

Dear Editor,
ATP1A3 mutations span a wide range of neurological disorders, including alternating hemiplegia of childhood (AHC), rapid-onset dystonia-parkinsonism, cerebellar ataxia, areflexia, pes cavus, optic atrophy, and sensorineural hearing loss syndrome, with an expanding phenotypic spectrum involving rare presentations like early infantile epilepsy with encephalopathy and recurrent encephalopathy with cerebellar ataxia. This case outlines a 19-year-old female’s journey with early-onset epilepsy, unresponsive to conventional treatments, ultimately attributed to an ATP1A3 gene mutation. Notably, cannabidiol (CBD) therapy showed promising results. Understanding ATP1A3 mutations’ role in sodium–potassium ATPase dysfunction is crucial for diagnosing early-onset developmental and epileptic encephalopathies. Further research is vital for comprehending the prevalence and characteristics of ATP1A3-related disorders, particularly in the Indian context, shaping future diagnostic and therapeutic strategies.
A 19-year-old female with subnormal intelligence and global developmental delay presented with focal onset seizures since the age of 2. She had multiple episodes of right oculocephalic version with tonic–clonic movements of right-sided limbs, each episode lasting between 2 and 3 h. Neuroimaging was not done initially. She was managed conservatively with multiple antiseizure medications (ASMs). However, she continued to have one to two seizures every year till 14 years of age and was seizure free thereafter till the present admission. She presented now with tonic–clonic movements of the left upper limb with left head version and orofacial dyskinesias. She had frequent seizures (up to six episodes per day). Examination revealed dysmorphic facies, generalized hypotonia, and right hemiparesis. Her magnetic resonance imaging of the brain showed asymmetric bilateral cerebral atrophy, left more involved than right. There was bilateral caudate atrophy and thinning of the corpus callosum [Figure 1]. Ictal electroencephalogram (EEG) showed right hemispheric electrographic seizures, and interictal EEG was characterized by multifocal epileptiform discharges from both hemispheres. Despite having multiple ASMs, she continued to have seizures and had to be initiated on midazolam for seizure control. In view of focal drug-refractory seizures and hemiparesis with cerebral hemiatrophy, Rasmussen’s encephalitis was suspected, for which intravenous immunoglobulin was administered followed by rituximab. Though hemiconvulsion–hemiplegia–epilepsy syndrome has a similar presentation, the disease course and absence of fever preceding onset were unusual. However, her seizures did not cease. New-onset focal seizure originating in the unaffected side and radiological features of cerebellar atrophy, corpus callosal thinning, and focal microgyria prompted us to consider rare causes for epilepsy, genetic in particular. Her whole exome sequencing revealed a heterozygous mutation in exon 16 of ATP1A3 gene (c.2149_2151del p.Lys717del). A trial of flunarizine was given, but demonstrated no response. Incorporation of keto diet also failed to control seizures. CBD was started off-label as it was beneficial in various drug-resistant epilepsies like Dravet and Lennox–Gastaut syndromes. Her seizure frequency came down and she was successfully weaned off midazolam. Two of her ASMs were tapered off. On a 6-month follow-up, her seizures were well controlled (three to four episodes in the past 6 months). No drug-related adverse events were noted.
Figure 1.
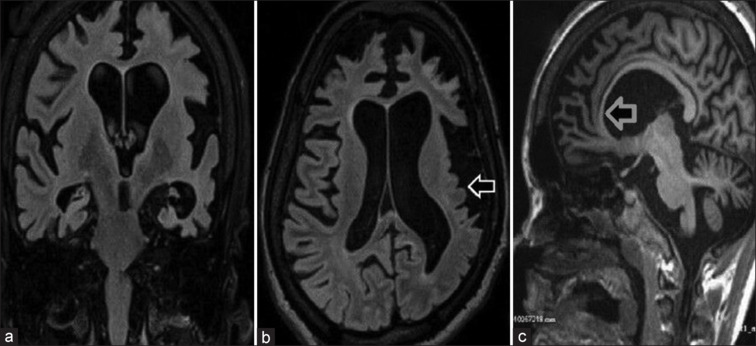
Coronal (a) and axial (b) sections show atrophy of bilateral cerebral hemispheres, predominantly affecting the left cerebral hemisphere with microgyria involving the left parietal and temporal lobes (white arrow). There is asymmetric dilatation of both the lateral ventricles (left is larger than right) as seen in coronal (a) and axial (b) sections. Sagittal section (c) shows thinning of the corpus callosum (gray arrow)
ATP1A3 gene encodes the α3 subunit of sodium–potassium ATPase pumps. Na–K ATPase pumps play an important role in exchange of ions across neuronal membranes and regulate its function. Neurons with ATP1A3 mutations have increased excitability due to decreased Na–K ATPase activity and increased intracellular sodium, resulting in hypersynchronous neuronal firing causing catastrophic epilepsy.[1] ATP1A3-related disorders were first described in 2004 by de Carvalho Aguiar et al.[2] in a series of patients with rapid-onset dystonia and parkinsonism. Increased expression in the basal ganglia and cerebellum gives rise to the prototypical phenotypes described in ATP1A3 mutations. Though previously thought to be rare, the last decade has shown the expanding clinical spectrum of neurological disorders associated with ATP1A3 mutations. Disorders exhibit an autosomal dominant pattern of inheritance with variable expressivity and reduced penetrance.[2] Disorders with early onset have a more severe presentation. In a study by Vetro et al.,[3] 21 of 22 patients with ATP1A3-related Developmental and Epileptic Encephalopathy (DEE) had neonatal and infantile onset and 45% presented with status epilepticus. Focal onset seizures are the predominant type. Our case had no features of the classical phenotypes, except for the paroxysmal nature of attacks, early onset and severe intellectual disability. In a study of 24 patients with ATP1A3 mutations, all had cognitive impairment and 70% of patients had significant impairment.[4] In a study by Vezyroglou et al.,[4] abnormal neuroimaging was reported in 60% of patients with ATP1A3 mutations. Cerebellar atrophy was the most common neuroimaging finding in up to 40% of patients, followed by hemispheric atrophy. Rapidly progressive brain atrophy has been reported previously in association with microcephaly. Polymicrogyria (PMG) has been described in ATP1A3 mutations and seen in up to 45% of patients with DEE.[3] Lack of adequate neuroimaging may have resulted in underreporting of PMG in previous studies. PMG typically involves the perisylvian cortex and correlates with severity of seizures.[5] Focal PMG involving the left parietotemporal region, as seen in our case, is a novel observation [see Figure 1]. Seizures are drug refractory, and no ASM has provided more than 50% seizure reduction. In a study of 638 patients with AHC, 64.5% of patients showed response to flunarizine.[6] However, no such response was observed in our case. Our patient showed a dramatic response after initiation of CBD. The possibility that the clinical response to CBD was incidental cannot be completely ruled out. CBD being a multitarget molecule modulates neuronal excitability and thus controls seizures. Surgical methods are beneficial in drug-refractory cases. Corpus callosotomy was extremely effective in a case of early-onset DEE with ATP1A3 variant.[7] In those patients not fit for palliative surgical treatment, CBD can be attempted.
It is important to screen for ATP1A3 mutation in early-onset developmental and epileptic encephalopathy, alongside routine ionic channel assessments. Further research is crucial to understand ATP1A3-related disorders in the Indian population, with a focus on targeting ATP1A3 regulators. Neuroradiological findings may not always be evident early in the disease. CBD shows promise in refractory cases, improving quality of life for patients and caregivers.
Declaration of informed consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Zou S, Lan YL, Gong Y, Chen Z, Xu C. The role of ATP1A3 gene in epilepsy: We need to know more. Front Cell Neurosci. 2023;17:1143956. doi: 10.3389/fncel.2023.1143956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Carvalho Aguiar P, Sweadner KJ, Penniston JT, Zaremba J, Liu L, Caton M, et al. Mutations in the Na+/K+-ATPase α3 gene ATP1A3 are associated with rapid-onset dystonia parkinsonism. Neuron. 2004;43:169–75. doi: 10.1016/j.neuron.2004.06.028. [DOI] [PubMed] [Google Scholar]
- 3.Vetro A, Nielsen HN, Holm R, Hevner RF, Parrini E, Powis Z, et al. ATP1A2- and ATP1A3-associated early profound epileptic encephalopathy and polymicrogyria. Brain J Neurol. 2021;144:1435–50. doi: 10.1093/brain/awab052. [DOI] [PubMed] [Google Scholar]
- 4.Vezyroglou A, Akilapa R, Barwick K, Koene S, Brownstein CA, Holder-Espinasse M, et al. The phenotypic continuum of ATP1A3-related disorders. Neurology. 2022;99:e1511–26. doi: 10.1212/WNL.0000000000200927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miyatake S, Kato M, Kumamoto T, Hirose T, Koshimizu E, Matsui T, et al. De novo ATP1A3 variants cause polymicrogyria. Sci Adv. 2021;7:eabd2368. doi: 10.1126/sciadv.abd2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kusunoki S, Kido J, Momosaki K, Sawada T, Kashiki T, Matsumoto S, et al. Effect of flunarizine on alternating hemiplegia of childhood in a patient with the p.E815K mutation in ATP1A3: A case report. Case Rep Neurol. 2020;12:299–306. doi: 10.1159/000509287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moriyama K, Mizuno T, Suzuki T, Inaji M, Maehara T, Fujita A, et al. ATP1A3-related early childhood onset developmental and epileptic encephalopathy responding to corpus callosotomy: A case report. Brain Dev. 2023;45:77–81. doi: 10.1016/j.braindev.2022.08.009. [DOI] [PubMed] [Google Scholar]