

Abstract
Optic neuritis (ON) is an inflammatory condition that affects the optic nerve and may be associated with various central nervous system demyelinating conditions, infectious diseases, and systemic autoimmune syndromes. This manuscript sheds light on the epidemiologic patterns and diverse clinical features of ON, emphasizing the importance of early detection and prompt treatment. Various studies have revealed geographic and ethnic variations across ON subtypes, which are likely related to the incidence and prevalence of co-associated disorders. Distinguishing ON subtypes may be challenging and requires use of paraclinical tools. Treatment strategies differ depending on the etiology, further highlighting the importance of accurately identifying specific ON subtypes in a timely manner.
Keywords: Optic Neuritis (ON), multiple sclerosis (MS), myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD), neuromyelitis optica spectrum disorders (NMOSD), magnetic resonance imaging (MRI), optical coherence tomography (OCT)
Introduction
Optic neuritis (ON) refers broadly to any inflammatory process involving the optic nerve, the causes of which are protean. ON has been linked to over 60 systemic, neurologic, or infectious disorders.[1] Affected individuals often experience pain, subacute-onset vision loss, dyschromatopsia (color vision dysfunction), visual field deficits, and variable recovery. The disease course is more likely to be monophasic in ON subtypes that are not autoimmune in origin, whereas immune-mediated syndromes may cause either monophasic or recurrent ON episodes.[1] It is important to bear in mind that disease associations depend on the geographic location. For example, in some parts of the world, ON is the heralding feature of infections such as tuberculosis (TB), whereas in other regions, primary disorders of the central nervous system (CNS), including multiple sclerosis (MS), are more common related conditions. Since the prognosis may be poor for some ON subtypes, early diagnostic precision is needed to optimize visual recovery. Accordingly, treatments should be tailored to target the underlying mechanism of optic nerve dysfunction. Clinicians must appreciate the full spectrum of ON subtypes to best facilitate early diagnosis, refine treatment protocols, and support visual recovery.
Epidemiologic Considerations
There are epidemiologic differences related to the worldwide incidence of ON, with historical estimates generally ranging from as low as 0.83 per 100,000 population per year in Singapore, for instance, to 5.1 per 100,000 population per year in the USA (Minnesota, 1935–1991).[2,3] These variances may partly reflect differences in the population prevalence of MS and other immune-mediated inflammatory diseases,[3] yet geographic disparities may also arise when diagnostic criteria and data-capturing techniques differ among studies.
Recently, Braithwaite et al.[3] reported an ON incidence of 3.7 per 100,000 person-years. The strongest co-association was with MS, and weaker links were noted with other systemic disorders such as Behçet’s disease, sarcoidosis, and Sjögren’s syndrome (SjS).[3] In contrast, a 5-year longitudinal, population-based Taiwanese study documented a total cumulative incidence of 1.33 per 1000.[4] In this report, the incidence of ON peaked among the subjects aged 40–59 years, women were more likely to be affected than men (1.00 vs. 1.67 for male vs. female subjects; P < 0.001, χ2 test), and there was a low rate of conversion to MS.
In Canada, Marrie et al.[2] performed a population-based study using administrative data from the province of Manitoba and reported an annual ON incidence (2011–2013) of 75.9 per 100,000 person-years (95% confidence interval [CI]: 72.8–79.1). While this incidence rate dwarfs some of the aforementioned reports from Europe and Asia, it is noteworthy that Manitoba has one of the highest rates of MS in the world, which may be a contributing factor to the high incidence of ON in this province.
Use of population-based administrative data arguably provides a cost-effective means of conducting chronic disease surveillance[5]; yet, the accuracy for identifying ON using coding systems remains unclear.[5] Muro-Fuentes et al.[5] used a medical record research repository (1998–2019) to retrospectively evaluate patients with International Classification of Diseases (ICD) codes 9/10 for ON, who also underwent magnetic resonance imaging (MRI) of the brain and orbits (within 2 months of the earliest initial ICD code). In this study involving 251 patients, there was an accuracy of only 25% for acute ON, 41% for acute or prior ON, and 48% for acute, prior, and considered ON.[5] These findings suggest that there are perils to relying on ICD codes to identify ON cases and emphasize the need for standardized, reliable diagnostic criteria to counter inaccuracies in estimated incidence and prevalence rates.
ON: Diagnostic Challenges
The diagnosis of ON is challenging to render in real-world settings because many clinicians may struggle with examination techniques, such as performing reliable ophthalmoscopy or examining for a relative afferent pupillary defect (RAPD). These issues, potentially confounded by a desire to not overlook an important diagnosis, may contribute to the phenomenon of “overdiagnosis.” In 2018, Stunkel et al.[6] retrospectively evaluated 122 patients referred with acute ON for neuro-ophthalmic evaluation. Of these, 73 patients (approximately 60%) declared another diagnosis. Overdiagnosis of ON was linked to errors in capturing details of the patient’s history, considering other potential cases, and interpreting the examination.[6] In light of the established challenges related to diagnostic accuracy in suspected cases of ON, there is a need for paraclinical tests to reduce the risk of misdiagnosis, provide insights regarding disease course, and help determine the need for long-term pharmacologic management.[1]
ON: Suggested Diagnostic and Classification Criteria
In 2022, international experts published findings from a validated Delphi process-based approach. This group aimed to develop criteria for the diagnosis and classification of ON that relied upon clinical and paraclinical features.[1] Per their recommendations as summarized below, the diagnosis of definite ON requires evidence of the following:
monocular vision loss
orbital pain
reduced contrast/color vision function
RAPD in the affected eye, and
-
at least one of the following paraclinical tests:
Optical coherence tomography (OCT): Showing evidence of optic disc edema acutely (elevated peripapillary retinal nerve fiber layer [pRNFL] values) or an inter-eye difference in the macular ganglion cell–inner plexiform layer thickness of >4% or >4 μm or in the pRNFL of >5% or >5 μm within 3 months of symptom onset.
MRI: Acutely, there may be contrast enhancement of the symptomatic optic nerve and sheaths. Alternatively, there may be hyperintense signal changes within 3 months.
Biomarker: Including serum aquaporin-4 (AQP4)-IgG (cell-based assay [CBA]), serum myelin oligodendrocyte glycoprotein (MOG)-IgG (CBA), serum collapsin response mediator protein 5 (CRMP5) antibodies, or intrathecal cerebrospinal fluid (CSF) IgG (oligoclonal bands).
For patients with all clinical features of definite ON except pain, two paraclinical tests are needed to confirm a definite ON diagnosis. Moreover, patients presenting with clinical features of definite ON (with or without pain) who manifest bilateral optic nerve involvement (RAPD may be unreliable) need supporting MRI evidence plus at least one other paraclinical test to confirm a diagnosis of definite ON.
The diagnosis of possible ON may be rendered for individuals who have acute symptoms and signs consistent with definite ON, demonstrate expected fundus findings, but lack available clinical tests. Patients with a history consistent with definite ON who have supportive OCT, MRI, and biomarker findings may also be diagnosed with possible ON. In contrast, individuals with vision loss who have clinical features and paraclinical testing results indicative of an alternative pathology (genetic optic neuropathy, compressive lesion) should be deemed as not having ON.
The criteria proposed by Petzold et al.[1] may help improve the diagnostic accuracy for ON, which is much needed in both clinical and research settings. Certainly, there are disadvantages to underdiagnosing ON, as patients with MS, for instance, may experience delays in treatment for this condition.[7] However, there are also perils to overdiagnosis because patients may be subjected to inappropriate tests and treatments. Moreover, misdiagnosis may result in patients being exposed to unnecessary agents while missing out on therapies they actually need.
ON Subtypes
Differentiating ON subtypes [Table 1] is a challenging yet important process that requires consideration of several factors. These include patient demographics, such as age, sex, and ethnicity; clinical presentation, including the course of disease, laterality, and severity of vision loss; the extent of optic disc swelling; neuroimaging findings; serologic data; and response to treatment.
Table 1.
| ON subtype | Epidemiology | Etiology | ON features | Other ocular features | Fluid markers | Acute treatment | Prognosis |
|---|---|---|---|---|---|---|---|
| Infections: As reported by Petzold et al.,[1] causes of infectious and postinfectious ON include but are not limited to Bartonella, Brucella, chikungunya fever, CMV, coronavirus, Coxiella burnetii, dengue, EBV, echovirus, ehrlichiosis, hepatitis B and C, herpes simplex, Histoplasma, HIV, herpesviruses, Inoue-Melnick virus, leprosy, Lyme disease, measles, mumps, Mycoplasma pneumoniae, neurotoxocariasis, ocular cat scratch disease, post-vaccination ON, rubella, streptococcus, syphilis, tick-borne encephalitis, toxoplasmosis, TB, typhus, VZV, West Nile virus, Whipple disease, and Zika virus[1] | |||||||
|
| |||||||
| Syphilis | Worldwide infection. In 2022, the WHO estimated that there were 8 million adults with syphilis aged 15–49 years | Caused by Treponema pallidum and transmitted through oral, anal, or vaginal sex or contact with infectious lesions. Syphilis can also be transmitted in pregnancy. | Normal, optic disc edema, optic disc atrophy | Uveitis, neuroretinitis, retinal vasculitis, perineuritis, chorioretinopathy | Positive serum and CSF VDRL and FTA-ABS | IV aqueous penicillin 2–4 million units every 4 h for 2 weeks | Variable |
| TB | In 2022, it was estimated that 10.6 million had TB, with the largest number of new cases in South-East Asia, Africa, and the Western Pacific | Caused by Mycobacterium tuberculosis. Airborne transmission | Normal, optic disc edema, or pale optic nerves | Uveitis, neuroretinitis, perineuritis, compressive lesions (tuberculomas) | Serum QuantiFERON testing. Growth of tuberculous bacteria by smear or culture | Isonicotinic acid hydrazide, ethambutol, rifampin, streptomycin, and pyrazinamide (may vary between immune-competent and immune-compromised hosts) | Variable |
| Lyme disease | Most common tick-borne infection in the USA and Europe | Caused by Borrelia burgdorferi. Transmitted to humans by tick bites. | Normal, optic disc edema, or pale optic nerves | Neuroretinitis, cranial neuropathies | ELISA IgG and IgM, western blot assay, intrathecal antibodies | IV ceftriaxone 2 g/day or doxycycline 100 mg orally twice daily | Variable |
| Primary CNS inflammatory disorders | |||||||
| Multiple sclerosis | Incidence is highest in Europe and America with a 2 to 3:1 female to male ratio | Unknown | Acutely, the optic nerve often looks normal or shows mild optic disc edema | Usually normal retinal examination | No specific serum antibody | High-dose corticosteroids and PLEX therapy | Favorable |
| MOGAD | Worldwide with involvement of children and adults | Unknown | Acutely, the optic nerve may show severe swelling or appear normal | The retina is usually normal; perineural enhancement with longitudinal intraorbital lesions may be seen on MRI. Orbital inflammation has been described. Uveitis is rare. | MOG-IgG | High-dose corticosteroids, PLEX, and IVIG | Mostly favorable with treatment |
| NMOSD | Highest in Afro-Caribbean and Asian countries with a high (9:1) female to male ratio | Unknown | Acutely, the optic nerve may show severe swelling or appear normal | The retina is usually normal. Rarely, uveitis occurs. Longitudinal intracranial optic nerve lesions are seen on MRI | Serum AQP4-IgG | High-dose corticosteroids, PLEX, and IVIG | Poor |
| GFAP | Unknown | Unknown, but may be associated with underlying cancers | Unilateral or bilateral optic disc edema | Iritis or uveitis may be seen | CSF GFAP-IgG | High-dose corticosteroids followed by a slow taper | These patients tend to have good vision despite optic disc edema |
| CRMP5 | Unknown | May be paraneoplastic | Optic disc edema is often seen | Vitritis, retinitis, macular exudates, and iritis | CV2/CRMP5 autoantibodies in serum and/or CSF IgG | Tumor removal and corticosteroids | Variable |
| Systemic diseases: As reported by Petzold et al.,[1] causes include but are not limited to allergic granulomatous angiitis, ANCA-associated vasculitis, ankylosing spondylitis, Behçet’s disease, Churg–Strauss disease, Cogan syndrome, giant cell arteritis, GPA, IgG4-related disease, Kawasaki disease, microscopic polyangiitis, polyarteritis nodosa, primary antiphospholipid syndrome, rheumatic disease, sarcoidosis, Sjögren’s syndrome, SLE, Susac syndrome, systemic sclerosis, Takayasu arteritis, and ulcerative colitis | |||||||
| Sarcoid | Sarcoidosis may be more common in people of African or Caribbean extraction and may affect women more than men | An autoimmune-mediated disorder characterized by noncaseating granulomas affecting one or multiple organs | Normal, edematous, or atrophic optic nerve | Conjunctival nodules, keratitic precipitates, iris nodules, uveitis, perineuritis, and lacrimal gland enlargement | Chest imaging, ACE, gallium scan, 24-h urine calcium, PET study, tissue diagnosis | Corticosteroids and long-term immunosuppression as needed | Variable |
| SLE | More commonly affects women relative to men, with a ratio of 9:1, typically affecting young adults (20–30 years) | An autoimmune-mediated condition that upregulates antibody production and causes deposition of immune complexes in tissues | Optic nerves may be normal, may show, atrophy, or may manifest edema | Retinal vasculitis, perineuritis, retinal arterial occlusions | ANA, anti-double stranded DNA, anti-Smith nuclear antigen, urinalysis | Corticosteroids and long-term immunosuppression as needed | Variable, often poor |
| GPA | The incidence rate has been estimated at 12.8 per million person-years with a slight female predominance | A form of vasculitis involving small and medium-sized arteries, primarily in the lung and kidneys | Optic nerves may be normal, may show atrophy, or may manifest edema | Orbital signs and lacrimal gland involvement | Cytoplasmic antineutrophil cytoplasmic antibodies, tissue diagnosis | Corticosteroids and long-term immune suppression | Variable |
| IgG4 disease | In Japan, the incidence has been estimated to be 0.28-1.08 per 100,000 population. Subjects tend to be aged 50–70 years (predilection for male sex) | IgG4 plasma cells infiltrate various organs and anatomic sites | Optic nerves may be normal, may show atrophy, or may manifest edema | Orbital signs and lacrimal gland involvement | The diagnosis is based on clinical, histopathologic, and serologic findings including increased IgG4 serum concentration, infiltration of IgG4 cells | Corticosteroids and immune suppression as needed | Variable |
| Sjögren’s syndrome | Sjögren’s syndrome affects 1–2 million people worldwide, with a female to male ratio of 9:1 | An autoimmune disease affecting the lacrimal and salivary glands | Normal, edematous, and atrophic optic nerves | Keratoconjunctivitis and dry eyes | Rheumatoid factor (50% of patients) and antibodies against antigens known as Ro (SSA) and La (SSB) | Corticosteroids with or without immune suppression | Variable |
| Medications | |||||||
| ICIs | Our understanding of the incidence of ICI-related ON is still evolving, with most reports associated with ipilimumab and pembrolizumab | ICIs overactivate the immune system | Normal, edematous, and atrophic optic nerves | Uveitis, endo-ophthalmitis | Increased NMDA receptor, GFAP, CRMP5 antibodies may be detected in serum and CSF | Consider drug cessation and corticosteroids | Variable |
| TNF-alpha blockers | The incidence of ON among users of TNF-alpha blockers (in patients without prior CNS demyelination) has been estimated to be 5–10 per 100,000 persons | These agents suppress the body’s natural response to TNF | Normal, edematous, and atrophic optic nerves | Uveitis | None | Drug cessation and corticosteroids | Variable |
ACE: angiotensin converting enzyme, ANA: antinuclear antigen, ANCA: anti-neutrophil cytoplasmic antibodies, AQP4: aquaporin-4, CMV: cytomegalovirus, CNS: central nervous system, CRMP5: collapsin response mediator protein 5, CSF: cerebrospinal fluid, ELISA: enzyme-linked immunosorbent assay, GFAP: glial fibrillary acidic protein, GPA: granulomatosis with polyangiitis, HIV: human immunodeficiency virus, ICIs: immune checkpoint inhibitors, IV: intravenous, IVIG: intravenous immunoglobulin, MOGAD: myelin oligodendrocyte glycoprotein antibody-associated disease, MRI: magnetic resonance imaging, NMDA: N-methyl-d-aspartate, NMOSD: neuromyelitis optica spectrum disorders, ON: optic neuritis, PLEX: plasma exchange, TB: tuberculosis, TNF: tumor necrosis factor, VDRL: venereal disease research laboratory, VZV: varicella zoster virus, WHO: World Health Organization, EVB: Epstein–Barr virus, FTA-ABS: Fluorescent treponemal antibody absorption test, CV2: Cerebellar Veil 2
ON Associated with Primary CNS Inflammatory Syndromes
Idiopathic and MS-associated ON
The Optic Neuritis Treatment Trial (ONTT) and subsequent follow-up studies have significantly informed our understanding of the natural history and treatment strategies for idiopathic ON and MS-related ON subtypes.[8,18] MS is an inflammatory CNS demyelinating disease commonly diagnosed in young Caucasian females between 20 and 40 years of age who live in northern geographic regions. However, MS may affect individuals of any sex, age, ethnicity, or geographic location.[19,20] ON is one of the most frequent manifestations of MS, with approximately 25% of MS patients initially presenting with ON and 50% experiencing at least one episode during their disease course.[21]
On average, MS ON presents with unilateral, subacute, painful, mild to moderate vision loss that worsens over days to a week, before starting to improve.[7,8] While optic atrophy develops in the weeks following the initial presentation, over 90% of MS ON patients will have high-contrast visual acuity (HCVA) of 20/40 or better 1 year after their presentation.[7,18] In addition to visual acuity reduction, 92% of patients in ONTT reported ocular discomfort and 70% noted pain on extraocular movement (with 40% reporting pain that preceded vision loss).[7,18] Ophthalmoscopy in patients with idiopathic or MS ON often shows a normal-appearing optic disc without overt swelling, peripapillary hemorrhage, or vitritis in the acute phase.[22]
Myelin oligodendrocyte glycoprotein antibody-associated disease and ON
Myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD) is an antibody-mediated CNS inflammatory demyelinating disease that is distinct from MS.[9] Epidemiologically, MOGAD may not have a clear sex or ethnic predilection.[8] However, this ON subtype is relatively common in children, accounting for 20%–30% of inflammatory CNS syndromes in pediatric populations compared to about 5% in adults.[9] Recently, Banwell et al.[23] proposed the diagnostic criteria for MOGAD, defining ON, acute disseminated encephalomyelitis, myelitis, cerebral mono- or multi-focal deficit, brainstem or cerebellar syndromes, and cerebral cortical encephalitis as the core clinical features of the disease. Serum MOG-IgG testing via CBA is highly specific for MOGAD; however, low titers should be interpreted with caution and judged in the clinical context of the patient.[9,23,24] It is also important to exclude other diagnoses that might better explain the patient’s clinical presentation and to repeat MOG-IgG testing after the initial diagnosis to both confirm the diagnosis and monitor disease activity.[23]
Clinically, ON is the presenting symptom approximately 50% of the time in MOGAD patients, often leading to severe vision loss. Despite this, most patients experience excellent visual recovery, with only 5%–15% having a final best-corrected visual acuity (BCVA) of 20/200 or worse.[7,8] Patients frequently present with optic disc swelling (70%–80% of cases), which is more frequent when compared to MS and neuromyelitis optica spectrum disorders (NMOSD) patients.[7,8] Pain is a common symptom, often preceding vision loss. MOGAD-related ON can be bilateral with either simultaneous or sequential involvement of the fellow eye.[7,9] Approximately half of MOGAD patients have a monophasic course of the disease, with the optic nerve being the most common site of recurrence for the other half.[25] Imaging features may include longitudinal intraorbital optic nerve involvement and perineural enhancement, sometimes extending into the periorbital fat on MRI of orbits.[9]
NMOSD-associated ON
Clinically, NMOSD presents with a wide range of symptoms, including ON, acute myelitis, and syndromes affecting the area postrema, brainstem, diencephalon, and various cerebral regions. Accordingly, patients with NMOSD may develop narcolepsy, myopathy, neuropsychiatric symptoms, and myeloradiculopathy.[26] It is noteworthy that NMOSD-associated ON often results in severe vision loss, with 75% of patients having an HCVA of 20/200 or worse at nadir and poor visual recovery.[7]
Epidemiologically, NMOSD more frequently affects middle-aged women of African or Asian descent, but can occur in patients across various ages and with different ethnic backgrounds.[26] This condition has stronger female predilection (female to male ratio is 9:1) compared to MS (2:1), with a mean age of onset around 40 years.[26] NMOSD ON may be bilateral and recurrent,[26] and optic disc edema is less common when compared to MOGAD ON.[7]
The discovery of autoantibodies against AQP4 by Lennon et al.[27,28] differentiated NMOSD from MS and distinguished NMOSD as an autoimmune astrocytopathy.[26] This observation fueled much research, leading to refined diagnostic and treatment criteria.[26] Recent data indicate that 70%–90% of NMOSD patients are AQP-IgG seropositive, with 99.8% specificity when using CBA of serum.[29] Serum sampling is more reliable than CSF as AQP4-IgG is synthesized outside the CNS.[26] Orbital MRI typically shows extensive lesions involving the intracranial region of the optic nerve, with isolated optic tract or chiasm involvement being more common relative to MOGAD and MS ON neuritis subtypes.[26] Understanding the unique epidemiology, clinical presentation, imaging, and immunologic profile of NMOSD ON is crucial for early diagnosis and timely treatment, given its poor prognosis if untreated.[26] Since NMOSD disability is relapse driven, long-term immunosuppressive agents are the mainstay of therapy for patients.
Glial fibrillary acidic protein-associated ON
Optic neuropathy can occur in patients with autoantibodies targeting glial fibrillary acidic protein (GFAP), typically detected in their serum and CSF.[30] In addition to optic neuropathy, which affects 25%–40% of these patients, individuals with GFAP meningoencephalitis may also manifest myelitis and ataxia.[30] Epidemiologically, GFAP ON is commonly diagnosed in middle-aged individuals and does not show clear ethnic or sex predilection.[23] Clinically, most GFAP ON patients present with painless, bilateral optic disc swelling and relatively normal visual function, with a presenting BCVA of 20/30 or better.[7] The disease course is often monophasic, though there are reports of relapsing cases.[7]
Infectious ON Subtypes (Bacterial and Viral)
Infectious diseases represent another cause of ON. These infections can be bacterial, viral, parasitic, or fungal, involving different anatomic locations of the optic nerve. Accordingly, patients may present with papillitis [Case 1], retrobulbar ON, neuroretinitis, or optic perineuritis.[31] Infectious ON may manifest with monocular or binocular (acute or subacute) vision loss that varies from mild to severe, depending on the infection type.[31] Vision loss can result directly from the infection or can be secondary to associated inflammation, compressive (tuberculomas) and/or vascular responses [Case 1]. The prevalence of infectious ON depends largely on the patient population. While certain infectious agents are more common in underdeveloped areas, the era of globalization, increased travel, and higher rates of immunosuppression necessitate considering infectious etiologies regardless of a patient’s geographic location.[32] This subsection will briefly explore some of the common infectious etiologies of ON.
Case 1.
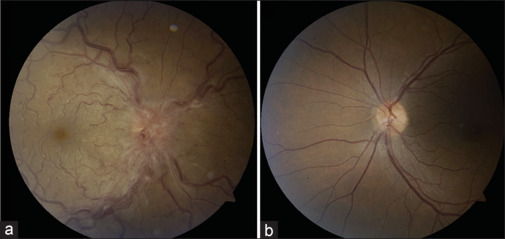
Case 1: This patient presented with right-sided TB-related optic neuritis and secondary central retinal vein occlusion (CRVO). Examination revealed an edematous optic nerve and CRVO in the right eye, as shown in the fundus photograph (a), while the left fundus photograph (b) was unremarkable. Serum QuantiFERON-TB testing was positive. A chest CT scan showed evidence of granulomatous disease with multiple calcified nodules. A cranial MRI, with appropriate orbital views, appeared normal. The patient was treated empirically with rifampin, isoniazid, and pyrazinamide. His right eye was treated with bevacizumab, which led to good visual recovery. CT: computed tomography, MRI: magnetic resonance imaging
Tuberculosis
TB has caused more human deaths over the last two centuries than any other infectious disease. It has a tremendous global impact, with an estimated 25% of the world population harboring latent TB and 10 million new cases reported annually.[32] It is caused by Mycobacterium tuberculosis, an obligate aerobe and acid-fast bacillus. Infections are typically acquired by inhaling aerosolized droplets containing M. tuberculosis cells.[32] While these bacterial cells are phagocytized by alveolar macrophages, a subset can survive, replicate, and spread through the lymphatics or circulation to other organs. The adaptive immune system responds by forming calcified encapsulated granulomas that typically contain latent but viable bacterial cells.[32] Most patients with latent TB never develop clinical disease, while approximately 5% experience reactivation of TB infection, especially if they are immunocompromised.[32]
Ocular TB may resemble other inflammatory, infectious, and neoplastic conditions, with the majority (90%) of cases presenting with uveitis.[33] TB-associated uveitis is typically bilateral, presenting with mutton-fat keratic precipitates, posterior synechiae, and Koeppe nodules.[32,34] TB-related ON does not usually present alone, but in association with retinal and choroidal involvement. A retrospective chart review of 49 patients with TB-related ON reported several common presenting features including papillitis (51.6%), neuroretinitis (14.5%), and optic nerve tubercles (11.3%), while compressive lesions and retrobulbar ON were encountered less commonly.[34] Of these patients, 85.7% reported decreased visual acuity and 26.5% had retrobulbar pain. Presenting BCVA was 20/50 or worse in 63% of the patients, with 77% achieving BCVA of 20/40 or better 1 year after their diagnosis.[32,34]
Diagnosing TB ON remains challenging as there are no specific diagnostic criteria and tissue sampling is often impractical.[31] Therefore, clinicians must maintain a high index of suspicion and order appropriate serologic testing, ophthalmic investigations, and neuroimaging.[31] In addition to a detailed history (TB contact history, travel history, and demographic profile) and examination, patients should undergo appropriate ophthalmic imaging including OCT and fundus fluorescein angiography in cases with associated retinal vasculitis.[31] The workup may include purified protein derivative skin testing or interferon-gamma release assays (e.g., QuantiFERON-TB Gold test), chest X-ray, and a biopsy of any accessible granuloma.[31] In many cases, the diagnosis of TB is presumed, and appropriate anti-TB therapy is initiated empirically [Case 1].
Neuroretinitis and cat scratch disease
Cat scratch disease is caused by Bartonella henselae, a gram-negative bacterium typically found in the saliva or nail beds of animals, especially cats.[32] The disease usually presents systemically with fever, lymphadenopathy, and a pustule at the site of injury. Ophthalmic involvement is reported in 5%–10% of patients, with unilateral neuroretinitis often manifesting approximately 2–4 weeks following the initial infection. In these cases, optic disc edema with or without associated peripapillary hemorrhage may precede the appearance of the classic macular star.[32] It is important to emphasize that while neuroretinitis is a classic manifestation of ocular B. henselae infection, it is a very nonspecific clinical finding that could result from other infectious or inflammatory etiologies.[32]
In addition to neuroretinitis, other ocular manifestations may include Parinaud oculoglandular syndrome, papillitis, uveitis, vasculitis, or retinitis.[31] Once B. henselae infection is suspected, serologic tests, such as indirect fluorescent assay or enzyme-linked immunosorbent assay (ELISA), should be requested to detect antibodies against Bartonella species. A polymerase chain reaction assay of intraocular tissue samples can also be used to confirm the diagnosis.[31] Fortunately, this disease is self-limiting for most patients, and visual function recovers even without therapy within a few weeks. However, most clinicians elect to treat with antibiotics (typically doxycycline or azithromycin) as the treatment is well tolerated and thought to be efficacious.[32]
Syphilis
Syphilis is a chronic multiorgan infection caused by Treponema pallidum. This infection is primarily transmitted through sexual contact or passed from a mother to her child during pregnancy.[32,35] Emerging epidemiologic data show a resurgence of syphilis rates globally, especially in populations with high-risk sexual behavior [Table 1] and in those infected with human immunodeficiency virus (HIV).[32,35] Clinically, syphilis is subdivided into four stages: primary, secondary, latent, and tertiary. The primary stage is characterized by painless skin chancre at the site of infection, which heals within 3–6 weeks. If untreated, the disease progresses to the secondary stage, marked by painless and nonpruritic rash on the soles and palms and accompanied by systemic symptoms including fever, myalgia, headache, and alopecia.[32] If syphilis remains untreated, the patient enters a latent, asymptomatic stage, where most untreated patients stay. A small proportion of patients with latent syphilis develop tertiary disease, potentially developing cardiac, hepatic, and ocular manifestations, even decades later.[32]
Syphilis patients may develop ocular manifestations at any stage of the disease, with uveitis and papillitis being the most common [Case 2].[35] Syphilitic uveitis can present as granulomatous or nongranulomatous inflammation, manifesting as anterior, posterior, or panuveitis.[32,35] Due to its broad range of ocular manifestations, syphilis is known as the “great masquerader,” necessitating a high index of suspicion when examining patients with ocular pathology. Syphilitic ON can manifest as papillitis, anterior or retrobulbar ON, neuroretinitis, or ischemic optic neuropathy secondary to obliterative endarteritis in tertiary syphilis.[32] In addition, neurosyphilis may lead to other neuro-ophthalmic dysfunction affecting the efferent and afferent pathways, including cranial neuropathy, supranuclear ocular motor dysfunction, and visual field defects, depending on the site of infection.[32]
Case 2.
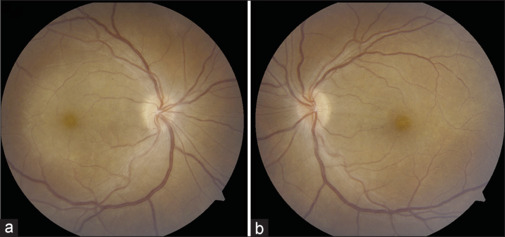
Case 2: This patient presented with unilateral vision loss and photopsia in the right eye, along with slight blurry vision in the left eye. Best-corrected visual acuity was 20/100 in the right eye and 20/30 in the left eye, with mild right pupillary escape. Fundus examination revealed optic disc edema and posterior placoid chorioretinitis, more pronounced in the right eye (a) than in the left eye (b). Cranial and orbital MRI scans were unremarkable. Serum aquaporin-4-IgG and MOG-IgG tests were negative. However, serum syphilis serology (rapid plasma reagin and Treponema pallidum particle agglutination tests) was positive. The patient was treated with intravenous penicillin G, leading to full resolution of symptoms after 2 weeks. IV: intravenous, MOG: myelin oligodendrocyte glycoprotein, MRI: magnetic resonance imaging
Patients with suspected ocular syphilis require a combination of systemic and ophthalmic investigations. Ophthalmic testing may include fundus photography (and autofluorescence), spectral domain-OCT (SD-OCT), and fluorescein angiography (FA).[35] SD-OCT imaging may reveal outer retinal changes including disruption of the ellipsoid zone and multinodular hyperreflectivity of retinal pigment epithelium.[35] In addition, SD-OCT can reveal complications associated with syphilis infection, including optic disc swelling, intraretinal fluid, and epiretinal membrane formation.[35] FA may identify retinal vasculitis, presenting as vascular occlusion, staining, leakage, or nonperfusion.[35] Systemically, serologic assays used for the diagnosis of syphilis patients include the rapid plasma reagin (RPR) test and the venereal disease research laboratory (VDRL) test, which may be positive 1–4 weeks after the appearance of syphilitic chancre.[35] Treponemal tests detect antibodies against T. pallidum that appear earlier in the disease, which can provide an advantage compared to RPR/VDRL. However, treponemal tests are not useful for monitoring treatment, as the antibodies remain positive even with successful antibiotic therapy.[32,35] Most clinicians screen patients with nontreponemal tests and may request confirmatory treponemal testing for suspected false-positive RPR/VDRL results.[32] Ocular involvement in syphilis should be considered neurosyphilis, which may require admission and treatment with intravenous (IV) or intramuscular penicillin G, depending on patient compliance.[32]
Lyme disease
Lyme is a multisystem disease caused by an infection with the Borrelia species, primarily Borrelia burgdorferi in North America, typically spread by Ixodes ticks.[32] Following transmission, patients first develop “erythema migrans,” a red maculopapular rash. This is commonly followed by a disseminated disease with multisystem manifestations, including myalgia, neurologic, and cardiac symptoms.[32] Approximately 15% of patients with Lyme disease experience neurologic manifestations, such as meningitis, radiculoneuritis, cranial nerve dysfunction, and encephalitis.[36] Although rare, Lyme neuroborreliosis is associated with various optic neuropathies, including neuroretinitis, ON, and ischemic optic neuropathy.[36]
Patients suspected of Lyme disease can be screened with ELISA, which can be followed by confirmatory western blot to detect antibodies against Borrelia species.[36] Patients with early-stage disease can be effectively treated with doxycycline, while IV antibiotics like ceftriaxone are reserved for those with late neuroborreliosis.[36] Patients with optic nerve involvement presumed to be secondary to Borrelia infection have been treated with both oral doxycycline and IV antibiotics, with no strong evidence favoring one strategy over the other.
Varicella zoster virus
Primary varicella zoster virus (VZV) infection typically occurs in childhood, leading to varicella (chickenpox). After the primary infection, the virus remains dormant in the dorsal root ganglia and may reactivate in adulthood, especially in elderly and immunocompromised individuals, manifesting as herpes zoster (“shingles”).[31,32] Optic nerve involvement may manifest with or without keratitis, uveitis, elevated intraocular pressure, or acute retinal necrosis. Patients with VZV-related ON may complain of ocular pain, vision loss, and visual field deficits. Orbital MRI usually shows T2 hyperintensity with postcontrast enhancement of the optic nerve.[31] In addition to ophthalmic imaging, systemic investigations may include serologic testing and CSF analysis to confirm the diagnosis and rule out other etiologies.[31] Patients with isolated optic neuropathy or cranial nerve palsy can be treated with oral antivirals, while those with clinical evidence of retinal or neurologic involvement will require IV acyclovir therapy.[31]
Cytomegalovirus
Cytomegalovirus (CMV) is a member of the herpes virus family and is quite prevalent, affecting over 50% of adults and often remaining dormant. It is considered an opportunistic infection, usually displaying ophthalmic manifestations in immunocompromised individuals, especially in patients with HIV and low CD4 counts.[31,37] Ocular CMV infections generally present with CMV necrotizing retinitis, hemorrhagic retinopathy, and optic neuropathy.[31] CMV optic neuropathy typically presents with papillitis accompanied by retinitis (most common) or isolated papillitis (less common). Treatment with IV foscarnet or ganciclovir has been shown to improve visual acuity,[37] with valganciclovir rising as a promising oral alternative. In addition, it is crucial to address the coexisting condition leading to severe immunosuppression, such as poorly controlled HIV with a low CD4 count. Finally, maintenance antiviral therapy may be required depending on patient’s immune system status.[31]
Human immunodeficiency virus
HIV infections can lead to various ocular manifestations, including optic neuropathy, presenting as papillitis, retrobulbar optic neuropathy, ischemic optic neuropathy, and chorioretinitis. Most commonly, optic neuropathy in HIV-positive patients is secondary to opportunistic infections or neoplasms, and it can be challenging to differentiate these etiologies from the rarer HIV optic neuropathy, which is thought to be autoimmune in nature.[38] Jindahra et al.[38] noted that HIV patients with opportunistic infections had low CD4 counts and high viral loads during the ON attack. As it is important not to miss an opportunistic infection, autoimmune-mediated HIV optic neuropathy is often considered a diagnosis of exclusion after ruling out infections[31,32] and is treated with steroids in addition to the gold standard highly active antiretroviral therapy.
Systemic Disorders Associated with ON
Several autoimmune conditions have been associated with acute ON, including granulomatosis with polyangiitis (GPA), systemic lupus erythematosus (SLE), SjS, sarcoidosis, and IgG4 disease, among others [Table 1].[8,22] Furthermore, antinuclear antibodies are found in approximately in 50% of NMOSD, 28% of MOGAD, and 15% of MS patients.[8] For this reason, patients with SjS or SLE who present with acute ON or other NMOSD clinical manifestations should undergo AQP-IgG testing, as these conditions can co-associate.[26]
In the absence of NMOSD, SLE ON is uncommon and typically presents with unilateral, acute to subacute severe vision loss associated with retrobulbar pain and poor visual prognosis in most cases.[22] These cases are thought to respond relatively well to high-dose steroid treatment (and potentially plasma exchange [PLEX]) if initiated in a timely manner.[8] GPA is another systemic autoimmune condition associated with ON.[22] While GPA ON often presents in patients with known diagnosis or classical symptoms of GPA, ON can be the presenting complaint in patients without other systemic manifestations.[39] These patients typically present with severe vision loss and retrobulbar pain on extraocular movement. Most are positive for proteinase 3 or myeloperoxidase anti-neutrophil cytoplasmic antibodies.[39] In addition to GPA and SLE, acute ON is also associated with SjS. Associated ON shows variability in presentation, with either unilateral or bilateral optic neve involvement, sometimes with associated retrobulbar pain. Patients with suspicious SjS symptoms, such as sicca, and ON should be investigated for anti-La/SSB and anti-Ro/SSA autoantibodies.[8]
Diagnostic “Toolbox” Considerations
The investigative approach for ON is influenced by demographic factors, severity of vision loss, examination findings, and concern for a vision- or life-threatening co-associated diagnosis.[8] To this end, the diagnostic “toolbox” has been fortified in recent years with MRI, visual evoked potentials (VEPs), OCT, and fluid markers.
MRI of the brain and orbits may help identify patients with MS or those who are at risk for the same.[8] Yet, it is important to carefully interpret MRI studies since there may be significant overlap in the radiologic features of ON associated with MS, MOGAD, and NMOSD.[11] To optimize MRI sensitivity, incorporating coronal short tau inversion or fat-suppressed T2 images, along with post-gadolinium fat-suppressed T1 images of the optic nerve with thin section slices (<2 mm up to the chiasm) has been recommended,[40] with newer imaging modalities on the horizon.
VEP testing captures evidence of demyelination and subsequent remyelination in patients with ON. Specifically, a delayed VEP 100 waveform suggests optic nerve demyelination.[41] Unfortunately, VEP P100 waveform latencies and decreased amplitudes do not always reliably differentiate between different ON subtypes.[7]
OCT-measured pRNFL thickness is used as a surrogate marker of afferent visual pathway axonal health, and the macular ganglion layer analysis provides an indirect measure of neuronal integrity.[42] For cases of acute ON, pRNFL values may be normal or elevated, depending on the extent of the optic disc swelling, while ganglion layer measures are typically normal.[42] Regardless of the cause of ON, pRNFL and ganglion layer thinning develop throughout follow-up, usually plateauing over 3–6 months.[8]
CSF constituents may help identify patients with infectious ON subtypes [Table 1]. The presence of oligoclonal bands is common in MS patients, but not specific for this condition. Other fluid biomarkers such as MOG-IgG, AQP4-IgG, GFAP-IgG, and CRMP5-IgG may be used to distinguish the ON subtypes related to specific CNS inflammatory disorders.
Acute Treatment
The treatment approach for ON should be tailored to the patient based on the subtype and etiology of ON. Accurate and timely diagnosis of the etiology of ON can be crucial for visual recovery and outcomes. One of the most important steps for a clinician is to rule out infectious etiology, since immunosuppression in patients with infectious ON may significantly worsen their infection and visual outcome. Treatments for immune-mediated ON subtypes have been influenced by ONTT, which has demonstrated that high-dose corticosteroids may hasten, but otherwise not change overall recovery for idiopathic and MS ON subtypes. Most clinicians use IV methylprednisolone (IVMP) 1 g/day for 5 days, followed by a slow taper.[43,44] Although tapering over 1–2 months is generally recommended to prevent relapses, the optimal taper remains unclear.[24] Interestingly, Morrow et al.[45] reported that the oral bioequivalent, oral prednisone 1250 mg/day, is noninferior to the 1 g/day IVMP, and thus may be offered as a practical alternative.
In contrast to MS ON, the natural history and visual outcomes associated with NMOSD- and MOGAD-associated ON may be relatively poor if not treated promptly with high-dose corticosteroids.[8] MOGAD ON patients often respond well to steroid treatment.[7] Unfortunately, NMOSD ON patients usually do not respond as well to high-dose corticosteroid treatment; however, early treatment with high-dose IVMP is thought to accelerate visual recovery.[7,8,26] In addition to ON secondary to MOGAD and NMOSD, high-dose steroid is considered the first-line treatment for other noninfectious and non-MS ON subtypes, including ON related to chronic relapsing inflammatory optic neuritis (CRION), GFAP, and CRMP-5-IgG syndrome.[8] In cases where acute attacks do not respond to high-dose corticosteroids, PLEX or immunoadsorption (IA) is typically utilized with varying degrees of success.[7,9,26] For NMOSD ON refractory to high-dose corticosteroids, early initiation of PLEX or IA within the first 5–7 days of symptoms onset is crucial for improved visual outcomes.[7] Recently, a multicenter retrospective study conducted by Chen et al.[46] reviewed the clinical outcomes of 317 patients with acute ON secondary to MS (108), MOGAD (92), and NMOSD (109) who received PLEX therapy.[46] A delay in PLEX treatment was associated with worse visual outcomes. A prospective randomized clinical trial will further delineate the role of PLEX in the treatment of acute ON.
In addition to PLEX and IA, IV immunoglobulin G (IVIG) has been proposed as another treatment option for acute ON attacks, either alone or in combination with high-dose corticosteroids.[8] Indeed, a retrospective study by Li et al.[47] presented some evidence that combining IVIG at a dose of 0.4 g/kg/day over five consecutive days with high-dose IVMP may be more effective in treating acute NMOSD attacks than IVMP alone. Ongoing studies are further investigating the role of IVIG in the treatment of acute ON attacks.
Beyond acute therapies, maintenance therapy, though not the focus of this review, is an evolving area of interest, particularly for MS, MOGAD, and NMOSD patients. Maintenance therapy is generally undertaken to prevent relapses. For instance, approximately 50% of MOGAD patients experience a recurrent course and therefore require long-term immunosuppression. While all patients diagnosed with NMOSD require maintenance immunosuppression to prevent severe neurologic disability. The specific maintenance therapies, regimens, and their efficacy are beyond the scope of this review.
Conclusions
ON subtypes fall into a diverse and complex spectrum that continues to expand. Phenotypic features and causes of ON vary around the globe. It is important to know the population prevalence of related disorders across different geographic regions to render accurate diagnoses and tailor effective therapies. Moving forward, the diagnostic toolbox for ON will continue to expand. With early detection and treatment, the potential for visual recovery will improve, across the entire spectrum of ON subtypes.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Petzold A, Fraser CL, Abegg M, Alroughani R, Alshowaeir D, Alvarenga R, et al. Diagnosis and classification of optic neuritis. Lancet Neurol. 2022;21:1120–34. doi: 10.1016/S1474-4422(22)00200-9. [DOI] [PubMed] [Google Scholar]
- 2.Marrie RA, Ekuma O, Wijnands JMA, Kingwell E, Zhu F, Zhao Y, et al. Identifying optic neuritis and transverse myelitis using administrative data. Mult Scler Relat Disord. 2018;25:258–64. doi: 10.1016/j.msard.2018.08.013. [DOI] [PubMed] [Google Scholar]
- 3.Braithwaite T, Subramanian A, Petzold A, Galloway J, Adderley NJ, Mollan SP, et al. Trends in optic neuritis incidence and prevalence in the UK and association with systemic and neurologic disease. JAMA Neurol. 2020;77:1514. doi: 10.1001/jamaneurol.2020.3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Woung L-C, Lin C-H, Tsai C-Y, Tsai M-T, Jou J-R, Chou P. Optic neuritis among National Health Insurance Enrollees in Taiwan, 2000–2004. Neuroepidemiology. 2007;29:250–4. doi: 10.1159/000112858. [DOI] [PubMed] [Google Scholar]
- 5.Muro-Fuentes EA, Villarreal Navarro SE, Moss HE. Accuracy of International Classification of Diseases Codes for identifying acute optic neuritis. J Neuroophthalmol. 2023;43:317–22. doi: 10.1097/WNO.0000000000001805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stunkel L, Kung NH, Wilson B, McClelland CM, Van Stavern GP. Incidence and causes of overdiagnosis of optic neuritis. JAMA Ophthalmol. 2018;136:76–81. doi: 10.1001/jamaophthalmol.2017.5470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bennett JL, Costello F, Chen JJ, Petzold A, Biousse V, Newman NJ, et al. Optic neuritis and autoimmune optic neuropathies: Advances in diagnosis and treatment. Lancet Neurol. 2023;22:89–100. doi: 10.1016/S1474-4422(22)00187-9. [DOI] [PubMed] [Google Scholar]
- 8.Al-Ani A, Costello F. Elsevier; 2024. Optic neuritis---The dawn of a new era. In: Reference Module in Neuroscience and Biobehavioral Psychology; pp. 1–18. [Google Scholar]
- 9.Al-Ani A, Chen JJ, Costello F. Myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD): Current understanding and challenges. J Neurol. 2023;270:4132–50. doi: 10.1007/s00415-023-11737-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Petzold A, Wattjes MP, Costello F, Flores-Rivera J, Fraser CL, Fujihara K, et al. The investigation of acute optic neuritis: A review and proposed protocol. Nat Rev Neurol. 2014;10:447–58. doi: 10.1038/nrneurol.2014.108. [DOI] [PubMed] [Google Scholar]
- 11.De Lott LB, Bennett JL, Costello F. The changing landscape of optic neuritis: A narrative review. J Neurol. 2022;269:111–24. doi: 10.1007/s00415-020-10352-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walton C, King R, Rechtman L, Kaye W, Leray E, Marrie RA, et al. Rising prevalence of multiple sclerosis worldwide: Insights from the Atlas of MS, third edition. Mult Scler J. 2020;26:1816–21. doi: 10.1177/1352458520970841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Winges KM, Gordon LK. Neuro-ophthalmic complications of immune checkpoint inhibitor therapy: Current status and future directions. Front Ophthalmol. 2022;2:1–11. doi: 10.3389/fopht.2022.1044904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Floreani A, Okazaki K, Uchida K, Gershwin ME. IgG4-related disease: Changing epidemiology and new thoughts on a multisystem disease. J Transl Autoimmun. 2021;4:100074. doi: 10.1016/j.jtauto.2020.100074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsuboi M, Evans J, Davies EP, Rowley J, Korenromp EL, Clayton T, et al. Prevalence of syphilis among men who have sex with men: A global systematic review and meta-analysis from 2000–20. Lancet Glob Health. 2021;9:e1110–8. doi: 10.1016/S2214-109X(21)00221-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.World Health Organization Syphilis. (Fact Sheets) 2024 Available from: https://www.who.int/news-room/fact-sheets/detail/syphilis . [Last accessed on 2024 Aug 28] [Google Scholar]
- 17.World Health Organization Tuberculosis. (Fact sheets) 2023 Available from: https://www.who.int/news-room/fact-sheets/detail/tuberculosis . [Last accessed on 2024 Aug 28] [Google Scholar]
- 18.Beck RW, Cleary PA, Anderson MM, Keltner JL, Shults WT, Kaufman DI, et al. A randomized, controlled trial of corticosteroids in the treatment of acute optic neuritis. N Engl J Med. 1992;326:581–8. doi: 10.1056/NEJM199202273260901. [DOI] [PubMed] [Google Scholar]
- 19.Wade BJ. Spatial analysis of global prevalence of multiple sclerosis suggests need for an updated prevalence scale. Multiple sclerosis international. 2014;2014:124578. doi: 10.1155/2014/124578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thompson AJ, Banwell BL, Barkhof F, Carroll WM, Coetzee T, Comi G, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17:162–73. doi: 10.1016/S1474-4422(17)30470-2. [DOI] [PubMed] [Google Scholar]
- 21.Toosy AT, Mason DF, Miller DH. Optic neuritis. Lancet Neurol. 2014;13:83–99. doi: 10.1016/S1474-4422(13)70259-X. [DOI] [PubMed] [Google Scholar]
- 22.Costello F. Inflammatory optic neuropathies. Contin Lifelong Learn Neurol. 2014;20:816–37. doi: 10.1212/01.CON.0000453316.60013.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Banwell B, Bennett JL, Marignier R, Kim HJ, Brilot F, Flanagan EP, et al. Diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease: International MOGAD Panel proposed criteria. Lancet Neurol. 2023;22:268–82. doi: 10.1016/S1474-4422(22)00431-8. [DOI] [PubMed] [Google Scholar]
- 24.Burton JM, Youn S, Al-Ani A, Costello F. Patterns and utility of myelin oligodendrocyte glycoprotein (MOG) antibody testing in cerebrospinal fluid. J Neurol. 2024;271:2662–71. doi: 10.1007/s00415-024-12213-7. [DOI] [PubMed] [Google Scholar]
- 25.Cobo‐Calvo A, Ruiz A, Rollot F, Arrambide G, Deschamps R, Maillart E, et al. Clinical features and risk of relapse in children and adults with myelin oligodendrocyte glycoprotein antibody–associated disease. Ann Neurol. 2021;89:30–41. doi: 10.1002/ana.25909. [DOI] [PubMed] [Google Scholar]
- 26.Costello F. Neuromyelitis optica spectrum disorders. Contin Lifelong Learn Neurol. 2022;28:1131–70. doi: 10.1212/CON.0000000000001168. [DOI] [PubMed] [Google Scholar]
- 27.Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K, et al. A serum autoantibody marker of neuromyelitis optica: Distinction from multiple sclerosis. Lancet (London, England) 2004;364:2106–12. doi: 10.1016/S0140-6736(04)17551-X. [DOI] [PubMed] [Google Scholar]
- 28.Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005;202:473–7. doi: 10.1084/jem.20050304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prain K, Woodhall M, Vincent A, Ramanathan S, Barnett MH, Bundell CS, et al. AQP4 antibody assay sensitivity comparison in the era of the 2015 diagnostic criteria for NMOSD. Front Neurol. 2019;10:1028. doi: 10.3389/fneur.2019.01028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fang B, McKeon A, Hinson SR, Kryzer TJ, Pittock SJ, Aksamit AJ, et al. Autoimmune glial fibrillary acidic protein astrocytopathy: A Novel meningoencephalomyelitis. JAMA Neurol. 2016;73:1297–307. doi: 10.1001/jamaneurol.2016.2549. [DOI] [PubMed] [Google Scholar]
- 31.Ambika S, Lakshmi P. Infectious optic neuropathy (ION), how to recognise it and manage it. Eye (Lond) 2024;38:2302–11. doi: 10.1038/s41433-024-03152-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eggenberger ER. Infectious optic neuropathies. Contin Lifelong Learn Neurol. 2019;25:1422–37. doi: 10.1212/CON.0000000000000777. [DOI] [PubMed] [Google Scholar]
- 33.Gupta V, Gupta A, Rao NA. Intraocular tuberculosis—An update. Surv Ophthalmol. 2007;52:561–87. doi: 10.1016/j.survophthal.2007.08.015. [DOI] [PubMed] [Google Scholar]
- 34.Davis EJ, Rathinam SR, Okada AA, Tow SL, Petrushkin H, Graham EM, et al. Clinical spectrum of tuberculous optic neuropathy. J Ophthalmic Inflamm Infect. 2012;2:183–9. doi: 10.1007/s12348-012-0079-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Furtado JM, Simões M, Vasconcelos-Santos D, Oliver GF, Tyagi M, Nascimento H, et al. Ocular syphilis. Surv Ophthalmol. 2022;67:440–62. doi: 10.1016/j.survophthal.2021.06.003. [DOI] [PubMed] [Google Scholar]
- 36.Träisk F, Lindquist L. Optic nerve involvement in Lyme disease. Curr Opin Ophthalmol. 2012;23:485–90. doi: 10.1097/ICU.0b013e328358b1eb. [DOI] [PubMed] [Google Scholar]
- 37.Patel SS, Rutzen AR, Marx JL, Thach AB, Chong LP, Rao NA. Cytomegalovirus papillitis in patients with acquired immune deficiency syndrome. Ophthalmology. 1996;103:1476–82. doi: 10.1016/s0161-6420(96)30480-6. [DOI] [PubMed] [Google Scholar]
- 38.Jindahra P, Phuphuakrat A, Tangjaisanong T, Siriyotha S, Padungkiatsagul T, Vanikieti K, et al. Clinical characteristics of HIV-associated optic neuritis. Int Med Case Rep J. 2020;13:609–16. doi: 10.2147/IMCRJ.S267867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clément M, Néel A, Toulgoat F, Weber M, Godmer P, Hutin P, et al. Inflammatory optic neuropathy in granulomatosis with polyangiitis can mimick isolated idiopathic optic neuritis. Eur J Ophthalmol. 2021;31:245–51. doi: 10.1177/1120672119889008. [DOI] [PubMed] [Google Scholar]
- 40.Traboulsee A, Simon JH, Stone L, Fisher E, Jones DE, Malhotra A, et al. Revised recommendations of the consortium of ms centers task force for a standardized MRI protocol and clinical guidelines for the diagnosis and follow-up of multiple sclerosis. Am J Neuroradiol. 2016;37:394–401. doi: 10.3174/ajnr.A4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen JJ, Sotirchos ES, Henderson AD, Vasileiou ES, Flanagan EP, Bhatti MT, et al. OCT retinal nerve fiber layer thickness differentiates acute optic neuritis from MOG antibody-associated disease and Multiple Sclerosis. Mult Scler Relat Disord. 2022;58:103525. doi: 10.1016/j.msard.2022.103525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Costello F, Chen JJ. The role of optical coherence tomography in the diagnosis of afferent visual pathway problems: A neuroophthalmic perspective. Handb Clin Neurol. 2021;178:97–113. doi: 10.1016/B978-0-12-821377-3.00007-6. [DOI] [PubMed] [Google Scholar]
- 43.Healy S, Elhadd KT, Gibbons E, Whittam D, Griffiths M, Jacob A, et al. Treatment of myelin oligodendrocyte glycoprotein immunoglobulin G–associated disease. Clin Exp Neuroimmunol. 2021;12:22–41. [Google Scholar]
- 44.Whittam DH, Karthikeayan V, Gibbons E, Kneen R, Chandratre S, Ciccarelli O, et al. Treatment of MOG antibody associated disorders: Results of an international survey. J Neurol. 2020;267:3565–77. doi: 10.1007/s00415-020-10026-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morrow SA, Fraser JA, Day C, Bowman D, Rosehart H, Kremenchutzky M, et al. Effect of treating acute optic neuritis with bioequivalent oral vs intravenous corticosteroids. JAMA Neurol. 2018;75:690–6. doi: 10.1001/jamaneurol.2018.0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen JJ, Flanagan EP, Pittock SJ, Stern NC, Tisavipat N, Bhatti MT, et al. Visual outcomes following plasma exchange for optic neuritis: An International multicenter retrospective analysis of 395 optic neuritis attacks. Am J Ophthalmol. 2023;252:213–24. doi: 10.1016/j.ajo.2023.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li X, Tian D-C, Fan M, Xiu Y, Wang X, Li T, et al. Intravenous immunoglobulin for acute attacks in neuromyelitis optica spectrum disorders (NMOSD) Mult Scler Relat Disord. 2020;44:102325. doi: 10.1016/j.msard.2020.102325. [DOI] [PubMed] [Google Scholar]