chem 24:6429–6439. [PubMed] [DOI] [PubMed] [Google Scholar]
- 11.Kreizinger Z, Makrai L, Helyes G, Magyar T, Erdélyi K, Gyuranecz M. 2013. Antimicrobial susceptibility of Francisella tularensis subsp. holarctica strains from Hungary, Central Europe. J Antimicrob Chemother 68:370–373. [PubMed] [DOI] [PubMed] [Google Scholar]
- 12.Kiliç S, Celebi B, Acar B, Ataş M. 2013. In vitro susceptibility of isolates of Francisella tularensis from Turkey. Scand J Infect Dis 45:337–341. [PubMed] [DOI] [PubMed] [Google Scholar]
- 13.Gonzalo X, Casali N, Broda A, Pardieu C, Drobniewski F. 2015. Combination of amikacin and doxycycline against multidrug-resistant and extensively drug-resistant tuberculosis. Int J Antimicrob Agents 45:406–412. [PubMed] [DOI] [PubMed] [Google Scholar]
- 14.Ji B, Lefrançois S, Robert J, Chauffour A, Truffot C, Jarlier V. 2006. In vitro and in vivo activities of rifampin, streptomycin, amikacin, moxifloxacin, R207910, linezolid, and PA-824 against Mycobacterium ulcerans. Antimicrob Agents Chemother 50:1921–1926. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xie J, Talaska AE, Schacht J. 2011. New developments in aminoglycoside therapy and ototoxicity. Hear Res 281:28–37. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Avent ML, Rogers BA, Cheng AC, Paterson DL. 2011. Current use of aminoglycosides: indications, pharmacokinetics and monitoring for toxicity. Intern Med J 41:441–449. [PubMed] [DOI] [PubMed] [Google Scholar]
- 17.Simonsen KA, Anderson-Berry AL, Delair SF, Davies HD. 2014. Early-onset neonatal sepsis. Clin Microbiol Rev 27:21–47. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boulanger LL, Ettestad P, Fogarty JD, Dennis DT, Romig D, Mertz G. 2004. Gentamicin and tetracyclines for the treatment of human plague: review of 75 cases in New Mexico, 1985-1999. Clin Infect Dis 38:663–669. [PubMed] [DOI] [PubMed] [Google Scholar]
- 19.Inglesby TV, Dennis DT, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Fine AD, Friedlander AM, Hauer J, Koerner JF, Layton M, McDade J, Osterholm MT, O’Toole T, Parker G, Perl TM, Russell PK, Schoch-Spana M, Tonat K, Working Group on Civilian Biodefense. 2000. Plague as a biological weapon: medical and public health management. JAMA 283:2281–2290. [PubMed] [DOI] [PubMed] [Google Scholar]
- 20.Hepburn MJ, Simpson AJ. 2008. Tularemia: current diagnosis and treatment options. Expert Rev Anti Infect Ther 6:231–240. [PubMed] [DOI] [PubMed] [Google Scholar]
- 21.Grill MF, Maganti RK. 2011. Neurotoxic effects associated with antibiotic use: management considerations. Br J Clin Pharmacol 72:381–393. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vandewalle A, Farman N, Morin JP, Fillastre JP, Hatt PY, Bonvalet JP, Gastineau M, Wanstok F. 1981. Gentamicin incorporation along the nephron: autoradiographic study on isolated tubules. Kidney Int 19:529–539. [PubMed] [DOI] [PubMed] [Google Scholar]
- 23.Dai CF, Mangiardi D, Cotanche DA, Steyger PS. 2006. Uptake of fluorescent gentamicin by vertebrate sensory cells in vivo. Hear Res 213:64–78. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang M, Karasawa T, Steyger PS. 2017. Aminoglycoside-induced cochleotoxicity: a review. Front Cell Neurosci 11:308. 10.3389/fncel.2017.00308. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lopez-Novoa JM, Quiros Y, Vicente L, Morales AI, Lopez-Hernandez FJ. 2011. New insights into the mechanism of aminoglycoside nephrotoxicity: an integrative point of view. Kidney Int 79:33–45. [PubMed] [DOI] [PubMed] [Google Scholar]
- 26.Meyer RD. 1986. Risk factors and comparisons of clinical nephrotoxicity of aminoglycosides. Am J Med 80(6B):119–125. [DOI] [PubMed] [Google Scholar]
- 27.Mingeot-Leclercq MP, Tulkens PM. 1999. Aminoglycosides: nephrotoxicity. Antimicrob Agents Chemother 43:1003–1012. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nagai J, Takano M. 2014. Entry of aminoglycosides into renal tubular epithelial cells via endocytosis-dependent and endocytosis-independent pathways. Biochem Pharmacol 90:331–337. [PubMed] [DOI] [PubMed] [Google Scholar]
- 29.Fabre J, Rudhardt M, Blanchard P, Regamey C. 1976. Persistence of sisomicin and gentamicin in renal cortex and medulla compared with other organs and serum of rats. Kidney Int 10:444–449. [PubMed] [DOI] [PubMed] [Google Scholar]
- 30.Sandoval R, Leiser J, Molitoris BA. 1998. Aminoglycoside antibiotics traffic to the Golgi complex in LLC-PK1 cells. J Am Soc Nephrol 9:167–174. [PubMed] [DOI] [PubMed] [Google Scholar]
- 31.Garinis AC, Cross CP, Srikanth P, Carroll K, Feeney MP, Keefe DH, Hunter LL, Putterman DB, Cohen DM, Gold JA, Steyger PS. 2017. The cumulative effects of intravenous antibiotic treatments on hearing in patients with cystic fibrosis. J Cyst Fibros 16:401–409. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ariano RE, Zelenitsky SA, Kassum DA. 2008. Aminoglycoside-induced vestibular injury: maintaining a sense of balance. Ann Pharmacother 42:1282–1289. [PubMed] [DOI] [PubMed] [Google Scholar]
- 33.O’Sullivan ME, Perez A, Lin R, Sajjadi A, Ricci AJ, Cheng AG. 2017. Towards the prevention of aminoglycoside-related hearing loss. Front Cell Neurosci 11:325. 10.3389/fncel.2017.00325. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wong J, Brown G. 1996. Does once-daily dosing of aminoglycosides affect neuromuscular function? J Clin Pharm Ther 21:407–411. [PubMed] [DOI] [PubMed] [Google Scholar]
- 35.Barrons RW. 1997. Drug-induced neuromuscular blockade and myasthenia gravis. Pharmacotherapy 17:1220–1232. [PubMed] [PubMed] [Google Scholar]
- 36.Soloviev VN, Firsov AA, Dolgova GV, Berezhinskaya VV, Fishman VM. 1977. Relationship between the neuromuscular blocking effect of gentamicin and streptomycin and their concentration in blood. Acta Biol Med Ger 36:1307–1314. [PubMed] [PubMed] [Google Scholar]
- 37.Lee SI, Lee JH, Lee SC, Lee JM, Lee JH. 2008. Calcium and neostigmine antagonize gentamicin, but augment clindamycin-induced tetanic fade in rat phrenic nerve-hemidiaphragm preparations. J Anesth 22:385–390. [PubMed] [DOI] [PubMed] [Google Scholar]
- 38.Paradelis AG, Triantaphyllidis CJ, Mironidou M, Crassaris LG, Karachalios DN, Giala MM. 1988. Interaction of aminoglycoside antibiotics and calcium channel blockers at the neuromuscular junctions. Methods Find Exp Clin Pharmacol 10:687–690. [PubMed] [PubMed] [Google Scholar]
- 39.Le J, McKee B, Srisupha-Olarn W, Burgess DS. 2011. In vitro activity of carbapenems alone and in combination with amikacin against KPC-producing Klebsiella pneumoniae. J Clin Med Res 3:106–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rhodes A, Evans LE, Alhazzani W, Levy MM, Antonelli M, Ferrer R, Kumar A, Sevransky JE, Sprung CL, Nunnally ME, Rochwerg B, Rubenfeld GD, Angus DC, Annane D, Beale RJ, Bellinghan GJ, Bernard GR, Chiche JD, Coopersmith C, De Backer DP, French CJ, Fujishima S, Gerlach H, Hidalgo JL, Hollenberg SM, Jones AE, Karnad DR, Kleinpell RM, Koh Y, Lisboa TC, Machado FR, Marini JJ, Marshall JC, Mazuski JE, McIntyre LA, McLean AS, Mehta S, Moreno RP, Myburgh J, Navalesi P, Nishida O, Osborn TM, Perner A, Plunkett CM, Ranieri M, Schorr CA, Seckel MA, Seymour CW, Shieh L, Shukri KA, et al. 2017. Surviving sepsis campaign: international guidelines for management of sepsis and septic shock: 2016. Crit Care Med 45:486–552. [PubMed] [DOI] [PubMed] [Google Scholar]
- 41.Tamma PD, Cosgrove SE, Maragakis LL. 2012. Combination therapy for treatment of infections with gram-negative bacteria. Clin Microbiol Rev 25:450–470. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sick AC, Tschudin-Sutter S, Turnbull AE, Weissman SJ, Tamma PD. 2014. Empiric combination therapy for gram-negative bacteremia. Pediatrics 133:e1148–e1155. [PubMed] [DOI] [PubMed] [Google Scholar]
- 43.May AK. 2016. An argument for the use of aminoglycosides in the empiric treatment of ventilator-associated pneumonia. Surg Infect (Larchmt) 17:329–333. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nicolau DP, Belliveau PP, Nightingale CH, Quintiliani R, Freeman CD. 1995. Implementation of a once-daily aminoglycoside program in a large community-teaching hospital. Hosp Pharm 30:674–676, 679–680. [PubMed] [PubMed] [Google Scholar]
- 45.Drusano GL, Ambrose PG, Bhavnani SM, Bertino JS, Nafziger AN, Louie A. 2007. Back to the future: using aminoglycosides again and how to dose them optimally. Clin Infect Dis 45:753–760. [PubMed] [DOI] [PubMed] [Google Scholar]
- 46.Stankowicz MS, Ibrahim J, Brown DL. 2015. Once-daily aminoglycoside dosing: an update on current literature. Am J Health Syst Pharm 72:1357–1364. [PubMed] [DOI] [PubMed] [Google Scholar]
- 47.De Waele JJ, De Neve N. 2014. Aminoglycosides for life-threatening infections: a plea for an individualized approach using intensive therapeutic drug monitoring. Minerva Anestesiol 80:1135–1142. [PubMed] [PubMed] [Google Scholar]
- 48.Davis BD. 1987. Mechanism of bactericidal action of aminoglycosides. Microbiol Rev 51:341–350. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mingeot-Leclercq MP, Glupczynski Y, Tulkens PM. 1999. Aminoglycosides: activity and resistance. Antimicrob Agents Chemother 43:727–737. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Taber HW, Mueller JP, Miller PF, Arrow AS. 1987. Bacterial uptake of aminoglycoside antibiotics. Microbiol Rev 51:439–457. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Anand N, Davis BD. 1960. Damage by streptomycin to the cell membrane of Escherichia coli. Nature 185:22–23. [PubMed] [DOI] [PubMed] [Google Scholar]
- 52.Anand N, Davis BD, Armitage AK. 1960. Uptake of streptomycin by Escherichia coli. Nature 185:23–24. [PubMed] [DOI] [PubMed] [Google Scholar]
- 53.Bryan LE, Van Den Elzen HM. 1977. Effects of membrane-energy mutations and cations on streptomycin and gentamicin accumulation by bacteria: a model for entry of streptomycin and gentamicin in susceptible and resistant bacteria. Antimicrob Agents Chemother 12:163–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bryan LE, Van den Elzen HM. 1976. Streptomycin accumulation in susceptible and resistant strains of Escherichia coli and Pseudomonas aeruginosa. Antimicrob Agents Chemother 9:928–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stratton C. 2005. Molecular mechanisms of action for antimicrobial agents: general principles and mechanisms for selected classes of antibiotics, p 532–563. In Lorian V (ed). Antibiotics in Laboratory Medicine, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 56.Ramirez MS, Tolmasky ME. 2010. Aminoglycoside modifying enzymes. Drug Resist Updat 13:151–171. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schlessinger D. 1988. Failure of aminoglycoside antibiotics to kill anaerobic, low-pH, and resistant cultures. Clin Microbiol Rev 1:54–59. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jana S, Deb JK. 2006. Molecular understanding of aminoglycoside action and resistance. Appl Microbiol Biotechnol 70:140–150. [PubMed] [DOI] [PubMed] [Google Scholar]
- 59.Martin WJ, Gardner M, Washington JA II. 1972. In vitro antimicrobial susceptibility of anaerobic bacteria isolated from clinical specimens. Antimicrob Agents Chemother 1:148–158. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kislak JW. 1972. The susceptibility of Bacteroides fragilis to 24 antibiotics. J Infect Dis 125:295–299. [PubMed] [DOI] [PubMed] [Google Scholar]
- 61.Nichols WW, Young SN. 1985. Respiration-dependent uptake of dihydrostreptomycin by Escherichia coli. Its irreversible nature and lack of evidence for a uniport process. Biochem J 228:505–512. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Davis BD, Chen LL, Tai PC. 1986. Misread protein creates membrane channels: an essential step in the bactericidal action of aminoglycosides. Proc Natl Acad Sci USA 83:6164–6168. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yoshimura F, Nikaido H. 1985. Diffusion of beta-lactam antibiotics through the porin channels of Escherichia coli K-12. Antimicrob Agents Chemother 27:84–92. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Harder KJ, Nikaido H, Matsuhashi M. 1981. Mutants of Escherichia coli that are resistant to certain beta-lactam compounds lack the ompF porin. Antimicrob Agents Chemother 20:549–552. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hirai K, Aoyama H, Irikura T, Iyobe S, Mitsuhashi S. 1986. Differences in susceptibility to quinolones of outer membrane mutants of Salmonella typhimurium and Escherichia coli. Antimicrob Agents Chemother 29:535–538. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Thanassi DG, Suh GS, Nikaido H. 1995. Role of outer membrane barrier in efflux-mediated tetracycline resistance of Escherichia coli. J Bacteriol 177:998–1007. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Delcour AH. 2009. Outer membrane permeability and antibiotic resistance. Biochim Biophys Acta 1794:808–816. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pagès JM, James CE, Winterhalter M. 2008. The porin and the permeating antibiotic: a selective diffusion barrier in Gram-negative bacteria. Nat Rev Microbiol 6:893–903. [PubMed] [DOI] [PubMed] [Google Scholar]
- 69.Fei Y, Ma V, Maerkl N, You P. 2012. The down regulation of E. coli OmpF in response to sub-inhibitory concentrations of kanamycin is not mediated by MarA. J ExpMicrobiol Immunol 16:101–107. [Google Scholar]
- 70.Foulds J, Chai TJ. 1978. New major outer membrane proteins found in an Escherichia coli tolF mutant resistant to bacteriophage TuIb. J Bacteriol 133:1478–1483. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nakae R, Nakae T. 1982. Diffusion of aminoglycoside antibiotics across the outer membrane of Escherichia coli. Antimicrob Agents Chemother 22:554–559. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Laursen BS, Sørensen HP, Mortensen KK, Sperling-Petersen HU. 2005. Initiation of protein synthesis in bacteria. Microbiol Mol Biol Rev 69:101–123. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lodish H, Berk A, Zipursky AL, Matsudaira P, Baltimore D, Darnell J. 2000. Molecular Cell Biology, 4th ed. W. H. Freeman, New York, NY. [Google Scholar]
- 74.Green R, Noller HF. 1997. Ribosomes and translation. Annu Rev Biochem 66:679–716. [PubMed] [DOI] [PubMed] [Google Scholar]
- 75.Kotra LP, Haddad J, Mobashery S. 2000. Aminoglycosides: perspectives on mechanisms of action and resistance and strategies to counter resistance. Antimicrob Agents Chemother 44:3249–3256. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fourmy D, Recht MI, Blanchard SC, Puglisi JD. 1996. Structure of the A site of Escherichia coli 16S ribosomal RNA complexed with an aminoglycoside antibiotic. Science 274:1367–1371. [PubMed] [DOI] [PubMed] [Google Scholar]
- 77.Fourmy D, Yoshizawa S, Puglisi JD. 1998. Paromomycin binding induces a local conformational change in the A-site of 16 S rRNA. J Mol Biol 277:333–345. [PubMed] [DOI] [PubMed] [Google Scholar]
- 78.Carter AP, Clemons WM, Brodersen DE, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. 2000. Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature 407:340–348. [PubMed] [DOI] [PubMed] [Google Scholar]
- 79.Davies J, Anderson P, Davis BD. 1965. Inhibition of protein synthesis by spectinomycin. Science 149:1096–1098. [PubMed] [DOI] [PubMed] [Google Scholar]
- 80.Wilson DN. 2014. Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nat Rev Microbiol 12:35–48. [PubMed] [DOI] [PubMed] [Google Scholar]
- 81.Mehta R, Champney WS. 2002. 30S ribosomal subunit assembly is a target for inhibition by aminoglycosides in Escherichia coli. Antimicrob Agents Chemother 46:1546–1549. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wallace BJ, Tai PC, Herzog EL, Davis BD. 1973. Partial inhibition of polysomal ribosomes of Escherichia coli by streptomycin. Proc Natl Acad Sci USA 70:1234–1237. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tai PC, Wallace BJ, Davis BD. 1973. Actions of aurintricarboxylate, kasugamycin, and pactamycin on Escherichia coli polysomes. Biochemistry 12:616–620. [PubMed] [DOI] [PubMed] [Google Scholar]
- 84.Roth H, Amos H, Davis BD. 1960. Purine nucleotide excretion by Escherichia coli in the presence of streptomycin. Biochim Biophys Acta 37:398–405. [DOI] [PubMed] [Google Scholar]
- 85.Tai PC, Wallace BJ, Davis BD. 1978. Streptomycin causes misreading of natural messenger by interacting with ribosomes after initiation. Proc Natl Acad Sci USA 75:275–279. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Magnet S, Blanchard JS. 2005. Molecular insights into aminoglycoside action and resistance. Chem Rev 105:477–498. [PubMed] [DOI] [PubMed] [Google Scholar]
- 87.Doi Y, Wachino JI, Arakawa Y. 2016. Aminoglycoside resistance: the emergence of acquired 16S ribosomal RNA methyltransferases. Infect Dis Clin North Am 30:523–537. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rather PN, Orosz E, Shaw KJ, Hare R, Miller G. 1993. Characterization and transcriptional regulation of the 2′-N-acetyltransferase gene from Providencia stuartii. J Bacteriol 175:6492–6498. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cundliffe E. 1989. How antibiotic-producing organisms avoid suicide. Annu Rev Microbiol 43:207–233. [PubMed] [DOI] [PubMed] [Google Scholar]
- 90.Rather PN. 1998. Origins of the aminoglycoside modifying enzymes. Drug Resist Updat 1:285–291. [DOI] [PubMed] [Google Scholar]
- 91.Beauclerk AA, Cundliffe E. 1987. Sites of action of two ribosomal RNA methylases responsible for resistance to aminoglycosides. J Mol Biol 193:661–671. [DOI] [PubMed] [Google Scholar]
- 92.Garneau-Tsodikova S, Labby KJ. 2016. Mechanisms of resistance to aminoglycoside antibiotics: overview and perspectives. MedChemComm 7:11–27. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kim JY, Park YJ, Kwon HJ, Han K, Kang MW, Woo GJ. 2008. Occurrence and mechanisms of amikacin resistance and its association with beta-lactamases in Pseudomonas aeruginosa: a Korean nationwide study. J Antimicrob Chemother 62:479–483. [PubMed] [DOI] [PubMed] [Google Scholar]
- 94.Castanheira M, Deshpande L, Hubler C, Mendes R, Serio A, Krause K, Flamm R. 2017. Activity of plazomicin against Enterobacteriaceae isolates collected in the United States including isolates carrying aminoglycoside-modifying enzymes detected by whole genome sequencing, abstr. 1235. IDWeek 2017, San Diego, CA, 4 to 8 October, 2017. [Google Scholar]
- 95.López Díaz M, Ríos E, Rodríguez-Avial I, Simaluiza RJ, Picazo JJ, Culebras E. 2017. In-vitro activity of several antimicrobial agents against methicillin-resistant Staphylococcus aureus (MRSA) isolates expressing aminoglycoside-modifying enzymes: potency of plazomicin alone and in combination with other agents. Int J Antimicrob Agents 50:191–196. [PubMed] [DOI] [PubMed] [Google Scholar]
- 96.Matsumoto T. 2014. Arbekacin: another novel agent for treating infections due to methicillin-resistant Staphylococcus aureus and multidrug-resistant Gram-negative pathogens. Clin Pharmacol 6:139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kondo S, Hotta K. 1999. Semisynthetic aminoglycoside antibiotics: development and enzymatic modifications. J Infect Chemother 5:1–9. [PubMed] [DOI] [PubMed] [Google Scholar]
- 98.Cox G, Ejim L, Stogios PJ, Koteva K, Bordeleau E, Evdokimova E, Sieron AO, Savchenko A, Serio AW, Krause KM, Wright GD. 2018. Plazomicin retains antibiotic activity against most aminoglycoside modifying enzymes. ACS Infect Dis 4:980–987. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ramirez MS, Nikolaidis N, Tolmasky ME. 2013. Rise and dissemination of aminoglycoside resistance: the aac(6′)-Ib paradigm. Front Microbiol 4:121. 10.3389/fmicb.2013.00121. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Robicsek A, Strahilevitz J, Jacoby GA, Macielag M, Abbanat D, Park CH, Bush K, Hooper DC. 2006. Fluoroquinolone-modifying enzyme: a new adaptation of a common aminoglycoside acetyltransferase. Nat Med 12:83–88. [PubMed] [DOI] [PubMed] [Google Scholar]
- 101.Castanheira M, Doyle T, Woosley L, Serio A, Krause K, Flamm R. 2017. Plazomicin activity against European Enterobacteriaceae isolates carrying aminoglycoside-modifying enzymes and 16S rRNA methylases, abstr. 3611. ECCMID, Vienna, Austria, 22 to 25 April 2017. [Google Scholar]
- 102.Castanheira M, Woosley L, Doyle T, Serio A, Krause K, Flamm R. 2017. Aminoglycoside-resistance genes among 2014-2015 US carbapenem-resistant enterobacteriaceae isolates and activity of plazomicin against characterized isolates. ASM Microbe 2017, New Orleans, LA, 1 to 5 June 2017. [Google Scholar]
- 103.Almaghrabi R, Clancy CJ, Doi Y, Hao B, Chen L, Shields RK, Press EG, Iovine NM, Townsend BM, Wagener MM, Kreiswirth B, Nguyen MH. 2014. Carbapenem-resistant Klebsiella pneumoniae strains exhibit diversity in aminoglycoside-modifying enzymes, which exert differing effects on plazomicin and other agents. Antimicrob Agents Chemother 58:4443–4451. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Holbrook SYL, Garneau-Tsodikova S. 2017. Evaluation of aminoglycoside and carbapenem resistance in a collection of drug-resistant Pseudomonas aeruginosa clinical isolates. Microb Drug Resist 24:1020–1030. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Aghazadeh M, Rezaee MA, Nahaei MR, Mahdian R, Pajand O, Saffari F, Hassan M, Hojabri Z. 2013. Dissemination of aminoglycoside-modifying enzymes and 16S rRNA methylases among Acinetobacter baumannii and Pseudomonas aeruginosa isolates. Microb Drug Resist 19:282–288. [PubMed] [DOI] [PubMed] [Google Scholar]
- 106.Liu Z, Ling B, Zhou L. 2015. Prevalence of 16S rRNA methylase, modifying enzyme, and extended-spectrum beta-lactamase genes among Acinetobacter baumannii isolates. J Chemother 27:207–212. [PubMed] [DOI] [PubMed] [Google Scholar]
- 107.Miró E, Grünbaum F, Gómez L, Rivera A, Mirelis B, Coll P, Navarro F. 2013. Characterization of aminoglycoside-modifying enzymes in enterobacteriaceae clinical strains and characterization of the plasmids implicated in their diffusion. Microb Drug Resist 19:94–99. [PubMed] [DOI] [PubMed] [Google Scholar]
- 108.Tsai SF, Zervos MJ, Clewell DB, Donabedian SM, Sahm DF, Chow JW. 1998. A new high-level gentamicin resistance gene, aph(2′)-Id, in Enterococcus spp. Antimicrob Agents Chemother 42:1229–1232. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Perumal N, Murugesan S, Krishnan P. 2016. Distribution of genes encoding aminoglycoside-modifying enzymes among clinical isolates of methicillin-resistant staphylococci. Indian J Med Microbiol 34:350–352. [PubMed] [DOI] [PubMed] [Google Scholar]
- 110.Mahdiyoun SM, Kazemian H, Ahanjan M, Houri H, Goudarzi M. 2016. Frequency of aminoglycoside-resistance genes in methicillin-resistant Staphylococcus aureus (MRSA) isolates from hospitalized patients. Jundishapur J Microbiol 9:e35052. 10.5812/jjm.35052. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wright GD. 1999. Aminoglycoside-modifying enzymes. Curr Opin Microbiol 2:499–503. [DOI] [PubMed] [Google Scholar]
- 112.Wachino J, Arakawa Y. 2012. Exogenously acquired 16S rRNA methyltransferases found in aminoglycoside-resistant pathogenic Gram-negative bacteria: an update. Drug Resist Updat 15:133–148. [PubMed] [DOI] [PubMed] [Google Scholar]
- 113.Livermore DM, Mushtaq S, Warner M, Zhang JC, Maharjan S, Doumith M, Woodford N. 2011. Activity of aminoglycosides, including ACHN-490, against carbapenem-resistant Enterobacteriaceae isolates. J Antimicrob Chemother 66:48–53. [PubMed] [DOI] [PubMed] [Google Scholar]
- 114.Thompson J, Skeggs PA, Cundliffe E. 1985. Methylation of 16S ribosomal RNA and resistance to the aminoglycoside antibiotics gentamicin and kanamycin determined by DNA from the gentamicin-producer, Micromonospora purpurea. Mol Gen Genet 201:168–173. [PubMed] [DOI] [PubMed] [Google Scholar]
- 115.Skeggs PA, Thompson J, Cundliffe E. 1985. Methylation of 16S ribosomal RNA and resistance to aminoglycoside antibiotics in clones of Streptomyces lividans carrying DNA from Streptomyces tenjimariensis. Mol Gen Genet 200:415–421. [PubMed] [DOI] [PubMed] [Google Scholar]
- 116.Yokoyama K, Doi Y, Yamane K, Kurokawa H, Shibata N, Shibayama K, Yagi T, Kato H, Arakawa Y. 2003. Acquisition of 16S rRNA methylase gene in Pseudomonas aeruginosa. Lancet 362:1888–1893. [DOI] [PubMed] [Google Scholar]
- 117.Wachino J, Shibayama K, Kurokawa H, Kimura K, Yamane K, Suzuki S, Shibata N, Ike Y, Arakawa Y. 2007. Novel plasmid-mediated 16S rRNA m1A1408 methyltransferase, NpmA, found in a clinically isolated Escherichia coli strain resistant to structurally diverse aminoglycosides. Antimicrob Agents Chemother 51:4401–4409. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Al Sheikh YA, Marie MA, John J, Krishnappa LG, Dabwab KH. 2014. Prevalence of 16S rRNA methylase genes among β-lactamase-producing Enterobacteriaceae clinical isolates in Saudi Arabia. Libyan J Med 9:24432. 10.3402/ljm.v9.24432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Potron A, Poirel L, Nordmann P. 2015. Emerging broad-spectrum resistance in Pseudomonas aeruginosa and Acinetobacter baumannii: mechanisms and epidemiology. Int J Antimicrob Agents 45:568–585. [PubMed] [DOI] [PubMed] [Google Scholar]
- 120.Zhou Y, Yu H, Guo Q, Xu X, Ye X, Wu S, Guo Y, Wang M. 2010. Distribution of 16S rRNA methylases among different species of Gram-negative bacilli with high-level resistance to aminoglycosides. Eur J Clin Microbiol Infect Dis 29:1349–1353. [PubMed] [DOI] [PubMed] [Google Scholar]
- 121.Strateva T, Markova B, Markovska R, Marteva-Proevska Y, Ivanova D, Mitov I. 2011. Emergence of 16s rRNA methylase-producing nosocomial Acinetobacter baumannii isolates in a university hospital in Bulgaria. J Chemother 23:374–375. [PubMed] [DOI] [PubMed] [Google Scholar]
- 122.Poole K. 2011. Pseudomonas aeruginosa: resistance to the max. Front Microbiol 2:65. 10.3389/fmicb.2011.00065. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zacharczuk K, Piekarska K, Szych J, Jagielski M, Hidalgo L, San Millán A, Gutiérrez B, Rastawicki W, González-Zorn B, Gierczynski R. 2011. Plasmid-borne 16S rRNA methylase ArmA in aminoglycoside-resistant Klebsiella pneumoniae in Poland. J Med Microbiol 60:1306–1311. [PubMed] [DOI] [PubMed] [Google Scholar]
- 124.Zacharczuk K, Piekarska K, Szych J, Zawidzka E, Sulikowska A, Wardak S, Jagielski M, Gierczynski R. 2011. Emergence of Klebsiella pneumoniae coproducing KPC-2 and 16S rRNA methylase ArmA in Poland. Antimicrob Agents Chemother 55:443–446. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Park YJ, Lee S, Yu JK, Woo GJ, Lee K, Arakawa Y. 2006. Co-production of 16S rRNA methylases and extended-spectrum beta-lactamases in AmpC-producing Enterobacter cloacae, Citrobacter freundii and Serratia marcescens in Korea. J Antimicrob Chemother 58:907–908. [PubMed] [DOI] [PubMed] [Google Scholar]
- 126.Bercovier H, Kafri O, Sela S. 1986. Mycobacteria possess a surprisingly small number of ribosomal RNA genes in relation to the size of their genome. Biochem Biophys Res Commun 136:1136–1141. [DOI] [PubMed] [Google Scholar]
- 127.Schwartz JJ, Gazumyan A, Schwartz I. 1992. rRNA gene organization in the Lyme disease spirochete, Borrelia burgdorferi. J Bacteriol 174:3757–3765. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Honoré N, Marchal G, Cole ST. 1995. Novel mutation in 16S rRNA associated with streptomycin dependence in Mycobacterium tuberculosis. Antimicrob Agents Chemother 39:769–770. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Meier A, Kirschner P, Bange FC, Vogel U, Böttger EC. 1994. Genetic alterations in streptomycin-resistant Mycobacterium tuberculosis: mapping of mutations conferring resistance. Antimicrob Agents Chemother 38:228–233. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Finken M, Kirschner P, Meier A, Wrede A, Böttger EC. 1993. Molecular basis of streptomycin resistance in Mycobacterium tuberculosis: alterations of the ribosomal protein S12 gene and point mutations within a functional 16S ribosomal RNA pseudoknot. Mol Microbiol 9:1239–1246. [PubMed] [DOI] [PubMed] [Google Scholar]
- 131.Prammananan T, Sander P, Brown BA, Frischkorn K, Onyi GO, Zhang Y, Böttger EC, Wallace RJ Jr. 1998. A single 16S ribosomal RNA substitution is responsible for resistance to amikacin and other 2-deoxystreptamine aminoglycosides in Mycobacterium abscessus and Mycobacterium chelonae. J Infect Dis 177:1573–1581. [PubMed] [DOI] [PubMed] [Google Scholar]
- 132.Criswell D, Tobiason VL, Lodmell JS, Samuels DS. 2006. Mutations conferring aminoglycoside and spectinomycin resistance in Borrelia burgdorferi. Antimicrob Agents Chemother 50:445–452. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Li XZ, Plésiat P, Nikaido H. 2015. The challenge of efflux-mediated antibiotic resistance in Gram-negative bacteria. Clin Microbiol Rev 28:337–418. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Poole K. 2004. Efflux-mediated multiresistance in Gram-negative bacteria. Clin Microbiol Infect 10:12–26. [PubMed] [DOI] [PubMed] [Google Scholar]
- 135.Islam S, Jalal S, Wretlind B. 2004. Expression of the MexXY efflux pump in amikacin-resistant isolates of Pseudomonas aeruginosa. Clin Microbiol Infect 10:877–883. [PubMed] [DOI] [PubMed] [Google Scholar]
- 136.Magnet S, Courvalin P, Lambert T. 2001. Resistance-nodulation-cell division-type efflux pump involved in aminoglycoside resistance in Acinetobacter baumannii strain BM4454. Antimicrob Agents Chemother 45:3375–3380. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Rosenberg EY, Ma D, Nikaido H. 2000. AcrD of Escherichia coli is an aminoglycoside efflux pump. J Bacteriol 182:1754–1756. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Moore RA, DeShazer D, Reckseidler S, Weissman A, Woods DE. 1999. Efflux-mediated aminoglycoside and macrolide resistance in Burkholderia pseudomallei. Antimicrob Agents Chemother 43:465–470. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Poole K. 2005. Efflux-mediated antimicrobial resistance. J Antimicrob Chemother 56:20–51. [PubMed] [DOI] [PubMed] [Google Scholar]
- 140.Muller C, Plésiat P, Jeannot K. 2011. A two-component regulatory system interconnects resistance to polymyxins, aminoglycosides, fluoroquinolones, and β-lactams in Pseudomonas aeruginosa. Antimicrob Agents Chemother 55:1211–1221. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Singh M, Yau YCW, Wang S, Waters V, Kumar A. 2017. MexXY efflux pump overexpression and aminoglycoside resistance in cystic fibrosis isolates of Pseudomonas aeruginosa from chronic infections. Can J Microbiol 63:929–938. [PubMed] [DOI] [PubMed] [Google Scholar]
- 142.Yoon EJ, Courvalin P, Grillot-Courvalin C. 2013. RND-type efflux pumps in multidrug-resistant clinical isolates of Acinetobacter baumannii: major role for AdeABC overexpression and AdeRS mutations. Antimicrob Agents Chemother 57:2989–2995. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Coyne S, Courvalin P, Périchon B. 2011. Efflux-mediated antibiotic resistance in Acinetobacter spp. Antimicrob Agents Chemother 55:947–953. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zhao ZP, Liu TT, Zhang L, Luo M, Nie X, Li ZX, Pan Y. 2014. High-grade mutant OmpF induces decreased bacterial survival rate. Acta Biochim Pol 61:369–373. [PubMed] [PubMed] [Google Scholar]
- 145.Bryan LE, Kowand SK, Van Den Elzen HM. 1979. Mechanism of aminoglycoside antibiotic resistance in anaerobic bacteria: Clostridium perfringens and Bacteroides fragilis. Antimicrob Agents Chemother 15:7–13. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lin YT, Huang YW, Liou RS, Chang YC, Yang TC. 2014. MacABCsm, an ABC-type tripartite efflux pump of Stenotrophomonas maltophilia involved in drug resistance, oxidative and envelope stress tolerances and biofilm formation. J Antimicrob Chemother 69:3221–3226. [PubMed] [DOI] [PubMed] [Google Scholar]
- 147.Buroni S, Pasca MR, Flannagan RS, Bazzini S, Milano A, Bertani I, Venturi V, Valvano MA, Riccardi G. 2009. Assessment of three resistance-nodulation-cell division drug efflux transporters of Burkholderia cenocepacia in intrinsic antibiotic resistance. BMC Microbiol 9:200. 10.1186/1471-2180-9-200. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Jassem AN, Zlosnik JE, Henry DA, Hancock RE, Ernst RK, Speert DP. 2011. In vitro susceptibility of Burkholderia vietnamiensis to aminoglycosides. Antimicrob Agents Chemother 55:2256–2264. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bador J, Amoureux L, Blanc E, Neuwirth C. 2013. Innate aminoglycoside resistance of Achromobacter xylosoxidans is due to AxyXY-OprZ, an RND-type multidrug efflux pump. Antimicrob Agents Chemother 57:603–605. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Li XZ, Zhang L, McKay GA, Poole K. 2003. Role of the acetyltransferase AAC(6′)-Iz modifying enzyme in aminoglycoside resistance in Stenotrophomonas maltophilia. J Antimicrob Chemother 51:803–811. [PubMed] [DOI] [PubMed] [Google Scholar]
- 151.Costa Y, Galimand M, Leclercq R, Duval J, Courvalin P. 1993. Characterization of the chromosomal aac(6′)-Ii gene specific for Enterococcus faecium. Antimicrob Agents Chemother 37:1896–1903. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Galimand M, Schmitt E, Panvert M, Desmolaize B, Douthwaite S, Mechulam Y, Courvalin P. 2011. Intrinsic resistance to aminoglycosides in Enterococcus faecium is conferred by the 16S rRNA m5C1404-specific methyltransferase EfmM. RNA 17:251–262. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Cloutier D, Miller L, Komirenko A, Cebrik D, Keepers T, Krause K, Connolly L, Wagenlehner F. 2017. Evaluating once-daily plazomicin versus meropenem for the treatment of complicated urinary tract infection (cUTI) and acute pyelonephritis (AP): results from a phase 3 study (EPIC). ASM Microbe 2017, New Orleans, LA, 1 to 5 June 2017. [Google Scholar]
- 154.ZEMDRI™ (plazomicin). 2018. Full Prescribing Information. Achaogen Inc., South San Francisco, CA. [Google Scholar]
- 155.Connolly LE, Jubb A, O’Keeffe B, Serio A, Smith A, Gall J, Riddle V, Krause K, McKinnell J, Zakynthinos E, Daikos G. 2017. Plazomicin (PLZ) associated with improved survival and safety compared to colistin (CST) in the treatment of serious infections due to carbapenem-resistant Enterobacteriaceae (CRE): results of the CARE study. ASM Microbe 2017, New Orleans, LA, 1 to 5 June 2017. [Google Scholar]
- 156.Zárate G, De la Cruz Claure ML, Benito-Arenas R, Revuelta J, Santana AG, Bastida A. 2018. Overcoming aminoglycoside enzymatic resistance: design of novel antibiotics and inhibitors. Molecules 23:E284. 10.3390/molecules23020284. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Hobbie SN, Kalapala SK, Akshay S, Bruell C, Schmidt S, Dabow S, Vasella A, Sander P, Böttger EC. 2007. Engineering the rRNA decoding site of eukaryotic cytosolic ribosomes in bacteria. Nucleic Acids Res 35:6086–6093. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Mandhapati AR, Shcherbakov D, Duscha S, Vasella A, Böttger EC, Crich D. 2014. Importance of the 6′-hydroxy group and its configuration for apramycin activity. ChemMedChem 9:2074–2083. [PubMed] [DOI] [PubMed] [Google Scholar]
- 159.Perez-Fernandez D, Shcherbakov D, Matt T, Leong NC, Kudyba I, Duscha S, Boukari H, Patak R, Dubbaka SR, Lang K, Meyer M, Akbergenov R, Freihofer P, Vaddi S, Thommes P, Ramakrishnan V, Vasella A, Böttger EC. 2014. 4′-O-substitutions determine selectivity of aminoglycoside antibiotics. Nat Commun 5:3112. 10.1038/ncomms4112. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Prokhorova I, Altman RB, Djumagulov M, Shrestha JP, Urzhumtsev A, Ferguson A, Chang CT, Yusupov M, Blanchard SC, Yusupova G. 2017. Aminoglycoside interactions and impacts on the eukaryotic ribosome. Proc Natl Acad Sci USA 114:E10899–E10908. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Greber BJ, Bieri P, Leibundgut M, Leitner A, Aebersold R, Boehringer D, Ban N. 2015. Ribosome. The complete structure of the 55S mammalian mitochondrial ribosome. Science 348:303–308. [PubMed] [DOI] [PubMed] [Google Scholar]
- 162.Chandrika NT, Garneau-Tsodikova S. 2016. A review of patents (2011-2015) towards combating resistance to and toxicity of aminoglycosides. MedChemComm 7:50–68. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Houghton JL, Green KD, Chen W, Garneau-Tsodikova S. 2010. The future of aminoglycosides: the end or renaissance? ChemBioChem 11:880–902. [PubMed] [DOI] [PubMed] [Google Scholar]
- 164.Dozzo P, Moser HE. 2010. New aminoglycoside antibiotics. Expert Opin Ther Pat 20:1321–1341. 10.1517/13543776.2010.506189. [PubMed] [DOI] [PubMed] [Google Scholar]
- 165.Takahashi Y, Umemura E, Kobayashi Y, Murakami S, Nawa T, Morinaka A, Miyake T, Shibasaki M. 2018. Discovery of 2-hydroxyarbekacin, a new aminoglycoside antibiotic with reduced nephrotoxicity. J Antibiot (Tokyo) 71:345–347. [PubMed] [DOI] [PubMed] [Google Scholar]
- 166.Sonousi A, Sarpe VA, Brilkova M, Schacht J, Vasella A, Böttger EC, Crich D. 2018. Effects of the 1-N-(4-Amino-2 S-hydroxybutyryl) and 6′-N-(2-Hydroxyethyl) substituents on ribosomal selectivity, cochleotoxicity, and antibacterial activity in the sisomicin class of aminoglycoside antibiotics. ACS Infect Dis 4:1114–1120. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Sati GC, Shcherbakov D, Hobbie SN, Vasella A, Böttger EC, Crich D. 2017. N6′, N6″, and O4′ Modifications to neomycin affect ribosomal selectivity without compromising antibacterial activity. ACS Infect Dis 3:368–377. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Duscha S, Boukari H, Shcherbakov D, Salian S, Silva S, Kendall A, Kato T, Akbergenov R, Perez-Fernandez D, Bernet B, Vaddi S, Thommes P, Schacht J, Crich D, Vasella A, Böttger EC. 2014. Identification and evaluation of improved 4′-O-(alkyl) 4,5-disubstituted 2-deoxystreptamines as next-generation aminoglycoside antibiotics. MBio 5:e01827-14. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Umemura E, Takahashi Y, Igarashi M, Hayashi C, Shibasaki M, Yamada K, Ida T, Yonezawa M, Ago K. TS3112, a novel aminoglycoside antibiotic active against multidrug-resistant pathogens producing 16S rRNA methyltransferases: synthesis and structure-activity relationships. ASM Microbe 2017, New Orleans, LA, 1 to 5 June 2017. [Google Scholar]
- 170.Yamada K, Takata T, Takayama Y, Senju N, Tabata Y, Ida T, Yonezawa M, Ago K. 2017. TS3112, a novel aminoglycoside antibiotic: in vitro and in vivo activity against multiple drug-resistant gram-positive and gram-negative pathogens. ASM Microbe 2017, New Orleans, LA, 1 to 5 June 2017. [Google Scholar]
- 171.Krause KM, Serio AW, Kane TR, Connolly LE. 2016. Aminoglycosides: an overview. Cold Spring Harb Perspect Med 6:a027029. 10.1101/cshperspect.a027029. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]