

Dear Editor,
Epileptic encephalopathy is considered when refractory seizures are associated with developmental delay or regression and severe electroencephalography (EEG) abnormalities. The major causes of epileptic encephalopathy are structural, metabolic, and genetic. Among these, metabolic causes can be cured with treatment. Hyperprolinemia is one such metabolic disorder that results from deficiency of enzymes involved in the metabolism of proline. Neurologic manifestations, including seizures and neuropsychiatric illness, are common in patients with hyperprolinemia.
A 7-month-old boy, born to third-degree consanguineous parents, with no perinatal issues and with normal development had seizures in the form of epileptic spasms, lasting continuously for 2 h in clusters. He was managed with injection adrenocorticotropic hormone, levetiracetam (20 mg/kg/day) and valproate (30 mg/kg/day) at the referral center. The epileptic spasms subsided in 4 days. However, 3 days later, he developed multifocal clonic movements of all four limbs and was encephalopathic with loss of neck control. This prompted referral to our facility for further management.
On admission, the child was hemodynamically stable; however, he had altered sensorium. His Glasgow Coma Scale score was E2V2M4. There were no neurocutaneous markers or facial dysmorphism. The child’s weight was 7 kg and head circumference was 41 cm (<3rd percentile). The infant had a normal eye examination. Severe head lag with hypotonia in all four limbs was present. He was moving all limbs against gravity, and deep tendon reflexes were exaggerated with bilateral flexor plantars. There were no focal neurologic deficits, and no signs of raised intracranial pressure were observed.
Pyridoxine and biotin were added, optimized doses of levetiracetam (40mg/kg/day), topiramate (5mg/kg/day) and stopped valproic acid. The sensorium gradually improved, and he remained seizure free. During the hospital stay, the child developed dyskinesias involving the perioral area and limbs (proximal and distal), which were managed symptomatically, and there was no recurrence.
EEG demonstrated diffuse slowing with no interictal epileptiform discharges and no sleep markers [Figure 1]. Magnetic resonance imaging (MRI) of the brain was normal. Tandem mass spectrometry showed elevated proline levels- 2478 μmol against normal range of 31–349 µmol, indicating hyperprolinemia type 1 or 2. This was confirmed with repeated testing. Whole exome sequencing confirmed the diagnosis as hyperprolinemia type 2 having homozygous mutation with pathogenic variant of ALDH4A1 gene in intron 3 c.249+1G>A [Figure 2]. The child was continued with pyridoxine along with antioxidants vitamin E and vitamin C.
Figure 1.
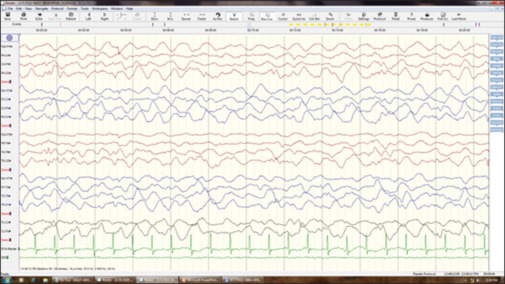
EEG showing diffuse slowing and no sleep markers. EEG: electroencephalography
Figure 2.
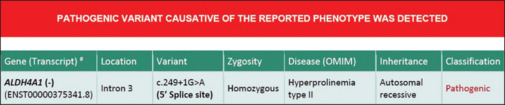
Homozygous mutation in ALDH4A1 gene
At 2 years of age follow-up, the child was seizure free while receiving treatment with pyridoxine and antioxidants. He achieved age-appropriate developmental milestones. Follow-up EEG was normal.
This case highlights the importance of considering metabolic causes of epilepsy, particularly in children with consanguineous parents. Hyperprolinemia is a rare metabolic disorder that can present with epileptic encephalopathy and caused by mutations in genes involved in proline metabolism, for example, ALDH4A1.[1]
Hyperprolinemia type 2 is caused by a deficiency of ALDH4A1-encoded Δ-1-pyrroline-5-carboxylate dehydrogenase, which is the enzyme responsible for the second step in the degradation of proline. Conversion of l-glutamate 5-semialdehyde, which is in equilibrium with its cyclic counterpart Δ-1-pyrroline-5-carboxylate (P5C), to glutamic acid is one key component for neurologic manifestation [Figure 3]. It results in the accumulation of P5C, which inhibits pyridoxine. This secondary pyridoxine deficiency results in refractory epilepsy with altered sensorium. As described in the previous literature,[2,3] the child responded well to pyridoxine and antioxidants vitamin E and vitamin C, as the role of oxidative stress is emerging.[4]
Figure 3.
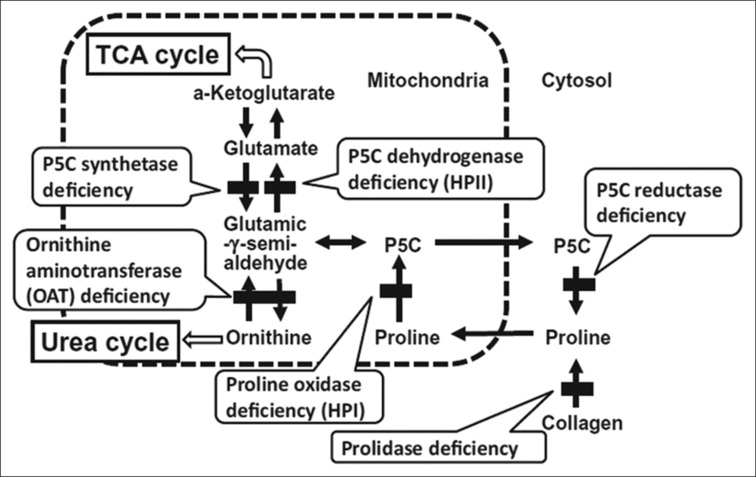
Proline Metabolism Pathway. HPI, Hyperprolinemia type I; HPII, Hyperprolinemia type II; P5C, Δ-1- pyrroline-5-carboxylate; TCA, Tricarboxylic acid. Reproduced with permission from Pediatrics International (2014) 56, 492–496
Hyperprolinemia type 2 can present from neonate to adult age group, with the oldest case report of a 64-year-old patient with symptom onset from 54 years of age.[5] Its incidence was estimated to be 1:70,000 in newborn screening programs.[6]
One case from North India was reported with similar clinical characteristics including acute-onset seizures (clonic seizures, spasms) followed by encephalopathy, which responded to pyridoxine.[3] EEG showed diffuse delta slowing with burst suppression pattern, while in our patient, EEG showed only diffuse delta slowing with no sleep markers. Early childhood cases present with identical features,[2,7] whereas late childhood cases and adults have intellectual impairment, neuropsychiatric issues, and epilepsy.[5]
Hyperprolinemia type 2 is one of the epilepsy syndromes that respond to pyridoxine.[8,9] The other epilepsy syndromes with good response to pyridoxine are ALDH7A1 mutation, pyridoxamine-5’-phosphate oxidase deficiency, nonketotic hyperglycinemia, and KCNQ2 mutation.[8]
In conclusion, this case report shows the importance of considering metabolic causes of epilepsy in children with consanguineous parents, particularly in the absence of neurocutaneous markers and dysmorphic facial features. The presence of diffuse high-voltage slowing with relative paucity of epileptiform discharges and sudden loss of acquired milestones with normal MRI should raise the possibility of hyperprolinemia. This should be considered in the differential diagnosis of epileptic encephalopathy with acute presentation, and tandem mass spectrometry and whole exome sequencing are essential for confirming the diagnosis. Lifelong pyridoxine supplementation is an effective treatment for hyperprolinemia type 2, and early diagnosis and treatment could lead to better outcomes.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient’s guardian has given consent for the child’s images and other clinical information to be reported in the journal. The patient’s guardian understands that the child’s name and initial will not be published, and due efforts will be made to conceal the identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Valle D, Goodman SI, Harris SC, Phang JM. Genetic evidence for a common enzyme catalyzing the second step in the degradation of proline and hydroxyproline. J Clin Invest. 1979;64:1365–70. doi: 10.1172/JCI109593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Onenli-Mungan N, Yüksel B, Elkay M, Topaloğlu AK, Baykal T, Ozer G. Type II hyperprolinemia: A case report. Turk J Pediatr. 2004;46:167–9. [PubMed] [Google Scholar]
- 3.Kaur R, Paria P, Saini AG, Suthar R, Bhatia V, Attri SV. Metabolic epilepsy in hyperprolinemia type II due to a novel nonsense ALDH4A1 gene variant. Metab Brain Dis. 2021;36:1413–7. doi: 10.1007/s11011-021-00757-w. [DOI] [PubMed] [Google Scholar]
- 4.Ferreira AGK, Stefanello FM, Cunha AA, Da Cunha MJ, Pereira TCB, Bonan CD, et al. Role of antioxidants on Na+, K+-ATPase activity and gene expression in cerebral cortex of hyperprolinemic rats. Metab Brain Dis. 2011;26:141–7. doi: 10.1007/s11011-011-9243-0. [DOI] [PubMed] [Google Scholar]
- 5.Motte J, Fisse AL, Grüter T, Schneider R, Breuer T, Lücke T, et al. Novel variants in a patient with late-onset hyperprolinemia type II: Diagnostic key for status epilepticus and lactic acidosis. BMC Neurol. 2019;19:345. doi: 10.1186/s12883-019-1583-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van De Ven S, Gardeitchik T, Kouwenberg D, Kluijtmans L, Wevers R, Morava E. Long-term clinical outcome, therapy and mild mitochondrial dysfunction in hyperprolinemia. J Inherit Metab Dis. 2014;37:383–90. doi: 10.1007/s10545-013-9660-9. [DOI] [PubMed] [Google Scholar]
- 7.Flynn MP, Martin MC, Moore PT, Stafford JA, Fleming GA, Phang JM. Type II hyperprolinaemia in a pedigree of Irish travellers (nomads) Arch Dis Child. 1989;64:1699–707. doi: 10.1136/adc.64.12.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clayton PT. B6-responsive disorders: A model of vitamin dependency. J Inherit Metab Dis. 2006;29:317–26. doi: 10.1007/s10545-005-0243-2. [DOI] [PubMed] [Google Scholar]
- 9.Mastrangelo M, Gasparri V, Bernardi K, Foglietta S, Ramantani G, Pisani F. Epilepsy phenotypes of vitamin B6-dependent diseases: An updated systematic review. Children (Basel) 2023;10:553. doi: 10.3390/children10030553. [DOI] [PMC free article] [PubMed] [Google Scholar]