

Abstract
The transition element molybdenum (Mo) is of primordial importance for biological systems, because it is required by enzymes catalyzing key reactions in the global carbon, sulfur, and nitrogen metabolism. To gain biological activity, Mo has to be complexed by a special cofactor. With the exception of bacterial nitrogenase, all Mo-dependent enzymes contain a unique pyranopterin-based cofactor coordinating a Mo atom at their catalytic site. Various types of reactions are catalyzed by Mo-enzymes in prokaryotes including oxygen atom transfer, sulfur or proton transfer, hydroxylation, or even nonredox reactions. Mo-enzymes are widespread in prokaryotes and many of them were likely present in the Last Universal Common Ancestor. To date, more than 50 – mostly bacterial – Mo-enzymes are described in nature. In a few eubacteria and in many archaea, Mo is replaced by tungsten bound to the same unique pyranopterin. How Mo-cofactor is synthesized in bacteria is reviewed as well as the way until its insertion into apo-Mo-enzymes.
INTRODUCTION
The transition element molybdenum (Mo) is of essential importance for biological systems because it is required by enzymes catalyzing key reactions in the global carbon, sulfur, and nitrogen metabolism (for recent reviews, see references 1 and 2). But tungsten is also biologically important. Both elements have a very rich redox chemistry, which might explain why they are the only members of the second (molybdenum) and third (tungsten) transition series with known biological functions. Molybdenum is very abundant in the oceans in the form of the molybdate anion (MoO42–), whereas the tungsten concentration (as tungstate WoO42–) is 100-fold lower (3). Under anaerobic conditions and high sulfur concentrations that prevail in deep-sea hydrothermal vents, molybdenum occurs as MoS2 and thus becomes unavailable for biological systems. This is the site where tungsten-using extremophilic bacteria (archaea) were found. In soils, the oxidation state of molybdenum varies from II to VI, but only the soluble molybdate anion is bioavailable.
In order to gain biological activity, Mo has to be complexed by a special cofactor. With the exception of bacterial nitrogenase, all Mo-dependent enzymes utilize a molybdenum cofactor (Moco) consisting of a mononuclear Mo atom coordinated via a cis-dithiolene moiety to the organic molecule pyranopterin (PPT) (formerly called molybdopterin or MPT) at their catalytic site (4). Accordingly, an extreme conservation of the Moco biosynthetic machinery (see below) is observed. More than 50 different Mo/W enzymes have been described in nature so far. The molybdoproteome might even be more diverse than previously recognized in prokaryotes as suggested by the study of Cvetkovic et al. (5) revealing the existence of several Mo-binding proteins with unrelated sequence homology to any known Mo-enzymes. In a few eubacteria and in many archaea, Mo is replaced by tungsten bound to the same unique pterin (see for instance, reference 6). With the exception of nitrogenase, Mo or W ions in prokaryotes are coordinated by either one or two pyranopterins allowing categorization to three families based on the distinct coordination pattern of the metal ion: the sulfite oxidase (SO) family, the xanthine oxidase (XO) family, and the dimethyl sulfoxide (DMSO) reductase family. For enzymes of the SO family, the Mo is coordinated by a single PPT molecule. A clear distinction with members of the XO family is the additional presence of an inorganic sulfur at the Mo center further coordinated by a PPT or a cytosine-substituted PPT. Finally, members of the DMSO reductase family have a Mo atom coordinated by two guanosine-substituted PPTs. These latter enzymes are extraordinarily diverse in terms of structure and/or subunit composition, but were named, referring to the first representative that was crystallized (7), as members of the DMSO reductase family. However, the latter enzyme represents an exception rather than the rule among this family in terms of structure and subunit composition, and this denomination furthermore refers to a precise enzymatic activity (2). The name Complex Iron–Sulfur Molybdoenzyme (CISM) superfamily was later introduced for heterotrimeric enzymes harboring a Mo/W-bisPGD cofactor in their catalytic subunit and containing both a FeS subunit and a membrane anchor one (8). Again, today, exceptions are more frequent than the rule. We therefore recommend the use of the “Mo/W-bisPGD” denomination instead of “DMSO reductase” for a safe description of the actual recognized diversity of this large enzyme family exclusively found in prokaryotes (2, 9).
In this context, Escherichia coli represents a unique situation in having members of all three families (Fig. 1). Most Mo-enzymes belong to the Mo/W-bisPGD family, and, only a few years ago, members of the SO (i.e., YedY) or XO families (i.e., PaoABCD, XdhABC, and XdhD) were identified.
Figure 1.
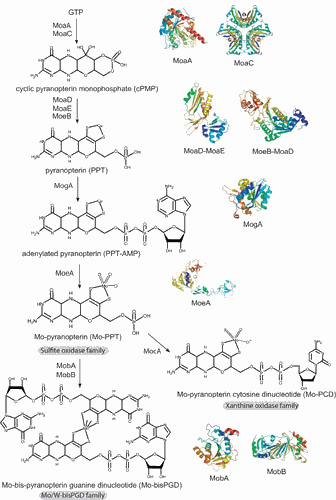
Molybdenum cofactor biosynthesis in E. coli. Shown are the known biosynthetic intermediates dividing the whole pathway into four steps and giving rise to the different forms of cofactor found in the three distinct Mo-enzyme families: the Mo/W-bisPGD family, the sulfite oxidase family, and the xanthine oxidase family. Ribbon representation of the crystal structures of the Moco biosynthetic proteins are shown: MoaA (56), MoaC (58), MoaD-MoaE complex (63), MoeB-MoaD complex (68), MogA (91), MoeA (222), MobA (111, 112), and MobB (114). Individual figures were generated with PYMOL (223) using the deposited coordinates from the protein structure data base. doi:10.1128/ecosalplus.ESP-0006-2013.f1
In this article, we will review how Moco is synthesized and follow the way until its insertion into apo-Mo-enzymes. For the W-enzymes it is assumed that the formation of the W cofactor follows the same principles as outlined for bacterial Moco (6). At the same time, very restrictive reactions should enable a clear discrimination between these related metals because they are both bioavailable and known to often represent antagonists for each other (10). As such, exquisitely discriminating systems have evolved at the levels of metal uptake into bacterial cells, metal insertion into the cofactor, and the possible interplay of Mo-enzyme specific chaperones with cofactor incorporation.
MOLYBDENUM COFACTOR BIOSYNTHESIS IN E. COLI
Genetics of Molybdenum Cofactor Biosynthesis
The investigation of Mo metabolism started with the genetic analysis of mutants of the filamentous fungus Aspergillus nidulans (11) that were defective in nitrate reductase. Cove and Pateman (12) isolated nitrate reductase-deficient mutants that showed concurrently the loss of two Mo-dependent enzymes, nitrate reductase and xanthine dehydrogenase. Because Mo was the only link between these two otherwise very different enzymes, it was suggested that both enzymes should share a common Mo-related cofactor, named molybdenum cofactor. Later, Johnson et al. (13) demonstrated that the organic compound of Moco from different Mo-enzymes is a unique pterin, which they called molybdopterin.
In parallel to the achievements of fungal biochemical genetics, also in E. coli, Moco mutants had been isolated by using the same selection principle: growth of mutagenized cells in the presence of high concentrations of chlorate. Mutants selected for chlorate resistance (chl) do not reduce chlorate to the toxic chlorite because they have lost chlorate reductase activity, which appears to be a nonphysiological catalytic activity of the Moco-dependent nitrate reductase (14, 15). The chlorate-resistant phenotype reflects the lack of nitrate reductase activity either due to a mutation in the corresponding structural genes or to a loss of Moco. It is noteworthy that most of the Moco-dependent enzymes are involved in multiple anaerobic respiratory pathways (see below) and none of them are essential for the growth of E. coli. Accordingly, Moco deficiency is nonlethal, allowing isolation of pleiotropic mutants. It turned out that the loci chlA, chlB, chlD, chlE, and chlG were all essential for Moco biosynthesis, and Stewart and MacGregor (16) isolated a large series of Mu-phage insertion mutants for all these loci. In 1992, the Moco-specific chl loci were renamed in mo loci (17).
In pregenomic times, a detailed mutant characterization had already contributed significantly to our understanding of the genetics and biochemistry of Moco biosynthesis in bacteria, plants, fungi, and humans. Investigations such as phenotype suppression by external molybdate or reconstitution experiments mixing cell-free protein extracts of different complementation groups provided evidence for two intermediates of the biosynthetic pathway. As a next step, the defects in molybdate uptake and processing could be assigned to specific mutants. As in all organisms, several different genetic complementation groups were identified, and the existence of a conserved multistep biosynthetic pathway of Moco was proposed. Comprehensive analyses of these mutants involving molecular, genetic, and biochemical studies by several laboratories led to a detailed picture of Moco biosynthesis in E. coli. Five operons (moaABCDE, mobAB, modABC, modEF, moeAB, and mogA) are required for Moco biosynthesis encoding 15 proteins, and details for these genes/proteins are summarized in Table 1.
Table 1.
Genetic and biochemical characteristics of the proteins involved in Moco biosynthesis in E. coli
| Gene | Localization (min) | Operon | Phenotype of the mutant | Protein | Molecular mass and quaternary architecture | Property/functiona | 3D structure | Biosynthesis step | Interacting partner |
|---|---|---|---|---|---|---|---|---|---|
| moaA | 17.6 | moa | Moco deficient | MoaA | 37 kDa, α | Conversion of GTP to cPMP | Yes | Step 1: cPMP biosynthesis | MoaC? |
| moaB | 17.6 | moa | No phenotype | MoaB | 18.5 kDa, α6 | ND | Yes | ND | |
| moaC | 17.6 | moa | Moco deficient | MoaC | 17.3 kDa, α | Conversion of GTP to cPMP | Yes | Step 1: cPMP biosynthesis | MoaA? |
| moaD | 17.6 | moa | Moco deficient | MoaD | 8.6 kDa, α | Small subunit of the PPT synthase | Yes | Step 2: PPT biosynthesis | MoaE, MoeB |
| moaE | 17.6 | moa | Moco deficient | MoaE | 17 kDa, α | Large subunit of the PPT synthase | Yes | Step 2: PPT biosynthesis | MoaD |
| moeA | 18.6 | moe | Moco deficient | MoeA | 44 kDa, α2 | Metal addition and hydrolysis of PPT-AMP | Yes | Step 4: metal addition | MogA, MobA, MobB |
| moeB | 18.6 | moe | Moco deficient | MoeB | 26.5 kDa, α | PPT synthase sulfurase | Yes | Step 2: PPT biosynthesis | MoaD |
| mogA | 0.2 | Moco deficient | MogA | 21 kDa, α3 | Adenylylation of PPT | Yes | Step 3: PPT-AMP biosynthesis | MoeA, MoaDE, MobB | |
| mobA | 87 | mob | Moco deficient | MobA | 21.5 kDa, α | Guanosine nucleotide addition | Yes | Step 4: nucleotide addition | MoeA, MobB |
| mobB | 87 | mob | No phenotype | MobB | 18.7 kDa, α2 | ND | Yes | ND | MogA, MoeA, MobA |
| modA | 17 | modABCD | Mo repairable | ModA | 27 kDa, α | Periplasmic Mo binding protein | Yes | Molybdate transport | ModB, ModC |
| modB | 17 | modABCD | Mo repairable | ModB | 25 kDa, α2 | Membrane-bound component of the ABC transporter | Yes | Molybdate transport | ModA, ModC |
| modC | 17 | modABCD | Mo repairable | ModC | 39 kDa, α | ATPase component of the ABC transporter | Yes | Molybdate transport | ModA, ModB |
| modE | 17 | modEF | ModE | 28 kDa, α2 | Mo-ModE acts as repressor of the modABCDE operon | Yes | |||
| modF | 17 | modEF | No phenotype | ModF | 54 kDa | ND | No | ND | ND |
ND, not determined.
Molybdenum Uptake
When we follow the way that Mo takes from entry into the cell until its final position within the Mo-enzyme’s catalytic center, the first step is the active uptake of Mo in the form of its molybdate anion (see for review, reference 18). The analysis of Moco biosynthesis started with the identification of mutants exhibiting a so-called molybdate-repairable phenotype (19). Those mutants were found in all organisms where Moco deficiency was studied and they are characterized as mutants with partially or completely restored Mo-enzyme activity after growth on unphysiologically high concentrations of molybdate (up to 1 mM). Mo uptake requires specific systems to scavenge molybdate in the presence of competing anions. E. coli cells grown aerobically in 10 nM molybdate contain 1 µM molybdate (20, 21). In E. coli and other bacteria, high-affinity ABC-type molybdate transporters (encoded by the modABC operon) are described, consisting of three protein components and requiring ATP hydrolysis for operation (18). The periplasmic molybdate-binding protein ModA specifically binds molybdate and tungstate with a very high affinity (KD 20 to 50 nM). ModB is the dimeric membrane-integral translocation component, and ModC is the cytoplasmic membrane-associated protein that couples ATP hydrolysis with molybdate translocation through the membrane into the cytoplasm (Table 1). Diverging from the E. coli modABC operon is another one encoding ModE and ModF proteins (22). ModF is homologous to ModC and has no defined function. ModE is a transcriptional regulator member of the LysR family present in a few bacteria in which it is found in the molybdate transport locus. The ModE regulator protein comprises two domains: a DNA-binding domain and a molybdate-binding domain with a similar KD of 0.8 µM for molybdate or tungstate (23, 24, 25). Binding of the oxyanions strongly increases the ModE affinity for DNA because of extensive conformational changes within the molecule (26). The molybdate-bound form of the ModE protein activates transcription of genes for several Mo-enzymes, dimethyl sulfoxide reductase (dmsABC) (27), formate hydrogenlyase (hyc and fdhF), and nitrate reductase (narGHJI) (28), and for molybdenum cofactor biosynthesis (moaABCD) (22, 29) (see below). Moreover, ModE-Mo represses the expression of the modABC operon as might be expected if Mo is available (30). While ModA proteins cannot discriminate between molybdate and tungstate, tungsten-specific transporters have been identified in Eubacterium acidaminophilum (of the Tup-type) (31, 32) or in Pyrococcus furiosus (of the Wtp-type) (33). In addition to the high-affinity transport system, two low-affinity transport systems are operating for molybdate and tungstate, such as the CysPTWA sulfate-thiosulfate permease, and can also transport sulfate or selenate (34, 35).
Regulation of Molybdenum Cofactor Biosynthesis
In E. coli, Moco biosynthesis is enhanced under anaerobiosis already through the moa operon, which encodes the enzymes required for the first step of Moco synthesis. Expression of moa is enhanced under anaerobic growth conditions through transcriptional activation by Fumarate and Nitrate reduction (FNR) regulator but is repressed in strains able to synthesize active molybdenum cofactor (36). Molybdate acts as a major positive regulator of moa and its action requires the ModE protein (29). Transcription of moa is controlled at two sigma-70-type promoters immediately upstream of the moaA gene. The distal promoter is the site of the anaerobic enhancement that is FNR dependent. The molybdate induction of moa is exerted at the proximal promoter. The molybdate activation of moa, however, is revealed only in a molybdenum cofactor-deficient background, since moa is effectively repressed in molybdenum cofactor-sufficient strains. Interestingly, tungstate is also able to relieve the repression of the moa operon (29). This ensures that Moco is synthesized under conditions of anaerobicity (FNR) and sufficient molybdate (ModE), while preventing overproduction by binding free Moco. A new aspect of molybdenum cofactor biosynthesis regulation has been the discovery of a highly conserved RNA motif located upstream of the moaA gene in E. coli (37). This RNA aptamer located in the untranslated region of the moaA gene controls the moa operon in response to cofactor production by binding Moco. Most interestingly, such a riboswitch is also located upstream of genes encoding Mo transport and Mo-enzymes in various prokaryotes, including Gamma- and Deltaproteobacteria, clostridia, actinobacteria, and deinococcus, and, as such, may regulate the initiation of translation or transcription elongation (38). Recently, regulation of the moa operon has been further complicated by the discovery that CsrA, a posttranscriptional regulator that affects translation of its gene targets by binding mRNAs, positively controls expression of the moaA gene (39). While CsrA is well-known to regulate genes that function in aerobic carbon metabolism (see for review, reference 40), RNA-seq analyses identified many mRNA targets of CsrA that are necessary for anaerobic respiration, including the uptake of molybdate (modA), biosynthesis of Moco (moaA and moeA), and the production of Moco-dependent enzymes (narL, narGHJI, fdoGHI, bisC, dmsABC, fdnGHI, napA, and torZ) (41). Interestingly, the above-mentioned RNA aptamer located upstream of the moaA gene serves as a target for posttranscriptional regulation, not only by Moco, but also by CsrA, which binds the moaA leader with high affinity and specificity (39). As such, CsrA should enhance the Moco biosynthesis under conditions of high metabolic demand. The exact understanding of such an intricate regulation awaits further studies.
Also, the moe operon is regulated, and it was found that its expression is independent of genes coding for Mo transport and for PPT synthesis; instead, anaerobic conditions as well as nitrate stimulate moe expression via the transcriptional factors NarL and ArcA, respectively (42). Earlier, the authors reported that the product of the MoeA-catalyzed reaction is required for Mo-dependent control of genes coding for E. coli Mo-enzymes (43). Apparently, the bacterium coordinates Moco biosynthesis with apoprotein synthesis at the level of moe operon transcription. Posttranscriptional regulation by CsrA as suggested by the RNA-seq analyses needs to be confirmed. Finally, the mob locus appears to be constitutively expressed (44).
Biosynthesis of the Molybdenum Cofactor
Most of our knowledge about Moco biosynthesis was obtained from studies in E. coli, and this work was pioneered by Rajagopalan, Johnson, and coworkers (45, 46). Moco biosynthesis proceeds in four steps, and these steps are defined by the following biosynthetic intermediates: cyclic pyranopterin monophosphate (cPMP, formerly precursor Z), pyranopterin (PPT), adenylated pyranopterin, and Mo-bound pyranopterin (Fig. 1). In prokaryotes, a nucleotide is added during a fifth step, thus forming the Mo-bisPGD (i.e., pyranopterin guanine dinucleotide) or Mo-PCD (i.e., pyranopterin cytosine dinucleotide). However, we will describe steps four and five as one combined last step (Fig. 1).
Step 1: conversion of GTP to cPMP
PPT is the only pterin known to be substituted with a four-carbon side chain, while several other pteridines such as biopterin have three-carbon side chains. Two pathways are known for the synthesis of pteridines (47) and flavins (48) that start with the conversion of GTP by the enzymes cyclohydrolase I and II, respectively, whereas Moco synthesis depends on a third route also starting with GTP. Based on labeling studies in E. coli, it was determined that a guanosine derivative is the initial precursor for cPMP formation, the first stable intermediate of Moco biosynthesis (49, 50). Originally, this first intermediate was named precursor Z (51). In 2004, its chemical structure was clarified by mass spectrometry and 1H nuclear magnetic resonance (NMR) spectroscopy (52). It was demonstrated that the molecule is a pyranopterin, similar to Moco, and carries a geminal diol at the C1′ position of the side chain (53). Therefore, precursor Z was renamed cPMP for cyclic pyranopterin monophosphate.
In E. coli, two proteins MoaA and MoaC were identified as essential for cPMP synthesis (Fig. 1 and Table 1). MoaA contains two oxygen-sensitive [Fe-S] clusters that are bound via three highly conserved cysteine residues (54) and shows sequence similarities to a number of proteins, including biotin synthase or thiamine synthase. MoaA and all homologues belong to the family of S-adenosylmethionine (SAM)-dependent radical enzymes. Members of this large family catalyze the formation of protein and/or substrate radicals by reductive cleavage of SAM by a [4Fe-4S] cluster (55). The structure of MoaA was determined in the apo-state as well as with the cosubstrate SAM or the 5′-GTP (56, 57). These data were of considerable value because they provided insights into the radical reaction underlying the conversion of 5′-GTP to cPMP. Indeed, MoaA is not able itself to catalyze the release of pyrophosphate, which indicates that, during catalysis, 5′-GTP and its reactive radical intermediates are tightly anchored by the triphosphate moiety to prevent their escape. MoaC appears to be responsible for pyrophosphate release. The X-ray crystallographic structure of E. coli MoaC reveals that it forms a homohexamer (58) with a hypothetical active site made up by several strictly conserved residues at the interface of two MoaC monomers. Nevertheless, complete understanding of this reaction step, which most likely involves multistep reactions, must await further studies. Also, the detailed function played by MoaC remains enigmatic.
Recently, it has been reported that the chemical synthesis of a tricyclic pyranopterin intermediate can be successfully converted in vitro into a functional Moco (59). These studies appear most promising for the treatment of human patients suffering from a Moco deficiency as already demonstrated by cPMP curation (60, 61).
Step 2: conversion of cPMP to PPT
During the second step of Moco biosynthesis, the PPT dithiolate is formed by incorporating two sulfur atoms into cPMP (Figs. 1 and 2). This reaction is catalyzed by PPT synthase, a heterotetrameric complex of two small (MoaD) and two large (MoaE) subunits that stoichiometrically convert cPMP into PPT (Fig. 2 and Table 1). Biochemical studies using in vitro assembled PPT synthase from individually expressed and purified subunits demonstrated that the C terminus of MoaD carries the sulfur as thiocarboxylate (62). The functional importance of this thiocarboxylate was also demonstrated in the crystal structure of the E. coli PPT synthase, which shows that the C terminus of MoaD is deeply inserted into the large subunit MoaE to form the active site (63). The heterotetramer is formed by dimerization of two large subunits forming two clearly separated active sites. Interestingly, although the thiocarboxylate moiety on the MoaD subunit is essential for PPT synthase activity, it is not required for formation of the synthase heterotetramer. Detailed investigations of the thermodynamic properties of the interaction between MoaD and MoaE in PPT synthase revealed an increased binding affinity of MoaD-SH to MoaE consistent with the proposed reaction mechanism (64). Moreover, the solvent-accessible surface area buried on formation of the heterotetramer was considerably increased on activation of the protein, changing from 2,376Å to 4,117Å. In 2008, the crystal structure of the PPT synthase in complex with cPMP provided insights into the mechanism and the delineation of a model for conversion of cPMP to PPT (65). Because each small subunit of PPT synthase carries a single sulfur atom, a two-step mechanism for the formation of PPT dithiolate has been proposed, which involves the formation of a mono-sulfurated intermediate (62, 65, 66). Together with this two-step mechanism is the proposed existence of an intermediate in which the MoaD C terminus is covalently linked to the substrate via a thioester linkage, further resolved by a water molecule (65).
Figure 2.
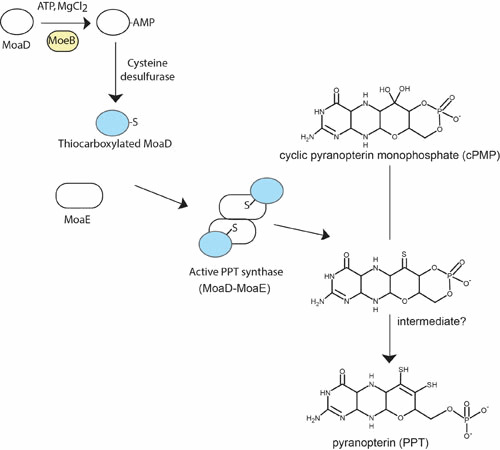
Conversion of cPMP to PPT (step 2). See the text for a detailed description of the reaction mechanism leading to the two-step conversion of cPMP to PPT. doi:10.1128/ecosalplus.ESP-0006-2013.f2
After PPT synthase has transferred the two sulfurs to cPMP, it has to be resulfurated in a separate reaction. This resulfuration is catalyzed by MoeB involving an adenylation of the small subunit MoaD in a MoaD-MoeB complex by using Mg2+-ATP as substrate followed by sulfur transfer (67) (Fig. 2). The crystal structures of MoeB in complex with MoaD have been determined in the apo-, ATP-bound, and MoaD-adenylate form (68), depicting a conserved mechanism of acyl-adenylate formation in ubiquitin-dependent protein degradation and in the synthesis of Moco. MoaD and homologous proteins harbor in their C-terminal region a conserved double-glycine motif also found in ubiquitin, a crucial protein in eukaryotic protein degradation (69). Only the terminal glycine appears to be essential for proper function of MoaD (70). Besides the homology of MoaD and MoeB to the ubiquitin-activating system, similarities between Moco biosynthesis and thiamin biosynthesis can also be seen in E. coli. The proteins ThiF, ThiS, and ThiI participate in the synthesis of the thiazole moiety. Begley and coworkers (71, 72, 73) have shown that ThiS is thiocarboxylated by ThiF (homologous to MoeB) and ThiI (sulfurtransferase) (for review, see reference 74). A similar way of sulfur activation (i.e., adenylation of the sulfur transfer protein MoaD or ThiS followed by exchange of AMP for sulfur) has been proposed for the synthesis of biotin and lipoic acid (see for review, reference 75).
MoeB alone is not sufficient to reactivate carboxylated MoaD. Conversion of the acyl-adenylate of MoaD to a thiocarboxylate by sulfur transfer is made through the action of the major pyridoxal phosphate-dependent E. coli L-cysteine desulfurase IscS (76). One has to mention that IscS serves as a major sulfur donor for a number of different processes through direct protein interaction with various sulfur-accepting proteins (77). A further participant to the sulfur transfer process between IscS and MoeB has been discovered with YnjE in E. coli, thus mediating sulfur transfer and directing IscS toward Moco biosynthesis (78). However, YnjE is not essential for Moco biosynthesis, an effect being observed only upon deletion of both ynjE and iscS genes. Recently, studies aimed at understanding the pleiotropic effect of the deletion of the tusA gene encoding a sulfur mediator protein between IscS and TusBCD in thiomodification of tRNAs (79) have reported a pronounced effect on the E. coli transcriptome (80). In particular, in the absence of TusA, sulfur transfer for FeS biosynthesis is increased while the activity of several Mo-enzymes is drastically reduced. It has been proposed that TusA is involved in regulating the IscS pool and shifting it away from IscU and FeS biosynthesis, thereby making IscS available for sulfur transfer for PPT biosynthesis. At this stage, the actual interplay of YnjE and TusA in Moco biosynthesis remains to be clarified.
Step 3: adenylylation of PPT
After synthesis of PPT, the chemical backbone is ready to bind and coordinate the molybdenum atom. Mo has to be taken up into the cell in the form of molybdate followed by the coordination to PPT. In this context, it is worth mentioning that the high-affinity ModABC transporter is subjected to molybdate-dependent gene regulation by the Mo-ModE complex (22). The Mo-loaded ModE protein also enhances the expression of the moa operon (29) as well as several Mo-enzymes (27, 28). Once inside the cell, a key question of Moco biosynthesis resides in whether the molybdate serves as a donor for insertion of Mo into PPT or whether it has to undergo intracellular processing before insertion. In the following, we will discuss the existence of an additional intermediate in Moco biosynthesis, adenylated PPT (PPT-AMP), preceding the Mo insertion step (Fig. 3).
Figure 3.
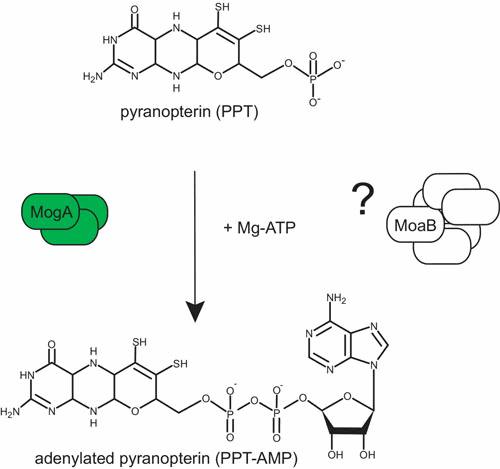
Adenylylation of PPT (step 3). See the text for a description of the reaction mechanism leading to MPT adenylylation. doi:10.1128/ecosalplus.ESP-0006-2013.f3
In bacteria, two proteins, MogA and MoeA, are involved in Mo insertion (Table 1), while during evolution to eukaryotes, these two proteins were fused to a two-domain protein (see for review, reference 81). Whereas it had earlier been postulated that one protein should be essential for PPT binding, the other being in charge of generating an activated form of Mo, the exact mechanism was initially uncovered in plants where the protein Cnx1 catalyzes this step (82). The C-terminal domain (Cnx1-G) known to complement a mogA mutant was shown to tightly bind PPT (83). The crystal structure of Cnx1-G in complex with PPT confirmed the proposed binding of PPT (84). Further, the Cnx1-G active site was mapped by structure-based mutagenesis and functional analysis to a large surface depression with a clear discrimination between PPT binding and catalysis (85, 86). Unexpectedly, the structure of a variant (S583A) with a gain of function revealed a novel intermediate in Moco biosynthesis as an adenosine moiety was covalently bound via a pyrophosphate bound to the C4′ carbon of PPT, thereby forming adenylated PPT (84). Subsequently, it has been demonstrated that Cnx1-G adenylates PPT in a Mg2+- and ATP-dependent way and forms PPT-AMP that remains bound to Cnx1-G (82) (Fig. 3). This intermediate appears to be mechanistically relevant because it serves as a substrate for the subsequent Mg2+-dependent Mo insertion reaction by MoeA (or the equivalent Cnx1-E domain) (87) (Fig. 4). Based on the ability of Cnx1-G to reconstitute mogA mutants and on their nearly identical X-ray structures, one can conclude that both proteins catalyze the PPT adenylation reaction, which is essential for and takes place before metal insertion. Overall, the process can be described as follows in E. coli: before Mo addition by MoeA, PPT is activated by MogA-dependent adenylation under physiological Mo concentrations, because this activation step appears unnecessary at high molybdate concentrations (> 1 mM) (88).
Figure 4.
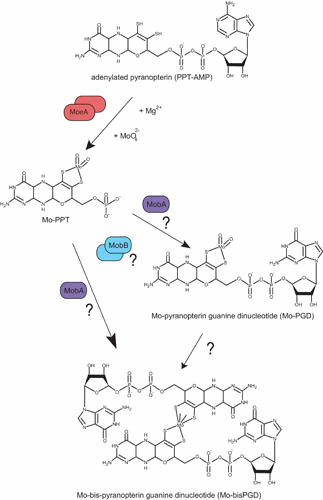
Mo insertion and nucleotide addition (step 4). See the text for description of the reaction mechanism leading to Mo addition and of the different postulated pathways for the nucleotide addition step leading to the Mo-bisPGD molecule. doi:10.1128/ecosalplus.ESP-0006-2013.f4
Within the moa operon, the second open reading frame encodes for a protein, MoaB, with significant sequence similarity to MogA (Table 1). Crystal structures of MoaB (89, 90) and MogA (91) confirmed their strong structural similarity, supporting the idea of a conserved function. However, the function played by MoaB in E. coli remains enigmatic as mogA mutants show Moco deficiency, while moaB mutants do not affect the activity of Mo-dependent enzymes (92). Careful examination of the MoaB structure reveals the absence of the catalytically essential residues present in MogA, pointing toward a loss of function of MoaB in E. coli. Such bioinformatic analyses have been confirmed experimentally (93). In archaea, MoaB proteins could have a MogA-like function in the biosynthesis of W-cofactors since MoaB homologous proteins were predominantly found (93).
Step 4: Mo insertion and nucleotide addition
Considering that adenylated PPT is an intermediate of Moco biosynthesis in bacteria as well, two further steps need to be accomplished for synthesis of the active form of Moco found in most prokaryotic Mo-enzymes. Indeed, the vast majority of these enzymes that make use of Mo do so through nucleotide-substituted Moco. In E. coli, this cofactor contains a nucleotide, mostly a guanosine monophosphate, covalently linked to PPT via a pyrophosphate bond resulting in the PPT dinucleotide cofactor (Fig. 1). A cytosine monophosphate was also shown to be an alternative to GMP in E. coli and to be present exclusively in a specific group of Mo-enzymes belonging to the XO family (94, 95). Other prokaryotic variants of the cofactor containing CMP, AMP, or IMP linked to the PPT were identified as well in bacteria (45). It is important to notice that, in addition to the nucleotide moiety, another difference compared with eukaryotes is the formation of bis-PPT-based cofactors where one Mo (or W) atom is coordinated by two dithiolenes of two PPT molecules. Therefore, in addition to Mo insertion, dinucleotide formation has to be performed in the final stages of Moco biosynthesis (Fig. 4). There is cumulative evidence that these two steps are linked to each other, so that we will discuss both steps as a fourth step of Moco biosynthesis.
For a long time, the exact mechanism underlying Mo incorporation remained one of the most enigmatic aspects of Moco biosynthesis. Leimkuhler et al. (96) reported that the activity of the PPT-dependent xanthine dehydrogenase in a Rhodobacter capsulatus moeA mutant could be recovered on growth with 1 mM sodium molybdate indicating that MoeA is involved in Mo incorporation. Later on, Nichols and Rajagopalan (97) clearly demonstrated that, while mutations in either mogA or moeA have no effect on PPT biosynthesis, they completely abolish the ligation of Mo to PPT. Interestingly, Kuper et al. (84) reported copper coordination by the adenylated PPT bound to MogA, copper having higher affinity for the dithiolene group than molybdate and suspected of having a protective role of the intermediate. Moreover, copper, cadmium, or arsenite ions can all insert nonspecifically into PPT without the aid of MoeA or MogA (98).
As mentioned above, the vast majority of E. coli Mo-enzymes contain a bisPPT-type cofactor and moeA mutants show no molybdate-repairable phenotype with respect to the activity of those enzymes (99). One can argue that a bisPPT-based cofactor should require a different metal insertion process than a mono-PPT-based cofactor, as found in all eukaryotes as well as in some bacterial enzymes. Interestingly, a novel PPT-type oxidoreductase (YedY) was identified and characterized in E. coli that belongs to the eukaryotic SO family of Mo-enzymes (100, 101). Finally, three E. coli enzymes belonging to the XO family appear to bind Mo-PCD as cofactor (94, 102, 103), further expanding the list of Moco types in this organism. These data indicate that both mono- and bis-PPT forms of Moco do exist in E. coli. In this context, it is important to notice that the function of E. coli moeA cannot be reconstituted by Cnx1-E as seen on the basis of bis-PPT-dependent enzyme activity. These observations provide further support for functional diversity between bacterial and eukaryotic Mo insertion processes. In vitro studies have shown that MogA stimulates, in an ATP-dependent manner, the activity played by MoeA in mediating Mo incorporation to PPT using eukaryotic PPT-dependent aposulfite oxidase as reporter enzyme (104). Future investigations aimed at testing the effect of these purified proteins on the activity of a bis-PPT-type cofactor-dependent enzyme are thus of substantial importance.
Because of its intrinsic instability, Moco has to remain bound to proteins during the whole biosynthetic process until its final delivery to apo-Mo-enzymes. Interestingly, the use of an in vivo approach, the bacterial two-hybrid system, was proven to be valuable in determining the conditions required for visualization of the interaction between proteins involved in the late stages of Moco biosynthesis (Fig. 5). Indeed, PPT appears to be of crucial importance for the interaction between MogA and MoeA (105). Based on the conserved fusion event occurring between eukaryotic MogA and MoeA and on the observed interactions between E. coli counterparts in the presence of PPT, Magalon et al. (105) suggested that during evolution it became important to facilitate substrate-product flow by the existence of a Moco-biosynthetic multienzyme complex. Formation of such complexes would ensure both the fast and protected transfer of reactive and oxygen-sensitive intermediates within the reaction sequence from PPT to Mo-PPT. These data pointed to a concerted mechanistic action of MogA and MoeA, and this concept was later fully supported by biochemical analyses (82, 87), as seen below. Previous biochemical studies had indicated that newly formed PPT remains tightly bound to the PPT synthase complex until its transfer to MogA by direct protein interaction (88). The same applies to the newly synthesized adenylated PPT remaining associated with Cnx1-G (equivalent to MogA) (82). Later on, Llamas et al. (87) demonstrated that PPT-AMP and molybdate bind with high affinity in a cooperative and equimolar manner to Cnx1E. Once transferred from Cnx1G to Cnx1E, PPT-AMP is rapidly hydrolyzed in the presence of Mg2+ or Zn2+ in a molybdate-dependent manner with rates that are several orders of magnitude higher than PPT-AMP synthesis (82). Therefore, PPT-AMP synthesis seems to be the rate-limiting step in Cnx1 reaction. Recently, establishment of a fully defined in vitro system with the full-length gephyrin protein (equivalent to the plant Cnx1 and corresponding to a MogA-MoeA fusion) allowed researchers to confirm that the intimate interaction between MogA and MoeA domains significantly enhances the overall catalytic efficiency (106). Overall, these results substantiate the idea according to which substrate-product channeling is operating within the reaction sequence from PPT to Mo-PPT and may be responsible for the functional origin of a domain fusion in eukaryotes exemplified by Cnx1 or gephyrin. In summary, MogA and MoeA are both essential for the two-step reaction leading to metal transfer to PPT (Fig. 4).
Figure 5.
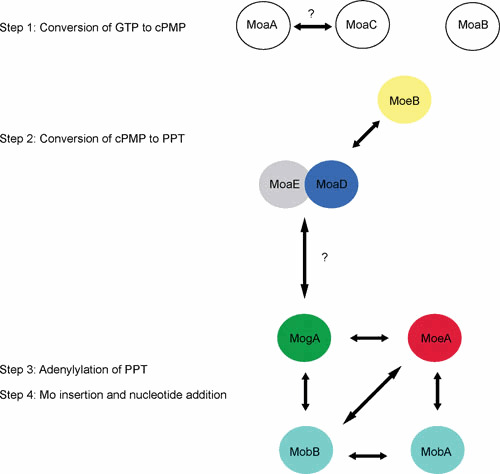
Interactions network among Moco biosynthetic proteins in E. coli during the four-step process. The arrows represent the interactions as detected by using bacterial two-hybrid methodology, TAP-Tag, or biochemical assays (see the text for details and references). doi:10.1128/ecosalplus.ESP-0006-2013.f5
While the reaction mechanism that leads to the formation of Mo-PPT is known, in which order, however, must those steps take place that lead not only to nucleotide addition, but also to the formation of a bis-PPT-type cofactor? At first, the mobAB locus is responsible for nucleotide attachment in E. coli Moco biosynthesis (Fig. 4 and Table 1). MobA catalyzes the conversion of PPT and GTP to PGD (107), whereas MobB, a GTP-binding protein, is not absolutely required for PGD synthesis (44, 108). Using a fully defined in vitro system, Temple and Rajagopalan (109) demonstrated that MobA alone, when incubated with GTP, Mg2+, and a source of PPT, catalyzes the formation of PGD, indicating that it is both necessary and sufficient for GMP attachment. Specific protein-protein interactions have already been shown to play a central role in the early stages of Moco biosynthesis (110). In the same way, Magalon et al. (105), with the use of a bacterial two-hybrid approach, were able to demonstrate that MobA interacts with MoeA and MobB in vivo (Fig. 5). In particular, the interaction between MobA and MoeA strictly depends on the presence of PPT. The crystal structure of MobA was solved and indicated an overall α/β architecture and a nucleotide-binding Rossmann fold within the N-terminal half (111, 112). The active site was defined by highly conserved residues as well as by cocrystallization of MobA with GTP that is bound in the N-terminal half (111). An important finding was that MobA can also be copurified along with PPT and PGD, demonstrating a tight binding of both its substrate and product (113).
Consistent with its ability to bind GTP, the amino acid sequence of MobB reveals a putative nucleotide-binding motif, the Walker A motif. Crystal structure of MobB indicated a dimeric state of the protein and confirmed the lack of structural elements required to interact with and efficiently bind a nucleotide base (114). Structural homologues of MobB include a number of nucleotide-binding proteins. Based on the observation that MobA and MobB interact in vivo, McLuskey et al. (114) proposed a model in which the formation of a MobA-MobB complex enhances the efficiency of conversion of PPT to PGD through better GTP binding and utilization.
Interestingly, MobB not only interacts with MobA, but also with MogA and MoeA (105) (Fig. 5). The functional significance for such interactions is not yet understood. It is worth mentioning that MobB does exist in some organisms as a fusion protein with MoeA in Gammaproteobacteria such as Vibrio species and Shewanella oneidensis (Fig. 6). In this case, MoeA exists in two forms, fused with MobA or not, both forms harboring the essential catalytic residues. In other bacteria such as Chlorobium species or Geobacter metallireducens, MobB is fused with MobA. An extensive network of protein interactions has thus been revealed among proteins involved in the final stages of Moco biosynthesis. Although clear understanding of these steps has not yet been attained, these data provide further evidence that the processes of Mo insertion and of dinucleotide attachment are strongly linked. Clearly, an understanding of the exact function played by MobB in Moco biosynthesis awaits further studies.
Figure 6.
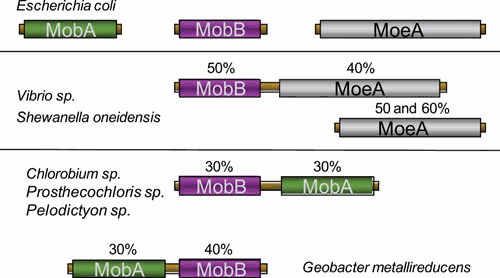
Schematic representation of the fusion proteins involving MobB. Identity percentages are indicated by using E. coli proteins as reference. doi:10.1128/ecosalplus.ESP-0006-2013.f6
Recently, members of the XO family have been identified in E. coli with XdhABC, XdhD, and PaoABC (94, 102, 103). All three enzymes bind Mo-PCD (i.e., pyranopterin cytosine dinucleotide) as a cofactor, thereby requiring an alternative factor to MobA for nucleotide addition during PCD biosynthesis. Indeed, PCD formation is catalyzed by MocA sharing nearly 20% of sequence identity with MobA (95). MocA exhibits a CTP:molybdopterin cytidylyl transferase activity thanks to a range of amino acid substitutions allowing high affinity toward the pyrimidine nucleotide CTP (115). Interestingly, two separate domains have been defined in both MobA and MocA and are associated with distinct functions. While the N-terminal domain defines specificity toward nucleotide binding, the C-terminal domain allows specific interaction with the target Mo-enzymes in conjunction with their requirement for either a PCD- or PGD-type cofactor.
Further Modifications of the Cofactor
Apart from the nucleotide addition in prokaryotes, a distinct modification of the cofactor consists in the exchange of a Mo oxygen ligand with a sulfur atom. Sulfurated Moco is the characteristic form of the cofactor present in all members of the XO family (116) but also in a few members of the Mo/W-bisPGD family (117, 118, 119, 120). To date, genetic and biochemical studies have provided evidence for the participation of a chaperone protein dedicated for its target Mo-enzyme and which is involved in the sulfuration step. The current working model states that these proteins not only ensure protected sulfuration of the Mo cofactor, but also its subsequent incorporation into target enzymes timely with other metal insertion and folding processes. This has been illustrated by XdhC for the xanthine oxidoreductase in R. capsulatus (121). XdhC specifically promotes the sulfuration of Mo-PPT by interaction with a cysteine desulfurase, which transfers the sulfur to Moco bound to XdhC. XdhC protects the sulfurated form of Moco from oxidation before its transfer into apoXdhAB (122). Importantly, to prevent all available Mo-PPT in the cell from being converted to Mo-bisPGD and to guarantee a Mo-PPT supply for XdhAB, XdhC interacts with MoeA and MobA proteins involved in the final stages of Moco synthesis (123). Whereas interaction with MoeA allows Mo-PPT transfer to XdhC, its interaction with MobA prevents Mo-bisPGD formation.
Recently, Thome et al. (124) reported that FdhD, a chaperone dedicated to the formation of active formate dehydrogenases in E. coli, is a sulfurtransferase between the major cysteine desulfurase IscS and FdhF. In particular, the interaction of IscS with FdhD results in a sulfur transfer in the form of persulfides bound by conserved cysteine residues in FdhD. Furthermore, formate dehydrogenase activity of FdhF is sulfur dependent and critically depends on the integrity of the conserved cysteine residues of FdhD. Overall, these results strongly suggest that formate dehydrogenases in E. coli harboring Mo-bisPGD as cofactor may require sulfuration of the Mo atom for their reactivity. It is noteworthy that the X-ray crystal structure of the tungsten-containing formate dehydrogenase from Desulfovibrio gigas sharing high structural similarity with FdhF reveals the presence of an additional sulfur atom at the tungsten coordination sphere (117). Furthermore, reinterpretation of the X-ray data of E. coli FdhF shows that sulfur refines better than oxygen at the apical position of the molybdenum coordination sphere (118). In a more recent study, Arnoux et al. (125) reported that E. coli FdhD binds Mo-bisPGD in vivo and has submicromolar affinity for GDP, used as a surrogate of the Moco’s nucleotide moieties. The crystal structure of E. coli FdhD shows that each monomer folds into two domains, an N-terminal domain (NTD; residues 1 to 97) that is unique to the FdhD family, and a C-terminal domain (CTD; residues 142 to 277) bearing some structural homology with the cytidine deaminase fold. A long loop connects the NTD to the CTD in FdhD (residues 98 to 146). This loop is disordered, in part, in each monomer with residues 113 to 131 not visible in the electron density map. Remarkably, this disordered part of the loop contains the pair of cysteine residues (Cys121–Cys124) shown to be functionally important (124). It is more interesting that this crystal structure of E. coli FdhD was obtained in complex with GDP at 2.8 Å, revealing two symmetrical binding sites located on the same face of the dimer (Fig. 7). These binding sites are connected via a tunnel-like cavity to the opposite face of the dimer where two dynamic loops, each harboring two functionally important cysteine residues, are present. A striking feature in the crystal structure of the FdhD/GDP complex is the distance that separates the two GDP moieties in the dimeric complex. Indeed, this distance approximately corresponds to the distance observed in Mo-bisPGD-containing enzymes. For example, the distance between the two N2 atoms of the guanine base is on average 32.9 ± 1.2 Å in a selection of 13 independent Mo-bisPGD-containing enzymes, while it corresponds to 28.8 Å in FdhD. Furthermore, the large solvent exposed pocket that is present between the two GDP molecules in the FdhD/GDP complex could accommodate such a large molecule as shown by molecular modeling. The current working model anticipates a sophisticated mechanism by which the sulfur is transported from IscS on one side of the FdhD dimer to the molybdenum cofactor on the other side before its final insertion into formate dehydrogenases. Therefore, Mo cofactor sulfuration step goes beyond the biosynthesis itself; rather, it is part of a process by which the synthesized cofactor is trafficked to the target enzymes by dedicated chaperones.
Figure 7.
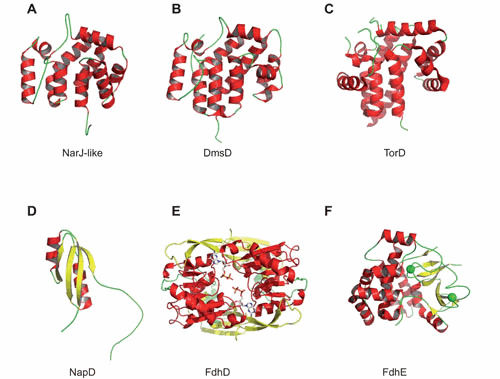
Space-filling structure of different REMPs for Mo/W-bisPGD enzymes. (A) NarJ-like from Archaeoglobus fulgidus (PDB ID code 2o9x). (B) DmsD from Escherichia coli (PDB ID code 3efp). (C) TorD monomer from Shewanella massilia (PDB ID code 1n1c). (D) NapD from E. coli (PDB ID code 2jsx). (E) FdhD dimer from Escherichia coli in complex with GDP (PDB ID code 4PDE). (F) FdhE from Pseudomonas aeruginosa (PDB ID code 2fiy). NarJ-like, DmsD, and TorD belong to the Pfam PF02613 family. Individual figures were generated with PYMOL (223) by using the deposited coordinates from the protein structure database. The proteins are represented in cartoon with α-helices colored in red and β-sheets colored in yellow. Two GDPs are cocrystallized with FdhD, while two iron atoms are coordinated by FdhE (represented by green spheres). doi:10.1128/ecosalplus.ESP-0006-2013.f7
ASSEMBLY OF MO-ENZYMES AND INSERTION OF MOCO
Prokaryotic Mo-enzymes constitute a large and diverse group of metalloproteins harboring several metal cofactors (hemes b and c, [Fe-S] clusters) in addition to the Moco (2, 126). X-ray crystallographic studies of all known Mo-enzymes revealed that the Moco is not located at the surface of the protein, but it is buried deeply within the enzyme and, in some cases, in close proximity to [Fe-S] clusters (see for review, references 127 and 128). This observation suggests that Moco insertion is intimately connected to protein folding and subunit assembly.
Another consideration is that, once Moco is liberated from the holoenzyme, it loses Mo and undergoes rapid and irreversible loss of function due to oxidation. Consequently, the lability of the cofactor has limited elucidation of the biosynthesis of Mo-bisPGD and its incorporation into Mo-enzymes. To date, it is assumed that there is no free Moco present in the cell and that Moco occurs permanently protein bound in the cell. Since the availability of sufficient amounts of Moco is essential for the bacterial cell to meet its changing demand for synthesizing Mo-enzymes, the existence of a Moco-storage protein would be a good way to buffer supply and demand of Moco. In contrast to the green algae Chlamydomonas reinhardtii (129, 130, 131) or the plant Arabidopsis thaliana (132) where Moco carrier proteins (MCP) shuttle synthesized Moco from the biosynthetic machinery to the apo-Mo-enzymes, a complex of proteins involved in the last steps of Moco synthesis is in charge of Moco delivery to different apo-Mo-enzymes in E. coli (105, 133). This process appears to be highly regulated and assisted by enzyme-specific chaperones (also named REMPs for redox enzyme maturation proteins [134]). These proteins are in charge of coordinating the metal insertion and folding processes and are not part of the final structures.
Early work in Giordano’s group has shown that a lesion in the narJ gene present in the nar operon, encoding for the nitrate reductase A complex, blocks the generation of an active enzyme complex (135). Similarly, the fdhD and fdhE genes flanking the fdo operon encoding for the aerobic formate dehydrogenase-O complex in E. coli have been shown to be essential for synthesis of both active formate dehydrogenase-O and -N (136, 137). Because of its essential character, the NarJ protein has constituted the prototype of an accessory protein for Mo-enzymes in prokaryotes. Later on, many groups reported the implication of similar proteins in the synthesis of active Mo-enzymes, such as trimethylamine-N-oxide (TMAO) reductase (138), dimethyl sulfoxide (DMSO) reductase (139, 140, 141), periplasmic nitrate reductase (142, 143, 144), putative tetrathionate reductase (145), or xanthine dehydrogenase (121, 146). This list considerably expanded with the genomic era, and numerous operons encoding for prokaryotic Mo-enzymes have revealed the existence of additional genes, each encoding for a putative enzyme-specific chaperone (see for review, reference 126).
As recently reviewed by Grimaldi et al. (2), the modular organization of many Mo-enzymes can involve multiple subunits and can be tethered to the cytoplasmic membrane often through b-or c-type cytochromes. The membrane subunits connect the cytoplasmic or periplasmic redox reactions with electron transport to or from the respiratory quinone/quinol pool (see for review, reference 147). In addition to the Sec machinery, membrane insertion may require the help of the accessory protein YidC (148). These enzymes can form subcomplexes of cytoplasmic subunits in the absence of the membrane-anchoring subunits, and these subcomplexes can retain oxidoreductase activity, although this activity is uncoupled from the electron transfer chain. This suggests that the attachment of the enzymes to the membrane by their membrane anchor subunits is the last step in complex assembly. Within the prokaryotic cell, successful synthesis and assembly of Mo-enzymes is thus an intricate process that requires several steps such as the synthesis of the different subunits in the cytoplasm, their assembly, the incorporation of various types of metal or organic cofactors, and the anchoring of the complex to the membrane. In the case of periplasmic or outer-membrane Mo-enzymes, the assembly and metal cofactor incorporation steps takes place in the cytoplasm before translocation across the inner membrane via the Tat apparatus (149). Importantly, enzyme-specific chaperones often assist formation of active Mo-enzymes. In this context, Li et al. (150) reported the interaction of the enzyme-specific chaperone DmsD with a number of general chaperones illustrating their more general participation in metalloenzyme maturation. Altogether, these enzyme-specific and general chaperones may function to stabilize the substrates against misfolding and proteolysis, such that a certain level of structure is acquired before Moco insertion can proceed, as well as to help escort Tat substrates to the translocon while preventing early engagement with the Tat machinery. Bacterial Tat systems export folded proteins, including [Fe-S]-containing proteins, partner subunits of most exported Mo-enzymes, but also proofread these substrates. When Tat substrates are misfolded, they are subjected to proteolysis, likely through a Tat-independent process (151, 152, 153). Although it is most likely that all these events occur in a coordinate fashion to yield a final functional multimeric metalloprotein, information about how this coordination is performed is scarce. Most of the available information concerns members of the Mo/W-bisPGD and XO families and will be extensively described below. Concerning the maturation of aldehyde oxidoreductase and sulfite oxidase family enzymes, no enzyme-specific chaperones have yet been reported while the presence of several cofactors within the catalytic subunit or the periplasmic location of the enzyme precludes involvement of enzyme-specific chaperones (see below).
Enzyme-specific chaperones have been grouped in a much larger classification encompassing other metalloproteins than Mo/W-enzymes, the REMPs (134). Those proteins, in common, interact with specific partner subunits either to prevent premature folding or to induce a proper folding but not being part of the final structures. When considering the REMPs associated with Mo/W-enzymes, a broad range of functions can now be assigned to them thanks to extensive biochemical studies during the past two decades. Functions ranged from binding to the N-terminal signal peptide, or to a remnant one, to FeS binding, Moco binding, or its sulfuration (10). This list is far from being exhaustive because the exact function of a number of them is still unclear. At this stage, as exemplified below, one can conclude that a salient feature of those REMPs associated with Mo/W-enzymes is their multifunctionality, making it difficult to classify them according to their function. On the contrary, these proteins can be divided into subfamilies according to structural data. X-ray structures of several REMPs associated with Mo/W-enzymes revealed the existence of single-domain proteins sharing a common all-helical fold (145, 154, 155, 156, 157, 158) and allowing the description of a new family of chaperones (Pfam PF02613) to which NarJ, DmsD, and TorD belong (Fig. 7). It has to be mentioned that their limited size (less than 250 residues) precluded any reliable structure-guided phylogenetic analysis. Furthermore, distinct folding is associated with all other REMPs, such as NapD, FdhD, FdhE, and XdhC, sharing no sequence homology. As developed below, these four proteins are involved in distinct functions. The actual view is that a single folding state of the REMPs as deduced from X-ray data can hardly be reconciled with their multifunctional character. A better understanding of this structure-function relationship would be most welcome and may be attained through more systematic consideration of their structural flexibility, which bestows their multifunctionality, and a structure-guided search of domains associated with a specific function.
Maturation of Mo/W-bisPGD Enzymes
The archetypal NarGHI-NarJ couple
The first step of the bacterial denitrification pathway is catalyzed by the cytoplasmically oriented membrane-bound NarGHI-type nitrate reductase (i.e., nNar) which is directly coupled to the generation of a pmf to sustain cell growth (Fig. 8). The nNar is largely conserved among all nitrate-respiring bacteria. The enzyme has been extensively studied in mesophilic nitrate-reducing bacteria, such as the ammonifier E. coli and the denitrifiers Paracoccus and Pseudomonas species. The E. coli nNarGHI enzyme has been crystallized (159, 160), and, apart from serving as the prototype in our understanding of the structure and function of respiratory nitrate reductases in other bacteria, it was also one of the first Mo-enzymes whose maturation pathway was intensively studied. In the early 1990s, initial biochemical and genetic studies indicated that the NarJ protein encoded by the narGHJI operon plays an essential role in nitrate reductase activity, promoting correct assembly of the enzyme complex without being part of the final structure (135). Based on these properties, a role as a private or system-specific chaperone was proposed (135, 161, 162). E. coli synthesizes a second nitrate reductase complex, the nNarZYV isoenzyme, whose maturation involves the NarW protein, a NarJ homologue, which is interchangeable with NarJ (163).
Figure 8.
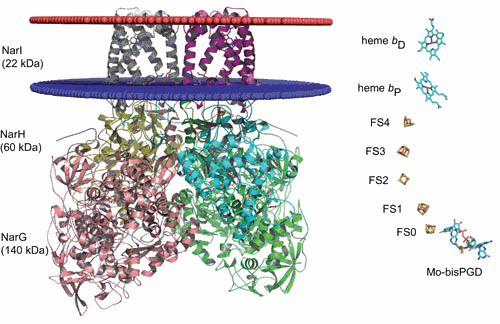
Schematic representation of the membrane-bound nitrate reductase from E. coli. The NarGHI complex is surface-represented (PDB ID code 1q16) and represented as a homodimer. NarG is colored in pink and in green, NarH in yellow and in cyan, and NarI in gray and fuchsia. The cytoplasmic membrane is represented as two ellipses, one colored in red at the interface with the periplasm and the other one colored in blue at the interface with the cytoplasm. Metal centers are shown on the left. The Mo-bisPGD is buried in NarG close to the [Fe-S] cluster (FS0). NarH harbors 4 [Fe-S] clusters: FS1, FS2, FS3, and FS4. NarI harbors two b-type hemes: bP (P as proximal) and bD (D as distal). doi:10.1128/ecosalplus.ESP-0006-2013.f8
Recent progress in the functions associated with the NarJ chaperone has provided significant insights into this process (164, 165, 166) (Fig. 9). Two distinct NarJ-binding sites were mapped on the NarG catalytic subunit, one of them corresponding to the N terminus (167). NarJ binding to this region represents part of a chaperone-mediated quality control process preventing membrane anchoring of the soluble and cytoplasmic NarGH complex before all maturation events have been completed. In particular, NarJ ensures complete maturation of the b-type cytochrome NarI by proper timing for membrane anchoring of the cytoplasmic NarGH complex (164). Indeed, the absence of the proximal heme bP in a narJ strain has been interpreted as the result of a loss of coordination between maturation of the NarI and NarGH components since both hemes are inserted in the absence of the catalytic dimer and NarJ. This process strongly resembles the “Tat proofreading” of periplasmic metalloproteins, of which the best-studied example relates to EcTorA (168). This idea is further supported by the similarity between the Tat signal peptides and the N terminus of the nonexported nNarG (134, 169). Moreover, in archaea and some bacteria, the NarG sequence harbors a typical Tat signal peptide responsible for the periplasmic localization of the NarGH complex (see for review, reference 2). A second yet undefined NarJ-binding site within the catalytic subunit NarG is responsible for sequential insertion of the FeS cluster (FS0) followed by Mo-bisPGD (164). The exact function of NarJ in this process is unclear. Nevertheless, NarJ is an indispensable component of the Moco insertional process in authorizing the interaction of the apoenzyme with a complex made up of several cofactor biosynthetic proteins in charge of Moco delivery (167).
Figure 9.
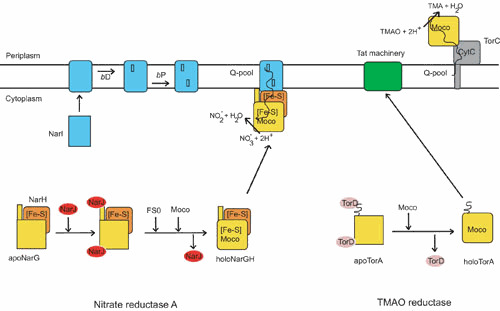
Biogenesis model of the E. coli nitrate reductase A (NarGHI) and the TMAO reductase (TorCA) complexes. NarG and NarH constitute the catalytic dimer, while NarI is the b-type membrane anchor subunit of the complex. NarI maturation takes place in the inner membrane where the two b-type bD and bP hemes are sequentially inserted. Concomitantly, the apoNarGH complex retained by the enzyme-specific chaperone NarJ in the cytoplasm is maturated. First, [Fe-S] clusters are inserted in the NarH subunit through the action of one of the [Fe-S] biosynthetic machineries. Second, both Moco and its proximal [Fe-S] cluster, FS0, are inserted in the catalytic subunit NarG in a NarJ-dependent manner. On complete maturation of the NarGH complex, NarJ dissociates, allowing membrane anchoring of the NarGH dimer. TorA constitutes the catalytic subunit of the TMAO reductase system and harbors a twin-arginine signal peptide at the N terminus. Early interaction of the enzyme-specific chaperone TorD on apoTorA facilitates Moco insertion. Subsequently, mature TorA is exported to the periplasm through the Tat translocase. TorC, a pentahemic membrane-bound cytochrome c, constitutes the electron donor to TorA. Whatever the considered system, Moco insertion proceeds as the action of a multiprotein complex of Moco biosynthetic proteins and chaperones (70, 125). doi:10.1128/ecosalplus.ESP-0006-2013.f9
A combination of biophysical approaches allowed for the recognition mode between NarJ and the N terminus of NarG to be deciphered. NMR structural analysis demonstrated that the N terminus of NarG adopts a helical conformation in solution that remains largely unchanged upon NarJ binding (165). NarJ recognizes and binds the helical NarG(1–15) peptide within a highly conserved and elongated groove, mostly via hydrophobic interactions (165, 166). Mechanistically, isothermal titration calorimetry and BIAcore experiments support a model whereby the protonated state of the chaperone controls the time dependence of peptide interaction (165). Protonation of a specific residue of NarJ increases the affinity toward the N terminus of NarG, in particular, via the lifespan of the complex. A tentative model for the physiological NarJ chaperone cycle can be deduced from these data and initiates with the rapid binding of the N terminus of NarG by the protonated chaperone at high affinity (KD ∼3 nM) followed by its release at a later stage of the process by a deprotonated chaperone at lower affinity (KD ∼3 µM) and reduced lifespan once cofactor loading and protein folding are complete. The nature of the signal that may trigger dissociation of the complex remains unclear; however, preliminary results from structural analysis of the apo-Mo-NarGH complex produced in the absence of NarJ are consistent with a substantial conformational change of NarG allowing access to the interior of the protein to metal cofactors (Magalon A, unpublished data). Structural modifications associated with metal cofactor insertion within NarG may thus be responsible for NarJ dissociation from the N terminus.
A second yet undefined NarJ-binding site within the catalytic subunit NarG is responsible for sequential insertion of the proximal [Fe-S] cluster (FS0) and of the Mo-bisPGD (164). Moreover, NarJ-assisted FS0 insertion constitutes a prerequisite for Mo-bisPGD insertion (Fig. 9). This was later confirmed by X-ray structural analysis of specific mutants of the FS0 coordination sphere (170). Overall, while the lack of Mo-bisPGD does not preclude FS0 insertion, substitution of cysteine ligands of the FS0 cluster or the absence of NarJ prevents insertion both of FS0 and of Mo-bisPGD. The exact function of NarJ in this process is unclear. Nevertheless, NarJ is an indispensable component of the Mo-bisPGD insertional process in authorizing the interaction of apoNarGH with a complex made up of several cofactor biosynthetic proteins in charge of Mo-bisPGD delivery (133).
The multifunctional character of this enzyme-specific chaperone raises the question, at a structural level, of how this can be achieved. Indeed, as mentioned above, NarJ belongs to a new family of proteins sharing a common fold (Pfam PF02613). It is tempting to speculate that the ability of NarJ to recognize and interact with distinct sites of a metalloprotein partner or with several metalloprotein partners is based on a structural flexibility. Initial NMR studies conducted on E. coli NarJ reported a global change of the 1H,15N-HSQC spectrum of NarJ upon peptide binding, which has been interpreted as the result of a conformational change upon complex formation. Recently, the combination of site-directed spin-labeling electron paramagnetic resonance spectroscopy and ion mobility mass spectrometry has unearthed conformational substates of the dedicated chaperone NarJ and during the partner binding process, revealed distinct molecular species and conformational dynamics (166). In particular, NarJ is represented by at least three discrete conformations in equilibrium. Furthermore, evidence was provided for the existence of a conformational selection mechanism operating during binding of the N terminus of NarG by NarJ. Therefore, the binding of the N terminus of NarG results from the selection of an accessible conformation of the NarJ chaperone and its further rearrangement induced upon protein recognition. It may be anticipated that a structural flexibility of the chaperone is at play for other members and even is a common feature as deduced from the observation of several disordered regions (154, 155, 171). In support of this idea, different folding forms of the EcTorA-specific chaperone TorD are associated with different biological activities (172). The function of these chaperone proteins is not restricted to the recognition and binding of the N terminus of the nascent metalloprotein, but includes their participation in metal cofactor insertion processes through additional contacts with their specific partner. However, no information is available concerning the binding interface for the multiple partners of the REMPs involved in metal center delivery during the assembly process of the cognate metalloprotein. In this context, stabilization of a specific conformer of NarJ with NarG(1–15) peptide binding and thus redistribution of the protein conformational ensembles recalls allostery, a mechanism by which binding of the ligand at one site can affect binding of others through a propagated change in the protein shape (173). Overall, the shift in population resulting from binding the N terminus of the cognate Mo-enzyme could be one of the keys to facilitate subsequent binding of additional partners at yet unidentified sites of NarJ.
In the absence of NarJ, a global defect in metal incorporation into NarGHI is observed. In addition to both metal cofactors of the catalytic subunit NarG, the proximal heme bP is absent because of the loss of coordination between maturation of the NarI and NarGH components (164). Finally, the absence of NarJ did not affect the stability or the cellular distribution of the apoenzyme that remains largely associated to the cytoplasmic membrane (167, 174). An explanation may derive from the analysis of the apoNarGHI structures lacking either the Mo-bisPGD (PDB ID code 1siw) (175) or both FS0 and Mo-bisPGD (PDB ID code 3ir6) (170). In both cases, GDP moieties can be inserted into positions corresponding to GDP-P and GDP-D, thus conferring a relative structural stability to the enzymatic complex. Such a situation has also been encountered in the case of the CO dehydrogenase from Hydrogenophaga pseudoflava expressed from tungstate-grown cells (176) or in the case of the Rhodobacter sphaeroides DorA protein heterologously expressed in a Mo-bisPGD deficient E. coli strain (109). One could envision that the chaperone either catalyzes the rapid exchange of the nucleotides for Mo-bisPGD or prevents nucleotide insertion through direct contacts with the metalloenzyme.
Altogether, these data demonstrated that NarJ orchestrates metal cofactor insertion, subunit assembly, and membrane-anchoring steps during the maturation of the NarGHI complex (Fig. 8). NarJ proofreads metal center insertion within the catalytic subunit before membrane anchoring through binding to a remnant Tat signal peptide. The underlying mechanism described herein is comparable to the one that occurs during translocation of Tat substrates. Importantly, it can be inferred from comparison with other multimeric Mo-enzymes that the multiple functions played by NarJ in the biogenesis process of the nitrate reductase complex will be extended to other related systems (Fig. 9). Indeed, the phylogenetic tree from sequence comparison of the catalytic subunit of all known archaeal and bacterial Mo-enzymes belonging to the Mo/W-bisPGD family indicates that all contain a [Fe-S] cluster in close proximity to the Mo/W-bisPGD with the exception of a small clade including DorA/TorA/BisC enzymes only present in proteobacteria (8, 9). This taxonomic distribution indicates that the entire group of DorA/TorA/BisC enzymes arose at a late stage on the microbial evolutionary timescale. Accordingly, their REMPs (with TorD as prototype) may have lost the function associated with the strict requirement in FS0 insertion prior to Mo-bisPGD, as seen in NarJ.
The periplasmic and multimeric Mo-bisPGD enzymes
The DMSO reductase DmsABC and formate dehydrogenase FdnGHI, two periplasmically oriented and membrane-bound heterotrimeric complexes in E. coli, share strong similarities in terms of subunit and redox cofactor composition with the NarGHI complex and may follow the same folding and assembly pathways and be assisted by a system-specific chaperone sharing functional similarities to NarJ.
Oresnik et al. reported that DmsD is essential for the assembly of a fully active DmsABC complex (140). The dmsD gene (formerly ynfI) is part of the ynf operon encoding putative Tat targeted selenate reductases (177, 178). Interestingly, DmsD ensures the folding and assembly of both the DmsABC and the two putative selenate reductases, which contrasts with the usual exquisite specificity displayed by the so-called enzyme-specific chaperones (178). The strong sequence similarity of the corresponding catalytic subunits may explain such behavior. X-ray structural analysis of DmsD from E. coli (PDB ID codes 3efp and 3cw0) (157, 158) and Salmonella enterica serovar Typhimurium (PDB ID code 1s9u) (156) indicate an all-helical architecture as in NarJ and TorD. DmsD displays a structural plasticity, as revealed by the existence of different folding forms (171) or a disordered loop in the X-ray data (156), which may be beneficial for interaction with multiple partners as suggested above for NarJ. Furthermore, DmsD was isolated as a protein binding the Tat signal peptide of DmsA with a KD ∼220 nM (140, 171). Its ability to interact with two components of the Tat translocase, namely TatB and TatC located in the inner membrane, suggests a role in delivering the DmsAB substrate to the TatBC receptor complex (179, 180). More recently, DmsD has been shown to interact with general chaperones and with several Moco biosynthesis proteins extending its interactome (150). Combining genetic, biochemical, and structural analysis on EcDmsD, a pocket of surface residues important for signal peptide binding was modeled and is made up of hydrophobic sections of three conserved loops (158, 181). Microcalorimetric analysis of peptide binding by either NarJ, DmsD, or TorD chaperones provide further support to the hydrophobic character of the interaction (165, 171, 182). Finally, deletion of the DmsA signal peptide results in the formation of a less stable but soluble and cytoplasmically active DmsAB complex (183). Accordingly, the DmsA variant has likely benefited from the action of DmsD to a second unidentified binding site for metal cofactor acquisition. Concerted action of DmsD on both the signal peptide and most likely on a second site of the DmsA protein, in the same way as NarJ, is thus required for productive synthesis of DmsABC. For instance, Weiner’s group recently reached the same conclusion to the NarGHI enzyme (164) by showing that FS0 insertion in the catalytic subunit DmsA is a prerequisite for Mo-bisPGD insertion (184). In the case of NarGHI, NarJ chaperone has been shown to be essential for FS0 insertion.
A number of periplasmic or periplasmically oriented multimeric molybdo-enzymes of the Mo/W-bisPGD family have been genetically or biochemically characterized in different bacteria or archaea, such as the selenate (185), nitrate (186), chlorate (187), or perchlorate (188) reductases and the ethylbenzene (189) or dimethyl sulfide (190) dehydrogenases. Considering their relatedness to the NarGHI or DmsABC complexes and the existence of an additional gene encoding for a NarJ/TorD/DmsD-like chaperone protein in the corresponding operons that include similar phylogenetic traits (9), it is tempting to speculate that the folding and assembly of these Mo-enzymes will follow the same trend (Fig. 9). Exceptions are the periplasmic and multimeric arsenite oxidase (Aro), polysulfide (Psr), and arsenate reductase (Arr) enzymes that do not possess in their respective operons an additional gene encoding for a REMP (191, 192). However, the presence of a Tat signal peptide and of a [Fe-S] cluster together with the Mo-bisPGD in the catalytic subunit support the hypothesis that their maturation pathway will need assistance by an as yet unidentified REMP that may be located elsewhere in the genome. Examination of the phylogenetic tree that can be inferred from comparative sequence analysis of the catalytic subunit of those members of the Mo/W-bisPGD family discloses the singular situation of the periplasmic nitrate reductases (Nap) and formate dehydrogenases forming two separate but closely related phyla and distant from the others (9). As developed below, peculiar situations are encountered in those enzymes with the participation of REMPs that do not belong to the NarJ family (Pfam PF02613).
E. coli synthesizes three formate dehydrogenases. Two of these are periplasmically oriented membrane-bound respiratory complexes, namely, the nitrate-inducible FdnGHI and the cryptic FdoGHI (193). Typical Tat signal peptides are present on the FdnG and FdoG catalytic subunits. The third formate dehydrogenase, composed of a single cytoplasmic subunit (FdhF), is part of the formate hydrogenlyase complex (194). All three isoenzymes harbor a [4Fe-4S] cluster in addition to the Mo-bisPGD cofactor in their catalytic subunit. Genetic studies have demonstrated that both fdhD and fdhE genes, located astride the fdo operon, are involved in the formation of active formate dehydrogenase enzymes, fdhE being restricted to periplasmic ones (136, 153, 195, 196, 197). Interestingly, FdhD and FdhE do not share any structural similarity with other REMPs such as NarJ/TorD/DmsD. FdhD contains several cysteine residues and displays a mostly helical architecture as revealed by the crystal structure of FdhD from E. coli (PDB ID code 4PDE) (Fig. 7) (125). Careful analysis of the X-ray crystal structure of E. coli FdhD in complex with GDP shows the presence of a structural motif known to interact with nucleotides: a α/β-Rossmann fold. FdhE is an iron-binding rubredoxin that possesses four conserved CX2C motifs essential both for FdhE stability and biological function (198). As disclosed by the crystal structure at 2.1 Å of FdhE from Pseudomonas aeruginosa PAO1 (PDB ID code 2fiy), each two pairs of CX2C motifs located in disordered loops coordinate an iron atom with an as-yet unclear role (Fig. 7). It has to be mentioned that, contrary to FdhD, FdhE is only required for the activity of periplasmically located formate dehydrogenases, surmising its involvement in the translocation event. However, if FdhE was shown to interact both with the FdnG and FdoG catalytic subunits, a direct role for a Tat proofreading chaperone has not yet been demonstrated (198). Recently, Thome et al (124) reported that FdhD functions as a sulfurtransferase between the major cysteine desulfurase IscS and FdhF. As described above, Arnoux et al. (125) reported Mo-bisPGD binding in vivo and proposed a working model where FdhD ensures sulfuration of Moco and its subsequent protected transfer to apo-formate dehydrogenases, thereby combining sulfurtransferase and chaperone activities. Such a mechanistic model not only explains the presence of inorganic sulfur at the metal ion coordination sphere of Mo- or W-formate dehydrogenases (117, 118), but also its essential character for the reactivity.
As in the case of the E. coli formate dehydrogenases, folding and assembly of the periplasmic nitrate reductase (NapAB) involves two cytoplasmic proteins, NapD and NapF, which do not display any sequence or structure similarity with other REMPs. Maillard et al. (199) reported that E. coli NapD displayed a ferredoxin-type fold and is involved in Tat signal peptide binding. Recently, NMR studies reported that the Tat signal peptide of NapA adopts a helical conformation during complex formation with NapD and binds mostly through hydrophobic contacts (200). Despite the unrelated structures of NarJ and NapD, a similar situation has been observed with a helical conformation of the remnant signal peptide of EcNarG (165). Moreover, detailed analysis of the binding process by ITC revealed the existence of two distinct populations of NapD, a minor one (35%) having an apparent KD of ∼3 nM and a major one (64%) having a much higher KD of ∼140 nM (200). While no molecular explanation for such a phenomenon in NapD is provided, protonation of a specific residue within the binding pocket of NarJ was shown to be responsible for such variation of the binding constants. Altogether, these similarities may point toward a common mode of action. On the contrary, NapF interacts directly with the catalytic subunit NapA, and may be involved in NapA folding and assembly prior to export via the Tat translocon (201). NapF harbors four conserved tetracysteine motifs coordinating labile [Fe-S] clusters. Interestingly, NapF from R. sphaeroides was shown to be required for [Fe-S] cluster insertion within the catalytic subunit NapA (202). Recently, Kern and Simon reported that a napF knockout mutant accumulates the inactive cytoplasmic NapA precursor in Wolinella succinogenes (203). However, there are many examples of organisms that lack napF such as Campylobacter species. One may envision that the napF gene could have been lost during evolution if it had been functionally redundant in the cell.
In addition to formate dehydrogenases, the Mo/W-bisPGD family includes other members for which an additional sulfur ligand of the Mo atom is encountered at the catalytic site. The X-ray crystal structure of the periplasmic nitrate reductase (Nap) of Cupriavidus necator was resolved at 1.5 Å and shows the presence of a terminal sulfur ligand at the molybdenum coordination sphere (119). These results confirm X-ray data obtained on the homologous NapA from Desulfovibrio desulfuricans ATCC 27774, which revealed a unique coordination sphere of six sulfur ligands bound to the Mo atom (120). Although the resolution was not sufficient to ascertain the identity of the Mo ligands in two other Nap structures (204, 205), altogether, these observations raise the question of the sulfuration of the Moco being a general feature of this group of enzymes. The mechanism could be enzyme-specific, as described for FdhD or for the R. capsulatus xanthine dehydrogenase that requires XdhC for sulfuration (121, 122) (see below). Alternatively, the sulfuration mechanism for Nap could involve a sulfurtransferase dedicated to several enzymes, as is the case for ABA3, the A. thaliana Moco sulfurase of all members of the XO family. ABA3 carries L-cysteine desulfurase activity and interacts with the Moco for sulfuration of both xanthine oxidase and aldehyde oxidase (206, 207, 208). In this regard, further work is required for the identification of the sulfurtransferase that would be required for the sulfuration and thus enzymatic activity of periplasmic nitrate reductases.
The periplasmic and monomeric Mo-bisPGD enzymes
While biogenesis of multimeric Mo-enzymes harboring a [Fe-S] cluster and a Mo-bisPGD in the catalytic subunit appears to be intricate and requires a REMP, what is the situation in those monomeric Mo-enzymes (TorA/DorA/BisC) harboring Mo-bisPGD as a sole prosthetic group? Among them, BisC is the unique nonexported Mo-enzyme, and it is unclear whether Moco insertion is assisted by a REMP. Conversely, formation of an active and periplasmically localized TorA/DorA enzyme relies on the action of REMPs.
In E. coli, reduction of TMAO is mainly ensured by a respiratory system encoded by the inducible torCAD operon (209, 210). The terminal reductase, TorA, is a periplasmic Mo-enzyme of 97 kDa harboring Mo-bisPGD cofactor as the sole prosthetic group as disclosed by the X-ray structure of the Shewanella massilia counterpart (211). The last gene of the tor operon, torD, encodes a cytoplasmic protein of 23 kDa behaving as a private chaperone of TorA (138). Interestingly, the TorD chaperone from S. massilia forms multiple and stable oligomeric species and has been crystallized as a dimer with domain swapping between the two monomers having an all-helical fold (PDB ID code 1n1c) (Fig. 7) (154, 212). Nevertheless, the biological significance of these oligomers is still under debate, because no gain of function in terms of binding affinity, usually encountered through domain swapping, has been observed on dimerization. Pommier et al. (138) reported that a torD knockout mutant produces twofold less but still active and correctly localized TorA. Furthermore, the produced enzyme in a torD strain is fully degraded under thermal stress conditions, Moco deficiency, or molybdenum starvation (213). Instability of the apo-Mo-enzyme is also observed with RcDorA, in the absence of the REMP DorD (139). These observations differ markedly from the multimeric nitrate reductase where the absence of the NarJ chaperone does not significantly affect the stability and the cellular localization of the enzyme complex (167, 174). An explanation can be provided by the initial formation of a stable apo-NarGH complex prior to the action of NarJ in comparison with the monomeric DorA/TorA enzymes.
As in the case of NarJ or DmsD, TorD interacts on two distinct sites of the TorA enzyme; one of them is the Tat signal peptide located at the N terminus (182, 214). This property is a general principle for this new class of chaperones (Pfam PF02613). Considering the periplasmic location of the TorA enzyme and the fact that Moco incorporation is a strictly cytoplasmic event, it is important that export is prevented until all assembly processes are complete. One of the mechanisms that ensures proper coordination between cofactor insertion and export is based on the ability of the chaperone to shield the signal peptide from interaction with the Tat translocase until Moco insertion proceeds (168). This mechanism is reminiscent of the protection of the N terminus of NarG by NarJ to allow proper timing for membrane anchoring of the soluble and cytoplasmic NarGH complex (164, 167). Interestingly, while TorD has a weak affinity for GTP (KD ∼ 370 µM), which is enhanced by signal peptide binding (182), only the dimeric form displays a low intrinsic magnesium-dependent GTP hydrolysis activity despite the absence of classical nucleotide-binding motifs (172). The role of nucleotide binding or hydrolysis in modulating the interaction between the signal peptide and TorD remains unclear. Alternatively, Genest et al. reported TorA signal peptide protection by TorD regardless of the presence of Moco and or the Tat translocase (215). At this stage, it is not clear whether TorD binding to the signal peptide monitors the folding and assembly of the substrate, or alternatively, retards the export kinetics sufficiently to allow completion of the Moco insertion process.
A second TorD-binding site was established on the core of the TorA enzyme and is responsible for a conformational change of the latter (138). With the use of an in vitro system, Mo-bisPGD insertion within apo-TorA was shown to be strongly facilitated by the presence of TorD with no significant effect upon removal of the signal peptide as judged by TorA activity (216, 217). In this context, SAXS experiments have been conducted in E. coli on a complex made between TorD and apoTorA with or without its Tat signal peptide (218). These studies further confirm that a stable complex between the chaperone and its target can be made independently of the signal peptide. Binding to the core of TorA is mediated by helix 5 of TorD and appears to be necessary for Mo-bisPGD insertion (214). In this context, TorD was shown to interact with Moco biosynthesis components, including MobA, and Mo-PPT (214). The authors suggest that TorD, while being dispensable, acts as a platform connecting the last step of the synthesis of the Mo-bisPGD with its insertion into TorA. The immediate question raised by these observations is whether this role is shared by other REMPs. An answer to this question awaits further studies on various chaperones.
Maturation of Mo-PPT Enzymes
While E. coli has been considered for a long time to only synthesize Mo-bisPGD cofactor, a Mo-PPT-dependent enzyme (YedYZ) belonging to the SO family (101), and members of the XO family using Mo-PCD as cofactor (XdhABC, XdhD and PaoABC), as well, were also identified in this organism (94, 102, 103). A clear distinction between the sulfite oxidases and xanthine oxidoreductases is the presence of a sulfur atom on the Mo coordination sphere in the latter. A conserved posttranslational mechanism from bacteria to eukaryotes for Moco sulfuration has been studied in detail during the past decade where a Moco-binding protein, through direct interaction with a cysteine desulfurase, ensures both the sulfuration step of the cofactor and its protected transfer to the apo-Mo-enzymes. Whereas, in eukaryotes, the two components are fused into a single polypeptide, also called Moco sulfurase (206), this is not the case in prokaryotes where a system-specific chaperone is operating together with a cysteine desulfurase (121, 122).
We will review the incorporation of sulfurated Mo-PPT using R. capsulatus xanthine dehydrogenase as the model. This enzyme consists of a cytoplasmic heterodimeric complex (α2β2) that catalyzes the hydroxylation of hypoxanthine and xanthine, the last two steps in purine degradation (128). The XdhA subunit contains two [2Fe-2S] clusters in addition to flavin adenine dinucleotide (FAD), while the XdhB subunit binds Mo-PPT (219) as confirmed by the crystal structure (220). Functional synthesis of the R. capsulatus XdhAB complex requires the presence of an additional protein, XdhC (33 kDa), encoded by the xdhABC operon, which entails binding and sulfuration of Mo-PPT and participation in the insertional process into XdhB subunit (146). Considering the structure of the heterotetrameric enzyme and the presence of three different metal centers, biochemical and biophysical studies revealed that assembly of XdhAB is a multistep process that occurs in an ordered manner. It involves the synthesis and interaction of both subunits, followed by [Fe-S] cluster and FAD insertion within XdhA, dimerization of the XdhAB complex, and, finally, insertion of sulfurated Moco into XdhB, resulting in an active enzyme (221). Such a model is reminiscent of the nitrate reductase complex where insertion of [Fe-S] clusters precedes Moco insertion (164). Furthermore, scrutiny of the crystal structure of the bovine xanthine oxidase allowed Hille et al (128) to envision that rotation of a small conserved domain of the protein is sufficient to provide access to the interior of the complex as well as defining a new interacting motif for the Moco insertion machinery. Clearly, experimental investigations of the Moco insertion process into a complex enzyme structure, which may have to accommodate other metal centers beforehand, are most required.
On the other extreme, it would be interesting to compare the situation with a Mo-PPT enzyme of the sulfite oxidase family that does not require sulfuration of the cofactor. To date, no REMPs have been reported to be involved in maturation of such enzymes. In particular, E. coli YedY is a soluble periplasmic oxidoreductase that contains Mo-PPT as the unique redox cofactor as disclosed by the crystal structure (101).
CONCLUDING REMARKS
Our understanding of the biological role and the function of Mo is progressing rapidly. Now that most of the relevant genes are cloned and the principles of Moco biosynthesis are known, research concentrates on the steps beyond Moco biosynthesis, i.e., studying the release of Moco from the biosynthetic complex, its transfer and possible storage, its modification via sulfuration, and insertion of Moco into diverse target enzymes. The latter can be grouped into two classes. Class 1 is represented by enzymes harboring Moco as the sole redox cofactor in their catalytic subunit. This class groups essentially monomeric enzymes of the Mo/W-bisPGD family (Tor, Dor, Bis), but also members of the SO and XO family being either monomeric (YedY) or multimeric (XdhA, XdhD, PaoC). Class 2 represents those enzymes (Nar, Fdn, Nap, and Dms) that are multimeric and harbor a [Fe-S] cluster in addition to Moco in their catalytic subunit. Genomic analyses indicate that most of prokaryotic Mo-enzymes fall into class 2 and belong to the Mo/W-bisPGD family. Accordingly, their REMPs are markedly different: chaperones specific for class 1 enzymes only facilitate Moco insertion (with TorD as the prototype) for Tor/Dor/Bis enzymes or add a sulfuration step before its insertion for XO enzymes, while those specific for class 2 enzymes (with NarJ as the prototype) orchestrate sequential incorporation of both the [Fe-S] cluster and Moco into the catalytic subunit. In the specific case of formate dehydrogenases and periplasmic nitrate reductases, the associated REMPs would ensure sulfuration of the cofactor and its subsequent insertion in a coordinated fashion into a catalytic subunit harboring a FeS cluster. There are several open questions to address. How is the multienzyme complex for Moco biosynthesis organized? What is the detailed mechanism of Moco insertion into target enzymes? In particular, how is Moco trafficked from the multienzyme complex to the different apoenzymes, and what is the exact function played by class 2 REMPs in this process? How is Moco sulfurated for enzymes that need a terminal sulfur ligand in the Mo center? How is Moco biosynthesis regulated to meet the changing demands of the cell for Moco? The coming years will bring insight into the integration and (perhaps unexpected) regulatory connections of Moco biosynthesis and Mo-enzymes within the metabolic and physiological network of the cell.
ACKNOWLEDGMENTS
We would like to apologize to all those whose important contributions to the field could not be cited because of space limitations. A.M. acknowledges support from the Centre National de la Recherche Scientifique, Aix-Marseille University, and the Agence Nationale de la Recherche. R.R.M. acknowledges support from the Deutsche Forschungsgemeinschaft.
REFERENCES
- 1.Mendel RR, Kruse T. 2012. Cell biology of molybdenum in plants and humans. Biochim Biophys Acta 1823:1568–1579. [PubMed] 10.1016/j.bbamcr.2012.02.007 [DOI] [PubMed] [Google Scholar]
- 2.Grimaldi S, Schoepp-Cothenet B, Ceccaldi P, Guigliarelli B, Magalon A. 2013. The prokaryotic Mo/W-bisPGD enzymes family: a catalytic workhorse in bioenergetic. Biochim Biophys Acta 1827:1048–1085. [PubMed] 10.1016/j.bbabio.2013.01.011 [DOI] [PubMed] [Google Scholar]
- 3.Williams RJ, Frausto da Silva JJ. 2002. The involvement of molybdenum in life. Biochem Biophys Res Commun 292:293–299. [PubMed] 10.1006/bbrc.2002.6518 [DOI] [PubMed] [Google Scholar]
- 4.Hille R. 2002. Molybdenum enzymes containing the pyranopterin cofactor: an overview. Met Ions Biol Syst 39:187–226. [PubMed] 10.1201/9780203909331.ch6 [DOI] [PubMed] [Google Scholar]
- 5.Cvetkovic A, Menon AL, Thorgersen MP, Scott JW, Poole FL, 2nd, Jenney FE, Jr., Lancaster WA, Praissman JL, Shanmukh S, Vaccaro BJ, Trauger SA, Kalisiak E, Apon JV, Siuzdak G, Yannone SM, Tainer JA, Adams MW. 2010. Microbial metalloproteomes are largely uncharacterized. Nature 466:779–782. [PubMed] 10.1038/nature09265 [DOI] [PubMed] [Google Scholar]
- 6.Bevers LE, Hagedoorn PL, Hagen WR. 2009. The bioinorganic chemistry of tungsten. Coord Chem Rev 253:269–290. 10.1016/j.ccr.2008.01.017 [DOI] [Google Scholar]
- 7.Schneider F, Lowe J, Huber R, Schindelin H, Kisker C, Knablein J. 1996. Crystal structure of dimethyl sulfoxide reductase from Rhodobacter capsulatus at 1.88 A resolution. J Mol Biol 263:53–69. [PubMed] 10.1006/jmbi.1996.0555 [DOI] [PubMed] [Google Scholar]
- 8.Rothery RA, Workun GJ, Weiner JH. 2008. The prokaryotic complex iron-sulfur molybdoenzyme family. Biochim Biophys Acta 1778:1897–1929. [PubMed] 10.1016/j.bbamem.2007.09.002 [DOI] [PubMed] [Google Scholar]
- 9.Schoepp-Cothenet B, van Lis R, Philippot P, Magalon A, Russell MJ, Nitschke W. 2012. The ineluctable requirement for the trans-iron elements molybdenum and/or tungsten in the origin of life. Sci Rep 2:263. 10.1038/srep00263 [PubMed] 10.1038/srep00263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Magalon A, Fedor JG, Walburger A, Weiner JH. 2011. Molybdenum enzymes in bacteria and their maturation. Coord Chem Rev 255:1159–1178. 10.1016/j.ccr.2010.12.031 [DOI] [Google Scholar]
- 11.Pateman JA, Cove DJ, Rever BM, Roberts DB. 1964. A common co-factor for nitrate reductase and xanthine dehydrogenase which also regulates the synthesis of nitrate reductase. Nature 201:58–60. [PubMed] 10.1038/201058a0 [DOI] [PubMed] [Google Scholar]
- 12.Cove DJ, Pateman JA. 1963. Independently segregating genetic loci concerned with nitrate reductase activity in Aspergillus nidulans. Nature 198:262–263. [PubMed] 10.1038/198262a0 [DOI] [PubMed] [Google Scholar]
- 13.Johnson JL, Hainline BE, Rajagopalan KV. 1980. Characterization of the molybdenum cofactor of sulfite oxidase, xanthine, oxidase, and nitrate reductase. Identification of a pteridine as a structural component. J Biol Chem 255:1783–1786. [PubMed] [PubMed] [Google Scholar]
- 14.Pichinoty F, Puig J, Chippaux M, Bigliardi-Rouvier J, Gendre J. 1969. [Studies of bacterial mutants that have lost catalytic activities associated with nitrate reductase A. II. Behavior toward chlorate and chlorite]. Ann Inst Pasteur (Paris) 116:409–432. [PubMed] [Google Scholar]
- 15.Piechaud M, Pichinoty F, Azoulay E, Couchoud-Beaumont P, Gendre J. 1969. [Study of bacterial mutants that have lost catalytic activity associated with nitrate reductase A. I. Description of isolation methods]. Ann Inst Pasteur (Paris) 116:276–287. [PubMed] [PubMed] [Google Scholar]
- 16.Stewart V, MacGregor CH. 1982. Nitrate reductase in Escherichia coli K-12: involvement of chlC, chlE, and chlG loci. J Bacteriol 151:788–799. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shanmugam KT, Stewart V, Gunsalus RP, Boxer DH, Cole JA, Chippaux M, DeMoss JA, Giordano G, Lin EC, Rajagopalan KV. 1992. Proposed nomenclature for the genes involved in molybdenum metabolism in Escherichia coli and Salmonella typhimurium. Mol Microbiol 6:3452–3454. [PubMed] 10.1111/j.1365-2958.1992.tb02215.x [DOI] [PubMed] [Google Scholar]
- 18.Hagen W. 2011. Cellular uptake of molybdenum and tungsten. Coord Chem Rev 255:1117–1128. 10.1016/j.ccr.2011.02.009 [DOI] [Google Scholar]
- 19.Glaser JH, DeMoss JA. 1971. Phenotypic restoration by molybdate of nitrate reductase activity in chlD mutants of Escherichia coli. J Bacteriol 108:854–860. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scott D, Amy NK. 1989. Molybdenum accumulation in chlD mutants of Escherichia coli. J Bacteriol 171:1284–1287. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pau RN, Lawson DM. 2002. Transport, homeostasis, regulation, and binding of molybdate and tungstate to proteins. Met Ions Biol Syst 39:31–74. [PubMed] 10.1201/9780203909331.ch2 [DOI] [PubMed] [Google Scholar]
- 22.McNicholas PM, Rech SA, Gunsalus RP. 1997. Characterization of the ModE DNA-binding sites in the control regions of modABCD and moaABCDE of Escherichia coli. Mol Microbiol 23:515–524. [PubMed] 10.1046/j.1365-2958.1997.d01-1864.x [DOI] [PubMed] [Google Scholar]
- 23.Anderson LA, Palmer T, Price NC, Bornemann S, Boxer DH, Pau RN. 1997. Characterisation of the molybdenum-responsive ModE regulatory protein and its binding to the promoter region of the modABCD (molybdenum transport) operon of Escherichia coli. Eur J Biochem 246:119–126. [PubMed] 10.1111/j.1432-1033.1997.00119.x [DOI] [PubMed] [Google Scholar]
- 24.Hall DR, Gourley DG, Duke EM, Leonard GA, Anderson LA, Pau RN, Boxer DH, Hunter WN. 1999. Two crystal forms of ModE, the molybdate-dependent transcriptional regulator from Escherichia coli. Acta Crystallogr D Biol Crystallogr 55(Pt 2):542–543. [PubMed] 10.1107/S0907444998011354 [DOI] [PubMed] [Google Scholar]
- 25.Gourley DG, Schuttelkopf AW, Anderson LA, Price NC, Boxer DH, Hunter WN. 2001. Oxyanion binding alters conformation and quaternary structure of the c-terminal domain of the transcriptional regulator mode. Implications for molybdate-dependent regulation, signaling, storage, and transport. J Biol Chem 276:20641–20647. [PubMed] 10.1074/jbc.M100919200 [DOI] [PubMed] [Google Scholar]
- 26.Schuttelkopf AW, Boxer DH, Hunter WN. 2003. Crystal structure of activated ModE reveals conformational changes involving both oxyanion and DNA-binding domains. J Mol Biol 326:761–767. [PubMed] 10.1016/S0022-2836(02)01358-X [DOI] [PubMed] [Google Scholar]
- 27.McNicholas PM, Chiang RC, Gunsalus RP. 1998. Anaerobic regulation of the Escherichia coli dmsABC operon requires the molybdate-responsive regulator ModE. Mol Microbiol 27:197–208. [PubMed] 10.1046/j.1365-2958.1998.00675.x [DOI] [PubMed] [Google Scholar]
- 28.Self WT, Grunden AM, Hasona A, Shanmugam KT. 1999. Transcriptional regulation of molybdoenzyme synthesis in Escherichia coli in response to molybdenum: ModE-molybdate, a repressor of the modABCD (molybdate transport) operon is a secondary transcriptional activator for the hyc and nar operons. Microbiology 145(Pt 1):41–55. [PubMed] 10.1099/13500872-145-1-41 [DOI] [PubMed] [Google Scholar]
- 29.Anderson LA, McNairn E, Leubke T, Pau RN, Boxer DH. 2000. ModE-dependent molybdate regulation of the molybdenum cofactor operon moa in Escherichia coli. J Bacteriol 182:7035–7043. [PubMed] 10.1128/JB.182.24.7035-7043.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grunden AM, Ray RM, Rosentel JK, Healy FG, Shanmugam KT. 1996. Repression of the Escherichia coli modABCD (molybdate transport) operon by ModE. J Bacteriol 178:735–744. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Makdessi K, Andreesen JR, Pich A. 2001. Tungstate uptake by a highly specific ABC transporter in Eubacterium acidaminophilum. J Biol Chem 276:24557–24564. [PubMed] 10.1074/jbc.M101293200 [DOI] [PubMed] [Google Scholar]
- 32.Andreesen JR, Makdessi K. 2008. Tungsten, the surprisingly positively acting heavy metal element for prokaryotes. Ann N Y Acad Sci 1125:215–229. [PubMed] 10.1196/annals.1419.003 [DOI] [PubMed] [Google Scholar]
- 33.Bevers LE, Hagedoorn PL, Krijger GC, Hagen WR. 2006. Tungsten transport protein A (WtpA) in Pyrococcus furiosus: the first member of a new class of tungstate and molybdate transporters. J Bacteriol 188:6498–6505. [PubMed] 10.1128/JB.00548-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grunden AM, Shanmugam KT. 1997. Molybdate transport and regulation in bacteria. Arch Microbiol 168:345–354. [PubMed] 10.1007/s002030050508 [DOI] [PubMed] [Google Scholar]
- 35.Rosentel JK, Healy F, Maupin-Furlow JA, Lee JH, Shanmugam KT. 1995. Molybdate and regulation of mod (molybdate transport), fdhF, and hyc (formate hydrogenlyase) operons in Escherichia coli. J Bacteriol 177:4857–4864. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baker KP, Boxer DH. 1991. Regulation of the chlA locus of Escherichia coli K12: involvement of molybdenum cofactor. Mol Microbiol 5:901–907. [PubMed] 10.1111/j.1365-2958.1991.tb00764.x [DOI] [PubMed] [Google Scholar]
- 37.Regulski EE, Moy RH, Weinberg Z, Barrick JE, Yao Z, Ruzzo WL, Breaker RR. 2008. A widespread riboswitch candidate that controls bacterial genes involved in molybdenum cofactor and tungsten cofactor metabolism. Mol Microbiol 68:918–932. [PubMed] 10.1111/j.1365-2958.2008.06208.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Serganov A, Patel DJ. 2009. Amino acid recognition and gene regulation by riboswitches. Biochim Biophys Acta 1789:592–611. [PubMed] 10.1016/j.bbagrm.2009.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Patterson-Fortin LM, Vakulskas CA, Yakhnin H, Babitzke P, Romeo T. 2013. Dual posttranscriptional regulation via a cofactor-responsive mRNA leader. J Mol Biol 425:3662–3677. [PubMed] 10.1016/j.jmb.2012.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Timmermans J, Van Melderen L. 2010. Post-transcriptional global regulation by CsrA in bacteria. Cell Mol Life Sci 67:2897–2908. [PubMed] 10.1007/s00018-010-0381-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Edwards AN, Patterson-Fortin LM, Vakulskas CA, Mercante JW, Potrykus K, Vinella D, Camacho MI, Fields JA, Thompson SA, Georgellis D, Cashel M, Babitzke P, Romeo T. 2011. Circuitry linking the Csr and stringent response global regulatory systems. Mol Microbiol 80:1561–1580. [PubMed] 10.1111/j.1365-2958.2011.07663.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hasona A, Self WT, Shanmugam KT. 2001. Transcriptional regulation of the moe (molybdate metabolism) operon of Escherichia coli. Arch Microbiol 175:178–188. [PubMed] 10.1007/s002030100252 [DOI] [PubMed] [Google Scholar]
- 43.Hasona A, Self WT, Ray RM, Shanmugam KT. 1998. Molybdate-dependent transcription of hyc and nar operons of Escherichia coli requires MoeA protein and ModE-molybdate. FEMS Microbiol Lett 169:111–116. [PubMed] 10.1111/j.1574-6968.1998.tb13306.x [DOI] [PubMed] [Google Scholar]
- 44.Iobbi-Nivol C, Palmer T, Whitty PW, McNairn E, Boxer DH. 1995. The mob locus of Escherichia coli K12 required for molybdenum cofactor biosynthesis is expressed at very low levels. Microbiology 141(Pt 7):1663–1671. [PubMed] 10.1099/13500872-141-7-1663 [DOI] [PubMed] [Google Scholar]
- 45.Rajagopalan KV, Johnson JL. 1992. The pterin molybdenum cofactors. J Biol Chem 267:10199–10202. [PubMed] [PubMed] [Google Scholar]
- 46.Rajagopalan KV, Johnson JL, Wuebbens MM, Pitterle DM, Hilton JC, Zurick TR, Garrett RM. 1993. Chemistry and biology of the molybdenum cofactors. Adv Exp Med Biol 338:355–362. [PubMed] 10.1007/978-1-4615-2960-6_74 [DOI] [PubMed] [Google Scholar]
- 47.Thony B, Auerbach G, Blau N. 2000. Tetrahydrobiopterin biosynthesis, regeneration and functions. Biochem J 347(Pt 1):1–16. [PubMed] 10.1042/0264-6021:3470001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bacher A, Eberhardt S, Eisenreich W, Fischer M, Herz S, Illarionov B, Kis K, Richter G. 2001. Biosynthesis of riboflavin. Vitam Horm 61:1–49. [PubMed] 10.1016/s0083-6729(01)61001-x [DOI] [PubMed] [Google Scholar]
- 49.Wuebbens MM, Rajagopalan KV. 1995. Investigation of the early steps of molybdopterin biosynthesis in Escherichia coli through the use of in vivo labeling studies. J Biol Chem 270:1082–1087. [PubMed] 10.1074/jbc.270.3.1082 [DOI] [PubMed] [Google Scholar]
- 50.Rieder C, Eisenreich W, O’Brien J, Richter G, Gotze E, Boyle P, Blanchard S, Bacher A, Simon H. 1998. Rearrangement reactions in the biosynthesis of molybdopterin–an NMR study with multiply 13C/15N labelled precursors. Eur J Biochem 255:24–36. [PubMed] 10.1046/j.1432-1327.1998.2550024.x [DOI] [PubMed] [Google Scholar]
- 51.Wuebbens MM, Rajagopalan KV. 1993. Structural characterization of a molybdopterin precursor. J Biol Chem 268:13493–13498. [PubMed] [PubMed] [Google Scholar]
- 52.Santamaria-Araujo JA, Fischer B, Otte T, Nimtz M, Mendel RR, Wray V, Schwarz G. 2004. The tetrahydropyranopterin structure of the sulfur-free and metal-free molybdenum cofactor precursor. J Biol Chem 279:15994–15999. [PubMed] 10.1074/jbc.M311815200 [DOI] [PubMed] [Google Scholar]
- 53.Santamaria-Araujo JA, Wray V, Schwarz G. 2012. Structure and stability of the molybdenum cofactor intermediate cyclic pyranopterin monophosphate. J Biol Inorg Chem 17:113–122. [PubMed] 10.1007/s00775-011-0835-2 [DOI] [PubMed] [Google Scholar]
- 54.Hanzelmann P, Hernandez HL, Menzel C, Garcia-Serres R, Huynh BH, Johnson MK, Mendel RR, Schindelin H. 2004. Characterization of MOCS1A, an oxygen-sensitive iron-sulfur protein involved in human molybdenum cofactor biosynthesis. J Biol Chem 279:34721–34732. [PubMed] 10.1074/jbc.M313398200 [DOI] [PubMed] [Google Scholar]
- 55.Sofia HJ, Chen G, Hetzler BG, Reyes-Spindola JF, Miller NE. 2001. Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: functional characterization using new analysis and information visualization methods. Nucleic Acids Res 29:1097–1106. [PubMed] 10.1093/nar/29.5.1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hanzelmann P, Schindelin H. 2004. Crystal structure of the S-adenosylmethionine-dependent enzyme MoaA and its implications for molybdenum cofactor deficiency in humans. Proc Natl Acad Sci USA 101:12870–12875. [PubMed] 10.1073/pnas.0404624101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hanzelmann P, Schindelin H. 2006. Binding of 5′-GTP to the C-terminal FeS cluster of the radical S-adenosylmethionine enzyme MoaA provides insights into its mechanism. Proc Natl Acad Sci USA 103:6829–6834. [PubMed] 10.1073/pnas.0510711103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wuebbens MM, Liu MT, Rajagopalan K, Schindelin H. 2000. Insights into molybdenum cofactor deficiency provided by the crystal structure of the molybdenum cofactor biosynthesis protein MoaC. Structure Fold Des 8:709–718. [PubMed] 10.1016/S0969-2126(00)00157-X [DOI] [PubMed] [Google Scholar]
- 59.Clinch K, Watt DK, Dixon RA, Baars SM, Gainsford GJ, Tiwari A, Schwarz G, Saotome Y, Storek M, Belaidi AA, Santamaria-Araujo JA. 2013. Synthesis of cyclic pyranopterin monophosphate, a biosynthetic intermediate in the molybdenum cofactor pathway. J Med Chem 56:1730–1738. [PubMed] 10.1021/jm301855r [DOI] [PubMed] [Google Scholar]
- 60.Schwarz G, Santamaria-Araujo JA, Wolf S, Lee HJ, Adham IM, Grone HJ, Schwegler H, Sass JO, Otte T, Hanzelmann P, Mendel RR, Engel W, Reiss J. 2004. Rescue of lethal molybdenum cofactor deficiency by a biosynthetic precursor from Escherichia coli. Hum Mol Genet 13:1249–1255. [PubMed] 10.1093/hmg/ddh136 [DOI] [PubMed] [Google Scholar]
- 61.Veldman A, Santamaria-Araujo JA, Sollazzo S, Pitt J, Gianello R, Yaplito-Lee J, Wong F, Ramsden CA, Reiss J, Cook I, Fairweather J, Schwarz G. 2010. Successful treatment of molybdenum cofactor deficiency type A with cPMP. Pediatrics 125:e1249–e1254. [PubMed] 10.1542/peds.2009-2192 [DOI] [PubMed] [Google Scholar]
- 62.Gutzke G, Fischer B, Mendel RR, Schwarz G. 2001. Thiocarboxylation of molybdopterin synthase provides evidence for the mechanism of dithiolene formation in metal-binding pterins. J Biol Chem 276:36268–36274. [PubMed] 10.1074/jbc.M105321200 [DOI] [PubMed] [Google Scholar]
- 63.Rudolph MJ, Wuebbens MM, Rajagopalan KV, Schindelin H. 2001. Crystal structure of molybdopterin synthase and its evolutionary relationship to ubiquitin activation. Nat Struct Biol 8:42–46. [PubMed] 10.1038/87531 [DOI] [PubMed] [Google Scholar]
- 64.Tong Y, Wuebbens MM, Rajagopalan KV, Fitzgerald MC. 2005. Thermodynamic analysis of subunit interactions in Escherichia coli molybdopterin synthase. Biochemistry 44:2595–2601. [PubMed] 10.1021/bi047762h [DOI] [PubMed] [Google Scholar]
- 65.Daniels JN, Wuebbens MM, Rajagopalan KV, Schindelin H. 2008. Crystal structure of a molybdopterin synthase-precursor Z complex: insight into its sulfur transfer mechanism and its role in molybdenum cofactor deficiency. Biochemistry 47:615–626. [PubMed] 10.1021/bi701734g [DOI] [PubMed] [Google Scholar]
- 66.Wuebbens MM, Rajagopalan KV. 2003. Mechanistic and mutational studies of Escherichia coli molybdopterin synthase clarify the final step of molybdopterin biosynthesis. J Biol Chem 278:14523–14532. [PubMed] 10.1074/jbc.M300453200 [DOI] [PubMed] [Google Scholar]
- 67.Leimkuhler S, Wuebbens MM, Rajagopalan KV. 2001. Characterization of Escherichia coli MoeB and its involvement in the activation of molybdopterin synthase for the biosynthesis of the molybdenum cofactor. J Biol Chem 276:34695–34701. [PubMed] 10.1074/jbc.M102787200 [DOI] [PubMed] [Google Scholar]
- 68.Lake MW, Wuebbens MM, Rajagopalan KV, Schindelin H. 2001. Mechanism of ubiquitin activation revealed by the structure of a bacterial MoeB-MoaD complex. Nature 414:325–329. [PubMed] 10.1038/35104586 [DOI] [PubMed] [Google Scholar]
- 69.Hershko A, Ciechanover A. 1998. The ubiquitin system. Annu Rev Biochem 67:425–479. [PubMed] 10.1146/annurev.biochem.67.1.425 [DOI] [PubMed] [Google Scholar]
- 70.Schmitz J, Wuebbens MM, Rajagopalan KV, Leimkuhler S. 2007. Role of the C-terminal Gly-Gly motif of Escherichia coli MoaD, a molybdenum cofactor biosynthesis protein with a ubiquitin fold. Biochemistry 46:909–916. [PubMed] 10.1021/bi062011w [DOI] [PubMed] [Google Scholar]
- 71.Taylor SV, Kelleher NL, Kinsland C, Chiu HJ, Costello CA, Backstrom AD, McLafferty FW, Begley TP. 1998. Thiamin biosynthesis in Escherichia coli. Identification of ThiS thiocarboxylate as the immediate sulfur donor in the thiazole formation. J Biol Chem 273:16555–16560. [PubMed] 10.1074/jbc.273.26.16555 [DOI] [PubMed] [Google Scholar]
- 72.Wang C, Xi J, Begley TP, Nicholson LK. 2001. Solution structure of ThiS and implications for the evolutionary roots of ubiquitin. Nat Struct Biol 8:47–51. [PubMed] 10.1038/83041 [DOI] [PubMed] [Google Scholar]
- 73.Lehmann C, Begley TP, Ealick SE. 2006. Structure of the Escherichia coli ThiS-ThiF complex, a key component of the sulfur transfer system in thiamin biosynthesis. Biochemistry 45:11–19. [PubMed] 10.1021/bi051502y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jurgenson CT, Ealick SE, Begley TP. 2009. Biosynthesis of thiamin pyrophosphate. In Kaper JB (ed), EcoSal PLus 10.1128/ecosalplus.3.6.3.7. 10.1128/ecosalplus.3.6.3.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cronan JE, Jr. 2014. Biotin and Lipoic acid: synthesis, attachment, and regulation. In Kaper JB (ed), EcoSal Plus 10.1128/ecosalplus.3.6.3.5. 10.1128/ecosalplus.3.6.3.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang W, Urban A, Mihara H, Leimkuhler S, Kurihara T, Esaki N. 2010. IscS functions as a primary sulfur-donating enzyme by interacting specifically with MoeB and MoaD in the biosynthesis of molybdopterin in Escherichia coli. J Biol Chem 285:2302–2308. [PubMed] 10.1074/jbc.M109.082172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shi R, Proteau A, Villarroya M, Moukadiri I, Zhang L, Trempe JF, Matte A, Armengod ME, Cygler M. 2010. Structural basis for Fe-S cluster assembly and tRNA thiolation mediated by IscS protein-protein interactions. PLoS Biol 8:e1000354. 10.1371/journal.pbio.1000354. 10.1371/journal.pbio.1000354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dahl JU, Urban A, Bolte A, Sriyabhaya P, Donahue JL, Nimtz M, Larson TJ, Leimkuhler S. 2011. The identification of a novel protein involved in molybdenum cofactor biosynthesis in Escherichia coli. J Biol Chem 286:35801–35812. [PubMed] 10.1074/jbc.M111.282368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ikeuchi Y, Shigi N, Kato J, Nishimura A, Suzuki T. 2006. Mechanistic insights into sulfur relay by multiple sulfur mediators involved in thiouridine biosynthesis at tRNA wobble positions. Mol Cell 21:97–108. [PubMed] 10.1016/j.molcel.2005.11.001 [DOI] [PubMed] [Google Scholar]
- 80.Dahl JU, Radon C, Buhning M, Nimtz M, Leichert LI, Denis Y, Jourlin-Castelli C, Iobbi-Nivol C, Mejean V, Leimkuhler S. 2013. The sulfur carrier protein TusA has a pleiotropic role in Escherichia coli that also affects molybdenum cofactor biosynthesis. J Biol Chem 288:5426–5442. [PubMed] 10.1074/jbc.M112.431569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schwarz G, Mendel RR, Ribbe MW. 2009. Molybdenum cofactors, enzymes and pathways. Nature 460:839–847. [PubMed] 10.1038/nature08302 [DOI] [PubMed] [Google Scholar]
- 82.Llamas A, Mendel RR, Schwarz G. 2004. Synthesis of adenylated molybdopterin: an essential step for molybdenum insertion. J Biol Chem 279:55241–55246. [PubMed] 10.1074/jbc.M409862200 [DOI] [PubMed] [Google Scholar]
- 83.Schwarz G, Boxer DH, Mendel RR. 1997. Molybdenum cofactor biosynthesis. The plant protein Cnx1 binds molybdopterin with high affinity. J Biol Chem 272:26811–26814. [PubMed] 10.1074/jbc.272.43.26811 [DOI] [PubMed] [Google Scholar]
- 84.Kuper J, Llamas A, Hecht HJ, Mendel RR, Schwarz G. 2004. Structure of the molybdopterin-bound Cnx1G domain links molybdenum and copper metabolism. Nature 430:803–806. [PubMed] 10.1038/nature02681 [DOI] [PubMed] [Google Scholar]
- 85.Kuper J, Winking J, Hecht HJ, Mendel RR, Schwarz G. 2003. The active site of the molybdenum cofactor biosynthetic protein domain Cnx1G. Arch Biochem Biophys 411:36–46. 10.1016/S0003-9861(02)00714-2 [DOI] [PubMed] [Google Scholar]
- 86.Schwarz G, Schrader N, Mendel RR, Hecht HJ, Schindelin H. 2001. Crystal structures of human gephyrin and plant Cnx1 G domains: comparative analysis and functional implications. J Mol Biol 312:405–418. [PubMed] 10.1006/jmbi.2001.4952 [DOI] [PubMed] [Google Scholar]
- 87.Llamas A, Otte T, Multhaup G, Mendel RR, Schwarz G. 2006. The Mechanism of nucleotide-assisted molybdenum insertion into molybdopterin. A novel route toward metal cofactor assembly. J Biol Chem 281:18343–18350. [PubMed] 10.1074/jbc.M601415200 [DOI] [PubMed] [Google Scholar]
- 88.Leimkuhler S, Rajagopalan KV. 2001. In vitro incorporation of nascent molybdenum cofactor into human sulfite oxidase. J Biol Chem 276:1837–1844. [PubMed] 10.1074/jbc.M007304200 [DOI] [PubMed] [Google Scholar]
- 89.Bader G, Gomez-Ortiz M, Haussmann C, Bacher A, Huber R, Fischer M. 2004. Structure of the molybdenum-cofactor biosynthesis protein MoaB of Escherichia coli. Acta Crystallogr D Biol Crystallogr 60:1068–1075. [PubMed] 10.1107/S0907444904007164 [DOI] [PubMed] [Google Scholar]
- 90.Sanishvili R, Beasley S, Skarina T, Glesne D, Joachimiak A, Edwards A, Savchenko A. 2004. The crystal structure of Escherichia coli MoaB suggests a probable role in molybdenum cofactor synthesis. J Biol Chem 279:42139–42146. [PubMed] 10.1074/jbc.M407694200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu MT, Wuebbens MM, Rajagopalan KV, Schindelin H. 2000. Crystal structure of the gephyrin-related molybdenum cofactor biosynthesis protein MogA from Escherichia coli. J Biol Chem 275:1814–1822. [PubMed] 10.1074/jbc.275.3.1814 [DOI] [PubMed] [Google Scholar]
- 92.Kozmin SG, Schaaper RM. 2013. Genetic characterization of moaB mutants of Escherichia coli. Res Microbiol 164:689–694. [PubMed] 10.1016/j.resmic.2013.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bevers LE, Hagedoorn PL, Santamaria-Araujo JA, Magalon A, Hagen WR, Schwarz G. 2008. Function of MoaB proteins in the biosynthesis of the molybdenum and tungsten cofactors. Biochemistry 47:949–956. [PubMed] 10.1021/bi7020487 [DOI] [PubMed] [Google Scholar]
- 94.Neumann M, Mittelstadt G, Iobbi-Nivol C, Saggu M, Lendzian F, Hildebrandt P, Leimkuhler S. 2009. A periplasmic aldehyde oxidoreductase represents the first molybdopterin cytosine dinucleotide cofactor containing molybdo-flavoenzyme from Escherichia coli. FEBS J 276:2762–2774. [PubMed] 10.1111/j.1742-4658.2009.07000.x [DOI] [PubMed] [Google Scholar]
- 95.Neumann M, Mittelstadt G, Seduk F, Iobbi-Nivol C, Leimkuhler S. 2009. MocA is a specific cytidylyltransferase involved in molybdopterin cytosine dinucleotide biosynthesis in Escherichia coli. J Biol Chem 284:21891–21898. [PubMed] 10.1074/jbc.M109.008565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Leimkuhler S, Angermuller S, Schwarz G, Mendel RR, Klipp W. 1999. Activity of the molybdopterin-containing xanthine dehydrogenase of Rhodobacter capsulatus can be restored by high molybdenum concentrations in a moeA mutant defective in molybdenum cofactor biosynthesis. J Bacteriol 181:5930–5939. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nichols J, Rajagopalan KV. 2002. Escherichia coli MoeA and MogA. Function in metal incorporation step of molybdenum cofactor biosynthesis. J Biol Chem 277:24995–25000. [PubMed] 10.1074/jbc.M203238200 [DOI] [PubMed] [Google Scholar]
- 98.Neumann M, Leimkuhler S. 2008. Heavy metal ions inhibit molybdoenzyme activity by binding to the dithiolene moiety of molybdopterin in Escherichia coli. FEBS J 275:5678–5689. [PubMed] 10.1111/j.1742-4658.2008.06694.x [DOI] [PubMed] [Google Scholar]
- 99.Hasona A, Ray RM, Shanmugam KT. 1998. Physiological and genetic analyses leading to identification of a biochemical role for the moeA (molybdate metabolism) gene product in Escherichia coli. J Bacteriol 180:1466–1472. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Brokx SJ, Rothery RA, Zhang G, Ng DP, Weiner JH. 2005. Characterization of an Escherichia coli sulfite oxidase homologue reveals the role of a conserved active site cysteine in assembly and function. Biochemistry 44:10339–10348. [PubMed] 10.1021/bi050621a [DOI] [PubMed] [Google Scholar]
- 101.Loschi L, Brokx SJ, Hills TL, Zhang G, Bertero MG, Lovering AL, Weiner JH, Strynadka NC. 2004. Structural and biochemical identification of a novel bacterial oxidoreductase. J Biol Chem 279:50391–50400. [PubMed] 10.1074/jbc.M408876200 [DOI] [PubMed] [Google Scholar]
- 102.Xi H, Schneider BL, Reitzer L. 2000. Purine catabolism in Escherichia coli and function of xanthine dehydrogenase in purine salvage. J Bacteriol 182:5332–5341. [PubMed] 10.1128/JB.182.19.5332-5341.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kozmin SG, Schaaper RM. 2007. Molybdenum cofactor-dependent resistance to N-hydroxylated base analogs in Escherichia coli is independent of MobA function. Mutat Res 619:9–15. [PubMed] 10.1016/j.mrfmmm.2006.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nichols JD, Rajagopalan KV. 2005. In vitro molybdenum ligation to molybdopterin using purified components. J Biol Chem 280:7817–7822. [PubMed] 10.1074/jbc.M413783200 [DOI] [PubMed] [Google Scholar]
- 105.Magalon A, Frixon C, Pommier J, Giordano G, Blasco F. 2002. In vivo interactions between gene products involved in the final stages of molybdenum cofactor biosynthesis in Escherichia coli. J Biol Chem 277:48199–48204. [PubMed] 10.1074/jbc.M205806200 [DOI] [PubMed] [Google Scholar]
- 106.Belaidi AA, Schwarz G. 2013. Metal insertion into the molybdenum cofactor: product-substrate channelling demonstrates the functional origin of domain fusion in gephyrin. Biochem J 450:149–157. [PubMed] 10.1042/BJ20121078 [DOI] [PubMed] [Google Scholar]
- 107.Palmer T, Vasishta A, Whitty PW, Boxer DH. 1994. Isolation of protein FA, a product of the mob locus required for molybdenum cofactor biosynthesis in Escherichia coli. Eur J Biochem 222:687–692. [PubMed] 10.1111/j.1432-1033.1994.tb18913.x [DOI] [PubMed] [Google Scholar]
- 108.Palmer T, Santini CL, Iobbi-Nivol C, Eaves DJ, Boxer DH, Giordano G. 1996. Involvement of the narJ and mob gene products in distinct steps in the biosynthesis of the molybdoenzyme nitrate reductase in Escherichia coli. Mol Microbiol 20:875–884. [PubMed] 10.1111/j.1365-2958.1996.tb02525.x [DOI] [PubMed] [Google Scholar]
- 109.Temple CA, Rajagopalan KV. 2000. Mechanism of assembly of the bis(molybdopterin guanine dinucleotide)molybdenum cofactor in Rhodobacter sphaeroides dimethyl sulfoxide reductase. J Biol Chem 275:40202–40210. [PubMed] 10.1074/jbc.M007407200 [DOI] [PubMed] [Google Scholar]
- 110.Rizzi M, Schindelin H. 2002. Structural biology of enzymes involved in NAD and molybdenum cofactor biosynthesis. Curr Opin Struct Biol 12:709–720. [PubMed] 10.1016/S0959-440X(02)00385-8 [DOI] [PubMed] [Google Scholar]
- 111.Lake MW, Temple CA, Rajagopalan KV, Schindelin H. 2000. The crystal structure of the Escherichia coli MobA protein provides insight into molybdopterin guanine dinucleotide biosynthesis. J Biol Chem 275:40211–40217. [PubMed] 10.1074/jbc.M007406200 [DOI] [PubMed] [Google Scholar]
- 112.Stevenson CE, Sargent F, Buchanan G, Palmer T, Lawson DM. 2000. Crystal structure of the molybdenum cofactor biosynthesis protein MobA from Escherichia coli at near-atomic resolution. Struct Fold Des 8:1115–1125. [PubMed] 10.1016/S0969-2126(00)00518-9 [DOI] [PubMed] [Google Scholar]
- 113.Guse A, Stevenson CE, Kuper J, Buchanan G, Schwarz G, Giordano G, Magalon A, Mendel RR, Lawson DM, Palmer T. 2003. Biochemical and structural analysis of the molybdenum cofactor biosynthesis protein MobA. J Biol Chem 278:25302–25307. [PubMed] 10.1074/jbc.M302639200 [DOI] [PubMed] [Google Scholar]
- 114.McLuskey K, Harrison JA, Schuttelkopf AW, Boxer DH, Hunter WN. 2003. Insight into the role of Escherichia coli MobB in molybdenum cofactor biosynthesis based on the high resolution crystal structure. J Biol Chem 278:23706–23713. [PubMed] 10.1074/jbc.M301485200 [DOI] [PubMed] [Google Scholar]
- 115.Neumann M, Seduk F, Iobbi-Nivol C, Leimkuhler S. 2011. Molybdopterin dinucleotide biosynthesis in Escherichia coli: identification of amino acid residues of molybdopterin dinucleotide transferases that determine specificity for binding of guanine or cytosine nucleotides. J Biol Chem 286:1400–1408. [PubMed] 10.1074/jbc.M110.155671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hille R. 1996. The mononuclear molybdenum enzymes. Chem Rev 96:2757–2816. [PubMed] 10.1021/cr950061t [DOI] [PubMed] [Google Scholar]
- 117.Raaijmakers H, Macieira S, Dias JM, Teixeira S, Bursakov S, Huber R, Moura JJ, Moura I, Romao MJ. 2002. Gene sequence and the 1.8 A crystal structure of the tungsten-containing formate dehydrogenase from Desulfovibrio gigas. Structure 10:1261–1272. [PubMed] 10.1016/S0969-2126(02)00826-2 [DOI] [PubMed] [Google Scholar]
- 118.Raaijmakers HC, Romao MJ. 2006. Formate-reduced E. coli formate dehydrogenase H: the reinterpretation of the crystal structure suggests a new reaction mechanism. J Biol Inorg Chem 11:849–854. [PubMed] 10.1007/s00775-006-0129-2 [DOI] [PubMed] [Google Scholar]
- 119.Coelho C, Gonzalez PJ, Moura JG, Moura I, Trincao J, Joao Romao M. 2011. The crystal structure of Cupriavidus necator nitrate reductase in oxidized and partially reduced states. J Mol Biol 408:932–948. [PubMed] 10.1016/j.jmb.2011.03.016 [DOI] [PubMed] [Google Scholar]
- 120.Najmudin S, Gonzalez PJ, Trincao J, Coelho C, Mukhopadhyay A, Cerqueira NM, Romao CC, Moura I, Moura JJ, Brondino CD, Romao MJ. 2008. Periplasmic nitrate reductase revisited: a sulfur atom completes the sixth coordination of the catalytic molybdenum. J Biol Inorg Chem 13:737–753. [PubMed] 10.1007/s00775-008-0359-6 [DOI] [PubMed] [Google Scholar]
- 121.Neumann M, Schulte M, Junemann N, Stocklein W, Leimkuhler S. 2006. Rhodobacter capsulatus XdhC is involved in molybdenum cofactor binding and insertion into xanthine dehydrogenase. J Biol Chem 281:15701–15708. [PubMed] 10.1074/jbc.M601617200 [DOI] [PubMed] [Google Scholar]
- 122.Neumann M, Stocklein W, Walburger A, Magalon A, Leimkuhler S. 2007. Identification of a Rhodobacter capsulatus L-cysteine desulfurase that sulfurates the molybdenum cofactor when bound to XdhC and before its insertion into xanthine dehydrogenase. Biochemistry 46:9586–9595. [PubMed] 10.1021/bi700630p [DOI] [PubMed] [Google Scholar]
- 123.Neumann M, Stocklein W, Leimkuhler S. 2007. Transfer of the molybdenum cofactor synthesized by Rhodobacter capsulatus MoeA to XdhC and MobA. J Biol Chem 282:28493–28500. [PubMed] 10.1074/jbc.M704020200 [DOI] [PubMed] [Google Scholar]
- 124.Thome R, Gust A, Toci R, Mendel R, Bittner F, Magalon A, Walburger A. 2012. A sulfurtransferase is essential for activity of formate dehydrogenases in Escherichia coli. J Biol Chem 287:4671–4678. [PubMed] 10.1074/jbc.M111.327122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Arnoux P, Ruppelt C, Oudouhou F, Lavergne J, Siponen MI, Toci R, Mendel RR, Bittner F, Pignol D, Magalon A, Walburger A. 2015. Sulphur shuttling across a chaperone during molybdenum cofactor maturation. Nat Commun 6:6148. 10.1038/ncomms7148. [PubMed] 10.1038/ncomms7148 [DOI] [PubMed] [Google Scholar]
- 126.Magalon A, Fedor JG, Walburger A, Weiner JH. 2011. Molybdenum enzymes in bacteria and their maturation. Coord Chem Rev 255:1159–1178. 10.1016/j.ccr.2010.12.031 [DOI] [Google Scholar]
- 127.Dobbek H. 2011. Structural aspects of mononuclear Mo/W enzymes. Coord Chem Rev 255:1104–1116. 10.1016/j.ccr.2010.11.017 [DOI] [Google Scholar]
- 128.Hille R, Nishino T, Bittner F. 2011. Molybdenum enzymes in higher organisms. Coord Chem Rev 255:1179–1205. [PubMed] 10.1016/j.ccr.2010.11.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Aguilar M, Kalakoutskii K, Cardenas J, Fernandez E. 1992. Direct transfer of molybdopterin cofactor to aponitrate reductase from a carrier protein in Chlamydomonas reinhardtii. FEBS Lett 307:162–163. 10.1016/0014-5793(92)80758-9 [DOI] [PubMed] [Google Scholar]
- 130.Witte CP, Igeno MI, Mendel R, Schwarz G, Fernandez E. 1998. The Chlamydomonas reinhardtii MoCo carrier protein is multimeric and stabilizes molybdopterin cofactor in a molybdate charged form. FEBS Lett 431:205–209. 10.1016/S0014-5793(98)00756-X [DOI] [PubMed] [Google Scholar]
- 131.Fischer K, Llamas A, Tejada-Jimenez M, Schrader N, Kuper J, Ataya FS, Galvan A, Mendel RR, Fernandez E, Schwarz G. 2006. Function and structure of the molybdenum cofactor carrier protein from Chlamydomonas reinhardtii. J Biol Chem 281:30186–30194. [PubMed] 10.1074/jbc.M603919200 [DOI] [PubMed] [Google Scholar]
- 132.Kruse T, Gehl C, Geisler M, Lehrke M, Ringel P, Hallier S, Hansch R, Mendel RR. 2010. Identification and biochemical characterization of molybdenum cofactor-binding proteins from Arabidopsis thaliana. J Biol Chem 285:6623–6635. [PubMed] 10.1074/jbc.M109.060640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Vergnes A, Gouffi-Belhabich K, Blasco F, Giordano G, Magalon A. 2004. Involvement of the molybdenum cofactor biosynthetic machinery in the maturation of the Escherichia coli nitrate reductase A. J Biol Chem 279:41398–41403. [PubMed] 10.1074/jbc.M407087200 [DOI] [PubMed] [Google Scholar]
- 134.Turner RJ, Papish AL, Sargent F. 2004. Sequence analysis of bacterial redox enzyme maturation proteins (REMPs). Can J Microbiol 50:225–238. [PubMed] 10.1139/w03-117 [DOI] [PubMed] [Google Scholar]
- 135.Blasco F, Pommier J, Augier V, Chippaux M, Giordano G. 1992. Involvement of the narJ or narW gene product in the formation of active nitrate reductase in Escherichia coli. Mol Microbiol 6:221–230. [PubMed] 10.1111/j.1365-2958.1992.tb02003.x [DOI] [PubMed] [Google Scholar]
- 136.Schlindwein C, Giordano G, Santini CL, Mandrand MA. 1990. Identification and expression of the Escherichia coli fdhD and fdhE genes, which are involved in the formation of respiratory formate dehydrogenase. J Bacteriol 172:6112–6121. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Schlindwein C, Mandrand MA. 1991. Nucleotide sequence of the fdhE gene involved in respiratory formate dehydrogenase formation in Escherichia coli K-12. Gene 97:147–148. [PubMed] 10.1016/0378-1119(91)90023-5 [DOI] [PubMed] [Google Scholar]
- 138.Pommier J, Mejean V, Giordano G, Iobbi-Nivol C. 1998. TorD, a cytoplasmic chaperone that interacts with the unfolded trimethylamine N-oxide reductase enzyme (TorA) in Escherichia coli. J Biol Chem 273:16615–16620. [PubMed] 10.1074/jbc.273.26.16615 [DOI] [PubMed] [Google Scholar]
- 139.Shaw AL, Leimkuhler S, Klipp W, Hanson GR, McEwan AG. 1999. Mutational analysis of the dimethylsulfoxide respiratory (dor) operon of Rhodobacter capsulatus. Microbiology 145(Pt 6):1409–1420. [PubMed] 10.1099/13500872-145-6-1409 [DOI] [PubMed] [Google Scholar]
- 140.Oresnik IJ, Ladner CL, Turner RJ. 2001. Identification of a twin-arginine leader-binding protein. Mol Microbiol 40:323–331. [PubMed] 10.1046/j.1365-2958.2001.02391.x [DOI] [PubMed] [Google Scholar]
- 141.Ray N, Oates J, Turner RJ, Robinson C. 2003. DmsD is required for the biogenesis of DMSO reductase in Escherichia coli but not for the interaction of the DmsA signal peptide with the Tat apparatus. FEBS Lett 534:156–160. [PubMed] 10.1016/S0014-5793(02)03839-5 [DOI] [PubMed] [Google Scholar]
- 142.Liu HP, Takio S, Satoh T, Yamamoto I. 1999. Involvement in denitrification of the napKEFDABC genes encoding the periplasmic nitrate reductase system in the denitrifying phototrophic bacterium Rhodobacter sphaeroides f. sp. denitrificans. Biosci Biotechnol Biochem 63:530–536. [PubMed] 10.1271/bbb.63.530 [DOI] [PubMed] [Google Scholar]
- 143.Potter LC, Cole JA. 1999. Essential roles for the products of the napABCD genes, but not napFGH, in periplasmic nitrate reduction by Escherichia coli K-12. Biochem J 344(Pt 1):69–76. [PubMed] 10.1042/0264-6021:3440069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kern M, Mager AM, Simon J. 2007. Role of individual nap gene cluster products in NapC-independent nitrate respiration of Wolinella succinogenes. Microbiology 153:3739–3747. [PubMed] 10.1099/mic.0.2007/009928-0 [DOI] [PubMed] [Google Scholar]
- 145.Coulthurst SJ, Dawson A, Hunter WN, Sargent F. 2012. Conserved signal peptide recognition systems across the prokaryotic domains. Biochemistry 51:1678–1686. [PubMed] 10.1021/bi201852d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Leimkuhler S, Klipp W. 1999. Role of XDHC in Molybdenum cofactor insertion into xanthine dehydrogenase of Rhodobacter capsulatus. J Bacteriol 181:2745–2751. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Unden G, Dünnwald P. 2008. The Aerobic and anaerobic respiratory chain of Escherichia coli and Salmonella enterica: enzymes and energetics. In Curtiss RI, Kaper JB, Squires CL, Karp PD, Neidhardt FC, Slauch JM (ed), EcoSal. ASM Press, Washington, DC. 10.1128/ecosal.3.2.2 [DOI] [PubMed] [Google Scholar]
- 148.Price CE, Driessen AJ. 2008. YidC is involved in the biogenesis of anaerobic respiratory complexes in the inner membrane of Escherichia coli. J Biol Chem 283:26921–26927. [PubMed] 10.1074/jbc.M804490200 [DOI] [PubMed] [Google Scholar]
- 149.Sargent F. 2007. The twin-arginine transport system: moving folded proteins across membranes. Biochem Soc Trans 35:835–847. [PubMed] 10.1042/BST0350835 [DOI] [PubMed] [Google Scholar]
- 150.Li H, Chang L, Howell JM, Turner RJ. 2010. DmsD, a Tat system specific chaperone, interacts with other general chaperones and proteins involved in the molybdenum cofactor biosynthesis. Biochim Biophys Acta 1804:1301–1309. [PubMed] 10.1016/j.bbapap.2010.01.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Matos CF, Robinson C, Di Cola A. 2008. The Tat system proofreads FeS protein substrates and directly initiates the disposal of rejected molecules. EMBO J 27:2055–2063. [PubMed] 10.1038/emboj.2008.132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Matos CF, Di Cola A, Robinson C. 2009. TatD is a central component of a Tat translocon-initiated quality control system for exported FeS proteins in Escherichia coli. EMBO Rep 10:474–479. [PubMed] 10.1038/embor.2009.34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lindenstrauss U, Matos CF, Graubner W, Robinson C, Bruser T. 2010. Malfolded recombinant Tat substrates are Tat-independently degraded in Escherichia coli. FEBS Lett 584:3644–3648. [PubMed] 10.1016/j.febslet.2010.07.039 [DOI] [PubMed] [Google Scholar]
- 154.Tranier S, Iobbi-Nivol C, Birck C, Ilbert M, Mortier-Barriere I, Mejean V, Samama JP. 2003. A novel protein fold and extreme domain swapping in the dimeric TorD chaperone from Shewanella massilia. Structure (Camb) 11:165–174. [PubMed] 10.1016/S0969-2126(03)00008-X [DOI] [PubMed] [Google Scholar]
- 155.Kirillova O, Chruszcz M, Shumilin IA, Skarina T, Gorodichtchenskaia E, Cymborowski M, Savchenko A, Edwards A, Minor W. 2007. An extremely SAD case: structure of a putative redox-enzyme maturation protein from Archaeoglobus fulgidus at 3.4 A resolution. Acta Crystallogr D Biol Crystallogr 63:348–354. [PubMed] 10.1107/S0907444906055065 [DOI] [PubMed] [Google Scholar]
- 156.Qiu Y, Zhang R, Binkowski TA, Tereshko V, Joachimiak A, Kossiakoff A. 2008. The 1.38 A crystal structure of DmsD protein from Salmonella typhimurium, a proofreading chaperone on the Tat pathway. Proteins 71:525–533. [PubMed] 10.1002/prot.21828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ramasamy SK, Clemons WM, Jr. 2009. Structure of the twin-arginine signal-binding protein DmsD from Escherichia coli. Acta Crystallogr Sect F Struct Biol Cryst Commun 65:746–750. [PubMed] 10.1107/S1744309109023811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Stevens CM, Winstone TM, Turner RJ, Paetzel M. 2009. Structural analysis of a monomeric form of the twin-arginine leader peptide binding chaperone Escherichia coli DmsD. J Mol Biol 389:124–133. [PubMed] 10.1016/j.jmb.2009.03.069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Bertero MG, Rothery RA, Palak M, Hou C, Lim D, Blasco F, Weiner JH, Strynadka NC. 2003. Insights into the respiratory electron transfer pathway from the structure of nitrate reductase A. Nat Struct Biol 10:681–687. [PubMed] 10.1038/nsb969 [DOI] [PubMed] [Google Scholar]
- 160.Jormakka M, Richardson D, Byrne B, Iwata S. 2004. Architecture of NarGH reveals a structural classification of Mo-bisMGD enzymes. Structure (Camb) 12:95–104. [PubMed] 10.1016/j.str.2003.11.020 [DOI] [PubMed] [Google Scholar]
- 161.Dubourdieu M, DeMoss JA. 1992. The narJ gene product is required for biogenesis of respiratory nitrate reductase in Escherichia coli. J Bacteriol 174:867–872. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Liu X, DeMoss JA. 1997. Characterization of NarJ, a system-specific chaperone required for nitrate reductase biogenesis in Escherichia coli. J Biol Chem 272:24266–24271. [PubMed] 10.1074/jbc.272.39.24266 [DOI] [PubMed] [Google Scholar]
- 163.Blasco F, Nunzi F, Pommier J, Brasseur R, Chippaux M, Giordano G. 1992. Formation of active heterologous nitrate reductases between nitrate reductases A and Z of Escherichia coli. Mol Microbiol 6:209–219. [PubMed] 10.1111/j.1365-2958.1992.tb02002.x [DOI] [PubMed] [Google Scholar]
- 164.Lanciano P, Vergnes A, Grimaldi S, Guigliarelli B, Magalon A. 2007. Biogenesis of a respiratory complex is orchestrated by a single accessory protein. J Biol Chem 282:17468–17474. [PubMed] 10.1074/jbc.M700994200 [DOI] [PubMed] [Google Scholar]
- 165.Zakian S, Lafitte D, Vergnes A, Pimentel C, Sebban-Kreuzer C, Toci R, Claude JB, Guerlesquin F, Magalon A. 2010. Basis of recognition between the NarJ chaperone and the N-terminus of the NarG subunit from Escherichia coli nitrate reductase. FEBS J 277:1886–1895. [PubMed] 10.1111/j.1742-4658.2010.07611.x [DOI] [PubMed] [Google Scholar]
- 166.Lorenzi M, Sylvi L, Gerbaud G, Mileo E, Halgand F, Walburger A, Vezin H, Belle V, Guigliarelli B, Magalon A. 2012. Conformational selection underlies recognition of a molybdoenzyme by its dedicated chaperone. PLoS One 7:e49523. 10.1371/journal.pone.0049523 [PubMed] 10.1371/journal.pone.0049523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Vergnes A, Pommier J, Toci R, Blasco F, Giordano G, Magalon A. 2006. NarJ chaperone binds on two distinct sites of the aponitrate reductase of Escherichia coli to coordinate molybdenum cofactor insertion and assembly. J Biol Chem 281:2170–2176. [PubMed] 10.1074/jbc.M505902200 [DOI] [PubMed] [Google Scholar]
- 168.Jack RL, Buchanan G, Dubini A, Hatzixanthis K, Palmer T, Sargent F. 2004. Coordinating assembly and export of complex bacterial proteins. EMBO J 23:3962–3972. [PubMed] 10.1038/sj.emboj.7600409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ize B, Coulthurst SJ, Hatzixanthis K, Caldelari I, Buchanan G, Barclay EC, Richardson DJ, Palmer T, Sargent F. 2009. Remnant signal peptides on non-exported enzymes: implications for the evolution of prokaryotic respiratory chains. Microbiology 155:3992–4004. [PubMed] 10.1099/mic.0.033647-0 [DOI] [PubMed] [Google Scholar]
- 170.Rothery RA, Bertero MG, Spreter T, Bouromand N, Strynadka NC, Weiner JH. 2010. Protein crystallography reveals a Role for the FS0 cluster of Escherichia coli nitrate reductase A (NarGHI) in enzyme maturation. J Biol Chem 285:8801–8807. [PubMed] 10.1074/jbc.M109.066027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Winstone TL, Workentine ML, Sarfo KJ, Binding AJ, Haslam BD, Turner RJ. 2006. Physical nature of signal peptide binding to DmsD. Arch Biochem Biophys 455:89–97. [PubMed] 10.1016/j.abb.2006.08.009 [DOI] [PubMed] [Google Scholar]
- 172.Guymer D, Maillard J, Agacan MF, Brearley CA, Sargent F. 2010. Intrinsic GTPase activity of a bacterial twin-arginine translocation proofreading chaperone induced by domain swapping. FEBS J 277:511–525. [PubMed] 10.1111/j.1742-4658.2009.07507.x [DOI] [PubMed] [Google Scholar]
- 173.Tzeng SR, Kalodimos CG. 2011. Protein dynamics and allostery: an NMR view. Curr Opin Struct Biol 21:62–67. [PubMed] 10.1016/j.sbi.2010.10.007 [DOI] [PubMed] [Google Scholar]
- 174.Blasco F, Dos Santos JP, Magalon A, Frixon C, Guigliarelli B, Santini CL, Giordano G. 1998. NarJ is a specific chaperone required for molybdenum cofactor assembly in nitrate reductase A of Escherichia coli. Mol Microbiol 28:435–447. [PubMed] 10.1046/j.1365-2958.1998.00795.x [DOI] [PubMed] [Google Scholar]
- 175.Rothery RA, Bertero MG, Cammack R, Palak M, Blasco F, Strynadka NC, Weiner JH. 2004. The Catalytic subunit of Escherichia coli nitrate reductase a contains a novel [4Fe-4S] cluster with a high-spin ground state. Biochemistry 43:5324–5333. [PubMed] 10.1021/bi049938l [DOI] [PubMed] [Google Scholar]
- 176.Hanzelmann P, Dobbek H, Gremer L, Huber R, Meyer O. 2000. The effect of intracellular molybdenum in Hydrogenophaga pseudoflava on the crystallographic structure of the seleno-molybdo-iron-sulfur flavoenzyme carbon monoxide dehydrogenase. J Mol Biol 301:1221–1235. [PubMed] 10.1006/jmbi.2000.4023 [DOI] [PubMed] [Google Scholar]
- 177.Weiner JH, Bilous PT, Shaw GM, Lubitz SP, Frost L, Thomas GH, Cole JA, Turner RJ. 1998. A novel and ubiquitous system for membrane targeting and secretion of cofactor-containing proteins. Cell 93:93–101. [PubMed] 10.1016/S0092-8674(00)81149-6 [DOI] [PubMed] [Google Scholar]
- 178.Guymer D, Maillard J, Sargent F. 2009. A genetic analysis of in vivo selenate reduction by Salmonella enterica serovar Typhimurium LT2 and Escherichia coli K12. Arch Microbiol 191:519–528. [PubMed] 10.1007/s00203-009-0478-7 [DOI] [PubMed] [Google Scholar]
- 179.Papish AL, Ladner CL, Turner RJ. 2003. The twin-arginine leader-binding protein, DmsD, interacts with the TatB and TatC subunits of the Escherichia coli twin-arginine translocase. J Biol Chem 278:32501–32506. [PubMed] 10.1074/jbc.M301076200 [DOI] [PubMed] [Google Scholar]
- 180.Kostecki JS, Li H, Turner RJ, DeLisa MP. 2010. Visualizing interactions along the Escherichia coli twin-arginine translocation pathway using protein fragment complementation. PLoS One 5:e9225. 10.1371/journal.pone.0009225. 10.1371/journal.pone.0009225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Chan CS, Winstone TM, Chang L, Stevens CM, Workentine ML, Li H, Wei Y, Ondrechen MJ, Paetzel M, Turner RJ. 2008. Identification of residues in DmsD for twin-arginine leader peptide binding, defined through random and bioinformatics-directed mutagenesis. Biochemistry. 47:2749–2759. [PubMed] 10.1021/bi702138a [DOI] [PubMed] [Google Scholar]
- 182.Hatzixanthis K, Clarke TA, Oubrie A, Richardson DJ, Turner RJ, Sargent F. 2005. Signal peptide-chaperone interactions on the twin-arginine protein transport pathway. Proc Natl Acad Sci USA 102:8460–8465. [PubMed] 10.1073/pnas.0500737102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Sambasivarao D, Turner RJ, Simala-Grant JL, Shaw G, Hu J, Weiner JH. 2000. Multiple roles for the twin arginine leader sequence of dimethyl sulfoxide reductase of Escherichia coli. J Biol Chem 275:22526–22531. [PubMed] 10.1074/jbc.M909289199 [DOI] [PubMed] [Google Scholar]
- 184.Tang H, Rothery RA, Voss JE, Weiner JH. 2011. Correct assembly of iron-sulfur cluster FS0 into Escherichia coli dimethyl sulfoxide reductase (DmsABC) is a prerequisite for molybdenum cofactor insertion. J Biol Chem 286:15147–15154. [PubMed] 10.1074/jbc.M110.213306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Schroder I, Rech S, Krafft T, Macy JM. 1997. Purification and characterization of the selenate reductase from Thauera selenatis. J Biol Chem 272:23765–23768. [PubMed] 10.1074/jbc.272.38.23765 [DOI] [PubMed] [Google Scholar]
- 186.Martinez-Espinosa RM, Dridge EJ, Bonete MJ, Butt JN, Butler CS, Sargent F, Richardson DJ. 2007. Look on the positive side! The orientation, identification and bioenergetics of ‘Archaeal’ membrane-bound nitrate reductases. FEMS Microbiol Lett 276:129–139. [PubMed] 10.1111/j.1574-6968.2007.00887.x [DOI] [PubMed] [Google Scholar]
- 187.Thorell HD, Stenklo K, Karlsson J, Nilsson T. 2003. A gene cluster for chlorate metabolism in Ideonella dechloratans. Appl Environ Microbiol 69:5585–5592. [PubMed] 10.1128/AEM.69.9.5585-5592.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Bender KS, Shang C, Chakraborty R, Belchik SM, Coates JD, Achenbach LA. 2005. Identification, characterization, and classification of genes encoding perchlorate reductase. J Bacteriol 187:5090–5096. [PubMed] 10.1128/JB.187.15.5090-5096.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Kniemeyer O, Heider J. 2001. Ethylbenzene dehydrogenase, a novel hydrocarbon-oxidizing molybdenum/iron-sulfur/heme enzyme. J Biol Chem 276:21381–21386. [PubMed] 10.1074/jbc.M101679200 [DOI] [PubMed] [Google Scholar]
- 190.McDevitt CA, Hugenholtz P, Hanson GR, McEwan AG. 2002. Molecular analysis of dimethyl sulphide dehydrogenase from Rhodovulum sulfidophilum: its place in the dimethyl sulphoxide reductase family of microbial molybdopterin-containing enzymes. Mol Microbiol 44:1575–1587. [PubMed] 10.1046/j.1365-2958.2002.02978.x [DOI] [PubMed] [Google Scholar]
- 191.Stolz JF, Basu P, Santini JM, Oremland RS. 2006. Arsenic and selenium in microbial metabolism. Annu Rev Microbiol 60:107–130. [PubMed] 10.1146/annurev.micro.60.080805.142053 [DOI] [PubMed] [Google Scholar]
- 192.Duval S, Ducluzeau AL, Nitschke W, Schoepp-Cothenet B. 2008. Enzyme phylogenies as markers for the oxidation state of the environment: the case of respiratory arsenate reductase and related enzymes. BMC Evol Biol 8:206. [PubMed] 10.1186/1471-2148-8-206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Jormakka M, Tornroth S, Byrne B, Iwata S. 2002. Molecular basis of proton motive force generation: structure of formate dehydrogenase-N. Science 295:1863–1868. [PubMed] 10.1126/science.1068186 [DOI] [PubMed] [Google Scholar]
- 194.Boyington JC, Gladyshev VN, Khangulov SV, Stadtman TC, Sun PD. 1997. Crystal structure of formate dehydrogenase H: catalysis involving Mo, molybdopterin, selenocysteine, and an Fe4S4 cluster. Science 275:1305–1308. [PubMed] 10.1126/science.275.5304.1305 [DOI] [PubMed] [Google Scholar]
- 195.Mandrand-Berthelot MA, Couchoux-Luthaud G, Santini CL, Giordano G. 1988. Mutants of Escherichia coli specifically deficient in respiratory formate dehydrogenase activity. J Gen Microbiol 134:3129–3139. [PubMed] 10.1099/00221287-134-12-3129 [DOI] [PubMed] [Google Scholar]
- 196.Stewart V, Lin JT, Berg BL. 1991. Genetic evidence that genes fdhD and fdhE do not control synthesis of formate dehydrogenase-N in Escherichia coli K-12. J Bacteriol 173:4417–4423. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Pommier J, Mandrand MA, Holt SE, Boxer DH, Giordano G. 1992. A second phenazine methosulphate-linked formate dehydrogenase isoenzyme in Escherichia coli. Biochim Biophys Acta 1107:305–313. [PubMed] 10.1016/0005-2736(92)90417-K [DOI] [PubMed] [Google Scholar]
- 198.Luke I, Butland G, Moore K, Buchanan G, Lyall V, Fairhurst SA, Greenblatt JF, Emili A, Palmer T, Sargent F. 2008. Biosynthesis of the respiratory formate dehydrogenases from Escherichia coli: characterization of the FdhE protein. Arch Microbiol 190:685–696. [PubMed] 10.1007/s00203-008-0420-4 [DOI] [PubMed] [Google Scholar]
- 199.Maillard J, Spronk CA, Buchanan G, Lyall V, Richardson DJ, Palmer T, Vuister GW, Sargent F. 2007. Structural diversity in twin-arginine signal peptide-binding proteins. Proc Natl Acad Sci USA 104:15641–15646. 10.1073/pnas.0703967104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Grahl S, Maillard J, Spronk CA, Vuister GW, Sargent F. 2012. Overlapping transport and chaperone-binding functions within a bacterial twin-arginine signal peptide. Mol Microbiol 83:1254–1267. [PubMed] 10.1111/j.1365-2958.2012.08005.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Nilavongse A, Brondijk TH, Overton TW, Richardson DJ, Leach ER, Cole JA. 2006. The NapF protein of the Escherichia coli periplasmic nitrate reductase system: demonstration of a cytoplasmic location and interaction with the catalytic subunit, NapA. Microbiology 152:3227–3237. [PubMed] 10.1099/mic.0.29157-0 [DOI] [PubMed] [Google Scholar]
- 202.Olmo-Mira MF, Gavira M, Richardson DJ, Castillo F, Moreno-Vivian C, Roldan MD. 2004. NapF is a cytoplasmic iron-sulfur protein required for Fe-S cluster assembly in the periplasmic nitrate reductase. J Biol Chem 279:49727–49735. [PubMed] 10.1074/jbc.M406502200 [DOI] [PubMed] [Google Scholar]
- 203.Kern M, Simon J. 2009. Periplasmic nitrate reduction in Wolinella succinogenes: cytoplasmic NapF facilitates NapA maturation and requires the menaquinol dehydrogenase NapH for membrane attachment. Microbiology 155:2784–2794. [PubMed] 10.1099/mic.0.029983-0 [DOI] [PubMed] [Google Scholar]
- 204.Arnoux P, Sabaty M, Alric J, Frangioni B, Guigliarelli B, Adriano JM, Pignol D. 2003. Structural and redox plasticity in the heterodimeric periplasmic nitrate reductase. Nat Struct Biol 10:928–934. [PubMed] 10.1038/nsb994 [DOI] [PubMed] [Google Scholar]
- 205.Jepson BJ, Mohan S, Clarke TA, Gates AJ, Cole JA, Butler CS, Butt JN, Hemmings AM, Richardson DJ. 2007. Spectropotentiometric and structural analysis of the periplasmic nitrate reductase from Escherichia coli. J Biol Chem 282:6425–6437. [PubMed] 10.1074/jbc.M607353200 [DOI] [PubMed] [Google Scholar]
- 206.Bittner F, Oreb M, Mendel RR. 2001. ABA3 is a molybdenum cofactor sulfurase required for activation of aldehyde oxidase and xanthine dehydrogenase in Arabidopsis thaliana. J Biol Chem 276:40381–40384. [PubMed] 10.1074/jbc.C100472200 [DOI] [PubMed] [Google Scholar]
- 207.Heidenreich T, Wollers S, Mendel RR, Bittner F. 2005. Characterization of the NifS-like domain of ABA3 from Arabidopsis thaliana provides insight into the mechanism of molybdenum cofactor sulfuration. J Biol Chem 280:4213–4218. [PubMed] 10.1074/jbc.M411195200 [DOI] [PubMed] [Google Scholar]
- 208.Wollers S, Heidenreich T, Zarepour M, Zachmann D, Kraft C, Zhao Y, Mendel RR, Bittner F. 2008. Binding of sulfurated molybdenum cofactor to the C-terminal domain of ABA3 from Arabidopsis thaliana provides insight into the mechanism of molybdenum cofactor sulfuration. J Biol Chem 283:9642–9650. [PubMed] 10.1074/jbc.M708549200 [DOI] [PubMed] [Google Scholar]
- 209.Mejean V, Iobbi-Nivol C, Lepelletier M, Giordano G, Chippaux M, Pascal MC. 1994. TMAO anaerobic respiration in Escherichia coli: involvement of the tor operon. Mol Microbiol 11:1169–1179. [PubMed] 10.1111/j.1365-2958.1994.tb00393.x [DOI] [PubMed] [Google Scholar]
- 210.Silvestro A, Pommier J, Pascal MC, Giordano G. 1989. The inducible trimethylamine N-oxide reductase of Escherichia coli K12: its localization and inducers. Biochim Biophys Acta 999:208–216. [PubMed] 10.1016/0167-4838(89)90220-3 [DOI] [PubMed] [Google Scholar]
- 211.Czjzek M, Dos Santos JP, Pommier J, Giordano G, Mejean V, Haser R. 1998. Crystal structure of oxidized trimethylamine N-oxide reductase from Shewanella massilia at 2.5 A resolution. J Mol Biol 284:435–447. [PubMed] 10.1006/jmbi.1998.2156 [DOI] [PubMed] [Google Scholar]
- 212.Tranier S, Mortier-Barriere I, Ilbert M, Birck C, Iobbi-Nivol C, Mejean V, Samama JP. 2002. Characterization and multiple molecular forms of TorD from Shewanella massilia, the putative chaperone of the molybdoenzyme TorA. Protein Sci 11:2148–2157. [PubMed] 10.1110/ps.0202902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Genest O, Ilbert M, Mejean V, Iobbi-Nivol C. 2005. TorD, an essential chaperone for TorA molybdoenzyme maturation at high temperature. J Biol Chem 280:15644–15648. [PubMed] 10.1074/jbc.M501119200 [DOI] [PubMed] [Google Scholar]
- 214.Genest O, Neumann M, Seduk F, Stocklein W, Mejean V, Leimkuhler S, Iobbi-Nivol C. 2008. Dedicated metallochaperone connects apoenzyme and molybdenum cofactor biosynthesis components. J Biol Chem 283:21433–21440. [PubMed] 10.1074/jbc.M802954200 [DOI] [PubMed] [Google Scholar]
- 215.Genest O, Seduk F, Theraulaz L, Mejean V, Iobbi-Nivol C. 2006. Chaperone protection of immature molybdoenzyme during molybdenum cofactor limitation. FEMS Microbiol Lett 265:51–55. [PubMed] 10.1111/j.1574-6968.2006.00468.x [DOI] [PubMed] [Google Scholar]
- 216.Ilbert M, Mejean V, Giudici-Orticoni MT, Samama JP, Iobbi-Nivol C. 2003. Involvement of a mate chaperone (TorD) in the maturation pathway of molybdoenzyme TorA. J Biol Chem 278:28787–28792. [PubMed] 10.1074/jbc.M302730200 [DOI] [PubMed] [Google Scholar]
- 217.Ilbert M, Mejean V, Iobbi-Nivol C. 2004. Functional and structural analysis of members of the TorD family, a large chaperone family dedicated to molybdoproteins. Microbiology 150:935–943. [PubMed] 10.1099/mic.0.26909-0 [DOI] [PubMed] [Google Scholar]
- 218.Dow JM, Gabel F, Sargent F, Palmer T. 2013. Characterization of a pre-export enzyme-chaperone complex on the twin-arginine transport pathway. Biochem J 452:57–66. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Leimkuhler S, Kern M, Solomon PS, McEwan AG, Schwarz G, Mendel RR, Klipp W. 1998. Xanthine dehydrogenase from the phototrophic purple bacterium Rhodobacter capsulatus is more similar to its eukaryotic counterparts than to prokaryotic molybdenum enzymes. Mol Microbiol 27:853–869. [PubMed] 10.1046/j.1365-2958.1998.00733.x [DOI] [PubMed] [Google Scholar]
- 220.Truglio JJ, Theis K, Leimkuhler S, Rappa R, Rajagopalan KV, Kisker C. 2002. Crystal structures of the active and alloxanthine-inhibited forms of xanthine dehydrogenase from Rhodobacter capsulatus. Structure (Camb) 10:115–125. 10.1016/S0969-2126(01)00697-9 [DOI] [PubMed] [Google Scholar]
- 221.Schumann S, Saggu M, Moller N, Anker SD, Lendzian F, Hildebrandt P, Leimkuhler S. 2008. The mechanism of assembly and cofactor insertion into Rhodobacter capsulatus xanthine dehydrogenase. J Biol Chem 283:16602–16611. [PubMed] 10.1074/jbc.M709894200 [DOI] [PubMed] [Google Scholar]
- 222.Xiang S, Nichols J, Rajagopalan KV, Schindelin H. 2001. The crystal structure of Escherichia coli MoeA and its relationship to the multifunctional protein gephyrin. Structure (Camb) 9:299–310. 10.1016/S0969-2126(01)00588-3 [DOI] [PubMed] [Google Scholar]
- 223.DeLano WL. 2002. The PyMOL Molecular Graphics System. DeLano Scientific, San Carlos, CA. [Google Scholar]